Preotact 100 Microgramos Polvo Y Disolvente Para Solucion Inyectable
ANEXO I
RESUMEN DE LAS CARACTERISTICAS DEL PRODUCTO

NOMBRE DEL MEDICAMENTO
1.
Preotact 100 microgramos polvo y disolvente para solución inyectable en una pluma precargada.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada pluma precargada contiene 1,61 mg de hormona paratiroidea correspondiente a 14 dosis.
Tras la reconstitución, cada dosis de 71,4 microlitos contiene 100 microgramos de hormona paratiroidea elaborada utilizando una cepa de Escherichia coli modificada mediante tecnología de ADN recombinante.
Excipientes:
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo de color blanco a color hueso, y disolvente claro e incoloro.
jf
<#
Preotact está indicado para el tratamiento de la osteoporosis en mujeres postmenopáusicas con alto
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
riesgo de fracturas (ver sección 5.1).
Se ha demostrado una reducción significativa en la incidencia de fracturas vertebrales, pero no de fracturas de cadera.
Xp
nistr a
4.2 Posología y forma de administr ación Posología
La dosis recomendada es de 100 microgramos de hormona paratiroidea administrados una vez al día.
Las pacientes recibirán complemento de calcio y vitamina D en caso de que el aporte de la dieta sea insuficiente.
Los datos apoyan el tratamiento continuo con Preotact durante un máximo de 24 meses (ver sección 4.4).
Después del tratamiento con Preotact las pacientes pueden ser tratadas con un bisfosfonato para aumentar la densidad mineral ósea (ver sección 5.1).
Poblaciones especiales Insuficiencia renal
No es necesario ningún ajuste de dosis en pacientes con insuficiencia renal leve a moderada (aclaramiento de creatinina 30 a 80 ml/min). No se dispone de datos en pacientes con insuficiencia renal severa. Por lo tanto, no se utilizará Preotact en pacientes con insuficiencia renal severa (ver sección 4.3).
Insuficiencia hepática
No es necesario ningún ajuste de dosis en pacientes con insuficiencia hepática leve o moderada (puntuación total de 7 a 9 en la escala de Child-Pugh). No se dispone de datos en pacientes con insuficiencia hepática severa. En consecuencia, no se utilizará Preotact en pacientes con insuficiencia hepática severa (ver sección 4.3).
Población pediátrica
No se ha estudiado la seguridad y eficacia de Preotact en pacientes menores de 18 años. No hay un uso relevante de Preotact en pacientes pediátricos para el tratamiento de la osteoporosis con alto riesgo de fracturas.
Pacientes de edad avanzada.
No se requiere ajuste de dosis en función de la edad (ver sección 5.2).
Forma de administración
La dosis se administra como inyección subcutánea en el abdomen.
Se debe formar a los pacientes sobre las técnicas de inyección adecuadas (ver sección 6.6). En el envase se incluye un manual de usuario para instruir a los pacientes sobre el correcto uso de la pluma. Precauciones que deben tomarse antes de manipular o administrar el medicamento Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
n¡2T
4.3 Contraindicaciones
Preotact está contraindicado en pacientes
• con hipersensibilidad a la hormona paratiroidea o a alguno de los excipientes incluidos en la sección 6.1.
• que están recibiendo o han recibido previamente radioterapia ósea
• con tumores óseos o metástasis óseas.
• con hipercalcemia preexistente y otras alteraciones en el metabolismo del fósforo-calcio.
• con osteopatías metabólicas diferentes a la osteoporosis primaria (incluyendo hiperparatiroidismo y enfermedad ósea de Paget
• con elevaciones no explicadas de la fosfatasa alcalina específica del hueso
• con insuficiencia renal severa
• con insuficiencia hepática severa
4.4 Advertencias y precauciones especiales de empleo
Monitorización de los pacientes durante el tratamiento
En las pacientes que han iniciado el tratamiento con Preotact se controlará la concentración de calcio urinario y/o sérico en los meses 1, 3 y 6. No se recomienda el seguimiento después de 6 meses en las pacientes en las que el calcio sérico total esté dentro de los límites normales a los 6 meses.
Se ha observado elevación del calcio sérico durante el tratamiento con Preotact. Las concentraciones de calcio sérico alcanzan un máximo entre 6 y 8 horas después de la dosis y normalmente vuelven al estado basal en 20 a 24 horas después de cada una de las administraciones de hormona paratiroidea. Por lo tanto, en caso de que se obtengan muestras de sangre de una paciente para controlar las concentraciones de calcio, deberá hacerse al menos 20 horas después de la inyección más reciente.
Tratamiento con el calcio sérico elevado
En las pacientes con calcio sérico elevado de forma persistente (por encima del límite normal superior) se evaluará la presencia de enfermedades subyacentes (por ejemplo, hiperparatiroidismo). Si no se descubre ninguna enfermedad subyacente, se seguirán los siguientes procedimientos:
• Deberá retirarse el complemento de calcio y vitamina D.
• Deberá cambiarse la frecuencia de la administración de Preotact a 100 microgramos en días alternos
• Si continúan las concentraciones elevadas, se suspenderá el tratamiento con Preotact y se controlará a la paciente hasta que los valores anómalos vuelvan a estar dentro del intervalo normal.
Se debe tener precaución en:
Pacientes con hipercalciuria pre-existente
Se ha estudiado Preotact en pacientes con hipercalciuria preexistente. En estos pacientes, Preotact tuvo una mayor probabilidad de agravar su hipercalciuria subyacente.
Pacientes con urolitiasis
Preotact no se ha estudiado en pacientes con urolitiasis activa. Preotact se utilizará con precaución en pacientes con urolitiasis previa o activa.
Pacientes que reciben glucósidos cardíacos
Se tendrá precaución en pacientes tratados con glucósidos cardíacos debido al riesgo de toxicidad digitálica si se desarrolla hipercalcemia (ver sección 4.5).
&
Duración del tratamiento
&
e interacción
Los estudios en ratas indican una mayor incidencia de osteosarcoma con la administración a largo plazo de Preotact (ver sección 5.3). El osteosarcoma apareció únicamente a dosis que produjeron exposiciones sistémicas >27 veces superiores a las observadas en seres humanos con la dosis de 100 microgramos. Hasta que se disponga de más datos clínicos no se superará el tiempo de tratamiento recomendado de 24 meses.
4.5 Interacción con otros medicamentos y otras formas
La hormona paratiroidea es un péptido natural que no se metaboliza por las enzimas microsómicas hepáticas que metabolizan los fármacos, y no las inhibe (por ejemplo, isoenzima citocromo P450). Además, la hormona paratiroidea no se une a las proteínas y tiene un bajo volumen de distribución. En consecuencia, no se prevén interacciones con otros medicamentos y no se han realizado estudios específicos de interacciones medicamentosas. No se han identificado posibles interacciones medicamentosas en el programa clínico.
cP
Del mecanismo de acción conocido se deduce que la asociación de Preotact con glucósidos cardíacos puede predisponer a las pacientes a toxicidad por digitálicos en caso de que aparezca hipercalcemia.( ver sección 4.4)
4.6
Fertilidad, embarazo y lactancia
No se dispone de datos del uso de hormona paratiroidea en mujeres en edad fértil, durante el embarazo y lactancia materna. Los estudios en animales sobre la toxicidad reproductiva son incompletos (ver sección 5.3).
No se utilizará hormona paratiroidea en mujeres en edad fértil, durante el embarazo o lactancia.
4.7 Efectos sobre la capacidad para conducir y utilizar maquinas
No se han realizado estudios sobre la capacidad para conducir y utilizar máquinas. Como se han descrito algunos episodios de mareos en pacientes tratados con Preotact, las pacientes no conducirán vehículos ni utilizarán maquinaria hasta que hayan desaparecido los síntomas.
4.8 Reacciones adversas
Los siguientes datos de reacciones adversas (RAM) se basan en dos estudios comparativos con placebo en 2.642 mujeres osteoporóticas posmenopáusicas de las cuales 1.341 recibieron hormona paratiroidea. Aproximadamente 71,4% de las pacientes tratadas con hormona paratiroidea notificaron al menos una RAM.
La hipercalcemia y/o hipercalciuria reflejan la actividad farmacodinámica conocida de la hormona paratiroidea en el aparato digestivo, riñón y hueso. Se notificó hipercalcemia en el 25,3% e hipercalciuria en el 39,3% de las pacientes tratadas con Preotact. La hipercalcemia fue transitoria y se detectó más frecuentemente en los primeros 3 meses de tratamiento. Durante el programa clínico, se trató la hipercalcemia monitorizando los parámetros de laboratorio y utilizando un algoritmo de tratamiento pre-especificado (ver las secciones 4.3, 4.4 y 5.1).
La otra única RAM muy frecuentemente notificada fue náuseas.
La tabla siguiente muestra una visión general de las RAMs donde la incidencia es al menos 0,5% superior en el grupo tratado con hormona paratiroidea, comparado con el grupo tratado con placebo. Se han utilizado las siguientes categorías para clasificar las reacciones adversas por frecuencia de aparición: Muy frecuentes (> 1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); y muy raras (<1/10.000), incluyendo las notificaciones aisladas.
Hormona Paratiroid ea N=1341 (%)
JP
0,5
Clase de sistema orgánico
Infecciones e infestaciones
Poco Frecuentes Gripe
Trastornos del metabolismo y de la nutrición
Muy frecuentes Hipercalcemia
Frecuentes
Aumento de los niveles de calcio en sangre Poco frecuentes , , jsP.
Aumento de los niveles de fosfatasa alcalina en sangre Anorexia
Aumento de los niveles de ácido úi
Trastornos del sistema nervioso
Frecuentes Cefalea Mareos
Poco frecuentes Disgeusia Parosmia
rico en sangre
25,3
3,1
0,8
0,6
0,6
9,3
3,9
0,8
0,7
Trastornos cardíacos
Frecuentes
Palpitaciones_1,0
|
Trastornos gastrointestinales | |
|
Muy frecuentes | |
|
13,5 | |
|
Náuseas | |
|
Frecuentes | |
|
Vómitos |
2,5 |
|
Estreñimiento |
1,8 |
|
Dispepsia |
1,3 |
Diarrea 1.0
Poco frecuentes
Dolor abdominal_0,8.
|
Trastornos musculoesqueléticos y del tejido conectivo Frecuentes | |
|
Calambres musculares |
1,1 |
|
Dolor en extremidades |
1,1 |
|
Dolor de espalda |
1,0 |
|
Trastornos renales y urinarios Muy frecuentes Hipercalciuria Frecuentes |
39,3 |
|
Aumento del ratio calcio/creatinina en orina |
2,9 |
|
Aumento del calcio en orina |
2,2 |
Trastornos generales y alteraciones en el lugar de administración
&
Frecuentes
Eritema en la zona de la inyección 2,6
1,2
Astenia
Poco frecuentes
Irritación en la zona de inyección 0,9
Preotact aumenta las concentraciones séricas de ácido úrico. De todas las pacientes que recibieron 100 microgramos de hormona paratiroidea, se notificó elevación del ácido úrico en sangre en 8 pacientes (0,6%) e hiperuricemia en 5 pacientes (0,4%). Aunque se han notificado casos de gota, artralgias y nefrolitiasis como RAMs, no se ha establecido por completo una relación causal entre las elevaciones de ácido úrico y la administración de Preotact.
aOnoaaaroda
En un estudio clínico extenso fase III, se detectaron anticuerpos frente a la hormona paratiroidea en el 3% de las mujeres tratadas con Preotact, comparado con el 0,2% de las mujeres tratadas con placebo. En estas mujeres con un título positivo, no se observaron pruebas de reacciones de hipersensibilidad, reacciones alérgicas, efectos sobre la respuesta de la densidad mineral ósea o efectos sobre el calcio sérico.
4.9 Sobredosi s
Signos y síntomas
En el programa clínico de Preotact se ha notificado sobredosis accidental.
Preotact se ha administrado en dosis únicas de hasta 5 microgramos/kg, en dosis repetidas de hasta 3 microgramos/kg/día durante 3 días y hasta 2,5 microgramos/kg/día durante 7 días. Los efectos de sobredosis que pueden esperarse incluyen hipercalcemia retardada, náuseas, vómitos, mareos y cefalea.
Tratamiento de la sobredosis
No existe un antídoto específico de Preotact. El tratamiento de la sobredosis sospechada incluye la suspensión temporal de Preotact, el control del calcio sérico y la implementación de medidas de apoyo adecuadas, tal como la hidratación. Debido a la duración relativamente breve de la actividad farmacológica de Preotact, no son necesarias otras medidas.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Homeostasis del calcio, hormonas paratiroideas y análogos. Código ATC: H05 AA03
Mecanismo de acción
Preotact contiene hormona paratiroidea humana recombinante que es idéntica al polipéptido de 84 aminoácidos natural intacto.
La actividad fisiológica de la hormona paratiroidea incluye el estímulo de la formación de hueso por medio de efectos directos sobre las células formadoras de hueso (osteoblastos), aumentando indirectamente la absorción intestinal de calcio y aumentado la reabsorción tubular de calcio y la excreción de fosfatos por el riñón.
Efectos farmacodinámicos
Los efectos de la hormona paratiroidea sobre el hueso dependen de las características de la exposición sistémica. Las elevaciones transitorias de las concentraciones de hormona paratiroidea después de la inyección subcutánea de Preotact estimulan la formación de nuevo hueso en las superficies óseas trabecular y cortical (perióstica y/o endóstica) por medio de la estimulación preferencial de la actividad osteoblástica sobre la actividad osteoclástica.
Efectos sobre las concentraciones séricas de calcio ■:é>,..... .
La hormona paratiroidea es el principal regulador de la homeostasis del calcio sérico. En respuesta a las administraciones subcutáneas de Preotact (100 microgramos de hormona paratiroidea), aumentan gradualmente las concentraciones del calcio sérico total y alcanzan la concentración máxima (aumento medio en 129 pacientes, 0,15 mmol/l) en aproximadamente 6 a 8 horas después de la administración. En general, las concentraciones de calcio sérico regresan al nivel basal 24 horas después de la administración.
En dos estudios comparativos con placebo con 2.642 mujeres osteoporóticas postmenopáusicas, se observó hipercalcemia en el 25,3% de las pacientes tratadas con Preotact comparado con 4,3% de las pacientes tratadas con placebo. La hipercalcemia fue transitoria y se detectó más frecuentemente en los primeros 3 meses de tratamiento. Durante el programa clínico, se trató la hipercalcemia monitorizando los parámetros de laboratorio y utilizando un algoritmo de tratamiento pre-especificado (ver secciones 4.3 y 4.4).
Eficacia clínica ver
Efecto sobre la incidencia de fracturas
El estudio fundamental fue un estudio de 18 meses, doble ciego, comparativo con placebo y fase III (TOP) del efecto de Preotact sobre la incidencia de fractura en mujeres con osteoporosis posmenopáusica.
Se aleatorizó a un total de 2.532 pacientes (1286 Preotact y 1246 placebo), de edades comprendidas entre 45 y 94 años (8,1% 45-54 años y 11,4% > 75 años), a recibir 100 microgramos/día o placebo con complemento diario de calcio (700 mg) y vitamina D (400 UI).
En general, aproximadamente el 19% de las pacientes en cada grupo de tratamiento tenía al menos 1 fractura vertebral sin consolidar en el periodo basal. La puntuación T lumbar basal media fue de aproximadamente -3,0 en cada grupo de tratamiento.
De las 2.532 pacientes aleatorizadas en el grupo por intención de tratar (ITT), un total de 59 pacientes experimentó al menos una nueva fractura vertebral, placebo 42 (3,37%) - Preotact 17 (1,32%), p = 0,001. Las pacientes del grupo de tratamiento con Preotact tuvieron una reducción del riesgo relativo de sufrir una nueva fractura vertebral del 61% en el mes 18, comparado con las pacientes del grupo placebo.
Para evitar una o más fracturas vertebrales nuevas, 48 mujeres de la población total debieron ser tratadas durante una media de 18 meses. Para las pacientes con fracturas preexistentes, el número necesario de tratar (NNT) fue de 21 pacientes.
No se observó una diferencia significativa entre los grupos de tratamiento en cuanto a la incidencia de fracturas clínicas no vertebrales, 5,52% con Preotact frente a 5,86% con placebo.
La reducción de fractura mas relevante se observó en pacientes de alto riesgo de fracturas tales como pacientes con fracturas previas y en pacientes con una puntuación T de la columna lumbar < -3.
Se incluyeron relativamente pocos pacientes en el estudio fase III de menos de 5 años postmenopáusicas y entre 45 y 54 años de edad (2-3%). Los resultados para estos sujetos no fueron diferentes de los resultados globales del estudio.
Efecto sobre la densidad mineral ósea (DMO)
En el estudiopivotal, Preotact aumentó en un 6,5% la densidad mineral ósea (DMO) en la columna lumbar después de 18 meses de tratamiento, comparado con -0,3% con placebo (p<0,001). Se observaron aumentos significativos en la densidad mineral ósea (DMO) de la cadera (total, cuello del fémur, trocánter) al final del estudio; 1,0, 1,8 y 1,0%, respectivamente, con Preotact comparado con -1,1, -0,7 y -0,6% con placebo (p<0,001).
• ‘cT
El tratamiento mantenido durante un máximo de 24 meses en una extensión abierta de este estudio tuvo como resultado un aumento mantenido en la DMO. El aumento en la DMO de la columna lumbar y cuello del fémur con respecto al periodo basal fue de 6,8% y 2,2%, respectivamente, en los pacientes tratados con Preotact.
Se evaluaron los efectos de Preotact sobre la estructura ósea utilizando tomografía computarizada cuantitativa (QCT) y periférica QCT. La DMO trabecular volumétrica en la columna lumbar aumentó un 38% con respecto al periodo basal tras 18 m íeses. De igual forma, la DMO trabecular volumétrica en la cadera total aumentó un 4,7%. Se produjeron aumentos similares en el cuello del fémur, trocánter e intertrocánter. El tratamiento con Preotact redujo la DMO volumétrica del hueso cortical (determinada en la mitad de la diáfisis de la tibia y en el radio distal), mientras se mantenían la circunferencia perióstica o los índices de resistencia del hueso cortical.
En el estudio de 24 meses de tratamiento con asociación de alendronato (PaTH), se evaluaron los efectos de Preotact sobre la estructura ósea utilizando QCT. La DMO trabecular volumétrica en la columna lumbar aumentó en 26, 13 y 11% (Preotact, Preotact y alendronato, y alendronato, respectivamente) con respecto al valor basal tras 12 meses. De forma similar, la DMO trabecular volumétrica en la cadera total aumentó en el 9, 6 y 2%, respectivamente, en los 3 grupos.
Tratamiento de la osteoporosis con terapia combinada y secuencial
El estudio PaTH fue un estudio promovido por el Instituto Nacional de Salud (INS)aleatorizado, comparativo con placebo, de 2 años, multicéntrico y doble ciego de Preotact y alendronato en monoterapia y en asociación para el tratamiento de la osteoporosis postmenopáusica. Los criterios de inclusión fueron mujeres entre 55 y 85 años con puntuación T de DMO inferior a -2,5, o inferior a -2 con al menos otro factor de riesgo de fractura. Todas las mujeres recibieron complemento de calcio (400-500 mg) y vitamina D (400 UI).
Se asignó aleatoriamente a un total de 238 mujeres postmenopáusicas, a uno de los siguientes grupos de tratamiento; Preotact (100 microgramos de hormona paratiroidea), alendronato (10 mg) o la asociación de ambos, y se mantuvieron en observación durante 12 meses. En el segundo año de estudio, se asignó aleatoriamente a las mujeres del grupo original de Preotact a recibir alendronato o placebo, y las pacientes de los otros dos grupos recibieron alendronato.
En el periodo basal un total de 165 pacientes (69%) tenían una puntuación T inferior a -2,5, y 112 (47%) habían sufrido al menos una fractura después de la menopausia.
Después de un año de tratamiento se observaron los siguientes resultados: los aumentos en la DMO de la columna lumbar sobre el valor basal fueron similares en los grupos de tratamiento de Preotact y de tratamiento combinado (6,3 y 6,1% respectivamente), pero fueron algo inferiores en el grupo de alendronato (4,6%). Los aumentos en la DMO de la cadera total fueron 1,9, 0,3 y 3,0% en los 3 grupos, respectivamente.
Al finalizar el segundo año (12 meses después de suspender Preotact), se observó un aumento medio de 12% en la DMO de la columna por absorciometría dual de rayos X (DXA) en las pacientes tratadas con alendronato durante el segundo año. En las pacientes que recibieron placebo durante el segundo año, el aumento porcentual medio fue de 4% comparado con el periodo basal, pero había disminuido ligeramente con respecto al valor observado al finalizar 12 meses de tratamiento con Preotact. En la DMO de la cadera, se observó un aumento del 4,5% con respecto al periodo basal en un año de tratamiento con alendronato, comparado con una disminución del 0,1% después de un año de placebo.
THS únicamente (7,1 % comparado con 1,1%, p<0,001). La asociación fue efectiva con independencia
no lo orlon foco Knool no moomniA oca/v o I i \ /II i Koonl
Preotact asociado a terapia hormonal sustitutiva (TSH) en 180 pacientes posmenopáusicas ha demostrado aumentar significativamente la DMO de la columna lumbar en 12 mese s, comparado con
de la edad, tasa basal de recambio óseo o DMO basal.
5.2 Propiedades farmacocinéticas
Absorción
La administración subcutánea de 100 microgramos de hormona paratiroidea en el abdomen produce un aumento rápido de las concentraciones plasmáticas de hormona paratiroidea y produce una concentración máxima en 1 a 2 horas después de la administración. La semivida promedio es de aproximadamente 1,5 horas. La biodisponibilidad absoluta de 100 pg de hormona paratiroidea tras su administración por vía subcutánea en el abdomen es de 55%.
Distribución
El volumen de distribución en situación de equilibrio después de la administración por vía intravenosa es de aproximadamente 5,4 l. La variabilidad interindividual en el volumen de distribución de la hormona paratiroidea es de aproximadamente 40%.
ti?
Biotransformación
La hormona paratiroidea se elimina de manera eficiente de la sangre por un proceso mediado por un receptor en el hígado y se hidroliza en péptidos de menor tamaño. Los fragmentos con grupos amino terminales se degradan en la célula mientras que los fragmentos con grupos carboxílicos terminales pasan de nuevo a la sangre y se eliminan en el riñón. Se considera que estos fragmentos con grupos carboxílicos terminales intervienen en la regulación de la actividad de la hormona paratiroidea. En condiciones fisiológicas normales, la hormona paratiroidea intacta (1-84) constituye tan sólo el 5-30% de las formas circulantes de la molécula, mientras que el 70-95% está presente como fragmentos carboxi terminales. Después de una administración subcutánea de Preotact, los fragmentos carboxi terminales suponen aproximadamente el 60-90% de las formas circulantes de la molécula. El aclaramiento sistémico de la hormona paratiroidea (45,3 l/hora) después de una administración intravenosa está próximo al flujo plasmático hepático normal y es coherente con el amplio metabolismo hepático del principio activo. La variabilidad interindividual en el aclaramiento sistémico es de aproximadamente el 15%.
Eliminación
La hormona paratiroidea se metaboliza en el hígado y en menor medida en el riñón. La hormona paratiroidea no se excreta en su forma intacta. Los fragmentos carboxi-terminales circulantes se filtran en el riñón, pero se metabolizan posteriormente a fragmentos aún más pequeños durante la reabsorción tubular.
Insuficiencia hepática
Hubo un incremento moderado de aproximadamente 20% sobre la media basal corregida de la exposición sistémica (AUC) a la hormona paratiroidea en un estudio realizado en 6 hombres y 6 mujeres con insuficiencia hepática moderada comparado con un grupo de referencia de 12 sujetos con función hepática normal.
No se han realizado estudios en pacientes con insuficiencia hepática severa.
Insuficiencia renal
La exposición global y C max de la hormona paratiroidea fueron ligeramente mayores (22% y 56% respectivamente) en un grupo de sujetos de 8 hombres y 8 mujeres con insuficiencia renal leve a moderada (aclaramiento de creatinina de 30 a 80 ml/min) comparado con un grupo de referencia de 16 sujetos con función renal normal.
No se han investigado las características farmacocinéticas de la hormona paratiroidea en pacientes con insuficiencia renal severa (aclaramiento de creatinina inferior a 30 ml/min).
Ancianos
No se han observado diferencias en las características farmacocinéticas de Preotact con respecto a la edad (intervalo 47-88 años). No se requieren ajustes en la dosis en función de la edad.
Sexo
El producto medicamentoso se ha investigado tan sólo en mujeres postmenopáusicas.
♦Jcr
5.3 Datos preclínicos sobre seguridad
Los datos en los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales sobre farmacología de seguridad, toxicidad de dosis repetidas, genotoxicidad, potencial carcinogénico, toxicidad para la reproducción.
En monos tratados con dosis subcutáneas diarias durante 6 meses, se observó un aumento en la mineralización tubular renal en niveles de exposición inferiores a la exposición clínica.
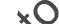
Ratas tratadas con inyecciones diarias durante casi toda su vida tuvieron una formación ósea desmesurada dependiente de la dosis y una mayor incidencia de tumores óseos, incluso osteosarcoma, probablemente debido a un mecanismo epigenético. Debido a las diferencias en la fisiología ósea en ratas y seres humanos, la importancia clínica de estos datos es probablemente menor. No se han observado osteosarcomas en los estudios clínicos.
io estui
<2T
No se han realizado estudios de toxicidad fetal, en el desarrollo, perinatal o posnatal. Se desconoce si la hormona paratiroidea recombinante humana se excreta por la leche de los animales en periodo de lactancia.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo
Manitol
Ácido cítrico monohidratado Cloruro sódico
Ácido clorhídrico, diluido (para el ajuste del pH) Hidróxido de sodio (para el ajuste del pH).
Disolvente
Meta-cresol
Agua para inyección.
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
30 meses.
Solución reconstituida: Se ha demostrado la estabilidad química y física en uso durante 28 días entre 2°C y 8°C. Durante un periodo de 28 días la solución reconstituida puede almacenarse un máximo de 7 días a temperaturas por debajo de 25 °C.
6.4 Precauciones especiales de conservación
No conservar a Ta superior a 25°C. No congelar. Mantener el producto protegido de la luz.
Solución reconstituida: Conservar en nevera (entre 2°C y 8 °C). No congela vez que el producto se ha reconstituido puede conservarse fuera de la nevera a temperaturas por debajo de 25 °C un máximo de 7 días durante el periodo de uso de 28 días (ver sección 6. 3).
6.5 Naturaleza y contenido del envase
El medicamento se suministra en una pluma precargada que contiene un cartucho de doble cámara.
vmenert
El sistema de cierre del envase se compone de un cartucho de cámara doble, un tapón central, una tapa ondulada (con un sello de caucho) sellando la primera cámara con el polvo liofilizado y un tapón
terminal sellando la segunda cámara con el disolvente para el mezclado. Cartucho: De vidrio Tipo I.
*o
caucho, color gris.
o°
solv
x0
Tapón (central y terminal): De bromobutil
Tapa ondulada (con un sello de caucho). De aluminio. el sello de caucho está elaborado de bromobutil caucho.
gr
Dentro de la pluma precargada, cada cartucho contiene 1,61 mg de hormona paratiroidea y 1,13 ml de disolvente (14 dosis).
Preotact se presenta en envases de 2 plumas precargadas.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Preotact se inyecta utilizando una pluma precargada. Cada pluma se debe usar para un solo paciente. En cada inyección se debe usar una nueva aguja estéril. La pluma puede utilizarse con agujas de inyección estándar. El contenido del cartucho se reconstituye en la pluma. Después de la reconstitución el líquido deberá ser claro e incoloro.
NO AGITAR; la agitación puede causar la desnaturalización del principio activo.
Si la solución reconstituida está turbia, tiene color o contiene partículas, no se debe usar Preotact. Para el uso de la pluma, ver el manual de instrucciones.
La eliminación de los productos no utilizados o de los envases se establecerá de acuerdo con las exigencias locales.
TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
7.
NPS Pharma Holdings Limited Grand Canal House 1 Grand Canal Street Upper Dublin 4 Irlanda
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/06/339/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 24.04.2006 Fecha de la última revalidación: 24.04.2011
10. FECHA DE LA REVISIÓN DEL TEXTO


La información detallada de este medicamen a Agencia
Europea de Medicamentos (EMA) http://www
O

ANEXO II
A.
J3
&
B.
C.
FABRICANTE DEL PRINCIPIO BIOLÓGICO ACTIVO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
CONDICIONES O RESTRICCIONES DE SUMINISTRO DE USO
OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
cr

FABRICANTE DEL PRINCIPIO BIOLÓGICO ACTIVO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
A.
Nombre y dirección del fabricante de la sustancia biológica activa
Boehringer-Ingelheim Austria GmbH Dr. Boehringer Gasse 5-11 1211 Vienna Austria
Nombre o razón social del fabricante responsable de la liberación de los lotes
Nycomed Danmark ApS Langebjerg 1, 4000 Roskilde Dinamarca
B.
CONDICIONES O RESTRICCIONES DE SUMINISTRO
DE U
SO
CONDICIONES O RESTRICCIONES RELATIVAS AL SUMINISTRO Y USO IMPUESTAS AL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Medicamento sujeto a prescripción médica.
• CONDICIONES O RESTRICCIÓN DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN RESPECTO A UN USO SEGURO Y EFICAZ DEL MEDICAMENTO
No aplicable
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Sistema de Farmacovigilancia
El Titular de la Autorización de Comercialización (TAC) debe asegurar que el Sistema de Farmacovigilancia, incluido en el Módulo 1.8.1. de la Autorización de Comercialización, esté instaurado y en funcionamiento antes de que el medicamento se comercialice y durante el tiempo que permanezca en el mercado.
Plan de Gestión de Riesgos (pgr)
El TAC se compromete a realizar los estudios y las actividades adicionales de farmacovigilancia detalladas en el Plan de Farmacovigilancia, de acuerdo con la versión 03 del Plan de Gestión de Riesgos (PGR) incluido en el Módulo 1.8.2. de la Autorización de Comercialización y cualquier actualización posterior del PGR acordada por el Comité de Medicamentos de Uso Humano (CHMP).
De acuerdo con la Directriz del CHMP sobre Sistemas de Gestión de Riesgos para medicamentos de uso humano, el PGR actualizado se debe presentar junto con el siguiente Informe Periódico de Seguridad (IPS).
Además, se debe presentar un PGR actualizado:
• Cuando se reciba nueva información que pueda afectar a las especificaciones de seguridad vigentes, al Plan de Farmacovigilancia o a las actividades de minimización de riesgos
• Dentro de los 60 días posteriores a la consecución de un hito importante (farmacovigilancia o minimización de riesgos)
• A petición de la Agencia Europea de Medicamentos.
&
ANEXO III ETIQUETADO Y PROSPECTO
/
■O*

A. ETIQUETADO
<#
&

INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR Caja exterior (2 plumas precargadas)
1. NOMBRE DEL MEDICAMENTO
Preotact 100 microgramos polvo y disolvente para solución inyectable en pluma precargada. Hormona paratiroidea
2. PRINCIPIO(S) ACTIVOS(S)
Cada pluma precargada contiene 1,61 mg de hormona paratiroidea, correspondiendo a 14 dosis. Tras la reconstitución, cada dosis de 71,4 microlitros contiene 100 microgramos de hormona paratiroidea.

Mantener fuera de la vista y del alcance de los niños.
7. OTRAS ADVERTENCIAS ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Solución reconstituida: 28 días.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Pluma precargada (antes de la reconstitución): No conservar a temperatura superior a 25 °C. No congelar. Conservar la pluma precargada en el embalaje exterior para protegerlo de la luz.
Pluma precargada (después de la reconstitución): Conservar en nevera (entre 2°C y 8 °C). No congelar. No agitar. Una vez que se ha reconstituido el cartucho puede conservarse a temperaturas por debajo de 25 °C un máximo de 7 días durante el periodo de uso de 28 días.

10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL PRODUCTO NO
UTILIZADO O DE LOS MATERIALES QUE ESTÉN EN CONTACTO DIRECTO CON EL PRODUCTO (CUANDO CORRESPONDA)_
16. INFORMACIÓN EN BRAILLE
Preotact
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
Pluma precargada_
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Preotact 100 microgramos polvo y disolvente para solución inyectable. Hormona paratiroidea Vía subcutánea
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes del uso.
|
3. |
FECHA DE CADUCIDAD | |
|
CAD |
_. _ | |
|
4. |
NÚMERO DE LOTE | |
|
Lote | ||
|
5. |
CONTENIDO EN PESO, VOLUMEN O EN UNIDADES | |
|
1,61 mg de hormona paratiroidea y 1,13 ml de disolvente (14 dosis). _ | ||
|
6. |
OTROS | |
B.PROSPECTO
&
&

Prospectoinformación para el usuario:
Preotact 100 microgramos polvo y disolvente para solución inyectable en una pluma precargada.
Hormona paratiroidea
- Lea todo el prospecto detenidamente antes de empezar a usar el medicamento, porque contiene información importante para usted.Conserve este prospecto. Puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas de enfermedad que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico. Esto incluye cualquier posible efecto adverso que no aparezca en este prospecto.
Contenido del prospecto:
1. Qué es Preotact y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Preotact
&
3. Cómo usar Preotact
4. Posibles efectos adversos
5. Conservación de Preotact
6. Contenido del envase e información adicional
1
Qué es Preotact y para qué se utiliza
Preotact se utiliza para tratar la osteoporosis en mujeres postmenopáusicas con alto riesgo de fracturas. La osteoporosis es una enfermedad que hace que los huesos se vuelvan finos y frágiles. Es especialmente frecuente en las mujeres después de la menopausia. La enfermedad progresa gradualmente por lo que es posible que no experimente ningún síntoma al principio. Sin embargo, si usted sufre osteoporosis tiene mayor probabilidad de sufrir fracturas en los huesos, especialmente en la columna vertebral, cadera y muñecas. También pude causarle dolor de espalda, pérdida de estatura y curvatura en la espalda.
Preotact reduce el riesgo de fracturas en los huesos de la columna debido a que aumenta su resistencia y calidad.
No se ha demostrado que Preotact reduzca el riesgo de fractura de cadera.
2. Qué necesita saber antes de empezar a usar Preotact
No use Preotact:
• si es alérgico a la hormona paratiroidea o a cualquiera de los demás componentes de este medicamento(indicados en la sección 6);
• si ha recibido o recibe radioterapia en los huesos;
• si tiene cáncer de los huesos;
• si tiene niveles altos de calcio en sangre u otros desórdenes en el metabolismo del fósforo-calcio;
• si sufre otra enfermedad ósea (tal como hiperparatiroidismo o enfermedad de Paget);
• si tiene niveles altos de fosfatasa alcalina;(una enzima producida por el organismo: puede indicar ciertas condiciones médicas relacionadas con los huesos y el hígado);
• si sufre problemas renales severos;
• si sufre alguna enfermedad hepática severa;
Advertencias y precauciones:
Consulte a su médico o farmacéutico antes de usar Preotact si:
• tiene un elevado nivel de calcio en orina
• sufre de cálculos renales
• recibe medicamentos para el corazón (ej. digoxina también conocido como digitálico)
Control de los niveles de calcio en sangre y/o en orina
Su médico comprobará su respuesta al tratamiento a intervalos regulares. Su médico le realizará análisis de sangre y/o orina para medir los niveles de calcio en sangre y/o orina en los meses 1, 3 y 6 después de empezar su tratamiento con Preotact.
Niños y adolescentes
No se utilizará Preotact en niños o adolescentes menores de 18 años.
Otros medicamentos y Preotact
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o vaha tomar otros medicamentos.
Debe usar Preotact con precaución si recibe algún tratamiento para el corazón (ej: digoxina también conocida como digitálico).
Embarazo y lactancia
Consulte a su médico o farmacéutico antes de tomar cualquier medicamento.
No utilice Preotact si está embarazada o en periodo de lactancia.
Si se siente mareada, no conduzca o utilice máquinas hasta que se encuentre bien.
Preotact contiene menos de 1 mmol de sodio (23 mg) por dosis.
Esto significa que esencialmente está libre de sodio.
3.
o°
sodi
Siga exactamente las instrucciones de administración de Preotact de su médico. Consulte a su médico o farmacéutico si tiene dudas.
♦ C;
Dosis
La dosis recomendada de Preotact es de 100 microgramos por día.
Su médico puede prescribirle un complemento de calcio y vitamina D. En ese caso le indicará la cantidad que debe tomar cada día.
Vía de administración
Antes de utilizarla por primera vez, la medicación en la pluma precargada de Preotact debe ser mezclada (por favor consultar el apartado “instrucciones de uso”).
En ese momento, la pluma precargada de Preotact está lista para su uso y la medicación está lista para ser inyectada en el abdomen (bajo la piel).
Cuando no esté utilizando la pluma precargada, póngala de nuevo en la nevera.
Información importante de uso de Preotact
• Aplíquese la inyección de Preotact poco después de que haya sacado la pluma precargada del refrigerador.
• Introduzca la pluma precargada Preotact de nuevo en el refrigerador inmediatamente después de que la haya utilizado.
• No agitar la pluma precargada Preotact (ni antes ni después de la inyección) debido a que puede anular el efecto de la medicación.
• Utilice una nueva aguja en cada inyección y tírela después de cada uso.
• No guarde nunca su pluma precargada Preotact con la aguja puesta.
• Coloque siempre una nueva aguja antes de usar.
• No comparta nunca su pluma precargada Preotact con nadie.
Para instrucciones de cómo utilizar su pluma precargada, por favor lea el apartado “Instrucciones de uso”.
Duración del tratamiento
Utilice Preotact durante el tiempo que le haya prescrito el médico, normalmente no más de 24 meses. Si Vd. usa más Preotact del que debiera
Si se inyecta accidentalmente más de una dosis de Preotact en un día, contacte inmediatamente con su médico o farmacéutico.
Si olvidó usar Preotact
Si olvida administrarse Preotact (o no puede hacerlo en el momento indicado), hágalo lo antes posible en ese día.
No se inyecte nunca más de una dosis en el mismo día.
No se aplique una dosis doble para compensar la dosi olvidada.
Si interrumpe la administración de Preotact
Informe a su médico si considera dejar el tratamiento con Preotact antes de finalizar el periodo de prescripción.
Si tiene cualquier otra duda sobre el uso de este producto, pregunte a su médico o farmacéutico.
4. POSIBLES EFECTOS ADVERSOS
n.
Al igual que todos los medicamentos, Preotact puede tener efectos adversos, aunque no todas las personas los sufran.
Muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
• aumento del nivel de calcio en la sangre,
• aumento del nivel de calcio en la orina,
• náuseas.
Frecuentes (pueden afectar a 1 de cada 10 personas):
• dolor de espalda,
• estreñimiento, diarrea,
• disminución del tono muscular, calambres musculares,mareos,
• enrojecimiento de la piel (eritema) en lugar de la inyección,
• latidos cardíacos rápidos o irregulares,
• dolor de cabeza,
• dolor en brazos y piernas (extremidades),
• malestar de estómago, vómitos,
• cansancio.
Poco frccucntcs(pucdcn afectar a 1 de cada 100 personas):
• dolor abdominal,
• gripe,
• aumento de los niveles de ácido úrico en sangre,
• aumento del nivel de fosfatasa alcalina en sangre,
• irritación de la piel en la zona de la inyección,
• pérdida del apetito,
• alteraciones del olfato y del gusto.
Si nota alguno de los efectos adversos, comuníqueselo a su médico o farmacéutico. Esto incluye efectos adversos no mencionados en este prospecto.
5. Conservación de Preotact
Mantener este medicamento fuera de la vista y del alcance de los niños.
&
No use Preotact después de la fecha de caducidad que aparece en el cartucho y en la caja después de CAD. La fecha de caducidad es el último día del mes que se indica.
Antes de la mezcla
• No conservar a temperatura superior a 25 °C.
• No congelar.
• Conservar Preotact en el embalaje original, para protegerlo de la luz.
Después de la mezcla
• Conservar en nevera (entre 2°C y 8 °C).
• No congelar. 1
6
<$
Composición de Preotact
-El principio activo es hormona paratiroidca. Cada cartucho contiene 1,61 mg de hormona paratiroidca equivalente a 14 dosis. Tras reconstitución, cada dosis de 71,4 microlitros contiene 100 microgramos de hormona paratiriodca.
Los demás componentes son:
El polvo contiene:
• cloruro sódico,
• manitol,
• ácido cítrico monohidratado,
• ácido clorhídrico,
• hidróxido de sodio.
El disolvente contiene:
• meta-cresol,
• agua para inyección
meta-cresol y agua para inyección.
Aspecto de Preotact y tamaño del envase
Preotact es un polvo y disolvente para solución inyectable en una pluma precargada.
Preotact se suministra en una pluma precargada que contiene un cartucho. La primera cámara contiene 1,61 mg de hormona paratiroidea en polvo y la segunda cámara contiene 1,13 ml de disolvente.
Preotact se presenta en un envase con 2 plumas precargadas.
Titular de la autorización de comercialización
NPS Pharma Holdings Limited Grand Canal House 1 Grand Canal Street Upper Dublin 4 Irlanda
Responsable de la fabricación
Nycomed Danmark ApS Langebjerg 1 DK-4000 Roskilde Dinamarca
Este prospecto fue aprobado en MM/AAAA
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos (EMA) http://www.ema.europa.eu
O?
■O1

PLUMA PRECARGADA
La pluma precargada Preotact® está especialmente diseñada para facilitar la administración de su tratamiento para la osteoporosis.
Antes de realizar su primera inyección con una nueva pluma precargada, deberá colocar una aguja y mezclar la medicación de acuerdo con las instrucciones de este prospecto. Mezcle sólo una pluma cada vez.
La pluma precargada contiene medicación para 14 días.

PREOTACT®
INSTRUCCIONES DE USO
Cada día tendrá que comprobar que la medicación está transparente, colocar una aguja nueva, administrarse la inyección en el abdomen y, a continuación, desechar la aguja antes de guardar su pluma precargada en el frigorífico (2 °C - 8 °C).
Pluma precargada antes de realizar la mezcla:

Lea con atención la información de estos recuadros, ya que contienen información importante para usted.
botón de liberación contador de dosis
botón de inyección
cuerpo de la plum; ¡o?
ma
parte frontal
tapa
Pasos que debe seguir con una pluma precargada nueva:
• Coloque una aguja
• Mezcle la medicación
• Retire el aire residual (purgue la pluma)
• Adminístrese su dosis diaria o guarde la pluma precargada
Pasos que debe seguir para cada una de las 14 inyecciones:
• Coloque una aguja
• Póngase la inyección diaria
• Guarde la pluma precargada
Aguja
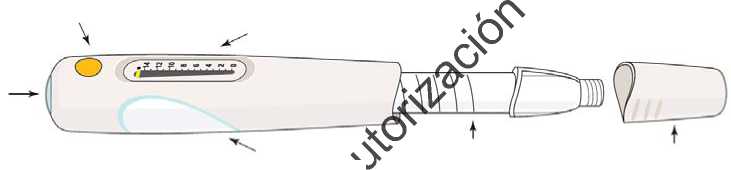
Contador de dosis
indicador de dosis
número de dosis restantes
la pluma precargada está vacía
la pluma precargada está llena
Cuando recibe su pluma precargada, el contador de dosis está situado en • p ara mostrar que está llena. Cuando el contador de dosis está a 0, la pluma precargada está vacía y debe utilizar una pluma precargada nueva.

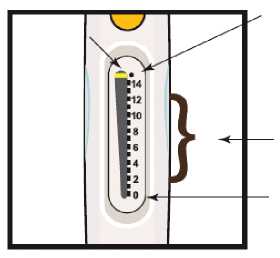
Colocación de una aguja
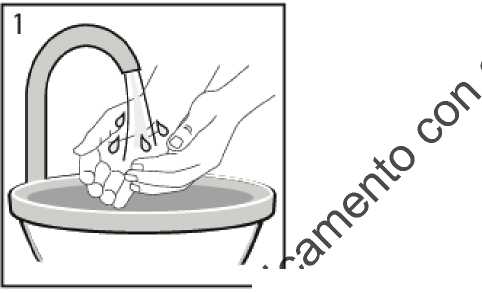
Lávese las manos con agua y jabón antes de manipular la pluma ST precargada.
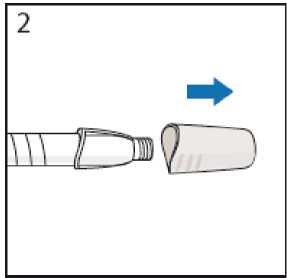
Separe la tapa del extremo frontal de la pluma precargada tirando hacia fuera.
Retire el papel protector del protector extemo de la aguja.
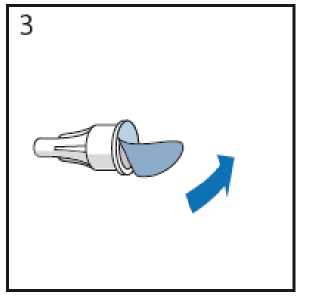
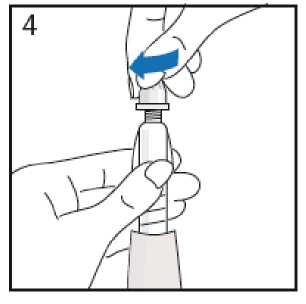
Sujete el extremo transparente de la pluma precargada y, enrosque la aguja hasta el final de ese extremo transparente de la pluma precargada.
Tenga cuidado de no pulsar el botón de liberación amarillo al manipular la pluma precargada. Si lo hace por error, el botón de inyección azul saltará.
No trate de empujarlo hacia dentro hasta que se lo indique en este manual.
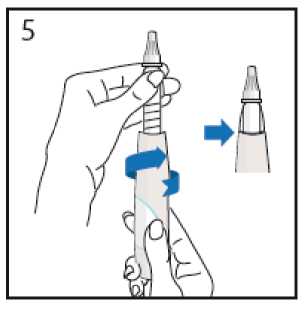
”clic”
Cómo mezclar la medicación 2
• Mueva con suavidad la pluma precargada hacia delante y hacia atrás varias veces para mezclar la medicación.
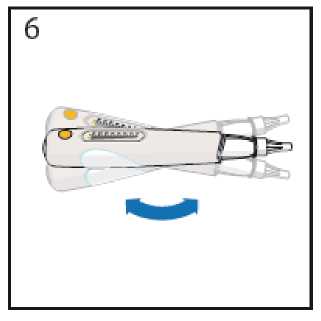
No agite la pluma precargada
• Deje que repose durante un minuto hasta que la medicación esté mezclada por completo.
• Compruebe que la medicación está transparente.

Preparación de la nueva pluma precargada para su uso' extracción del aire (purgado)
&
<&>u
íP
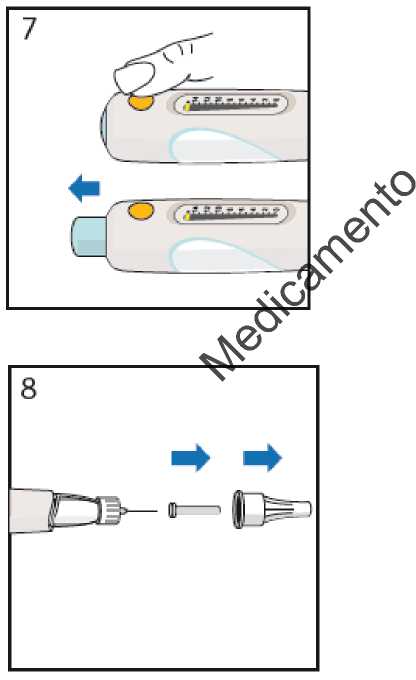
'ulse el botón de liberación amarillo para liberar el ■O2 botón de inyección azul.
Retire los dos protectores de la aguja. Guarde el protector externo de la aguja, lo necesitará para extraer la aguja después de la inyección.
Ponga la pluma apuntando con la aguja hacia arriba y pulse el botón de inyección azul hasta el final.
Oirá un ”clic“ (ver la imagen).
Esto eliminará la mayor parte del aire que haya en la pluma precargada y a esto se le denomina “purgado”.
El purgado debe realizarse cada vez que mezcle una nueva pluma precargada. Puede salir algo de medicamento, esto es normal.
Puede quedar una pequeña burbuja de aire en la pluma precargada, esto es normal.
&
«J-
■O*
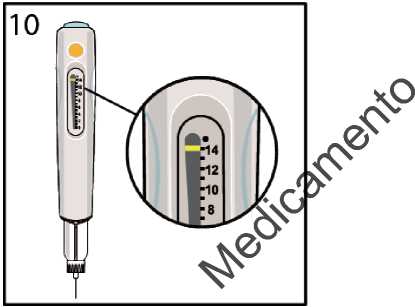
El contador de dosis ahora mostrará 14 dosis y la pluma precargada está lista para su uso. Puede optar por continuar y administrarse su dosis diaria ahora o guardar la pluma precargada en el frigorífico, según se describe en el apartado “Información práctica” al final de estas instrucciones de uso.
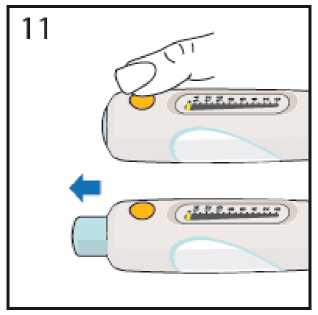
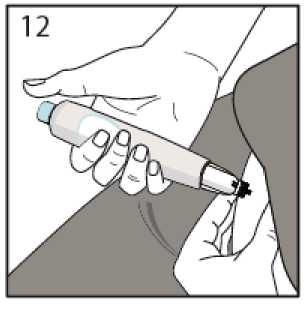
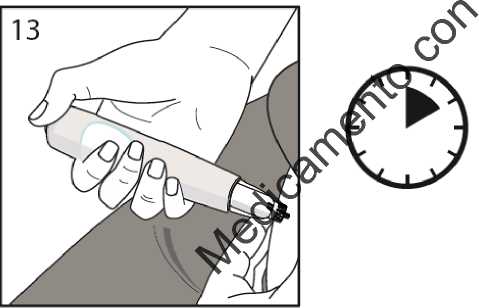
10 secs.
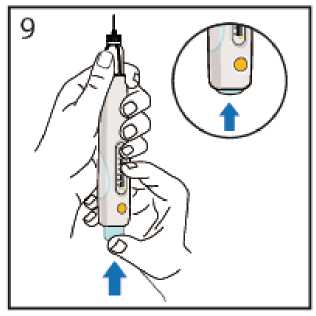
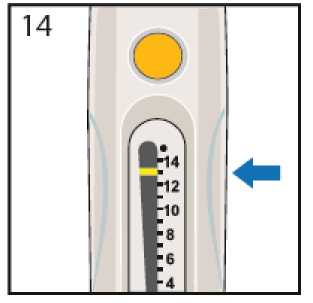
Presione el botón de inyección azul todo el recorrido hasta que se bloquee por completo - después cuente lentamente hasta 10 y retire la aguja de su piel.
10 secs. = 10 segundos
• Asegúrese de que tiene una aguja en la pluma precargada, (ver 2 imágenes 3 y 4). Si acaba de mezclar una nueva pluma precargada, puede usar la aguja ya colocada en la pluma.
• Pulse el botón de liberación amarillo para liberar el botón de inyección azul.
Coja un pellizco de piel del abdomen y adminístrese la inyección formando un ángulo de 90 grados, tal y como le ha indicado su médico o enfermera.
«j-
El contador de dosis ahora habrá bajado una posición.
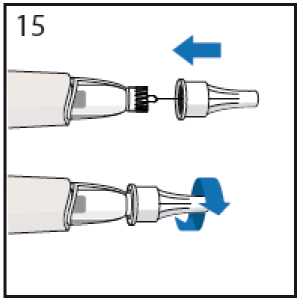
• Ponga el protector extemo de la aguja.
• Desenrosque la aguja.
• Deseche la aguja según las instrucciones de su médico o enfermera.
Use cada aguja sólo una vez
Mormajiónn
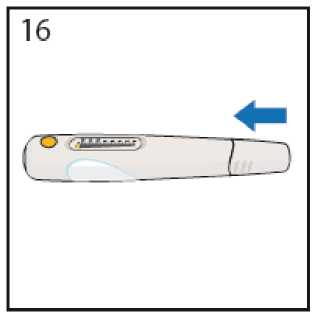
Ponga la tapa en la pluma precargada y guárdela en el frigorífico.
uáctica
La pluma precargada tiene una fecha de caducidad impresa; no use la medicación después de esa ^ 1
fecha.
La medicación no deberá usarse durante más de 28 días después de haberse mezclado.
Puede almacenar la pluma precargada sin mezclar a una temperatura entre 2 °C y 25 °C.
Retire la aguja después de cada inyección diaria y vuelva a guardar la pluma precargada en el frigorífico entre 2 °C y 8 °C.
Puede guardar la pluma precargada mezclada hasta 7 días a temperatura ambiente entre 2 °C y 25 °C.
Proteja la medicación y la pluma precargada de la luz solar directa.
No use la medicación si esta turbia o coloreada (si no está transparente). No guarde la pluma precargada con la aguja puesta.
No comparta su medicación con otras personas.
Si se le cae la pluma precargada, deberá sustituirla por otra.
Se podrá conservar la pluma prccargada mezclada durante un máximo de 28 días en el refrigerador. No use esta medicina durante más de 28 días después de que haya sido mezclada.
• Puede conservar la pluma prccargada mezclada durante 7 días fuera del refrigerador (por debajo de 25°C) durante los 28 días del período de uso.
• No utilice este medicamento ¡ si no ha sido conservado correctamente, incluso si no ha sido utilizado.
• No utilice esta medicina si nota que se ha vuelto turbia o coloreada.
Ponga la pluma con el extremo de la aguja apuntando hacia arriba.
• Enrosque la pluma precargada hasta que el extremo transparente se encuentre con el cuerpo de la pluma. Oirá y sentirá un clic al final.