Perjeta 420 Mg Concentrado Para Solucion Para Perfusion
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo información sobre su seguridad. Se invita a los profesionales reacciones adversas. Ver la sección 4.8, en la que se incluye
que agilizará la detección de nueva sanitarios a notificar las sospechas de información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Perjeta 420 mg concentrado para solución para perfusión
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un vial de 14 ml de concentrado contiene 420 mg de pertuzumab a una concentración de 30 mg/ml. Después de la dilución, un ml de solución contiene aproximadamente 3,02 mg de pertuzumab para la dosis inicial y aproximadamente 1,59 mg de pertuzumab para la dosis de mantenimiento (ver sección 6.6).
Pertuzumab es un anticuerpo monoclonal IgG1 humanizado producido en células de mamífero (ovario de hámster chino) por tecnología recombinante de ADN.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Concentrado para solución para perfusión.
Líquido de transparente a ligeramente opalescente, de incoloro a amarillo pálido.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Cáncer de mama metastásico
Perjeta está indicado en combinación con trastuzumab y docetaxel para el tratamiento de pacientes adultos con cáncer de mama HER2 positivo localmente recidivante irresecable o metastásico, que no han recibido tratamiento previo anti-HER2 o quimioterapia para la enfermedad metastásica.
Tratamiento neoadyuvante del cáncer de mama
Perjeta está indicado en combinación con trastuzumab y quimioterapia para el tratamiento neoadyuvante de pacientes adultos con cáncer de mama HER2-positivo, localmente avanzado, inflamatorio, o en estadio temprano con alto riesgo de recaída (ver sección 5.1).
4.2 Posología y forma de administración
Perjeta está sujeto a prescripción médica restringida y el tratamiento sólo debe iniciarse bajo la supervisión de un médico con experiencia en la administración de fármacos antineoplásicos. Perjeta debe ser administrado por un profesional sanitario preparado para manejar la anafilaxia y en un lugar donde se pueda disponer inmediatamente de equipos de reanimación.
Las pacientes tratadas con Perjeta deberán tener un tumor positivo para HER2, definido con una puntuación de 3+ mediante inmunohistoquímica (IHQ) y/o un cociente> 2,0 mediante hibridación in situ (HIS) determinado por un ensayo validado.
Para asegurar resultados exactos y reproducibles, la determinación debe ser realizada en un laboratorio especializado, que pueda asegurar la validación de los procedimientos de ensayo. Para unas
instrucciones completas sobre la realización e interpretación del ensayo, por favor consulte el prospecto del procedimiento de ensayo HER2 validado.
Posología
La dosis de carga inicial recomendada de Perjeta es de 840 mg, administrados en una perfusión intravenosa de 60 minutos, seguida luego cada 3 semanas de una dosis de mantenimiento de 420 mg administrada durante un periodo de 30 a 60 minutos.
Cuando se administre con Perjeta la dosis de carga inicial recomendada de trastuzumab es 8 mg/kg de peso corporal administrada en perfusión intravenosa seguida luego cada 3 semanas de una dosis de mantenimiento de 6 mg/kg de peso corporal.
Cuando se administre con Perjeta la dosis inicial recomendada de docetaxel es de 75 mg/m2, administrada en lo sucesivo en una pauta cada 3 semanas. La dosis de docetaxel puede aumentarse a 100 mg/m2 en los ciclos posteriores si la dosis inicial se tolera bien (la dosis de docetaxel no se debe aumentar cuando se utilice en combinación con carboplatino, trastuzumab y Perjeta).
Los medicamentos deben administrarse secuencialmente y no mezclarse en la misma bolsa de perfusión. Perjeta y trastuzumab se pueden dar en cualquier orden. Cuando el paciente vaya a recibir docetaxel, éste debe administrarse después de Perjeta y trastuzumab. Se recomienda un periodo de observación de 30 a 60 minutos después de cada perfusión de Perjeta y antes del comienzo de la perfusión posterior de trastuzumab o docetaxel (ver sección 4.4).
Cáncer de mama metastásico
Las pacientes deben ser tratadas con Perjeta y trastuzumab hasta que se produzca progresión de la enfermedad o toxicidad incontrolable.
Tratamiento neoadyuvante del cáncer de mama
Perjeta deber ser administrado durante 3 a 6 ciclos en combinación con trastuzumab y quimioterapia en neoadyuvancia, como parte de un régimen de tratamiento para cáncer de mama precoz. Después de la cirugía, los pacientes deben ser tratados con trastuzumab en adyuvancia para completar un año de tratamiento (ver sección 5.1).
Retrasos u omisiones de dosis
Si el intervalo entre dos perfusiones consecutivas es inferior a 6 semanas, la dosis de 420 mg de Perjeta debe administrarse lo antes posible independientemente de la siguiente dosis prevista.
Si el intervalo entre dos perfusiones consecutivas es de 6 semanas o más, se debe volver a administrar la dosis de carga inicial de 840 mg de Perjeta en una perfusión intravenosa de 60 minutos seguida cada 3 semanas en lo sucesivo de una dosis de mantenimiento de 420 mg administrada durante un periodo de 30 a 60 minutos.
Modificación de la dosis
No se recomienda reducir la dosis de Perjeta.
Las pacientes pueden continuar el tratamiento durante periodos de mielosupresión reversible inducida por quimioterapia pero deben ser vigilados estrechamente por si hay complicaciones debidas a la neutropenia durante este tiempo. Para las modificaciones de la dosis de docetaxel y otras quimioterapias, ver la correspondiente ficha técnica o resumen de las características del producto (RCP).
No se recomienda reducir la dosis de trastuzumab; ver la ficha técnica o resumen de las características del producto (RCP) de trastuzumab.
Si se interrumpe el tratamiento con trastuzumab, se debe interrumpir el tratamiento con Perjeta.
Si se interrumpe el uso de docetaxel, puede continuar el tratamiento con Perjeta y trastuzumab hasta que se produzca progresión de la enfermedad o toxicidad incontrolable en la enfermedad metastásica.
Disfunción ventricular izquierda
La administración de Perjeta y trastuzumab debe retrasarse durante al menos 3 semanas en caso de:
- signos y síntomas que sugieran insuficiencia cardíaca congestiva (Perjeta se debe interrumpir si se confirma fallo cardíaco sintomático)
- un descenso de la fracción de eyección del ventrículo izquierdo (FEVI) a menos del 40 %
- un valor de FEVI del 40-45 % asociado con un descenso >10 % puntos por debajo de los valores previos al inicio del tratamiento.
Perjeta y trastuzumab pueden reanudarse si la FEVI se ha recuperado a > 45 % o a un valor del 40-45 % asociado con <10 % por debajo del valor previo al inicio del tratamiento.
Si tras una nueva valoración en aproximadamente 3 semanas, la FEVI no ha mejorado o ha disminuido aún más, debe considerarse seriamente la interrupción de Perjeta y trastuzumab, a menos que se considere que los efectos beneficiosos para la paciente superan a los riesgos (ver sección 4.4).
Reacciones a la perfusión
Si la paciente sufre una reacción a la perfusión, se puede disminuir la velocidad de la perfusión o interrumpirse ésta (ver sección 4.8). La perfusión puede reanudarse si los síntomas disminuyen. El tratamiento con oxígeno, agonistas beta, antihistamínicos, fluidos i.v. rápidos y antipiréticos pueden también ayudar a aliviar los síntomas.
Reacciones de hipersensibilidad/anafilaxis
Debe interrumpirse de inmediato la perfusión si la paciente tiene una reacción NCI-CTCAE (anafilaxia) de grado 4, broncoespasmo o síndrome de sufrimiento respiratorio agudo (ver sección 4.4).
Pacientes de edad avanzada
Se dispone de pocos datos sobre la seguridad y eficacia de Perjeta en pacientes > 65 años de edad. No se observaron diferencias significativas en la seguridad y eficacia de Perjeta entre pacientes de edad avanzada de 65 a 75 años de edad y pacientes adultas < 65 años de edad. No es necesario ajustar la dosis en pacientes de edad avanzada > 65 años de edad. Se dispone de muy pocos datos en pacientes > 75 años de edad.
Pacientes con insuficiencia renal
No es necesario ajustar la dosis de Perjeta en pacientes con insuficiencia renal leve o moderada. No se puede hacer una recomendación de las dosis en pacientes con insuficiencia renal grave debido a que existen pocos datos de farmacocinética disponibles (ver sección 5.2).
Pacientes con insuficiencia hepática
No se ha estudiado la seguridad y la eficacia de Perjeta en pacientes con insuficiencia hepática. No se puede hacer una recomendación específica de las dosis.
Población pediátrica
No se ha establecido la seguridad ni la eficacia de Perjeta en niños y adolescentes menores de 18 años. No existe una recomendación de uso específica para Perjeta en la población pediátrica para la indicación de cáncer de mama.
Forma de administración
Perjeta se administra por vía intravenosa mediante perfusión. No debe administrarse en inyección intravenosa rápida o bolo. Para consultar las instrucciones de dilución de Perjeta antes de la administración, ver secciones 6.2 y 6.6.
El período de perfusión recomendado de la dosis inicial es de 60 minutos. Si la primera perfusión se tolera bien, las perfusiones posteriores pueden administrarse en periodos de 30 minutos a 60 minutos (ver sección 4.4).
4.3 Contraindicaciones
Hipersensibilidad a pertuzumab o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Para mejorar la trazabilidad de los medicamentos biológicos, debe consignarse (o indicarse) claramente el nombre y el número de lote del medicamento administrado en la historia clínica del paciente.
Disfunción ventricular izquierda (incluida insuficiencia cardíaca congestiva)
Se han notificado descensos de la FEVI con fármacos que antagonizan la actividad HER2, incluido Perjeta. Las pacientes que hayan recibido previamente antraciclinas o radioterapia en la región torácica pueden tener un mayor riesgo de disminución de la FEVI. En el ensayo pivotal CLEOPATRA en pacientes con cáncer de mama metastásico, Perjeta en combinación con trastuzumab y docetaxel no se asoció con una mayor incidencia de disfunción sistólica ventricular izquierda (DVI) sintomática ni con descensos de la FEVI en comparación con placebo y trastuzumab y docetaxel (ver sección 4.8).
En neoadyuvancia (NEOSPHERE) la incidencia de DVI fue mayor en los grupos tratados con Perjeta que en los que no recibieron Perjeta. También se observó un incremento en la incidencia de descensos de la FEVI en pacientes tratados con Perjeta en combinación con trastuzumab y docetaxel; la FEVI se recuperó hasta >50% en todos los pacientes.
Perjeta no se ha estudiado en pacientes con: un valor de FEVI antes del inicio del tratamiento <50 %; antecedentes de insuficiencia cardíaca congestiva (ICC); descensos de la FEVI a <50 % durante el tratamiento adyuvante previo con trastuzumab; o procesos que puedan alterar la función del ventrículo izquierdo como hipertensión no controlada, infarto de miocardio reciente, arritmia cardíaca grave que precise tratamiento o una exposición previa a antraciclinas acumulada > 360 mg/m2 de doxorubicina o su equivalente.
Hay que valorar la FEVI antes de iniciar con Perjeta y durante el tratamiento con Perjeta (cada 3 ciclos en el contexto metastásico y cada 2 ciclos en el contexto neoadyuvante) para asegurarse de que la FEVI está dentro de los límites normales del centro hospitalario. Si la FEVI es < 40 % o del 40%-45 % y asociado con > 10 % puntos por debajo del valor previo al inicio del tratamiento, debe aplazarse la administración de Perjeta y trastuzumab y realizarse una nueva valoración de la FEVI en aproximadamente 3 semanas. Si la FEVI no ha mejorado o ha descendido aún más, debe considerarse seriamente la interrupción de Perjeta y trastuzumab, a menos que se considere que los beneficios para la paciente concreta superan a los riesgos (ver sección 4.2).
Se debe considerar detenidamente el riesgo cardiaco y balancear frente a la necesidad clínica de cada paciente antes de utilizar Perjeta con una antraciclina. Hay datos de seguridad limitados disponibles del ensayo TRYPHAENA relativos a la administración secuencial o concomitante de Perjeta con epirubicina, como parte de un régimen FEC (ver secciones 4.8 y 5.1). No hay datos de seguridad disponibles relativos al uso de Perjeta con doxorubicina.
De acuerdo a la acción farmacológica de pertuzumab y de las antraciclinas, se puede esperar un aumento del riesgo de toxicidad cardiaca con el uso concomitante de estos fármacos comparado con el uso secuencial, aunque esto no se ha observado en el ensayo TRYPHAENA. En este ensayo, los pacientes fueron tratados con dosis acumulativas bajas de epirubicina, es decir, hasta 300 mg/m2, y se trataba de pacientes sin tratamiento previo con quimioterapia y que no recibieron quimioterapia adicional tras la cirugía.
Reacciones a la perfusión
Perjeta se ha asociado con reacciones a la perfusión (ver sección 4.8). Se recomienda la observación estrecha de la paciente durante la primera perfusión y en los 60 minutos siguientes a ella y durante las perfusiones posteriores de Perjeta y en los 30-60 minutos siguientes a ellas. Si se produce una reacción a la perfusión importante, se debe reducir la velocidad de perfusión o interrumpirse ésta, y administrarse el tratamiento médico apropiado. Hay que evaluar y vigilar estrechamente a las pacientes hasta la resolución completa de los signos y síntomas. Se debe considerar la interrupción permanente de Perjeta en pacientes con reacciones a la perfusión graves. Esta valoración clínica se debe basar en la gravedad de la reacción precedente y en la respuesta al tratamiento administrado para la reacción adversa (ver sección 4.2).
Reacciones de hipersensibilidad/anafilaxis
Los pacientes deben ser observados estrechamente en cuanto a reacciones de hipersensibilidad. En ensayos clínicos con Perjeta se ha observado hipersensibilidad grave, incluyendo anafilaxis (ver sección 4.8). Los medicamentos para tratar tales reacciones, así como el equipo de emergencia, deben estar disponibles para su uso inmediato. Se debe suspender permanentemente el tratamiento con Perjeta en caso de reacciones de hipersensibilidad (anafilaxia) Grado 4 NCI-CTCAE, broncoespasmo o síndrome de sufrimiento respiratorio agudo (ver sección 4.2). Perjeta está contraindicado en pacientes con hipersensibilidad conocida a pertuzumab o a cualquiera de sus excipientes (ver sección 4.3).
Neutropenia febril
Las pacientes tratadas con Perjeta, trastuzumab y docetaxel tienen mayor riesgo de neutropenia febril comparado con las pacientes tratadas con placebo, trastuzumab y docetaxel, especialmente durante los 3 primeros ciclos de tratamiento (ver sección 4.8). En el ensayo CLEOPATRA en cáncer de mama metastásico, el recuento más bajo de neutrófilos fue similar en las pacientes tratadas con Perjeta y las pacientes tratadas con placebo. La incidencia mayor de neutropenia febril en las pacientes tratadas con Perjeta se asoció a la incidencia mayor de mucositis y diarrea en estas pacientes. Se debe considerar el tratamiento sintomático para la mucositis y la diarrea. No se notificaron acontecimientos de neutropenia febril después de la suspensión del docetaxel.
Diarrea
Pertuzumab puede producir diarrea grave. En caso de aparición de diarrea grave, se debe iniciar un tratamiento antidiarreico y se debe considerar la interrupción del tratamiento con pertuzumab si no se obtiene mejoría. Cuando la diarrea esté bajo control se puede restablecer el tratamiento con pertuzumab.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han observado interacciones farmacocinéticas (FC) entre pertuzumab y trastuzumab, o entre pertuzumab y docetaxel en un subestudio en 37 pacientes del ensayo pivotal aleatorizado CLEOPATRA en cáncer de mama metastásico. Además, en el análisis FC de la población, no se ha demostrado una evidencia de interacción fármaco-fármaco entre pertuzumab y trastuzumab o entre pertuzumab y docetaxel. Esta ausencia de interacción fármaco-fármaco fue confirmada por los datos farmacocinéticos del ensayo NEOSPHERE en neoadyuvancia.
Se han evaluado en cuatro estudios los efectos de pertuzumab sobre la FC de los fármacos citotóxicos, docetaxel, gemcitabina, erlotinib y capecitabina, administrados de forma concomitante. No se observaron indicios de ninguna interacción FC entre pertuzumab y cualquiera de estos fármacos. La FC de pertuzumab en estos estudios fue similar a la observada en los estudios en monoterapia.
4.6 Fertilidad, embarazo y lactancia
Anticoncepción
Las mujeres en edad fértil deben usar métodos anticonceptivos eficaces durante el tratamiento con Perjeta y hasta 6 meses después de la última dosis de Perjeta
Embarazo
Hay datos limitados sobre el uso de pertuzumab en mujeres embarazadas.
Los estudios en animales han demostrado toxicidad reproductiva (ver sección 5.3).
Perjeta no se recomienda durante el embarazo y en mujeres en edad fértil que no usen métodos anticonceptivos.
Lactancia
Dado que la IgG humana se excreta en la leche materna y se desconoce el potencial de absorción y daño al lactante, debe decidirse si se interrumpe la lactancia o el tratamiento teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento con Perjeta para la mujer (ver sección 5.2).
Fertilidad
No se han realizado estudios específicos de fertilidad en animales para evaluar el efecto de pertuzumab. Sólo hay disponibles datos muy limitados de los estudios de toxicidad de dosis repetidas con respecto al riesgo de reacciones adversas en el sistema reproductor masculino. No se observaron reacciones adversas en hembras de monos cynomolgus sexualmente maduras que habían sido expuestas a pertuzumab.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
En base a las reacciones adversas notificadas, no se espera que Perjeta tenga influencia sobre la capacidad para conducir y utilizar máquinas. Se debe aconsejar a las pacientes que tengan reacciones a la perfusión que no conduzcan ni utilicen máquinas hasta que los síntomas desaparezcan.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Se ha evaluado la seguridad de Perjeta en más de 1.600 pacientes en los ensayos aleatorizados CLEOPATRA (n=808), NEOSPHERE (n=417) y TRYPHAENA (n=225) y en ensayos Fase I y fase II realizados en pacientes con diversas neoplasias malignas tratados predominantemente con Perjeta en combinación con otros fármacos antineoplásicos. En general, la seguridad de Perjeta en los ensayos
Fase I y Fase II fue coherente con la observada en los ensayos CLEOPATRA, NEOSPHERE y TRYPHAENA, aunque la incidencia y las reacciones adversas a medicamentos (RAMs) más frecuentes variaron en función de si Perjeta se administraba en monoterapia o de manera concomitante con fármacos antineoplásicos.
Cáncer de mama metastásico
En el ensayo clínico pivotal CLEOPATRA, 408 pacientes recibieron al menos una dosis de Perjeta en combinación con trastuzumab y docetaxel. Las RAMs más frecuentes (> 50 %) observadas con Perjeta en combinación con trastuzumab y docetaxel fueron diarrea, alopecia y neutropenia. Las RAMs de Grado 3-4 del NCI-CTCAE v.3 más frecuentes (> 10 %) fueron neutropenia, neutropenia febril y leucopenia, y las reacciones adversas más graves fueron neutropenia febril, neutropenia y diarrea. Las muertes relacionadas con el tratamiento ocurrieron en 1,2 % de las pacientes del grupo tratado con Perjeta y en 1,5 % de las pacientes del grupo tratado con placebo y fueron principalmente debido a la neutropenia febril y/o infección.
En el ensayo pivotal CLEOPATRA, después de la interrupción del tratamiento con docetaxel se notificaron RAMs con menos frecuencia. Después de la interrupción del tratamiento con docetaxel, las RAMs en el grupo tratado con Perjeta y trastuzumab ocurrieron en 10% de los pacientes a excepción de diarrea (28,1%), infección del tracto respiratorio superior (18,3%), erupción (18,3%), cefalea (17,0%), fatiga (13,4%), nasofaringitis (17,0%), astenia (13,4%), prurito (13,7%), artralgia (11,4%), náuseas (12,7%), dolor en una extremidad (13,4%), dolor de espalda (12,1%) y tos (12,1%).
Tratamiento neoadyuvante del cáncer de mama
En el ensayo en neoadyuvancia NEOSPHERE, las RAMs más frecuentes (>50%) observadas con Perjeta en combinación con trastuzumab y docetaxel fueron alopecia y neutropenia. La RAM de Grado 3-4 del NCI-CTCAE v.3 más frecuente (>10%) fue neutropenia.
En el ensayo en neoadyuvancia TRYPHAENA, cuando Perjeta se administró en combinación con trastuzumab y FEC (5-fluorouracilo, epirubicina, ciclofosfamida) durante 3 ciclos seguidos de 3 ciclos de Perjeta, trastuzumab y docetaxel, las RAMs más frecuentes (>50%) fueron neutropenia, diarrea y náuseas. Las RAMs Grado 3-4 del NCI-CTCAE v.3 más frecuentes (>10%) fueron neutropenia, neutropenia febril y leucopenia. Cuando Perjeta se administró en combinación con trastuzumab y docetaxel durante 3 ciclos seguidos de 3 ciclos de FEC (5-fluorouracilo, epirubicina, ciclofosfamida), las RAMs más frecuentes (>50%) fueron diarrea, náuseas y alopecia. Las RAMs de Grado 3-4 del NCI-CTCAE v.3 más frecuentes (>10%) fueron neutropenia y leucopenia. De manera similar, cuando Perjeta se administró en combinación con TCH (docetaxel, carboplatino y trastuzumab) durante 6 ciclos, las RAMs más frecuentes (>50%) fueron diarrea y alopecia. Las RAMs de Grado 3-4 del NCI-CTCAE v.3 más frecuentes (>10%) fueron neutropenia, neutropenia febril, anemia, leucopenia y diarrea. No se ha establecido la seguridad de Perjeta administrado durante más de 6 ciclos en neoadyuvancia.
Tabla de reacciones adversas
En la tabla 1 se resumen las RAMs del ensayo pivotal CLEOPATRA, en el que se administró Perjeta combinado con docetaxel y trastuzumab a pacientes con cáncer de mama metastásico, y de los ensayos en neoadyuvancia NEOSPHERE y TRYPHAENA, en los que se administró Perjeta combinado con trastuzumab y quimioterapia a pacientes con cáncer de mama precoz. Como Perjeta se utiliza en combinación con trastuzumab y quimioterapia, es difícil determinar la relación causal de un acontecimiento adverso con un fármaco concreto.
Las RAMs se enumeran a continuación por clase de órgano y sistema del MedDRA y por categoría de frecuencia:
Muy frecuentes (> 1/10)
Frecuentes (> 1/100 a < 1/10)
Poco frecuentes (> 1/1.000 a < 1/100)
Raras (> 1/10.000 a < 1/1.000)
Muy raras (< 1/10.000)
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
Dentro de cada grupo de frecuencia y clasificación por órgano y sistema, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 1 Resumen de las RAMs en pacientes tratados con Perjeta en enfermedad metastásica y en neoadyuvancia
|
Sistema de Clasificación de órganos |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Infecciones e infestaciones |
Infección de las vías respiratorias superiores Nasofaringitis |
Paroniquia | |
|
Trastornos de la sangre y del sistema linfático |
Neutropenia febril* Neutropenia Leucopenia Anemia | ||
|
Trastornos del sistema inmunológico |
Hipersensibilidad/reacción anafiláctica° Reacción a la perfusión/síndrome de liberación de citoquina°° | ||
|
Trastornos del metabolismo y de la nutrición |
Disminución del apetito f | ||
|
Trastornos psiquiátricos |
Insomnio | ||
|
Trastornos del sistema nervioso |
Neuropatía periférica Cefalea f Disgeusia |
Neuropatía periférica sensitiva Mareo | |
|
Trastornos oculares |
Aumento de lagrimeo | ||
|
Trastornos cardíacos |
Disfunción ventricular izquierda f (que incluye insuficiencia cardíaca congestiva)** | ||
|
Trastornos respiratorios, torácicos y mediastínicos |
Tos f |
Derrame pleural Disnea f |
Enfermedad pulmonar intersticial |
|
Trastornos gastrointestinales |
Diarrea f Vómitos f Estomatitis Náuseas f Estreñimiento f Dispepsia | ||
|
Trastornos de la piel y del tejido subcutáneo |
Alopecia Exantema f Alteraciones de las uñas |
Prurito Piel seca |
|
Sistema de Clasificación de órganos |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Mialgia Artralgia | ||
|
Trastornos generales y alteraciones en el lugar de administración |
Mucositis/Inflamación de las mucosas Dolor f Edema f Fiebre Fatiga f Astenia f |
Escalofríos |
Tabla 1 muestra el conjunto de datos de todo el periodo de tratamiento en CLEOPATRA (corte de datos 11 febrero 2014; la mediana del número de ciclos de Perjeta fue 24); y del periodo del tratamiento en neoadyuvancia en NEOSPHERE (la mediana del número de ciclos de Perjeta fue 4, en todos los grupos de tratamiento) y TRYPHAENA (la mediana del número de ciclos de Perjeta fue 3-6 en todos los grupos de tratamiento)
* Se incluyen reacciones adversas con un desenlace mortal.
** Para el periodo de tratamiento completo durante los 3 estudios.
f Excepto neutropenia febril, neutropenia, leucopenia, aumento del lagrimeo, enfermedad pulmonar intersticial, paroniquia y alopecia, todos los acontecimientos de esta tabla se notificaron también en al menos un 1 % de los pacientes que participaban en ensayos con Perjeta en monoterapia, aunque no necesariamente el investigador los consideró relacionados causalmente con Perjeta. Los acontecimientos muy frecuentes (notificados en > 10 % de las pacientes tratadas con Perjeta en monoterapia) incluidos en la tabla esta marcados con f.
° Reacción anafiláctica/hipersensibilidad se basa en un grupo de condiciones.
°° Reacción a la perfusión/sindrome de liberación de citoquina incluye un rango de condiciones distintas dentro del mismo intervalo de tiempo, ver abajo “Descripción de reacciones adversas seleccionadas”.
Descripción de reacciones adversas seleccionadas
Disfunción ventricular izquierda
En el ensayo pivotal CLEOPATRA en cáncer de mama metastásico, la incidencia de DVI durante el tratamiento de estudio fue mayor en el grupo tratado con placebo que en el grupo tratado con Perjeta (8,6% y 6,6%, respectivamente). La incidencia de DVI sintomática fue también menor en el grupo tratado con Perjeta (1,8% en el grupo tratado con placebo frente a 1,5% en el grupo tratado con Perjeta) (ver sección 4.4).
En el ensayo en neoadyuvancia NEOSPHERE, en el que los pacientes recibieron 4 ciclos de Perjeta como tratamiento neoadyuvante, la incidencia de DVI (durante todo el periodo de tratamiento) fue mayor en el grupo tratado con Perjeta, trastuzumab y docetaxel (7,5%), en comparación con el grupo tratado con trastuzumab y docetaxel (1,9%). Hubo un caso de DVI sintomática en el grupo tratado con Perjeta y trastuzumab.
En el ensayo en neoadyuvancia TRYPHAENA, la incidencia de DVI (durante todo el periodo de tratamiento) fue 8,3% en el grupo tratado con Perjeta más trastuzumab y FEC (seguido de Perjeta más trastuzumab y docetaxel); 9,3% en el grupo tratado con Perjeta más trastuzumab y docetaxel seguido de FEC; y 6,6% en el grupo tratado con Perjeta en combinación con TCH. La incidencia de DVI sintomática (insuficiencia cardiaca congestiva) fue del 1,3% en el grupo tratado con Perjeta más trastuzumab y docetaxel seguido de FEC (esto excluye a un paciente que experimentó DVI sintomática durante el tratamiento con FEC antes de recibir Perjeta más trastuzumab y docetaxel) y también del 1,3% en el grupo tratado con Perjeta en combinación con TCH. Ningún paciente del grupo tratado con Perjeta más trastuzumab y FEC seguido de Perjeta más trastuzumab y docetaxel experimentó DVI sintomática.
Reacciones a la perfusión
En el ensayo pivotal CLEOPATRA en cáncer de mama metastásico, se definió la reacción a la perfusión como cualquier acontecimiento (notificado como reacción de hipersensibilidad, anafiláctica, reacción aguda a la perfusión o síndrome de liberación de citoquina ocurrido durante una perfusión o en el mismo día que la perfusión. En el ensayo pivotal CLEOPATRA, la dosis inicial de Perjeta se administró el día antes que trastuzumab y docetaxel para permitir la revisión de las reacciones asociadas con Perjeta. En el primer día cuando sólo se administró Perjeta, la frecuencia total de reacciones a la perfusión fue del 9,8 % en el grupo tratado con placebo y del 13,2 % en el grupo tratado con Perjeta, y la mayoría de las reacciones a la perfusión fueron leves o moderadas. Las reacciones a la perfusión más frecuentes (> 1,0 %) en el grupo tratado con Perjeta fueron fiebre, escalofríos, fatiga, cefalea, astenia, hipersensibilidad y vómitos.
Durante el segundo ciclo cuando todos los fármacos se administraron en el mismo día, las reacciones a la perfusión más frecuentes (> 1,0 %) en el grupo tratado con Perjeta fueron fatiga, disgeusia, hipersensibilidad al medicamento, mialgia y vómitos (ver sección 4.4).
En los ensayos NEOSPHERE Y TRYPHAENA en neoadyuvancia, Perjeta se administró el mismo día que los otros medicamentos del ensayo en todos los ciclos. Las reacciones a la perfusión fueron coherentes con las observadas en CLEOPATRA en los ciclos en los que Perjeta se administró el mismo día que trastuzumab y docetaxel, con la mayoría de las reacciones de leves a moderadas.
Reacciones de hipersensibilidad/anafilaxis
En el ensayo pivotal CLEOPATRA en cáncer de mama metastásico, la frecuencia total de acontecimientos de hipersensibilidad/anafilaxia notificados por el investigador durante el periodo entero de tratamiento fue del 9,3 % en el grupo tratadas con placebo y del 11,3 % en el grupo tratadas con Perjeta; de ellas, el 2,5 % y el 2,0 % fueron de Grado 3-4 del NCI-CTCAE, respectivamente. En total, 2 pacientes del grupo tratado con placebo y 4 pacientes del grupo tratado con Perjeta tuvieron acontecimientos descritos como anafilaxia por el investigador (ver sección 4.4).
En general, la mayoría de las reacciones de hipersensibilidad fueron de intensidad leve o moderada y se resolvieron en el curso del tratamiento. Basándose en las modificaciones del tratamiento del estudio realizadas, la mayoría de las reacciones se determinaron como consecuentes a las perfusiones de docetaxel.
En los ensayos NEOSPHERE Y TRYPHAENA en neoadyuvancia, los acontecimientos de hipersensibilidad/anafilaxis fueron coherentes con los observados en CLEOPATRA. En NEOSPHERE, dos pacientes del grupo tratado con Perjeta y docetaxel experimentaron anafilaxis. En TRYPHAENA, la frecuencia total de hipersensiblidad/anafilaxis fue mayor en el grupo tratado con Perjeta y TCH (13,2%), de los cuales el 2,6% fueron de Grado 3-4 del NCI-CTCAE v.3.
Neutropenia febril
En el ensayo pivotal CLEOPATRA, la mayoría de las pacientes de ambos grupos de tratamiento tuvieron al menos un acontecimiento de leucopenia (63,0 % de las pacientes tratadas en el grupo de Perjeta y 58,3 % de las pacientes tratadas en el grupo del placebo), de los cuales la mayoría fueron acontecimientos de neutropenia. La neutropenia febril ocurrió en el 13,7 % de las pacientes tratadas con Perjeta y 7,6 % de las pacientes tratadas con placebo. En ambos grupos de tratamiento, el porcentaje más elevado de pacientes que tuvieron una neutropenia febril fue en el primer ciclo de tratamiento y a partir de ahí disminuyó regularmente. Se observó un aumento en la incidencia de neutropenia febril entre los pacientes asiáticos en ambos grupos de tratamiento comparados con pacientes de otras razas y de otras regiones geográficas. Entre los pacientes asiáticos, la incidencia de neutropenia febril fue mayor en el grupo tratado con Perjeta (25,8 %) que en el grupo tratado con placebo (11,3 %).
En el ensayo NEOSPHERE, el 8,4% de los pacientes tratados en neoadyuvancia con Perjeta, trastuzumab y docetaxel experimentaron neutropenia febril en comparación con el 7,5% de los pacientes que fueron tratados con trastuzumab y docetaxel. En el ensayo TRYPHAENA, se produjo neutropenia febril en un 17,1% de los pacientes tratados en neoadyuvancia con Perjeta + TCH, y en un 9,3% de los pacientes tratados en neoadyuvancia con Perjeta, trastuzumab y docetaxel seguido de FEC. En TRYPHAENA, la incidencia de neutropenia febril fue mayor en los pacientes que recibieron seis ciclos de Perjeta en comparación con los pacientes que recibieron tres ciclos de Perjeta, independientemente de la quimioterapia administrada. Al igual que en el ensayo CLEOPATRA, se observó una mayor incidencia de neutropenia y neutropenia febril entre pacientes asiáticos en comparación con otros pacientes en ambos ensayos en neoadyuvancia. En NEOSPHERE, un 8,3% de los pacientes asiáticos tratados en neoadyuvancia con Perjeta, trastuzumab y docetaxel experimentaron neutropenia febril en comparación con el 4,0% de los pacientes asiáticos tratados en neoadyuvancia con trastuzumab y docetaxel.
Diarrea
En el ensayo pivotal CLEOPATRA en cáncer de mama metastásico, la diarrea ocurrió en un 68,4% de las pacientes tratadas con Perjeta y en un 48,7 % de las pacientes tratadas con placebo. La mayoría de los acontecimientos fueron de intensidad leve a moderada y ocurrieron en los primeros ciclos de tratamiento. La incidencia de diarrea de Grado 3-4 según el NCI-CTCAE fue del 9,3 % en las pacientes tratadas con Perjeta vs. 5,1 % en las pacientes tratadas con placebo. La mediana de duración del episodio más largo de diarrea fue de 18 días en las pacientes tratadas con Perjeta y de 8 días en las pacientes tratadas con placebo. Los acontecimientos de diarrea respondieron bien al uso proactivo de fármacos antidiarreicos.
En el ensayo NEOSPHERE, la diarrea ocurrió en un 45,8% de los pacientes tratados en neoadyuvancia con Perjeta, trastuzumab y docetaxel en comparación con el 33,6% de los pacientes tratados con trastuzumab y docetaxel. En el ensayo TRYPHAENA, la diarrea ocurrió en un 72,3% de los pacientes tratados en neoadyuvancia con Perjeta+TCH y en un 61,4% de los pacientes tratados con Perjeta, trastuzumab y docetaxel en neoadyuvancia seguido de FEC. En ambos ensayos la mayoría de los acontecimientos fueron de intensidad leve a moderada.
Erupción
En el ensayo pivotal CLEOPATRA en cáncer de mama metastásico, la erupción ocurrió en un 51,7 % de las pacientes tratadas con Perjeta, comparado con un 38,9 % de las pacientes tratadas con placebo. La mayoría de los acontecimientos fueron de Grado 1 ó 2 de gravedad, ocurrieron en los 2 primeros ciclos y respondieron a los tratamientos estándar tales como tratamiento tópico u oral del acné.
En el ensayo NEOSPHERE, la erupción ocurrió en el 40,2% de los pacientes tratados en neoadyuvancia con Perjeta, trastuzumab y docetaxel en comparación con el 29,0% de los pacientes tratados con trastuzumab y docetaxel. En el ensayo TRYPHAENA, la erupción ocurrió en el 36,8% de los pacientes tratados en neoadyuvancia con Perjeta + TCH y en el 20,0% de los pacientes tratados en neoadyuvancia con Perjeta, trastuzumab y docetaxel seguido de FEC. La incidencia de erupción fue mayor en pacientes que recibieron seis ciclos de Perjeta en comparación con pacientes que recibieron tres ciclos de Perjeta, independientemente de la quimioterapia administrada.
Anomalías analíticas
En el ensayo pivotal CLEOPATRA en cáncer de mama metastásico, la incidencia de neutropenia de Grado 3-4 del NCI-CTCAE v.3 fue similar en los dos grupos de tratamiento (86,3 % de las pacientes tratadas con Perjeta y 86,6 % de las pacientes tratadas con placebo, que incluyeron un 60,7 % y
64,8 % de neutropenia de Grado 4, respectivamente).
En el ensayo NEOSPHERE, la incidencia de neutropenia de Grado 3-4 del NCI-CTCAE v.3 fue del 74,5% en pacientes tratados en neoadyuvancia con Perjeta, trastuzumab y docetaxel en comparación con el 84,5% de los pacientes tratados con trastuzumab y docetaxel, incluyendo un 50,9% y 60,2% de neutropenia de Grado 4, respectivamente. En el ensayo TRYPHAENA, la incidencia de neutropenia de Grado 3-4 del NCI-CTCAE v.3 fue del 85,3% en pacientes tratados en neoadyuvancia con Perjeta + TCH y del 77,0% en pacientes tratados en neoadyuvancia con Perjeta, trastuzumab y docetaxel seguido de FEC, incluyendo un 66,7% y 59,5% de neutropenia de Grado 4, respectivamente.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V
Sobredosis
4.9
No se ha determinado la dosis máxima tolerada de Perjeta. En los ensayos clínicos no se han evaluado dosis únicas superiores a 25 mg/kg (1.727 mg).
En caso de sobredosis, deberá vigilarse estrechamente a las pacientes en busca de signos o síntomas de reacciones adversas e instaurarse el tratamiento sintomático apropiado.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: agentes antineoplásicos, anticuerpos monoclonales, código ATC: L01XC13. Mecanismo de acción
Perjeta es un anticuerpo monoclonal humanizado recombinante dirigido específicamente contra el dominio de dimerización extracelular (subdominio II) de la proteína receptor 2 del factor de crecimiento epidérmico humano (HER2), por lo que bloquea la heterodimerización dependiente de ligando de HER2 con otros miembros de la familia HER, como EGFR, HER3 y HER4. Como resultado, Perjeta inhibe la señalización intracelular iniciada por ligando a través de dos vías de señalización importantes, la proteincinasa activada por mitógenos (MAP) y la fosfoinositido 3-cinasa (PI3K). La inhibición de estas vías de señalización puede originar detención del crecimiento y apoptosis de las células, respectivamente. Además, Perjeta media en la citotoxicidad celular dependiente de anticuerpos (CCDA).
Aunque Perjeta administrado en monoterapia, inhibía la proliferación de células tumorales humanas, la combinación de Perjeta y trastuzumab potenciaba notablemente la actividad antitumoral en modelos de xenoinjerto que sobreexpresaban HER2.
Eficacia clínica y seguridad
La eficacia de Perjeta en el cáncer de mama HER2-positivo está avalada por un ensayo comparativo de fase III aleatorizado en cáncer de mama metastásico y por dos ensayos de fase II (un ensayo de un solo grupo en cáncer de mama metastásico y un ensayo aleatorizado comparativo en un contexto neoadyuvante).
Cáncer de mama metastásico
Perjeta combinado con trastuzumab y docetaxel
CLEOPATRA (WO20698) es un ensayo clínico de fase III multicéntrico, aleatorizado, doble ciego y controlado con placebo realizado en 808 pacientes con cáncer de mama localmente recidivante irresecable o metastásico HER2 positivo. Las pacientes con factores de riesgo cardíacos de importancia clínica no se incluyeron (ver sección 4.4). Debido a la exclusión de pacientes con metástasis cerebrales no existen datos disponibles de la acción de Perjeta sobre las metástasis cerebrales. Hay datos muy limitados disponibles en pacientes con enfermedad localmente recidivante irresecable. Las pacientes fueron aleatorizadas 1:1 para recibir placebo + trastuzumab + docetaxel o Perjeta + trastuzumab + docetaxel.
Se dio una dosis estándar de Perjeta y trastuzumab en una pauta cada 3 semanas. Se trató a las pacientes con Perjeta y trastuzumab hasta que se producía progresión de la enfermedad, retirada del consentimiento o toxicidad incontrolable. El docetaxel se administró en una dosis inicial de 75 mg/m2 en perfusión intravenosa cada tres semanas durante al menos 6 ciclos. La dosis de docetaxel podía aumentarse a 100 mg/m2 a criterio del investigador si la dosis inicial se toleraba bien.
La variable principal del estudio fue la supervivencia libre de progresión (SLP), valorada por un centro de revisión independiente (CRI) y definida como el tiempo desde la fecha de aleatorización hasta la fecha de progresión de la enfermedad o de fallecimiento (por cualquier causa) si la muerte se producía en las 18 semanas siguientes a la última valoración del tumor. Las variables de eficacia secundarias fueron supervivencia global (SG), SLP (evaluada por el investigador), tasa de respuesta objetiva (TRO), duración de la respuesta y tiempo hasta progresión de los síntomas según el cuestionario FACT B Calidad de Vida.
Alrededor de la mitad de las pacientes de cada grupo de tratamiento tenían enfermedad con receptores hormonales positivos (es decir, con receptores de estrógenos (ER) positivos y/o receptores de progesterona (PgR) positivos) y alrededor de la mitad de las pacientes de cada grupo de tratamiento habían recibido tratamiento adyuvante o neoadyuvante previo. La mayoría de las pacientes habían recibido previamente tratamiento con antraciclinas y un 11 % de todas las pacientes habían recibido previamente trastuzumab. Un total de 43 % de pacientes de ambos grupos de tratamiento habían recibido previamente radioterapia. La mediana de la FEVI de los pacientes al inicio fue de 65,0% (rango 50 % - 88 %) en ambos grupos.
Los resultados de eficacia del ensayo CLEOPATRA están resumidos en la tabla 2. Se demostró una mejoría estadísticamente significativa de la SLP valorada por el CRI en el grupo tratado con Peijeta comparado con el grupo tratado con placebo. Los resultados de la SLP valorada por el investigador fueron similares a los de la SLP valorada por el CRI.
Tabla 2. Resumen de la eficacia del ensayo CLEOPATRA
|
Parámetro |
Placebo + trastuzumab + docetaxel n=406 |
Perjeta + trastuzumab + docetaxel n=402 |
HR (IC del 95 %) |
Valor de p |
|
Supervivencia libre de progresión (revisión independiente) - variable principal* N° de pacientes con un evento Mediana, meses |
242 (59 %) 12,4 |
191 (47,5 %) 18,5 |
0,62 [0,51;0,75] |
<0,0001 |
|
Supervivencia global - variable secundaria** N° de pacientes con un evento Mediana, meses |
221 (54,4%) 40,8 |
168 (41,8%) 56,5 |
0,68 [0,56;0,84] |
0,0002 |
|
Tasa de respuesta objetiva (TRO)A -variable secundaria N° de pacientes con enfermedad medible Pacientes con respuesta*** IC del 95 % para la TRO Respuesta completa (RC) Respuesta parcial (RP) Enfermedad estable (EE) Progresión de la enfermedad (PE) |
336 233 (69,3 %) [64,1; 74,2] 14 (4,2 %) 219 (65,2 %) 70 (20,8 %) 28 (8,3 %) |
343 275 (80,2 %) [75,6; 84,3] 19 (5,5 %) 256 (74,6 %) 50 (14,6 %) 13 (3,8 %) |
Diferencia en TRO 10,8 % [4,2;17,5]% |
0,0011 |
|
Duración de la respuestafA n= Mediana, semanas IC del 95 % para la mediana |
233 54,1 [46;64] |
275 87,6 [71;106] |
* Análisis primario de supervivencia libre de progresión, fecha de corte 13 de mayo de 2011. ** Análisis final de supervivencia global, fecha de corte 11 de febrero de 2014.
*** Pacientes con una mejor respuesta global de RC o RP confirmadas según RECIST. f Valorada en las pacientes con mejor respuesta global de RC o RP
A La tasa de respuesta objetiva y la duración de la respuesta se basan en las valoraciones del tumor por el CRI
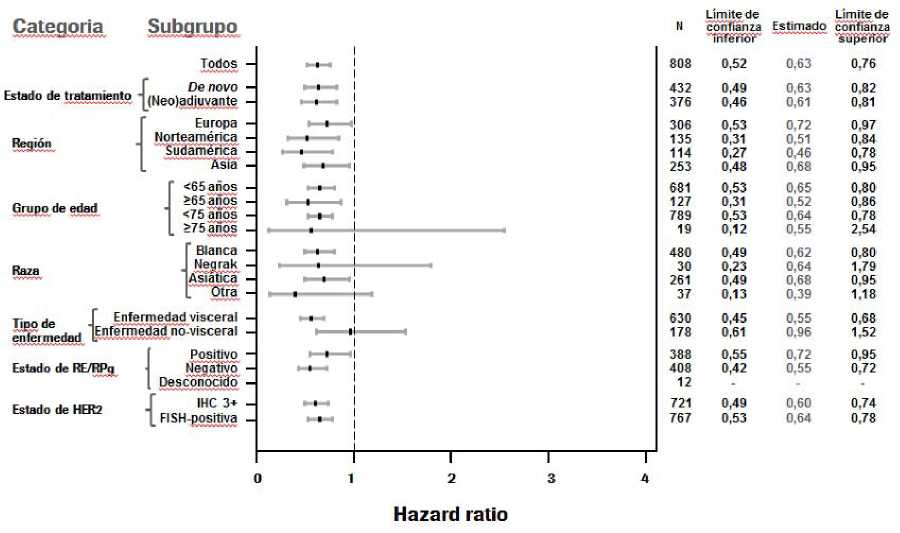
Se observaron resultados constantes en todos los subgrupos de pacientes preestablecidos, incluidos los subgrupos basados en los factores de estratificación de la región geográfica y el tratamiento adyuvante/neoadyuvante previo o de novo del cáncer de mama metastásico (ver Figura 1). Un análisis exploratorio adicional posterior mostró que en los pacientes que habían recibido trastuzumab previamente (n=88), el hazard ratio para la SLP valorada por el CRI fue de 0,62 (IC 95% 0,35; 1,07) comparado con 0,60 (IC 95% 0,43; 0,83) en los pacientes que habían recibido tratamiento previo que no incluía trastuzumab (n=288).
Figura 1 SLP valorada por el CRI por subgrupo de pacientes
El análisis final de SG se realizó cuando habían fallecido 389 pacientes (221 en el grupo tratado con placebo y 168 en el grupo tratado con Perjeta). Se mantuvo el beneficio en la SG estadísticamente significativo a favor del grupo tratado con Perjeta (HR 0,68; p=0,0002 test log-rank), previamente observado en un análisis intermedio de SG (realizado un años después del análisis primario). El tiempo medio hasta la muerte fue 40,8 meses en el grupo tratado con placebo y 56,5 meses en el grupo tratado con Perjeta (ver Tabla 2, Figura 2).
Figura 2 Curva de Kaplan-Meier de supervivencia global
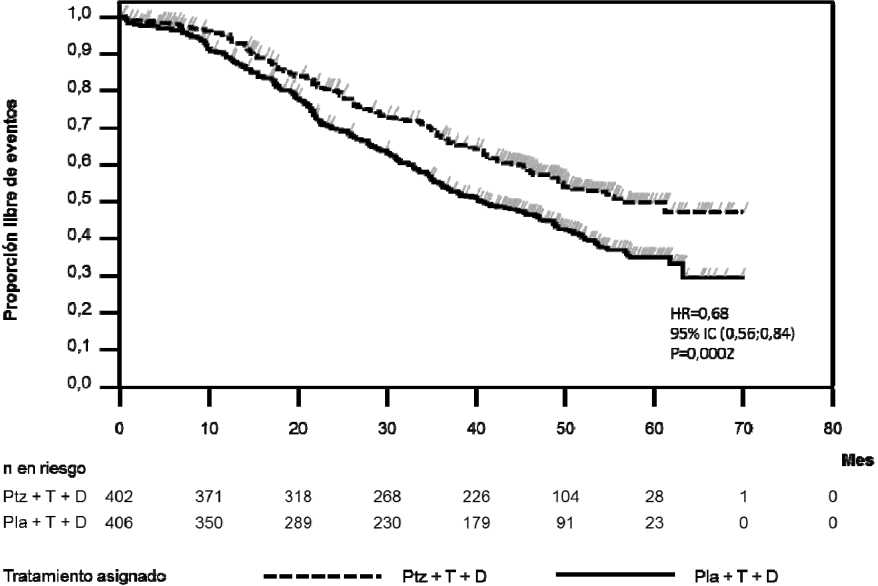
HR= hazard ratio; IC= intervalo de confianza; Pla= placebo; Ptz= pertuzumab (Perjeta); T= trastuzumab
(Herceptin); D= docetaxel.
No se hallaron diferencias estadísticamente significativas entre los dos grupos de tratamiento en la Calidad de Vida relacionada con la Salud valorada mediante las puntuaciones TOI-SFB del FACT-B.
Información adicional de apoyo procedente de ensayos clínicos
BO17929 - ensayo de un solo grupo en cáncer de mama metastásico
El BO17929 fue un ensayo de fase II, no aleatorizado realizado en pacientes con cáncer de mama metastásico cuyos tumores habían progresado durante el tratamiento con trastuzumab. El tratamiento con Perjeta y trastuzumab tuvo una tasa de respuesta del 24,2 %, y además un 25,8% de las pacientes experimentaron una estabilización de la enfermedad que duró al menos 6 meses, indicando que Perjeta es activo tras la progresión a trastuzumab.
Tratamiento neoadyuvante del cáncer de mama
En neoadyuvancia, el cáncer de mama localmente avanzado e inflamatorio son considerados de alto riesgo independientemente del estado del receptor hormonal. En el cáncer de mama en estadio temprano, el tamaño del tumor, el grado, el estado del receptor hormonal y la metástasis en los ganglios linfáticos deben tenerse en cuenta en la evaluación de riesgos.
La indicación del tratamiento neoadyuvante del cáncer de mama se basa en la demostración de una mejora en la tasa de respuesta patológica completa, y las tendencias de mejora en la supervivencia libre de enfermedad que, sin embargo, no establece o mide con precisión un beneficio con respecto a los resultados a largo plazo, tales como la supervivencia global o la supervivencia libre de enfermedad.
NEOSPHERE (WO20697)
NEOSPHERE es un ensayo de fase II multicéntrico, internacional, aleatorizado, controlado con Perjeta y se realizó en 417 pacientes mujeres adultas con cáncer de mama localmente avanzado, inflamatorio o precoz HER2 positivo diagnosticado de novo (T2-4d; tumor primario 2cm de diámetro) que no habían recibido tratamiento previo con trastuzumab, quimioterapia o radioterapia.
Las pacientes con metástasis, cáncer de mama bilateral, factores de riesgo cardíacos clínicamente importantes (ver sección 4.4) o FEVI 55% no estaban incluidas. La mayoría de las pacientes eran menores de 65 años.
Las pacientes fueron aleatorizadas para recibir uno de los siguientes tratamientos en neoadyuvancia durante 4 ciclos antes de la cirugía:
• Trastuzumab más docetaxel
• Perjeta más trastuzumab y docetaxel
• Perjeta más trastuzumab
• Perjeta más docetaxel
La aleatorización se estratificó por tipo de cáncer de mama (operable, localmente avanzado o inflamatorio) y por positividad ER o PgR.
Perjeta fue administrado por vía intravenosa con una dosis inicial de 840mg, seguida de 420 mg cada tres semanas. Trastuzumab fue administrado por vía intravenosa con una dosis inicial de 8 mg/kg, seguida de 6 mg/kg cada tres semanas. Docetaxel fue administrado por vía intravenosa con una dosis inicial de 75 mg/m2 seguida de 75 mg/m2 o 100 mg/m2 (si es tolerada) cada 3 semanas. Después de la cirugía, todas las pacientes recibieron 3 ciclos de 5-fluorouracilo (600 mg/m2), epirubicina (90 mg/m2), ciclofosfamida (600 mg/m2) (FEC) administrados por vía intravenosa cada tres semanas, y trastuzumab administrado por vía intravenosa cada tres semanas hasta completar un año de tratamiento. Los pacientes que sólo recibieron Perjeta más trastuzumab antes de la cirugía, posteriormente recibieron tanto FEC como docetaxel tras la cirugía.
La variable principal del estudio fue la tasa de respuesta patológica completa (RpC) en mama (ypT0/is). Las variables secundarias de eficacia fueron la tasa de respuesta clínica, la tasa de cirugía conservadora de la mama (sólo tumores T2-3), la supervivencia libre de enfermedad (SLE), ySLP. Tasas de RpC exploratorias adicionales incluyeron el estado ganglionar (ypT0/isN0 y ypT0N0).
Los datos demográficos estaban bien equilibrados (la edad media era 49-50 años, la mayoría eran caucásicas (71%) y todas las pacientes eran mujeres. En general, el 7% de las pacientes tenía cáncer de mama inflamatorio, el 32% tenía cáncer de mama localmente avanzado y el 61% tenía cáncer de mama operable. Aproximadamente la mitad de las pacientes en cada grupo de tratamiento presentaba receptor hormonal positivo (definido como ER positivo y/o PgR positivo).
Los resultados de eficacia se muestran en la Tabla 3. Se observó una mejoría estadísticamente significativa de la tasa de RpC (ypT0/is) en pacientes que recibieron Perjeta más trastuzumab y docetaxel en comparación con pacientes que recibieron trastuzumab y docetaxel (45,8% frente a 29,0%, valor de p=0.0141). Se observó un patrón consistente de los resultados independientemente de la definición de RpC. Se considera probable que la diferencia en la tasa de RpC se traduzca en una diferencia clínicamente significativa en los resultados a largo plazo y está apoyado por la tendencia positiva en SLP (HR 0,69, IC del 95% 0,34, 1,40) y SLE (HR 0,60, IC del 95% 0,28, 1,27).
Las tasas de RpC, así como la magnitud del beneficio con Perjeta (Perjeta más trastuzumab y docetaxel comparado con pacientes que recibían trastuzumab y docetaxel), fueron inferiores en el subgrupo de pacientes con tumores con receptores hormonales positivos (diferencia del 6% en RpC en mama) que en los pacientes con tumores con receptores hormonales negativos (diferencia del 26,4% en RpC en mama). Las tasas de RpC fueron similares en las pacientes con enfermedad operable que en las que tenían enfermedad localmente avanzada. Hubo muy pocas pacientes con cáncer de mama inflamatorio para establecer conclusiones firmes, pero la tasa de RpC fue mayor en las pacientes que recibieron Perjeta más trastuzumab y docetaxel.
TRYPHAENA (BO22280)
TRYPHAENA es un ensayo clínico de fase II multicéntrico, aleatorizado, realizado en 225 pacientes mujeres adultas con cáncer de mama HER2-positivo localmente avanzado, operable o inflamatorio (T2-4d; tumor primario 2cm de diámetro) que anteriormente no hubieran recibido trastuzumab, quimioterapia o radioterapia. Las pacientes con metástasis, cáncer de mama bilateral, factores de riesgo cardíacos clínicamente importantes (ver sección 4.4) o FEVI 55% no estaban incluidas. La mayoría de las pacientes eran menores de 65 años. Las pacientes fueron aleatorizadas para recibir uno de los tres siguientes tratamientos en neoadyuvancia antes de la cirugía:
• 3 ciclos de FEC seguidos de 3 ciclos de docetaxel, todos administrados simultáneamente con Perjeta y trastuzumab
• 3 ciclos de FEC solo seguidos de 3 ciclos de docetaxel, con trastuzumab y Perjeta administrados simultáneamente
• 6 ciclos de THC en combinación con Perjeta.
La aleatorización se estratificó por tipo de cáncer de mama (operable, localmente avanzado o inflamatorio) y por positividad ER y/o PgR.
Perjeta fue administrado por vía intravenosa con una dosis inicial de 840mg, seguida de 420 mg cada tres semanas. Trastuzumab fue administrado por vía intravenosa con una dosis inicial de 8 mg/kg, seguida de 6 mg/kg cada tres semanas. FEC (5-fluorouracilo [500mg/m2], epirubicina [100mg/m2], ciclofosfamida [600mg/m2] fueron administrados por vía intravenosa cada tres semanas durante 3 ciclos. Docetaxel fue administrado con una dosis inicial de 75 mg/m2 en perfusión intravenosa cada tres semanas, con la opción de aumentar a 100 mg/m2 a elección del investigador si la dosis inicial era bien tolerada. Sin embargo, en el grupo tratado con Perjeta en combinación con TCH, docetaxel fue administrado por vía intravenosa en 75 mg/m2 (no se permitió aumento) y carboplatino (AUC 6) fue administrado por vía intravenosa cada tres semanas. Después de la cirugía, todos los pacientes recibieron trastuzumab para completar un año de tratamiento.
La variable principal de este estudio fue la seguridad cardíaca durante el periodo del tratamiento neoadyuvante del estudio. Las variables secundarias de eficacia fueron la tasa de RpC en mama (ypT0/is), SLE, SLP y SG.
Los datos demográficos estaban bien equilibrados entre los grupos (la edad media era 49-50 años, la mayoría eran caucásicas [77%]) y todas las pacientes eran mujeres. En general, el 6% de las pacientes tenía cáncer de mama inflamatorio, el 25% tenía cáncer de mama localmente avanzado y el 69% tenía cáncer de mama operable. Aproximadamente la mitad de las pacientes en cada grupo de tratamiento tenía la enfermedad con ER positivo y/o PgR positivo.
En comparación con datos publicados de regímenes similares sin pertuzumab, se observó un aumento de las tasas de RpC en los 3 grupos de tratamiento (ver tabla 3). Se observó un patrón consistente de los resultados independientemente de la definición de RpC. Las tasas de RpC fueron inferiores en el subgrupo de pacientes con tumores con receptores hormonales positivos (rango 46,2% a 50,0%) que en las pacientes con tumores con receptores hormonales negativos (rango 65,0% a 83,8%).
Las tasas de RpC fueron similares en las pacientes con enfermedad operable que en las que tenían enfermedad localmente avanzada. Hubo muy pocas pacientes con cáncer de mama inflamatorio para establecer conclusiones firmes.
Tabla 3 NEOSPHERE (WO20697) y THYPHAENA (BO22280): Datos de eficacia (población por intención de tratar)__
|
NEOSPHERE (WO20697) |
TRYPHAENA (BO22280) | ||||||
|
Parámetro |
Trastuzum ab +Docetaxel N=107 |
Perjeta+ Trastuzuma b+ Docetaxel N=107 |
Perjeta+ Trastuzuma b N=107 |
Perjeta +Docetaxel N=96 |
Perjeta+ Trastuzumab+ FEC^ Perjeta+ Trastuzumab + Docetaxel N=73 |
FEC^ Perjeta+ Trastuzumab + Docetaxel N=75 |
Perjeta +TCH N=77 |
|
Tasa de RpC en la mama (ypT0/is) n (%) [IC del 95%]1 |
31 (29,0%) [20,6; 38,5] |
49 (45,8%) [36,1; 55,7] |
18 (16,8%) [10,3; 25,3] |
23 (24,0%) [15,8; 33,7] |
45 (61,6%) [49,5; 72,8] |
43 (57,3%) [45,4; 68,7] |
51 (66,2%) [54,6; 76,6] |
|
Diferencia en tasas de RpC2 [IC del 95%]3 |
+16,8 % [3,5; 30,1] |
-12,2 % [-23,8; -0,5] |
-21,8 % [-35,1; -8,5] |
NA |
NA |
NA | |
|
Valor de p (con correl. de Simes para la prueba CMH)4 |
0,0141 (vs. Trastuzumab +Docetaxel) |
0,0198 (vs. Trastuzumab +Docetaxel) |
0,0030 (vs Perjeta+ Trastuzumab+ Docetaxel) |
NA |
NA |
NA | |
|
Tasa de RpC en la mama y ganglio linfático (ypT0/is N0) n (%) [IC del 95%] |
23 (21,5%) [14,1; 30.5] |
42 (39,3%) [30,3; 49,2] |
12 (11,2%) [5,9; 18,8] |
17 (17,7%) [10,7; 26,8] |
41 (56,2%) [44,1; 67,8] |
41 (54,7%) [42,7; 66,2] |
49 (63,6%) [51,9; 74,3] |
|
ypT0 N0 n (%) [IC del 95%] |
13 (12,1%) [6.6; 19,9] |
35 (32,7%) [24,0; 42,5] |
6 (5,6%) [2,1; 11,8] |
13 (13,2%) [7,4; 22,0] |
37 (50,7%) [38,7; 62,6] |
34 (45,3%) [33,8; 57,3] |
40 (51,9%) [40,3; 63,5] |
|
NEOSPHERE (WO20697) |
TRYPHAENA (BO22280) | ||||||
|
Parámetro |
Trastuzum ab +Docetaxel N=107 |
Perjeta+ Trastuzuma b+ Docetaxel N=107 |
Perjeta+ Trastuzuma b N=107 |
Perjeta +Docetaxel N=96 |
Perjeta+ Trastuzumab+ FEC^ Perjeta+ Trastuzumab + Docetaxel N=73 |
FEC^ Perjeta+ Trastuzumab + Docetaxel N=75 |
Perjeta +TCH N=77 |
|
Respuesta clínica |
79 (79,8%) |
89 (88,1%) |
69 (67,6%) |
65 (71,4%) |
67 (91,8%) |
71 (94,7%) |
69 (89,6%) |
FEC: 5-fluorouracilo, epirubicina, ciclofosfamida; TCH: docetaxel, carboplatino y trastuzumab, CMH: Cochran-Mantel-Haenszel
1. IC del 95% para una distribución binomial de la muestra utilizando el método de Pearson Clopper.
2. Tratamiento Perjeta+Trastuzumab+Docetaxel y Perjeta+Trastuzumab son comparados con Trastuzumab+Docetaxel mientras que Perjeta+Docetaxel es comparado con Perjeta+Trastuzumab+Docetaxel.
3. IC del 95% aproximado para la diferencia entre dos tasas de respuesta utilizando el método de Hauck-Anderson.
4. Valor de la p de la prueba de Cochran-Mantel-Haenszel con ajuste por multiplicidad de Simes.
5. La respuesta clínica representa a las pacientes con una mejor respuesta general de CR o PR durante el período neoadyuvante (en lesiones de mama primarias).
Inmunogenicidad
Las pacientes del ensayo pivotal CLEOPATRA se sometieron a análisis de anticuerpos antiterapéuticos (ATA) frente a Perjeta en varios momentos. Aproximadamente el 2,8 % (11/386 pacientes) de las pacientes tratadas con Perjeta y el 6,2 % (23/372 pacientes) de las pacientes tratadas con placebo dieron positivo para ATA. De estas 34 pacientes, ninguna tuvo reacciones graves (Grado 4 NCI-CTCAE) a la perfusión o reacciones de hipersensibilidad (anafilaxia) claramente relacionadas con ATA. Sin embargo, las reacciones de hipersensibilidad de Grado 3 relacionadas con ATA detectables ocurrieron en 2 de 366 pacientes tratadas con Perjeta (0,5 %) en ensayos de fase I y II. Actualmente no existen datos suficientes para evaluar los efectos de ATA en la eficacia de Perjeta en combinación con trastuzumab y docetaxel.
Población pediátrica
La Agencia Europea del Medicamento ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Perjeta en los diferentes grupos de la población pediátrica en cáncer de mama (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 Propiedades farmacocinéticas
El análisis de farmacocinética poblacional se realizó con datos de 481 pacientes de diferentes ensayos clínicos (fase I, II y III) con varios tipos de tumores avanzados que habían recibido Perjeta en monoterapia o en combinación a dosis con un rango entre 2 y 25 mg/kg administrados en perfusión intravenosa de 30-60 minutos cada 3 semanas.
Absorción
Perjeta se administra en perfusión intravenosa. No se han realizado estudios en otras vías de administración.
Distribución
Entre todos los ensayos clínicos, el volumen de distribución del compartimento central (Vc) y periférico (Vp) de un paciente típico, fue de 3,11 litros y de 2,46 litros, respectivamente.
Biotransformación
El metabolismo de Perjeta no se ha estudiado directamente. Los anticuerpos se eliminan principalmente por catabolismo.
Eliminación
La mediana del aclaramiento (CL) de Perjeta fue de 0,235 litros/día y la mediana de la semivida fue de 18 días.
Linealidad/No linealidad
Se obtuvo una farmacocinética lineal con Perjeta dentro del rango de dosis recomendado.
Pacientes de edad avanzada
En base al análisis de farmacocinética poblacional, no se observó una diferencia significativa de la farmacocinética de Perjeta entre los pacientes < 65 años (n=306) y los pacientes > 65 años (n=175).
Pacientes con insuficiencia renal
No se ha realizado un ensayo específico de Perjeta en insuficiencia renal. En base a los resultados del análisis de farmacocinética poblacional, la exposición de Perjeta en pacientes con insuficiencia renal leve (aclaramiento de creatinina [CLcr] 60 a 90 ml/min, n=200) y con insuficiencia renal moderada (CLcr 30 a 60 ml/min, n=71) fue similar a la de los pacientes con función renal normal (CLcr mayor de 90 ml/min, n=200). No se observó relación entre el CLcr y la exposición de Perjeta en el rango de CLcr (27 a 244 ml/min).
Otras poblaciones especiales
El análisis de FC poblacional no indicó diferencias FC basadas en la edad, el sexo y la raza (japoneses frente a no japoneses). La albúmina basal y el peso corporal magro fueron las covariables más importantes que influían en el CL. El CL descendió en pacientes con concentraciones de albúmina basales más altas y aumentó en pacientes con mayor peso corporal magro. Sin embargo, los análisis de sensibilidad realizados con la dosis y la pauta recomendadas de Perjeta mostraron que en los valores extremos de estas dos covariables no había una repercusión importante sobre la capacidad de alcanzar las concentraciones diana en estado de equilibrio identificadas en los modelos de xenoinjertos tumorales preclínicos. Por consiguiente, basándose en estas covariables no es necesario ajustar la posología de Perjeta.
Los resultados FC de pertuzumab en el ensayo NEOSPHERE son coherentes con las predicciones del anterior modelo de FC poblacional.
5.3 Datos preclínicos sobre seguridad
No se han realizado estudios específicos de fertilidad en animales para evaluar el efecto de pertuzumab. De los estudios de toxicidad de dosis repetidas realizados en monos cynomolgus no se puede obtener una conclusión definitiva de las reacciones adversas sobre los órganos reproductores masculinos.
Se han realizado estudios de toxicología para la reproducción en monos cynomolgus embarazadas (desde el día de gestación (DG) 19 hasta el DG 50) con dosis iniciales de 30 a 150 mg/kg seguidas de dosis de 10 a 100 mg/kg dos veces por semana. En base a la Cmax, estos niveles de dosis produjeron exposiciones clínicamente relevantes de 2,5 a 20 veces mayor que con la dosis recomendada en humanos. La administración intravenosa de pertuzumab desde el DG 19 hasta el DG 50 (periodo de organogénesis) fue embriotóxica, con aumentos dosis dependiente de las muertes embriofetal entre el DG 25 y el DG 70. Las incidencias de perdidas embriofetales fueron de 33, 50, y 85 % para las hembras de monos embarazadas tratadas dos veces por semana con dosis de pertuzumab de 10, 30 y 100 mg/kg, respectivamente (en base a la Cmax son 2,5 a 20 veces mayores que con la dosis recomendada en humanos). En la cesárea del DG 100, en todos los grupos que recibieron dosis de pertuzumab se identificaron oligohidramnios, disminución relativa del peso del pulmón y de los riñones y evidencia microscópica de hipoplasia renal consecuente con el retraso del desarrollo renal. Además, consecuentemente con las limitaciones en el crecimiento fetal, también se observaron asociado a oligohidramnios, hipoplasia pulmonar (1 de 6 en el grupo de 30 mg/kg y 1 de 2 en el grupo de 100 mg/kg), comunicación intraventricular (1 de 6 en el grupo de 30 mg/kg), estrechamiento de la pared del ventrículo (1 de 2 en el grupo de 100 mg/kg) y anomalías menores en el esqueleto (externo - 3 de 6 en el grupo de 30 mg/kg). La exposición a Pertuzumab se notificó en la descendencia de todos los grupos de tratamiento, con valores del 29 % al 40 % de los niveles de suero materno en el DG 100.
En monos cynomolgus, la administración IV semanal de pertuzumab en dosis de hasta 150 mg/kg/dosis se toleró bien en general. Con dosis de 15 mg/kg y superiores, se observó diarrea asociada con el tratamiento leve e intermitente. En un subgrupo de monos, la administración crónica (7 a 26 dosis semanales) originó episodios de diarrea secretoria grave. La diarrea se controló (a excepción de la eutanasia de un animal, 50 mg/kg/dosis) con terapia de soporte incluyendo tratamiento de reposición de líquido intravenoso.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Ácido acético glacial L-histidina Sacarosa Polisorbato 20
Agua para preparaciones inyectables
6.2 Incompatibilidades
No se han observado incompatibilidades entre Perjeta y las bolsas de cloruro de polivinilo (PVC) o de poliolefina sin PVC incluyendo de polietileno. No debe utilizarse solución de glucosa (5 %) para diluir Perjeta porque es química y físicamente inestable en estas soluciones.
Este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección 6.6.
6.3 Periodo de validez
Vial sin abrir 2 años.
Solución diluida
Se ha demostrado la estabilidad química y física en uso durante 24 horas a 30°C.
Desde el punto de vista microbiológico, el producto debe usarse inmediatamente. Si no se utiliza de inmediato, los tiempos y condiciones de conservación antes del uso son responsabilidad del usuario, y normalmente no deben superar las 24 horas entre 2°C y 8°C, a menos que la dilución se haya realizado en condiciones asépticas validadas y controladas.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8 °C).
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Para las condiciones de conservación del medicamento diluido, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Un vial (vidrio de tipo I) con tapón (caucho butílico) que contiene 14 ml de solución.
Envase de 1 vial.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Perjeta no contiene conservantes antimicrobianos. Por consiguiente, hay que tener cuidado de garantizar la esterilidad de la solución preparada para perfusión y debe prepararla un profesional sanitario.
Perjeta es de un solo uso y se administra por vía intravenosa mediante perfusión.
No se debe agitar el vial. Se deben extraer 14 ml de concentrado de Perjeta del vial y diluirlo en bolsas para perfusión de PVC o de poliolefina sin PVC de 250 ml con una solución para perfusión de cloruro de sodio de 9 mg/ml (0,9 %). Después de la dilución, un ml de la solución contiene aproximadamente
3,02 mg de pertuzumab (840 mg/278 ml) para la dosis inicial en la que se necesitan dos viales y aproximadamente 1,59 mg de pertuzumab (420 mg/264 ml) para la dosis de mantenimiento en la que se necesita un vial.
La bolsa debe invertirse suavemente para mezclar la solución a fin de evitar la formación de espuma.
Los medicamentos de uso parenteral deben inspeccionarse visualmente antes de la administración en busca de partículas y cambios de color. Si se observan partículas o cambios de color, no se debe usar la solución. Una vez preparada la perfusión, debe administrarse de inmediato (ver sección 6.3).
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/813/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 04/Marzo/2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
A. FABRICANTE DEL DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante del principio activo biológico(s).
Genentech, Inc.
1000 New Horizons Way Vacaville, CA 95688-9431 Estados Unidos
Nombre y dirección del fabricante responsable de la liberación de los lotes
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Whylen Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad
El Titular de la Autorización de Comercialización (TAC) presentará el primer informe periódico de seguridad para este medicamento en un plazo de 6 meses después de la autorización. Posteriormente, el titular de la autorización de comercialización presentará informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2. de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado anualmente hasta la renovación:
Cuando coincida la presentación de un informe periódico de seguridad con la actualización del PGR, ambos documentos deberán presentarse conjuntamente.
Además, se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
• Obligación de llevar a cabo medidas posautorización
El TAC deberá llevar a cabo, dentro del plazo establecido, las siguientes medidas:
|
Descripción |
Fecha límite |
|
MO28047 (PERUSE) Ensayo multicéntrico, abierto, con un solo grupo de tratamiento de pertuzumab en combinación con trastuzumab y un taxano como tratamiento de primera línea de pacientes con cáncer de mama avanzado (metastásico o localmente recidivante) HER2-positivo. |
Septiembre 2020 |
|
Estudio posautorización de eficacia (EPAE): Para proporcionar datos de eficacia a largo plazo en términos de SLE y SG, el TAC debe presentar los resultados del ensayo BO25126 (APHINITY), un ensayo multicéntrico aleatorizado, doble ciego, controlado con placebo donde se compara quimioterapia más trastuzumab más placebo frente a quimioterapia más trastuzumab más pertuzumab como terapia adyuvante en pacientes con cáncer de mama primario HER2-positivo operable |
Mayo 2017 |
|
Estudio posautorización de seguridad (EPAS): Para evaluar la seguridad cardiaca y proporcionar más resultados de eficacia en neoadyuvancia, el TAC debe presentar los resultados del ensayo WO29217 (BERENICE), un ensayo de Fase II multicéntrico, internacional, para evaluar pertuzumab en combinación con trastuzumab y quimioterapia neoadyuvante estándar basada en antraciclinas en pacientes con cáncer de mama HER2-positivo, localmente avanzado, inflamatorio o en estadio temprano |
Mayo 2017 |
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
Perjeta 420 mg concentrado para solución para perfusión pertuzumab
Un vial de 14 ml contiene 420 mg de pertuzumab a una concentración de 30 mg/ml.
Ácido acético glacial, L-histidina, Sacarosa y Polisorbato 20. Agua para preparaciones inyectables
Concentrado para solución para perfusión 420 mg/14 ml 1 x 14 ml
Vía intravenosa tras dilución No agitar
Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
Conservar en nevera (2°C - 8°C)
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
EU/1/13/813/001
Lote
Medicamento sujeto a prescripción médica
Se acepta la justificación para no incluir la información en Braille
<Incluido el código de barras 2D que lleva el identificador único.>
IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC:
SN:
NN:
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETA DEL VIAL
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Perjeta 420 mg concentrado para solución para perfusión pertuzumab
IV
2. FORMA DE ADMINISTRACIÓN
Vía intravenosa tras dilución
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
420 mg/14 ml
6. OTROS
B. PROSPECTO
Prospecto: Información para el usuario
Perjeta 420 mg concentrado para solución para perfusión
pertuzumab
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
• Conserve este prospecto, ya que puede tener que volver a leerlo.
• Si tiene alguna duda, consulte a su médico o enfermero.
• Si experimenta efectos adversos, consulte a su médico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Perjeta y para qué se utiliza
2. Qué necesita saber antes de que le administren Perjeta
3. Cómo se le administra Perjeta
4. Posibles efectos adversos
5. Conservación de Perjeta
6. Contenido del envase e información adicional 1. Qué es Perjeta y para qué se utiliza
cáncer
para
Perjeta contiene el principio activo pertuzumab y se utiliza para tratar a pacientes adultos con de mama cuando:
• Se ha identificado que el cáncer es del tipo “HER2-positivo” - su médico le hará pruebas averiguar si es así.
• El cáncer se ha extendido a otras partes del cuerpo (ha metastatizado) y no ha sido tratado previamente con un medicamento para el cáncer (quimioterapia) u otros medicamentos que se unen al HER2, o bien si el cáncer ha progresado después de un tratamiento previo.
• El cáncer no se ha diseminado a otras partes del cuerpo y el tratamiento se va a llevar a cabo antes de la cirugía (el tratamiento antes de la cirugía se llama terapia neoadyuvante)
Además de Perjeta, recibirá también trastuzumab y el medicamento quimioterápico docetaxel. Si está recibiendo Perjeta antes de la cirugía, puede recibir también otra quimioterapia como parte de su tratamiento completo. La información sobre estos medicamentos se describe en prospectos independientes. Pida al médico o enfermero que le dé información sobre estos otros medicamentos.
Cómo actúa Perjeta
Perjeta es un tipo de medicamento llamado “anticuerpo monoclonal” que se une a objetivos específicos dentro del cuerpo y a las células cancerosas.
Perjeta reconoce y se une a un objetivo denominado “receptor del factor de crecimiento epidérmico humano 2”, (HER2). HER2 se encuentra en grandes cantidades en la superficie de algunas células cancerosas, donde estimula su crecimiento. Cuando Perjeta se une a las células cancerosas HER2, puede retrasar o detener el crecimiento de las células cancerosas, o puede destruirlas.
No deben administrarle Perjeta:
• Si es alérgico al pertuzumab o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Si no está seguro, hable con su médico o enfermero antes de que le administren Perjeta.
Advertencias y precauciones
Consulte a su médico o enfermero antes de que le administren Perjeta si:
• Ha tenido alguna vez problemas de corazón (como insuficiencia cardíaca, tratamiento por latidos cardíacos irregulares graves, hipertensión no controlada, ataque al corazón reciente); su médico hará pruebas para comprobar si su corazón funciona bien.
• Ha tenido alguna vez problemas cardíacos durante el tratamiento previo con trastuzumab.
• Ha recibido alguna vez un medicamento de quimioterapia del grupo llamado antraciclinas como por ejemplo, doxorubicina o epirubicina; estos medicamentos pueden dañar el músculo cardíaco y aumentar el riesgo de sufrir problemas cardíacos con Perjeta.
Si le ha sucedido algo de lo anterior (o no está seguro), consulte a su médico o enfermero antes de que le administren Perjeta.
Reacciones a la perfusión
Pueden ocurrir reacciones a la perfusión, reacciones alérgicas o anafilácticas (más graves que las alérgicas). Su médico o enfermero comprobará si aparecen efectos adversos durante la perfusión y en los 30 a 60 minutos siguientes a ella. Si tuviera cualquier reacción grave, su médico interrumpirá el tratamiento con Perjeta. Ver en el apartado de los “Efectos adversos graves” de la sección 4 más detalles sobre las reacciones a la perfusión que hay que vigilar durante y después de la perfusión.
Problemas cardíacos
El tratamiento con Perjeta puede afectar al corazón. Por consiguiente, se comprobará el funcionamiento de su corazón antes y durante el tratamiento con Perjeta. Ver en el apartado de los “Efectos adversos graves” de la sección 4 más detalles sobre los signos de problemas cardíacos que hay que vigilar.
Neutropenia febril (nivel bajo de glóbulos blancos sanguíneos y fiebre)
Cuando se administra Perjeta con otros tratamientos para el cáncer (trastuzumab y docetaxel), el número de glóbulos blancos en sangre puede disminuir y puede aparecer fiebre (aumento de la temperatura). Si tiene inflamación del tubo digestivo (ej. dolor de boca o diarrea) puede tener más probabilidad de sufrir este efecto adverso.
Diarrea
El tratamiento con Perjeta puede causar diarrea grave. La diarrea es un trastorno en el cual el cuerpo produce más heces líquidas de lo normal. Si experimenta diarrea grave mientras esté recibiendo su tratamiento para el cáncer, su médico puede iniciar un tratamiento antidiarreico y puede que interrumpa su tratamiento con Perjeta hasta que la diarrea esté bajo control.
Uso en niños y adolescentes
No se recomienda Perjeta a pacientes menores de 18 años porque no se dispone de información sobre su eficacia en este grupo de edad.
Uso de Perjeta con otros medicamentos
Informe a su médico o enfermero si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento. Esto incluye los medicamentos obtenidos sin receta y los medicamentos a base de plantas.
Embarazo y lactancia
Antes de empezar el tratamiento, debe informar a su médico o enfermero si está embarazada o en periodo de lactancia, o si cree que podría estar embarazada o tiene intención de quedarse embarazada. Se le informará de los beneficios y los riesgos para usted y su hijo de la administración de Perjeta durante el embarazo.
• Informe inmediatamente a su médico si se queda embarazada durante el tratamiento con Perjeta o en los 6 meses siguientes a la interrupción del tratamiento.
• Pregunte a su médico si puede dar el pecho durante o después del tratamiento con Perjeta.
Perjeta puede dañar al feto. Debe utilizar un método anticonceptivo eficaz durante el tratamiento con Perjeta y en los 6 meses siguientes a la interrupción del tratamiento. Consulte con su médico el mejor método anticonceptivo para usted.
Conducción y uso de máquinas
Es poco probable que Perjeta afecte a su capacidad para conducir o usar máquinas. Sin embargo, si tiene reacciones a la perfusión, reacciones alérgicas o anafilácticas, espere a que estas desaparezcan para conducir o usar máquinas.
3. Cómo se le administra Perjeta Administración de este medicamento
Un médico o enfermero le administrará Perjeta en un hospital o clínica.
• Se administra mediante goteo en una vena (perfusión intravenosa) una vez cada tres semanas.
• La cantidad de medicamento que se le administre y la duración de la perfusión serán diferentes durante la primera dosis y las siguientes dosis.
• El número de perfusiones que le administren dependerá de cómo responda al tratamiento y de si está recibiendo tratamiento antes de la cirugía (terapia neoadyuvante) o si su enfermedad se ha extendido.
• Perjeta se administra con otros tratamientos para el cáncer (trastuzumab y docetaxel).
En la primera perfusión:
• Se le administrarán 840 mg de Perjeta en 60 minutos. Su médico o enfermero comprobará si aparecen efectos adversos durante la perfusión y en los 60 minutos siguientes a ella.
• Le administrarán también trastuzumab y docetaxel.
En todas las perfusiones siguientes, si se toleró bien la primera perfusión:
• Se le administrarán 420 mg de Perjeta en 30 a 60 minutos. Su médico o enfermero comprobará si aparecen efectos adversos durante la perfusión y en los 30 a 60 minutos siguientes a ella
• Le administrarán también trastuzumab y docetaxel
Para más información sobre la administración de trastuzumab y docetaxel (que también pueden causar efectos adversos), ver el prospecto de estos productos para comprender el uso de estos medicamentos. Si tiene dudas sobre estos medicamentos, consulte a su médico o enfermero.
Si olvidó usar Perjeta
Si olvida o no acude a su cita para recibir Perjeta, pida otra cita lo antes posible. Si han pasado 6 semanas o más desde la última visita se le administrará una dosis más alta de Perjeta de 840 mg.
Si interrumpe el tratamiento con Perjeta
No deje de usar este medicamento sin hablar antes con su médico. Es importante que se le administren todas las dosis que están recomendadas.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o enfermero.
Posibles efectos adversos
4.
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Efectos adversos graves
Informe a un médico o enfermero de inmediato si advierte cualquiera de los efectos adversos siguientes:
• Los efectos adversos más frecuentes que pueden ocurrir en 2 de cada 3 pacientes son diarrea, caída del cabello y disminución del número de glóbulos blancos (mostrado en un análisis de sangre) con o sin fiebre.
• En aproximadamente 13 de cada 100 pacientes pueden ocurrir reacciones a la perfusión que incluyen náuseas, fiebre, escalofríos, cansancio, dolor de cabeza, pérdida de apetito.
Pueden ocurrir reacciones alérgicas y anafilácticas (más graves que las alérgicas) en 1 de cada 10 pacientes. Entre ellas se incluyen hinchazón de la cara y la garganta con dificultad en la respiración..
• Los síntomas de problemas cardíacos (insuficiencia cardíaca) se han observado en 5 de cada 100 pacientes y pueden ser tos, dificultad respiratoria al dormir en posición horizontal e hinchazón (retención de líquidos) en piernas o brazos.
Informe a un médico o enfermero inmediatamente si observa cualquiera de los efectos adversos anteriores.
Otros efectos adversos
Muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
• Fiebre
• Imposibilidad de dormir
• Descenso del número de glóbulos rojos, mostrado en un análisis de sangre
• Dolor de garganta, enrojecimiento, dolor o moqueo nasal, síntomas seudogripales y fiebre
• Sensaciones de debilidad, entumecimiento, hormigueo o pinchazos que afectan sobre todo a los pies y las piernas
• Problemas en las uñas
• Pérdida o alteración del gusto
• Náuseas o vómitos
• Disminución del apetito
• Estreñimiento
• Erupción
• Dolor articular o muscular, debilidad muscular
• Dolor (dolor de huesos, cuello, pecho, abdomen)
• Inflamación del tubo digestivo (ej. dolor de boca)
• Hinchazón de tobillos u otras partes del cuerpo por la retención de una cantidad excesiva de agua
Frecuentes (pueden afectar hasta 1 de cada 10 personas):
• Sensación de mareo
• Dificultad respiratoria
• Mayor producción de lágrimas
• Sequedad, picor o acné en la piel
• Líquido en los pulmones que causa dificultad para respirar
• Inflamación del lecho de las uñas en su unión con la piel
• Un proceso en el que el ventrículo izquierdo del corazón está funcionalmente alterado con o sin síntomas
Poco frecuentes (pueden afectar hasta 1 de cada 100 personas):
• Síntomas en el pecho como tos seca o dificultad al respirar (signos posibles de enfermedad
pulmonar intersticial, una enfermedad de daño en los tejidos de alrededor de los sacos de aire en los pulmones)
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
Si sufre alguno de los síntomas anteriores después de que se haya interrumpido el tratamiento con Perjeta, debe consultar a su médico de inmediato e informarle de que se le ha tratado previamente con Perjeta.
Algunos de los efectos adversos que sufra pueden deberse a su cáncer de mama. Si se le administra Perjeta con trastuzumab y docetaxel al mismo tiempo, algunos efectos adversos también pueden deberse a estos otros medicamentos.
5. Conservación de Perjeta
Perjeta lo conservarán los profesionales sanitarios en el hospital o la clínica. Los detalles sobre la
conservación son los siguientes:
• Mantener este medicamento fuera de la vista y del alcance de los niños.
• No utilice este medicamento después de la fecha de caducidad que aparece en el envase después de CAD. La fecha de caducidad es el último día del mes que se indica.
• Conservar en nevera (2°C - 8°C).
• No congelar.
• Conservar el vial en el embalaje exterior para protegerlo de la luz.
• No utilice este medicamento si observa partículas en el líquido o tiene un color raro (ver la sección 6).
• Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de Perjeta
• El principio activo es el pertuzumab. Cada vial contiene un total de 420 mg de pertuzumab a una concentración de 30 mg/ml.
• Los demás componentes son ácido acético glacial, L-histidina, sacarosa, polisorbato 20 y agua para preparaciones inyectables.
Aspecto de Perjeta y contenido del envase
Perjeta es un concentrado para solución para perfusión. Es un líquido de transparente a ligeramente
perlado (opalescente), de incoloro a amarillo pálido. Se suministra en un vial de vidrio que contiene
14 ml de concentrado.
Cada envase contiene un vial.
Titular de la autorización de comercialización
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
Responsable de la fabricación
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien N.V. Roche S.A. Tél/Tel: +32 (0) 2 525 82 11 |
Lietuva UAB “Roche Lietuva” Tel: +370 5 2546799 |
|
Btnrapun Pom Etarapua EOOfl Tea: +359 2 818 44 44 |
Luxembourg/Luxemburg (Voir/siehe Belgique/Belgien) |
|
Ceská republika Roche s. r. o. Tel: +420 - 2 20382111 |
Magyarország Roche (Magyarország) Kft. Tel: +36 - 23 446 800 |
|
Danmark Roche a/s Tlf: +45 - 36 39 99 99 |
Malta (See United Kingdom) |
|
Deutschland Roche Pharma AG Tel: +49 (0) 7624 140 |
Nederland Roche Nederland B.V. Tel: +31 (0) 348 438050 |
|
Eesti Roche Eesti OÜ Tel: + 372 - 6 177 380 |
Norge Roche Norge AS Tlf: +47 - 22 78 90 00 |
|
ELLáóa Roche (Hellas) A.E. T^k: +30 210 61 66 100 |
Osterreich Roche Austria GmbH Tel: +43 (0) 1 27739 |
|
España Roche Farma S.A. Tel: +34 - 91 324 81 00 |
Polska Roche Polska Sp.z o.o. Tel: +48 - 22 345 18 88 |
|
France Roche Tél: +33 (0)1 47 61 40 00 |
Portugal Roche Farmacéutica Química, Lda Tel: +351 - 21 425 70 00 |
|
Hrvatska Roche d.o.o. Tel: + 385 1 47 22 333 |
Romania Roche Romania S.R.L. Tel: +40 21 206 47 01 |
|
Ireland Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700 |
Slovenija Roche farmacevtska druzba d.o.o Tel: +386 - 1 360 26 00 |
|
Ísland Roche a/s c/o Icepharma hf Sími: +354 540 8000 |
Slovenská republika Roche Slovensko, s.r.o. Tel: +421 - 2 52638201 |
|
Italia Roche S.p.A. Tel: +39 - 039 2471 |
Suomi/Finland Roche Oy Puh/Tel: +358 (0) 10 554 500 |
|
Kúnpoq r.A.Exapáxn? & Era AxS. Ttfk: +357 - 22 76 62 76 |
Sverige Roche AB Tel: +46 (0) 8 726 1200 |
|
Latvija Roche Latvija SIA Tel: +371 - 6 7039831 |
United Kingdom Roche Products Ltd. Tel: +44 (0) 1707 366000 |
Fecha de la última revisión de este prospecto:
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
41