Opsumit 10 Mg Comprimidos Recubiertos Con Pelicula
ANEXO I
RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Opsumit 10 mg comprimidos recubiertos con película.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 10 mg de macitentan.
Excipientes con efecto conocido
Cada comprimido recubierto con película contiene aproximadamente 37 mg de lactosa (en forma de monohidrato) y aproximadamente 0,06 mg de lecitina (soja) (E322).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido).
Comprimidos recubiertos con película de color blanco a blanquecino, redondos, biconvexos y de
5,5 mm con el grabado “10” en una cara.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Opsumit, en monoterapia o en combinación, está indicado para el tratamiento a largo plazo de la hipertensión arterial pulmonar (HAP) en pacientes adultos clasificados como clase funcional (CF) II a III de la Organización Mundial de la Salud (OMS).
Se ha demostrado su eficacia en una población con HAP, incluidos HAP idiopática o heredable, HAP asociada a trastornos del tejido conjuntivo e HAP asociada a cardiopatía congénita corregida simple (ver sección 5.1).
4.2 Posología y forma de administración
El tratamiento debe ser iniciado y supervisado únicamente por un médico con experiencia en el tratamiento de la HAP.
Posología
Opsumit se debe tomar por vía oral en una dosis de 10 mg una vez al día, con o sin alimentos. Los comprimidos recubiertos con película no se deben romper y se deben tragar enteros, con agua.
Opsumit se debe tomar cada día sobre la misma hora. Si el paciente olvida una dosis de Opsumit, deberá tomársela lo antes posible y tomar la siguiente dosis a la hora habitual. El paciente debe ser advertido de que no podrá tomar una dosis doble si se olvida de tomar una.
Edad avanzada
No se requieren ajustes de la dosis en pacientes mayores de 65 años (ver sección 5.2). Existe una experiencia clínica limitada en pacientes mayores de 75 años. Por tanto, Opsumit se debe utilizar con precaución en esta población (ver sección 4.4).
Insuficiencia hepática
Según los datos de farmacocinética, no se precisan ajustes de la dosis en pacientes con insuficiencia hepática leve, moderada o severa (ver secciones 4.4 y 5.2). Sin embargo, no existe experiencia clínica con el uso de macitentan en pacientes con HAP e insuficiencia hepática moderada o severa. Opsumit no se debe iniciar en pacientes con insuficiencia hepática severa, o elevación clínicamente significativa de las aminotransferasas hepáticas (3 veces por encima del Límite Superior de Normalidad (>3 x LSN); ver secciones 4.3 y 4.4).
Insuficiencia renal
Según los datos de farmacocinética, no se precisa ajuste de la dosis en pacientes con insuficiencia renal. No existe experiencia clínica con el uso de macitentan en pacientes con HAP e insuficiencia renal severa. No se recomienda el uso de Opsumit en pacientes sometidos a diálisis (ver secciones 4.4 y 5.2).
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de macitentan en niños.
4.3 Contraindicaciones
• Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
• Embarazo (ver la sección 4.6).
• Mujeres en edad fértil que no utilizan métodos anticonceptivos fiables (ver secciones 4.4 y 4.6).
• Lactancia (ver sección 4.6).
• Pacientes con insuficiencia hepática severa (con o sin cirrosis) (ver sección 4.2).
• Valores de aminotransferasas hepáticas (aspartato aminotransferasa(AST) y/o alanina aminotransferasa (ALT) > 3 x LSN) (ver secciones 4.2 y 4.4).
4.4 Advertencias y precauciones especiales de empleo
No se ha establecido el balance beneficio/riesgo de macitentan en pacientes con hipertensión arterial pulmonar en clase funcional I de la OMS.
Función hepática
Las elevaciones en las aminotransferasas hepáticas (AST, ALT) se han asociado a HAP y a los antagonistas de los receptores de la endotelina (AREs). No se debe iniciar tratamiento con Opsumit en pacientes con insuficiencia hepática severa o niveles elevados de aminotransferasas (> 3 x LSN) (ver secciones 4.2 y 4.3), y no está recomendado en pacientes con insuficiencia hepática moderada. Se debe realizar una determinación de los niveles de enzimas hepáticas antes de iniciar el tratamiento con Opsumit.
Se debe monitorizar los signos de lesión hepática de los pacientes y se recomienda controlar mensualmente la ALT y la AST. En caso de que se produzcan elevaciones clínicamente relevantes e inexplicables de aminotransferasas, o si las elevaciones se acompañan de un aumento en la bilirrubina > 2 x LSN, o de síntomas clínicos de daño hepático (p. ej., ictericia), se debe suspender el tratamiento con Opsumit.
Podrá considerarse la reanudación del tratamiento con Opsumit una vez los niveles de enzimas hepáticas hayan retornado al intervalo normal en pacientes que no han experimentado síntomas clínicos de daño hepático. Se recomienda el asesoramiento de un hepatólogo.
Concentración de hemoglobina
Como sucede con otros AREs, el tratamiento con macitentan se ha asociado a una reducción en la concentración de hemoglobina (ver sección 4.8). En estudios controlados con placebo, las reducciones relacionadas con macitentan en la concentración de hemoglobina no fueron progresivas, se estabilizaron después de las primeras 4-12 semanas de tratamiento y permanecieron estables durante el tratamiento crónico. Se han notificado casos de anemia que precisaron transfusiones de sangre con macitentan y otros AREs. No se recomienda el inicio de Opsumit en pacientes con anemia severa. Se recomienda medir las concentraciones de hemoglobina antes del inicio del tratamiento y repetir las determinaciones durante el tratamiento según esté clínicamente indicado.
Enfermedad venooclusiva pulmonar
Se han notificado casos de edema pulmonar con vasodilatadores (principalmente prostaciclinas) cuando se han utilizado en pacientes con enfermedad venooclusiva pulmonar. En consecuencia, si se producen signos de edema pulmonar con la administración de macitentan en pacientes con HAP, se debe considerar la posibilidad de que exista una enfermedad venooclusiva pulmonar.
Uso en mujeres en edad fértil
El tratamiento con Opsumit sólo se debe iniciar en mujeres en edad fértil cuando se ha descartado el embarazo, se les ha aconsejado adecuadamente sobre métodos anticonceptivos y se utilice un método anticonceptivo fiable (ver secciones 4.3 y 4.6). Las mujeres no se deben quedar embarazadas hasta después de 1 mes de suspender el tratamiento con Opsumit. Se recomienda realizar pruebas de embarazo mensuales durante el tratamiento con Opsumit para facilitar la detección precoz del embarazo.
Uso concomitante con inductores potentes del CYP3A4
En presencia de inductores potentes del CYP3A4 puede producirse una reducción de la eficacia de macitentan. Se debe evitar la combinación de macitentan con inductores potentes del CYP3A4 (p. ej., rifampicina, hierba de San Juan, carbamazepina y fenitoína) (ver sección 4.5).
Uso concomitante con inhibidores potentes del CYP3A4
Se debe tener precaución cuando macitentan se administra de forma concomitante con inhibidores potentes del CYP3A4 (p. ej., itraconazol, ketoconazol, voriconazol, claritromicina, telitromicina, nefazodona, ritonavir y saquinavir) (ver sección 4.5).
Insuficiencia renal
Los pacientes con insuficiencia renal pueden presentar un mayor riesgo de hipotensión y anemia durante el tratamiento con macitentan. Por tanto, se debe considerar el control de la presión arterial y la hemoglobina. No existe experiencia clínica con el uso de macitentan en pacientes con HAP e insuficiencia renal severa. Se recomienda precaución en esta población. No hay experiencia en el uso de macitentan en pacientes sometidos a diálisis, por lo que no se recomienda el uso de Opsumit en esta población (ver secciones 4.2 y 5.2).
Edad avanzada
Existe una experiencia clínica limitada con macitentan en pacientes mayores de 75 años, por lo que Opsumit se debe utilizar con precaución en esta población (ver sección 4.2).
Excipientes
Los comprimidos de Opsumit contienen lactosa. Los pacientes con problemas hereditarios raros de intolerancia a galactosa, insuficiencia de lactasa de Lapp o malabsorción de glucosa-galactosa no deben tomar este medicamento.
Los comprimidos de Opsumit contienen lecitina derivada de la soja. En caso de hipersensibilidad a la soja, no se debe utilizar Opsumit (ver sección 4.3).
4.5 Interacción con otros medicamentos y otras formas de interacción
Estudios in vitro
Las enzimas CYP3A4, CYP2C8, CYP2C9 y CYP2C19 del citocromo P450 intervienen en el metabolismo de macitentan y la formación de sus metabolitos (ver sección 5.2). Macitentan y su metabolito activo no tienen efectos inhibidores o inductores clínicamente relevantes en las enzimas del citocromo P450.
Macitentan y su metabolito activo no son inhibidores de los transportadores de la captación hepática o renal a concentraciones clínicamente relevantes, incluidos los polipéptidos transportadores de aniones orgánicos (OATP1B1 y OATP1B3). Macitentan y su metabolito activo no son sustratos relevantes de OATP1B1 y OATP1B3, penetrando en el hígado mediante difusión pasiva.
Macitentan y su metabolito activo no son inhibidores de las bombas de eflujo hepático o renal a concentraciones clínicamente relevantes, incluidas la proteína de resistencia a multifármacos (P-gp, MDR-1) y los transportadores de expulsión de toxinas y multifármacos (MATE1 y MATE2-K). Macitentan inhibe la proteína de resistencia en cáncer de mama (BCRP) a concentraciones intestinales clínicamente relevantes. Macitentan no es un sustrato de la P-gp/MDR-1.
A concentraciones clínicamente relevantes, macitentan y su metabolito activo no interactúan con proteínas implicadas en el transporte de sales biliares hepáticas, es decir, la bomba de exportación de sales biliares (BSEP) y el polipéptido cotransportador de sodio taurocolato (NTCP).
Estudios in vivo
Los estudios de interacciones se han realizado solo en adultos.
Warfarina: Macitentan administrado como dosis múltiples de 10 mg una vez al día no tuvo efectos sobre la exposición a S-warfarina (sustrato del CYP2C9) o R-warfarina (sustrato del CYP3A4) después de una dosis única de 25 mg de warfarina. El efecto farmacodinámico de warfarina en el cociente normalizado internacional (INR) no se vio afectado por macitentan. La farmacocinética de macitentan y su metabolito activo no se vieron afectados por el efecto de la warfarina.
Sildenafilo: En el estado estacionario, la exposición a sildenafilo 20 mg tres veces al día se incrementó en un 15% durante la administración concomitante de macitentan 10 mg una vez al día. Sildenafilo, un sustrato del CYP3A4, no afectó a la farmacocinética de macitentan, mientras que se produjo una reducción del 15% en la exposición al metabolito activo de macitentan. Estos cambios no se consideran clínicamente relevantes. En un ensayo controlado con placebo en pacientes con HAP, se demostró la eficacia y la seguridad de macitentan en combinación con sildenafilo.
Ketoconazol: En presencia de ketoconazol 400 mg una vez al día, un inhibidor potente del CYP3A4, la exposición a macitentan se incrementó en aproximadamente 2 veces. El aumento previsto fue de aproximadamente 3 veces en presencia de ketoconazol 200 mg dos veces al día con un modelo farmacocinético basado en la fisiología (FCBF). Se deben considerar las incertidumbres de dicho modelado. La exposición al metabolito activo de macitentan se redujo en un 26%. Se debe tener precaución cuando macitentan se administre de forma concomitante con inhibidores potentes del CYP3A4 (ver sección 4.4).
Ciclosporina A: El tratamiento concomitante con ciclosporina A 100 mg dos veces al día, un inhibidor combinado del CYP3A4 y OATP, no alteró de forma clínicamente relevante la exposición en equilibrio a macitentan y su metabolito activo.
Inductores potentes del CYP3A4: El tratamiento concomitante con rifampicina 600 mg diarios, un inductor potente del CYP3A4, redujo la exposición en el estado estacionario a macitentan en un 79%, pero no afectó a la exposición al metabolito activo. Se debe considerar la reducción de la eficacia de macitentan en presencia de un inductor potente del CYP3A4 como la rifampicina. Se debe evitar la combinación de macitentan con inductores potentes del CYP3A4 (ver sección 4.4).
Anticonceptivos hormonales: Una dosis diaria de 10 mg de macitentan no afectó a la farmacocinética de un anticonceptivo oral (1 mg de noretisterona y 35 pg de etinilestradiol).
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos relativos al uso de macitentan en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección 5.3). El riesgo potencial en humanos aún se desconoce. Opsumit está contraindicado durante el embarazo y en mujeres en edad fértil que no utilizan métodos anticonceptivos fiables (ver sección 4.3).
Uso en mujeres en edad fértil
El tratamiento con Opsumit solo se debe iniciar en mujeres en edad fértil cuando se haya confirmado la ausencia de embarazo, se haya proporcionado asesoramiento adecuado sobre la anticoncepción y se utilicen métodos anticonceptivos fiables (ver secciones 4.3 y 4.4). Las mujeres no se deben quedar embarazadas durante el mes posterior a la suspensión de Opsumit. Se recomienda realizar pruebas de embarazo mensuales durante el tratamiento con Opsumit para una detección temprana de embarazo.
Lactancia
Se desconoce si macitentan se excreta en la leche materna. En ratas, macitentan y sus metabolitos se excretan en la leche durante la lactancia (ver sección 5.3). No se puede excluir el riesgo para los lactantes. El uso de Opsumit está contraindicado durante la lactancia (ver sección 4.3).
Fertilidad masculina
Se observó atrofia tubular testicular en animales macho después del tratamiento con macitentan (ver sección 5.3). Se desconoce la relevancia de este hallazgo para los humanos, aunque no puede descartarse un deterioro de la espermatogénesis.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Macitentan tiene una influencia leve en la capacidad para conducir y utilizar máquinas. Se debe tener en cuenta el estado clínico del paciente y el perfil de reacciones adversas de macitentan (como cefalea, hipotensión) a la hora de considerar la capacidad del paciente para conducir y utilizar máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Las reacciones adversas al fármaco notificadas con mayor frecuencia son nasofaringitis (14,0%), cefalea (13,6%) y anemia (13,2%, ver sección 4.4). La mayoría de las reacciones adversas que se produjeron fueron de intensidad leve a moderada.
Tabla de reacciones adversas
La seguridad de macitentan se ha evaluado en un ensayo controlado con placebo a largo plazo en 742 pacientes con HAP sintomática. La media de la duración del tratamiento fue de 103,9 semanas en el grupo de macitentan 10 mg, y de 85,3 semanas en el grupo de placebo. En la tabla siguiente se muestran las reacciones adversas asociadas a macitentan obtenidas a partir de este estudio clínico.
Las frecuencias se definen de la siguiente manera: muy frecuentes (> 1/10), frecuentes (de > 1/100 a < 1/10), poco frecuentes (de > 1/1.000 a < 1/100), raras (de > 1/10.000 a < 1/1.000), muy raras (< 1/10.000).
|
Sistema de Clasificación de Órganos |
Frecuencia |
Reacción adversa |
|
Infecciones e infestaciones |
Muy frecuentes |
Nasofaringitis |
|
Muy frecuentes |
Bronquitis | |
|
Frecuentes |
Faringitis | |
|
Frecuentes |
Gripe | |
|
Frecuentes |
Infección urinaria | |
|
Trastornos de la sangre y del sistema linfático |
Muy frecuentes |
Anemia |
|
Trastornos del sistema inmunológico |
Raras |
Reacciones de hipersensibilidad (p.ej., angioedema, prurito, erupción cutánea)* |
|
Trastornos del sistema nervioso |
Muy frecuentes |
Cefalea |
|
Trastornos vasculares |
Frecuentes |
Hipotensión** |
|
Trastornos respiratorios, torácicos y mediastínicos |
Frecuentes |
Congestión nasal* |
|
Trastornos generales y alteraciones en el lugar de administración |
Muy frecuentes |
Edema, retención de líquidos1 |
* Información obtenida del análisis de datos acumulados de estudios controlados con placebo.
Descripción de reacciones adversas seleccionadas
** La hipotensión se ha asociado al uso de ARE. En un estudio doble ciego a largo plazo en pacientes con HAP, la hipotensión se notificó en el 7,0% y el 4,4% de los pacientes de macitentan 10 mg y placebo, respectivamente. Estos datos corresponden a 3,5 acontecimientos/100 pacientes-año entre los tratados con macitentan 10 mg frente a 2,7 acontecimientos/100 pacientes-año entre los tratados con placebo.
Aminotransferasas hepáticas
La incidencia de elevaciones de aminotransferasas (ALT/AST) > 3 * LSN fue del 3,4% con macitentan 10 mg y del 4,5% con placebo en un estudio doble ciego en pacientes con HAP. Se produjeron elevaciones > 5 * LSN en el 2,5% de los pacientes de macitentan 10 mg frente al 2% de los pacientes con placebo.
Hemoglobina
En un estudio doble ciego en pacientes con HAP, macitentan 10 mg se asoció a una reducción media en la hemoglobina frente a placebo de 1 g/dl. Se notificó una reducción en la concentración de hemoglobina desde el inicio hasta menos de 10 g/dl en el 8,7% de los pacientes tratados con macitentan 10 mg y en el 3,4% de los pacientes tratados con placebo.
Leucocitos
En un estudio doble ciego en pacientes con HAP, macitentan 10 mg se asoció a una reducción de 0,7 * 102 3 4 5 6/l en el recuento leucocitario medio respecto al inicio frente a la ausencia de cambio en los pacientes tratados con placebo.
Trombocitos
En un estudio doble ciego en pacientes con HAP, macitentan 10 mg se asoció a una reducción en el recuento medio de plaquetas de 17 * 106/l frente a una reducción media de 11 * 106/l en pacientes tratados con placebo.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de macitentan en niños.
Notificación de sospechas de reacciones adversas
Es importante notificar las sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Macitentan se ha administrado en una dosis única de hasta 600 mg en sujetos sanos. Se observaron reacciones adversas de cefalea, náuseas y vómitos. En caso de sobredosis, se deben adoptar medidas habituales de soporte, según proceda. Debido al alto grado de unión a proteínas de macitentan, es improbable que la diálisis resulte efectiva.
como la HAP, el sistema local de la ET está aumentado e interviene en la hipertrofia vascular y el daño orgánico.
Macitentan es un antagonista potente de los receptores de la endotelina ETA y ETB, activo por vía oral y aproximadamente 100 veces más selectivo para ETA comparado con ETB in vitro. Macitentan presenta gran afinidad y ocupación prolongada de los receptores de ET en células del músculo liso de la arteria pulmonar humana. Esto previene la activación mediada por la endotelina de otros sistemas de segundos mensajeros que dan lugar a vasoconstricción y proliferación de células del músculo liso.
Eficacia y seguridad clínicas
Eficacia en pacientes con hipertensión arterial pulmonar
Se llevó a cabo un estudio multicéntrico, doble ciego, controlado con placebo, de grupos paralelos, basado en eventos y de fase 3 (AC-055-302/SERAPHIN) en 742 pacientes con HAP sintomática, aleatorizados a tres grupos de tratamiento (placebo [N = 250], 3 mg [N = 250] o 10 mg [N = 242] de macitentan una vez al día), para evaluar el efecto a largo plazo sobre la morbilidad o la mortalidad.
En el periodo basal, la mayoría de los pacientes incluidos (64%) estaban recibiendo tratamiento con una dosis estable de terapia específica para la HAP, como inhibidores de la fosfodiesterasa (61%) y/o prostanoides inhalados/orales (6%).
El criterio principal de valoración fue el tiempo hasta la primera incidencia de un evento de morbilidad o mortalidad, hasta el final del tratamiento doble ciego, definido como la muerte, o septostomía auricular, o trasplante de pulmón o inicio de prostanoides intravenosos (i.v.) o subcutáneos (s.c.), u otro empeoramiento de la HAP. Otro empeoramiento de la HAP se definió como la presencia de los tres componentes siguientes: una reducción mantenida en la distancia recorrida en 6 minutos (TM6M) de al menos el 15% respecto a basal, un deterioro de los síntomas de HAP (deterioro de la CF de la OMS o insuficiencia cardíaca derecha) y la necesidad de un nuevo tratamiento para la HAP. Todos los eventos fueron adjudicados y confirmados por un comité ciego de eventos clínicos independiente.
Se realizó un seguimiento de todos los pacientes hasta el final del estudio (FdE) para determinar el estado vital. El FdE se declaró cuando se alcanzó el número predefinido de eventos del criterio de valoración principal. En el período entre el final del tratamiento (FdT) y el FdE, los pacientes pudieron recibir macitentan 10 mg en régimen abierto o un tratamiento alternativo para la HAP. La mediana global de la duración del tratamiento doble ciego fue de 115 semanas (hasta un máximo de 188 semanas con macitentan).
La media de edad de todos los pacientes fue de 46 años (rango de 12 a 85 años, incluidos 20 pacientes menores de 18 años, 706 pacientes de entre 18 y 74 años y 16 pacientes de 75 años o más) siendo la mayoría de los sujetos de raza blanca (55%) y mujeres (77%). Aproximadamente el 52%, 46% y 2% de los pacientes presentaban CF II, III y IV de la OMS, respectivamente.
La HAP idiopática o heredable fue la etiología más frecuente de la población del estudio (57%), seguida de HAP debida a trastornos del tejido conjuntivo (31%), HAP asociada a cardiopatía congénita corregida simple (8%) e HAP asociada a otras etiologías (fármacos y toxinas [3%] y VIH [1%]).
Criterios de valoración
El tratamiento con macitentan 10 mg dio lugar a una reducción del riesgo del 45% (hazard ratio [HR] 0,55; IC del 97,5%: 0,39 a 0,76;p de la prueba del logaritmo del rango < 0,0001) del criterio de valoración compuesto de morbilidad y mortalidad hasta el FdT frente a placebo [Figura 1 y Tabla 1]. El efecto terapéutico se estableció de forma precoz y se mantuvo.
La eficacia de macitentan 10 mg en el criterio de valoración principal fue consistente en todos los subgrupos de edad, sexo, origen étnico, región geográfica, etiología, uso en monoterapia o en combinación con otro tratamiento para la HAP y CF de la OMS (I/II y III/IV).
Figura 1 Resultados de Kaplan-Meier del primer evento de morbilidad-mortalidad en SERAPHIN
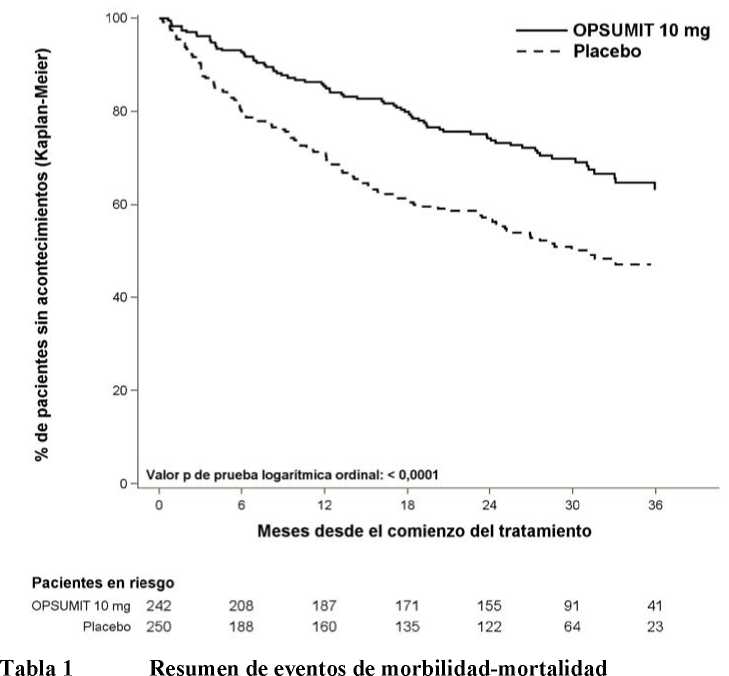
|
Pacientes con eventos |
Comparación de tratamientos: | |||||
|
Macitentan 10 mg |
frente a placebo | |||||
|
Criterios de |
Placebo (N = 250) |
Macitenta n 10 mg (N = 242) |
Reducción |
p de la prueba del logaritmo del rango | ||
|
valoración y estadística |
Reducción de riesgo absoluta |
de riesgo relativo (IC del 97,5%) |
CRI a (IC del 97,5%) | |||
|
Eventos de |
45% (24%; 61%) |
0,55 | ||||
|
morbilidad-mortalidad b |
53% |
37% |
16% |
(0,39; 0,76) |
< 0,0001 | |
|
Muerte c n (%) |
19 (7,6%) |
14 (5,8%) |
2% |
36% (-42%; 71%) |
0,64 (0,29; 1,42) |
0,20 |
|
Deterioro de la HAP n (%) |
93 (37,2%) |
59 (24,4%) |
13% |
49% |
0,51 | |
|
Inicio de |
(27%, 65%) |
(0,35; |
< 0,0001 | |||
|
prostanoides i.v./s.c. nf%) |
6 (2,4%) |
1 (0,4%) |
2% |
0,73) | ||
a = basado en el modelo de riesgos proporcionales de Cox
b = % de pacientes con un evento a los 36 meses = 100 x (1 - cálculo de KM)
c= muerte por cualquier causa hasta el FdT independientemente del deterioro previo
El número de muertes por cualquier causa hasta el FdE con macitentan 10 mg fue de 35 frente a 44 con placebo (CRI 0,77; IC del 97,5% 0,46 a 1,28).
El riesgo de muerte u hospitalización relacionada con la HAP hasta el FdT se redujo en un 50%
(HR 0,50; IC del 97,5% 0,34 a 0,75; p de la prueba del logaritmo del rango < 0,0001) en pacientes tratados con macitentan 10 mg (50 eventos) respecto a placebo (84 eventos). A los 36 meses, el 44,6% de los pacientes tratados con placebo y el 29,4% de los tratados con macitentan 10 mg (Reducción del Riesgo Absoluto = 15,2%) habían sido hospitalizados por HAP o habían muerto por una causa relacionada con la HAP.
Criterios de valoración sintomáticos
La capacidad de realizar ejercicio se evaluó como variable secundaria. El tratamiento con macitentan 10 mg a los 6 meses dio lugar a un aumento medio corregido para placebo en el TM6M de 22 metros (IC del 97,5% 3 a 41; p = 0,0078). La evaluación del TM6M en función de la clase funcional dio lugar a un aumento medio corregido para placebo entre basal y el mes 6 en los pacientes con CF III/IV de 37 metros (IC del 97,5% 5 a 69) y de 12 metros en la CF I/II (IC del 97,5% -8 a 33). El aumento en el TM6M alcanzado con macitentan se mantuvo durante todo el estudio.
El tratamiento con macitentan 10 mg a los 6 meses dio lugar a una probabilidad un 74% mayor de mejora de la CF de la OMS respecto a placebo (cociente de riesgo de 1,74; IC del 97,5% 1,10 a 2,74; p = 0,0063).
Macitentan 10 mg mejoró la calidad de vida según la evaluación del cuestionario SF-36.
Criterios de valoración hemodinámicos
Se evaluaron los parámetros hemodinámicos en un subconjunto de pacientes (placebo [N = 67], macitentan 10 mg [N = 57]) después de 6 meses de tratamiento. Los pacientes tratados con macitentan 10 mg alcanzaron una reducción media del 36,5% (IC del 97,5% 21,7 a 49,2%) en la resistencia vascular pulmonar y un aumento de 0,58 l/min/m2 (IC del 97,5% 0,28 a 0,93 l/min/m2) en el índice cardíaco en comparación con placebo.
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con macitentan en todos los subconjuntos de la población pediátrica para la HAP (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
La farmacocinética de macitentan y su metabolito activo se han documentado principalmente en sujetos sanos. La exposición a macitentan en pacientes con HAP fue aproximadamente 1,2 veces superior que en sujetos sanos. La exposición al metabolito activo, que es aproximadamente 5 veces menos potente que macitentan, fue aproximadamente 1,3 veces superior respecto a los sujetos sanos. La farmacocinética de macitentan en los pacientes con HAP no se vio influenciada por la gravedad de la enfermedad.
Después de la administración repetida, la farmacocinética de macitentan es dosis-proporcional hasta los 30 mg, inclusive.
Absorción
Las concentraciones plasmáticas máximas de macitentan se alcanzan aproximadamente 8 horas después de la administración. A partir de entonces, las concentraciones plasmáticas de macitentan y su metabolito activo se reducen lentamente, con una semivida de eliminación aparente de aproximadamente 16 horas y 48 horas, respectivamente.
En sujetos sanos, la exposición a macitentan y su metabolito activo permanece inalterada en presencia de alimentos y, por tanto, macitentan se puede tomar con o sin alimentos.
Distribución
Macitentan y su metabolito activo se unen de forma importante a las proteínas plasmáticas (> 99%), principalmente a la albúmina y, en menor medida, a la alfa-1-glucoproteína ácida. Macitentan y su metabolito activo ACT-132577 se distribuyen bien en los tejidos tal como indica un volumen de distribución (Vss/F) aparente de aproximadamente 50 L y 40 L para macitentan y ACT-132577, respectivamente.
Biotransformación
Macitentan tiene cuatro vías metabólicas principales. La despropilación oxidativa de la sulfamida proporciona un metabolito farmacológicamente activo. Esta reacción depende del sistema del citocromo P450, principalmente CYP3A4 (aproximadamente el 99%) con contribuciones mínimas de CYP2C8, CYP2C9 y CYP2C19. El metabolito activo circula en el plasma humano y puede contribuir al efecto farmacológico. Otras vías metabólicas proporcionan productos sin actividad farmacológica. Varios miembros de la familia CYP2C, como CYP2C8, CYP2C9 y CYP2C19, así como CYP3A4, intervienen en la formación de estos metabolitos.
Eliminación
Macitentan solo se excreta después de un amplio metabolismo. La principal vía de excreción es a través de la orina, que representa aproximadamente la eliminación del 50% de la dosis.
Poblaciones especiales
No hay un efecto clínicamente relevante de la edad, el sexo o el origen étnico en la farmacocinética de macitentan y su metabolito activo.
Insuficiencia renal
La exposición a macitentan y su metabolito activo se incrementó en 1,3 y 1,6 veces, respectivamente, en pacientes con insuficiencia renal severa. Este aumento no se considera clínicamente relevante (ver secciones 4.2 y 4.4).
Insuficiencia hepática
La exposición a macitentan se redujo en un 21%, 34% y 6% y, la del metabolito activo en un 20%, 25% y 25% en sujetos con insuficiencia hepática leve, moderada o severa, respectivamente. Esta reducción no se considera clínicamente relevante (ver secciones 4.2 y 4.4).
5.3 Datos preclínicos sobre seguridad
En perros, macitentan redujo la presión arterial con exposiciones similares a la exposición terapéutica humana. Se observó un engrasamiento de la íntima de las arterias coronarias con una exposición 17 veces superior a la exposición en humanos después de 4 a 39 semanas de tratamiento. Debido a la sensibilidad específica de la especie y al margen de seguridad, este hallazgo no se considera relevante para los humanos.
Se observó aumento del peso hepático e hipertrofia hepatocelular en ratones, ratas y perros después del tratamiento con macitentan. Estos cambios revirtieron en gran medida y se consideraron adaptaciones de tipo no adverso del hígado al aumento de la demanda metabólica.
Macitentan indujo hiperplasia mucosa entre mínima y ligera, así como infiltración inflamatoria en la submucosa de la cavidad nasal en el estudio de carcinogenicidad de ratones en todas las dosis. No se observaron hallazgos en la cavidad nasal en el estudio de toxicidad a 3 meses en ratones o en estudios de ratas y perros.
Macitentan no fue genotóxico en una batería estándar de ensayos in vitro e in vivo. Macitentan no fue fototóxico in vivo después de una dosis única con exposiciones de hasta 24 veces la exposición en humanos.
Estudios de carcinogenicidad a 2 años no mostraron un potencial carcinogénico con exposiciones 18 y 116 veces superiores a la exposición en humanos en ratas y ratones, respectivamente.
Se observó dilatación tubular testicular en estudios de toxicidad crónica con ratas y perros macho con márgenes de seguridad de 11,6 y 5,8, respectivamente. La dilatación tubular fue totalmente reversible. Después de 2 años de tratamiento, se observó atrofia tubular testicular en ratas con una exposición 4 veces superior a la humana. Se observó hipoespermatogénesis en el estudio de carcinogenicidad de larga duración en ratas y en estudios de toxicidad a dosis repetidas en perros tratados con dosis que proporcionaron márgenes de seguridad de 9,7 en ratas y de 23 en perros. Los márgenes de seguridad para la fertilidad fueron 18 para las ratas macho y de 44 para las ratas hembras. No se observaron hallazgos testiculares en ratones después del tratamiento de hasta 2 años. Se desconoce el efecto de macitentan en la fertilidad masculina humana (sección 4.6).
Macitentan fue teratogénico en conejos y ratas en todas las dosis analizadas. En ambas especies, hubo anomalías cardiovasculares y de fusión del arco mandibular.
La administración de macitentan a ratas hembra desde el final del embarazo y hasta la lactancia con exposiciones 5 veces superiores a la exposición en humanos provocó una reducción de la supervivencia de los cachorros y alteración de la capacidad reproductiva de la descendencia, expuesta a macitentan durante la vida intrauterina final y a través de la leche durante el período de lactancia.
El tratamiento de ratas jóvenes entre el día 4 y el 114 posnatales provocó una reducción del aumento del peso que dio lugar a efectos secundarios en el desarrollo (ligero retraso del descenso testicular, reducción reversible de la longitud de los huesos largos y prolongación del ciclo estrogénico). Se observaron un ligero aumento de la pérdida pre y postimplantación, reducción del número medio de cachorros y reducción del peso de los testículos y el epidídimo con exposiciones 7 veces superiores a la exposición en humanos. Se registraron atrofia tubular testicular y efectos mínimos en las variables reproductivas y la morfología espermática con exposiciones 3,8 veces superiores a la exposición en humanos.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido Lactosa monohidrato Celulosa microcristalina (E460i) Almidón glicolato de sodio tipo A Povidona
Estearato de magnesio (E572) Polisorbato 80 (E433)
Recubrimiento
Alcohol polivinílico (E1203) Dióxido de titanio (E171) Talco (E553b)
Lecitina, soja (E322)
Goma xantana (E415)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
4 años.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30 °C.
6.5 Naturaleza y contenido del envase
Blísteres de PVC/PE/PVdC/aluminio blancos, opacos en cajas de 15 o 30 comprimidos recubiertos con película.
Frascos de polietileno blanco de alta densidad con desecante de gel de sílice en cajas de 30 comprimidos recubiertos con película.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Ninguna especial.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Actelion Registration Ltd Chiswick Tower 13th Floor 389 Chiswick High Road Londres W4 4AL Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/893/001
EU/1/13/893/002
EU/1/13/893/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 20 de diciembre de 2013.
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante responsable de la liberación de los lotes
Actelion Manufacturing GmbH Emil-Barrel-Strasse 7 79639 Grenzach-Wyhlen Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
• Medidas adicionales de minimización de riesgos
El TAC deberá acordar los detalles del kit del prescriptor y un sistema de distribución controlada con la Autoridad Nacional Competente y implementarlo antes de su lanzamiento en dicho Estado Miembro. El TAC se asegurará de que antes de la prescripción, todos los profesionales sanitarios que tengan la intención de prescribir y/o dispensar Opsumit están provistos de un kit del prescriptor que contenga lo siguiente:
• El Resumen de las Características del Producto Opsumit;
• Listas de control de la prescripción;
• Folleto para el Profesional Sanitario con información sobre Opsumit;
• Tarjetas de alerta para el paciente.
La lista de control de la prescripción debe recordar a los prescriptores las contraindicaciones, advertencias y precauciones, así como los siguientes puntos clave:
• Que se proporcione a los pacientes la información adecuada sobre el uso seguro del producto;
• Que se asegure que las mujeres en edad fértil no están embarazadas y están utilizando métodos anticonceptivos fiables antes de comenzar el tratamiento con Opsumit;
• Que se proporcione a los pacientes la Tarjeta de paciente;
• Que es necesario realizar pruebas de embarazo basales y mensuales y del seguimiento de los niveles de hemoglobina y de la función hepática.
El folleto
para el Profesional Sanitario deberá contener los siguientes puntos clave:
Que los pacientes deben ser capaces de cumplir con los requisitos para el uso seguro de Opsumit;
Que existe riesgo de anemia, hepatotoxicidad y teratogenicidad y que es necesario una anticoncepción fiable;
Que es necesario un control basal y de:
• pruebas de embarazo mensuales;
• control periódico de los niveles de hemoglobina;
• control periódico de la función hepática;
• la importancia de informar a los pacientes que deben notificar de inmediato cualquier posible embarazo que ocurra durante el uso de Opsumit.
La tarjeta de alerta para los pacientes en tratamiento con Opsumit debe incluir los siguientes puntos clave:
• Que Opsumit es teratogénico en animales;
• Que las mujeres embarazadas no deben tomar Opsumit;
• Que las mujeres en edad fértil deben utilizar métodos anticonceptivos fiables;
• Que es necesario realizar pruebas de embarazo mensuales;
• Que es necesario realizar análisis de sangre regulares debido a que Opsumit causa
una disminución de los niveles de hemoglobina.
• Que es necesario monitorizar la función hepática debido a que Opsumit tiene un potencial efecto hepatotóxico.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
Opsumit 10 mg comprimidos recubiertos con película. macitentan
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 10 mg de macitentan.
3. LISTA DE EXCIPIENTES
Los comprimidos recubiertos con película también contienen lactosa y lecitina (soja) (E322). Para mayor información consultar el prospecto
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
15 comprimidos recubiertos con película 30 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 30 °C.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Actelion Registration Ltd Chiswick Tower 13th Floor 389 Chiswick High Road Londres W4 4AL Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/893/001
EU/1/13/893/002
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Opsumit 10 mg
BLÍSTERS
|
1. |
NOMBRE DEL MEDICAMENTO |
Opsumit 10 mg comprimidos macitentan
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Actelion
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE, CÓDIGO DE DONACIÓN Y DEL PRODUCTO_
Lot
"5 OTROS
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CAJA EXTERIOR/FRASCOS
1. NOMBRE DEL MEDICAMENTO
Opsumit 10 mg comprimidos recubiertos con película. macitentan
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 10 mg de macitentan.
3. LISTA DE EXCIPIENTES
Cada comprimido recubierto con película también contiene lactosa y lecitina (soja) (E322). Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
30 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 30 °C.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Actelion Registration Ltd Chiswick Tower 13th Floor 389 Chiswick High Road Londres W4 4AL Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/893/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Opsumit 10 mg
INFORMACIÓN MÍNIMA A INCLUIR EN FRASCOS FRASCOS_
1. NOMBRE DEL MEDICAMENTO_
Opsumit 10 mg comprimidos recubiertos con película. macitentan
2. PRINCIPIO(S) ACTIVO(S)
Cada comprimido recubierto con película contiene 10 mg de macitentan.
3. LISTA DE EXCIPIENTES
Cada comprimido recubierto con película también contiene lactosa.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
30 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 30 °C.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Actelion Registration Ltd Chiswick Tower 13th Floor 389 Chiswick High Road Londres W4 4AL Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/893/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Opsumit 10 mg
Tarjeta de paciente
|
Página 1 (anverso) |
Página 2 |
|
Para el tratamiento de la hipertensión arterial pulmonar |
Es importante que informe inmediatamente al médico prescriptor de cualquier embarazo o efecto adverso que pueda producirse durante el |
|
Esta tarjeta contiene información de seguridad importante que necesita conocer si recibe tratamiento con OPSUMIT. Lleve siempre consigo esta tarjeta y muéstresela al médico que le atienda. |
tratamiento con Opsumit. Centro de tratamiento: |
|
Opsumit 10 mg macitentan Comprimidos recubiertos con película |
Nombre del médico prescriptor: Número de teléfono del médico prescriptor: |
|
ES |
▼ Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. Ver el Prospecto en el que se incluye información sobre cómo comunicar estos efectos adversos. |
|
Página 3 (interior izquierdo) |
Página 4 (interior centro) |
|
Embarazo Opsumit puede afectar de forma negativa al desarrollo del feto. Por tanto, no debe tomar Opsumit si está embarazada y tampoco debe quedarse embarazada mientras tome Opsumit. Además, si padece hipertensión arterial pulmonar, el embarazo puede deteriorar en gran medida los síntomas de su enfermedad. |
Debe realizar pruebas de embarazo antes de iniciar el tratamiento con Opsumit y mensualmente durante el tratamiento, incluso si piensa que no está embarazada. Del mismo modo que otros medicamentos de esta clase, Opsumit puede inducir anemia (reducción del número de glóbulos rojos) y puede afectar |
|
Anticoncepción Deberá utilizar un método anticonceptivo fiable para el control de la natalidad (anticoncepción) durante el tratamiento con Opsumit. Consulte con su médico ante cualquier duda que pueda tener. |
al hígado. El médico le realizará análisis de sangre antes de que comience el tratamiento con Opsumit y durante el tratamiento para determinar: • Si tiene anemia (reducción del número de glóbulos rojos) • Si el hígado funciona correctamente |
|
Página 5 (interior derecho) |
Página 6 (reverso) |
|
Los signos indicativos de que el hígado puede no estar funcionando correctamente son: • Náuseas (necesidad de vomitar) • Vómitos • Fiebre (temperatura elevada) • Dolor de estómago (abdomen) • Ictericia (coloración amarillenta de la piel o el blanco de los ojos) • Orina de color oscuro • Picor en la piel • Letargo o fatiga (cansancio o agotamiento inusuales) • Síndrome pseudogripal (dolor articular o muscular con fiebre) |
La dosis recomendada de Opsumit es de un comprimido de 10 mg una vez al día. Trague el comprimido entero, con un vaso de agua y no lo mastique ni lo rompa. Opsumit puede tomarse con o sin alimentos. Si olvida tomar Opsumit, tome una dosis en cuanto lo recuerde y, a partir de entonces, siga tomando los comprimidos a las horas habituales. No tome una dosis doble para compensar el comprimido olvidado. Para obtener más información sobre Opsumit, lea detenidamente la hoja de información al paciente. Si tiene alguna duda sobre su tratamiento, consulte a su médico o farmacéutico. ©2013 Actelion Pharmaceuticals Ltd Opsumit es una marca comercial de Actelion Pharmaceuticals Ltd |
|
inmediatamente. |
B. PROSPECTO
Prospecto: Información para el usuario
Opsumit 10 mg comprimidos recubiertos con película
macitentan
'VEste medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Opsumit y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Opsumit
3. Cómo tomar Opsumit
4. Posibles efectos adversos
5. Conservación de Opsumit
6. Contenido del envase e información adicional
1. Qué es Opsumit y para qué se utiliza
Opsumit contiene el principio activo macitentan, que pertenece a la clase de medicamentos denominados “antagonistas de los receptores de la endotelina”.
Opsumit se utiliza para el tratamiento a largo plazo de la hipertensión arterial pulmonar (HAP) en adultos; puede utilizarse solo o con otros medicamentos para la HAP. La HAP es la presión arterial elevada en los vasos sanguíneos que llevan sangre del corazón a los pulmones (arterias pulmonares). En personas con HAP, estas arterias pueden estrecharse, por lo que el corazón tiene que esforzarse más para bombear sangre a través de ellas. Como consecuencia, los afectados se sienten cansados, mareados y con dificultad para respirar.
Opsumit ensancha las arterias pulmonares, con lo que facilita que el corazón bombee sangre a través de ellas. De este modo, se reduce la presión arterial, se alivian los síntomas y mejora la evolución de la enfermedad.
2. Qué necesita saber antes de empezar a tomar Opsumit
No tome Opsumit:
• si es alérgico a macitentan o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
• si está embarazada o planea quedarse embarazada, o si pudiera quedarse embarazada porque no utiliza un método anticonceptivo fiable. Lea la información de “Embarazo”.
• si está dando el pecho. Por favor lea la información detallada en "Lactancia".
• si tiene una enfermedad hepática o si tiene los niveles de enzimas hepáticas muy elevados en sangre. Consulte con su médico, quién decidirá si el medicamento es adecuado para usted.
Si cumple alguno de los puntos anteriores, informe al médico.
Advertencias y precauciones
Tenga especial cuidado con Opsumit:
Si tiene anemia (reducción del número de glóbulos rojos).
Necesitará someterse a análisis de sangre, según las indicaciones del médico:
El médico le realizará análisis de sangre antes de que comience el tratamiento con Opsumit y durante el tratamiento para determinar:
• Si tiene anemia (reducción del número de glóbulos rojos)
• Si el hígado funciona correctamente
Los signos indicativos de que el hígado puede no estar funcionando correctamente son:
• Ganas de vomitar (náuseas)
• Vómitos
• Fiebre
• Dolor de estómago (abdomen)
• Coloración amarillenta de la piel o el blanco de los ojos (ictericia)
• Orina de color oscuro
• Picor en la piel
• Cansancio o agotamiento inusuales (letargo o fatiga)
• Síndrome pseudogripal (dolor articular o muscular con fiebre)
Si experimenta cualquiera de estos signos, informe al médico inmediatamente.
Si tiene problemas de riñón, hable con el médico antes de utilizar Opsumit. Macitentan dará lugar a una mayor reducción de la presión arterial y disminución de la hemoglobina en pacientes con problemas de riñón.
Niños y adolescentes
No administrar este medicamento a niños menores de 18 años.
Edad avanzada
La información de Opsumit en pacientes mayores de 75 años es limitada. Opsumit se debe utilizar con precaución en este grupo de edad.
Uso de Opsumit con otros medicamentos
Opsumit puede afectar a otros medicamentos.
Si toma Opsumit junto con otros medicamentos, incluidos los que se indican a continuación, los efectos de Opsumit u otros medicamentos pueden verse afectados. Hable con el médico o farmacéutico si está tomando cualquiera de los medicamentos siguientes:
• Rifampicina, claritromicina, telitromicina (antibióticos utilizados para el tratamiento de infecciones),
• Fenitoína (medicamento utilizado para el tratamiento de las convulsiones),
• Carbamazepina (utilizado para el tratamiento de la depresión y la epilepsia),
• Hierba de San Juan (medicamento a base de plantas utilizado para tratar la depresión),
• Ritonavir, saquinavir (utilizados para tratar la infección por VIH),
• Nefazodona (utilizado para el tratamiento de la depresión),
• Ketoconazol (excepto champú), itraconazol, voriconazol (medicamentos utilizados frente a las infecciones por hongos)
Consulte con su médico o farmacéutico si está tomando, ha tomado recientemente o pudiera tomar otro medicamento.
Embarazo
Si está embarazada o en periodo de lactancia, o cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento.
Opsumit puede dañar a los fetos concebidos antes, durante o poco después del tratamiento.
• Si es posible que pueda quedarse embarazada, utilice un método anticonceptivo fiable mientras está tomando Opsumit. Hable con el médico al respecto.
• No tome Opsumit si está embarazada o tiene previsto quedarse embarazada.
• Si se queda embarazada o cree que puede haberse quedado embarazada durante el tratamiento con Opsumit, acuda al médico inmediatamente.
Si es usted una mujer en edad fértil, el médico le pedirá que se realice una prueba de embarazo antes de empezar a tomar Opsumit y de forma periódica (una vez al mes) durante el tratamiento.
Lactancia
Se desconoce si Opsumit pasa a la leche materna. No dé el pecho durante el tratamiento con Opsumit. Hable con el médico al respecto.
Conducción y uso de máquinas
Opsumit puede provocar efectos adversos como dolores de cabeza (indicados en la sección 4) y los síntomas de la enfermedad también pueden hacer que sea menos apto para conducir.
Información importante sobre algunos ingredientes de Opsumit
Los comprimidos de Opsumit contienen pequeñas cantidades de un azúcar denominado lactosa. Si tiene intolerancia a la lactosa o cualquier otro azúcar, contacte con el médico antes de tomar Opsumit.
Los comprimidos de Opsumit contienen lecitina derivada de la soja. Si es usted alérgico a la soja, no tome este medicamento (ver sección 2 ‘No tome Opsumit’).
3. Cómo tomar Opsumit
Opsumit únicamente debe recetarlo un médico con experiencia en el tratamiento de la hipertensión arterial pulmonar.
Siga exactamente las instrucciones de administración indicadas por su médico. En caso de duda, consulte de nuevo a su médico.
La dosis recomendada de Opsumit es de un comprimido de 10 mg una vez al día. Trague el comprimido entero, con un vaso de agua y no lo mastique ni lo rompa. Opsumit puede tomarse con o sin alimentos. Lo mejor es tomar el comprimido a la misma hora cada día.
Si toma más Opsumit del que debe
Si toma más comprimidos de los indicados, solicite asesoramiento al médico.
Si olvidó tomar Opsumit
Si olvida tomar Opsumit, tome una dosis en cuanto lo recuerde y, a partir de entonces, siga tomando los comprimidos a las horas habituales. No tome una dosis doble para compensar las dosis olvidadas.
Si interrumpe el tratamiento con Opsumit
Opsumit es un tratamiento que deberá seguir tomando para el control de la HAP. No deje de tomar Opsumit a menos que así lo haya acordado con el médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
• Anemia (número reducido de glóbulos rojos) o disminución de la hemoglobina
• Dolor de cabeza
• Bronquitis (inflamación de vías respiratorias)
• Nasofaringitis (inflamación de la garganta y de los conductos nasales)
• Edema (hinchazón), especialmente en tobillos y pies
Efectos adversos frecuentes (pueden afectar a hasta 1 de cada 10 personas):
• Faringitis (inflamación de la garganta)
• Gripe
• Infección urinaria (infección de la vejiga)
• Hipotensión (presión arterial baja)
• Congestión nasal (nariz taponada)
Efectos adversos raros (pueden afectar a hasta 1 de cada 100 personas):
• Reacciones de hipersensibilidad (inflamación alrededor de los ojos, cara, labios, lengua o garganta, picor y/o eritema cutáneo)
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Opsumit
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice Opsumit después de la fecha de caducidad que aparece en el envase después de “CAD”. La fecha de caducidad es el último día del mes que se indica.
No conservar a temperatura superior a 30 °C.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Opsumit
- El principio activo es macitentan. Cada comprimido contiene 10 mg de macitentan.
- Los demás componentes del comprimido son lactosa monohidrato, celulosa microcristalina (E460i), povidona, almidón glicolato de sodio de tipo A, estearato de magnesio (E572),
polisorbato 80 (E433), alcohol polivinílico (E1203), dióxido de titanio (E171), talco (E553b), lecitina de soja (E322) y goma de xantano (E415).
Aspecto del producto y contenido del envase
Los comprimidos de Opsumit 10 mg son comprimidos recubiertos con película, de color blanco a blanquecino, redondos y con el grabado “10” en un lado.
Opsumit se presenta como comprimidos recubiertos de 10 mg en envases tipo blíster de 15 o 30 comprimidos, o en frascos de 30 comprimidos.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Actelion Registration Ltd Chiswick Tower 13th Floor 389 Chiswick High Road Londres W4 4AL Reino Unido Tel: +44 20 8987 3320
medicamento dirigiéndose al representante local del
Responsable de la fabricación
Actelion Manufacturing GmbH Emil-Barrel-Strasse 7 79639 Grenzach-Wyhlen Alemania
Pueden solicitar más información respecto a este titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Actelion Pharmaceuticals Belgium N.V.
Tél/Tel: +32-(0)15 284 777
Etarapna
AKBaXHM Afl Tea.: +359 2 807 50 00
Ceská republika
Actelion Pharmaceuticals CZ, s.r.o.
Tel: +420 221 968 006
Danmark
Actelion Danmark,
Filial af Actelion Pharmaceuticals Sverige AB, Sverige
Tlf: +45 3694 45 95 Deutschland
Actelion Pharmaceuticals Deutschland GmbH Tel: +49 761 45 64 0
Eesti
Algol Pharma OÜ Tel: +372 605 6014
Lietuva
UAB ALGOL PHARMA Tel: +370 37 40 86 81
Luxembourg/Luxemburg
Actelion Pharmaceuticals Belgium N.V. Tél/Tel: +32-(0)15 284 777
Magyarország
Actelion Pharmaceuticals Hungaria Kft. Tel: +36 1 413 3270
Malta
Actelion Pharmaceuticals UK Ltd Tel: +44 208 987 3333
Nederland
Actelion Pharmaceuticals Nederland B.V. Tel: +31 (0)348 435950
Norge
Actelion Pharmaceuticals Sverige AB,
Filial Norge
Tlf: +47 22480370
EXláSa
Actelion Pharmaceuticals E^Mg A.E. Tn^: +30 210 675 25 00
España
Actelion Pharmaceuticals España S.L. Tel.: +34 93 366 4399
France
Actelion Pharmaceuticals France SAS Tél: +33 (0)1 58 62 32 32
Hrvatska
Medis Adria d.o.o.
Tel: +385 (0) 1 2303 446
Ireland
Actelion Pharmaceuticals UK Ltd Tel: +44 208 987 3333
Ísland
Actelion Pharmaceuticals Sverige AB Sími: +46 (0)8 544 982 50
Italia
Actelion Pharmaceuticals Italia S.r.l. Tel: +39 0542 64 87 40
Kúnpoq
Actelion Pharmaceuticals EAAág A.E. Tn^: +30 210 675 25 00
Latvija
Algol Pharma SIA Tel: +371 67 61 9365
Osterreich
Actelion Pharmaceuticals Austria GmbH Tel: +43 1 505 4527
Polska
Actelion Pharma Polska Sp. z o.o.
Tel: +48 (22) 262 31 00
Portugal
Actelion Pharmaceuticals Portugal Lda. Tel: +351 21 358 6120
Romania
Geneva Romfarm International SRL Tel: + 40 (021) 231 3561
Slovenija
Medis d.o.o.
Tel: +386-(0) 1 589 69 00
Slovenská republika
Actelion Pharmaceuticals SK, s.r.o.
Tel: +420 221 968 006
Suomi/Finland
Actelion Pharmaceuticals Sverige AB, Filial Finland
Puh/Tel: +358 9 2510 7720 Sverige
Actelion Pharmaceuticals Sverige AB Tel: +46 8 544 982 50
United Kingdom
Actelion Pharmaceuticals UK Ltd Tel: +44 208 987 3333
Fecha de la última revisión de este prospecto:
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
34
Se ha asociado el edema/retención de líquidos al uso de ARE. En un estudio doble ciego a largo plazo en pacientes con HAP, la incidencia de AA de edema en los grupos de macitentan 10 mg y placebo fue 21,9% y 20,5%, respectivamente. En un estudio doble ciego en pacientes con fibrosis pulmonar idiopática, la incidencia de AA de edema periférico en los grupos de tratamiento con macitentan y placebo fue 11,8% y 6,8% respectivamente. En dos ensayos clínicos doble ciego en pacientes con úlceras digitales asociadas a esclerosis sistémica, las incidencias de AA de edema periférico oscilaron en un rango de 13,4% a 16,1% en los grupos de macitentan 10 mg y de 6,2% a 4,5% en los grupos placebo.
PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Otros antihipertensivos, código ATC: C02KX04.
Mecanismo de acción
La endotelina (ET)-1 y sus receptores (ETA y ETB) median en diferentes efectos como vasoconstricción, fibrosis, proliferación, hipertrofia e inflamación. En condiciones de enfermedad