Pharmagrip Duo 650 Mg/8,2 Mg Polvo Oral
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
pharmagrip dúo 650 mg/8,2 mg polvo oral pharmagrip dúo forte 1000 mg/10 mg polvo oral
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
pharmagrip dúo 650 mg/8,2 mg polvo oral
Composición por sobre:
(equivalente a 10 mg de fenilefrina hidrocloruro)
Para consultar la lista completa de excipientes, ver sección 6.1. pharmagrip dúo forte 1000 mg/10 mg polvo oral
Composición por sobre:
(equivalente a 12,2 mg de fenilefrina hidrocloruro)
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA Polvo oral.
Se trata de una mezcla de polvo homogénea de color blanco anaranjado.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Alivio sintomático de los procesos catarrales o gripales que cursen con dolor leve o moderado, fiebre y congestión nasal.
4.2 Posología y forma de administración
pharmagrip dúo 650 mg/8,2 mg polvo oral
- Adultos: Administrar 1 sobre cada 6 horas. La dosis máxima es de 6 sobres en 24 horas.
- Jóvenes de 12 a 18 años: 1 sobre cada 6 horas. La dosis máxima es de 5 sobres en 24 horas.
- Uso en niños: no está recomendado para su uso en niños menores de 12 años. No hay experiencia en niños (ver sección 4.4).
pharmagrip dúo forte 1000 mg/10 mg polvo oral
- Adultos: Administrar 1 sobre 8 horas. La dosis máxima es de 4 sobres al día.
- Uso en niños: no está recomendado para su uso en niños y jóvenes menores de 18 años. No hay experiencia en niños (ver sección 4.4 ).
Insuficiencia renal grave (CLcr < 10 ml/min):: no se necesita un ajuste de dosis unitaria, pero se
recomienda que el intervalo entre dos tomas sea como mínimo de 8 horas (ver sección 4.4).
Insuficiencia renal moderada (CLcr de 10 a 50 ml/min): no se necesita un ajuste de dosis unitaria, pero se recomienda que el intervalo entre dos tomas sea como mínimo de 6 horas (ver sección 4.4).
Insuficiencia hepática): Está contraindicado su uso (ver sección 4.)3
Mayores de 65 años: No es necesario un ajuste de dosis (ver sección 4.4.
Con la toma concomitante de paracetamol y alimentos, el tiempo de absorción de paracetamol aumenta, debido a que los alimentos disminuyen la motilidad y el tiempo de transito gastrointestinal. Para un alivio rápido del dolor, tomar el medicamento sin comida, especialmente si ésta presenta un alto contenido en carbohidratos.
VIA ORAL
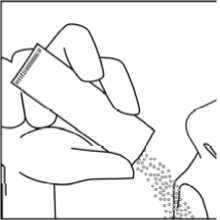
Vierta el contenido del sobre directamente en la lengua. El polvo oral se dispersa en la saliva antes de tragar por lo que no es necesaria la ingestión de líquidos en el momento de la toma.
Usar siempre la dosis menor que sea efectiva. La medicación debe iniciarse al aparecer los primeros síntomas. A medida que estos desaparezcan debe suspenderse esta medicación.
Si la fiebre persiste más de 3 días, el dolor más de 3 en niños o 5 días en adultos, o bien el dolor o la fiebre empeoran o aparecen otros síntomas, se deberá evaluar la situación clínica.
Si además hay dolor de garganta y no mejora, no se debe usar más de 2 días seguidos, sin evaluar la situación clínica.
No exceder las dosis recomendadas.
4.3 Contraindicaciones
- Hipersensibilidad a paracetamol, fenilefrina o alguno de los excipientes
- Enfermedades hepáticas (con insuficiencia hepática o sin ella) o hepatitis viral (aumenta el riesgo de hepatotoxicidad).
- Cardiopatía isquémica, enfermedad cardiovascular grave, hipertensión arterial no controlada o hipertiroidismo.
- Glaucoma
- Pacientes que estén o hayan estado en tratamiento con inhibidores de la monoaminooxidasa dentro de los 15 días anteriores a la toma de este medicamento.
4.4 Advertencias y precauciones especiales de empleo
Debido al contenido en fenilefrina se deberá administrar con precaución en las siguientes situaciones: Diabetes, síndrome de Raynaud, enfermedad cardiovascular de leve a moderada e hipertensión controlada.
Debido a su contenido en paracetamol:
la utilización de este medicamento en pacientes que consumen habitualmente alcohol (tres o más bebidas alcohólicas -cerveza, vino, licor, ...- al día) puede provocar daño hepático.
En alcohólicos crónicos, no se debe administrar más de 2 g (2 sobres) al día.
Se debe administrar con precaución, evitando tratamientos prolongados en pacientes con anemia,
afecciones cardíacas o pulmonares o con disfunción renal grave (en este último caso, el uso ocasional es aceptable, pero la administración prolongada de dosis elevadas puede aumentar el riesgo de aparición de efectos renales adversos).
Se recomienda precaución en pacientes asmáticos sensibles al ácido acetilsalicílico, debido a que se han descrito ligeras reacciones broncoespásticas con paracetamol (reacción cruzada) en estos pacientes, aunque sólo se manifestaron en menos del 5% de los ensayados.
El uso simultáneo de más de un medicamento que contiene paracetamol, puede dar lugar a cuadros de intoxicación (ver sección 4.9)
Los cuadros tóxicos asociados a paracetamol pueden producirse tanto por la ingesta de una sobredosis única o por varias tomas con dosis excesivas de paracetamol.
Se han producido comunicaciones de casos de hepatotoxicidad con dosis diarias inferiores a 4g. No se debe utilizar en caso de insuficiencia hepática crónica (ver sección 4.3).
Uso en niños: No usar pharmagrip dúo 650 mg/8,2 mg en niños menores de 12 años. No usar pharmagrip dúo forte 1000 mg/10 mg en menores de 18 años.
Uso en ancianos: Las personas ancianas pueden ser más susceptibles a padecer efectos adversos estimulantes del SNC incluso a la dosis usual para adultos.
Advertencias sobre excipientes:
Este medicamento puede ser perjudicial para personas con fenilcetonuria porque contiene aspartamo que es una fuente de fenialanina.
4.5 Interacción con otros medicamentos y otras formas de interacción
Interacciones debidas al paracetamol:
El paracetamol se metaboliza intensamente en el hígado, por lo que puede interaccionar con otros medicamentos que utilicen las mismas vías metabólicas o sean capaces de actuar, inhibiendo o induciendo, tales vías. Algunos de sus metabolitos son hepatotóxicos, por lo que la administración conjunta con potentes inductores enzimáticos (rifampicina, determinados anticonvulsivantes, etc) puede conducir a reacciones de hepatotoxicidad, especialmente cuando se emplean dosis elevadas de paracetamol.
Entre las interacciones potencialmente más relevantes pueden citarse las siguientes:
- Alcohol etílico: potenciación de la toxicidad del paracetamol, por posible inducción de la producción hepática de productos hepatotóxicos derivados del paracetamol.
- Anticoagulantes orales (acenocumarol, warfarina): posible potenciación del efecto anticoagulante, por inhibición de la síntesis hepática de factores de coagulación. No obstante, dada la aparentemente escasa relevancia clínica de esta interacción en la mayoría de los pacientes, se considera la alternativa terapéutica analgésica con salicilatos, cuando existe terapia con anticoagulantes. Sin embargo, la dosis y duración del tratamiento deben ser lo más bajo posibles, con monitorización periódica del INR.
- Anticolinérgicos (glicopirronio, propantelina): disminución en la absorción del paracetamol, con posible inhibición de su efecto, por la disminución de velocidad en el vaciado gástrico.
- Anticonceptivos hormonales/estrógenos: disminución de los niveles plasmáticos de paracetamol, con posible inhibición de su efecto, por posible inducción de su metabolismo.
- Anticonvulsivantes (fenitoína, fenobarbital, metilfenobarbital, primidona): disminución de la biodisponibilidad del paracetamol así como potenciación de la hepatotoxicidad a sobredosis, debido a la inducción del metabolismo hepático.
- Carbón activado: disminuye la absorción del paracetamol cuando se administra rápidamente tras una sobredosis
- Cloranfenicol: potenciación de la toxicidad del cloranfenicol, por posible inhibición de su metabolismo hepático.
Isoniazida: disminución del aclaramiento de paracetamol, con posible potenciación de su acción y/o toxicidad, por inhibición de su metabolismo hepático.
Lamotrigina: disminución de la biodisponibilidad de lamotrigina, con posible reducción de su efecto, por posible inducción de su metabolismo hepático.
Metoclopramida y domperidona: aumentan la absorción del paracetamol en el intestino delgado, por el efecto de estos medicamentos sobre el vaciado gástrico
Probenecid: incrementa la semivida plasmática del paracetamol, al disminuir la degradación y excreción urinaria de sus metabolitos
Propranolol: aumento de los niveles plasmáticos de paracetamol, por posible inhibición de su metabolismo hepático.
Resinas de intercambio iónico (colestiramina): disminución en la absorción del paracetamol, con posible inhibición de su efecto, por fijación del paracetamol en intestino.
Rifampicina: aumento del aclaramiento de paracetamol y formación metabolitos hepatotóxicos de éste, por posible inducción de su metabolismo hepático.
Zidovudina: aunque se han descrito una posible potenciación de la toxicidad de zidovudina (neutropenia, hepatotoxicidad) en pacientes aislados, no parece que exista ninguna interacción de carácter cinético entre ambos medicamentos.
Interacciones debidas a la fenilefrina:
Inhibidores de la monoaminooxidasa (IMAO): se debe evitar su administración simultánea o se debe separar la administración de fenilefrina un mínimo de 15 días después de interrumpir un tratamiento de este tipo (tiempo que se estima necesario para que las enzimas MAO se recuperen del efecto inhibidor que inducen los IMAO que hace que se reduzca el metabolismo de la fenilefrina), tanto antidepresivos como fenelzina, isocarboxacida, nialamida, tranilcipromina o moclobemida o para tratamiento de la enfermedad de Parkinson como selegilina, u otros como furazolidona; se pueden potenciar los efectos cardiacos y vasopresores, y el riesgo de crisis hipertensivas.
Bloqueantes alfa-adrenérgicos: no se recomienda su uso simultáneo con medicamentos con efectos similares como dihidroergotamina, metilergometrina, ergotaminas (medicamentos para la migraña), oxitocina (inductor al parto), porque se puede producir un aumento de los efectos vasoconstrictores. Además, los medicamentos alfa-bloqueantes antihipertensivos o para hiperplasia benigna de próstata, antagonizan los efectos de los alfa-receptores pero dejan los efectos mediados por los beta sin oposición, pudiendo causar un riesgo incrementado de hipotensión y taquicardia.
■ Bloqueantes beta-adrenérgicos: sus efectos terapéuticos pueden inhibirse, pudiéndose causar elevación de la tensión arterial. Asimismo, el bloqueo beta-adrenérgico puede dar lugar a actividad alfa-adrenérgica sin oposición, con riesgo de hipertensión y bradicardia excesiva.
■ Antidepresivos tricíclicos como amitriptilina, amoxapina, clomipramina, desipramina y doxepina
o tetracíclicos como maprotilina: su uso simultáneo puede potenciar los efectos presores de la fenilefrina.
■ Anestésicos volátiles, como ciclopropano o halotano: pueden aumentar el riesgo de arritmias.
■ Antihipertensivos, particularmente los que tienen relación con el sistema nervioso simpático: se requiere precaución ya que se pueden antagonizar sus efectos hipotensivos (como metildopa que es de acción central y guanetidina, antihipertensivo bloqueante adrenérgico neuronal), pudiéndose producir hipertensión grave.
■ Medicamentos que causan pérdida de potasio, como algunos diuréticos como furosemida: se puede potenciar la hipocaliemia y puede disminuir la sensibilidad arterial a los vasopresores como fenilefrina.
■ Medicamentos que afectan a la conducción cardiaca, como glucósidos cardiacos y antiarrítmicos: se requiere precaución.
■ Hormonas tiroideas: se requiere precaución.
■ Medicamentos bloqueantes de ambos receptores, alfa y beta-adrenérgicos como labetalol y carvedilol: puede haber complejas interacciones con el uso simultáneo de fenilefrina y se puede potenciar la toxicidad por producirse un antagonismo a nivel de receptores beta.
■ Atropina sulfato: bloquea la bradicardia refleja causada por fenilefrina y aumenta la respuesta presora a fenilefrina.
■ Se debe evitar la administración junto a otros simpaticomiméticos, incluso los de administración tópica (nasal u ocular), ya que se puede potenciar el efecto
Interferencias con pruebas de diagnóstico:
Debido a su contenido en paracetamol puede alterar los valores de las siguientes determinaciones analíticas:
- Sangre: aumento (biológico) de transaminasas (ALT y AST), fosfatasa alcalina, amoníaco, bilirrubina, creatinina, lactato-deshidrogenasa (LDH) y urea; aumento (interferencia analítica) de glucosa, teofilina y ácido úrico. Aumento del tiempo de protrombina (en pacientes con dosis de mantenimiento de warfarina, aunque sin significación clínica). Reducción (interferencia analítica) de glucosa cuando se utiliza el método de oxidasa-peroxidasa.
- Orina: pueden aparecer valores falsamente aumentados de metadrenalina y ácido úrico.
- Pruebas de función pancreática mediante la bentiromida: el paracetamol, como la bentiromida, se metaboliza también en forma de arilamina, por lo que aumenta la cantidad aparente de ácido paraaminobenzoico (PABA) recuperada; se recomienda interrumpir el tratamiento con paracetamol al menos tres días antes de la administración de bentiromida.
- Determinaciones del ácido 5-hidroxiindolacético (5-HIAA) en orina: en las pruebas cualitativas diagnósticas de detección que utilizan nitrosonaftol como reactivo, el paracetamol puede producir resultados falsamente positivos. Las pruebas cuantitativas no resultan alteradas.
4.6 Fertilidad, embarazo y lactancia
Los principios activos que componen: este medicamento atraviesan la barrera placentaria y mamaria. Por tanto, su uso durante el embarazo o lactancia no está recomendado.
No debe administrarse este medicamento durante el embarazo o lactancia salvo en los casos donde, a estricto juicio médico, esté justificado, debiéndose en todo caso valorar los posibles efectos beneficiosos frente a los riesgos potenciales para la madre y el feto, especialmente en el primer y tercer mes de embarazo.
Para el Paracetamol: no se han descrito problemas en humanos tras la administración del mismo pero no se han realizado estudios controlados en humanos durante el embarazo ni durante la lactancia, Aunque en la leche materna se han medido concentraciones máximas de 10 a 15 pg/ml (de 66,2 a 99,3 pmoles/l) al cabo de 1 ó 2 horas de la ingestión, por parte de la madre, de una dosis única de 650 mg de paracetamol, en la orina de los lactantes no se ha detectado paracetamol ni sus metabolitos. La vida media en la leche materna es de 1,35 a 3,5 horas. En estudios preclínicos en conejos, se ha observado la capacidad de la fenilefrina de reducir el flujo sanguíneo uterino durante el embarazo. No se han realizado estudios controlados en humanos.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se ha descrito ningún efecto en este sentido.
4.8 Reacciones adversas
Durante el periodo de utilización de paracetamol y fenilefrina se han notificado las siguientes
reacciones adversas cuya frecuencia no se ha podido establecer con exactitud:
Reacciones adversas relacionadas con el paracetamol:
- Trastornos de la sangre y del sistema linfático: trombocitopenia, leucopenia, pancitopenia, agranulocitosis , neutropenia y, anemia hemolítica, especialmente con la administración prolongada y a altas dosis.
- Trastornos respiratorios, torácicos y mediastínicos: asma y neumonía.
- Trastornos renales y urinarios: el uso excesivo de analgésicos, incluyendo el paracetamol, puede provocar nefropatía que a su vez puede evolucionar a un cuadro de insuficiencia renal grave, piuria estéril (orina turbia),efectos renales adversos .
- Trastornos de la piel y del tejido subcutáneo: prurito, exantema, dermatitis alérgica.
- Trastornos del metabolismo y de la nutrición: hipoglucemia.
- Trastornos del sistema inmunológico: edema de laringe, angioedema y reacciones anafilactoides.
- Trastornos hepatobiliares: aumento de transaminasas (ALT y AST), hepatotoxicidad (ictericia). Se puede producir hepatotoxicidad por la ingesta de 1 dosis tóxica o varias tomas de dosis excesivas de paracetamol.Muy raros: Trastornos de la piel y del tejido subcutáneo:
Se han notificado reacciones cutáneas graves.
Reacciones adversas relacionadas con la fenilefrina:
- Trastornos del sistema nervioso: inquietud, ansiedad, nerviosismo, debilidad, mareo, temblores, insomnio, irritabilidad, dolor de cabeza (con dosis altas y puede ser un síntoma de hipertensión); con dosis altas pueden producirse convulsiones, parestesias y psicosis con alucinaciones.
Trastornos vasculares: hipertensión (generalmente con dosis elevadas o en individuos susceptibles), vasoconstricción periférica con reducción del flujo de sangre a órganos vitales (los efectos vasoconstrictores pueden más probablemente suceder a pacientes hipovolémicos); frío en las extremidades, rubor, hipotensión. En uso prolongado se puede producir depleción del volumen plasmático.
- Trastornos cardiacos: dolor precordial o malestar, bradicardia grave, aumento del trabajo cardiaco por incremento de la resistencia arterial periférica que afecta especialmente a ancianos o pacientes con pobre circulación cerebral o coronaria, posible inducción o exacerbación de una insuficiencia cardiaca asociada a enfermedad cardiaca, palpitaciones (con altas dosis).
- Trastornos renales y urinarios: disminución de la perfusión renal y probablemente reducción de la cantidad de orina, retención urinaria.
- Trastornos respiratorios, torácicos y mediastínicos: disnea, distress respiratorio.
- Trastornos de la piel y del tejido subcutáneo: palidez en la piel, piloerección, sudoración incrementada.
- Trastornos gastrointestinales: vómitos (con altas dosis).
- Trastornos del metabolismo y la nutrición: hiperglucemia.
- Exploraciones complementarias: hipocaliemia, acidosis metabólica.
Reacciones adversas raras:
- Trastornos cardiacos: infarto de miocardio, arritmia ventricular.
- Trastornos respiratorios, torácicos y mediastínicos: edema pulmonar (a dosis elevadas generalmente o en individuos susceptibles).
- Trastornos vasculares: hemorragia cerebral (a dosis elevadas generalmente o en individuos susceptibles).
Notificación de sospechas de reacciones adversas:
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de medicamentos de Uso Humano: https: //www .notificaram.es.
4.9 Sobredosis
La sintomatología por sobredosis debida a paracetamol incluye confusión, excitabilidad, inquietud, nerviosismo, irritabilidad, mareos, vómitos, pérdida del apetito, ictericia, dolor abdominal e insuficiencia renal y hepática.
En los niños, estados de sopor, o alteraciones en la forma de andar.
Es especialmente importante la identificación precoz de la sobredosificación por paracetamol, debido a la gravedad del cuadro, así como, a la existencia de un posible tratamiento.
Si se ha ingerido una sobredosis de paracetamol debe tratarse rápidamente al paciente en un centro médico aunque no haya síntomas o signos significativos ya que, aunque estos pueden causar la muerte, a menudo no se manifiestan inmediatamente después de la ingestión, sino a partir del tercer día. Puede producirse la muerte por necrosis hepática. Asimismo, puede aparecer fallo renal agudo.
La sobredosis de paracetamol se evalúa en cuatro fases, que comienzan en el momento de la ingestión de la sobredosis:
FASE I (12-24 horas): náuseas, vómitos, diaforesis y anorexia.
FASE II (24-48 horas): mejoría clínica; comienzan a elevarse los niveles de AST, ALT, bilirrubina y protrombina.
FASE III (72-96 horas): pico de hepatotoxicidad; pueden aparecer valores de 20.000 para la AST. FASE IV (7-8 días): recuperación.
Puede aparecer hepatotoxicidad. La mínima dosis tóxica es 6 g en adultos y más de 100 mg/Kg de peso en niños. Dosis superiores a 20-25 g son potencialmente fatales. Los síntomas de la hepatotoxicidad incluyen náuseas, vómitos, anorexia, malestar, diaforesis, dolor abdominal y diarrea. La hepatotoxicidad no se manifiesta hasta pasadas 48-72 horas después de la ingestión. Si la dosis ingerida fue superior a 150 mg/Kg o no puede determinarse la cantidad ingerida, hay que obtener una muestra de paracetamol sérico a las 4 horas de la ingestión.
En el caso de que se produzca hepatotoxicidad, realizar un estudio de la función hepática y repetir el estudio con intervalos de 24 horas. El fallo hepático puede desencadenar encefalopatía, coma y muerte.
Niveles plasmáticos de paracetamol superiores a 300 pg/ml, encontrados a las 4 horas de la ingestión, se han asociado con el daño hepático producido en el 90% de los pacientes. Éste comienza a producirse cuando los niveles plasmáticos de paracetamol a las 4 horas son inferiores a 120 pg/ml o menores de 30
pg/ml a las 12 horas de la ingestión.
La ingestión crónica de dosis superiores a 4 g/día puede dar lugar a hepatotoxicidad transitoria. Los riñones pueden sufrir necrosis tubular, y el miocardio puede resultar lesionado.
Tratamiento: en todos los casos se procederá a aspiración y lavado gástrico, preferiblemente dentro de las 4 horas siguientes a la ingestión.
Existe un antídoto específico para la toxicidad producida por paracetamol: la N-acetilcisteína. Se recomiendan 300 mg/Kg de N-acetilcisteína (equivalentes a 1,5 ml/Kg de solución acuosa al 20%; pH: 6,5), administrados por vía I.V. durante un período de 20 horas y 15 minutos, según el siguiente esquema:
Adultos
Dosis de ataque: 150 mg/Kg (equivalentes a 0,75 ml/Kg de solución acuosa al 20% de N-acetilcisteína; pH: 6,5), lentamente por vía intravenosa o diluidos en 200 ml de dextrosa al 5%, durante 15 minutos Dosis de mantenimiento:
1.
a) Inicialmente se administrarán 50 mg/Kg (equivalentes a 0,25 ml/Kg de solución acuosa al 20% de N-acetilcisteína; pH: 6,5), en 500 ml de dextrosa al 5% en infusión lenta durante 4 horas
b) Posteriormente, se administrarán 100 mg/Kg (equivalentes a 0,50 ml/Kg de
solución acuosa al 20% de N-acetilcisteína; pH: 6,5), en 1000 ml de dextrosa al 5% en infusión lenta durante 16 horas
2. Niños
El volumen de la solución de dextrosa al 5% para la infusión debe ser ajustado en base a la edad y al peso del niño, para evitar congestión vascular pulmonar.
La efectividad del antídoto es máxima si se administra antes de que transcurran 8 horas tras la intoxicación. La efectividad disminuye progresivamente a partir de la octava hora, y es ineficaz a partir de las 15 horas de la intoxicación.
La administración de la solución acuosa de N-acetilcisteína al 20% podrá ser interrumpida cuando los resultados del examen de sangre muestren niveles hemáticos de paracetamo/ inferiores a 200 pg/ml.
En caso de convulsiones, administrar benzodiazepinas I.V, o rectal en función de la edad.
Efectos adversos de la N-acetilcisteína por vía IV: excepcionalmente, se han observado erupciones cutáneas y anafilaxia, generalmente en el intervalo entre 15 minutos y 1 hora desde el comienzo de la infusión.
Por vía oral, es preciso administrar el antídoto de N-acetilcisteína antes de que transcurran 10 horas desde la sobredosificación. La dosis de antídoto recomendada para los adultos es:
- una dosis inicial de 140 mg/Kg de peso corporal.
- 17 dosis de 70 mg/Kg de peso corporal, una cada 4 horas.
Cada dosis debe diluirse al 5% con una bebida de cola, zumo de uva, de naranja o agua, antes de ser administrada, debido a su olor desagradable y a sus propiedades irritantes o esclerosantes. Si la dosis se vomita en el plazo de una hora después de la administración, debe repetirse. Si resulta necesario, el antídoto (diluido con agua) puede administrarse mediante la intubación duodenal.
La sintomatología de sobredosis de fenilefrina produce excesiva estimulación del sistema nervioso simpático con efectos como ansiedad, temor, agitación, dolor de cabeza (puede ser síntoma de hipertensión), convulsiones, insomnio, confusión, irritabilidad, temblores, anorexia, nauseas, vómitos, psicosis con alucinaciones (más frecuentes en niños) y efectos sobre el sistema cardiovascular como hipertensión (a veces con hemorragia cerebral y edema pulmonar), arritmias, palpitaciones, vasoconstricción periférica y visceral, reducción del caudal de sangre a órganos vitales pudiendo disminuir la perfusión renal, con reducción de la producción de orina y acidosis metabólica; incremento del trabajo cardiaco por incremento de la resistencia arterial periférica; los efectos vasoconstrictores graves pueden más probablemente ocurrir en pacientes hipovolémicos, bradicardia grave. En uso prolongado se puede producir depleción del volumen plasmático.
El tratamiento de la sobredosis de fenilefrina es sintomático y de soporte.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
- Grupo farmacoterapéutico sistema respiratorio; otros preparados combinados para el resfriado.
- Código ATC: R05X.
El paracetamol es un fármaco analgésico que también posee propiedades antipiréticas. El mecanismo de la acción analgésica no está totalmente determinado. El paracetamol puede actuar predominantemente inhibiendo la síntesis de prostaglandinas a nivel del sistema nervioso central y en menor grado bloqueando la generación del impulso doloroso a nivel periférico. La acción periférica puede deberse también a la inhibición de la síntesis de prostaglandinas o a la inhibición de la síntesis o de la acción de otras sustancias que sensibilizan los nociceptores ante estímulos mecánicos o químicos.
Probablemente, el paracetamol produce el efecto antipirético actuando a nivel central sobre el centro hipotalámico regulador de la temperatura, para producir una vasodilatación periférica que da lugar a un aumento de sudoración y de flujo de sangre en la piel y pérdida de calor. La acción a nivel central probablemente está relacionada con la inhibición de la síntesis de prostaglandinas en el hipotálamo.
La fenilefrina es un simpaticomimético que actúa directamente sobre los receptores a-adrenérgicos. A dosis terapéuticas, no tiene efecto estimulante en los receptores p-adrenérgicos del corazón. Debido a su mecanismo de acción, produce constricción de los vasos sanguíneos de la mucosa nasal aliviando la congestión.
5.2 Propiedades farmacocinéticas
Por vía oral la biodisponibilidad del paracetamol es del 75-85%. Es absorbido amplia y rápidamente, las concentraciones plasmáticas máximas se alcanzan en función de la forma farmacéutica con un tiempo hasta la concentración máxima de 0,5-2 horas. La velocidad y el grado de absorción por vía rectal dependerán de la composición de la base del supositorio. El grado de unión a proteínas plasmáticas es de un 10%.
El tiempo que transcurre hasta lograr el efecto máximo es de 1 a 3 horas, y la duración de la acción es de 3 a 4 horas. El metabolismo del paracetamol experimenta un efecto de primer paso hepático, siguiendo una cinética lineal. Sin embargo, esta linealidad desaparece cuando se administran dosis superiores a 2 g. El paracetamol se metaboliza fundamentalmente en el hígado (90-95%), siendo eliminado mayoritariamente en la orina como un conjugado con el ácido glucurónico, y en menor proporción con el ácido sulfúrico y la cisteína; menos del 5% se excreta en forma inalterada. Su semivida de eliminación es de 1,5-3 horas (aumenta en caso de sobredosis y en pacientes con insuficiencia hepática, ancianos y niños). Dosis elevadas pueden saturar los mecanismos habituales de metabolización hepática, lo que hace que se utilicen vías metabólicas alternativas que dan lugar a metabolitos hepatotóxicos y posiblemente nefrotóxicos, por agotamiento de glutation.
La fenilefrina es absorbida amplia y rápidamente. A nivel de la pared intestinal, se transforma ampliamente en la forma sulfato-conjugada. A nivel del hígado y en menor proporción, se forman metabolitos por medio de la enzima monoamino oxidasa (MAO). Tras la administración oral, la descongestión nasal se produce a los 15-20 minutos y permanece durante 2-4 horas. La eliminación de fenilefrina y de sus metabolitos se produce fundamentalmente en la orina.
5.3 Datos preclínicos sobre seguridad
Los estudios de toxicidad crónica en animales demuestran que dosis elevadas de paracetamol producen atrofia testicular e inhibición de la espermatogénesis; se desconoce la importancia de este hecho para su uso en humanos.
6 . DATOS FARMACÉUTICOS 6.1 Lista de excipientes
Manitol (E- 421), ácido cítrico, ácido ascórbico, hidrógenocarbonato de sodio, aroma de naranja, etilcelulosa, aspartamo (E- 951), betacaroteno y povidona.
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
2 años
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30oC
6.5 Naturaleza y contenido del envase
Sobres monodosis.
Cada sobre está formado por tres capas unidas por termosellado que respectivamente desde el exterior al interior son: una primera capa de poliésteres termoplásticos, una segunda capa de aluminio y una tercera capa de polietileno de baja densidad.
Se presenta en cajas con 10 sobres
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local, o se procederá a su devolución a la farmacia.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
LABORATORIOS CINFA, S.A.
C/Olaz-Chipi, 10. Polígono Industrial Areta.
31620 HUARTE-PAMPLONA. (Navarra)
ESPAÑA
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN Julio 2008
10. FECHA DE LA REVISIÓN DEL TEXTO
Febrero 2016
10 de 10