Osseor 2 G Granulado Para Suspension Oral
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
OSSEOR 2 g granulado para suspensión oral.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada sobre contiene 2 g de ranelato de estroncio.
Excipiente con efecto conocido:
Cada sobre también contiene 20 mg de aspartamo (E951).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Granulado para suspensión oral. Gránulos de color amarillo.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento de la osteoporosis severa:
- en mujeres posmenopáusicas,
- en hombres adultos,
con alto riesgo de fracturas, para los que el tratamiento con otros medicamentos aprobados para el tratamiento de la osteoporosis no es posible debido a, por ejemplo, contraindicaciones o intolerancia. En mujeres posmenopáusicas, el ranelato de estroncio reduce el riesgo de fracturas vertebrales y de cadera (ver sección 5.1).
La decisión de prescribir ranelato de estroncio debe estar basada en la valoración individual de los riesgos globales de cada paciente (ver sección 4.3 y 4.4).
4.2 Posología y forma de administración
El tratamiento sólo debe ser iniciado por un médico con experiencia en el tratamiento de la osteoporosis.
Posología
La dosis recomendada consiste en un sobre de 2 g, una vez al día, por vía oral.
Dada la naturaleza de la enfermedad tratada, el ranelato de estroncio está destinado al uso a largo plazo.
La absorción del ranelato de estroncio disminuye con los alimentos, la leche y los productos lácteos, de modo que OSSEOR debe administrarse entre las comidas. Como OSSEOR se absorbe lentamente, debe tomarlo preferiblemente al acostarse, si es posible, dos horas después de cenar como mínimo (ver secciones 4.5 y 5.2).
Los pacientes tratados con ranelato de estroncio deben recibir suplementos de vitamina D y calcio, si la ingestión alimentaria resulta insuficiente.
Pacientes de edad avanzada
Se han constatado la eficacia y la seguridad del ranelato de estroncio en hombres adultos y mujeres posmenopáusicas con osteoporosis de una amplia franja de edad (pacientes de hasta 100 años en el momento de la inclusión). No es necesario el ajuste de dosis en función de la edad.
Insuficiencia renal
El ranelato de estroncio no se recomienda en pacientes con insuficiencia renal grave (aclaramiento de creatinina menor de 30 ml/min) (ver secciones 4.4 y 5.2). Los pacientes con insuficiencia renal leve o moderada (aclaramiento de creatinina de 30-70 ml/min) no precisan ningún ajuste de dosis (ver secciones 4.4 y 5.2).
Insuficiencia hepática
Los pacientes con insuficiencia hepática no precisan ningún ajuste de dosis (ver sección 5.2).
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de OSSEOR en niños menores de 18 años. No hay datos disponibles.
Forma de administración Para uso oral.
Los gránulos contenidos en los sobres deben tomarse en forma de suspensión en un vaso conteniendo un mínimo de 30 ml de agua (aproximadamente un tercio de un vaso estándar).
Aunque los estudios sobre su uso han demostrado la estabilidad del ranelato de estroncio en suspensión durante las 24 horas siguientes a su preparación, la suspensión debe beberse de inmediato una vez preparada.
4.3 Contraindicaciones
- Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
- Episodios de tromboembolismo venoso (TEV) actuales o previos, incluyendo trombosis venosa profunda y embolismo pulmonar.
- Inmovilización permanente o temporal debida p. ej. a recuperación post-quirúrgica o reposo prolongado en cama.
- Episodios actuales o antecedentes de cardiopatía isquémica, enfermedad arterial periférica y/o enfermedad cerebrovascular.
- Hipertensión arterial no controlada.
4.4 Advertencias y precauciones especiales de empleo
Cardiopatía isquémica
En estudios agrupados randomizados controlados con placebo en pacientes postmenopáusicas con osteoporosis, se ha observado un aumento significativo de infarto de miocardio en las pacientes tratadas con OSSEOR en comparación con placebo (ver sección 4.8).
Los pacientes deben ser evaluados con respecto al riesgo cardiovascular antes de comenzar el tratamiento.
Los pacientes con factores de riesgo significativos de eventos cardiovasculares (ej.: hipertensión, hiperlipidemia, diabetes mellitus, fumadores) deben ser tratados con ranelato de estroncio únicamente tras una cuidadosa consideración (ver sección 4.3 y 4.8).
Durante el tratamiento con OSSEOR, se deben evaluar estos riesgos cardiovasculares periódicamente, generalmente cada 6-12 meses.
El tratamiento debe interrumpirse si el paciente desarrolla cardiopatía isquémica, enfermedad arterial periférica, enfermedad cerebrovascular o si la hipertensión arterial no está controlada (ver sección 4.3).
Tromboembolia venosa
En los estudios de fase III, controlados con placebo, el tratamiento con ranelato de estroncio se asoció con una mayor incidencia anual de tromboembolia venosa (TEV), incluida la embolia pulmonar (ver sección 4.8). Se ignora la causa de este hallazgo. OSSEOR está contraindicado en pacientes con antecedentes de episodios de tromboembolismo venoso (ver sección 4.3) y debe utilizarse con precaución en pacientes con riesgo de TEV.
Cuando se trate a pacientes mayores de 80 años con riesgo de TEV, debe revaluarse la necesidad de continuar el tratamiento con OSSEOR.
Se debe interrumpir el tratamiento con OSSEOR tan pronto como sea posible en el caso de una enfermedad o un proceso que conlleve una inmovilización (ver sección 4.3) y tomar las medidas preventivas adecuadas. El tratamiento no debe reiniciarse hasta que la dolencia inicial se ha resuelto y el paciente ha recuperado totalmente la movilidad. Cuando se produce una TEV, el tratamiento con OSSEOR debe interrumpirse.
Uso en pacientes con insuficiencia renal
Al no disponer de datos sobre la seguridad ósea entre pacientes con insuficiencia renal grave tratados con ranelato de estroncio, se desaconseja el uso de OSSEOR si el aclaramiento de creatinina es inferior a 30 ml/min (ver sección 5.2). De acuerdo con la buena práctica clínica, se aconseja una evaluación periódica de la función renal de los pacientes con insuficiencia renal crónica. La continuación del tratamiento con OSSEOR por parte de los pacientes con insuficiencia renal grave se considerará de manera individual.
Reacciones cutáneas
Con el uso de OSSEOR se han notificado reacciones cutáneas con amenaza vital (síndrome de Stevens-Johnson (SSJ), necrólisis epidérmica tóxica (NET) y erupción cutánea con eosinofilia y síntomas sistémicos (DRESS)).
Se debe advertir a los pacientes de los signos y síntomas y llevar un control cuidadoso de las reacciones cutáneas. El mayor riesgo para la aparición de SSJ o NET está dentro de las primeras semanas de tratamiento y por lo general alrededor de las 3-6 semanas para DRESS.
Si aparecen síntomas o signos de SSJ o NET (ej.: erupción cutánea progresiva a menudo con ampollas o lesiones de la mucosa) o DRESS (ej.: erupción cutánea, fiebre, eosinofilia y afectación orgánica (ej.: adenopatía, hepatitis, nefropatía intersticial, enfermedad pulmonar intersticial)), se debe interrumpir inmediatamente el tratamiento con OSSEOR.
Los mejores resultados en el control del SSJ, NET o DRESS provienen de un diagnóstico precoz y de la interrupción inmediata de cualquier medicamento sospechoso. La retirada temprana se asocia con un mejor pronóstico. El desenlace clínico de DRESS es favorable en la mayoría de los casos tras la interrupción del tratamiento con OSSEOR y tras el inicio de terapia con corticosteroides. La recuperación podría ser lenta y se han notificado recidivas del síndrome en algunos casos tras suspender la terapia con corticosteroides.
Si el paciente ha desarrollado SSJ, NET o DRESS con el uso de OSSEOR, no se debe reiniciar en ningún momento el tratamiento con OSSEOR en este paciente.
Se ha notificado una mayor incidencia, aunque todavía rara, de reacciones de hipersensibilidad incluyendo erupciones cutáneas, SSJ o NET en pacientes de origen asiático (ver sección 4.8).
En un estudio farmacogenético, retrospectivo, caso-control se han identificado los alelos HLA-A*33:03 y HLA-B*58:01 como potenciales factores de riesgo genéticos para el SSJ/NET asociado al ranelato de estroncio en pacientes chinos Han. En los casos en los que sea posible, se puede considerar realizar un estudio de los alelos HLA-A*33:03 y HLA-B*58:01 antes de empezar el tratamiento con OSSEOR en pacientes de origen chino Han. Si los controles son positivos para uno o ambos alelos, no se debe iniciar el tratamiento con OSSEOR. Sin embargo, la ausencia de estos alelos tras el análisis del genotipo, no excluye que pueda aparecer SSJ/NET.
Interacción con pruebas analíticas
El estroncio interfiere los métodos colorimétricos para la determinación de las concentraciones sanguíneas y urinarias de calcio. Por eso, en la práctica clínica, para medir con exactitud las concentraciones sanguíneas y urinarias de calcio se requieren métodos de espectrometría de emisión atómica con plasma de acoplamiento inductivo o bien de espectrometría de absorción atómica.
Excipientes
OSSEOR contiene aspartamo, una fuente de fenilalanina que puede ser perjudicial para las personas con fenilcetonuria.
4.5 Interacción con otros medicamentos y otras formas de interacción
Los alimentos, la leche y los productos lácteos y los medicamentos que contienen calcio pueden reducir la biodisponibilidad del ranelato de estroncio en un 60-70%. Por eso, hay que separar la administración de OSSEOR y de dichos productos, como mínimo, dos horas (ver secciones 4.2 y 5.2).
Como los cationes divalentes pueden formar complejos en el tracto gastrointestinal con la tetraciclina (ej. doxiciclina) y las quinolonas (ej. ciprofloxacino) administradas por vía oral y, en consecuencia, podrían reducir su absorción, se desaconseja la administración simultánea del ranelato de estroncio con estos medicamentos. Como medida de precaución, el tratamiento con OSSEOR debe suspenderse mientras se administren la tetraciclina o las quinolonas por vía oral.
En un estudio de interacción clínica in vivo se comprobó que la administración de los hidróxidos de aluminio y magnesio, bien dos horas antes o junto con el ranelato de estroncio, reducía ligeramente la absorción del ranelato de estroncio (descenso de AUC del 20-25%), mientras que la absorción apenas se modificaba cuando el antiácido se administraba dos horas después del ranelato de estroncio. Por consiguiente, es preferible tomar los antiácidos, como mínimo, dos horas después de OSSEOR. No obstante, si esta pauta posológica no es factible, dada la recomendación de administrar OSSEOR al acostarse, se puede aceptar la ingestión concomitante.
No se ha encontrado ninguna interacción con los suplementos de vitamina D por vía oral.
En los ensayos clínicos no se apreció ningún indicio de interacción clínica o de aumento relevante de los valores sanguíneos de estroncio con los fármacos que es de esperar que se prescriban habitualmente junto con OSSEOR a la población destinataria. Estos medicamentos comprendían: antiinflamatorios no esteroideos (incluido el ácido acetilsalicílico), anilidas (como el paracetamol), antagonistas H2 inhibidores de la bomba de protones, diuréticos, digoxina y glucósidos digitálicos, nitratos orgánicos y otros vasodilatadores para las enfermedades cardíacas, antagonistas del calcio, betabloqueantes, IECA, antagonistas de la angiotensina II, agonistas selectivos de los receptores adrenérgicos beta-2, anticoagulantes orales, antiagregantes plaquetarios, estatinas, fibratos y benzodiazepinas.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No existen datos sobre la utilización de ranelato de estroncio en mujeres embarazadas.
En los estudios con animales, las dosis altas mostraron efectos óseos reversibles en la descendencia de las ratas y conejas tratadas durante la gestación (ver sección 5.3). Si se administrara OSSEOR involuntariamente durante el embarazo, se suspenderá el tratamiento.
Lactancia
Datos físico-químicos indican que el ranelato de Estroncio se excreta en la leche humana. OSSEOR no debe administrarse durante la lactancia.
Fertilidad
No se observaron efectos sobre la fertilidad de machos y hembras en estudios en animales.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de OSSEOR sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
OSSEOR se ha investigado en ensayos clínicos donde intervinieron casi 8.000 participantes. La seguridad a largo plazo se ha evaluado en estudios de fase III entre mujeres posmenopáusicas con osteoporosis que recibieron tratamiento con 2 g/día de ranelato de estroncio (n=3.352) o placebo (n=3.317) a lo largo de 60 meses, como máximo. La media de edad en el momento de la inclusión era de 75 años y el 23% de las pacientes reclutadas tenía entre 80 y 100 años.
En un análisis de datos agrupados de estudios aleatorizados controlados con placebo en pacientes osteoporóticas posmenopáusicas, las reacciones adversas más frecuentes consistieron en náuseas y diarrea que, por regla general, aparecieron al comienzo del tratamiento sin que luego se apreciaran grandes diferencias entre los grupos. La retirada del tratamiento obedeció, sobre todo, a las náuseas.
No hubo ninguna diferencia en la naturaleza de las reacciones adversas entre los diferentes grupos tratados, con independencia de que las pacientes tuvieran una edad inferior o superior a 80 años en el momento de la inclusión.
Tabla de reacciones adversas
Las siguientes reacciones adversas han sido notificadas durante los ensayos clínicos y/o durante la utilización post-comercialización con ranelato de estroncio.
Las reacciones adversas, se enumeran a continuación empleando la convención siguiente: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
|
Sistema de Clasificación de Órganos |
Frecuencia |
Reacción adversa |
|
Trastornos de la sangre y del sistema linfático |
Poco frecuentes |
Linfadenopatía (asociada con reacciones de hipersensibilidad cutánea) |
|
Raras |
Insuficiencia de la médula ósea# | |
|
Eosinofilia (asociada con reacciones de hipersensibilidad cutánea) | ||
|
Trastornos del metabolismo y de la nutrición |
Frecuentes |
Hipercolesterolemia |
|
Trastornos psiquiátricos |
Frecuentes |
Insomnio |
|
Poco frecuentes |
Estado de confusión | |
|
Trastornos del sistema nervioso |
Frecuentes |
Cefalea |
|
Trastornos de la consciencia | ||
|
Pérdida de memoria | ||
|
Mareo | ||
|
Parestesia | ||
|
Poco frecuentes |
Crisis convulsivas | |
|
Trastornos del oído y del laberinto |
Frecuentes |
Vértigo |
|
Trastornos cardiacos |
Frecuentes |
Infarto de miocardio |
|
Trastornos vasculares |
Frecuentes |
Tromboembolia venosa (TEV) |
|
Trastornos respiratorios, torácicos y mediastínicos |
Frecuentes |
Hiperreactividad bronquial |
|
Trastornos gastrointestinales |
Frecuentes |
Náuseas |
|
Diarrea y Heces blandas | ||
|
Vómitos | ||
|
Dolor abdominal | ||
|
Dolor gastrointestinal | ||
|
Reflujo gastroesofágico | ||
|
Dispepsia |
|
Estreñimiento | ||
|
Flatulencia | ||
|
Poco frecuentes |
Irritación de la mucosa oral (estomatitis y/o úlceras bucales) | |
|
Xerostomía | ||
|
Trastornos hepatobiliares |
Frecuentes |
Hepatitis |
|
Poco frecuentes |
Aumento de las transaminasas séricas (asociado con reacciones de hipersensibilidad cutánea) | |
|
Trastornos de la piel y del tejido subcutáneo |
Muy frecuentes |
Reacciones de hipersensibilidad cutánea (erupción cutánea, prurito, urticaria, angioedema) § |
|
Frecuentes |
Eccema | |
|
Poco frecuentes |
Dermatitis | |
|
Alopecia | ||
|
Raras |
Erupción Cutánea con Eosinofilia y Síntomas Sistémicos (DRESS) (ver sección 4.4)# | |
|
Muy raras |
Reacciones adversas cutáneas graves (SCARs): síndrome de Stevens- Johnson y necrólisis epidérmica tóxicac* (ver sección 4.4)# | |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Muy frecuentes |
Artromialgias (calambres musculares, mialgias, dolores óseos, artralgias y dolores en las extremidades)§ |
|
Trastornos generales y |
Frecuentes |
Edema periférico |
|
alteraciones en el lugar de |
Poco frecuentes |
Fiebre (asociada con reacciones de |
|
administración |
hipersensibilidad cutánea) | |
|
Malestar general | ||
|
Exploraciones complementarias |
Frecuentes |
Aumento de la Creatina-fosfocinasa sanguínea (CPK) a |
§ La frecuencia en los Ensayos Clínicos fue similar en el grupo tratado con ranelato de estroncio y en grupo placebo.
* Notificadas como raras en los países asiáticos
# Para reacciones adversas no observadas en los ensayos clínicos, el límite superior del intervalo de confianza del 95% no es mayor que el 3/X con X representando el tamaño de la muestra total obtenida de la suma de todos los ensayos clínicos y estudios.
a Fracción musculoesquelética >3 veces el límite superior de la normalidad. En la mayoría de los casos, estos valores revirtieron espontáneamente a la normalidad sin modificar el tratamiento.
Descripción de reacciones adversas seleccionadas
Tromboembolia venosa
En los estudios de fase III, la incidencia anual de tromboembolia venosa (TEV) observada a lo largo de 5 años se aproximó a 0,7%; el riesgo relativo para los pacientes tratados con ranelato de estroncio resultó de 1,4 en comparación con el del placebo (95% CI = [1,0; 2,0]) (ver sección 4.4).
Infarto de miocardio
En estudios agrupados randomizados controlados con placebo en pacientes postmenopáusicas con osteoporosis, se ha observado un aumento significativo de infarto de miocardio en las pacientes tratadas con ranelato de estroncio en comparación con placebo (1,7% versus 1,1%), con un riesgo relativo de 1,6 (95% CI = [1,07 ; 2,38]).
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Síntomas
En un estudio clínico donde se investigó la administración repetida de 4 g de ranelato de estroncio al día durante 25 días, las mujeres posmenopáusicas sanas toleraron bien la medicación. La administración aislada de dosis de hasta 11 g a varones voluntarios jóvenes y sanos no causó ningún síntoma especial.
Tratamiento
Tras los episodios de sobredosificación durante los ensayos clínicos (hasta 4 g/día durante un máximo de 147 días) no se observó ninguna complicación clínica.
La administración de leche o antiácidos podría reducir la absorción del principio activo. En el caso de una sobredosificación considerable, cabe plantear la inducción del vómito para eliminar el principio activo no absorbido.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Medicamentos para tratar las enfermedades óseas - Otros fármacos que modifican la estructura y la mineralización ósea, código ATC: M05BX03.
Mecanismo de acción
En condiciones in vitro, el ranelato de estroncio:
- aumenta la formación de hueso en los cultivos de tejido óseo así como la replicación de los precursores de osteoblastos y la síntesis de colágeno en los cultivos de células óseas.
- reduce la resorción ósea al disminuir la diferenciación de los osteoclastos y la actividad de resorción.
De esta manera, el balance del recambio óseo se inclina a favor de la formación de hueso.
La actividad del ranelato de estroncio se investigó en diversos modelos preclínicos. En concreto, el ranelato de estroncio aumenta la masa ósea trabecular, así como el número y el grosor de las trabéculas de ratas intactas. Con ello, mejora la fuerza ósea.
En el tejido óseo animal y humano tratados con estroncio, éste se adsorbe sobre todo en la superficie de los cristales y apenas reemplaza al calcio en los cristales de apatita del hueso recién formado. El ranelato de estroncio no modifica las características de los cristales óseos. Las biopsias de la cresta ilíaca, obtenidas hasta 60 meses después del tratamiento con 2 g/día de ranelato de estroncio en los ensayos de fase III no revelaron ningún efecto nocivo para la calidad o mineralización de los huesos.
Los efectos combinados de la distribución ósea del estroncio (ver sección 5.2) y la mayor absorción de los rayos X por el estroncio, en comparación con el calcio, explica el incremento de la densidad mineral ósea (DMO) medida por absorciometría de rayos X de doble fotón (DXA). Los datos disponibles señalan que estos factores explican casi la mitad del cambio de la DMO durante los tres años de tratamiento con 2 g/día de OSSEOR. Este dato debe tomarse en consideración al interpretar las variaciones de la DMO durante el tratamiento con OSSEOR. En los estudios de fase III, que pusieron de relieve la eficacia del tratamiento de OSSEOR frente a las fracturas, la DMO media hallada con OSSEOR aumentó (con respecto a la basal) casi en un 4% cada año en la columna lumbar y un 2% cada año en el cuello femoral, alcanzando entre el 13% y el 15% y entre el 5% y el 6%, respectivamente, a los 3 años, en función de los estudios respectivos.
En los estudios de fase III, los marcadores bioquímicos de la formación de hueso (fosfatasa alcalina específica del hueso y propéptido carboxiterminal del procolágeno de tipo I) aumentaron en comparación con el placebo y los de la resorción (C-telopéptido sérico y entrecruzamientos del N-telopéptido urinario) disminuyeron a partir del tercer mes de tratamiento hasta los tres años.
Las concentraciones séricas del calcio y de la hormona paratiroidea (PTH) disminuyen ligeramente mientras que las concentraciones sanguíneas de fósforo y la actividad de la fosfatasa alcalina total aumentan como consecuencia de los efectos farmacológicos del ranelato de estroncio, si bien no se observó ninguna secuela clínica.
Eficacia clínica
La osteoporosis se define como un valor de la DMO de la columna o de la cadera situado 2,5 DE o más por debajo del valor medio de una población joven y sana. Hay una serie de factores de riesgo asociados con la osteoporosis posmenopáusica, como la masa ósea reducida, la densidad mineral ósea baja, la menopausia prematura, los antecedentes de tabaquismo y los antecedentes familiares de osteoporosis. La secuela clínica de la osteoporosis son las fracturas. El riesgo de fracturas aumenta con el número de factores de riesgo.
Tratamiento de la osteoporosis posmenopáusica:
El programa de estudios de prevención de fracturas con OSSEOR se componía de dos ensayos de fase III controlados con placebo: los ensayos SOTI y TROPOS. En SOTI participaron 1649 mujeres posmenopáusicas con osteoporosis consolidada (DMO lumbar baja y fracturas vertebrales prevalentes) y una media de edad de 70 años. En TROPOS intervinieron 5091 mujeres posmenopáusicas con osteoporosis (DMO baja del cuello del fémury fracturas prevalentes en más de la mitad de ellas) y una media de edad de 77 años. En conjunto, en SOTI y TROPOS se reclutó a 1556 pacientes que tenían más de 80 años en el momento de la inclusión (23,1% de la población examinada). Además del tratamiento (2 g/día de ranelato de estroncio o del placebo), las pacientes recibieron suplementos ajustados de calcio y vitamina D en ambos estudios.
OSSEOR redujo el riesgo relativo de nuevas fracturas vertebrales en un 41% a lo largo de 3 años en el estudio SOTI (tabla 1). El efecto alcanzó significación a partir del primer año. Se observaron efectos beneficiosos parecidos entre las mujeres con varias fracturas iniciales. En cuanto a las fracturas vertebrales clínicas (definidas como las fracturas asociadas con dolor de espalda y/o una pérdida de talla de, al menos, 1 cm), el riesgo relativo disminuyó en un 38%. OSSEOR también redujo el número de pacientes con una pérdida de altura de 1 cm, como mínimo, en comparación con el placebo.
La evaluación de la calidad de vida con la escala específica QUALIOST y con el índice de percepción de salud general de la escala general SF-36 reveló el beneficio de OSSEOR en comparación con placebo.
La eficacia de OSSEOR a la hora de reducir el riesgo de nuevas fracturas vertebrales se confirmó en el estudio TROPOS, incluso para las pacientes con osteoporosis que no presentaban ninguna fractura por fragilidad ósea en condiciones basales.
Tabla 1: Incidencia de pacientes con fracturas vertebrales y reducción del riesgo relativo
|
Estudio |
Placebo |
OSSEOR |
Reducción del riesgo relativo frente al placebo (IC del 95%), valor p |
|
SOTI |
N=723 |
N=719 | |
|
Nueva fractura vertebral durante los 3 años |
32,8% |
20,9% |
41% (27-52), p<0,001 |
|
Nueva fractura vertebral durante el 1er año |
11,8% |
6,1% |
49% (26-64), p<0,001 |
|
Nueva fractura vertebral clínica durante los 3 años |
17,4% |
11,3% |
38% (17-53), p<0,001 |
|
TROPOS |
N=1823 |
N=1817 | |
|
Nueva fractura vertebral durante los 3 años |
20,0% |
12,5% |
39% (27-49), p<0,001 |
Entre las pacientes que tenían más de 80 años en el momento de la inclusión se efectuó un análisis global de los estudios SOTI y TROPOS; se comprobó que OSSEOR reducía el riesgo relativo de nuevas fracturas vertebrales en un 32% a lo largo de 3 años (incidencia del 19,1% con el ranelato de estroncio frente al 26,5% con el placebo).
En un análisis a posteriori de las pacientes de los estudios SOTI y TROPOS agrupados, que presentaban una DMO basal en la columna lumbar, en el cuello del fémur o en ambos en el intervalo osteopénico y ninguna fractura prevalente, pero con un factor de riesgo adicional de fractura, por lo menos, (N=176), OSSEOR redujo el riesgo de la primera fractura vertebral en un 72% a lo largo de 3 años (incidencia de fractura vertebral 3,6% con el ranelato de estroncio frente a 12,0% con el placebo).
Se realizó un análisis a posteriori de un subgrupo de pacientes del estudio TROPOS con un interés clínico especial y un alto riesgo de fractura [definida por un índice T de la DMO del cuello del fémur < -3 DE (el intervalo del fabricante correspondía a -2,4 DE basado en NHANES III) y una edad > 74 años (n=1977, es decir, 40% de la población del estudio TROPOS)]. En este grupo, OSSEOR redujo el riesgo de fractura de cadera en un 36%, con relación al placebo, durante los 3 años de tratamiento (tabla 2).
Tabla 2: incidencia de pacientes con fractura de cadera y reducción relativa del riesgo de las pacientes con DMO < -2,4 DE (NHANES III) y una edad > 74 años
|
Estudio |
Placebo |
OSSEOR |
Reducción del riesgo relativo frente al placebo |
|
(IC del 95%), valor p | |||
|
TROPOS |
N=995 |
N=982 | |
|
Fractura de cadera durante 3 años |
6,4% |
4,3% |
36% (0-59), p=0,046 |
Tratamiento de la osteoporosis en hombres:
La eficacia de OSSEOR se demostró en hombres con osteoporosis con un riesgo elevado de fractura (edad media 72,7 años; un índice T medio de la DMO lumbar de -2,6; 28% de fracturas vertebrales prevalentes), en un estudio controlado con placebo, doble ciego, de 2 años de duración, con un análisis principal después de un año en 243 pacientes (población con intención de tratar, 161 pacientes recibieron ranelato de estroncio).
Todos los pacientes recibieron suplementos diarios de calcio (1000 mg) y vitamina D (800 UI).
Tan solo a los 6 meses del inicio del tratamiento con OSSEOR ya se observaron aumentos estadísticamente significativos en la DMO en comparación con placebo.
A lo largo de 12 meses, se observó un aumento estadísticamente significativo en la DMO media de la columna lumbar, criterio principal de eficacia (E (SE) = 5,32% (0,75); IC 95% = [3,86; 6,79]; p <0,001), similar al observado en los estudios pivotales anti-fractura de fase III llevados a cabo en mujeres posmenopáusicas.
Tras 12 meses se observaron aumentos estadísticamente significativos en la DMO del cuello del fémur y en la DMO total de la cadera (p <0,001).
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con OSSEOR en los diferentes grupos de la población pediátrica en osteoporosis (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 Propiedades farmacocinéticas
El ranelato de estroncio se compone de dos átomos de estroncio estable y una molécula de ácido ranélico; el componente orgánico facilita un compromiso óptimo entre el peso molecular, la farmacocinética y la aceptabilidad del fármaco. Las farmacocinéticas del estroncio y del ácido ranélico se han comprobado entre varones jóvenes sanos y mujeres posmenopáusicas sanas y también durante la exposición prolongada en hombres con osteoporosis y mujeres con osteoporosis posmenopáusica, incluidas mujeres de edad avanzada.
Debido a su elevada polaridad, la absorción, distribución y unión a las proteínas plasmáticas del ácido ranélico son bajas. El ácido ranélico no se acumula ni tampoco se metaboliza en los animales o en la especie humana. El ácido ranélico absorbido se elimina rápidamente e inalterado por los riñones.
Absorción
La biodisponibilidad absoluta del estroncio se aproxima al 25% (intervalo: 19-27%) después de administrar una dosis oral de ranelato de estroncio de 2 g. Las concentraciones plasmáticas máximas se alcanzan de 3 a 5 horas después de una dosis única de 2 g. El estado estacionario se alcanza después de dos semanas de tratamiento. La ingestión del ranelato de estroncio con el calcio o los alimentos reduce la biodisponibilidad del estroncio aproximadamente en un 60-70%, en comparación con su administración 3 horas después de las comidas. Debido a la absorción relativamente lenta del estroncio, conviene evitar la ingestión de alimentos y de calcio tanto antes como después de administrar OSSEOR. Los suplementos de vitamina D por vía oral no afectan la exposición al estroncio.
Distribución
El estroncio posee un volumen de distribución aproximado de 1 l/kg. La unión del estroncio a las proteínas plasmáticas humanas es baja (25%); el estroncio muestra una gran afinidad por el tejido óseo. La medición de la concentración del estroncio en muestras de biopsia de la cresta ilíaca de pacientes tratadas hasta 60 meses con 2 g/día de ranelato de estroncio indicó que la concentración ósea de estroncio puede alcanzar una meseta al cabo de unos 3 años de tratamiento. No hay datos que revelen la cinética de eliminación ósea del estroncio después del tratamiento.
Biotransformación
El estroncio, como catión divalente, no se metaboliza. El ranelato de estroncio no inhibe las enzimas del citocromo P450.
Eliminación
La eliminación del estroncio no depende del tiempo ni de la dosis. La semivida eficaz del estroncio es de unas 60 horas. El estroncio se excreta por los riñones y el tracto gastrointestinal. Su depuración plasmática se acerca a 12 ml/min (CV 22%) y su depuración renal, 7 ml/min (CV 28%).
Farmacocinética en poblaciones especiales
Pacientes de edad avanzada
Los datos farmacocinéticos de población no revelaron ninguna relación entre la edad y la depuración aparente del estroncio en la población destinataria.
Insuficiencia renal
En pacientes con insuficiencia renal leve o moderada (aclaramiento de creatinina de 30-70 ml/min), la depuración del estroncio disminuye según lo hace el aclaramiento de creatinina (un descenso aproximado del 30% en un intervalo de aclaramiento de creatinina de 30 a 70 ml/min) y, en consecuencia, induce un aumento de los valores plasmáticos del estroncio. En los estudios de fase III, el 85% de las pacientes presentaba un aclaramiento de creatinina entre 30 y 70 ml/min y el 6%, inferior a 30 ml/min, en el momento de la inclusión; la media del aclaramiento de creatinina se aproximó a 50 ml/min. Por consiguiente, no es necesario ajustar la posología en pacientes con insuficiencia renal leve a moderada.
No hay datos farmacocinéticos en pacientes con insuficiencia renal grave (aclaramiento de creatinina inferior a 30 ml/min).
Insuficiencia hepática
No hay ningún dato farmacocinético en pacientes con insuficiencia hepática. En virtud de las propiedades farmacocinéticas del estroncio, no cabe esperar ningún efecto.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, genotoxicidad y potencial carcinogénico.
La administración oral crónica de ranelato de estroncio en dosis altas indujo anomalías óseas y dentales en roedores, principalmente fracturas espontáneas y retraso de la mineralización que revirtieron al suspender el tratamiento. Estos efectos aparecieron con valores óseos de estroncio dos a tres veces mayores que los valores óseos de estroncio en humanos que recibieron hasta tres años de tratamiento. Los datos de acumulación ósea tras exposición a ranelato de estroncio durante periodos más largos son limitados.
Los estudios sobre la toxicidad durante el desarrollo de ratas y conejos dieron como resultado anomalías óseas y dentales (p. ej., angulación de los huesos largos y costillas onduladas) en la descendencia. Los efectos en las ratas revirtieron a las 8 semanas de la interrupción del tratamiento.
Evaluación del Riesgo Medioambiental (ERA)
La evaluación del riesgo medioambiental del ranelato de estroncio se ha realizado de acuerdo con las guías europeas sobre ERA.
El ranelato de estroncio no presenta un riesgo para el medio ambiente.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Aspartamo (E951)
Maltodextrina Manitol (E421)
6.2 Incompatibilidades
No procede
6.3 Periodo de validez
- 3 años.
- Una vez reconstituida en agua, la suspensión es estable durante 24 horas. Sin embargo, se recomienda beber la suspensión inmediatamente después de su preparación (ver sección 4.2).
6.4 Precauciones especiales de conservación
Este medicamento no requiere condiciones especiales de conservación.
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Sobres de papel/polietileno/aluminio/polietileno.
Presentación
Cajas con 7, 14, 28, 56, 84 ó 100 sobres.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
Ninguna especial
LES LABORATOIRES SERVIER 50, rue Carnot 92284 Suresnes cedex Francia
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/04/287/001
EU/1/04/287/002
EU/1/04/287/003
EU/1/04/287/004
EU/1/04/287/005
EU/1/04/287/006
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 21/09/2004 Fecha de la última renovación: 22/05/2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
A. FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
Nombre y dirección del (de los) fabricante(s) responsable(s) de la liberación de los lotes
Les Laboratoires Servier Industrie 905, route de Saran 45520 Gidy Francia
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107ter, apartado 7, de la Directiva 2001/83/CE y cualquier actualización posterior publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva
información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
• Obligación de llevar a cabo medidas post-autorización
El TAC deberá llevar a cabo, dentro del plazo establecido, las siguientes medidas:
Descripción_
Estudio de seguridad observacional para evaluar la eficacia de las medidas de minimización de riesgos aplicadas, incluyendo una descripción de la población de pacientes tratados en la práctica clínica diaria, patrones de uso y riesgo cardiovascular.
Tras la autorización del protocolo, los informes anuales de este estudio deben ser presentados con el IPS hasta la presentación del informe final del estudio, que está previsto en Diciembre 2017._
Medidas adicionales de minimización de riesgo
En cada Estado Miembro donde OSSEOR está comercializado, el Titular de la Autorización de Comercialización (TAC) debe acordar el programa informativo final con la Autoridad Nacional Competente.
El TAC debe asegurarse de que, tras la discusión y el acuerdo con la Autoridad Nacional Competente en cada Estado Miembro donde OSSEOR está comercializado, todos los médicos que se espera que prescriban OSSEOR reciban el siguiente material educacional:
• Ficha Técnica
• Prospecto
• Información para el médico y lista de verificación
• Tarjeta de alerta para el paciente
La información para el médico y la lista de verificación debe contener los siguientes mensajes clave:
• OSSEOR sólo está indicado para el tratamiento de pacientes con osteoporosis severa con alto
riesgo de fracturas , para los que el tratamiento con otros medicamentos aprobados para el tratamiento de la osteoporosis no es posible debido a, por ejemplo, contraindicaciones o intolerancia.
• El inicio del tratamiento con OSSEOR debe estar basado en una valoración individual de los
riesgos globales de cada paciente.
• Todos los pacientes deben ser plenamente informados de que los riesgos cardiovasculares deben
ser evaluados periódicamente, generalmente cada 6-12 meses.
• La tarjeta de alerta para el paciente debe entregarse a cada paciente.
• OSSEOR está contraindicado y no debe utilizarse en pacientes con:
o Episodios actuales o antecedentes de cardiopatía isquémica, enfermedad arterial periférica y/o enfermedad cerebrovascular.
o Hipertensión arterial no controlada.
o Episodios de tromboembolismo venoso (TEV) actuales o previos, incluyendo trombosis venosa profunda y embolismo pulmonar.
o Inmovilización permanente o temporal debida p. ej. a recuperación post-quirúrgica o reposo prolongado en cama.
o Hipersensibilidad al principio activo (ranelato de estroncio) o a alguno de los excipientes.
• OSSEOR debe utilizase con precaución en:
o Pacientes con factores significativos de riesgo para episodios cardiovasculares como hipertensión, hiperlipidemia, diabetes mellitus o tabaquismo.
o Pacientes con riesgo de TEV. Cuando se trate a pacientes mayores de 80 años con riego de TEV, debe reevaluarse la necesidad de continuar el tratamiento con OSSEOR.
• El tratamiento debe interrumpirse o suspenderse en las siguientes situaciones:
o Si el paciente desarrolla una cardiopatía isquémica, enfermedad arterial periférica, enfermedad cerebrovascular o si tiene hipertensión no controlada, el tratamiento debe interrumpirse.
o En el caso de una enfermedad o un estado que conlleve una inmovilización, el tratamiento debe suspenderse lo antes posible.
o Si aparecen síntomas o signos del Síndrome de Stevens-Johnson (SSJ), Necrólisis Epidérmica Tóxica (NET) o Erupción Cutánea con Eosinofilia y Síntomas Sistémicos (DRESS) (ej.: erupción cutánea, fiebre, eosinofilia y afectación orgánica, ej.: adenopatía, hepatitis, nefropatía intersticial, enfermedad pulmonar intersticial) se debe interrumpir inmediatamente el tratamiento con OSSEOR. Si el paciente ha desarrollado SSJ, NET o DRESS con el uso de OSSEOR, el tratamiento con OSSEOR no debe reiniciarse.
• Dentro del Material Informativo para el médico prescriptor habrá una lista de verificación para
recordar a los médicos prescriptores las contraindicaciones, advertencias y precauciones antes de prescribir y para facilitar la evaluación periódica del riesgo cardiovascular.
La tarjeta de alerta para el paciente debe contener los siguientes mensajes clave:
• La importancia de mostrar la tarjeta de alerta a cualquier Profesional Sanitario implicado en su
tratamiento.
• Las contraindicaciones para el tratamiento con OSSEOR.
• Los signos y síntomas principales del infarto de miocardio, TEV y reacciones cutáneas graves.
• Cuándo consultar urgentemente con un médico.
• La importancia del seguimiento periódico del riesgo cardiovascular.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
OSSEOR 2 g granulado para suspensión oral. Ranelato de Estroncio.
2. PRINCIPIO(S) ACTIVO(S)
Cada sobre contiene 2 g de Ranelato de Estroncio.
3. LISTA DE EXCIPIENTES
También contiene aspartamo (E 951).
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Granulado para suspensión oral. 7 sobres.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
Semana
Lunes
Martes
Miércoles
Jueves
Viernes
Sábado
Domingo
7. OTRAS ADVERTENCIAS ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Si no se utiliza el medicamento inmediatamente después de su reconstitución, debe consumirse en 24 horas.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Les Laboratoires Servier 50, rue Carnot 92284 Suresnes cedex Francia
12. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/04/287/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
OSSEOR 2 g
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único.
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC:
SN:
NN:
1. NOMBRE DEL MEDICAMENTO
OSSEOR 2 g granulado para suspensión oral. Ranelato de Estroncio.
2. PRINCIPIO(S) ACTIVO(S)
Cada sobre contiene 2 g de Ranelato de Estroncio.
3. LISTA DE EXCIPIENTES
También contiene aspartamo (E 951).
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Granulado para suspensión oral. 14 sobres.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
Martes
Miércoles
Jueves
Viernes
Sábado
Domingo
7. OTRAS ADVERTENCIAS ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Si no se utiliza el medicamento inmediatamente después de su reconstitución, debe consumirse en 24 horas.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Les Laboratoires Servier 50, rue Carnot 92284 Suresnes cedex Francia
12. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/04/287/002
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
OSSEOR 2 g
Incluido el código de barras 2D que lleva el identificador único.
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC:
SN:
NN:
1. NOMBRE DEL MEDICAMENTO
OSSEOR 2 g granulado para suspensión oral. Ranelato de Estroncio.
2. PRINCIPIO(S) ACTIVO(S)
Cada sobre contiene 2 g de Ranelato de Estroncio.
3. LISTA DE EXCIPIENTES
También contiene aspartamo (E 951).
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Granulado para suspensión oral. 28 sobres.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.

7. OTRAS ADVERTENCIAS ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Si no se utiliza el medicamento inmediatamente después de su reconstitución, debe consumirse en 24 horas.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Les Laboratoires Servier 50, rue Carnot 92284 Suresnes cedex Francia
12. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/04/287/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
OSSEOR 2 g
Incluido el código de barras 2D que lleva el identificador único.
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC:
SN:
NN:
1. NOMBRE DEL MEDICAMENTO
OSSEOR 2 g granulado para suspensión oral. Ranelato de Estroncio.
2. PRINCIPIO(S) ACTIVO(S)
Cada sobre contiene 2 g de Ranelato de Estroncio.
3. LISTA DE EXCIPIENTES
También contiene aspartamo (E 951).
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Granulado para suspensión oral. 56 sobres 84 sobres 100 sobres
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRAS ADVERTENCIAS ESPECIALES, SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Si no se utiliza el medicamento inmediatamente después de su reconstitución, debe consumirse en 24 horas.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Les Laboratoires Servier 50, rue Carnot 92284 Suresnes cedex Francia
12. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/04/287/004 56 sobres EU/1/04/287/005 84 sobres (3 envases de 28) EU/1/04/287/006 100 sobres
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
OSSEOR 2 g
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único.
PC:
SN:
NN:
31
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
Sobre
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
OSSEOR 2 g granulado para suspensión oral. Ranelato de Estroncio.
Vía oral.
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
6. OTROS
Leer el prospecto antes de utilizar este medicamento.
B. PROSPECTO
Prospecto: información para el paciente
OSSEOR 2 g granulado para suspensión oral
Ranelato de estroncio
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es OSSEOR y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar OSSEOR
3. Cómo tomar OSSEOR
4. Posibles efectos adversos
5. Conservación de OSSEOR
6. Contenido del envase e información adicional
1. Qué es OSSEOR y para qué se utiliza
OSSEOR es un medicamento que sirve para tratar la osteoporosis severa:
- en mujeres posmenopáusicas,
- en hombres adultos,
con alto riesgo de fracturas, para los que otros tratamientos alternativos no son posibles. En mujeres posmenopáusicas, el ranelato de estroncio reduce el riesgo de fracturas vertebrales y de cadera.
Acerca de la osteoporosis
El organismo está constantemente descomponiendo el hueso antiguo y fabricando hueso nuevo. Si usted sufre osteoporosis, su organismo descompone el hueso con más velocidad de la que lo forma y, por eso, va perdiendo hueso gradualmente. El hueso se vuelve más fino y frágil, sobre todo después de la menopausia.
Muchas personas con osteoporosis no sufren síntomas e incluso ignoran su enfermedad. Sin embargo, la osteoporosis aumenta el riesgo de fracturas (rotura del hueso), sobre todo en la columna vertebral, las caderas y las muñecas.
Cómo actúa OSSEOR
OSSEOR, el cual contiene ranelato de estroncio como sustancia activa, pertenece a un grupo de medicamentos que sirve para tratar las enfermedades óseas.
OSSEOR reduce la descomposición ósea y estimula la nueva formación de hueso, y por tanto, disminuye el riesgo de fracturas. El hueso recién formado tiene una calidad normal.
2. Qué necesita saber antes de empezar a tomar OSSEOR No tome OSSEOR:
- si es alérgico al ranelato de estroncio o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- si tiene o ha tenido un coágulo sanguíneo (por ejemplo, en los vasos sanguíneos de sus piernas o pulmones).
- si está inmovilizado de forma permanente o durante algún tiempo, como por ejemplo si está en silla de ruedas, o tiene que permanecer en cama o si va a ser operado o se está recuperando de una operación. El riesgo de trombosis venosa (coágulos de sangre en las piernas o pulmones) puede aumentar en el caso de inmovilización prolongada.
- si padece una cardiopatía isquémica, o enfermedad cerebrovascular, ej. si le han diagnosticado un ataque al corazón, ictus, o ataque isquémico transitorio (reducción temporal del flujo de sangre al cerebro; también conocido como “mini-ictus”), angina de pecho, o bloqueo de los vasos sanguíneos al corazón o al cerebro.
- si tiene o ha tenido problemas de circulación sanguínea (enfermedad arterial periférica) o si ha sido operado de las arterias de sus piernas.
- si tiene hipertensión arterial no controlada por el tratamiento.
Advertencias y precauciones
Consulte a su médico o farmacéutico antes de empezar a tomar OSSEOR:
- si tiene riesgo de enfermedad cardiaca, esto incluye hipertensión arterial, colesterol alto, diabetes, ser fumador.
- si tiene riesgo de desarrollar coágulos de sangre.
- si sufre una enfermedad renal grave.
Su médico evaluará periódicamente el estado de su corazón y sus vasos sanguíneos, generalmente cada 6-12 meses durante el tiempo que tome OSSEOR.
Durante el tratamiento, si sufre una reacción alérgica (como inflamación de la cara, lengua o garganta, dificultad para respirar o tragar, erupción cutánea), debe interrumpir inmediatamente su tratamiento con OSSEOR y acudir al médico (ver sección 4).
Con el uso de OSSEOR se han observado erupciones cutáneas con potencial amenaza para la vida (síndrome de Stevens-Johnson, necrólisis epidérmica tóxica y reacciones de hipersensibilidad graves (DRESS)).
El mayor riesgo para la aparición de reacciones cutáneas graves está dentro de las primeras semanas de tratamiento para el síndrome de Stevens-Johnson y la necrólisis epidérmica tóxica y por lo general alrededor de 3-6 semanas para el DRESS.
Si desarrolla una erupción o síntomas cutáneos graves (ver sección 4), interrumpa el tratamiento con OSSEOR, consulte urgentemente con un médico e infórmele de que está tomando este medicamento.
Si ha desarrollado síndrome de Stevens-Johnson o necrólisis epidérmica tóxica o DRESS con el uso de OSSEOR, no debe reiniciar en ningún momento el tratamiento con OSSEOR.
Si usted es de origen asiático, puede tener un mayor riesgo de sufrir reacciones cutáneas.
El riesgo de estas reacciones cutáneas en pacientes de origen asiático, en particular chinos Han, se puede prever. Los pacientes que tienen los genes HLA-A*33:03 y/o HLA-B*58:01 tienen más probabilidades de desarrollar una reacción cutánea grave que aquellos que no tienen estos genes.
Su médico debe advertirle en caso de que fuera necesario realizarle un análisis de sangre antes de tomar OSSEOR.
Niños y adolescentes
OSSEOR no está dirigido al uso en niños y adolescentes (menores de 18 años).
Uso de OSSEOR con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Deberá interrumpir el tratamiento con OSSEOR si recibe por vía oral tetraciclinas, tales como doxiciclina, o quinolonas, tales como ciprofloxacino (dos tipos de antibiótico). Podrá reanudar el tratamiento con OSSEOR cuando termine de tomar estos antibióticos. Si tiene alguna duda sobre esto, consulte a su médico o farmacéutico.
Si está tomando medicamentos que contengan calcio, deberán transcurrir, como mínimo, 2 horas antes de ingerir OSSEOR.
Si toma antiácidos (medicamentos que alivian el ardor de estómago), deberá tomarlos como mínimo 2 horas después de tomar OSSEOR. Si esto no es posible, puede tomar los dos medicamentos a la vez. Si tienen que realizarle un análisis de sangre u orina para revisar sus niveles de calcio, deberá comunicar al laboratorio que está tomando OSSEOR ya que puede interferir con algunos métodos analíticos.
Toma de OSSEOR con alimentos y bebidas
Los alimentos, la leche y los productos lácteos reducen la absorción del ranelato de estroncio. Se recomienda tomar OSSEOR separado de las comidas, preferiblemente al acostarse y al menos 2 horas después de tomar alimentos, leche o productos lácteos o suplementos de calcio.
Embarazo y lactancia:
No tome OSSEOR durante el embarazo o la lactancia. Si usted lo toma, por accidente, durante el embarazo o la lactancia, déjelo de tomar de inmediato y consulte a su médico.
Conducción y uso de máquinas
Es improbable que OSSEOR afecte a la capacidad para conducir o utilizar máquinas.
OSSEOR contiene aspartamo (E951):
Si usted sufre fenilcetonuria (un trastorno metabólico hereditario raro), consulte a su médico antes de tomar este medicamento.
3. Cómo tomar OSSEOR
El tratamiento sólo debe ser iniciado por un médico con experiencia en el tratamiento de la osteoporosis.
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
OSSEOR debe administrarse por vía oral.
La dosis normal es de un sobre de 2 g al día.
Se aconseja tomar OSSEOR al acostarse, preferiblemente al menos 2 horas después de la cena. Si lo desea, puede acostarse inmediatamente después de ingerir OSSEOR.
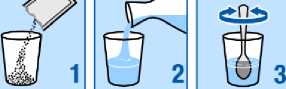
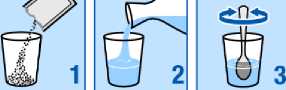
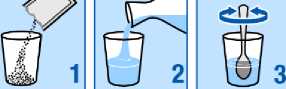
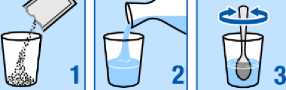
Tome los gránulos contenidos en los sobres como una suspensión en un vaso de agua que contenga un mínimo de 30 ml (aproximadamente un tercio de un vaso normal). Vea las instrucciones a continuación. OSSEOR puede interaccionar con la leche y los productos lácteos, por lo que resulta muy importante que sólo mezcle OSSEOR con agua para que actúe correctamente.


Vacíe los gránulos del sobre en un vaso
Añada agua
Remueva hasta que los gránulos se dispersen de forma uniforme dentro del agua.
Bébalo enseguida. No deben pasar más de 24 horas, desde que prepare la suspensión hasta que la tome. Si por alguna razón no puede tomar el medicamento de inmediato, vuelva a remover antes de tomarlo.
El médico le aconsejará si debe tomar suplementos de calcio o vitamina D, además de OSSEOR. No ingiera los suplementos de calcio al acostarse, es decir, a la misma hora que OSSEOR.
El médico le indicará cuánto tiempo debe tomar OSSEOR. El tratamiento de la osteoporosis suele ser prolongado. Conviene que tome OSSEOR durante todo el tiempo prescrito por su médico.
Si toma más OSSEOR del que debe
Si usted ingiere más sobres de OSSEOR que los recomendados por su médico, consulte a su médico o farmacéutico. Quizá, le aconsejen que beba leche o tome antiácidos para reducir la absorción del principio activo.
Si olvidó tomar OSSEOR
No tome una dosis doble para compensar las dosis olvidadas, tome la siguiente dosis a la hora prevista. Si interrumpe el tratamiento con OSSEOR:
Es importante que siga tomando OSSEOR durante el tiempo que su médico le haya recetado el medicamento. OSSEOR puede tratar su osteoporosis severa sólo si sigue su tratamiento.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no
todas las personas los sufran.
Si le ocurre lo siguiente, interrumpa el tratamiento con OSSEOR y hable con su médico
inmediatamente:
Frecuentes (pueden afectar hasta 1 de cada 10personas):
- Ataque al corazón: dolores opresivos repentinos en el pecho que pueden alcanzar su brazo izquierdo, mandíbula, estómago, espalda y/o hombros. Otros síntomas pueden ser nauseas/vómitos, sudoración, dificultad al respirar, palpitaciones, cansancio (extremo) y/o mareo. En pacientes con alto riesgo de enfermedad cardiaca puede ocurrir de forma frecuente un ataque al corazón. Su médico no le recetará OSSEOR en caso de que usted tenga una situación particular de riesgo.
- Coágulos de sangre en las venas: dolor, enrojecimiento, hinchazón de la pierna, dolor en el pecho repentino o dificultad para respirar.
Raros (pueden afectar hasta 1 de cada 1.000personas):
- Signos de reacciones de hipersensibilidad graves (DRESS): aparece inicialmente como síntomas de tipo gripal y una erupción cutánea en la cara, y después una erupción generalizada con temperatura elevada (poco frecuentes), aumento de los niveles de las enzimas hepáticas en los análisis de sangre (poco frecuentes) e incremento de un tipo de glóbulos blancos (eosinofilia) (raros) y aumento del tamaño de los ganglios linfáticos (poco frecuentes).
Muy raros (pueden afectar hasta 1 de cada 10.000personas):
- Signos de erupciones cutáneas potencialmente mortales (síndrome de Stevens-Johnson, necrólisis epidérmica tóxica): aparecen inicialmente como manchas rojizas en forma de diana o manchas circulares a menudo con ampollas centrales en el tronco. Otros signos adicionales pueden incluir úlceras en la boca, garganta, nariz, genitales y conjuntivitis (ojos rojos e hinchados). Estas erupciones cutáneas potencialmente mortales van a menudo acompañadas de
síntomas de tipo gripal. La erupción puede progresar a la formación de ampollas generalizadas o descamación de la piel.
Otros posibles efectos adversos
Muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
Picores, urticaria, erupción cutánea, angioedema (como inflamación de la cara, lengua o garganta, dificultad para respirar o tragar), dolor en huesos, extremidades, músculos y/o articulaciones, calambres musculares.
Frecuentes:
Vómitos, dolor abdominal, reflujo, indigestión, estreñimiento, flatulencia, dificultad para dormir, inflamación del hígado (hepatitis), inflamación de las extremidades, hiperreactividad bronquial (síntomas que incluyen respiración con pitos, dificultad al respirar y tos), niveles aumentados de una enzima muscular (Creatina-fosfocinasa), niveles aumentados de colesterol.
Náuseas, diarrea, dolor de cabeza, eczema, problemas de memoria, desvanecimientos, hormigueo, mareo, vértigo.
Sin embargo, estos efectos fueron leves y pasajeros y, por regla general, no obligaron a los pacientes a suspender el tratamiento. Consulte con su médico si nota algún efecto problemático o persistente.
Poco frecuentes (pueden afectar hasta 1 de cada 100 personas):
Convulsiones, irritación bucal (como úlceras en la boca e inflamación de las encías), pérdida de cabello, sensación de confusión, indisposición, sequedad de boca, irritación cutánea.
Raros:
Disminución en la formación de células sanguíneas en la médula ósea.
Si ha interrumpido el tratamiento debido a reacciones de hipersensibilidad, no tome OSSEOR de nuevo.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V.
Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de OSSEOR
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase y en el sobre después de CAD. La fecha de caducidad es el último día del mes que se indica.
Este medicamento no requiere condiciones especiales de conservación.
Una vez reconstituida en agua, la suspensión es estable durante 24 horas. Sin embargo, se recomienda beber la suspensión inmediatamente después de su preparación (ver sección 3).
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de OSSEOR
- El principio activo es ranelato de estroncio. Cada sobre contiene 2 g de ranelato de estroncio.
- Los demás componentes son aspartamo (E-951), maltodextrina y manitol (E-421).
Aspecto del producto y contenido del envase
OSSEOR se presenta en sobres que contienen gránulos de color amarillo para su suspensión y administración oral.
OSSEOR se suministra en cajas de 7, 14, 28, 56, 84 ó 100 sobres.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización
Les Laboratoires Servier 50, rue Carnot 92284 Suresnes cedex Francia
Responsable de la fabricación
Les Laboratoires Servier Industrie 905, route de Saran 45520 Gidy Francia
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización.
Belgie/Belgique/Belgien
S.A. Servier Benelux N.V. Tel: +32 (0)2 529 43 11
Etarapna
CepBne MegHKaa EOOfl Tea.: +359 2 921 57 00
Ceská republika
Servier s.r.o.
Tel: +420 222 118 111
Lietuva
UAB "SERVIER PHARMA" Tel: +370 (5) 2 63 86 28
Luxembourg/Luxemburg
S.A. Servier Benelux N.V. Tel: +32 (0)2 529 43 11
Magyarország
Servier Hungaria Kft.
Tel: +36 1 238 7799
Deutschland
Servier Deutschland GmbH Tel: +49 (0)89 57095 01
Eesti
Servier Laboratories OÜ Tel:+ 372 664 5040
Nederland
Servier Nederland Farma B.V. Tel: +31 (0)71 5246700
Norge
Servier Danmark A/S Tlf: +45 36 44 22 60
EXláóa Osterreich
IEPBIE EAAAI OAPMAKEYTIKH EnE Servier Austria GmbH
Tni: +30 210 939 1000 Tel: +43 (1) 524 39 99
Polska
Servier Polska Sp. z o.o. Tel: +48 (0) 22 594 90 00
España
Laboratorios Farmacéuticos Rovi, S.A. Tel: +34 91 375 62 30
|
France Les Laboratoires Servier Tel: +33 (0)1 55 72 60 00 |
Portugal Servier Portugal, Lda Tel.: +351 21 312 20 00 |
|
Hrvatska Servier Pharma, d. o. o. Tel.: +385 (0)1 3016 222 |
Romania Servier Pharma SRL Tel: +40 21 528 52 80 |
|
Ireland Servier Laboratories (Ireland) Ltd. Tel: +353 (0)1 6638110 |
Slovenija Servier Pharma d.o.o. Tel.: +386 (0)1 563 48 11 |
|
Ísland Servier Laboratories c/o Icepharma hf Sími: +354 540 8000 |
Slovenská republika Servier Slovensko spol. s r.o. Tel.:+421 (0)2 5920 41 11 |
|
Italia I.F.B. Stroder S.r.l. Tel: +39 06 669081 |
Suomi/Finland Servier Finland Oy P./Tel: +358 (0)9 279 80 80 |
|
Kórcpog C.A. Papaellinas Ltd. Tpk: +357 22741741 |
Sverige Servier Sverige AB Tel: +46 (0)8 522 508 00 |
|
Latvija SIA Servier Latvia Tel. +371 67502039 |
United Kingdom Servier Laboratories Ltd Tel: +44 (0)1753 666409 |
|
Fecha de la última revisión de este prospecto: |
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
40