Pergoveris 150 Ui/75 Ui Polvo Y Disolvente Para Solucion Inyectable
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Pergoveris 150 UI/75 UI polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un vial contiene 150 UI de folitropina alfa* (r-hFSH) (equivalente a 11 microgramos) y 75 UI de lutropina alfa* (r-hLH) (equivalente a 3 microgramos).
Tras la reconstitución, cada ml de solución contiene 150 UI de r-hFSH y 75 UI de r-hLH por mililitro. *producidas en células de ovario de hámster chino (CHO) modificadas por ingeniería genética.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: pastilla liofilizada blanca o blanquecina.
Disolvente: solución clara, incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Pergoveris está indicado para la estimulación del desarrollo folicular en mujeres adultas con déficit severo de LH y FSH.
En los ensayos clínicos, estas pacientes se definieron por un nivel sérico de LH endógena de < 1,2 UI/l.
4.2 Posología y forma de administración
El tratamiento con Pergoveris debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de problemas de fertilidad.
Posología
En mujeres con déficit de LH y FSH (hipogonadismo hipogonadotropo), el objetivo del tratamiento con Pergoveris es desarrollar un único folículo de Graaf maduro, a partir del cual se liberará el ovocito tras la administración de gonadotropina coriónica humana (hCG). Pergoveris debe administrarse como un ciclo de inyecciones diarias. Puesto que estas pacientes son amenorreicas y tienen una escasa secreción endógena de estrógenos, el tratamiento puede comenzar en cualquier momento.
El tratamiento debe adaptarse a la respuesta individual de la paciente, evaluada mediante el tamaño folicular determinado por ecografía y respuesta estrogénica. Una pauta recomendada comienza con un vial de Pergoveris al día. Si se administra menos de un vial de Pergoveris al día, la respuesta folicular puede que no sea satisfactoria porque la cantidad de lutropina alfa sea insuficiente (ver la sección 5.1).
Si se considera apropiado aumentar la dosis de FSH, el ajuste de dosis debe realizarse preferentemente a intervalos de 7-14 días, preferentemente con incrementos de 37,5-75 UI y utilizando una folitropina alfa autorizada. Puede aceptarse la prolongación del tiempo de estimulación en un ciclo determinado, hasta 5 semanas.
Cuando se obtiene una respuesta óptima, debe administrarse una inyección única de 250 microgramos de r-hCG ó 5.000 UI hasta 10.000 UI de hCG, 24-48 horas después de la última inyección de Pergoveris. Se recomienda a la paciente que realice el coito el mismo día de la administración de hCG, así como al día siguiente. De forma alternativa, se puede realizar inseminación intrauterina (IIU).
Puede considerarse la necesidad de apoyo de la fase lútea, ya que la falta de sustancias con actividad luteotropa (LH/hCG) después de la ovulación puede dar lugar a un fracaso prematuro del cuerpo lúteo.
Si se obtiene una respuesta excesiva, debe interrumpirse el tratamiento y no administrarse hCG. El tratamiento debe reiniciarse en el ciclo siguiente con una dosis de FSH más baja que la del ciclo previo.
Poblaciones especiales
Pacientes de edad avanzada
No existe una indicación de uso específica para Pergoveris en pacientes de edad avanzada. No se ha establecido la seguridad y eficacia de Pergoveris en los pacientes de edad avanzada.
Insuficiencia renal y hepática
No se ha establecido la seguridad, eficacia y farmacocinética de Pergoveris en pacientes con insuficiencia renal o hepática.
Población pediátrica
No existe una recomendación de uso específica para Pergoveris en la población pediátrica.
Forma de administración
Pergoveris está indicado para la administración por vía subcutánea. La primera inyección de Pergoveris debe realizarse bajo supervisión médica directa. El polvo debe ser reconstituido inmediatamente antes de su uso, con el disolvente suministrado. La autoadministración de Pergoveris solo debe ser realizada por pacientes que estén bien motivados, adecuadamente instruídos y que tengan acceso a los consejos de un profesional.
Para consultar las instrucciones adicionales de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Pergoveris está contraindicado en pacientes que presentan:
• hipersensibilidad a los principios activos o a alguno de los excipientes incluidos en la sección 6.1
• tumores del hipotálamo o de la hipófisis
• aumento del tamaño de los ovarios o presencia de quistes ováricos no relacionados con la enfermedad del ovario poliquístico y de origen desconocido
• hemorragias ginecológicas de origen desconocido
• carcinoma ovárico, uterino o mamario
Pergoveris no debe utilizarse cuando no pueda obtenerse una respuesta eficaz, en casos tales como:
• fallo ovárico primario
• malformaciones de los órganos sexuales incompatibles con el embarazo
• mioma uterino incompatible con el embarazo
4.4 Advertencias y precauciones especiales de empleo
Pergoveris contiene sustancias gonadotrópicas potentes capaces de causar reacciones adversas de leves a graves, y sólo debe utilizarse por médicos que estén muy familiarizados con los problemas de infertilidad y su tratamiento.
El tratamiento con gonadotropinas requiere cierta dedicación por parte de los médicos y profesionales del sector sanitario, además de disponer de instalaciones de monitorización apropiadas. Para un uso seguro y eficaz de Pergoveris en mujeres, se requiere monitorizar la respuesta ovárica mediante ecografías, solas o preferentemente combinadas con la determinación de los niveles séricos de estradiol, de manera regular. Puede existir cierto grado de variabilidad en la respuesta a la administración de FSH/LH entre las pacientes, presentando algunas pacientes escasa respuesta a la FSH/LH. En mujeres, se debe utilizar la mínima dosis efectiva para lograr el objetivo del tratamiento.
Porfiria
Las pacientes con porfiria o con historia familiar de porfiria deben controlarse estrechamente durante el tratamiento con Pergoveris. En estas pacientes, Pergoveris puede aumentar el riesgo de un ataque agudo. El deterioro de dicha enfermedad o su aparición por primera vez puede requerir la interrupción del tratamiento.
Tratamiento en mujeres
Antes de iniciar el tratamiento, debe valorarse adecuadamente el tipo de infertilidad de la pareja y la posible existencia de contraindicaciones para el embarazo. En particular, debe evaluarse la presencia de hipotiroidismo, insuficiencia suprarrenal e hiperprolactinemia, instaurando el tratamiento específico apropiado.
Las pacientes sometidas a estimulación del crecimiento folicular tienen un mayor riesgo de presentar hiperestimulación, debido a la posibilidad de una respuesta estrogénica excesiva y al desarrollo de múltiples folículos.
Síndrome de hiperestimulación ovárica (SHO)
Un efecto esperado de la estimulación ovárica controlada es cierto grado de incremento del tamaño del ovario. Se observa con mayor frecuencia en mujeres con síndrome del ovario poliquístico y, por lo general, remite sin tratamiento.
A diferencia del aumento del tamaño ovárico no complicado, el SHO es una afección que puede manifestarse con grados crecientes de gravedad. Incluye un aumento ovárico marcado, niveles séricos elevados de esteroides sexuales y un aumento de la permeabilidad vascular que puede dar lugar a un acúmulo de líquidos en la cavidad peritoneal, pleural y, raramente, pericárdica.
En los casos de SHO grave puede observarse la siguiente sintomatología: dolor abdominal, distensión abdominal, aumento importante del tamaño de los ovarios, aumento de peso, disnea, oliguria y síntomas gastrointestinales incluyendo náuseas, vómitos y diarrea.
La evaluación clínica puede revelar hipovolemia, hemoconcentración, desequilibrio electrolítico, ascitis, hemoperitoneo, derrames pleurales, hidrotórax o distrés respiratorio agudo y episodios tromboembólicos.
Muy raramente, el SHO grave puede complicarse con torsión del ovario o episodios tromboembólicos como embolia pulmonar, ictus isquémico o infarto de miocardio.
Algunos factores de riesgo independientes para presentar SHO incluyen edad joven, masa corporal escasa, síndrome del ovario poliquístico, dosis elevadas de gonadotropinas exógenas, concentraciones absolutas altas o en rápido aumento de estradiol en suero (> 900 pg/ml ó > 3.300 pmol/l en anovulación), episodios anteriores de SHO y una gran cantidad de folículos ováricos en desarrollo (3 folículos >14 mm de diámetro en anovulación).
El cumplimiento de la dosis recomendada de Pergoveris y FSH y la pauta de administración pueden minimizar el riesgo de hiperestimulación ovárica. Para la identificación temprana de los factores de riesgo, se recomienda la monitorización de los ciclos de estimulación mediante ecografías y determinaciones de estradiol.
Hay evidencia que indica que la hCG desempeña una función fundamental en el desencadenamiento del SHO y que el síndrome puede ser más grave y puede tener una duración más prolongada si se produce un embarazo. Por tanto, si se producen signos de SHO, por ejemplo, unos niveles séricos de estradiol > 5.500 pg/ml ó > 20.200 pmol/l y/o > 40 folículos en total, se recomienda no administrar hCG y se debe advertir a la paciente que no realice el coito o que utilice métodos anticonceptivos de barrera durante al menos cuatro días. El SHO puede progresar rápidamente (en menos de 24 horas) o en varios días hasta convertirse en un cuadro clínico grave. La mayoría de las veces se produce después de que el tratamiento hormonal se haya suspendido y alcanza su máxima intensidad aproximadamente de siete a diez días después del tratamiento. Habitualmente, el SHO se resuelve espontáneamente al comenzar la menstruación. Por lo tanto, debe seguirse a las pacientes durante al menos dos semanas tras la administración de hCG.
Si se produce SHO grave, debe interrumpirse el tratamiento con gonadotropinas si es que todavía continúa. Debe hospitalizarse a la paciente e iniciar el tratamiento específico del SHO. La incidencia de este síndrome es mayor en pacientes con poliquistosis ovárica.
Cuando se sospecha de que hay riesgo de SHO, debe considerarse la retirada del tratamiento.
Torsión ovárica
Se han comunicado casos de torsión ovárica después del tratamiento con otras gonadotropinas. Este hecho puede estar asociado con otros factores de riesgo, tales como SHO, embarazo, cirugía abdominal previa, antecedentes de torsión ovárica, quistes ováricos anteriores o actuales y síndrome del ovario poliquístico. Se pueden limitar los daños al ovario como consecuencia de la reducción del suministro de sangre mediante un diagnóstico precoz y una intervención médico-quirúrgica inmediata.
Embarazo múltiple
En pacientes sometidas a inducción de la ovulación, la incidencia de embarazos múltiples y nacimientos es más elevada que en los casos de concepción natural. La mayoría de embarazos múltiples son gemelares. El embarazo múltiple, especialmente si el número de fetos es alto, conlleva un mayor riesgo de complicaciones maternas y perinatales. Para minimizar el riesgo de embarazo múltiple, se recomienda una monitorización cuidadosa de la respuesta ovárica.
Antes de iniciar el tratamiento se debe informar a las pacientes del riesgo potencial de tener embarazos múltiples. Cuando se sospecha que hay riesgo de embarazos múltiples, debe considerarse la retirada del tratamiento.
Pérdida del embarazo
La incidencia de pérdida del embarazo debido a aborto espontáneo o provocado en pacientes sometidas a estimulación del desarrollo folicular para inducir la ovulación es superior a la observada en la población general.
Embarazo ectópico
Las mujeres con historia de enfermedad tubárica presentan riesgo de embarazo ectópico, tanto si el embarazo es por concepción espontánea como si se logra mediante tratamientos de fertilidad. Se ha notificado que la prevalencia de embarazo ectópico tras practicar TRA es mayor que en la población general.
Neoplasias del aparato reproductor
Se han notificado neoplasias de ovario y de otros órganos del aparato reproductor, tanto benignas como malignas, en mujeres sometidas a múltiples tratamientos de infertilidad. Todavía no está establecido si el tratamiento con gonadotropinas aumenta o no el riesgo de estos tumores en mujeres infértiles.
Malformaciones congénitas
La prevalencia de malformaciones congénitas tras TRA puede ser ligeramente superior a la observada tras la concepción natural. Esto se considera debido a diferencias en las características de los progenitores (por ejemplo, la edad de la madre o las características del semen) y a los embarazos múltiples.
Fenómenos tromboembólicos
En mujeres con enfermedad tromboembólica reciente o en curso o en mujeres con factores de riesgo generalmente reconocidos para presentar problemas tromboembólicos, tales como historia familiar o personal, trombofilia u obesidad grave (índice de masa corporal > 30 kg/m2), el tratamiento con gonadotropinas puede aumentar más el riesgo. En estas mujeres, los beneficios de la administración de gonadotropinas deben sopesarse frente a los riesgos. No obstante, hay que tener en cuenta que el embarazo por sí mismo, así como el SHO, también comportan un aumento del riesgo de fenómenos tromboembólicos.
Sodio
Pergoveris contiene menos de 1 mmol de sodio (23 mg) por dosis; esto es, esencialmente “exento de sodio”.
4.5 Interacción con otros medicamentos y otras formas de interacción
Pergoveris no debe mezclarse con otros medicamentos en la misma inyección, excepto con folitropina alfa, ya que los estudios correspondientes han demostrado que la co-administración de ambos fármacos no altera significativamente la actividad, estabilidad, ni las propiedades farmacocinéticas o farmacodinámicas de los principios activos.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No existe ninguna indicación para utilizar Pergoveris durante el embarazo. Los datos de un número limitado de embarazos expuestos indican que no hay reacciones adversas de la folitropina alfa y la lutropina alfa durante el embarazo, el desarrollo embrionario o fetal, el parto o el desarrollo postnatal tras una estimulación ovárica controlada. No se han reportado efectos teratógenos de estas gonadotropinas en estudios en animales. En caso de exposición durante el embarazo, los datos clínicos no son suficientes como para descartar que Pergoveris tenga efectos teratógenos.
Lactancia
Pergoveris no está indicado durante la lactancia.
Fertilidad
Pergoveris está indicado para su uso en la infertilidad (ver sección 4.1).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Pergoveris sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Las reacciones adversas notificadas con mayor frecuencia son cefalea, quistes ováricos y reacciones locales en el lugar de inyección (por ejemplo, dolor, eritema, hematoma, hinchazón y/o irritación en el lugar de la inyección).
Se ha reportado con frecuencia síndrome de hiperestimulación ovárica (SHO) leve o moderado y debe considerarse como un riesgo intrínseco del proceso de estimulación. El SHO grave es poco frecuente (ver sección 4.4).
En casos muy raros puede producirse tromboembolismo, asociado generalmente a un síndrome de hiperestimulación ovárica grave (ver sección 4.4).
Lista de reacciones adversas
Las reacciones adversas se enumeran según la clasificación de órganos del sistema MedDRA y la frecuencia. Las categorías de frecuencia utilizadas son: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100), raras (> 1/10.000 a < 1/1.000), muy raras (< 1/10.000)
Trastornos del sistema inmunológico
Muy raras: Reacciones de hipersensibilidad de leves a moderadas, incluyendo reacciones
anafilácticas y shock
Trastornos del sistema nervioso Muy frecuentes: cefalea
Trastornos vasculares
Muy raras: tromboembolismo, generalmente asociado a SHO grave
Trastornos respiratorios, torácicos y mediastínicos Muy raras: exacerbación o empeoramiento del asma
Trastornos gastrointestinales
Frecuentes: dolor abdominal, distensión abdominal, molestias abdominales, náuseas, vómitos,
diarrea
Trastornos del aparato reproductor y de la mama Muy frecuentes: quistes ováricos
Frecuentes: dolor mamario, dolor pélvico, SHO leve o moderado (incluida la sintomatología
relacionada)
Poco frecuentes: SHO grave (incluida la sintomatología relacionada) (ver sección 4.4)
Raras: complicación del SHO grave
Trastornos generales y alteraciones en el lugar de administración
Muy frecuentes: reacciones de leves a intensas en el lugar de inyección (por ejemplo, dolor, eritema, hematoma, cardenales, hinchazón y/o irritación en el lugar de inyección)
Notificación de sospechas de reacciones adversas
. Se invita a los sistema nacional
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento profesionales sanitarios a notificar las sospechas de reacciones adversas a través del de notificación incluido en el Anexo V.
4.9 Sobredosis
Síntomas
No se conocen los efectos de una sobredosis de Pergoveris. Sin embargo, existe la posibilidad de que se produzca un SHO lo cual se describe más ampliamente en la sección 4.4.
Atención terapéutica
El tratamiento va dirigido a los síntomas.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: hormonas sexuales y moduladores del sistema genital, gonadotropinas. Código ATC: G03GA30.
Pergoveris es un preparado de hormona foliculoestimulante y hormona luteinizante producidas en células de ovario de hámster chino (CHO), modificadas por ingeniería genética.
Mecanismo de acción
En los ensayos clínicos la eficacia de la combinación de folitropina alfa y lutropina alfa se ha demostrado en mujeres con hipogonadismo hipogonadotropo.
Durante la estimulación del desarrollo folicular en mujeres con anovulación y déficit de LH y FSH, el principal efecto debido a la administración de lutropina alfa es un aumento de la secreción de estradiol por los folículos, cuyo desarrollo es estimulado por la FSH.
Efectos farmacodinámicos
En los ensayos clínicos, las pacientes con déficit severo de FSH y LH se definieron por un nivel sérico de LH endógena de < 1,2 UI/l, medido en un laboratorio central. Sin embargo, debe tenerse en cuenta que existen variaciones entre las determinaciones de LH realizadas en diferentes laboratorios. En estos ensayos la tasa de ovulación por ciclo fue del 70-75%.
Eficacia clínica
En un ensayo clínico en pacientes con hipogonadismo hipogonadotropo y un nivel sérico de LH endógena menor de 1,2 UI/l se investigó la dosis adecuada de r-hLH (lutropina alfa). Una dosis de 75 UI de r-hLH administrada diariamente (en combinación con 150 UI de folitropina alfa (r-hFSH)) resultó adecuada para el desarrollo folicular y la producción de estrógenos. Una dosis de 25 UI de r-hLH administrada diariamente (en combinación con 150 UI de folitropina alfa) originó un desarrollo folicular insuficiente. Por tanto, la administración de menos de un vial de Pergoveris diariamente puede proporcionar una actividad de LH demasiado baja para asegurar un adecuado desarrollo folicular.
5.2 Propiedades farmacocinéticas
La combinación de folitropina alfa y lutropina alfa ha demostrado tener el mismo perfil farmacocinético que el de la folitropina alfa y lutropina alfa por separado.
Folitropina alfa
Distribución
Tras la administración intravenosa, la folitropina alfa se distribuye en el espacio extracelular con una semivida de distribución de unas 2 horas y con una semivida de eliminación, de alrededor 1 día. En equilibrio estacionario, el volumen de distribución es de 10 l.
Tras la administración subcutánea, la biodisponibilidad absoluta es de alrededor del 70%. Tras la administración de dosis repetidas de folitropina alfa, se produce una acumulación de 3 veces, alcanzando un equilibrio estacionario en un periodo de 3-4 días. En mujeres con supresión de la secreción endógena de gonadotrofinas, la folitropina alfa estimula adecuadamente el desarrollo folicular y la esteroidogénesis, a pesar de unos niveles indetectables de LH.
Eliminación
La eliminación total es de 0,6 l/h y la octava parte de la dosis de folitropina alfa se excreta en la orina. Lutropina alfa
Distribución
Tras la administración intravenosa, la lutropina alfa se distribuye rápidamente, con una semivida de distribución de aproximadamente una hora y se elimina del organismo con una semivida de eliminación de unas 10-12 horas. El volumen de distribución en equilibrio estacionario es de alrededor de 10-14 l. La farmacocinética de la lutropina alfa es lineal, ya que el AUC es directamente proporcional a la dosis administrada. Tras la administración subcutánea, la biodisponibilidad absoluta es aproximadamente del 60%; la semivida de eliminación es algo más prolongada. Tras la administración única y repetida de lutropina alfa, la farmacocinética de la lutropina alfa es comparable y su tasa de acumulación es mínima. El tiempo medio de permanencia es de aproximadamente 5 horas.
Eliminación
La eliminación total es de alrededor de 2 l/h, y menos del 5% de la dosis se excreta en la orina.
Relaciones farmacocinéticas/farmacodinámicas
No existe interacción farmacocinética con la folitropina alfa cuando se administran simultáneamente.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas y genotoxicidad.
6. DATOS FARMACÉUTICOS 6.1 Lista de excipientes
Polvo
Sacarosa Polisorbato 20 Metionina
Fosfato disódico dihidrato
Dihidrógeno fosfato de sodio
Acido fosfórico concentrado (para ajuste de pH)
Hidróxido sódico (para ajuste de pH)
Disolvente
Agua para preparaciones inyectables.
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección 6.6.
6.3 Periodo de validez Viales sin abrir
3 años.
Solución reconstituida
Pergoveris está destinado a uso único e inmediato tras la primera apertura y reconstitución, por lo que el producto no puede almacenarse una vez abierto y reconstituido.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 25°C.
Conservar en el embalaje original para protegerlo de la luz.
6.5 Naturaleza y contenido del envase
Polvo: viales de 3 ml (de vidrio tipo I) que se cierran con un tapón de bromobutilo protegidos por anillos de aluminio y cápsulas de cierre flip-off.
1 vial contiene 11 microgramos de r-hFSH y 3 microgramos de r-hLH.
Disolvente: viales de 3 ml (de vidrio tipo I) con un tapón de goma recubiertos de Teflon protegido por anillos de aluminio y cápsulas de cierre flip-off.
1 vial de disolvente contiene 1 ml de agua para preparaciones inyectables.
Tamaños de envases de 1, 3 y 10 viales, con el mismo número de disolvente (1, 3 y 10 viales).
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
Para uso único e inmediato tras la primera apertura y reconstitución.
Reconstitución
El pH de la solución reconstituida es de 6,5-7,5.
Pergoveris debe reconstituirse con el disolvente antes de su utilización, mediante rotación suave.
La solución reconstituida no debe administrarse si contiene partículas o no es nítida.
Pergoveris puede mezclarse con folitropina alfa y co-administrarse en una misma inyección.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Serano Europe Limited 56 Marsh Wall,
London E14 9TP,
Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/07/396/001
EU/1/07/396/002
EU/1/07/396/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 25/junio/2007 Fecha de la última renovación: 22/mayo/2012
10. FECHA DE LA REVISIÓN DEL TEXTO
{MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTES DE LOS PRINCIPIOS ACTIVOS BIOLÓGICOS Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTES DE LOS PRINCIPIOS ACTIVOS BIOLÓGICOS Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección de los fabricantes de los principios activos biológicos
Merck Serono S.A.
Zone Industrielle de l’Ouriettaz
CH-1170 Aubonne
Suiza
O
Merck S.L.
C/ Batanes 1 Tres Cantos E-28760 Madrid España
Nombre y dirección del fabricante responsable de la liberación de los lotes
Merck Serono S.p.A.
Via delle Magnolie 15 (Zona Industríale)
I-70026 Modugno (Bari)
Italia
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o
como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CAJA DE 1, 3 Y 10 VIALES DE POLVO Y DISOLVENTE
1. NOMBRE DEL MEDICAMENTO
Pergoveris 150 UI/75 UI polvo y disolvente para solución inyectable. Folitropina alfa/lutropina alfa.
2. PRINCIPIO(S) ACTIVO(S)
Un vial contiene 150 UI (equivalente a 11 microgramos) de folitropina alfa (r-hFSH) y 75 UI (equivalente a 3 microgramos) de lutropina alfa (r-hLH).
3. LISTA DE EXCIPIENTES
Demás componentes:
Polvo: fosfato disódico dihidrato, dihidrógeno fosfato de sodio, metionina, polisorbato 20, sacarosa. Disolvente: agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable.
1 vial de polvo (11 microgramos de r-hFSH y 3 microgramos de r-hLH). 1 vial de disolvente (1 ml).
3 viales de polvo.
3 viales de disolvente.
10 viales de polvo.
10 viales de disolvente.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 25°C. Conservar en el embalaje original para protegerlo de la luz.
Leer en el prospecto el periodo de validez del medicamento reconstituido.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
La eliminación del producto no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Serono Europe Limited 56 Marsh Wall,
London E14 9TP Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/07/396/001 1 vial de polvo para solución inyectable
1 vial de disolvente
|
EU/1/07/396/002 |
3 viales de polvo para solución inyectable |
|
3 viales de disolvente |
EU/1/07/396/003 10 viales de polvo para solución inyectable
10 viales de disolvente
13. NÚMERO DE LOTE
Lote
Lote de disolvente
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
pergoveris
PERGOVERIS 150 UI/75 UI ETIQUETA DEL VIAL
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Pergoveris 150 UI/75 UI polvo y disolvente para solución inyectable. Folitropina alfa/Lutropina alfa
S.C.
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
150 UI de r-hFSH/75 UI r-hLH
6. OTROS
VIAL DE DISOLVENTE
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Disolvente para Pergoveris Agua para preparaciones inyectables
|
2. |
FORMA DE ADMINISTRACIÓN |
|
3. |
FECHA DE CADUCIDAD |
|
CAD | |
|
4. |
NÚMERO DE LOTE |
|
Lote | |
|
5. |
CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES |
|
1 ml | |
|
6. |
OTROS |
B. PROSPECTO
Prospecto: información para el usuario
Pergoveris 150 UI/75 UI polvo y disolvente para solución inyectable
Folitropina alfa/Lutropina alfa
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Pergoveris y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Pergoveris
3. Cómo usar Pergoveris
4. Posibles efectos adversos
5. Conservación de Pergoveris
6. Contenido del envase e información adicional
1. Qué es Pergoveris y para qué se utiliza Qué es Pergoveris
Pergoveris contiene dos principios activos diferentes denominados “folitropina alfa” y “lutropina alfa”. Ambos pertenecen a la familia de hormonas llamadas “gonadotropinas”, las cuales están implicadas en la reproducción y la fertilidad.
Para qué se utiliza Pergoveris
Este medicamento se utiliza para estimular el desarrollo de los folículos (cada uno de los cuales contiene un óvulo) de los ovarios con el fin de ayudarla a quedarse embarazada. Está destinado al uso en mujeres adultas (18 años de edad o más) con niveles bajos (déficit severo) de “hormona foliculoestimulante” (FSH) y “hormona luteizante” (LH). Normalmente, estas mujeres son infértiles.
Cómo actúa Pergoveris
Los principios activos de Pergoveris son copias de las hormonas naturales FSH y LH. En el cuerpo:
• la FSH estimula la producción de óvulos
• la LH estimula la liberación de óvulos.
Al sustituir las hormonas ausentes, Pergoveris permite a las mujeres con niveles bajos de FSH y LH desarrollar un folículo, a partir del cual se liberará un óvulo, después de una inyección de la hormona “gonadotropina coriónica humana (hCG)”. Esto ayuda a las mujeres a quedarse embarazadas.
2. Qué necesita saber antes de empezar a usar Pergoveris
Antes de iniciar el tratamiento debe valorarse su fertilidad y la de su pareja por parte de un médico experimentado en el tratamiento de los trastornos de la fertilidad.
• si es alérgica a la hormona foliculoestimulante (FSH), hormona luteinizante (LH) o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
• si tiene un tumor cerebral (en el hipotálamo o en la hipófisis).
• si tiene ovarios grandes o bolsas de líquido en el interior de los ovarios (quistes ováricos) de origen desconocido.
• si tiene hemorragia vaginal inexplicable.
• si tiene cáncer de ovarios, de útero o de mama.
• si tiene una afección que imposibilitaría un embarazo normal, como menopausia precoz, malformaciones de los órganos sexuales o tumores benignos en el útero.
No utilice este medicamento si alguna de las condiciones anteriores le aplica a usted. Si no está segura, consulte a su médico, farmacéutico o enfermero antes de empezar a usar este medicamento.
Advertencias y precauciones
Consulte a su médico, farmacéutico o enfermero antes de empezar a usar Pergoveris.
Porfiria
Consulte a su médico antes de iniciar el tratamiento, si usted o cualquier miembro de su familia padece porfiria (una incapacidad para degradar las porfirinas que puede transmitirse de padres a hijos). Informe inmediatamente a su médico si:
• su piel se vuelve frágil y le salen ampollas con facilidad, especialmente en las zonas expuestas al sol con frecuencia.
• tiene dolor de estómago, de brazos o piernas.
En estos casos, su médico puede recomendarle interrumpir el tratamiento.
Síndrome de Hiperestimulación Ovárica (SHO)
Este medicamento estimula sus ovarios, lo que aumenta el riesgo de experimentar Síndrome de Hiperestimulación Ovárica o SHO. Esto ocurre cuando sus folículos se desarrollan demasiado y se convierten en quistes de gran tamaño. Si tiene dolor en la región pélvica, aumenta de peso rápidamente, tiene náuseas o vómitos o dificultad para respirar, consulte inmediatamente con su médico, quien puede ordenarle interrumpir el tratamiento (ver la sección 4, en “Efectos adversos más graves”).
En caso de que no ovule y si se respetan la dosis y la pauta posológica recomendadas, el SHO grave es menos probable que ocurra. El tratamiento con Pergoveris rara vez causa SHO grave. Esto es más probable si se administra el medicamento que se usa para la maduración folicular final (que contiene gonadotropina coriónica humana, hCG) (ver detalles en la sección 3, en “Qué cantidad se debe usar”). En caso de desarrollar SHO, su médico puede no recetarle hCG en este ciclo de tratamiento y aconsejarle que se abstenga de realizar el coito o que utilice métodos anticonceptivos de barrera durante al menos 4 días.
Su médico asegurará un control cuidadoso de la respuesta ovárica, mediante ecografías y análisis de sangre (determinaciones del estradiol), antes y durante el tratamiento.
Embarazo múltiple
Si usa Pergoveris, tiene un riesgo más alto de quedarse embarazada de más de un niño a la vez (“embarazo múltiple”, generalmente gemelos) que si se queda embarazada por concepción natural. El embarazo múltiple puede causar complicaciones médicas para usted y sus bebés. Usted puede reducir el riesgo de embarazo múltiple usando la dosis correcta de Pergoveris a las horas correctas.
Para minimizar el riesgo de embarazo múltiple, se recomienda realizar ecografías y análisis de sangre.
Aborto
Si se somete a estimulación de sus ovarios para producir óvulos, la probabilidad de tener un aborto es mayor que en el promedio de las mujeres.
Embarazo ectópico
Las mujeres que han sufrido alguna vez bloqueo o daños de las trompas de Falopio (enfermedad tubárica) presentan riesgo de embarazo con implantación del embrión fuera del útero (embarazo ectópico). Esto es así tanto si el embarazo es por concepción espontánea como si se logra mediante tratamientos de fertilidad.
Problemas de coagulación de la sangre (episodios tromboembólicos)
Consulte a su médico antes de usar Pergoveris si usted o algún miembro de su familia ha sufrido alguna vez coágulos de sangre en la pierna o en el pulmón, infarto de miocardio o ictus. Podría tener un riesgo más alto de sufrir coágulos de sangre graves o de empeoramiento de los coágulos existentes con el tratamiento con Pergoveris.
Tumores de los órganos sexuales
Se han comunicado tumores en los ovarios y en otros órganos sexuales, tanto benignos como malignos, en mujeres sometidas a múltiples tratamientos de infertilidad.
Reacciones alérgicas
Se han comunicado casos aislados de reacciones alérgicas no graves a Pergoveris. Si usted ha tenido alguna vez este tipo de reacción con un medicamento similar, informe a su médico antes de usar Pergoveris.
Niños y adolescentes
Pergoveris no debe utilizarse en niños y adolescentes menores de 18 años de edad.
Uso de Pergoveris con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento.
No use Pergoveris con otros medicamentos en la misma inyección, excepto con folitropina alfa, si se la prescribe su médico.
Embarazo y lactancia
No use Pergoveris si está embarazada o dando el pecho.
Conducción y uso de máquinas
No se espera que este medicamento afecte a su capacidad para conducir o utilizar máquinas. Información sobre el contenido de sodio en Pergoveris
Pergoveris contiene menos de 1 mmol de sodio (23 mg) por dosis. Esto es esencialmente “exento de sodio”.
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Uso de este medicamento
• Pergoveris está diseñado para ser inyectado justo debajo de la piel (por vía subcutánea). Para minimizar la irritación cutánea, seleccione un lugar de inyección diferente cada día.
• Se presenta en forma de polvo y líquido, que debe mezclar y usar de forma inmediata.
• Su médico o enfermero le enseñará a preparar e inyectarse este medicamento. Ellos supervisarán su primera inyección.
• Si están conformes con que pueda administrarse Pergoveris con seguridad, en adelante podrá preparar e inyectarse el medicamento usted mismo en casa. Cuando lo haga, lea y siga atentamente las instrucciones descritas a continuación en la sección “Cómo preparar y usar el polvo y disolvente de Pergoveris”.
Qué cantidad se debe usar
La dosis inicial habitual es de 1 vial de Pergoveris cada día.
• En función de la respuesta, su médico puede decidir añadir diariamente una dosis de folitropina alfa a la inyección de Pergoveris. En este caso, normalmente la dosis de folitropina alfa se incrementa cada 7 ó 14 días en 37,5-75 UI.
• El tratamiento continúa hasta que se obtenga la respuesta deseada. Esto sucede cuando ha desarrollado un folículo adecuado, evaluado mediante ecografías y análisis de sangre.
• Pueden ser necesarias hasta cinco semanas.
Cuando se obtenga la respuesta deseada, se le administrará una inyección única de gonadotropina coriónica humana (hCG) de 24 a 48 horas después de su última inyección de Pergoveris. El mejor momento para mantener relaciones sexuales es el mismo día de la inyección de hCG y al día siguiente. Como alternativa, también puede realizarse inseminación intrauterina (IIU).
Si se obtiene una respuesta excesiva, se interrumpirá su tratamiento y no le administrarán hCG (ver la sección 2, en “Síndrome de Hiperestimulación Ovárica”). En este caso, su médico le recetará una dosis de folitropina alfa más baja en el ciclo siguiente.
Cómo preparar y usar el polvo y disolvente de Pergoveris
Antes de comenzar la preparación, lea primero íntegramente estas instrucciones:
Póngase la inyección a la misma hora cada día.
1. Lávese las manos y busque un lugar limpio
• Es importante que sus manos y los materiales que utilice estén lo más limpios posible
• Un lugar adecuado es una mesa limpia o una superficie de la cocina
2. Reúna y disponga todo lo que vaya a necesitar
• 1 vial que contiene el polvo de Pergoveris
• 1 vial que contiene el agua para preparaciones inyectables (disolvente)
No se suministra en el envase:
• 2 torundas empapadas en alcohol
• 1 jeringa vacía para inyección
• 1 aguja para la preparación
• 1 aguja fina para la inyección debajo de la piel
un recipiente para objetos cortantes para desechar con precaución los envases de vidrio y las agujas
3. Preparación de la solución
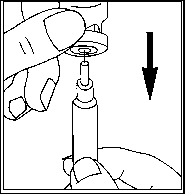
• Quite la cápsula de cierre protectora del vial lleno de agua (vial del disolvente).
• Coloque la aguja para la preparación en la jeringa vacía para inyección.
• Introduzca algo de aire en la jeringa tirando del émbolo aproximadamente hasta la marca de 1 ml.
• Introduzca la aguja en el vial, empuje el émbolo para expulsar el aire.
• Coloque el vial boca abajo y extraiga suavemente todo el agua (disolvente).
• Saque la jeringa del vial y déjela a un lado con cuidado. No toque la aguja y no permita que ésta entre en contacto con ninguna superficie.
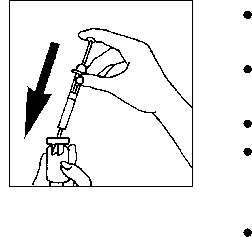
Quite la cápsula de cierre protectora del vial lleno de polvo de Pergoveris.
Coja su jeringa e inyecte lentamente el contenido de la jeringa en el vial de polvo.
Muévalo suavemente sin retirar la jeringa. No lo agite.
Una vez que el polvo se haya disuelto (lo que suele ocurrir inmediatamente), compruebe que la solución obtenida es límpida y no contiene ninguna partícula.
Ponga el vial boca abajo y cargue suavemente la solución en la jeringa. Compruebe que no hay partículas como antes y no utilice la solución si no es límpida.
4. Preparación de la jeringa para la inyección
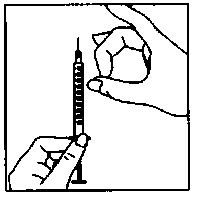
Cambie la aguja, colocando la aguja fina.
Elimine las posibles burbujas de aire: Si ve alguna burbuja de aire en la jeringa, tome ésta con la aguja hacia arriba y dé golpecitos en la jeringa suavemente hasta que el aire se reúna en la parte superior. Empuje el émbolo hasta que desaparezcan las burbujas de aire.
5. Inyección de la dosis
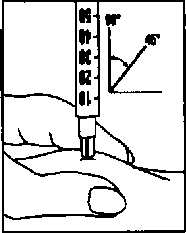
Inyecte inmediatamente la solución: su médico o enfermero le habrán indicado dónde debe ponerse la inyección (p. ej., en el vientre, en la parte delantera del muslo). Para reducir al mínimo la irritación de la piel, seleccione cada día un lugar de inyección diferente.
Limpie la zona elegida de la piel con un algodón empapado en alcohol, realizando un movimiento circular.
Pellizque enérgicamente la piel e introduzca la aguja con un ángulo de 45° a 90°, con un movimiento similar al de los dardos.
Inyecte debajo de la piel, según las instrucciones recibidas. No inyecte directamente en una vena.
Inyecte la solución presionando suavemente sobre el émbolo. Emplee todo el tiempo que necesite hasta inyectar la totalidad de la solución.
A continuación retire la aguja y limpie la piel con un algodón empapado en alcohol, realizando un movimiento circular.
6. Después de la inyección
Deseche todo el material: Una vez finalizada la inyección, deseche inmediatamente todas las agujas y viales vacíos en el recipiente para objetos cortantes. Debe desecharse cualquier porción de la solución no utilizada.
Si usa más Pergoveris del que debe
Los efectos de una sobredosis de Pergoveris son desconocidos; sin embargo, puede esperarse que se produzca un SHO. No obstante, esto sólo ocurrirá si se administra hCG (ver en la sección 2, en “Síndrome de Hiperestimulación Ovárica”).
Si olvidó usar Pergoveris
No tome una dosis doble para compensar las dosis olvidadas. Póngase en contacto con su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Efectos adversos más graves
Consulte a su médico inmediatamente si advierte alguno de los efectos adversos que se listan a continuación. El médico podría decirle que deje de usar Pergoveris.
Reacciones alérgicas
Las reacciones alérgicas, como erupción cutánea, enrojecimiento de la piel, ampollas, hinchazón de la cara con dificultad para respirar, a veces pueden ser graves. Este efecto adverso es muy raro.
Síndrome de Hiperestimulación Ovárica
• Dolor pélvico, acompañado de náuseas o vómitos. Pueden ser síntomas del Síndrome de Hiperestimulación Ovárica (SHO). Es posible que sus ovarios hayan reaccionado de forma excesiva al tratamiento y se hayan desarrollado quistes oválicos o bolsas de líquido de gran tamaño (ver también en la sección 2 en “Síndrome de Hiperestimulación Ovárica”). Este efecto adverso es frecuente. Si le ocurre esto, su médico tendrá que explorarla lo antes posible.
• El SHO puede agravarse con ovarios claramente aumentados de tamaño, disminución de la producción de orina, aumento de peso, dificultad para respirar y/o posible acumulación de líquido en el abdomen o en el pecho. Este efecto adverso es poco frecuente (puede afectar a hasta 1 de cada 100 personas).
• Las complicaciones del SHO como torsión ovárica o coagulación de la sangre se dan en raras ocasiones (pueden afectar a hasta 1 de cada 1.000 personas).
• Los problemas graves de la coagulación de la sangre (episodios tromboembólicos), normalmente con SHO grave, se dan muy raramente. Esto podría causar dolor en el pecho, sensación de falta de aire, ictus o infarto de miocardio. En casos raros esto también podría suceder independientemente del SHO (ver también la sección 2 en “Problemas de la coagulación de la sangre”).
Otros efectos adversos son:
Muy frecuentes (pueden afectar a más de 1 de cada 10 personas)
• bolsas de líquido en el interior de los ovarios (quistes ováricos)
• dolor de cabeza
• reacciones locales en el lugar de inyección como dolor, picor, moratones, hinchazón o irritación.
Frecuentes (pueden afectar hasta 1 de cada 10 personas)
• diarrea
• dolor en el pecho
• náuseas o vómitos
• dolor abdominal o pélvico
• calambres o distensión abdominal
Muy raros (pueden afectar hasta 1 de cada 10.000 personas) • el asma puede empeorar.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede
comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V
Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Pergoveris
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en los viales y en la caja después de CAD. La fecha de caducidad es el último día del mes que se indica.
No conservar a temperatura superior a 25°C. Conservar en el embalaje original para protegerlo de la luz.
El medicamento debe administrarse inmediatamente después de la reconstitución.
No utilice Pergoveris si observa signos visibles de deterioro.
La disolución no debe administrarse si contiene partículas o no es límpida.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Pergoveris
Los principios activos son folitropina alfa y lutropina alfa.
• Un vial contiene 150 UI de folitropina alfa (equivalente a 11 microgramos) y 75 UI de lutropina alfa (equivalente a 3 microgramos).
Tras la reconstitución, cada ml de la solución contiene 150 UI de folitropina alfa y 75 UI de lutropina alfa por mililitro.
• Los demás componentes son:
Polvo: sacarosa, fosfato disódico dihidrato, dihidrógeno fosfato de sodio, metionina, polisorbato 20, ácido fosfórico concentrado e hidróxido sódico.
Disolvente: agua para preparaciones inyectables. Cada vial contiene 1 ml.
Aspecto del producto y contenido del envase
• Pergoveris se presenta como polvo y disolvente para solución inyectable.
• El polvo es una pastilla liofilizada blanca o blanquecina en un vial de vidrio.
• El disolvente es un líquido claro e incoloro en un vial de vidrio.
• Pergoveris se presenta en envases que contienen 1, 3 y 10 viales de polvo junto con el mismo número de viales de disolvente (1, 3 y 10 viales). Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Merck Serono Europe Limited, 56 Marsh Wall, London E14 9TP, Reino Unido Responsable de la fabricación
Merck Serono S.p.A, Via delle Magnolie 15 (Zona industríale), I-70026 Modugno (Bari), Italia
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización.
Lietuva
Merck Serono UAB Tel: +370 37320603
Belgie/Belgique/Belgien
MERCK NV/SA Tél/Tel: +32-2-686 07 11
Etarapna
„MepK E'MrapHa“ EAfl Tea.: +359 24461 111
Luxembourg/Luxemburg
MERCK NV/SA, Belgique/Belgien Tél/Tel: +32-2-686 07 11
Ceská republika
Merck spol.s.r.o
Tel. +420 272084211
|
Danmark Merck A/S Tlf: +45 35253550 |
Malta Cherubino Ltd Tel: +356-21-343270/1/2/3/4 |
|
Deutschland Merck Serono GmbH Tel: +49-6151-6285-0 |
Nederland Merck BV Tel: +31-20-6582800 |
|
Eesti Merck Serono OÜ Tel: +372 682 5882 |
Norge Merck Serono Norge Tlf: +47 67 90 35 90 |
|
EAAáSa Merck A.E. T^A: +30-210-61 65 100 |
Osterreich Merck GesmbH. Tel: +43 1 57600-0 |
|
España Merck S.L. Línea de Información: 900 200 400 Tel: +34-91-745 44 00 |
Polska Merck Sp. z o.o. Tel.: +48 22 53 59 700 |
|
France Merck Serono s.a.s. Tél.: +33-4-72 78 25 25 Numéro vert : 0 800 888 024 |
Portugal Merck, s.a. Tel: +351-21-361 35 00 |
|
Hrvatska Merck d.o.o, Tel: +385 1 4864 111 |
Romania MERCK d.o.o., Slovenia Tel: +386 1 560 3 800 |
|
Íreland Merck Serono Ltd, United Kingdom Tel: +44-20 8818 7200 |
Slovenija MERCK d.o.o. Tel: +386 1 560 3 800 |
|
Ísland Icepharma hf Tel: + 354 540 8000 |
Slovenská republika Merck spol. s r.o. Tel: + 421 2 49 267 111 |
|
Italia Merck Serono S.p.A. Tel: +39-06-70 38 41 |
Suomi/Finland Merck Oy Puh/Tel: +358-9-8678 700 |
|
Kúnpoq Xp. r. nanaAorZou AtS T^A.: +357 22490305 |
Sverige Merck AB Tel: +46-8-562 445 00 |
|
Latvija Merck Serono SIA Tel: +371 67152500 |
United Kingdom Merck Serono Ltd Tel: +44-20 8818 7200 |
Fecha de la última revisión de este prospecto: {MM/AAAA}
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
30