Octanate Lv 200 Ui/Ml Polvo Y Disolvente Para Solucion Inyectable
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Octanate LV 200 Ul/ml
Polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Octanate LV 200 Ul/ml contiene nominalmente 1000 UI de factor VIII de la coagulación de plasma humano por vial.
El producto contiene aproximadamente 200 UI*/ml de factor VIII de la coagulación humano cuando se reconstituye con 5 ml de disolvente.
Producido a partir de plasma de donantes humanos.
El producto contiene aproximadamente < 120 UI/ml de Factor de von Willebrand (FVW:RCo).
Excipiente con efecto conocido:
Sodio hasta 1,75 mmol (40 mg) por dosis
Concentración de sodio tras la reconstitución: 250 - 350 mmol/l
Para consultar la lista completa de excipientes, ver sección 6.1.
*La potencia (UI) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La media de la actividad específica de Octanate LV es > 100 Ul/mg de proteína.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
El polvo es blanco o amarillo pálido o con apariencia de sólido friable.
El disolvente es un líquido claro e incoloro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de la hemorragia en pacientes con hemofilia A (deficiencia congénita del factor VIII).
Esta preparación no contiene factor de von Willebrand en cantidades farmacológicamente efectivas y, por lo tanto, no está indicada para la enfermedad de von Willebrand.
4.2 Posología y forma de administración
El tratamiento debe ser iniciado bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia.
Posología
La posología y la duración de la terapia de sustitución dependen de la gravedad del déficit de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades administradas del factor VIII se expresa en unidades internacionales (UI) las cuales están relacionadas con el estándar actual de la OMS para medicamentos con factor VIII. La actividad del factor VIII en plasma se expresa como un porcentaje (respecto al plasma humano normal) o en unidades internacionales (respecto a un Estándar Internacional para factor VIII en plasma).
Una unidad internacional (UI) de actividad del factor VIII es equivalente a la cantidad de factor VIII en un ml de plasma humano normal. El cálculo de la dosis requerida del factor VIII se basa en la observación empírica de que 1 UI de factor VIII por kg de peso corporal aumenta la
actividad del factor VIII plasmático en un 1,5% - 2% de la actividad normal. La dosis necesaria se determina utilizando la siguiente fórmula:
Unidades necesarias = peso corporal (kg) * aumento del factor VIII deseado (%) (Ul/dl) * 0,5
La cantidad a administrar y la frecuencia de administración debe estar siempre en concordancia a la efectividad clínica en el paciente individual.
En el caso de los siguientes episodios hemorrágicos la actividad del factor VIII no debe descender por debajo del nivel de actividad plasmática dado (en % de normal) dentro del período correspondiente.
La siguiente tabla se puede usar como una guía de dosificación en episodios hemorrágicos y cirugía:
|
Grado de hemorragia / Tipo de procedimiento quirúrgico |
Nivel necesario de factor VIII (%) (UI/dl) |
Frecuencia de la dosis (horas) / Duración de la terapia (días) |
|
Hemorragia: | ||
|
Hemartrosis incipiente, hemorragia muscular o hemorragia oral. |
20 - 40 |
Repetir cada 12 a 24 horas. Al menos 1 día, hasta que los episodios hemorrágicos, según lo indicado por el dolor, se resuelvan o se alcance la cicatrización. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma. |
30 - 60 |
Perfusión repetida cada 12 a 24 horas de 3 a 4 días o más hasta que el dolor y la discapacidad se hayan resuelto. |
|
Hemorragias con riesgo vital. |
60 - 100 |
Perfusión repetida cada 8 a 24 horas hasta que se supere el peligro. |
|
Cirugía: | ||
|
Menor incluyendo extracción dental |
30 - 60 |
Cada 24 horas, como mínimo 1 día, hasta que se alcance la cicatrización |
|
Mayor |
80 - 100 (pre- y postoperatorio) |
Perfusión repetida cada 8-24 horas hasta una adecuada cicatrización de la herida, seguida de una terapia como mínimo durante 7 días para mantener una actividad del F VIII del 30% al 60%. |
Durante el curso del tratamiento se aconseja una determinación adecuada de los niveles del factor VIII para orientar acerca de la dosis a administrar y la frecuencia de repetición de las perfusiones. En el caso de intervenciones quirúrgicas mayores en particular, es indispensable una monitorización precisa de la terapia de sustitución mediante análisis de la coagulación (actividad del factor VIII del plasma). Los pacientes pueden presentar una respuesta individual variable al factor VIII alcanzando diferentes niveles para la recuperación in-vivo y evidenciando diferentes vidas medias.
Para la profilaxis a largo plazo de hemorragia en pacientes con hemofilia A grave la dosis usual es de 20 a 40 UI de factor VIII por kg de peso corporal a intervalos de 2 a 3 días.
Población pediátrica
Un ensayo clínico realizado con 15 pacientes de 6 años de edad o menos, no identificó ningún requerimiento especial de dosis en niños.
Los datos clínicos sobre el uso de Octanate LV en pacientes no tratados previamente (PntP) son limitados (ver sección 4.8).
Los pacientes deben ser monitorizados para el desarrollo de inhibidores del factor VIII. Si no se alcanzan los niveles plasmáticos esperados de actividad del factor VIII o si no se controla la hemorragia con una dosificación adecuada debe realizarse un ensayo para determinar si hay un inhibidor del factor VIII. En pacientes con niveles elevados de inhibidor la terapia con el factor VIII puede no ser efectiva y deben considerarse otras opciones terapéuticas. Estas terapias deben estar dirigidas por médicos con experiencia en el tratamiento de pacientes con hemofilia. Ver también 4.4.
Forma de administración
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6. El producto debe administrarse por vía intravenosa. Se recomienda no administrar más de 2 - 3 ml por minuto.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
• Como con cualquier otro producto proteico intravenoso, son posibles las reacciones de hipersensibilidad de tipo alérgico. El producto contiene trazas de otras proteínas humanas además del factor VIII. Los pacientes deben ser informados de los signos tempranos de las reacciones de hipersensibilidad como eczema, urticaria generalizada, opresión en el pecho, dificultad para respirar, hipotensión y anafilaxis. Si estos síntomas aparecen debe aconsejarse a los pacientes interrumpir la utilización del producto inmediatamente y contactar con su médico.
En caso de shock, se aplicará el tratamiento estándar del shock.
• Una complicación conocida en el tratamiento de individuos con hemofilia A es la formación de anticuerpos neutralizantes (inhibidores) del factor VIII. Estos inhibidores son inmunoglobulinas IgG contra la actividad procoagulante del factor VIII la cual se cuantifica en función de las unidades Bethesda (UB) por ml de plasma utilizando el método de valoración modificado.
El riesgo de desarrollar inhibidores está relacionado con la exposición al factor VIII, siendo mayor el riesgo durante los primeros 20 días de exposición. Raramente, se desarrollan inhibidores después de los primeros 100 días de exposición. Los pacientes tratados con el factor VIII de la coagulación humano deben ser monitorizados cuidadosamente para el desarrollo de anticuerpos inhibidores mediante observación clínica adecuada y pruebas de laboratorio. Ver también el apartado 4.8. Reacciones adversas.
• Se han encontrado estudios en la literatura que muestran la relación entre la aparición de un inhibidor del factor VIII y reacciones alérgicas. Por lo tanto, si aparecen reacciones alérgicas, deberá investigarse la presencia de un inhibidor en el paciente. Los pacientes con inhibidores del factor VIII pueden tener un mayor riesgo de anafilaxis al seguir el tratamiento con el factor VIII. En consecuencia, la primera administración del factor VIII, de acuerdo con el criterio del médico responsable, debe ser realizada bajo supervisión médica donde pueda proporcionarse una atención médica adecuada para la aparición de reacciones alérgicas.
• Las medidas estándares para prevenir infecciones que son consecuencia del uso de medicamentos derivados de sangre o plasma humano incluyen la selección de los donantes, cribaje de las donaciones individuales y mezclas de plasma para marcadores específicos de infección, así como la inclusión de procedimientos efectivos para la inactivación/eliminación viral en el proceso de producción. A pesar de ello, cuando se
administran medicamentos derivados de sangre o plasma humano, no se puede descartar por completo la posibilidad de transmisión de enfermedades infecciosas. Esto también es aplicable a virus desconocidos o emergentes y otros patógenos.
• Estos procedimientos se consideran efectivos para virus envueltos como VIH, VHB y VHC, así como para el virus sin cubierta VHA. Estos procedimientos pueden tener un valor limitado frente a los virus sin cubierta tales como parvovirus B19. La infección por parvovirus B19 puede resultar seria en mujeres embarazadas (infección fetal) y en pacientes con inmunodeficiencia o con una producción de hematíes incrementada (p.ej. en anemia hemolítica).
• Se deberá considerar una vacunación adecuada (hepatitis A y B) para los pacientes que estén recibiendo de forma regular/repetida concentrados de factor VIII derivado de plasma.
• Es altamente recomendable que cada vez que se administre Octanate LV a un paciente se registre el nombre y el número de lote del producto con objeto de mantener una trazabilidad entre el paciente y el lote del producto.
• Este medicamento contiene hasta 1,75 mmol de sodio (40 mg) por dosis. Esto debe ser tomado en consideración por los pacientes con una dieta controlada en sodio.
4.5 Interacción con otros medicamentos y otras formas de interacción No se han realizado estudios de interacción.
No se conocen interacciones de productos de factor VIII de coagulación humano con otros medicamentos.
4.6 Fertilidad, embarazo y lactancia
No hay datos o estos son limitados relativos al uso de factor VIII en mujeres embarazadas.
No se han realizado estudios de reproducción en animales con el factor VIII. Debido a que son raros los casos de mujeres con hemofilia A, no se dispone de experiencia sobre la utilización del factor VIII durante el embarazo y la lactancia. Por lo tanto, el factor VIII debe ser utilizado durante el embarazo y la lactancia solo si está claramente indicado.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Octanate LV sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
• Se han observado de manera poco frecuente reacciones de hipersensibilidad o alérgicas (las cuales pueden incluir angioedema, irritación y escozor en el lugar de inyección, escalofríos, sofocos, urticaria generalizada, cefalea, eczemas, hipotensión, letargia, náuseas, ansiedad, taquicardia, opresión en el pecho, hormigueo, vómitos, disnea), y, en algunos casos, estas reacciones pueden progresar a anafilaxia grave (incluyendo shock).
• En casos raros se ha observado fiebre.
• Los pacientes con hemofilia A pueden desarrollar anticuerpos (inhibidores)
contra el factor VIII. Si aparecen estos inhibidores la condición se manifestará como una respuesta clínica insuficiente. En tales casos, se recomienda contactar con un centro especializado en hemofilia.
En un ensayo clínico en curso, en pacientes no tratados previamente (PntP), 3 de 39 (7,6%) PntP tratados con Octanate LV a demanda desarrollaron inhibidores con un título mayor de 5 UB. Un paciente desarrolló inhibidores con un título inferior a 5 UB.
35 PntP tuvieron una actividad basal de FVIII < 1% y 4 PntP tuvieron < 2% FVIII:C. En el momento del análisis provisional, había 34 pacientes que ya contaban con 20 o más días de exposición a Octanate LV y 30 pacientes con 50 o más días de exposición. No se observaron inhibidores en PntP en profilaxis con Octanate LV. Durante el estudio, 12 PntP fueron sometidos a 14 procedimientos quirúrgicos. La edad media en la primera exposición era de 7 meses (intervalo 3 días a 67 meses). El número medio de días de exposición en el ensayo clínico fue de 100 (intervalo 1-553).
|
Clasificación por óraanos v sistemas |
Raras |
Muv raras |
|
Trastornos del sistema inmune |
Reacciones de hipersensibilidad |
Shock anafiláctico |
|
Trastornos generales y alteraciones en el lugar de la administración |
Fiebre | |
|
Exploraciones complementarias |
Anticuerpos de factor VIII en sangre |
Raras (>1/10.000 a < 1/1.000)
Muy raras (<1/10.000).
Para información sobre seguridad viral ver 4.4.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaRAM.es
4.9 Sobredosis
No se ha comunicado ningún caso de sobredosis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos: factor VIII de la coagulación sanguínea.
Código ATC: B02BD02
El complejo factor VIII/ von Willebrand está formado por dos moléculas (FVIII y FvW) con distintas funciones fisiológicas. Cuando se administra a un paciente hemofílico el factor VIII se une al factor de von Willebrand en la circulación del paciente.
El factor VIII activado actúa como cofactor para el factor IX activado, acelerando la conversión del factor X a factor X activado. El factor X activado convierte la protrombina en trombina. A continuación, la trombina convierte el fibrinógeno en fibrina, produciendo la formación del coágulo.
La hemofilia A es una enfermedad hereditaria de la coagulación sanguínea ligada al sexo, debida a niveles disminuidos de factor VIII:C y da lugar de forma espontánea o como consecuencia de un trauma accidental o quirúrgico a hemorragias abundantes en articulaciones, músculos u órganos internos.
Con la terapia de sustitución se aumentan los niveles plasmáticos del factor VIII, de manera que temporalmente, se rectifica la deficiencia del factor y se corrige la tendencia hemorrágica. Octanate LV se está evaluando para la inducción de la inmunotolerancia (ITI) en un estudio clínico observacional en curso.
En un análisis provisional de los 69 pacientes tratados hasta ahora con Octanate LV en ITI, 49 pacientes han completado el estudio. En los pacientes en los que se erradicó el inhibidor con éxito, las tasas de hemorragia mensuales se redujeron significativamente.
5.2 Propiedades farmacocinéticas
El factor VIII de coagulación de plasma humano (del polvo) es un componente normal del plasma humano y actúa como el factor VIII endógeno. Después de la inyección del producto, aproximadamente entre dos tercios y tres cuartos del factor VIII permanecen en la circulación. El nivel de actividad del factor VIII alcanzado en plasma debe situarse entre el 80% y el 120% de la actividad esperada del mismo.
La actividad del factor VIII plasmático disminuye de modo exponencial en dos fases. En la fase inicial, la distribución ente los compartimentos intravascular y otros (fluidos corporales) tiene lugar con una vida media de eliminación del plasma de 3 a 6 horas. En la siguiente fase, más lenta, (la cual, probablemente, refleja el consumo del factor VIII) la vida media varía entre 8 y 20 horas con una media de 12 horas. Esta corresponde con la vida media biológica real.
Para Octanate LV se obtuvieron los siguientes resultados en dos estudios farmacocinéticos con 10 y 14 pacientes con hemofilia A, respectivamente:
|
Recuperación (% x UI-1 x kg) |
AUC*norm (% x h x UI-1 x kg) |
Vida media (h) |
MRT* (h) |
Aclaramiento 1 (ml x h- x kg) | |
|
Estudio 1, n= 10 Media ± DE* |
2,4 ± 0,36 |
45,5 ± 17,2 |
14,3 ± 4,01 |
19,6 ± 6,05 |
2,6 ± 1,21 |
|
Estudio 2, n= 14 Media ± DE* |
2,4 ± 0,25 |
33,4 ± 8,50 |
12,6 ± 3,03 |
16,6 ±3,73 |
3,2 ± 0,88 |
*AUC = área bajo la curva
*MRT = tiempo medio de residencia
*DE = desviación estándar
5.3 Datos preclínicos sobre seguridad
Los datos toxicológicos disponibles respecto al tri-n-butilfosfato (TNBP) y al polisorbato 80 (tween 80), los reactivos solvente/detergente utilizados en el método SD de inactivación viral durante la fabricación de Octanate LV, aunque son limitados para este último, indican que las reacciones adversas son poco probables en las exposiciones humanas previstas.
Incluso dosificaciones que multiplican la dosis humana recomendada por kilogramo de peso corporal de estos reactivos, no muestran efectos tóxicos en animales de laboratorio. No se observó potencial mutagénico para ninguna de las dos sustancias.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes Polvo:
. Citrato de sodio,
. Cloruro de sodio,
. Cloruro de calcio
. Glicina
Disolvente: agua para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe ser mezclado con otros medicamentos.
Solo debe utilizarse el equipo de inyección/perfusión suministrado, ya que el tratamiento puede fracasar debido a la adsorción del factor VIII de la coagulación humano en la superficie interna de ciertos equipos de inyección/perfusión.
6.3 Periodo de validez 2 años.
La solución reconstituida debe utilizarse inmediatamente y en una sola ocasión.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre +2 y +8°C).
No congelar.
Conservar los viales en el embalaje exterior para mantenerlos protegidos de la luz.
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3
6.5 Naturaleza y contenido del envase Un envase de Octanate LV contiene:
• Polvo en un vial (vidrio tipo I), con un tapón (goma de clorobutilo o bromobutilo) y una cápsula flip-off.
• 5 ml de disolvente en un vial (vidrio tipo I), con un tapón (goma de clorobutilo o bromobutilo), y una cápsula flip-off.
• Una jeringa desechable, un equipo de transferencia Mix2Vial®, un kit de inyección, dos apósitos con alcohol.
Un vial de Octanate LV contiene 500 UI de factor VIII de la coagulación humano.
6.6 Precauciones especiales de eliminación y otras manipulaciones
• Por favor lea todas las instrucciones y sígalas cuidadosamente.
• No utilice Octanate LV después de la fecha de caducidad que aparece en el envase.
• Durante el procedimiento descrito a continuación, debe mantenerse la esterilidad.
• Inspeccionar visualmente el medicamento reconstituido para comprobar si existen partículas o cambio de coloración antes de la administración. No inyectar soluciones turbias o que contengan sedimentos.
• Utilizar inmediatamente la solución preparada, para evitar una contaminación microbiana.
.w.
i**4
'ni®:
• Utilizar únicamente el equipo que se incluye. El uso de otro equipo de inyección/perfusión puede ocasionar un riesgo adicional y el fracaso del tratamiento.
instrucciones para preparar la solución:
1. No utilizar el producto directamente de la nevera. Dejar el disolvente y el polvo en los viales cerrados hasta alcanzar la temperatura ambiente.
2. Quitar las cápsulas flip-off de ambos viales y limpiar los tapones de goma con uno de los algodones impregnados en alcohol que se incluyen.
3. Colocar el vial de disolvente sobre una superficie plana y sujetarlo con firmeza. Coger el Mix2Vial® (Fig. 1) y darle la vuelta. Colocar la parte azul del Mix2Vial® sobre la parte superior del vial del disolvente y presionar con firmeza hasta que se oiga un chasquido (Fig. 2+3).
Fig. 1
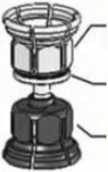
Adaptador del vial de polvo (transparente)
Filtro integrado
Adaptador del vial de disolvente (azul)
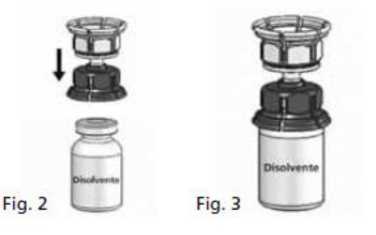

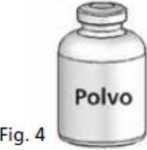
4. Colocar el vial del polvo sobre una superficie plana y sujetarlo con firmeza. Coger el vial de disolvente con el Mix2Vial® acoplado y darle la vuelta. Colocar la parte transparente sobre la parte superior del vial del polvo y presionar con firmeza hasta que se oiga un chasquido (Fig. 4). El disolvente fluye automáticamente al interior del vial del polvo.

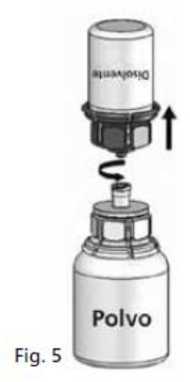
5. Con ambos viales todavía acoplados, agitar suavemente el vial del polvo hasta que el producto se haya disuelto.
La disolución es completa en menos de 10 minutos a temperatura ambiente. Podría aparecer una ligera espuma durante la preparación. Desenroscar las dos partes del Mix2Vial® (Fig. 5). Desaparecerá la espuma.
Desechar el vial de disolvente vacío con la parte azul del Mix2Vial®.
Instrucciones para la Inyección:
Como precaución debe medirse la velocidad del pulso antes y durante la inyección. Si se produce un marcado incremento en la velocidad del pulso, reducir la velocidad de la inyección o interrumpir la administración durante un breve periodo de tiempo.
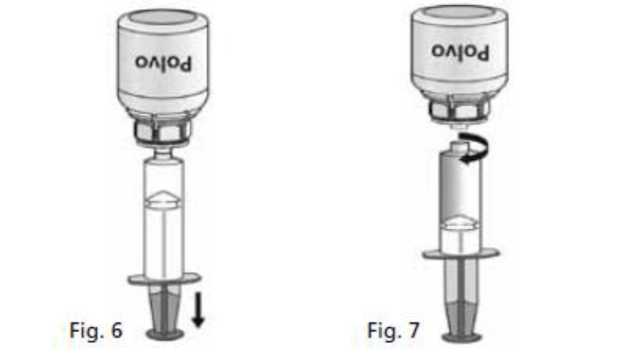
1. Acoplar la jeringa a la parte transparente del Mix2Vial®. Darle la vuelta al vial y extraer la solución al interior de la jeringa (Fig. 6). La solución en la jeringa debe ser límpida o ligeramente nacarada. Una vez que la solución ha sido transferida, sujetar con firmeza el émbolo de la jeringa (manteniéndolo hacia abajo) y extraer la jeringa del Mix2Vial®. (Fig. 7). Desechar el Mix2Vial® y el vial vacío.
2. Limpiar la zona donde se va a poner la inyección con uno de los apósitos con alcohol que se incluyen.
3. Acoplar el kit de inyección que se incluye a la jeringa.
4. Insertar la aguja de inyección en la vena escogida. Si ha empleado un torniquete para ver la vena con más facilidad, este torniquete debe ser liberado antes de empezar a inyectar Octanate LV.
No debe fluir sangre al interior de la jeringa debido al riesgo de formación de coágulos de fibrina.
5. Inyectar la solución dentro de la vena a una velocidad lenta, no superior a 2-3 ml por minuto.
Si usa más de un vial de polvo de Octanate LV para un tratamiento, puede utilizar el mismo kit de inyección y la misma jeringa. El Mix2Vial® es para un solo uso.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN Octapharma S.A.
Avda. Castilla, 2. (P.E. San Fernando)
Ed. Berlín, bajo
28830 San Fernando de Henares Madrid
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Abril 2015
10. FECHA DE LA REVISIÓN DEL TEXTO Noviembre 2014
10 de 10