Orfadin 2 Mg Capsulas Duras
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Orfadin 2 mg cápsulas duras Orfadin 5 mg cápsulas duras Orfadin 10 mg cápsulas duras Orfadin 20 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula contiene 2 mg de nitisinona.
Cada cápsula contiene 5 mg de nitisinona.
Cada cápsula contiene 10 mg de nitisinona.
Cada cápsula contiene 20 mg de nitisinona.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura.
Cápsulas blancas opacas (6x16 mm) con la estampación “NTBC 2mg” en negro en el cuerpo de la cápsula.
Cápsulas blancas opacas (6x16 mm) con la estampación “NTBC 5mg” en negro en el cuerpo de la cápsula.
Cápsulas blancas opacas (6x16 mm) con la estampación “NTBC 10mg” en negro en el cuerpo de la cápsula.
Cápsulas blancas opacas (6x16 mm) con la estampación “NTBC 20mg” en negro en el cuerpo de la cápsula.
Las cápsulas contienen un polvo entre blanco y blanquecino.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento de pacientes adultos y pediátricos (de cualquier intervalo de edad) con diagnóstico confirmado de tirosinemia hereditaria tipo I (TH-1) en combinación con dieta restrictiva de tirosina y fenilalanina.
4.2 Posología y forma de administración
El tratamiento con nitisinona debe ser iniciado y supervisado por un médico con experiencia en el tratamiento de pacientes con TH-1.
Posología
El tratamiento de todos los genotipos de la enfermedad deberá iniciarse lo antes posible para aumentar la supervivencia global y evitar complicaciones, como insuficiencia hepática, cáncer hepático y enfermedad renal. Conjuntamente con el tratamiento con nitisinona, se requiere una dieta pobre en fenilalanina y tirosina, que se monitorizará controlando los aminoácidos plasmáticos (ver secciones 4.4 y 4.8).
La dosis inicial recomendada en la población pediátrica y adulta es de 1 mg/kg de peso corporal al día y dividida en 2 tomas administradas por vía oral. La dosis de nitisinona se debe ajustar individualmente en cada caso.
Ajuste de la dosis
Durante la monitorización periódica se deben hacer determinaciones de la succinilacetona en orina, pruebas de la función hepática y medir las concentraciones de alfa-fetoproteína (ver la sección 4.4). Si un mes después de iniciar el tratamiento con nitisinona se sigue detectando succinilacetona en orina, deberá aumentarse la dosis de nitisinona hasta 1,5 mg/kg de peso corporal al día dividida en 2 tomas. Dependiendo de la evaluación de todos los parámetros bioquímicos, podría ser necesaria una dosis de 2 mg/kg de peso corporal al día. Esta dosis deberá considerarse como la dosis máxima para todos los pacientes.
Si la respuesta bioquímica es satisfactoria, deberá ajustarse la dosis sólo en función del aumento de peso corporal.
Sin embargo, además de las pruebas citadas más arriba, durante la iniciación del tratamiento o si existe deterioro, puede ser necesario controlar más exhaustivamente todos los parámetros bioquímicos (es decir, succinilacetona en plasma, 5-aminolevulinato (ALA) en orina y la actividad de la porfobilinógeno (PBG) sintasa en eritrocitos).
Poblaciones especiales
No existen recomendaciones de dosificación específicas para los pacientes de edad avanzada o los pacientes con insuficiencia renal o hepática.
Población pediátrica
La recomendación de dosificación en mg/kg de peso corporal es la misma en niños/niñas que en adultos.
Forma de administración
La cápsula puede abrirse y su contenido puede dispersarse en una pequeña cantidad de agua o suplemento dietético inmediatamente antes de tomarla.
Orfadin también está disponible como suspensión oral de 4 mg/ml para pacientes pediátricos que tienen dificultad para tragar cápsulas.
Se recomienda que, si se inicia el tratamiento de nitisinona con alimentos, se debe mantener de manera rutinaria (ver sección 4.5).
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Las madres que reciben nitisinona no deben amamantar a sus hijos (ver las secciones 4.6 y 5.3).
4.4 Advertencias y precauciones especiales de empleo Control de los niveles plasmáticos de tirosina
Se recomienda la exploración oftalmológica con lámpara de hendidura antes de iniciar el tratamiento con nitisinona. Cualquier paciente que muestre trastornos visuales durante el tratamiento con nitisinona deberá ser examinado inmediatamente por un oftalmólogo. Se establecen como condiciones la adherencia del paciente al régimen dietético y la determinación de la concentración plasmática de tirosina. En el caso de que la concentración de tirosina plasmática supere los 500 micromoles/l, deberá establecerse una dieta más restrictiva en tirosina y fenilalanina. No se recomienda reducir la concentración plasmática de tirosina reduciendo o suspendiendo la nitisinona, puesto que el defecto metabólico podría provocar el deterioro del estado clínico del paciente.
Control hepático
La función hepática deberá controlarse periódicamente mediante las pruebas de la función hepática y técnicas de imagen del hígado. También se recomienda controlar las concentraciones de alfa-fetoproteína sérica. El aumento de la concentración de alfa-fetoproteína sérica puede ser un signo de tratamiento inadecuado. Los pacientes con niveles crecientes de alfa-fetoproteína o con signos de nodulos hepáticos deberán ser evaluados para descartar un proceso hepático maligno.
Control de las plaquetas y leucocitos
Se recomienda controlar periódicamente el recuento de plaquetas y leucocitos, ya que se han observado algunos casos de trombocitopenia y leucopenia reversibles durante la exploración clínica.
Deberán realizarse visitas de revisión cada seis meses; en caso de producirse reacciones adversas se recomienda reducir los intervalos entre las visitas.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios formales de interacción con otros medicamentos.
La nitisinona se metaboliza in vitro por la CYP 3A4, por lo que puede ser necesario hacer un ajuste de la dosis cuando la nitisinona se administra conjuntamente con inhibidores o inductores de esta enzima.
A partir de los estudios realizados in vitro, no es previsible que la nitisinona inhiba el metabolismo mediado por CYP 1A2, 2C9, 2C19, 2D6, 2E1 o 3A4.
No se han realizado estudios formales de interacción con alimentos con Orfadin cápsulas duras. No obstante, la nitisinona se ha administrado junto con alimentos durante la generación de datos de eficacia y seguridad. Por lo tanto, se recomienda que, si se inicia el tratamiento de nitisinona con Orfadin cápsulas duras con alimentos, se debe mantener de manera rutinaria (ver sección 4.2).
4.6 Fertilidad, embarazo y lactancia
Embarazo
No existen datos suficientes sobre la utilización de nitisinona en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3). Se desconoce el riesgo potencial en seres humanos. No se debe utilizar Orfadin durante el embarazo a no ser que la situación clínica de la mujer requiera tratamiento con nitisinona.
Lactancia
Se desconoce si la nitisinona se excreta en la leche materna. Los estudios con animales han mostrado efectos adversos postnatales por la exposición a nitisinona en la leche. Por lo tanto, las madres que reciben nitisinona no deben amamantar a sus hijos ya que no puede descartarse el riesgo para los lactantes (ver secciones 4.3 y 5.3).
Fertilidad
No existen datos sobre si la nitisinona afecta a la fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Orfadin sobre la capacidad para conducir y utilizar máquinas es pequeña. Las reacciones adversas que afectan al ojo (ver sección 4.8) pueden afectar a la visión. Si la visión se ve afectada, el paciente no debe conducir ni utilizar máquinas hasta que el episodio haya remitido.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Por su mecanismo de acción la nitisinona aumenta los niveles de tirosina en todos los pacientes tratados. Por tanto, son frecuentes las reacciones adversas de carácter ocular, como conjuntivitis, opacidad corneal, queratitis, fotofobia y dolor ocular, relacionadas con niveles elevados de tisosina. Otras reacciones adversas frecuentes son trombocitopenia, leucopenia y granulocitopenia. Se puede producir dermatitis exfoliativa, aunque con poca frecuencia.
Tabla de reacciones adversas
Las reacciones adversas incluidas a continuación, según el sistema MedDRA de clasificación por órganos y sistemas y la frecuencia absoluta, se basan en los datos de un ensayo clínico y en el uso posterior a la comercialización. La frecuencia se define como muy frecuente (> 1/10), frecuente (de > 1/100 a < 1/10), poco frecuente (de > 1/1.000 a < 1/100), rara (de > 1/10.000 a < 1/1.000), muy rara (< 1/10.000), desconocida (no es posible realizar ninguna estimación a partir de los datos disponibles). Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de frecuencias.
|
Clasificación de órganos del sistema MedDRA |
Frecuencia |
Efecto adverso |
|
Trastornos de la sangre y del sistema linfático |
Frecuente |
Trombocitopenia, leucopenia, granulocitopenia |
|
Poco frecuente |
Leucocitosis | |
|
Trastornos oculares |
Frecuente |
Conjunctivitis, opacidad corneal, queratitis, fotofobia, dolor ocular |
|
Poco frecuente |
Blefaritis | |
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuente |
Dermatitis exfoliativa, exantema eritematoso, prurito |
|
Exploraciones complementarias |
Muy frecuente |
Concentraciones elevadas de tirosina |
Descripción de reacciones adversas seleccionadas
El tratamiento con nitisinona produce elevadas concentraciones de tirosina. Se ha asociado la presencia de elevadas concentraciones de tirosina con reacciones adversas de carácter ocular, como opacidad corneal y lesiones hiperqueratósicas. La restricción de tirosina y fenilalanina en la dieta debería limitar la toxicidad asociada a este tipo de tirosinemia al rebajar las concentraciones de tirosina (ver sección 4.4).
En los estudios clínicos, la granulocitopenia intensa (< 0,5x109/l) fue poco frecuente y no estuvo asociada a infecciones. Las reacciones adversas relacionadas con la categoría “Trastornos de la sangre y del sistema linfático” de la clasificación de órganos del sistemas MedDRA remitieron al continuar el tratamiento con nitisinona.
Población pediátrica
El perfil de seguridad está basado fundamentalmente en la población pediátrica, ya que el tratamiento con nitisinona se debe empezar tan pronto como se confirme el diagnóstico de tirosinemia hereditaria tipo I (TH-1). De los estudios clínicos y los datos posteriores a la comercialización no se desprende que el perfil de seguridad sea diferente en los distintos subgrupos de la población pediátrica, ni diferente del perfil de seguridad en los pacientes adultos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice VAnexo V.
4.9 Sobredosis
La ingestión accidental de nitisinona por individuos que toman dietas normales no restrictivas de tirosina y fenilalanina tendrá como consecuencia concentraciones elevadas de tirosina. Un nivel alto de tirosina se ha asociado con efectos tóxicos para los ojos, la piel y el sistema nervioso. La restricción de tirosina y fenilalanina en la dieta debería limitar la toxicidad asociada a este tipo de tirosinemia. No se dispone de información sobre el tratamiento específico de la sobredosis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Otros productos del tracto alimentario y metabólicos, varios productos del tracto alimentario y metabólicos, código ATC: A16A X04.
Mecanismo de acción
El defecto bioquímico en la tirosinemia hereditaria tipo 1 (TH-1) es un déficit de la fumarilacetoacetato hidrolasa, que es la enzima final de la ruta catabólica de la tirosina. La nitisinona es un inhibidor competitivo de la 4-hidroxilfenilpiruvato dioxigenasa, una enzima anterior a la fumarilacetoacetato hidrolasa en la ruta catabólica de la tirosina. Mediante la inhibición del catabolismo normal de la tirosina en pacientes con TH-1, la nitisinona impide la acumulación de los productos intermedios tóxicos maleilacetoacetato y fumarilacetoacetato. En pacientes con TH-1, estos productos intermedios se convierten en los metabolitos tóxicos succinilacetona y succinilacetoacetato. La succinilacetona inhibe la ruta de la síntesis de la porfirina y conduce a la acumulación del 5 -aminolevulinato.
Efectos farmacodinámicos
El tratamiento con nitisinona normaliza el metabolismo de la porfirina, la actividad de la porfobilinógeno sintasa eritrocítica y 5-aminolevulinato en orina son normales, se reduce la excreción urinaria de succinilacetona, aumenta la concentración plasmática de tirosina y aumenta la excreción urinaria de ácidos fenólicos. Los datos obtenidos en un estudio clínico indican que en más del 90% de los pacientes la succinilacetona en orina se normalizó durante la primera semana de tratamiento. Si la dosis de nitisinona está adecuadamente ajustada, no se debería detectar succinilacetona en plasma ni en orina.
Eficacia clínica y seguridad
Cuando se comparan con los datos de los controles históricos, se puede ver que el tratamiento con nitisinona junto con la dieta restrictiva aumenta la probabilidad de supervivencia en todos los fenotipos TH-1. Esto se puede ver en la siguiente tabla:
|
Edad al inicio del tratamiento o diagnóstico |
Probabilidad de supervivencia | |||
|
Tratamiento con nitisinona |
Control de la dieta | |||
|
5 años |
10 años |
5 años |
10 años | |
|
< 2 meses |
82 |
-- |
28 |
-- |
|
> 2-6 meses |
95 |
95 |
51 |
34 |
|
> 6 meses |
92 |
86 |
93 |
59 |
También se observó que el tratamiento con nitisinona reducía el riesgo de desarrollo de carcinoma hepatocelular (2,3 a 3,7 veces) en comparación con los datos históricos correspondientes al tratamiento con dieta restrictiva exclusivamente. Se observó que la iniciación temprana del tratamiento reducía adicionalmente el riesgo de desarrollo de carcinoma hepatocelular (13,5 veces cuando se iniciaba antes de los 12 meses).
5.2 Propiedades farmacocinéticas
No se han realizado estudios formales de absorción, distribución, metabolismo y eliminación de nitisinona. En 10 voluntarios sanos varones, después de la administración de una dosis única (1 mg/kg de peso corporal) de cápsulas de nitisinona, la semivida terminal (mediana) de nitisinona en plasma fue 54 horas (intervalo de 39 a 86 horas). El análisis farmacocinético poblacional se ha realizado en un grupo de 207 pacientes con TH-1. El aclaramiento y la semivida fueron de 0,0956 l/kg de peso corporal al día y 52,1 horas respectivamente.
En los estudios in vitro utilizando microsomas de hepatocito humano y la expresión de cADN de enzimas P450, se ha determinado el metabolismo mediado por CYP 3A4.
5.3 Datos preclínicos sobre seguridad
Se ha observado toxicidad embriofetal por nitisinona en ratón y conejo con dosis clínicamente importantes. En el conejo, la nitisinona produjo un aumento, dependiente de la dosis, de malformaciones (hernia umbilical y gastrosquisis) a partir de una dosis 2,5 veces superior al máximo de la dosis humana recomendada (2 mg/kg/día).
Un estudio de desarrollo prenatal y postnatal realizado en el ratón, mostró una reducción estadísticamente significativa de la supervivencia y del crecimiento de las crías durante el período de destete con unos niveles de exposición 125 y 25 veces superiores, respectivamente, que la dosis humana máxima recomendada, con tendencia hacia un efecto negativo en la supervivencia de las crías empezando desde la dosis de 5 mg/kg/día. En ratas, la exposición a través de la leche produjo en las crías una reducción del peso medio y lesiones corneales.
En los estudios in vitro no se observó actividad mutagénica, aunque sí una débil actividad clastogénica. No hubo evidencia de genotoxicidad in vivo (ensayo de micronúcleos en ratón y ensayo de síntesis de DNA no programada en hepatocito de ratón). La nitisinona no mostró un potencial carcinogénico en un estudio de carcinogenicidad de 26 semanas en ratones transgénicos (TgrasH2).
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Contenido de la cápsula Almidón, pregelatinizado (maíz)
Cubierta de la cápsula gelatina
dióxido de titanio (E 171)
Impresión
óxido de hierro negro (E 172) goma laca, propilenglicol ammoniumhydroksyd
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
18 meses.
Durante el período de validez, el paciente puede conservar las cápsulas durante un único período de 2 meses a una temperatura no superior a 25 °C, después del cual debe desechar el medicamento.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C).
6.5 Naturaleza y contenido del envase
Frasco de polietileno de alta densidad con una cápsula de cierre de polietileno de baja densidad con cierre inviolable. Cada frasco contiene 60 cápsulas.
Cada envase contiene 1 frasco.
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Swedish Orphan Biovitrum International AB
SE-112 76 Stockholm
Suecia
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/04/303/001
EU/1/04/303/002
EU/1/04/303/003
EU/1/04/303/004
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 21/02/2005 Fecha de la última renovación: 21/02/2010
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
1. NOMBRE DEL MEDICAMENTO
Orfadin 4 mg/ml suspensión oral
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
1 ml contiene 4 mg de nitisinona.
Excipientes con efecto conocido:
Cada ml contiene: sodio 0,7 mg (0,03 mmol) glicerol 500 mg benzoato de sodio 1 mg
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Suspensión oral.
Suspensión blanca, opaca y ligeramente viscosa.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento de pacientes adultos y pediátricos (de cualquier intervalo de edad) con diagnóstico confirmado de tirosinemia hereditaria tipo I (TH-1) en combinación con dieta restrictiva de tirosina y fenilalanina.
4.2 Posología y forma de administración
El tratamiento con nitisinona debe ser iniciado y supervisado por un médico con experiencia en el tratamiento de pacientes con TH-1.
Posología
El tratamiento de todos los genotipos de la enfermedad deberá iniciarse lo antes posible para aumentar la supervivencia global y evitar complicaciones, como insuficiencia hepática, cáncer hepático y enfermedad renal. Conjuntamente con el tratamiento con nitisinona, se requiere una dieta pobre en fenilalanina y tirosina, que se monitorizará controlando los aminoácidos plasmáticos (ver secciones 4.4 y 4.8).
La dosis inicial recomendada en la población pediátrica y adulta es de 1 mg/kg de peso corporal al día y dividida en 2 tomas administradas por vía oral. La dosis de nitisinona se debe ajustar individualmente en cada caso.
Ajuste de la dosis
Durante la monitorización periódica se deben hacer determinaciones de la succinilacetona en orina, pruebas de la función hepática y medir las concentraciones de alfa-fetoproteína (ver la sección 4.4). Si un mes después de iniciar el tratamiento con nitisinona se sigue detectando succinilacetona en orina, deberá aumentarse la dosis de nitisinona hasta 1,5 mg/kg de peso corporal al día dividida en 2 tomas. Dependiendo de la evaluación de todos los parámetros bioquímicos, podría ser necesaria una dosis de 2 mg/kg de peso corporal al día. Esta dosis deberá considerarse como la dosis máxima para todos los pacientes.
Si la respuesta bioquímica es satisfactoria, deberá ajustarse la dosis sólo en función del aumento de peso corporal.
Sin embargo, además de las pruebas citadas más arriba, durante la iniciación del tratamiento o si existe deterioro, puede ser necesario controlar más exhaustivamente todos los parámetros bioquímicos (es decir, succinilacetona en plasma, 5-aminolevulinato (ALA) en orina y la actividad de la porfobilinógeno (PBG) sintasa en eritrocitos).
Poblaciones especiales
No existen recomendaciones de dosificación específicas para los pacientes de edad avanzada o los pacientes con insuficiencia renal o hepática.
Población pediátrica
La recomendación de dosificación en mg/kg de peso corporal es la misma en niños/niñas que en adultos.
Forma de administración
La suspensión se administra sin diluir en la boca del paciente por medio de una jeringa para uso oral. En el envase se incluyen jeringas para uso oral de 1 ml, 3 ml y 5 ml para poder medir la dosis en mililitros (ml) conforme a la posología prescrita. Las jeringas para uso oral están graduadas en intervalos de 0,01 ml, 0,1 ml y 0,2 ml, respectivamente. La tabla siguiente muestra la conversión de dosis (mg-ml) para los tres tamaños de jeringas para uso oral.
Tablas de conversión de dosis para los tres tamaños respectivos de jeringas para uso oral:
Jeringa para uso oral de 1 ml (graduación de 0,01 ml)
|
Dosis de Orfadin | |
|
mg |
ml |
|
1,00 |
0,25 |
|
1,25 |
0,31 |
|
1,50 |
0,38 |
|
1,75 |
0,44 |
|
2,00 |
0,50 |
|
2,25 |
0,56 |
|
2,50 |
0,63 |
|
2,75 |
0,69 |
|
3,00 |
0,75 |
|
3,25 |
0,81 |
|
3,50 |
0,88 |
|
3,75 |
0,94 |
|
4,00 |
1,00 |
|
Dos Ori |
>is de 'adin |
|
mg |
ml |
|
4,5 |
1,1 |
|
5,0 |
1,3 |
|
5,5 |
1,4 |
|
6,0 |
1,5 |
|
6,5 |
1,6 |
|
7,0 |
1,8 |
|
7,5 |
1,9 |
|
8,0 |
2,0 |
|
8,5 |
2,1 |
|
9,0 |
2,3 |
|
9,5 |
2,4 |
|
10,0 |
2,5 |
|
10,5 |
2,6 |
|
11,0 |
2,8 |
|
11,5 |
2,9 |
|
12,0 |
3,0 |
|
Dos Ori |
>is de 'adin |
|
mg |
ml |
|
13,0 |
3,2 |
|
14,0 |
3,6 |
|
15,0 |
3,8 |
|
16,0 |
4,0 |
|
17,0 |
4,2 |
|
18,0 |
4,6 |
|
19,0 |
4,8 |
|
20,0 |
5,0 |
Jeringa para uso oral de 5 ml
(graduació n de 0,2 ml)
Jeringa para uso oral de 3 ml
(graduació n de 0,1 ml)
Información importante sobre las instrucciones de uso:
Es necesario volver a dispersar el producto antes de cada uso mediante agitación vigorosa. Antes de volver a dispersarlo, el medicamento puede aparecer como una torta sólida con un sobrenadante ligeramente opalescente. La dosis se debe extraer y administrar inmediatamente después de volver a dispersar el producto. Es importante seguir cuidadosamente las instrucciones facilitadas en la sección 6.6 sobre preparación y administración de la dosis para asegurar la exactitud de la dosificación.
Se recomienda que el profesional sanitario indique al paciente o cuidador la forma de utilizar las jeringas para uso oral a fin de garantizar que se administre el volumen correcto y que la prescripción se administre en ml.
Orfadin también se presenta en cápsulas de 2 mg, 5 mg, 10 mg y 20 mg, si se consideran más adecuadas para el paciente.
Se recomienda que la suspensión oral se tome con alimentos, ver sección 4.5.
Precauciones a tomar antes de manipular o administrar el medicamento
No se debe acoplar a la jeringa para uso oral ninguna aguja, tubo intravenoso o dispositivo para administración parenteral.
Orfadin es solo para administración por vía oral.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Las madres que reciben nitisinona no deben amamantar a sus hijos (ver las secciones 4.6 y 5.3).
4.4 Advertencias y precauciones especiales de empleo Control de los niveles plasmáticos de tirosina
Se recomienda la exploración oftalmológica con lámpara de hendidura antes de iniciar el tratamiento con nitisinona. Cualquier paciente que muestre trastornos visuales durante el tratamiento con nitisinona deberá ser examinado inmediatamente por un oftalmólogo. Se establecen como condiciones la adherencia del paciente al régimen dietético y la determinación de la concentración plasmática de tirosina. En el caso de que la concentración de tirosina plasmática supere los 500 micromoles/l, deberá establecerse una dieta más restrictiva en tirosina y fenilalanina. No se recomienda reducir la concentración plasmática de tirosina reduciendo o suspendiendo la nitisinona, puesto que el defecto metabólico podría provocar el deterioro del estado clínico del paciente.
Control hepático
La función hepática deberá controlarse periódicamente mediante las pruebas de la función hepática y técnicas de imagen del hígado. También se recomienda controlar las concentraciones de alfa-fetoproteína sérica. El aumento de la concentración de alfa-fetoproteína sérica puede ser un signo de tratamiento inadecuado. Los pacientes con niveles crecientes de alfa-fetoproteína o con signos de nódulos hepáticos deberán ser evaluados para descartar un proceso hepático maligno.
Control de las plaquetas y leucocitos
Se recomienda controlar periódicamente el recuento de plaquetas y leucocitos, ya que se han observado algunos casos de trombocitopenia y leucopenia reversibles durante la exploración clínica.
Deberán realizarse visitas de revisión cada seis meses; en caso de producirse reacciones adversas se recomienda reducir los intervalos entre las visitas.
Excipientes con efecto conocido:
Glicerol
Cada ml contiene 500 mg. Una dosis de 20 ml de suspensión oral (10 g de glicerol) o más puede provocar cefalea, molestias de estómago y diarrea.
Sodio
Cada ml contiene 0,7 mg (0,03 mmol).
Benzoato de sodio
Cada ml contiene 1 mg. El aumento de la bilirrubina tras su desplazamiento de la albúmina, causado por el ácido benzoico y sus sales, puede aumentar la ictericia en los neonatos ictéricos prematuros o nacidos a término y derivar en kemícterus (depósitos de bilirrubina no conjugada en el tejido encefálico). Tiene gran importancia, por tanto, el control estricto de los niveles plasmáticos de bilirrubina en el paciente neonato. Se deben medir los niveles de bilirrubina antes de iniciar el tratamiento; en caso de concentraciones plasmáticas marcadamente elevadas de bilirrubina, especialmente en pacientes prematuros con factores de riesgo como acidosis o bajo nivel de albúmina, se debe considerar el tratamiento con una parte debidamente pesada de una cápsula de Orfadin en lugar de la suspensión oral hasta que se normalicen las concentraciones plasmáticas de bilirrubina no conjugada.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios formales de interacción con otros medicamentos.
La nitisinona se metaboliza in vitro por la CYP 3A4, por lo que puede ser necesario hacer un ajuste de la dosis cuando la nitisinona se administra conjuntamente con inhibidores o inductores de esta enzima.
A partir de los estudios realizados in vitro, no es previsible que la nitisinona inhiba el metabolismo mediado por CYP 1A2, 2C9, 2C19, 2D6, 2E1 o 3A4.
Los alimentos no influyen en la biodisponibilidad de la nitisinona, pero la ingestión junto con alimentos reduce la tasa de absorción y, en consecuencia, ocasiona menores fluctuaciones de las concentraciones séricas dentro de un intervalo posológico. Por consiguiente, se recomienda que la suspensión oral se tome con alimentos, ver sección 4.2.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No existen datos suficientes sobre la utilización de nitisinona en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3). Se desconoce el riesgo potencial en seres humanos. No se debe utilizar Orfadin durante el embarazo a no ser que la situación clínica de la mujer requiera tratamiento con nitisinona.
Lactancia
Se desconoce si la nitisinona se excreta en la leche materna. Los estudios con animales han mostrado efectos adversos postnatales por la exposición a nitisinona en la leche. Por lo tanto, las madres que reciben nitisinona no deben amamantar a sus hijos ya que no puede descartarse el riesgo para los lactantes (ver secciones 4.3 y 5.3).
Fertilidad
No existen datos sobre si la nitisinona afecta a la fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Orfadin sobre la capacidad para conducir y utilizar máquinas es pequeña. Las reacciones adversas que afectan al ojo (ver sección 4.8) pueden afectar a la visión. Si la visión se ve afectada, el paciente no debe conducir ni utilizar máquinas hasta que el episodio haya remitido.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Por su mecanismo de acción la nitisinona aumenta los niveles de tirosina en todos los pacientes tratados. Por tanto, son frecuentes las reacciones adversas de carácter ocular, como conjuntivitis, opacidad corneal, queratitis, fotofobia y dolor ocular, relacionadas con niveles elevados de tisosina. Otras reacciones adversas frecuentes son trombocitopenia, leucopenia y granulocitopenia. Se puede producir dermatitis exfoliativa, aunque con poca frecuencia.
Tabla de reacciones adversas
Las reacciones adversas incluidas a continuación, según el sistema MedDRA de clasificación por órganos y sistemas y la frecuencia absoluta, se basan en los datos de un ensayo clínico y en el uso posterior a la comercialización. La frecuencia se define como muy frecuente (> 1/10), frecuente (de > 1/100 a < 1/10), poco frecuente (de > 1/1.000 a < 1/100), rara (de > 1/10.000 a < 1/1.000), muy rara (< 1/10.000), desconocida (no es posible realizar ninguna estimación a partir de los datos disponibles). Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de frecuencias.
|
Clasificación de órganos del sistema MedDRA |
Frecuencia |
Efecto adverso |
|
Trastornos de la sangre y del sistema linfático |
Frecuente |
Trombocitopenia, leucopenia, granulocitopenia |
|
Poco frecuente |
Leucocitosis | |
|
Trastornos oculares |
Frecuente |
Conjunctivitis, opacidad corneal, queratitis, fotofobia, dolor ocular |
|
Poco frecuente |
Blefaritis | |
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuente |
Dermatitis exfoliativa, exantema eritematoso, prurito |
|
Exploraciones complementarias |
Muy frecuente |
Concentraciones elevadas de tirosina |
Descripción de reacciones adversas seleccionadas
El tratamiento con nitisinona produce elevadas concentraciones de tirosina. Se ha asociado la presencia de elevadas concentraciones de tirosina con reacciones adversas de carácter ocular, como opacidad corneal y lesiones hiperqueratósicas. La restricción de tirosina y fenilalanina en la dieta debería limitar la toxicidad asociada a este tipo de tirosinemia al rebajar las concentraciones de tirosina (ver sección 4.4).
En los estudios clínicos, la granulocitopenia intensa (< 0,5x109/l) fue poco frecuente y no estuvo asociada a infecciones. Las reacciones adversas relacionadas con la categoría “Trastornos de la sangre y del sistema linfático” de la clasificación de órganos del sistemas MedDRA remitieron al continuar el tratamiento con nitisinona.
Población pediátrica
El perfil de seguridad está basado fundamentalmente en la población pediátrica, ya que el tratamiento con nitisinona se debe empezar tan pronto como se confirme el diagnóstico de tirosinemia hereditaria tipo I (TH-1). De los estudios clínicos y los datos posteriores a la comercialización no se desprende que el perfil de seguridad sea diferente en los distintos subgrupos de la población pediátrica, ni diferente del perfil de seguridad en los pacientes adultos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice VAnexo V.
4.9 Sobredosis
La ingestión accidental de nitisinona por individuos que toman dietas normales no restrictivas de tirosina y fenilalanina tendrá como consecuencia concentraciones elevadas de tirosina. Un nivel alto de tirosina se ha asociado con efectos tóxicos para los ojos, la piel y el sistema nervioso. La restricción de tirosina y fenilalanina en la dieta debería limitar la toxicidad asociada a este tipo de tirosinemia. No se dispone de información sobre el tratamiento específico de la sobredosis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Otros productos del tracto alimentario y metabólicos, varios productos del tracto alimentario y metabólicos, código ATC: A16A X04.
Mecanismo de acción
El defecto bioquímico en la tirosinemia hereditaria tipo 1 (TH-1) es un déficit de la fumarilacetoacetato hidrolasa, que es la enzima final de la ruta catabólica de la tirosina. La nitisinona es un inhibidor competitivo de la 4-hidroxilfenilpiruvato dioxigenasa, una enzima anterior a la fumarilacetoacetato hidrolasa en la ruta catabólica de la tirosina. Mediante la inhibición del catabolismo normal de la tirosina en pacientes con TH-1, la nitisinona impide la acumulación de los productos intermedios tóxicos maleilacetoacetato y fumarilacetoacetato. En pacientes con TH-1, estos productos intermedios se convierten en los metabolitos tóxicos succinilacetona y succinilacetoacetato. La succinilacetona inhibe la ruta de la síntesis de la porfirina y conduce a la acumulación del 5 -aminolevulinato.
Efectos farmacodinámicos
El tratamiento con nitisinona normaliza el metabolismo de la porfirina, la actividad de la porfobilinógeno sintasa eritrocítica y 5-aminolevulinato en orina son normales, se reduce la excreción urinaria de succinilacetona, aumenta la concentración plasmática de tirosina y aumenta la excreción urinaria de ácidos fenólicos. Los datos obtenidos en un estudio clínico indican que en más del 90% de los pacientes la succinilacetona en orina se normalizó durante la primera semana de tratamiento. Si la dosis de nitisinona está adecuadamente ajustada, no se debería detectar succinilacetona en plasma ni en orina.
Eficacia clínica y seguridad
Cuando se comparan con los datos de los controles históricos, se puede ver que el tratamiento con nitisinona junto con la dieta restrictiva aumenta la probabilidad de supervivencia en todos los fenotipos TH-1. Esto se puede ver en la siguiente tabla:
|
Edad al inicio del tratamiento o diagnóstico |
Probabilidad de supervivencia | |||
|
Tratamiento con nitisinona |
Control de la dieta | |||
|
5 años |
10 años |
5 años |
10 años | |
|
< 2 meses |
82 |
-- |
28 |
-- |
|
> 2-6 meses |
95 |
95 |
51 |
34 |
|
> 6 meses |
92 |
86 |
93 |
59 |
También se observó que el tratamiento con nitisinona reducía el riesgo de desarrollo de carcinoma hepatocelular (2,3 a 3,7 veces) en comparación con los datos históricos correspondientes al tratamiento con dieta restrictiva exclusivamente. Se observó que la iniciación temprana del tratamiento reducía adicionalmente el riesgo de desarrollo de carcinoma hepatocelular (13,5 veces cuando se iniciaba antes de los 12 meses).
5.2 Propiedades farmacocinéticas
No se han realizado estudios formales de absorción, distribución, metabolismo y eliminación de nitisinona. En 10 voluntarios sanos varones, después de la administración de una dosis única (1 mg/kg de peso corporal) de cápsulas de nitisinona, la semivida terminal (mediana) de nitisinona en plasma fue 54 horas (intervalo de 39 a 86 horas). El análisis farmacocinético poblacional se ha realizado en un grupo de 207 pacientes con TH-1. El aclaramiento y la semivida fueron de 0,0956 l/kg de peso corporal al día y 52,1 horas respectivamente.
En los estudios in vitro utilizando microsomas de hepatocito humano y la expresión de cADN de enzimas P450, se ha determinado el metabolismo mediado por CYP 3A4.
5.3 Datos preclínicos sobre seguridad
Se ha observado toxicidad embriofetal por nitisinona en ratón y conejo con dosis clínicamente importantes. En el conejo, la nitisinona produjo un aumento, dependiente de la dosis, de malformaciones (hernia umbilical y gastrosquisis) a partir de una dosis 2,5 veces superior al máximo de la dosis humana recomendada (2 mg/kg/día).
Un estudio de desarrollo prenatal y postnatal realizado en el ratón, mostró una reducción estadísticamente significativa de la supervivencia y del crecimiento de las crías durante el período de destete con unos niveles de exposición 125 y 25 veces superiores, respectivamente, que la dosis humana máxima recomendada, con tendencia hacia un efecto negativo en la supervivencia de las crías empezando desde la dosis de 5 mg/kg/día. En ratas, la exposición a través de la leche produjo en las crías una reducción del peso medio y lesiones corneales.
En los estudios in vitro no se observó actividad mutagénica, aunque sí una débil actividad clastogénica. No hubo evidencia de genotoxicidad in vivo (ensayo de micronúcleos en ratón y ensayo de síntesis de DNA no programada en hepatocito de ratón). La nitisinona no mostró un potencial carcinogénico en un estudio de carcinogenicidad de 26 semanas en ratones transgénicos (TgrasH2).
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Hipromelosa Glicerol Polisorbato 80 Benzoato de sodio (E211)
Ácido cítrico monohidrato Citrato de sodio Aroma de fresa (artificial)
Agua purificada
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
2 años.
Después de la primera apertura, la estabilidad en uso es de un único período de 2 meses a una temperatura no superior a 25 °C, después del cual se debe desechar.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C). No congelar.
Mantener en posición vertical.
Para las condiciones de conservación tras la primera apertura del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Frasco de 100 ml de vidrio de color topacio (tipo III) con un cierre de rosca blanco de seguridad de polietileno de alta densidad a prueba de niños y precinto antimanipulación. Cada frasco contiene 90 ml de suspensión oral.
Cada envase contiene un frasco, un adaptador para el frasco de polietileno de baja densidad y 3 jeringas para uso oral de polipropileno (PP) (de 1 ml, 3 ml y 5 ml).
6.6 Precauciones especiales de eliminacn y otras manipulaciones
Es necesario volver a dispersar el producto antes de cada uso mediante agitación vigorosa. Antes volver a dispersarlo, el medicamento puede aparecer como una torta sólida con un sobrenadante ligeramente opalescente. La dosis se debe extraer y administrar inmediatamente después de volver a dispersar el producto. Es importante seguir cuidadosamente las instrucciones facilitadas a continuación sobre preparación y administración de la dosis para asegurar la exactitud de la dosificación.
Se suministran jeringas para uso oral de 3 capacidades (1 ml, 3 ml y 5 ml) para una medición exacta de la dosis prescrita. Se recomienda que el profesional sanitario indique al paciente o cuidador la forma de utilizar las jeringas para uso oral a fin de garantizar que se administre el volumen correcto.
Cómo preparar un nuevo frasco del medicamento para utilizarlo por primera vez:
Antes de tomar la primera dosis, debe agitarse el frasco vigorosamente, ya que durante el almacenamiento prolongado las partículas forman una torta sólida en el fondo del frasco.
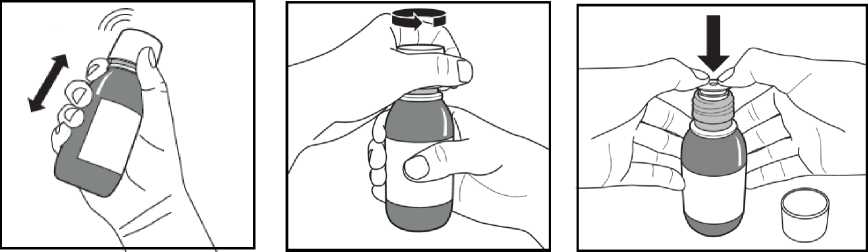
Figura A. Figura B. Figura C.
1. Sacar el frasco de la nevera y anotar la fecha en la etiqueta del frasco.
2. Agitar el frasco vigorosamente durante al menos 20 segundos hasta que la torta sólida que hay en el fondo del frasco se disperse por completo (Figura A).
3. Quitar el cierre de rosca a prueba de niños; para ello apretarlo hacia abajo con fuerza y girar en sentido contrario a las agujas del reloj (Figura B).
4. Colocar el frasco abierto derecho encima de la mesa y empujar con fuerza el adaptador de plástico dentro del cuello del frasco todo lo posible (Figura C). Cerrar el frasco con el cierre de rosca a prueba de niños.
Para las dosis posteriores consultar a continuación las instrucciones “Cómo preparar una dosis del
medicamento”
Cómo preparar una dosis del medicamento
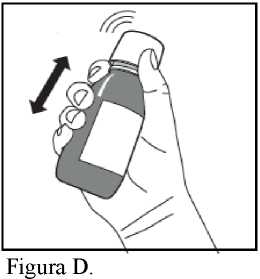
1. Agitar el frasco vigorosamente durante al menos 5 segundos (Figura D).
2. Inmediatamente, retirar el cierre de rosca a prueba de niños y abrir el frasco.
3. Empujar hasta el fondo el émbolo de la jeringa para uso oral.
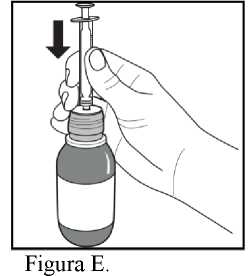
4. Mantener el frasco en posición vertical e insertar la jeringa para uso oral firmemente en el orificio del adaptador situado en la parte superior del frasco (Figura E).
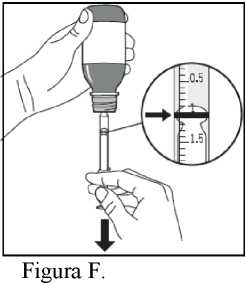
5. Invertir con cuidado el frasco sin extraer la jeringa para uso oral (Figura F).
6. Para obtener la dosis prescrita (ml), tirar del émbolo lentamente hacia abajo hasta que el borde superior del anillo negro quede nivelado exactamente con la línea que señala la dosis (Figura F). Si se observa alguna burbuja de aire dentro de la jeringa para uso oral llena, hacer retroceder el émbolo hasta que salgan las burbujas. Después volver a tirar del émbolo hacia abajo hasta que el borde superior del anillo negro quede nivelado exactamente con la línea que señala la dosis.
7. Colocar el frasco de nuevo en posición vertical y girar suavemente la jeringa para uso oral, tirando de ella para sacarla del frasco.
8. La dosis debe administrarse inmediatamente en la boca (sin diluir) para evitar que se forme un precipitado en la jeringa para uso oral. La jeringa para uso oral se debe vaciar lentamente para permitir que el paciente trague el producto; si el medicamento sale en un chorro rápido se puede provocar un atragantamiento.
9. Colocar inmediatamente el cierre de rosca de seguridad a prueba de niños. El adaptador del frasco no debe retirarse.
10. El frasco puede mantenerse a una temperatura no superior a 25 °C o en la nevera.
Limpieza:
Limpiar inmediatamente la jeringa para uso oral con agua. Separar el émbolo del cilindro, y
enjuagar ambos con agua. Sacudir el exceso de agua y dejar que la jeringa para uso oral se seque
desmontada hasta que tenga que volverse a montar para una nueva administración.
Eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Swedish Orphan Biovitrum International AB
SE-112 76 Stockholm
Suecia
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/04/303/005
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 21/02/2005 Fecha de la última renovación: 21/02/2010
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
A. FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
A.
Nombre y dirección del (de los) fabricantc(s) rcsponsablc(s) de la liberación de los lotes 2 mg, 5 mg, 10 mg y 20 mg cápsulas duras:
Apotek Produktion & Laboratorier AB
Prismavagen 2
SE-141 75 Kungens Kurva
Suecia
4 mg/ml suspensión oral:
Apotek Produktion & Laboratorier AB Celsiusgatan 43 SE-212 14 Malmo Suecia
El prospecto impreso del medicamento debe especificar el nombre y dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva
información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
Orfadin 2 mg cápsulas duras Orfadin 5 mg cápsulas duras Orfadin 10 mg cápsulas duras Orfadin 20 mg cápsulas duras Nitisinona
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula contiene 2 mg de nitisinona Cada cápsula contiene 5 mg de nitisinona Cada cápsula contiene 10 mg de nitisinona Cada cápsula contiene 20 mg de nitisinona
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
60 cápsulas duras
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Swedish Orphan Biovitrum International AB
SE-112 76 Stockholm
Sweden
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/04/303/001
EU/1/04/303/002
EU/1/04/303/003
EU/1/04/303/004
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
Orfadin 2 mg Orfadin 5 mg Orfadin 10 mg Orfadin 20 mg
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Orfadin 2 mg cápsulas duras Orfadin 5 mg cápsulas duras Orfadin 10 mg cápsulas duras Orfadin 20 mg cápsulas duras Nitisinona Vía oral
2. FORMA DE ADMINISTRACIÓN
3. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Swedish Orphan Biovitrum International AB
4. FECHA DE CADUCIDAD
EXP
5. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera. El medicamento se puede conservar durante un único período de 2 meses a una temperatura no superior a 25 °C, después del cual se debe desechar.
Fecha en la que se sacó de la nevera:
6. NÚMERO DE LOTE
Lot
7. CONTENIDO EN UNIDADES
60 cápsulas
1. NOMBRE DEL MEDICAMENTO
Orfadin 4 mg/ml suspensión oral Nitisinona
2. PRINCIPIO(S) ACTIVO(S)
1 ml contiene 4 mg de nitisinona.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Suspensión oral
1 frasco de 90 ml, 1 adaptador para el frasco, 3 jeringas para uso oral (1 ml, 3 ml, 5 ml).
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer detenidamente el prospecto antes de utilizar este medicamento. Solo por vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera.
No congelar.
Mantener en posición vertical.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Swedish Orphan Biovitrum International AB
SE-112 76 Stockholm
Sweden
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/04/303/005
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Orfadin 4 mg/ml
1. NOMBRE DEL MEDICAMENTO
Orfadin 4 mg/ml suspensión oral Nitisinona
2. PRINCIPIO(S) ACTIVO(S)
1 ml contiene 4 mg de nitisinona.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Suspensión oral 90 ml
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer cuidadosamente el prospecto antes de utilizar este medicamento. Solo por vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
EXP
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera.
No congelar.
Mantener en posición vertical.
El producto se puede conservar durante un único periodo de 2 meses a una temperatura no superior a 25 °C, después del cual, se debe desechar.
Fecha en la que se sacó de la nevera:
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Swedish Orphan Biovitrum International AB
SE-112 76 Stockholm
Sweden
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/04/303/005
13. NÚMERO DE LOTE
Lot
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
B. PROSPECTO
Prospecto: información para el usuario
Orfadin 2 mg cápsulas duras Orfadin 5 mg cápsulas duras Orfadin 10 mg cápsulas duras Orfadin 20 mg cápsulas duras
nitisinona
Lea todo el prospecto detenidamente antes de empezar a tomar el medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Orfadin y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Orfadin
3. Cómo tomar Orfadin
4. Posibles efectos adversos
5. Conservación de Orfadin
6. Contenido del envase e información adicional
1. Qué es Orfadin y para qué se utiliza
El principio activo de Orfadin es la nitisinona. Este medicamento se utiliza para el tratamiento de una enfermedad poco común denominada tirosinemia hereditaria tipo 1 en adultos, adolescentes y niños (de cualquier intervalo de edad).
En esta enfermedad, su organismo no puede degradar totalmente el aminoácido tirosina (los aminoácidos son los elementos fundamentales de las proteínas), formándose sustancias tóxicas. Estas sustancias se acumulan en su organismo. Orfadin bloquea la degradación de la tirosina, y las sustancias tóxicas no se forman.
Debe seguir una dieta especial mientras tome este medicamento, porque la tirosina seguirá estando en su organismo. Dicha dieta se basa en un bajo contenido de tirosina y fenilalanina (otro aminoácido).
2. Qué necesita saber antes de empezar a tomar Orfadin
No tome Orfadin
- si es alérgico a la nitisinona o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
No dé el pecho mientras esté tomando este medicamento (ver sección “Embarazo y lactancia”).
Advertencias y precauciones
Consulte a su médico o farmacéutico antes de empezar a tomar Orfadin,
- si advierte enrojecimiento de los ojos o si advierte cualquier otro efecto en los ojos. Póngase en contacto inmediatamente con su médico para que le realice una exploración oftalmológica. Los problemas oculares (ver sección 4) pueden ser un indicio de un control inadecuado de la dieta.
Durante el tratamiento se le extraerán muestras de sangre con el fin de controlar si el tratamiento es el adecuado y para asegurarse de que no existen efectos secundarios causantes de alteraciones sanguíneas.
Se le harán controles hepáticos periódicos porque la enfermedad afecta al hígado.
Su médico debe realizar un seguimiento cada 6 meses. Si sufre algún efecto adverso, es recomendable utilizar intervalos de tiempo más cortos.
Uso de Orfadin con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento.
Uso de Orfadin con alimentos
Si comienza el tratamiento tomándolo junto con alimentos, se recomienda seguir este régimen a lo largo de todo el tratamiento.
Embarazo y lactancia
No se ha estudiado la seguridad de este medicamento en mujeres embarazadas y en mujeres que amamantan.
Consulte a su médico si tiene previsto quedarse embarazada. Si se queda embarazada deberá consultar a su médico inmediatamente.
No dé el pecho mientras esté tomando este medicamento (ver sección “No tome Orfadin”). Conducción y uso de máquinas
La influencia de este medicamento sobre la capacidad para conducir y utilizar máquinas es pequeña. No obstante, si experimenta efectos adversos que afecten a la visión, no debe conducir ni utilizar máquinas hasta que su visión haya vuelto a la normalidad (ver sección 4 “Posibles efectos adversos”).
3. Cómo tomar Orfadin
Siga exactamente las instrucciones de administración del medicamento indicadas por su médico. En caso de duda, pregunte a su médico o farmacéutico.
El tratamiento con este medicamento debe ser iniciado y supervisado por un médico con experiencia en el tratamiento de la enfermedad (tirosinemia hereditaria tipo 1).
La dosis recomendada diaria es 1 mg/kg de peso corporal dividida en 2 tomas. Su médico ajustará la dosis individualmente.
Si tiene problemas para tragar las cápsulas, puede abrir las cápsulas y mezclar el polvo con una pequeña cantidad de agua o de suplemento dietético, antes de tomarlo.
Si toma más Orfadin del que debe
Si ha tomado más cantidad de este medicamento del que debiera, comuníqueselo inmediatamente a su médico o farmacéutico.
Si olvidó tomar Orfadin
No tome una dosis doble para compensar las dosis olvidadas. Si ha olvidado tomar una dosis, comuníqueselo a su médico o farmacéutico.
Si interrumpe el tratamiento con Orfadin
Si estima que la acción del medicamento no es la adecuada, comuníqueselo a su médico. No cambie la dosis ni suspenda el tratamiento sin hablar primero con su médico.
Si tiene cualquier duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Si aprecia cualquier efecto adverso relacionado con los ojos, comuníqueselo inmediatamente a su médico para que le realice una exploración oftalmológica. El tratamiento con nitisinona aumenta los niveles de tirosina en la sangre, que pueden causar síntomas relacionados con los ojos. Los efectos adversos oculares frecuentes (pueden afectar a más de 1 de cada 10 personas) debidos a los niveles más altos de tirosina son inflamación ocular (conjuntivitis), opacidad e inflamación de la córnea (queratitis), sensibilidad a la luz (fotofobia) y dolor de ojos. La inflamación de los párpados (blefaritis) es un efecto adversos poco frecuente (puede afectar hasta 1 de cada 100 personas).
Otros efectos adversos frecuentes
- Disminución del número de plaquetas (trombocitopenia) y glóbulos blancos (leucopenia), reducción de determinados tipos de glóbulos blancos (granulocitopenia).
Otros efectos adversos poco frecuentes
- aumento del número de glóbulos blancos (leucocitosis),
- picor (prurito), inflamación de la piel (dermatitis exfoliativa), sarpullido.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice VAnexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Orfadin
Mantener este medicamento fuera de la vista y el alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el bote y la caja después de “EXP”. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2 °C y 8 °C).
El medicamento se puede conservar durante un único período de 2 meses a una temperatura no superior a 25 °C, después del cual se debe desechar.
No olvide anotar en el frasco la fecha en la que lo sacó de la nevera.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico como deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composicion de Orfadin
- El principio activo es nitisinona.
Orfadin 2 mg: cada cápsula contiene 2 mg de nitisinona. Orfadin 5 mg: cada cápsula contiene 5 mg de nitisinona. Orfadin 10 mg: cada cápsula contiene 10 mg de nitisinona. Orfadin 20 mg: cada cápsula contiene 20 mg de nitisinona.
- Los demás componentes (excipientes) son:
Contenido de la cápsula: almidón, pregelatinizado (de maíz)
Cubierta de la cápsula: gelatina
dióxido de titanio (E 171)
Impresión:
óxido de hierro (E 172) goma laca propilenglicol hidróxido de amonio
Aspecto del producto y contenido del envase
Las cápsulas son duras, blancas, opacas, hechas de gelatina y tienen impreso “NTBC” y la dosis “2 mg”, “5 mg”, “10 mg” o “20 mg” en negro. La cápsula contiene un polvo que puede ser blanco o grisáceo.
Las cápsulas vienen envasadas en frascos de plástico con cierres inviolables. Cada frasco contiene 60 cápsulas.
Titular de la autorización de comercialización
Swedish Orphan Biovitrum International AB
SE-112 76 Stockholm
Suecia
Fabricante
Apotek Produktion & Laboratorier AB
Prismavágen 2
SE-141 75 Kungens Kurva
Suecia
Fecha de la última revisión de este prospecto:
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu. También existen enlaces a otras páginas web sobre enfermedades raras y medicamentos huérfanos.
Prospecto: información para el usuario
Orfadin 4 mg/ml suspensión oral
nitisinona
Lea todo el prospecto detenidamente antes de empezar a tomar el medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Orfadin y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Orfadin
3. Cómo tomar Orfadin
4. Posibles efectos adversos
5. Conservación de Orfadin
6. Contenido del envase e información adicional
1. Qué es Orfadin y para qué se utiliza
El principio activo de Orfadin es la nitisinona. Este medicamento se utiliza para el tratamiento de una enfermedad poco común denominada tirosinemia hereditaria tipo 1 en adultos, adolescentes y niños (de cualquier intervalo de edad).
En esta enfermedad, su organismo no puede degradar totalmente el aminoácido tirosina (los aminoácidos son los elementos fundamentales de las proteínas), formándose sustancias tóxicas. Estas sustancias se acumulan en su organismo. Orfadin bloquea la degradación de la tirosina, y las sustancias tóxicas no se forman.
Debe seguir una dieta especial mientras tome este medicamento, porque la tirosina seguirá estando en su organismo. Dicha dieta se basa en un bajo contenido de tirosina y fenilalanina (otro aminoácido).
2. Qué necesita saber antes de empezar a tomar Orfadin
No tome Orfadin
- si es alérgico a la nitisinona o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
No dé el pecho mientras esté tomando este medicamento (ver sección “Embarazo y lactancia”).
Advertencias y precauciones
Consulte a su médico o farmacéutico antes de empezar a tomar Orfadin,
- si advierte enrojecimiento de los ojos o si advierte cualquier otro efecto en los ojos. Póngase en contacto inmediatamente con su médico para que le realice una exploración oftalmológica. Los problemas oculares (ver sección 4) pueden ser un indicio de un control inadecuado de la dieta.
Durante el tratamiento se le extraerán muestras de sangre con el fin de controlar si el tratamiento es el
adecuado y para asegurarse de que no existen efectos secundarios causantes de alteraciones
sanguíneas.
Se le harán controles hepáticos periódicos porque la enfermedad afecta al hígado.
Su médico debe realizar un seguimiento cada 6 meses. Si sufre algún efecto adverso, es recomendable utilizar intervalos de tiempo más cortos.
Uso de Orfadin con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento.
Uso de Orfadin con alimentos
Se recomienda tomar la suspensión oral junto con alimentos.
Embarazo y lactancia
No se ha estudiado la seguridad de este medicamento en mujeres embarazadas y en mujeres que amamantan.
Consulte a su médico si tiene previsto quedarse embarazada. Si se queda embarazada deberá consultar a su médico inmediatamente.
No dé el pecho mientras esté tomando este medicamento (ver sección “No tome Orfadin”). Conducción y uso de máquinas
La influencia de este medicamento sobre la capacidad para conducir y utilizar máquinas es pequeña. No obstante, si experimenta efectos adversos que afecten a la visión, no debe conducir ni utilizar máquinas hasta que su visión haya vuelto a la normalidad (ver sección 4 “Posibles efectos adversos”).
Orfadin contiene sodio, glicerol y benzoato de sodio
Este medicamento contiene 0,7 mg (0,03 mmol) de sodio por ml.
Una dosis de 20 ml de suspensión oral (10 g de glicerol) o más puede provocar dolor de cabeza, molestias de estómago y diarrea.
El benzoato de sodio puede aumentar la ictericia (coloración amarilla de la piel y los ojos) en los neonatos ictéricos prematuros o nacidos a término y derivar en kernícterus (daño cerebral causado por depósitos de bilirrubina en el cerebro). En los recién nacidos, los niveles de bilirrubina en sangre (una sustancia que a altas concentraciones causa la coloración amarilla de la piel) se deben controlar estrictamente. Si los niveles son marcadamente más elevados de lo que debieran, especialmente en bebés prematuros con factores de riesgo como acidosis (pH de la sangre demasiado bajo) o baja concentración de albúmina (una proteína de la sangre), se debe considerar el tratamiento con Orfadin cápsulas en lugar de la suspensión oral hasta que las concentraciones plasmáticas de bilirrubina se hayan normalizado.
3. Cómo tomar Orfadin
Siga exactamente las instrucciones de administración del medicamento indicadas por su médico. En caso de duda, pregunte a su médico o farmacéutico.
Siga cuidadosamente las instrucciones facilitadas a continuación sobre preparación y administración de la dosis para asegurarse de que se administra la dosis correcta.
El tratamiento con este medicamento debe ser iniciado y supervisado por un médico con experiencia en el tratamiento de la enfermedad (tirosinemia hereditaria tipo 1).
La dosis recomendada diaria es 1 mg/kg de peso corporal dividida en 2 tomas. Su médico ajustará la dosis individualmente.
La suspensión oral se debe administrar directamente en la boca sin diluir mediante la jeringa para uso oral.
Orfadin no se debe inyectar. No acople una aguja a la jeringa.
Cómo preparar la dosis que se debe administrar
La dosis recetada por su médico se debe administrar en ml de suspensión y no en mg. Esto es así porque la jeringa para uso oral que se debe utilizar para extraer del frasco la dosis correcta del producto está marcada en ml. Si su receta está en mg, póngase en contacto con su médico o farmacéutico para que le aconseje.
El envase contiene un frasco de medicamento con una cápsula de cierre, un adaptador para el frasco y tres jeringas para uso oral (1 ml, 3 ml y 5 ml) Utilice siempre estas jeringas para uso oral para tomar el medicamento.
• La jeringa para uso oral de 1 ml (la más pequeña) está graduada entre 0,1 ml y 1 ml mediante pequeñas marcas de 0,01 ml. Se utiliza para medir dosis de hasta 1 ml.
• La jeringa para uso oral de 3 ml (la mediana) está graduada entre 1 ml y 3 ml mediante pequeñas marcas de 0,1 ml. Se utiliza para medir dosis de más de 1 ml y hasta 3 ml.
• La jeringa para uso oral de 5 ml (la más grande) está graduada entre 1 ml y 5 ml mediante pequeñas marcas de 0,2 ml. Se utiliza para medir dosis de más de 3 ml.
Es importante que utilice la jeringa para uso oral correcta para tomar el medicamento. Su médico, farmacéutico o enfermero le indicará cómo debe usarse la jeringa para uso oral en función de la dosis recetada.
Cómo preparar un nuevo frasco del medicamento para utilizarlo por primera vez:
Antes de tomar la primera dosis, se debe agitar el frasco vigorosamente, ya que durante el almacenamiento prolongado las partículas forman un precipitado sólido en el fondo del frasco. Siga las instrucciones descritas a continuación:
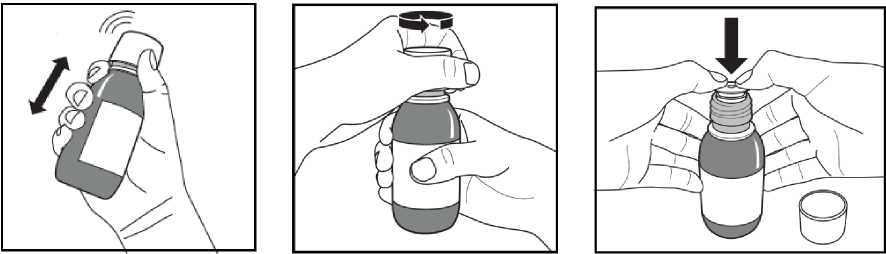
Figura A.
Figura B.
Figura C.
1. Saque el frasco de la nevera y anote la fecha en la etiqueta del frasco.
2. Agite el frasco vigorosamente durante al menos 20 segundos hasta que la torta sólida que hay en el fondo del frasco se disperse por completo (Figura A).
3. Quite el cierre de rosca a prueba de niños; para ello apriételo hacia abajo con fuerza y gírela en sentido contrario a las agujas del reloj (Figura B).
4. Coloque el frasco abierto derecho encima de la mesa. Empuje con fuerza el adaptador de plástico dentro del cuello del frasco todo lo que pueda (Figura C) y cierre el frasco con el cierre de rosca a prueba de niños.
Para las dosis posteriores consulte a continuación las instrucciones “Cómo preparar una dosis del medicamento”
Cómo preparar una dosis del medicamento
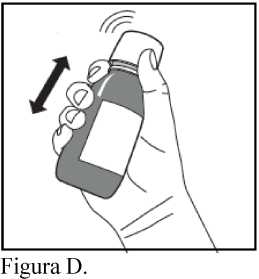
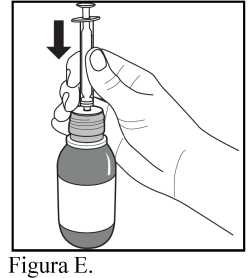
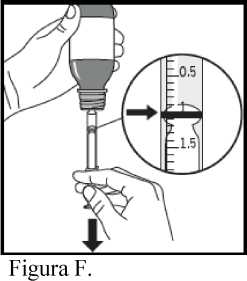
1. Agite el frasco vigorosamente durante al menos 5 segundos (Figura D).
2. Inmediatamente, retire el cierre de rosca a prueba de niños y abra el frasco.
3. Empuje hasta el fondo el émbolo de la jeringa para uso oral.
4. Mantenga el frasco en posición vertical e inserte la jeringa para uso oral firmemente en el orificio del adaptador situado en la parte superior del frasco (Figura E).
5. Invierta con cuidado el frasco sin extraer la jeringa para uso oral (Figura F).
6. Para obtener la dosis recetada (ml), tire del émbolo lentamente hacia abajo hasta que el borde superior del anillo negro quede nivelado exactamente con la línea que señala la dosis (Figura F). Si observa alguna burbuja de aire dentro de la jeringa para uso oral llena, haga retroceder el émbolo hasta que salgan las burbujas. Después vuelva a tirar del émbolo hacia abajo hasta que el borde superior del anillo negro quede nivelado exactamente con la línea que señala la dosis.
7. Coloque el frasco de nuevo en posición vertical. Gire suavemente la jeringa para uso oral tirando de ella para sacarla del frasco.
8. La dosis se debe administrar inmediatamente en la boca (sin diluir) para evitar que se forme un precipitado en la jeringa para uso oral. La jeringa para uso oral se debe vaciar lentamente para permitir que el paciente trague el producto; si el medicamento sale en un chorro rápido se puede provocar un atragantamiento.
9. Coloque inmediatamente el cierre de rosca a prueba de niños. El adaptador del frasco no debe retirarse.
10. El frasco puede mantenerse a temperatura ambiente (no superior a 25 °C).
Limpieza:
Limpie inmediatamente la jeringa para uso oral con agua. Separe el émbolo del cilindro de la jeringa y enjuague ambos con agua. Sacuda el exceso de agua y deje que la jeringa para uso oral se seque desmontada hasta que tenga que volver a montarla para una nueva administración.
Si toma más Orfadin del que debe
Si ha tomado más cantidad de este medicamento del que debiera, comuníqueselo inmediatamente a su
médico o farmacéutico.
Si olvidó tomar Orfadin
No tome una dosis doble para compensar las dosis olvidadas. Si ha olvidado tomar una dosis,
comuníqueselo a su médico o farmacéutico.
Si interrumpe el tratamiento con Orfadin
Si estima que la acción del medicamento no es la adecuada, comuníqueselo a su médico. No cambie la
dosis ni suspenda el tratamiento sin hablar primero con su médico.
Si tiene cualquier duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Si aprecia cualquier efecto adverso relacionado con los ojos, comuníqueselo inmediatamente a su médico para que le realice una exploración oftalmológica. El tratamiento con nitisinona aumenta los niveles de tirosina en la sangre, que pueden causar síntomas relacionados con los ojos. Los efectos adversos oculares frecuentes (pueden afectar a más de 1 de cada 10 personas) debidos a los niveles más altos de tirosina son inflamación ocular (conjuntivitis), opacidad e inflamación de la córnea (queratitis), sensibilidad a la luz (fotofobia) y dolor de ojos. La inflamación de los párpados (blefaritis) es un efecto adversos poco frecuente (puede afectar hasta 1 de cada 100 personas).
Otros efectos adversos frecuentes
- Disminución del número de plaquetas (trombocitopenia) y glóbulos blancos (leucopenia), reducción de determinados tipos de glóbulos blancos (granulocitopenia).
Otros efectos adversos poco frecuentes
- aumento del número de glóbulos blancos (leucocitosis),
- picor (prurito), inflamación de la piel (dermatitis exfoliativa), sarpullido.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice VAnexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Orfadin
Mantener este medicamento fuera de la vista y el alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el frasco y la caja después de “EXP”. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
Mantener el frasco en posición vertical.
Después de la primera apertura, el medicamento se puede conservar durante un único período de 2 meses a una temperatura no superior a 25 °C, después del cual se debe desechar.
No olvide anotar en el frasco la fecha en la que lo sacó de la nevera.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico como deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma ayudará a proteger el medio ambiente.
Contenido del envase e información adicional
6.
Composición de Orfadin
- El principio activo es nitisinona. Cada ml contiene 4 mg de nitisinona.
- Los demás componentes son hipromelosa, glicerol (ver sección 2), polisorbato 80, benzoato de sodio (E211) (ver sección 2), ácido cítrico monohidrato, citrato de sodio (ver sección 2), aroma de fresa (artificial) y agua purificada.
Aspecto del producto y contenido del envase
La suspensión oral es una suspensión blanca, opaca y ligeramente viscosa. Antes de agitar el frasco, puede aparecer como una torta sólida en el fondo con un sobrenadante ligeramente opalescente.
Se presenta en un frasco de 100 ml de color topacio con un cierre de rosca blanco a prueba de niños. Cada frasco contiene 90 ml de suspensión oral.
Cada envase contiene un frasco, un adaptador para el frasco y 3 jeringas para uso oral.
Titular de la autorización de comercialización
Swedish Orphan Biovitrum International AB
SE-112 76 Stockholm
Suecia
Fabricante
Apotek Produktion & Laboratorier AB Celsiusgatan 43 SE-212 14 Malmo Suecia
Fecha de la última revisión de este prospecto:
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu. También existen enlaces a otras páginas web sobre enfermedades raras y medicamentos huérfanos.
40