Recombinate 500 Ui Polvo Y Disolvente Para Solucion Inyectable
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Recombinate 250 UI polvo y disolvente para solución inyectable Recombinate 500 UI polvo y disolvente para solución inyectable Recombinate 1000 UI polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Octocog alfa 25 UI por ml de solución reconstituida.
Después de la reconstitución:
Un vial de 10 ml contiene 250 UI de octocog alfa.
Recombinate 250 UI contiene nominalmente 250 UI de octocog alfa, recombinante, factor VIII de coagulación por vial.
El producto contiene aproximadamente 25 UI/ml de octocog alfa, factor VIII de coagulación recombinante, cuando se reconstituye con 10 ml de agua para preparaciones inyectables.
Octocog alfa 50 UI por ml de solución reconstituida.
Después de la reconstitución:
Un vial de 10 ml contiene 500 UI de octocog alfa.
Recombinate 500 UI contiene nominalmente 500 UI de octocog alfa, recombinante, factor VIII de coagulación por vial.
El producto contiene aproximadamente 50 UI/ml de octocog alfa, factor VIII de coagulación recombinante, cuando se reconstituye con 10 ml de agua para preparaciones inyectables.
Octocog alfa 100 UI por ml de solución reconstituida.
Después de la reconstitución:
Un vial de 10 ml contiene 1000 UI de octocog alfa.
Recombinate 1000 UI contiene nominalmente 1000 UI de octocog alfa, recombinante, factor VIII de coagulación por vial.
El producto contiene aproximadamente 100 UI/ml de octocog alfa, factor VIII de coagulación recombinante, cuando se reconstituye con 10 ml de agua para preparaciones inyectables.
La potencia se determina utilizando el ensayo cromatográfico de la Farmacopea Europea en comparación con el Estándar Mega de la FDA que ha sido calibrado al estándar de la OMS.La actividad específica de Recombinate es, aproximadamente, de 4000-8000 UI/mg de proteína.
Recombinate contiene factor VIII de coagulación recombinante (INN: octocog alfa). El octocog alfa (factor VIII de coagulación recombinante) es una proteína purificada formada por 2332 aminoácidos. Contiene una secuencia de aminoácidos comparable al factor VIII y modificaciones post-translacionales que son similares a las de la molécula derivada del plasma. El factor VIII de coagulación recombinante es una glicoproteína que se obtiene por ingeniería genética de células mamarias derivadas de células de ovario de hámster chino.
Excipientes: para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo friable de color blanco a blanquecino. El disolvente (agua estéril para preparaciones inyectables) es un líquido transparente e incoloro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII).
Este producto no contiene factor von Willebrand y por lo tanto no está indicado en la enfermedad de von Willebrand.
Recombinate está indicado para todos los grupos de edad, desde neonatos hasta adultos.
4.2 Posología y forma de administración 4.2.1. Posología
La dosificación y duración de la terapia de sustitución depende de la gravedad del trastorno en la función hemostática, de la localización y extensión de los episodios hemorrágicos y de la condición clínica del paciente. El tratamiento debe llevarse a cabo en colaboración con un médico con experiencia en desórdenes hemorrágicos y un laboratorio con capacidad para la determinación de concentraciones de FAH en plasma.
El número de unidades de factor VIII administradas se expresa en Unidades Internacionales (UI), que se corresponden con el estándar actual de la OMS para medicamentos de factor VIII. La actividad del factor VIII en plasma se expresa bien como porcentaje (referido al plasma humano normal) o bien en Unidades Internacionales (referidas al estándar internacional de factor VIII en plasma). Una Unidad Internacional (UI) de actividad de factor VIII es equivalente a la cantidad de factor VIII existente en un ml de plasma humano normal.
El aumento pico in vivo esperado en el nivel de Recombinate expresado como UI/dl de plasma o porcentaje (%) del normal puede ser calculado multiplicando la dosis administrada por kg de peso corporal (UI/kg) por dos.
El método de cálculo se ilustra en los siguientes ejemplos:
unidades administradas x 2%/UI/Kg
Aumento de factor VIII esp erado (%) = ■
peso corp oral (Kg)
Ejemplo para un adulto de 70 Kg:
o bien
Dosis requerida (UI)
peso corporal (Kg) x aumento deseado de factor VIII (%)
2%/UI/Kg
Ejemplo para un niñ o de 40 Kg:
En casos de cirugía mayor o hemorragias que amenazan la vida, es especialmente importante un control cuidadoso de la terapia de sustitución.
Aunque la dosis puede estimarse por los cálculos dados anteriormente, se recomienda que, cuando sea posible, se realicen ensayos de laboratorio apropiados incluyendo ensayos seriados de FAH en el plasma del paciente a los intervalos necesarios para asegurar que los niveles de FAH adecuados se han alcanzado y se mantienen. Si el FAH del plasma del paciente no alcanza los niveles esperados o si la hemorragia no se puede controlar tras una dosis adecuada, debe sospecharse de la presencia de algún inhibidor. La presencia de inhibidor puede demostrarse mediante procedimientos de laboratorio adecuados y cuantificarse como Unidades Internacionales de FAH neutralizados por cada ml de plasma (Unidades Bethesda) o por el volumen total de plasma estimado. Si el inhibidor está presente a niveles menores de 10 Unidades Bethesda por ml, la administración de FAH adicional puede neutralizar el inhibidor. Después, la administración de Unidades Internacionales de FAH adicionales debe conseguir la respuesta prevista.
En estas situaciones, es necesario el control de los niveles de FAH mediante ensayos de laboratorio. Con títulos de inhibidor mayores de 10 Unidades Bethesda por ml, puede ser imposible o no práctico el control de la hemostasis con FAH por las grandes dosis requeridas.
La pauta de dosificación siguiente indicada en la Tabla 1 puede ser usada para adultos y niños. La cantidad a administrar y la frecuencia de aplicación deben siempre estar orientadas a la efectividad clínica en cada caso particular.
Recombinate puede ser también administrado como profilaxis (a corto o largo plazo) de hemorragias, según criterio médico, para cada caso concreto.
TABLA 1. PAUTA DE DOSIFICACIÓN
|
HEMORRAGIA | ||
|
Grado de hemorragia |
Actividad de FAH postperfusión en sangre requerida» (como % del normal o UI/dL plasma) |
FRECUENCIA DE LA PERFUSIÓN |
|
Hemartrosis incipiente o hemorragia oral o muscular |
20-40 |
Comenzar la perfusión cada 12-24 horas durante uno a tres días hasta que el episodio hemorrágico según indique el dolor se resuelva o se logre la curación |
|
Hemartrosis más extensa, hemorragia muscular o hematoma |
30-60 |
Repetir la perfusión cada 12-24 horas, normalmente durante tres días o más, hasta que cese el dolor y la incapacidad se resuelva |
|
Hemorragias que amenazan la vida, como hemorragia intracraneal, hemorragia en garganta, hemorragia abdominal grave |
60-100 |
Repetir la perfusión cada 8-24 horas hasta superar el peligro |
|
CIRUGÍA | ||
|
Tipo de operación | ||
|
Cirugía menor, incluyendo extracción dental |
30-60 |
Una única perfusión junto con terapia antifibrinolítica oral en una hora es suficiente en aproximadamente el 70% de los casos. Cada 24 horas, al menos 1 día, hasta la curación. |
|
Cirugía mayor |
80-100 (pre- y post-operación) |
Repetir la perfusión cada 8-24 horas dependiendo de la curación |
Esto representa la actividad pico de FAH para pacientes con la semi-vida promedio de factor VIII esperada. Si se considera necesario, la actividad pico puede ser medida dentro de la media hora siguiente a la administración. En pacientes con semi-vida de factor VIII relativamente corta, puede ser necesario aumentar la dosificación y/o la frecuencia de administración.
Cada vial de Recombinate, está etiquetado con el factor antihemofílico (Recombinante), actividad de Recombinate expresada en UI/vial. Esta asignación de potencia se hace con referencia al Estándar Internacional de la Organización Mundial de la Salud para Concentrado de factor VIII:C. Se ha demostrado experimentalmente que para obtener niveles de actividad exactos, el ensayo de potencia debe ser realizado con tubos de ensayo y pipetas de plástico y utilizar substrato con niveles normales de factor von Willebrand.
Para la profilaxis de larga duración frente a hemorragias en pacientes con hemofilia A grave, las dosis normales son de 20 a 40 UI de factor VIII por kg de peso corporal a intervalos de 2 a 3 días.
En los pacientes se debe controlar el desarrollo de inhibidores del factor VIII. Si no se logran los niveles plasmáticos de factor VIII esperados o si no se controla la hemorragia con una dosis adecuada, se debe realizar un análisis para determinar la presencia de inhibidores del factor VIII. En pacientes con niveles altos de inhibidor, la terapia con factor VIII puede no ser efectiva y deberían considerarse otras opciones terapéuticas. El manejo de estos pacientes deberá estar dirigido por médicos con experiencia en el tratamiento de la hemofilia.
Ver también sección 4.4.
Población pediátrica
Recombinate es adecuado para utilizarse en niños de todas las edades, incluyendo el recién nacido (se han realizado estudios de seguridad y eficacia tanto en niños tratados previamente como en niños no tratados; ver sección 5.1). Durante el tratamiento a demanda, la dosis en pacientes pediátricos no difiere de la de adultos. Para profilaxis a largo plazo de hemorragias en pacientes con hemofilia A grave, en algunos casos
puede ser necesario acortar los intervalos entre administraciones o utilizar dosis más elevadas que las normales de 20 a 40 UI de factor VIII por kg de peso a intervalos de 2 a 3 días.
4.2.2. Forma de administración
El preparado es para administración intravenosa después de su reconstitución con el disolvente que se acompaña (ver sección 6.6). El material reconstituido no debe ser refrigerado. Se recomienda administrar Recombinate a temperatura ambiente, no más tarde de las 3 horas siguientes a la reconstitución. La velocidad de administración debe ser la que asegure la comodidad del paciente, hasta un máximo de 10 ml/min. Antes y durante la administración de Recombinate, debe controlarse el pulso. Si hubiera un aumento significativo, al reducir el ritmo de administración o interrumpirla momentáneamente, los síntomas normalmente desaparecen pronto. (Ver secciones 4.4 y 4.8).
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes. Reacciones alérgicas conocidas a las proteínas bovina, de ratón o hámster.
4.4 Advertencias y precauciones especiales de empleo
Se han notificado reacciones alérgicas graves a Recombinate. Los pacientes con hipersensibilidad conocida a las proteínas de ratón, hámster o bovinas deben ser tratados con precaución. Se debe informar a los pacientes de los síntomas iniciales de las reacciones de hipersensibilidad incluyendo urticaria, urticaria generalizada, opresión del pecho, sibilancias, hipotensión y anafilaxis. Si se producen reacciones alérgicas o anafilácticas, debe interrumpirse la inyección/perfusión inmediatamente. Se debe disponer de medios para un adecuado tratamiento de choque.
Si los niveles de FAH en plasma no alcanzan los niveles esperados o si no se controla la hemorragia después de una dosis adecuada, deben realizarse ensayos de laboratorio apropiados para detectar la presencia de inhibidor.
La formación de anticuerpos neutralizantes (inhibidores) del factor VIII es una complicación conocida en el control de individuos con hemofilia A. Estos inhibidores son habitualmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en Unidades Bethesda (UB) por ml de plasma empleando el ensayo modificado Bethesda. El riesgo de desarrollar inhibidores se relaciona con el grado de exposición al factor VIII, siendo el riesgo mayor los primeros 20 días de exposición, y a otros factores ambientales y genéticos. De manera poco frecuente se desarrollan inhibidores después de los 100 primeros días de exposición.
Se han observado casos recurrentes de inhibidores (con título bajo), después de cambiar de un factor VIII recombinante a otro en los pacientes previamente tratados con más de 100 días de exposición y que tienen antecedentes de desarrollo de inhibidores.
En los pacientes tratados con un factor VIII de coagulación recombinante se debe monitorizar cuidadosamente el desarrollo de inhibidores por medio de observaciones clínicas apropiadas y ensayos de laboratorio. Ver también la sección 4.8.
En beneficio de los pacientes se recomienda que, siempre que sea posible, cada vez que se les administre Recombinate se deje constancia del nombre del medicamento y número de lote administrado.
Este medicamento contiene 1,5 mmol de sodio por dosis, lo que debe ser tenido en cuenta en pacientes con dietas pobres en sodio.
Población pediátrica
Las advertencias y precauciones especiales de empleo en pacientes pediátricos no difieren de las de adultos.
4.5 Interacción con otros medicamentos y otras formas de interacción No se han realizado estudios de interacciones con Recombinate.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción en animales con factor VIII. Dados los raros casos de hemofilia A en mujeres, no se dispone de experiencia del uso del factor VIII durante el embarazo y la lactancia. Por tanto, el factor VIII sólo debe utilizarse durante el embarazo y la lactancia si está claramente indicado.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han observado efectos sobre la capacidad para conducir y utilizar máquinas.
4.8 Reacciones adversas
Resumen tabulado de reacciones adversas
La tabla siguiente indica la frecuencia de pacientes con reacciones adversas aparecidas en los ensayos clínicos. Dentro de cada intervalo de frecuencia, las reacciones adversas se enumeran en orden decreciente de gravedad.
La frecuencia se ha evaluado utilizando el siguiente criterio: muy frecuentes (>1/10), frecuentes (>1/100,<1/10), poco frecuentes (>1/1000,<1/100), raras (>1/10000, <1/1000), muy raras (< 1/10000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
|
Base de datos de clasificación de órganos del sistema MedDRA |
Convenio MedDRA sobre frecuencia |
Término preferido del sitema MeDRa |
|
Infecciones e infestaciones |
Poco frecuentes |
Infección de oídos |
|
Trastornos de la sangre y del sistema linfático |
Frecuentes |
Inhibidores del factor VIII1 |
|
Trastornos del sistema inmunológico |
Frecuencia no conocida |
Reacción anafiláctica Hipersensibilidad2 |
|
Trastornos del sistema nervioso |
Poco frecuentes |
Mareo Temblores |
|
Frecuencia no conocida |
Pérdida de consciencia Síncope Dolor de cabeza Parestesia | |
|
Trastornos cardíacos |
Frecuencia no conocida |
Cianosis Taquicardia |
|
Trastornos vasculares |
Poco frecuentes |
Epistaxis Enrojecimiento Hematoma Hipotensión Palidez Frialdad en las extremidades |
|
Trastornos respiratorios, torácicos y mediastínicos |
Poco frecuentes |
Dolor faringolaringeo |
|
Frecuencia no conocida |
Dificultad para respirar Tos Sibilancias | |
|
Trastornos gastrointestinales |
Poco frecuentes |
Náuseas |
|
Frecuencia no conocida |
Vómitos Dolor abdominal | |
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuentes |
Hiperhidrosis Prurito Exantema Exantema maculopapular |
|
Frecuencia no conocida |
Angioedema Urticaria Exfoliación de la piel Eritema | |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Poco frecuentes |
Dolor en las extremidades |
|
Trastornos generales y alteraciones en el lugar de administración |
Frecuentes |
Escalofríos |
|
Poco frecuentes |
Fatiga Fiebre | |
|
Frecuencia no conocida |
Malestar general Reacciones en el lugar de la inyección Dolor en el pecho Malestar en el pecho | |
|
Exploraciones complementarias |
Poco frecuentes |
Test de estimulación acústica anormal |
1 En el ensayo clínico PTP (PTP= Pacientes Tratados Previamente), ninguno de los 71 pacientes desarrollaron anticuerpos de factor VIII de novo, pero 22 de los 72 evaluados en el protocolo PUPs (PUP= Pacientes no tratados Previamente) con Recombinate desarrollaron anticuerpos de factor VIII y la frecuencia se basó en los datos PUP. De los 22, 10 fueron de alto título (> 5 Unidades Bethesda) y 12 fueron de bajo título (<5 Unidades Bethesda).
2 Los signos iniciales de las reacciones de hipersensibilidad son urticaria, disnea, tos, opresión del pecho, sibilancias, anafilaxis, exantema, hipotensión, prurito, escalofríos, enrojecimiento, fiebre, cianosis, taquicardia, vómitos, síncope, dolor de cabeza. Se aconseja precaución en pacientes con reacciones alérgicas conocidas a los componentes del preparado (Ver secciones 4.3 y 4.4).
Descripción de algunas reacciones adversas
La formación de anticuerpos neutralizantes, inhibidores, de factor VIII es una complicación conocida en el control de pacientes con hemofilia A. Estos inhibidores son, invariablemente, inmunoglobulinas IgG, dirigidas contra la actividad procoagulante del factor VIII, que se expresan como Unidades Bethesda (U.B.) por ml de plasma.
El riesgo de desarrollar inhibidores se correlaciona con la exposición al factor VIII antihemofílico, siendo más alto dentro de los 20 primeros días de exposición. La incidencia de anticuerpos inhibitorios comunicada en pacientes con hemofilia A grave, que tienen mayor riesgo de desarrollar el inhibidor (por ejemplo, los pacientes no tratados previamente), se ha estimado en estudios ser del 31% para Recombinate, que está dentro del rango reportado para FAH derivado de plasma. Los pacientes tratados con Recombinate deben ser controlados cuidadosamente debido al desarrollo de anticuerpos inhibitorios mediante observaciones clínicas y ensayos de laboratorio apropiados.
Población pediátrica
Aparte del desarrollo de inhibidores en pacientes no tratados previamente (PUPs), no se observaron en los ensayos clínicos diferencias específicas de la edad en las reacciones adversas.
4.9 Sobredosis
No se conocen síntomas de sobredosis.
5. PROPIEDADES FARMACOLÓGICAS 5.1. Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos: factor VIII de coagulación. Código ATC: B02BD02.
El complejo del factor VIII /factor von Willebrand consiste en dos moléculas (factor VIII y factor von Willebrand) con funciones fisiológicas diferentes.
Cuando se perfunde en un paciente hemofílico, el factor VIII se une al factor von Willebrand en la circulación del paciente.
El factor VIII activado actúa como un cofactor para el factor IX activado, acelerando la conversión del factor X a factor X activado. El factor X activado convierte la protrombina en trombina. La trombina convierte el fibrinógeno en fibrina y se puede formar un coágulo. La hemofilia A es un transtorno hereditario de la coagulación sanguínea ligada al sexo y producida por la disminución de los niveles de factor VIII que da como resultado una hemorragia profusa en las articulaciones, músculos u órganos internos, bien de forma espontánea o bien como resultado de un trauma accidental o quirúrgico. Mediante terapia de sustitución, los niveles plasmáticos de factor VIII aumentan, consiguiendo una corrección temporal de la deficiencia de factor y una corrección de la tendencia hemorrágica.
Recombinate se ha estudiado en 71 niños no tratados previamente (PUPs). La mediana de edad de la cohorte en el momento de la primera perfusión de Recombinate fue de 10 meses (rango: entre 2 días y 50
meses). El medicamento fue bien tolerado y no se asoció a corto plazo con ningún efecto adverso significativo. Su eficacia clínica fue comparable a la de otras moléculas de FVIII de cadena larga tanto en el tratamiento de hemorragias agudas como en la profilaxis quirúrgica (10 sujetos tuvieron intervenciones quirúrgicas). El seguimiento a largo plazo de la cohorte reveló una incidencia de acontecimientos adversos relacionados con el medicamento de 0,86/1000 perfusiones, ninguna fue grave o amenazante para la vida.
5.2 Propiedades farmacocinéticas
Estudios farmacocinéticos en 69 pacientes previamente tratados revelaron una vida-media promedio circulante del Recombinate de 14,6 ± 4,9 horas (n= 67), cifra que no es significativamente diferente desde el punto de vista estadístico a la del Hemofil M, factor antihemofílico (Humano) derivado de plasma (FAHdp). La vida-media promedio de Hemofil M fue de 14,7 ± 5,1 horas (n=61). La recuperación de la línea base actual observada con Recombinate después de la perfusión de una dosis de 50 UI/Kg fue de
123,9 + 47,7 UI/dl (n=23), que es significativamente superior que la recuperación de la línea base actual de Hemofil M de 101,7 + 31,6 UI/dl (n=61). Sin embargo, la relación calculada de la recuperación actual respecto a la esperada (p.e. incremento del 2% en la actividad de factor VIII de 1 UI de FAHr/kg de peso corporal) con Recombinate (121,2 + 48,9 %) es similar a la de Hemofil M (123,4 + 16,4%).
Se obtuvieron un total de 494 estudios de recuperación de 68 pacientes no tratados previamente. Se realizaron doscientos doce estudios de recuperación cuando los pacientes eran tratados por hemorragias con una media + DE de recuperación actual de 70,0 + 37,9 UI/dl (N=208, por desviación se omitieron cuatro recuperaciones del análisis). La alta variabilidad es debida al amplio rango de la dosis actual dada, de 13,8 a 103,2 UI/kg (media + DE de 36,0 + 16,2 y mediana de 30,2 UI/kg). Se han calculado los porcentajes de recuperación actual pronosticados para contabilizar los intervalos de dosificación, dan como resultado una media de 1,0 + 0,3.
Se realizaron un total de 68 estudios de recuperación cuando los pacientes recibían una perfusión de seguimiento para el tratamiento continuado de una hemorragia pre-existente. Se corrigió el nivel de recuperación actual de FVIII para el nivel de pre-perfusión de FVIII. La media + DE de la recuperación actual fue de 88,6 + 38,2 UI/dl (N=66 con dos recuperaciones omitidas del análisis por desviación). De nuevo, el amplio rango de las dosis actuales dadas, de 18,5 a 85,7 UI/kg (media + DE de 38,6 + 15,9 y mediana de 32,1 UI/kg) da como resultado una variación sustancial de los niveles de recuperación observados. El porcentaje de recuperación actual pronosticado, media + DE, fue de 1,0 + 0,3 con una mediana de 1,0.
Se realizaron un total de 214 estudios de recuperación cuando los pacientes estaban en estado estable dando como resultado una recuperación actual media del 71,6 ± 29,7 U.I./dl (N=209 con 5 recuperaciones omitidas del análisis por desviación). Las dosis dadas se encontraban en el rango de 10,4 a 68,1 U.I./Kg (media ± DE de 38,0 ± 12,7 y mediana de 36,1 U.I./Kg). La relación entre la recuperación actual y la pronosticada, media ± DE, fue de 1,0 ±0,3.
5.3 Datos preclínicos sobre seguridad
Recombinate actúa como el factor VIII endógeno. Dosis varias veces superiores a las dosis por kilogramo de peso corporal recomendadas en humanos no mostraron ningún efecto tóxico en animales de laboratorio.Se analizó la mutagenicidad de Recombinate a dosis considerablemente mayores que las concentraciones plasmáticas de FAH in vitro y a dosis hasta diez veces las dosis clínicas máximas in vivo esperadas y no causó mutaciones reversas, aberraciones cromosómicas o aumento de micronúcleos en eritrocitos policromáticos de médula ósea. Como la experiencia clínica no proporciona evidencia de efectos
carcinogénicos ni mutagénicos, no se considera imperativo la realización de estudios a largo plazo en animales para evaluar el potencial carcinogénico.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo:
Albúmina Humana Cloruro de sodio Histidina Macrogol 3350 Cloruro de calcio dihidrato
Disolvente:
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad este medicamento no debe mezclarse con otros medicamentos o disolventes.
Solamente se debe utilizar el equipo de perfusión proporcionado ya que puede ocurrir un fallo del tratamiento como consecuencia de la adsorción del factor VIII de coagulación humano a las superficies internas de algún equipo de perfusión.
6.3 Periodo de validez
3 años.
Después de su reconstitución, Recombinate no debe ser refrigerado y se debe administrar dentro de las tres horas siguientes a la reconstitución
6.4 Precauciones especiales de conservación
Conservar en nevera (2°C-8°C).
No congelar.
Conservar en el embalaje exterior para protegerlo de la luz.
A lo largo de su periodo de validez, el producto puede almacenarse a 15°C-25°C antes de uso hasta seis meses.
No refrigerar de nuevo después de haber sido almacenado a 15-25°C.
Para las condiciones de conservación del medicamento reconstituido, ver sección 6.3
6.5 Naturaleza y contenido del envase
Un envase unitario contiene un vial de polvo, un vial de disolvente de 10 ml (ambos de vidrio Tipo I con tapones de caucho) y un equipo para reconstitución (BAXJECT II) + una jeringa estéril de plástico de un solo uso + un miniequipo de perfusión estéril + 2 toallitas impregnadas de alcohol + 2 apósitos adhesivos.
De manera alternativa al BAXJECT II, se puede suministrar un equipo de aguja para reconstitución que consta de una aguja de transferencia estéril (para transferir el disolvente dentro del vial de Recombinate) y una aguja de filtro estéril (para transferir la solución reconstituida dentro de la jeringa).
Envase unitario.
6.6 Precauciones especiales de eliminación y otras manipulaciones
La preparación debe administrarse por vía intravenosa después de la reconstitución con el agua para preparaciones inyectables suministrada Debe utilizarse la jeringa desechable de plástico que acompaña al producto.
• Utilizar dentro de las tres horas después de la reconstitución.
• No refrigerar la preparación después de la reconstitución.
• La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
La solución debe ser clara o ligeramente opalescente. No utilizar las soluciones que estén turbias o tengan depósitos. Los productos reconstituidos deben ser inspeccionados visualmente antes de su administración por si contienen alguna partícula o han perdido color.
No utilizar si el medicamento, su barrera de esterilidad o su envase está dañado o muestra cualquier signo de deterioro.
Reconstitución: Utilizar técnica aséptica:
Reconstitución con agujas
Reconstitución con BAXJECT II
1
1 Llevar Recombinate (polvo) y el agua esterilizada para preparaciones inyectables (disolvente) a 15-25°C.
2. Quitar los protectores de los viales de polvo y disolvente.
3 Limpiar los tapones con las toallitas impregnadas de alcohol. Colocar los viales en una superficie plana.
4. Abrir el envoltorio del equipo BAXJECT II
quitando la tapa de papel sin tocar el interior (Fig.
Llevar Recombinate (polvo) y el agua esterilizada para preparaciones inyectables (disolvente) a 15-25°C.
2. Quitar los protectores de los viales de polvo y disolvente.
3. Limpiar los tapones con solución germicida. Colocar los viales sobre una superficie plana.
4. Quitar el protector de uno de los extremos de la aguja de transferencia e insertar este extremo
a). No sacar el equipo del envoltorio.
5. Darle la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente. Coger el envoltorio por su extremo y sacar el equipo BAXJECT II de su envoltorio (Fig. b). No quitar el protector azul del equipo BAXJECT II.
6. Con el equipo BAXJECT II unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico blanca dentro del tapón de Recombinate. El vacío hará que el disolvente penetre en el vial de Recombinate (Fig. c).
de la aguja a través del tapón.
5. Quitar el protector del otro extremo de la aguja de transferencia. Invertir el vial de disolvente sobre la parte superior del vial de Recombinate, rápidamente insertar este extremo de la aguja a través del centro del tapón del vial de Recombinate. El vacío en el vial hará que penetre el disolvente.
6. Desconectar los dos viales retirando primero la aguja del tapón del vial de disolvente y luego la aguja del vial de Recombinate. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que Recombinate esté completamente disuelto, de otra manera el material activo no será extraido por la aguja filtro.
7. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que Recombinate esté completamente disuelto, de otra manera el material activo no pasará a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto).
Fig. c
Fig. a Fig. b



I t
/i r,

Administración: Utilizar técnica aséptica:
Se recomienda que la administración comience dentro de las tres horas después de su reconstitución. No refrigerar el producto reconstituido. Los medicamentos para administración parenteral deben inspeccionarse visualmente antes de su administración para verificar la ausencia de partículas, cuando la solución y el envase lo permitan. Una solución incolora o ligeramente amarilla es aceptable para Recombinate.
Se recomienda que la administración comience dentro de las tres horas después de su reconstitución. No refrigerar el producto reconstituido. Los medicamentos para administración parenteral deben inspeccionarse visualmente antes de su administración para verificar la ausencia de partículas, cuando la solución y el envase lo permitan. Una solución incolora o ligeramente amarilla es aceptable para Recombinate.
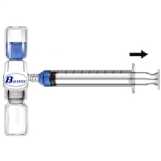
1. Quitar el protector azul del equipo BAXJECT II. NO INTRODUCIR AIRE EN LA JERINGA. Conectar la jeringa al equipo BAXJECT II (Fig. d).
1 Conectar la aguja filtro a la jeringa desechable y . tirar del émbolo hacia atrás para introducir aire en la jeringa.

2 Invertir el sistema (con el vial de concentrado en la parte superior). Introducir el concentrado en la jeringa, tirando del émbolo hacia atrás lentamente (Fig. e).
3 Desconectar la jeringa.
4. Conectar el equipo de administración a la jeringa. Inyectar intravenosamente. El preparado puede ser administrado a una velocidad de hasta 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de Recombinate. Si se observa una elevación significativa del pulso, reducir la velocidad de administración o interrumpir temporalmente la inyección suele hacer que los síntomas normalmente desaparezcan pronto (Ver secciones 4.4 y 4.8).
2 Insertar la aguja filtro en el vial reconstituido de . Recombinate.
3 Inyectar aire dentro del vial y extraer el
. producto reconstituido dentro de la jeringa.
4 Quitar y desechar la aguja filtro. Conectar el
. equipo de administración a la jeringa. Inyectar intravenosamente. El preparado puede ser administrado a una velocidad de hasta 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de Recombinate. Si se observa una elevación significativa del pulso, reducir la velocidad de administración o interrumpir temporalmente la inyección suele hacer que los síntomas normalmente desaparezcan pronto (Ver secciones 4.4 y 4.8).
Fig. d

Fig. e
5 Se deben usar agujas filtro sin utilizar para cada . una de las extracciones del vial reconstituido de Recombinate.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxalta Spain S.L.
Parque Empresarial San Fernando, Edificio Londres 28830 San Fernando de Henares (Madrid)
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
Recombinate 250 UI Recombinate 500 UI Recombinate 1000 UI
N° de Registro: 60.110 N° de Registro: 60.111 N° de Registro: 60.112
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
29 de Marzo de 1994
10. FECHA DE LA REVISIÓN DEL TEXTO
Febrero 2011
14 de 14