Nuwiq 1000 Ui Polvo Y Disolvente Para Solucion Inyectable
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Nuwiq 250 UI polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 250 UI de Factor VIII de coagulación humano (rDNA), simoctocog alfa.
Nuwiq contiene aproximadamente 100 UI/ml de Factor VIII de coagulación humano (rDNA), simoctocog alfa tras la reconstitución.
La potencia (UI) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de Nuwiq es de aproximadamente 9500 UI/mg de proteína.
El simoctocog alfa (factor VIII de coagulación humano (rDNA)) es una proteína purificada que contiene 1440 aminoácidos. La secuencia de aminoácidos es comparable a la forma de 90 + 80 kDa del factor VIII humano plasmático (esto es, con el dominio B suprimido). Nuwiq se ha producido a partir de tecnología de ADN recombinante de células embrionarias de riñón humano (HEK) modificadas genéticamente 293F. No se ha añadido ningún material derivado de seres humanos o animales durante el proceso de fabricación ni al medicamento final.
Excipiente(s) con efecto conocido:
7,35 mg de sodio por ml de solución reconstituida (18,4 mg de sodio por vial).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: polvo friable de color entre blanco y blanquecino.
Disolvente: agua para preparaciones inyectables, líquido transparente e incoloro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de hemorragias en pacientes con hemofilia A (deficiencia congénita del factor VIII).
Nuwiq se puede usar en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico experimentado en el tratamiento de la hemofilia.
Pacientes no tratados anteriormente
La seguridad y eficacia de Nuwiq en pacientes no tratados previamente no se ha establecido todavía. Posología
La dosis y duración del tratamiento de sustitución dependen de la gravedad de la deficiencia del factor VIII, así como de la localización y alcance de las hemorragias y del estado clínico del paciente.
El número de unidades administradas del factor VIII se expresa en Unidades Internacionales (UI) las cuales están relacionadas con el estándar actual de la OMS para medicamentos con factor VIII. La actividad del factor VIII en plasma se expresa como un porcentaje (respecto al plasma humano normal) o en Unidades Internacionales (respecto a un Estándar Internacional para factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII equivale a la cantidad de factor VIII en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis requerida de factor VIII se basa en el dato empírico de que 1 Unidad Internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad del factor VIII del plasma en aproximadamente un 2% de la actividad normal o 2 UI/dl. La dosis necesaria se determina utilizando la siguiente fórmula:
I. Unidades requeridas (UI) = peso corporal (kg) x aumento deseado de factor VIII (%)(UI/dl) x 0,5 (UI/kg por UI/dl)
II. Aumento previsto del factor VIII (% de lo normal) = 2 x UI administradas
peso corporal (kg)
La dosis a administrar y la frecuencia de administración debe estar siempre orientada a la eficacia clínica en cada caso individual.
En el caso de los episodios hemorrágicos siguientes, la actividad del factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % de lo normal o UI/dl) en el periodo correspondiente. La siguiente tabla se puede utilizar como guía de dosificación en cirugía y en episodios hemorrágicos.
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (%) (UI/dL) |
Frecuencia de las dosis (horas) / duración del tratamiento (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente, hemorragia muscular u oral |
20-40 |
Repetir cada 12 a 24 horas. Al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma |
30-60 |
Repetir la perfusión cada 12 a 24 horas, entre 3 y 4 días o más, hasta que cese el dolor y la discapacidad aguda. |
|
Hemorragia potencialmente mortal |
60-100 |
Repetir la perfusión cada 8 a 24 horas hasta que se supere el peligro |
|
Cirugía | ||
|
Cirugía menor Incluyendo extracción dental |
30-60 |
Cada 24 horas, al menos 1 día, hasta lograr la curación. |
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (%) (UI/dL) |
Frecuencia de las dosis (horas) / duración del tratamiento (días) |
|
Cirugía mayor |
80-100 (pre y postoperatorio) |
Repetir la perfusión cada 8-24 horas hasta que se consiga una cicatrización adecuada de la herida, y después al menos durante otros 7 días de tratamiento para mantener una actividad de factor VIII del 30% al 60% (UI/dL). |
Profilaxis
En la profilaxis a largo plazo de episodios hemorrágicos en pacientes con hemofilia A grave, las dosis habituales son de 20 a 40 UI de factor VIII por kg de peso corporal, en intervalos de 2 a 3 días. En algunos casos, especialmente en los pacientes más jóvenes, puede ser necesario un intervalo de dosis más corto o dosis mayores.
Durante el transcurso del tratamiento, se aconseja la determinación adecuada de los niveles de factor VIII como guía de la dosis a administrar y la frecuencia de repetición de las inyecciones. En el caso de intervenciones, cirugía mayor en particular, es indispensable la monitorización precisa del tratamiento de sustitución mediante el análisis de la coagulación (actividad del factor VIII del plasma). La respuesta al factor VIII puede variar en cada paciente, revelándose unos valores diferentes de semivida y recuperación.
Población pediátrica
La posología es la misma en adultos y niños. Sin embargo, hay que tener en cuenta que para los niños pueden ser necesarios intervalos de dosis más cortos o dosis mayores. Los datos actualmente disponibles se describen en las secciones 4.8, 5.1 y 5.2.
No se dispone de datos en niños menores de 2 años de edad.
Forma de administración
Vía intravenosa.
Se recomienda no administrar más de 4 ml por minuto.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo Hipersensibilidad
Como con cualquier producto proteínico vía intravenosa, es posible que se den reacciones de hipersensibilidad de tipo alérgico. Nuwiq contiene trazas de proteínas de células huésped humanas distintas al factor VIII. Si aparecen síntomas de hipersensibilidad, se debe aconsejar a los pacientes que interrumpan inmediatamente el uso del medicamento y contacten con su médico. Los pacientes deben ser informados acerca de los signos iniciales de las reacciones de hipersensibilidad, que incluyen sarpullido, urticaria generalizada, opresión en el pecho, sibilancias, hipotensión y anafilaxia.
En caso de shock, se debe aplicar el tratamiento médico habitual para el shock. Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) del factor VIII es una complicación conocida en el tratamiento de individuos con hemofilia A. Estos inhibidores son normalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en unidades Bethesda (UB) por ml de plasma usando el ensayo modificado. El riesgo de desarrollar inhibidores se relaciona con la exposición al factor VIII, siendo este riesgo más alto en los 20 primeros días de exposición. Rara vez se pueden desarrollar inhibidores transcurridos los primeros 100 días de exposición.
Se han observado casos de inhibidor recurrente (de bajo título) tras cambiar de un producto de factor VIII a otro en pacientes previamente tratados con un periodo de exposición de más de 100 días y con antecedentes de desarrollo de inhibidores. Por ello, se recomienda realizar un seguimiento meticuloso del desarrollo de inhibidores en todos los pacientes tras cualquier cambio de producto.
En general, todos los pacientes tratados con productos del factor de coagulación VIII deben ser vigilados cuidadosamente, de cara al desarrollo de inhibidores, mediante las observaciones clínicas y pruebas de laboratorio adecuadas. Si no se alcanzan los niveles plasmáticos de actividad del factor VIII esperados o si no se controla la hemorragia mediante una dosis adecuada, se debe realizar una prueba de presencia de inhibidor del factor VIII. En pacientes con altos niveles de inhibidor, es posible que el tratamiento con factor VIII no resulte eficaz y se deban considerar otras opciones terapéuticas, como la inducción de inmunotolerancia (ITI). El tratamiento de esos pacientes debe ser dirigido por médicos con experiencia en el tratamiento de la hemofilia y de los inhibidores del factor VIII.
Complicaciones asociadas a los catéteres
Si se requiere un dispositivo de acceso venoso central (CVAD (por sus siglas en inglés)), hay que tener en cuenta el riesgo de complicaciones asociadas al CVAD, incluidas las infecciones localizadas, la bacteremia y la trombosis en el lugar de implantación del catéter.
Se recomienda encarecidamente que cada vez que se administre Nuwiq a un paciente, se registre el nombre y número de lote del producto para mantener un vínculo entre el paciente y el lote del medicamento.
Población pediátrica
Las advertencias y precauciones enumeradas se refieren tanto a adultos como a niños.
Consideraciones relativas al excipiente (contenido de sodio)
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por vial.
Sin embargo, dependiendo del peso corporal y la posología, podría administrársele al paciente más de un vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones con Nuwiq.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción animal con Nuwiq.
Debido a la escasa frecuencia de la hemofilia A en mujeres, no se tiene experiencia sobre el uso del factor VIII durante el embarazo o periodo de lactancia. Por lo tanto, Nuwiq solo se debe usar durante el embarazo y la lactancia si está claramente indicado. No hay datos disponibles de fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Nuwiq no influye sobre la capacidad para conducir y utilizar máquinas.
4.8 Reacciones adversas Resumen del perfil de seguridad
Se han observado con poca frecuencia reacciones alérgicas o de hipersensibilidad con preparados de FVIII (que pueden incluir angioedema, quemazón y punzadas en el lugar de la inyección, escalofríos, rubefacción, urticaria generalizada, cefalea, sarpullido, hipotensión, letargia, náuseas, inquietud, taquicardia, opresión en el pecho, cosquilleo, vómitos, sibilancias) y en algunos casos pueden empeorar hasta convertirse en anafilaxia grave, (incluido el shock anafiláctico).
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) del factor VIII. Si aparecen dichos inhibidores, puede manifestarse en forma de una respuesta clínica insuficiente. En estos casos, se recomienda ponerse en contacto con un centro especializado en hemofilia.
Tabla de reacciones adversas
Durante los estudios clínicos con Nuwiq en pacientes pediátricos previamente tratados (2 a 11 años, n = 58), adolescentes (12 a 17 años, n = 3) y adultos (n = 74) con hemofilia A grave, se notificaron un total de 8 reacciones adversas a medicamentos (RAM) (6 en adultos, 2 en niños) en 5 pacientes (3 adultos, 2 niños).
La Tabla 1 que se presenta a continuación sigue la Clasificación de Órganos del y Sistemas MedDRA (COS y nivel de términos preferentes).
Las frecuencias se han evaluado conforme a la convención siguiente: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
En cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 1. Frecuencia de las reacciones adversas a medicamentos (RAM) en ensayos clínicos en 135 pacientes con hemofilia A grave previamente tratados__
|
Clasificación estándar de órganos del sistema MedDRA |
Reacciones adversas |
Frecuencia* |
|
Trastornos del sistema nervioso |
Parestesia Dolor de cabeza |
Poco frecuentes |
|
Trastornos del oído y del laberinto |
Vértigo |
Poco frecuentes |
|
Trastornos gastrointestinales |
Sequedad de boca |
Poco frecuentes |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor de espalda |
Poco frecuentes |
|
Trastornos generales y alteraciones en el lugar de administración |
Inflamación en la zona de inyección Dolor en la zona de inyección |
Poco frecuentes |
|
Exploraciones complementarias |
Positivo por anticuerpos no neutralizantes anti-factor VIII |
Poco frecuentes |
* Todas estas RAM solo se produjeron una vez. Como el número total de pacientes analizados es de 135, la frecuencia no puede ser menor que "poco frecuente" si se produce una RAM.
Descripción de las reacciones adversas seleccionadas
Se detectó un anticuerpo no neutralizante anti-Factor VIII en un paciente adulto (ver Tabla 1). Se analizó la muestra en el laboratorio central en ocho diluciones. El resultado fue positivo solo en la dilución factor 1 y el título de anticuerpos fue muy bajo. No se detectó en este paciente ninguna actividad inhibidora, según las mediciones del ensayo de Bethesda modificado. La eficacia clínica y la recuperación in vivo de Nuwiq no se vieron afectadas en este paciente.
Población pediátrica
Se asume que la frecuencia, tipo y gravedad de las reacciones adversas son las mismas en niños que en adultos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se ha notificado ningún caso de sobredosis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos, factor VIII de la coagulación sanguínea, código ATC: B02BD02.
El complejo factor VIII/factor von Willebrand consiste en dos moléculas (factor VIII y factor de von Willebrand) con distintas funciones fisiológicas. Cuando se inyecta en un paciente hemofílico, el factor VIII se une con el factor von Willebrand circulante del paciente. El factor VIII activado actúa como cofactor del factor IX activado, acelerando la conversión del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Seguidamente, la trombina convierte el fibrinógeno en fibrina y se puede formar un coágulo. La hemofilia A es una alteración hereditaria de la coagulación de la sangre vinculada al sexo, debida a niveles reducidos de factor VIII:C y da como resultado hemorragias profusas en articulaciones, músculos u órganos internos, ya sea de forma espontánea, o tras un accidente o un traumatismo quirúrgico. El tratamiento de sustitución aumenta los niveles plasmáticos de factor VIII, lo cual permite corregir temporalmente la deficiencia del factor VIII y la tendencia al sangrado.
Se ha evaluado la inmunogenicidad de Nuwiq en ensayos clínicos en 135 pacientes con hemofilia A grave previamente tratados (74 pacientes adultos y 61 pacientes pediátricos). Ninguno de los pacientes desarrollaron inhibidores.
En un estudio clínico en 32 pacientes adultos con hemofilia A grave, la mediana de consumo de Nuwiq para profilaxis fue de 468,7 UI/kg/mes. La mediana de la dosis para tratar episodios hemorrágicos intercurrentes fue de 33,0 UI/kg en aquellos pacientes en profilaxis. En otro estudio clínico, se trató a demanda a 22 pacientes adultos. En total, se trataron 986 episodios hemorrágicos con una mediana de dosis de 30,9 UI/kg. En general, los sangrados menores requirieron una dosis ligeramente menor, mientras que los sangrados más graves requirieron hasta tres veces la mediana de dosis.
Población pediátrica
Se han obtenido los datos de 29 niños previamente tratados con edades entre 2 y 5 años, 31 niños entre 6 y 12 años y un adolescente de 14 años. La mediana de la dosis por inyección profiláctica fue de 37,8 UI/kg. Veinte pacientes utilizaron una mediana de dosis mayor de 45 UI/kg. La mediana del consumo de Nuwiq para profilaxis por mes fue de 521,9 UI/kg. Se requirió una mediana de dosis de Nuwiq más alta para tratar las hemorragias en niños (43,9 Ul/kg) que en adultos (33,0 Ul/kg), y una mediana de dosis más alta para tratar las hemorragias de moderadas a importantes que para las menores (78,2 UI/kg frente a 41,7 UI/kg). Los niños de menor edad requirieron en general medianas de dosis más altas (de 6 a 12 años: 43,9 UI/kg; de 2 a 5 años: 52,6 UI/kg).
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Nuwiq en uno o más grupos de la población pediátrica en tratamiento de la Hemofilia A (deficiencia congénita del Factor VIII) (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas (PK)
Tabla 2. Parámetros PK para Nuwiq (dosis: 50 UI/kg) en pacientes adultos previamente tratados (de 18 a 65 años de edad) con hemofilia A grave (n = 20)_
|
Parámetro PK |
Ensayo cromogénico | |
|
Valor medio ± SD |
Mediana (rango) | |
|
AUC (h*UI/ml) |
22,6 ± 8,0 |
22,3 (8,4 - 38,1) |
|
Tm (h) |
14,7 ± 10,4 |
12,5 (5,4 -55,6) |
|
IVR (%/UI/kg) |
2,5 ± 0,4 |
2,5 (1,7 -3,2) |
|
Cl (ml/h/kg) |
3,0 ± 1,2 |
2,7 (1,5-6,4) |
AUC = Área bajo la curva (FVIII:C), Tm = Semivida terminal,
IVR = Recuperación incremental in vivo , Cl = Aclaramiento, SD = Desviación estándar
Tabla 3. Parámetros PK para Nuwiq (dosis: 50 UI/kg) en niños previamente tratados de entre 6 y 12 años de edad con hemofilia A grave (n = 12)_
|
Parámetro PK |
Ensayo cromogénico | |
|
Valor medio ± SD |
Mediana (rango) | |
|
AUC (h*UI/ml) |
13,2 ± 3,4 |
12,8 (7,8 - 19,1) |
|
Tm (h) |
10,0 ± 1,9 |
9,9 (7,6 -14,1) |
|
IVR (%/UI/kg) |
1,9 ± 0,4 |
1,9 (1,2 -2,6) |
|
Cl (ml/h/kg) |
4,3 ± 1,2 |
4,2 (2,8 - 6,9) |
AUC = Área bajo la curva (FVIII:C), Tm = Semivida terminal,
IVR = Recuperación incremental in vivo , Cl = Aclaramiento, SD = Desviación estándar
Tabla 4. Parámetros PK para Nuwiq (dosis: 50 UI/kg) en niños previamente tratados de entre 2 y 5 años de edad con hemofilia A grave (n = 13)_
|
Parámetro PK |
Ensayo cromogénico | |
|
Valor medio ± SD |
Mediana (rango) | |
|
AUC (h*UI/ml) |
11,7 ± 5,3 |
10,5 (4,9 -23,8) |
|
Tm (h) |
9,5 ± 3,3 |
8,2 (4,3 - 17,3) |
|
IVR (%/UI/kg) |
1,9 ± 0,3 |
1,8 (1,5 -2,4) |
|
Cl (ml/h/kg) |
5,4 ± 2,4 |
5,1 ( 2,3 -10,9) |
AUC = Área bajo la curva (FVIII:C), Tm = Semivida terminal,
IVR = Recuperación incremental in vivo , Cl = Aclaramiento, SD = Desviación estándar Población pediátrica
Según lo extraído de publicaciones médicas, la recuperación y semivida fueron menores en los niños más pequeños que en los adultos y el aclaramiento más alto, lo que se puede deber en parte al mayor volumen de plasma conocido por kilogramo de peso corporal en los pacientes más jóvenes.
Subgrupos de peso ajustado
Tabla 5. Parámetros PK de peso ajustado para Nuwiq (dosis: 50 Ul/kg) en pacientes adultos
|
previamente tratados (de 18 a 65 años ( |
e edad) con hemofilia A grave (n = 20) | |||
|
Parámetro PK |
Todos (n=20) |
Peso normal (n=14) |
Pre-adiposo (n=4) |
Adiposo (n=2) |
|
Ensayo cromogénico valor medio ± SD | ||||
|
AUC (h*UI/ml) |
22,6 ± 8,0 |
20,4 ± 6,9 |
24,9 ± 8,9 |
33,5 ± 6,5 |
|
T1/2 (h) |
14,7 ± 10,4 |
14,7 ± 12,1 |
13,4 ± 5,9 |
17,2 ± 4,8 |
|
IVR (%/UI/kg) |
2,5 ± 0,4 |
2,4 ± 0,4 |
2,7 ± 0,4 |
2,8 ± 0,3 |
|
Cl (ml/h/kg) |
3,0 ± 1,2 |
3,2 ± 1,3 |
2,6 ± 1,0 |
1,8 ± 0,4 |
|
Mediana ensayo cromogénico (rango) | ||||
|
AUC (h*UI/ml) |
22,3 (8,4 -38,1) |
21,2 (8,4 -32,6) |
23,3 (17,4 - 35,5) |
33,5 (28,9 -38,1) |
|
T1/2 (h) |
12,5 (5,4 -55,6) |
12,3 (5,4 -55,6) |
11,2 (9,3 -22,0) |
17,2 (13,8 -20,6) |
|
IVR (%/UI/kg) |
2,5 (1,7 -3,2) |
2,4 (1,7 -3,1) |
2,8 (2,3 - 3,2) |
2,8 (2,6 -3,0) |
|
Cl (ml/h/kg) |
2,7 (1,5 -6,4) |
2,8 (1,7 -6,4) |
2,5 (1,6 -3,7) |
1,8 (1,5 -2,0) |
Peso normal: IMC 18,5-25 kg/m2, pre-adiposo: IMC 25-30 kg/m2, adiposo: IMC > 30 kg/m2,
SD = Desviación estándar
5.3 Datos preclínicos sobre seguridad
En los estudios preclínicos, Nuwiq se utilizó para restablecer la hemostasia de manera segura y eficaz en perros con hemofilia. Los estudios toxicológicos demostraron que los animales de laboratorio (ratas y monos cynomolgus) toleran bien la administración vía intravenosa local y la exposición sistémica.
No se realizaron estudios específicos con Nuwiq con administración repetida a largo plazo como los de toxicidad para la reproducción, toxicidad crónica y carcinogenia debido a la respuesta inmune a proteínas heterólogas en todas las especies mamíferas no humanas.
No se realizaron estudios sobre el potencial mutagénico de Nuwiq.
Las evaluaciones ex vivo utilizando un kit comercial de ensayo para cuantificar la respuesta de la célula T a las proteínas terapéuticas, ponen de manifiesto un bajo riesgo de inmunogenicidad.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo:
Sacarosa Cloruro de sodio Cloruro de calcio dihidratado Clorhidrato de arginina Citrato de sodio dihidratado Poloxamer 188
Disolvente:
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
Solo deben utilizarse los kits de inyección suministrados, ya que el tratamiento puede fallar a consecuencia de la adsorción del factor VIII humano de coagulación a las superficies internas de otros productos de inyección.
6.3 Periodo de validez
2 años
Durante el periodo de validez, el medicamento se puede almacenar a temperatura ambiente (hasta 25°C) durante un único periodo no superior a 1 mes. Una vez que el medicamento se saca de la nevera, no se debe volver a refrigerar. Registrar la fecha de inicio del almacenamiento a temperatura ambiente en el embalaje del producto. Conservar el vial en el embalaje exterior para protegerlo de la luz.
Tras la reconstitución, se ha comprobado la estabilidad química y física del producto conservado a temperatura ambiente durante 24 horas.
Desde un punto de vista microbiológico, el producto se debe utilizar inmediatamente después de la reconstitución. Si no se usa de forma inmediata, los tiempos y condiciones de conservación previas a su uso son responsabilidad del usuario.
Mantener la solución reconstituida a temperatura ambiente. No refrigerar una vez reconstituida.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior de cartón para protegerlo de la luz.
Para consultar la información sobre almacenamiento a temperatura ambiente y condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Cada envase de Nuwiq 250 UI contiene:
- Polvo: 250 UI de polvo en vial de vidrio tipo 1 de 8 ml, cerrado con tapón de bromobutilo recubierto y sellado con cápsula de cierre de aluminio flip-off
- Disolvente: 2,5 ml de agua para preparaciones inyectables en una jeringa precargada de vidrio de borosilicato
- 1 adaptador de vial estéril para reconstitución con 1 aguja de mariposa y 2 toallitas con alcohol Tamaño de envase de 1 unidad.
Puede que solamente se comercialicen algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
El polvo se debe reconstituir únicamente con el disolvente incluido (2,5 ml de agua para preparaciones inyectables) utilizando el kit de inyección suministrado. Mover suavemente el vial en círculos hasta que todo el polvo se haya disuelto. Tras la reconstitución, transferir la solución a la jeringa.
Inspeccionar visualmente el medicamento para comprobar si existen partículas o cambio de coloración antes de la administración. El medicamento reconstituido es una solución transparente e incolora libre de partículas extrañas y tiene un pH de entre 6,5 y 7,5. No use soluciones turbias o con sedimentos.
Instrucciones para la preparación y administración
1. Deje que la jeringa de disolvente (agua para preparaciones inyectables) y el polvo alcancen la temperatura ambiente en el vial cerrado. Puede hacerlo sujetándolos con las manos hasta que tengan la misma temperatura que las manos. No caliente de ninguna otra manera el vial y la jeringa precargada. Esta temperatura debe mantenerse durante la reconstitución.
2. Retire la cápsula de cierre de plástico del vial de polvo para dejar al descubierto las partes centrales del tapón de goma. No retire el tapón gris ni la anilla metálica que rodea la parte superior del vial.
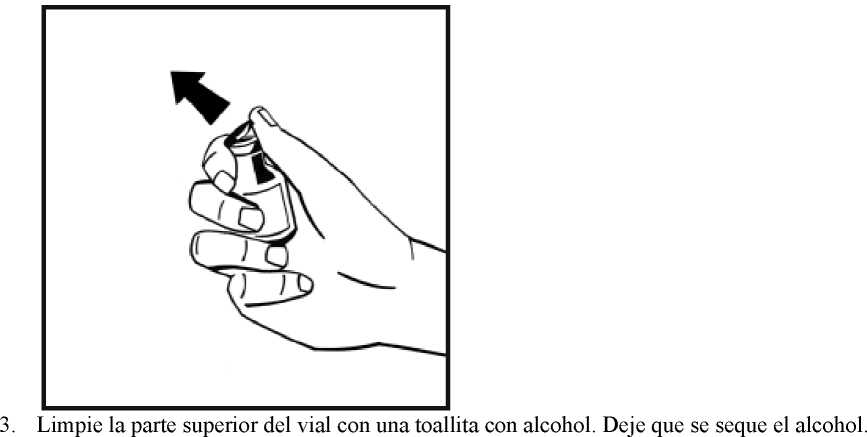
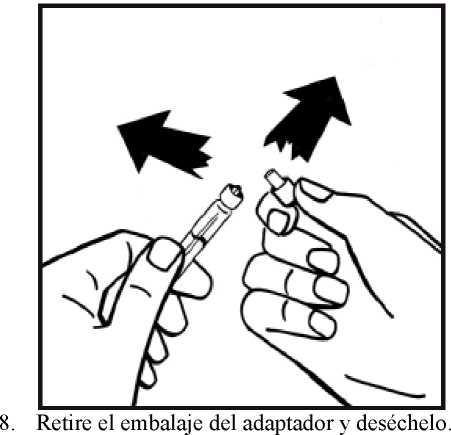
4. Retire la cubierta de papel del envase del adaptador del vial. No saque el adaptador de su envase.
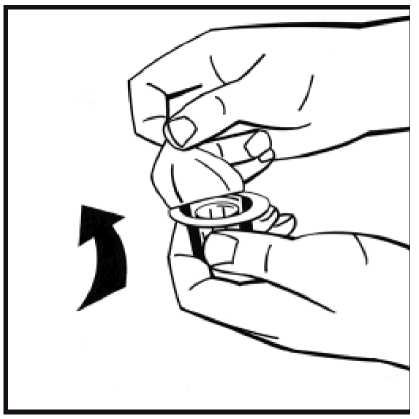
5. Coloque el vial de polvo en una superficie plana y sujételo. Tome el envase del adaptador y coloque el adaptador del vial sobre el centro del tapón de goma del vial de polvo. Presione el envase del adaptador firmemente hacia abajo hasta que la punta del adaptador atraviese el tapón de goma. El adaptador se acoplará al vial cuando esté hecho.
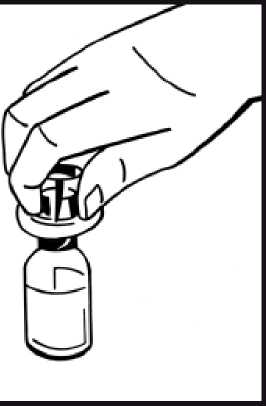
6. Retire la cubierta de papel del envase de la jeringa precargada. Sujete la varilla del émbolo de la jeringa por el extremo y no toque el eje. Fije el extremo con rosca de la varilla del émbolo al émbolo de la jeringa de disolvente. Gire la varilla del émbolo en el sentido de las agujas del reloj hasta que note una ligera resistencia.
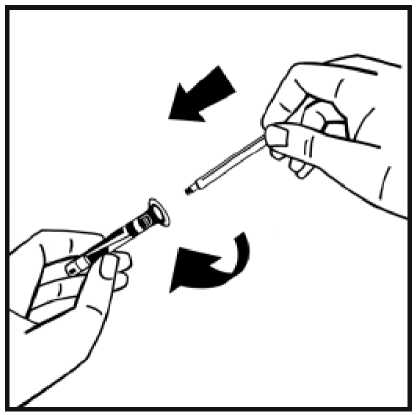
7. Rompa el precinto de la punta de plástico de protección de la jeringa de disolvente partiendo la perforación de la cápsula de cierre. No toque el interior de la cápsula de cierre ni la punta de la jeringa. En caso de no usar la solución inmediatamente, cierre la jeringa llena con la punta
de protección de plástico para almacenarla.
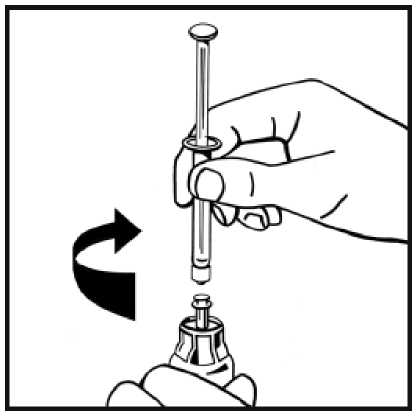
9. Acople firmemente la jeringa de disolvente al adaptador del vial girando en el sentido de las
agujas del reloj hasta que note una ligera resistencia.
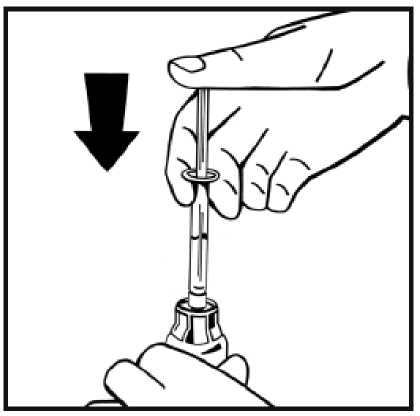
hacia abajo.
11. Sin retirar la jeringa, mueva suavemente o en círculos el vial unas cuantas veces para disolver el polvo. No agitar. Espere hasta que todo el polvo se disuelva completamente.
12. Fíjese en si la solución final tiene partículas antes de administrarla. La solución debe ser transparente e incolora, prácticamente libre de partículas visibles. No use soluciones turbias o con sedimentos.
13. Dé la vuelta al vial acoplado a la jeringa, y lentamente extraiga la solución a la jeringa. Asegúrese de transferir todo el contenido del vial a la jeringa.
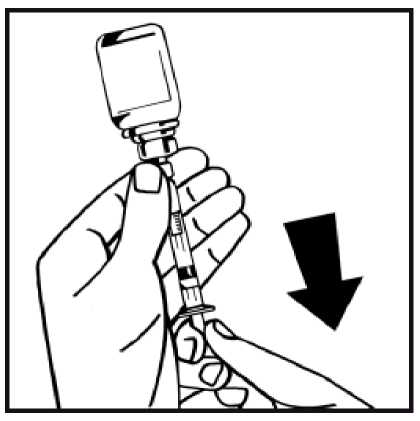
14. Separe la jeringa llena del adaptador del vial girando en sentido contrario a las agujas del reloj y deseche el vial vacío.
15. La solución estará preparada para su uso inmediato. No refrigerar.
16. Limpie la parte elegida para la inyección con una de las toallitas con alcohol suministradas.
17. Acople el kit de inyección suministrado a la jeringa.
Introduzca la aguja del kit de inyección en la vena elegida. Si ha utilizado un torniquete para hacer la vena más visible, deberá estar aflojado antes de empezar a inyectar la solución.
No deberá entrar sangre en la jeringa debido al riesgo de formación de coágulos de fibrina.
18. Inyecte la solución en la vena despacio, no más rápido de 4 ml por minuto.
Si utiliza más de un vial de polvo para un tratamiento, podrá usar la misma aguja de nuevo. El adaptador del vial y la jeringa son de un solo uso.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Octapharma AB Lars Forssells gata 23 112 75 Estocolmo Suecia
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/936/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 22 de julio de 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Nuwiq 500 UI polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 500 UI de Factor VIII de coagulación humano (rDNA), simoctocog alfa.
Nuwiq contiene aproximadamente 200 UI/ml de Factor VIII de coagulación humano (rDNA), simoctocog alfa tras la reconstitución.
La potencia (UI) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de Nuwiq es de aproximadamente 9500 UI/mg de proteína.
El simoctocog alfa (factor VIII de coagulación humano (rDNA)) es una proteína purificada que contiene 1440 aminoácidos. La secuencia de aminoácidos es comparable a la forma de 90 + 80 kDa del factor VIII humano plasmático (esto es, con el dominio B suprimido). Nuwiq se ha producido a partir de tecnología de ADN recombinante de células embrionarias de riñón humano (HEK) modificadas genéticamente 293F. No se ha añadido ningún material derivado de seres humanos o animales durante el proceso de fabricación ni al medicamento final.
Excipiente(s) con efecto conocido:
7,35 mg de sodio por ml de solución reconstituida (18,4 mg de sodio por vial).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: polvo friable de color entre blanco y blanquecino.
Disolvente: agua para preparaciones inyectables, líquido transparente e incoloro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de hemorragias en pacientes con hemofilia A (deficiencia congénita del factor VIII).
Nuwiq se puede usar en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico experimentado en el tratamiento de la hemofilia.
Pacientes no tratados anteriormente
La seguridad y eficacia de Nuwiq en pacientes no tratados previamente no se ha establecido todavía. Posología
La dosis y duración del tratamiento de sustitución dependen de la gravedad de la deficiencia del factor VIII, así como de la localización y alcance de las hemorragias y del estado clínico del paciente.
El número de unidades administradas del factor VIII se expresa en Unidades Internacionales (UI) las cuales están relacionadas con el estándar actual de la OMS para medicamentos con factor VIII. La actividad del factor VIII en plasma se expresa como un porcentaje (respecto al plasma humano normal) o en Unidades Internacionales (respecto a un Estándar Internacional para factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII equivale a la cantidad de factor VIII en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis requerida de factor VIII se basa en el dato empírico de que 1 Unidad Internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad del factor VIII del plasma en aproximadamente un 2% de la actividad normal o 2 UI/dl. La dosis necesaria se determina utilizando la siguiente fórmula:
I. Unidades requeridas (UI) = peso corporal (kg) x aumento deseado de factor VIII (%)(UI/dl) x 0,5 (UI/kg por UI/dl)
II. Aumento previsto del factor VIII (% de lo normal) = 2 x UI administradas
peso corporal (kg)
La dosis a administrar y la frecuencia de administración debe estar siempre orientada a la eficacia clínica en cada caso individual.
En el caso de los episodios hemorrágicos siguientes, la actividad del factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % de lo normal o UI/dl) en el periodo correspondiente. La siguiente tabla se puede utilizar como guía de dosificación en cirugía y en episodios hemorrágicos.
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (%) (UI/dL) |
Frecuencia de las dosis (horas) / duración del tratamiento (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente, hemorragia muscular u oral |
20-40 |
Repetir cada 12 a 24 horas. Al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma |
30-60 |
Repetir la perfusión cada 12 a 24 horas, entre 3 y 4 días o más, hasta que cese el dolor y la discapacidad aguda. |
|
Hemorragia potencialmente mortal |
60-100 |
Repetir la perfusión cada 8 a 24 horas hasta que se supere el peligro |
|
Cirugía | ||
|
Cirugía menor |
30-60 |
Cada 24 horas, al menos 1 día, hasta |
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (%) (UI/dL) |
Frecuencia de las dosis (horas) / duración del tratamiento (días) |
|
Incluyendo extracción dental |
lograr la curación. | |
|
Cirugía mayor |
80-100 (pre y postoperatorio) |
Repetir la perfusión cada 8-24 horas hasta que se consiga una cicatrización adecuada de la herida, y después al menos durante otros 7 días de tratamiento para mantener una actividad de factor VIII del 30% al 60% (UI/dL). |
Profilaxis
En la profilaxis a largo plazo de episodios hemorrágicos en pacientes con hemofilia A grave, las dosis habituales son de 20 a 40 UI de factor VIII por kg de peso corporal, en intervalos de 2 a 3 días. En algunos casos, especialmente en los pacientes más jóvenes, puede ser necesario un intervalo de dosis más corto o dosis mayores.
Durante el transcurso del tratamiento, se aconseja la determinación adecuada de los niveles de factor VIII como guía de la dosis a administrar y la frecuencia de repetición de las inyecciones. En el caso de intervenciones, cirugía mayor en particular, es indispensable la monitorización precisa del tratamiento de sustitución mediante el análisis de la coagulación (actividad del factor VIII del plasma). La respuesta al factor VIII puede variar en cada paciente, revelándose unos valores diferentes de semivida y recuperación.
Población pediátrica
La posología es la misma en adultos y niños. Sin embargo, hay que tener en cuenta que para los niños pueden ser necesarios intervalos de dosis más cortos o dosis mayores. Los datos actualmente disponibles se describen en las secciones 4.8, 5.1 y 5.2.
No se dispone de datos en niños menores de 2 años de edad.
Forma de administración
Vía intravenosa.
Se recomienda no administrar más de 4 ml por minuto.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo Hipersensibilidad
Como con cualquier producto proteínico vía intravenosa, es posible que se den reacciones de hipersensibilidad de tipo alérgico. Nuwiq contiene trazas de proteínas de células huésped humanas distintas al factor VIII. Si aparecen síntomas de hipersensibilidad, se debe aconsejar a los pacientes que interrumpan inmediatamente el uso del medicamento y contacten con su médico. Los pacientes deben ser informados acerca de los signos iniciales de las reacciones de hipersensibilidad, que incluyen sarpullido, urticaria generalizada, opresión en el pecho, sibilancias, hipotensión y anafilaxia.
En caso de shock, se debe aplicar el tratamiento médico habitual para el shock. Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) del factor VIII es una complicación conocida en el tratamiento de individuos con hemofilia A. Estos inhibidores son normalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en unidades Bethesda (UB) por ml de plasma usando el ensayo modificado. El riesgo de desarrollar inhibidores se relaciona con la exposición al factor VIII, siendo este riesgo más alto en los 20 primeros días de exposición. Rara vez se pueden desarrollar inhibidores transcurridos los primeros 100 días de exposición.
Se han observado casos de inhibidor recurrente (de bajo título) tras cambiar de un producto de factor VIII a otro en pacientes previamente tratados con un periodo de exposición de más de 100 días y con antecedentes de desarrollo de inhibidores. Por ello, se recomienda realizar un seguimiento meticuloso del desarrollo de inhibidores en todos los pacientes tras cualquier cambio de producto.
En general, todos los pacientes tratados con productos del factor de coagulación VIII deben ser vigilados cuidadosamente, de cara al desarrollo de inhibidores, mediante las observaciones clínicas y pruebas de laboratorio adecuadas. Si no se alcanzan los niveles plasmáticos de actividad del factor VIII esperados o si no se controla la hemorragia mediante una dosis adecuada, se debe realizar una prueba de presencia de inhibidor del factor VIII. En pacientes con altos niveles de inhibidor, es posible que el tratamiento con factor VIII no resulte eficaz y se deban considerar otras opciones terapéuticas, como la inducción de inmunotolerancia (ITI). El tratamiento de esos pacientes debe ser dirigido por médicos con experiencia en el tratamiento de la hemofilia y de los inhibidores del factor VIII.
Complicaciones asociadas a los catéteres
Si se requiere un dispositivo de acceso venoso central (CVAD (por sus siglas en inglés)), hay que tener en cuenta el riesgo de complicaciones asociadas al CVAD, incluidas las infecciones localizadas, la bacteremia y la trombosis en el lugar de implantación del catéter.
Se recomienda encarecidamente que cada vez que se administre Nuwiq a un paciente, se registre el nombre y número de lote del producto para mantener un vínculo entre el paciente y el lote del medicamento.
Población pediátrica
Las advertencias y precauciones enumeradas se refieren tanto a adultos como a niños.
Consideraciones relativas al excipiente (contenido de sodio)
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por vial.
Sin embargo, dependiendo del peso corporal y la posología, podría administrársele al paciente más de un vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones con Nuwiq.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción animal con Nuwiq.
Debido a la escasa frecuencia de la hemofilia A en mujeres, no se tiene experiencia sobre el uso del factor VIII durante el embarazo o periodo de lactancia. Por lo tanto, Nuwiq solo se debe usar durante el embarazo y la lactancia si está claramente indicado. No hay datos disponibles de fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Nuwiq no influye sobre la capacidad para conducir y utilizar máquinas.
4.8 Reacciones adversas Resumen del perfil de seguridad
Se han observado con poca frecuencia reacciones alérgicas o de hipersensibilidad con preparados de FVIII (que pueden incluir angioedema, quemazón y punzadas en el lugar de la inyección, escalofríos, rubefacción, urticaria generalizada, cefalea, sarpullido, hipotensión, letargia, náuseas, inquietud, taquicardia, opresión en el pecho, cosquilleo, vómitos, sibilancias) y en algunos casos pueden empeorar hasta convertirse en anafilaxia grave, (incluido el shock anafiláctico).
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) del factor VIII. Si aparecen dichos inhibidores, puede manifestarse en forma de una respuesta clínica insuficiente. En estos casos, se recomienda ponerse en contacto con un centro especializado en hemofilia.
Tabla de reacciones adversas
Durante los estudios clínicos con Nuwiq en pacientes pediátricos previamente tratados (2 a 11 años, n = 58), adolescentes (12 a 17 años, n = 3) y adultos (n = 74) con hemofilia A grave, se notificaron un total de 8 reacciones adversas a medicamentos (RAM) (6 en adultos, 2 en niños) en 5 pacientes (3 adultos, 2 niños).
La Tabla 1 que se presenta a continuación sigue la Clasificación de Órganos del y Sistemas MedDRA (COS y nivel de términos preferentes).
Las frecuencias se han evaluado conforme a la convención siguiente: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
En cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 1. Frecuencia de las reacciones adversas a medicamentos (RAM) en ensayos clínicos en 135 pacientes con hemofilia A grave previamente tratados__
|
Clasificación estándar de órganos del sistema MedDRA |
Reacciones adversas |
Frecuencia* |
|
Trastornos del sistema nervioso |
Parestesia Dolor de cabeza |
Poco frecuentes |
|
Trastornos del oído y del laberinto |
Vértigo |
Poco frecuentes |
|
Trastornos gastrointestinales |
Sequedad de boca |
Poco frecuentes |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor de espalda |
Poco frecuentes |
|
Trastornos generales y alteraciones en el lugar de administración |
Inflamación en la zona de inyección Dolor en la zona de inyección |
Poco frecuentes |
|
Exploraciones complementarias |
Positivo por anticuerpos no neutralizantes anti-factor VIII |
Poco frecuentes |
* Todas estas RAM solo se produjeron una vez. Como el número total de pacientes analizados es de 135, la frecuencia no puede ser menor que "poco frecuente" si se produce una RAM.
Descripción de las reacciones adversas seleccionadas
Se detectó un anticuerpo no neutralizante anti-Factor VIII en un paciente adulto (ver Tabla 1). Se analizó la muestra en el laboratorio central en ocho diluciones. El resultado fue positivo solo en la dilución factor 1 y el título de anticuerpos fue muy bajo. No se detectó en este paciente ninguna actividad inhibidora, según las mediciones del ensayo de Bethesda modificado. La eficacia clínica y la recuperación in vivo de Nuwiq no se vieron afectadas en este paciente.
Población pediátrica
Se asume que la frecuencia, tipo y gravedad de las reacciones adversas son las mismas en niños que en adultos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se ha notificado ningún caso de sobredosis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos, factor VIII de la coagulación sanguínea, código ATC: B02BD02.
El complejo factor VIII/factor von Willebrand consiste en dos moléculas (factor VIII y factor de von Willebrand) con distintas funciones fisiológicas. Cuando se inyecta en un paciente hemofílico, el factor VIII se une con el factor von Willebrand circulante del paciente. El factor VIII activado actúa como cofactor del factor IX activado, acelerando la conversión del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Seguidamente, la trombina convierte el fibrinógeno en fibrina y se puede formar un coágulo. La hemofilia A es una alteración hereditaria de la coagulación de la sangre vinculada al sexo, debida a niveles reducidos de factor VIII:C y da como resultado hemorragias profusas en articulaciones, músculos u órganos internos, ya sea de forma espontánea, o tras un accidente o un traumatismo quirúrgico. El tratamiento de sustitución aumenta los niveles plasmáticos de factor VIII, lo cual permite corregir temporalmente la deficiencia del factor VIII y la tendencia al sangrado.
Se ha evaluado la inmunogenicidad de Nuwiq en ensayos clínicos en 135 pacientes con hemofilia A grave previamente tratados (74 pacientes adultos y 61 pacientes pediátricos). Ninguno de los pacientes desarrollaron inhibidores.
En un estudio clínico en 32 pacientes adultos con hemofilia A grave, la mediana de consumo de Nuwiq para profilaxis fue de 468,7 UI/kg/mes. La mediana de la dosis para tratar episodios hemorrágicos intercurrentes fue de 33,0 UI/kg en aquellos pacientes en profilaxis. En otro estudio clínico, se trató a demanda a 22 pacientes adultos. En total, se trataron 986 episodios hemorrágicos con una mediana de dosis de 30,9 UI/kg. En general, los sangrados menores requirieron una dosis ligeramente menor, mientras que los sangrados más graves requirieron hasta tres veces la mediana de dosis.
Población pediátrica
Se han obtenido los datos de 29 niños previamente tratados con edades entre 2 y 5 años, 31 niños entre 6 y 12 años y un adolescente de 14 años. La mediana de la dosis por inyección profiláctica fue de 37,8 UI/kg. Veinte pacientes utilizaron una mediana de dosis mayor de 45 UI/kg. La mediana del consumo
de Nuwiq para profilaxis por mes fue de 521,9 Ul/kg. Se requirió una mediana de dosis de Nuwiq más alta para tratar las hemorragias en niños (43,9 UI/kg) que en adultos (33,0 UI/kg), y una mediana de dosis más alta para tratar las hemorragias de moderadas a importantes que para las menores (78,2 UI/kg frente a 41,7 UI/kg). Los niños de menor edad requirieron en general medianas de dosis más altas (de 6 a 12 años: 43,9 UI/kg; de 2 a 5 años: 52,6 UI/kg).
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Nuwiq en uno o más grupos de la población pediátrica en tratamiento de la Hemofilia A (deficiencia congénita del Factor VIII) (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas (PK)
Tabla 2. Parámetros PK para Nuwiq (dosis: 50 UI/kg) en pacientes adultos previamente tratados (de 18 a 65 años de edad) con hemofilia A grave (n = 20)_
|
Parámetro PK |
Ensayo cromogénico | |
|
Valor medio ± SD |
Mediana (rango) | |
|
AUC (h*UI/ml) |
22,6 ± 8,0 |
22,3 (8,4 - 38,1) |
|
T1/2 (h) |
14,7 ± 10,4 |
12,5 (5,4 -55,6) |
|
IVR (%/UI/kg) |
2,5 ± 0,4 |
2,5 (1,7 -3,2) |
|
Cl (ml/h/kg) |
3,0 ± 1,2 |
2,7 (1,5-6,4) |
AUC = Área bajo la curva (FVIII:C), T1/2 = Semivida terminal,
IVR = Recuperación incremental in vivo , Cl = Aclaramiento, SD = Desviación estándar
Tabla 3. Parámetros PK para Nuwiq (dosis: 50 UI/kg) en niños previamente tratados de entre 6 y 12 años de edad con hemofilia A grave (n = 12)_
|
Parámetro PK |
Ensayo cromogénico | |
|
Valor medio ± SD |
Mediana (rango) | |
|
AUC (h*UI/ml) |
13,2 ± 3,4 |
12,8 (7,8 - 19,1) |
|
T1/2 (h) |
10,0 ± 1,9 |
9,9 (7,6 -14,1) |
|
IVR (%/UI/kg) |
1,9 ± 0,4 |
1,9 (1,2 -2,6) |
|
Cl (ml/h/kg) |
4,3 ± 1,2 |
4,2 (2,8 - 6,9) |
AUC = Área bajo la curva (FVIII:C), T1/2 = Semivida terminal,
IVR = Recuperación incremental in vivo , Cl = Aclaramiento, SD = Desviación estándar
Tabla 4. Parámetros PK para Nuwiq (dosis: 50 UI/kg) en niños previamente tratados de entre 2 y 5 años de edad con hemofilia A grave (n = 13)_
|
Parámetro PK |
Ensayo cromogénico | |
|
Valor medio ± SD |
Mediana (rango) | |
|
AUC (h*UI/ml) |
11,7 ± 5,3 |
10,5 (4,9 -23,8) |
|
T1/2 (h) |
9,5 ± 3,3 |
8,2 (4,3 - 17,3) |
|
IVR (%/UI/kg) |
1,9 ± 0,3 |
1,8 (1,5 -2,4) |
|
Cl (ml/h/kg) |
5,4 ± 2,4 |
5,1 ( 2,3 -10,9) |
AUC = Área bajo la curva (FVIII:C), T1/2 = Semivida terminal,
IVR = Recuperación incremental in vivo , Cl = Aclaramiento, SD = Desviación estándar Población pediátrica
Según lo extraído de publicaciones médicas, la recuperación y semivida fueron menores en los niños más pequeños que en los adultos y el aclaramiento más alto, lo que se puede deber en parte al mayor volumen de plasma conocido por kilogramo de peso corporal en los pacientes más jóvenes.
Subgrupos de peso ajustado
Tabla 5. Parámetros PK de peso ajustado para Nuwiq (dosis: 50 Ul/kg) en pacientes adultos
|
previamente tratados (de 18 a 65 años ( |
e edad) con hemofilia A grave (n = 20) | |||
|
Parámetro PK |
Todos (n=20) |
Peso normal (n=14) |
Pre-adiposo (n=4) |
Adiposo (n=2) |
|
Ensayo cromogénico valor medio ± SD | ||||
|
AUC (h*UI/ml) |
22,6 ± 8,0 |
20,4 ± 6,9 |
24,9 ± 8,9 |
33,5 ± 6,5 |
|
T1/2 (h) |
14,7 ± 10,4 |
14,7 ± 12,1 |
13,4 ± 5,9 |
17,2 ± 4,8 |
|
IVR (%/UI/kg) |
2,5 ± 0,4 |
2,4 ± 0,4 |
2,7 ± 0,4 |
2,8 ± 0,3 |
|
Cl (ml/h/kg) |
3,0 ± 1,2 |
3,2 ± 1,3 |
2,6 ± 1,0 |
1,8 ± 0,4 |
|
Mediana ensayo cromogénico (rango) | ||||
|
AUC (h*UI/ml) |
22,3 (8,4 -38,1) |
21,2 (8,4 -32,6) |
23,3 (17,4 - 35,5) |
33,5 (28,9 -38,1) |
|
T1/2 (h) |
12,5 (5,4 -55,6) |
12,3 (5,4 -55,6) |
11,2 (9,3 -22,0) |
17,2 (13,8 -20,6) |
|
IVR (%/UI/kg) |
2,5 (1,7 -3,2) |
2,4 (1,7 -3,1) |
2,8 (2,3 - 3,2) |
2,8 (2,6 -3,0) |
|
Cl (ml/h/kg) |
2,7 (1,5 -6,4) |
2,8 (1,7 -6,4) |
2,5 (1,6 -3,7) |
1,8 (1,5 -2,0) |
Peso normal: IMC 18,5-25 kg/m2, pre-adiposo: IMC 25-30 kg/m2, adiposo: IMC > 30 kg/m2,
SD = Desviación estándar
5.3 Datos preclínicos sobre seguridad
En los estudios preclínicos, Nuwiq se utilizó para restablecer la hemostasia de manera segura y eficaz en perros con hemofilia. Los estudios toxicológicos demostraron que los animales de laboratorio (ratas y monos cynomolgus) toleran bien la administración vía intravenosa local y la exposición sistémica.
No se realizaron estudios específicos con Nuwiq con administración repetida a largo plazo como los de toxicidad para la reproducción, toxicidad crónica y carcinogenia debido a la respuesta inmune a proteínas heterólogas en todas las especies mamíferas no humanas.
No se realizaron estudios sobre el potencial mutagénico de Nuwiq.
Las evaluaciones ex vivo utilizando un kit comercial de ensayo para cuantificar la respuesta de la célula T a las proteínas terapéuticas, ponen de manifiesto un bajo riesgo de inmunogenicidad.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo:
Sacarosa Cloruro de sodio Cloruro de calcio dihidratado Clorhidrato de arginina Citrato de sodio dihidratado Poloxamer 188
Disolvente:
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
Solo deben utilizarse los kits de inyección suministrados, ya que el tratamiento puede fallar a consecuencia de la adsorción del factor VIII humano de coagulación a las superficies internas de otros productos de inyección.
6.3 Periodo de validez
2 años
Durante el periodo de validez, el medicamento se puede almacenar a temperatura ambiente (hasta 25°C) durante un único periodo no superior a 1 mes. Una vez que el medicamento se saca de la nevera, no se debe volver a refrigerar. Registrar la fecha de inicio del almacenamiento a temperatura ambiente en el embalaje del producto. Conservar el vial en el embalaje exterior para protegerlo de la luz.
Tras la reconstitución, se ha comprobado la estabilidad química y física del producto conservado a temperatura ambiente durante 24 horas.
Desde un punto de vista microbiológico, el producto se debe utilizar inmediatamente después de la reconstitución. Si no se usa de forma inmediata, los tiempos y condiciones de conservación previas a su uso son responsabilidad del usuario.
Mantener la solución reconstituida a temperatura ambiente. No refrigerar una vez reconstituida.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior de cartón para protegerlo de la luz.
Para consultar la información sobre almacenamiento a temperatura ambiente y condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Cada envase de Nuwiq 500 UI contiene:
- Polvo: 500 UI de polvo en vial de vidrio tipo 1 de 8 ml, cerrado con tapón de bromobutilo recubierto y sellado con cápsula de cierre de aluminio flip-off
- Disolvente: 2,5 ml de agua para preparaciones inyectables en una jeringa precargada de vidrio de borosilicato
- 1 adaptador de vial estéril para reconstitución con 1 aguja de mariposa y 2 toallitas con alcohol Tamaño de envase de 1 unidad.
Puede que solamente se comercialicen algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
El polvo se debe reconstituir únicamente con el disolvente incluido (2,5 ml de agua para preparaciones inyectables) utilizando el kit de inyección suministrado. Mover suavemente el vial en círculos hasta que todo el polvo se haya disuelto. Tras la reconstitución, transferir la solución a la jeringa.
Inspeccionar visualmente el medicamento para comprobar si existen partículas o cambio de coloración antes de la administración. El medicamento reconstituido es una solución transparente e incolora libre de partículas extrañas y tiene un pH de entre 6,5 y 7,5. No use soluciones turbias o con sedimentos.
Instrucciones para la preparación y administración
1. Deje que la jeringa de disolvente (agua para preparaciones inyectables) y el polvo alcancen la temperatura ambiente en el vial cerrado. Puede hacerlo sujetándolos con las manos hasta que tengan la misma temperatura que las manos. No caliente de ninguna otra manera el vial y la jeringa precargada. Esta temperatura debe mantenerse durante la reconstitución.
2. Retire la cápsula de cierre de plástico del vial de polvo para dejar al descubierto las partes centrales del tapón de goma. No retire el tapón gris ni la anilla metálica que rodea la parte superior del vial.
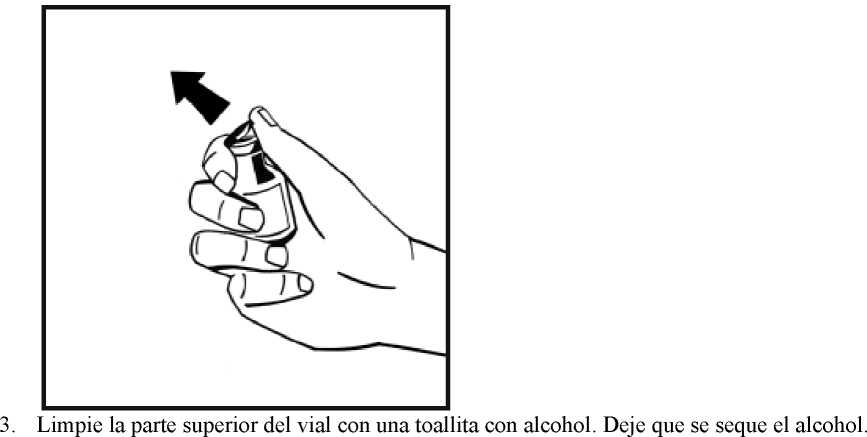
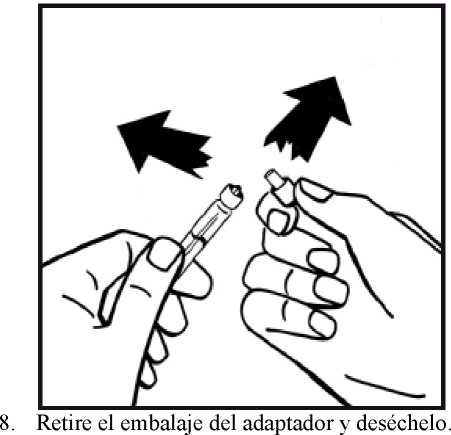
4. Retire la cubierta de papel del envase del adaptador del vial. No saque el adaptador de su envase.
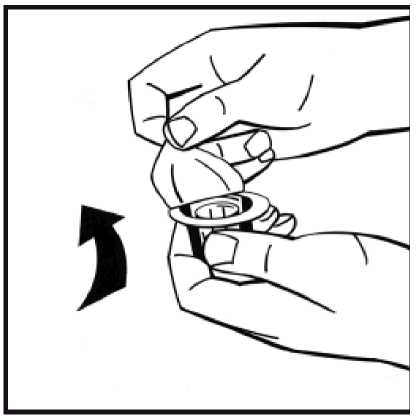
5. Coloque el vial de polvo en una superficie plana y sujételo. Tome el envase del adaptador y coloque el adaptador del vial sobre el centro del tapón de goma del vial de polvo. Presione el envase del adaptador firmemente hacia abajo hasta que la punta del adaptador atraviese el tapón de goma. El adaptador se acoplará al vial cuando esté hecho.
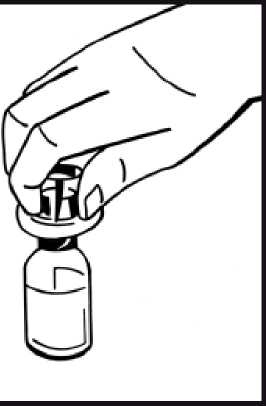
6. Retire la cubierta de papel del envase de la jeringa precargada. Sujete la varilla del émbolo de la jeringa por el extremo y no toque el eje. Fije el extremo con rosca de la varilla del émbolo al émbolo de la jeringa de disolvente. Gire la varilla del émbolo en el sentido de las agujas del reloj hasta que note una ligera resistencia.
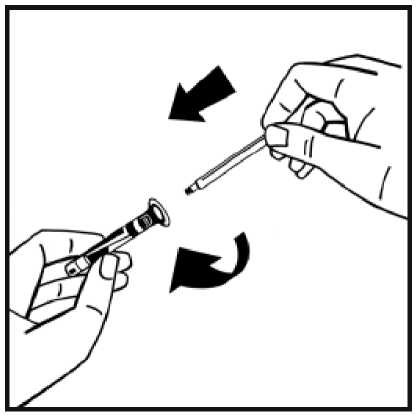
7. Rompa el precinto de la punta de plástico de protección de la jeringa de disolvente partiendo la perforación de la cápsula de cierre. No toque el interior de la cápsula de cierre ni la punta de la jeringa. En caso de no usar la solución inmediatamente, cierre la jeringa llena con la punta
de protección de plástico para almacenarla.
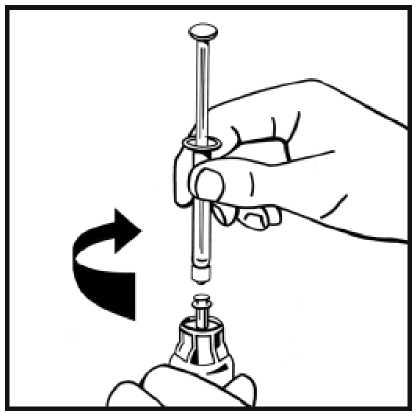
9. Acople firmemente la jeringa de disolvente al adaptador del vial girando en el sentido de las
agujas del reloj hasta que note una ligera resistencia.
hacia abajo.
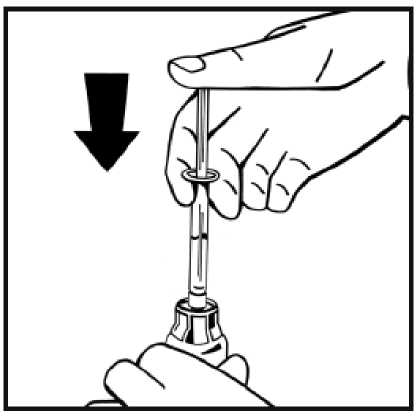
11. Sin retirar la jeringa, mueva suavemente o en círculos el vial unas cuantas veces para disolver el polvo. No agitar. Espere hasta que todo el polvo se disuelva completamente.
12. Fíjese en si la solución final tiene partículas antes de administrarla. La solución debe ser transparente e incolora, prácticamente libre de partículas visibles. No use soluciones turbias o con sedimentos.
13. Dé la vuelta al vial acoplado a la jeringa, y lentamente extraiga la solución a la jeringa. Asegúrese de transferir todo el contenido del vial a la jeringa.
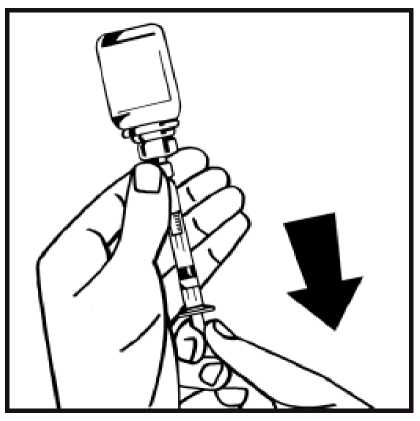
14. Separe la jeringa llena del adaptador del vial girando en sentido contrario a las agujas del reloj y deseche el vial vacío.
15. La solución estará preparada para su uso inmediato. No refrigerar.
16. Limpie la parte elegida para la inyección con una de las toallitas con alcohol suministradas.
17. Acople el kit de inyección suministrado a la jeringa.
Introduzca la aguja del kit de inyección en la vena elegida. Si ha utilizado un torniquete para hacer la vena más visible, deberá estar aflojado antes de empezar a inyectar la solución.
No deberá entrar sangre en la jeringa debido al riesgo de formación de coágulos de fibrina.
18. Inyecte la solución en la vena despacio, no más rápido de 4 ml por minuto.
Si utiliza más de un vial de polvo para un tratamiento, podrá usar la misma aguja de nuevo. El adaptador del vial y la jeringa son de un solo uso.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Octapharma AB Lars Forssells gata 23 112 75 Estocolmo Suecia
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/936/002
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 22 de julio de 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Nuwiq 1000 UI polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 1000 UI de Factor VIII de coagulación humano (rDNA), simoctocog alfa.
Nuwiq contiene aproximadamente 400 UI/ml de Factor VIII de coagulación humano (rDNA), simoctocog alfa tras la reconstitución.
La potencia (UI) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de Nuwiq es de aproximadamente 9500 UI/mg de proteína.
El simoctocog alfa (factor VIII de coagulación humano (rDNA)) es una proteína purificada que contiene 1440 aminoácidos. La secuencia de aminoácidos es comparable a la forma de 90 + 80 kDa del factor VIII humano plasmático (esto es, con el dominio B suprimido). Nuwiq se ha producido a partir de tecnología de ADN recombinante de células embrionarias de riñón humano (HEK) modificadas genéticamente 293F. No se ha añadido ningún material derivado de seres humanos o animales durante el proceso de fabricación ni al medicamento final.
Excipiente(s) con efecto conocido:
7,35 mg de sodio por ml de solución reconstituida (18,4 mg de sodio por vial).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: polvo friable de color entre blanco y blanquecino.
Disolvente: agua para preparaciones inyectables, líquido transparente e incoloro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de hemorragias en pacientes con hemofilia A (deficiencia congénita del factor VIII).
Nuwiq se puede usar en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico experimentado en el tratamiento de la hemofilia.
Pacientes no tratados anteriormente
La seguridad y eficacia de Nuwiq en pacientes no tratados previamente no se ha establecido todavía. Posología
La dosis y duración del tratamiento de sustitución dependen de la gravedad de la deficiencia del factor VIII, así como de la localización y alcance de las hemorragias y del estado clínico del paciente.
El número de unidades administradas del factor VIII se expresa en Unidades Internacionales (UI) las cuales están relacionadas con el estándar actual de la OMS para medicamentos con factor VIII. La actividad del factor VIII en plasma se expresa como un porcentaje (respecto al plasma humano normal) o en Unidades Internacionales (respecto a un Estándar Internacional para factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII equivale a la cantidad de factor VIII en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis requerida de factor VIII se basa en el dato empírico de que 1 Unidad Internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad del factor VIII del plasma en aproximadamente un 2% de la actividad normal o 2 UI/dl. La dosis necesaria se determina utilizando la siguiente fórmula:
I. Unidades requeridas (UI) = peso corporal (kg) x aumento deseado de factor VIII (%)(UI/dl) x 0,5 (UI/kg por UI/dl)
II. Aumento previsto del factor VIII (% de lo normal) = 2 x UI administradas
peso corporal (kg)
La dosis a administrar y la frecuencia de administración debe estar siempre orientada a la eficacia clínica en cada caso individual.
En el caso de los episodios hemorrágicos siguientes, la actividad del factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % de lo normal o UI/dl) en el periodo correspondiente. La siguiente tabla se puede utilizar como guía de dosificación en cirugía y en episodios hemorrágicos.
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (%) (UI/dL) |
Frecuencia de las dosis (horas) / duración del tratamiento (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente, hemorragia muscular u oral |
20-40 |
Repetir cada 12 a 24 horas. Al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma |
30-60 |
Repetir la perfusión cada 12 a 24 horas, entre 3 y 4 días o más, hasta que cese el dolor y la discapacidad aguda. |
|
Hemorragia potencialmente mortal |
60-100 |
Repetir la perfusión cada 8 a 24 horas hasta que se supere el peligro |
|
Cirugía | ||
|
Cirugía menor Incluyendo extracción dental |
30-60 |
Cada 24 horas, al menos 1 día, hasta lograr la curación. |
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (%) (UI/dL) |
Frecuencia de las dosis (horas) / duración del tratamiento (días) |
|
Cirugía mayor |
80-100 (pre y postoperatorio) |
Repetir la perfusión cada 8-24 horas hasta que se consiga una cicatrización adecuada de la herida, y después al menos durante otros 7 días de tratamiento para mantener una actividad de factor VIII del 30% al 60% (UI/dL). |
Profilaxis
En la profilaxis a largo plazo de episodios hemorrágicos en pacientes con hemofilia A grave, las dosis habituales son de 20 a 40 UI de factor VIII por kg de peso corporal, en intervalos de 2 a 3 días. En algunos casos, especialmente en los pacientes más jóvenes, puede ser necesario un intervalo de dosis más corto o dosis mayores.
Durante el transcurso del tratamiento, se aconseja la determinación adecuada de los niveles de factor VIII como guía de la dosis a administrar y la frecuencia de repetición de las inyecciones. En el caso de intervenciones, cirugía mayor en particular, es indispensable la monitorización precisa del tratamiento de sustitución mediante el análisis de la coagulación (actividad del factor VIII del plasma). La respuesta al factor VIII puede variar en cada paciente, revelándose unos valores diferentes de semivida y recuperación.
Población pediátrica
La posología es la misma en adultos y niños. Sin embargo, hay que tener en cuenta que para los niños pueden ser necesarios intervalos de dosis más cortos o dosis mayores. Los datos actualmente disponibles se describen en las secciones 4.8, 5.1 y 5.2.
No se dispone de datos en niños menores de 2 años de edad.
Forma de administración
Vía intravenosa.
Se recomienda no administrar más de 4 ml por minuto.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo Hipersensibilidad
Como con cualquier producto proteínico vía intravenosa, es posible que se den reacciones de hipersensibilidad de tipo alérgico. Nuwiq contiene trazas de proteínas de células huésped humanas distintas al factor VIII. Si aparecen síntomas de hipersensibilidad, se debe aconsejar a los pacientes que interrumpan inmediatamente el uso del medicamento y contacten con su médico. Los pacientes deben ser informados acerca de los signos iniciales de las reacciones de hipersensibilidad, que incluyen sarpullido, urticaria generalizada, opresión en el pecho, sibilancias, hipotensión y anafilaxia.
En caso de shock, se debe aplicar el tratamiento médico habitual para el shock. Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) del factor VIII es una complicación conocida en el tratamiento de individuos con hemofilia A. Estos inhibidores son normalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en unidades Bethesda (UB) por ml de plasma usando el ensayo modificado. El riesgo de desarrollar inhibidores se relaciona con la exposición al factor VIII, siendo este riesgo más alto en los 20 primeros días de exposición. Rara vez se pueden desarrollar inhibidores transcurridos los primeros 100 días de exposición.
Se han observado casos de inhibidor recurrente (de bajo título) tras cambiar de un producto de factor VIII a otro en pacientes previamente tratados con un periodo de exposición de más de 100 días y con antecedentes de desarrollo de inhibidores. Por ello, se recomienda realizar un seguimiento meticuloso del desarrollo de inhibidores en todos los pacientes tras cualquier cambio de producto.
En general, todos los pacientes tratados con productos del factor de coagulación VIII deben ser vigilados cuidadosamente, de cara al desarrollo de inhibidores, mediante las observaciones clínicas y pruebas de laboratorio adecuadas. Si no se alcanzan los niveles plasmáticos de actividad del factor VIII esperados o si no se controla la hemorragia mediante una dosis adecuada, se debe realizar una prueba de presencia de inhibidor del factor VIII. En pacientes con altos niveles de inhibidor, es posible que el tratamiento con factor VIII no resulte eficaz y se deban considerar otras opciones terapéuticas, como la inducción de inmunotolerancia (ITI). El tratamiento de esos pacientes debe ser dirigido por médicos con experiencia en el tratamiento de la hemofilia y de los inhibidores del factor VIII.
Complicaciones asociadas a los catéteres
Si se requiere un dispositivo de acceso venoso central (CVAD (por sus siglas en inglés)), hay que tener en cuenta el riesgo de complicaciones asociadas al CVAD, incluidas las infecciones localizadas, la bacteremia y la trombosis en el lugar de implantación del catéter.
Se recomienda encarecidamente que cada vez que se administre Nuwiq a un paciente, se registre el nombre y número de lote del producto para mantener un vínculo entre el paciente y el lote del medicamento.
Población pediátrica
Las advertencias y precauciones enumeradas se refieren tanto a adultos como a niños.
Consideraciones relativas al excipiente (contenido de sodio)
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por vial.
Sin embargo, dependiendo del peso corporal y la posología, podría administrársele al paciente más de un vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones con Nuwiq.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción animal con Nuwiq.
Debido a la escasa frecuencia de la hemofilia A en mujeres, no se tiene experiencia sobre el uso del factor VIII durante el embarazo o periodo de lactancia. Por lo tanto, Nuwiq solo se debe usar durante el embarazo y la lactancia si está claramente indicado. No hay datos disponibles de fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Nuwiq no influye sobre la capacidad para conducir y utilizar máquinas.
4.8 Reacciones adversas Resumen del perfil de seguridad
Se han observado con poca frecuencia reacciones alérgicas o de hipersensibilidad con preparados de FVIII (que pueden incluir angioedema, quemazón y punzadas en el lugar de la inyección, escalofríos, rubefacción, urticaria generalizada, cefalea, sarpullido, hipotensión, letargia, náuseas, inquietud, taquicardia, opresión en el pecho, cosquilleo, vómitos, sibilancias) y en algunos casos pueden empeorar hasta convertirse en anafilaxia grave, (incluido el shock anafiláctico).
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) del factor VIII. Si aparecen dichos inhibidores, puede manifestarse en forma de una respuesta clínica insuficiente. En estos casos, se recomienda ponerse en contacto con un centro especializado en hemofilia.
Tabla de reacciones adversas
Durante los estudios clínicos con Nuwiq en pacientes pediátricos previamente tratados (2 a 11 años, n = 58), adolescentes (12 a 17 años, n = 3) y adultos (n = 74) con hemofilia A grave, se notificaron un total de 8 reacciones adversas a medicamentos (RAM) (6 en adultos, 2 en niños) en 5 pacientes (3 adultos, 2 niños).
La Tabla 1 que se presenta a continuación sigue la Clasificación de Órganos del y Sistemas MedDRA (COS y nivel de términos preferentes).
Las frecuencias se han evaluado conforme a la convención siguiente: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
En cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 1. Frecuencia de las reacciones adversas a medicamentos (RAM) en ensayos clínicos en 135 pacientes con hemofilia A grave previamente tratados__
|
Clasificación estándar de órganos del sistema MedDRA |
Reacciones adversas |
Frecuencia* |
|
Trastornos del sistema nervioso |
Parestesia Dolor de cabeza |
Poco frecuentes |
|
Trastornos del oído y del laberinto |
Vértigo |
Poco frecuentes |
|
Trastornos gastrointestinales |
Sequedad de boca |
Poco frecuentes |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor de espalda |
Poco frecuentes |
|
Trastornos generales y alteraciones en el lugar de administración |
Inflamación en la zona de inyección Dolor en la zona de inyección |
Poco frecuentes |
|
Exploraciones complementarias |
Positivo por anticuerpos no neutralizantes anti-factor VIII |
Poco frecuentes |
* Todas estas RAM solo se produjeron una vez. Como el número total de pacientes analizados es de 135, la frecuencia no puede ser menor que "poco frecuente" si se produce una RAM.
Descripción de las reacciones adversas seleccionadas
Se detectó un anticuerpo no neutralizante anti-Factor VIII en un paciente adulto (ver Tabla 1). Se analizó la muestra en el laboratorio central en ocho diluciones. El resultado fue positivo solo en la dilución factor 1 y el título de anticuerpos fue muy bajo. No se detectó en este paciente ninguna
actividad inhibidora, según las mediciones del ensayo de Bethesda modificado. La eficacia clínica y la recuperación in vivo de Nuwiq no se vieron afectadas en este paciente.
Población pediátrica
Se asume que la frecuencia, tipo y gravedad de las reacciones adversas son las mismas en niños que en adultos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se ha notificado ningún caso de sobredosis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos, factor VIII de la coagulación sanguínea, código ATC: B02BD02.
El complejo factor VIII/factor von Willebrand consiste en dos moléculas (factor VIII y factor de von Willebrand) con distintas funciones fisiológicas. Cuando se inyecta en un paciente hemofílico, el factor VIII se une con el factor von Willebrand circulante del paciente. El factor VIII activado actúa como cofactor del factor IX activado, acelerando la conversión del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Seguidamente, la trombina convierte el fibrinógeno en fibrina y se puede formar un coágulo. La hemofilia A es una alteración hereditaria de la coagulación de la sangre vinculada al sexo, debida a niveles reducidos de factor VIII:C y da como resultado hemorragias profusas en articulaciones, músculos u órganos internos, ya sea de forma espontánea, o tras un accidente o un traumatismo quirúrgico. El tratamiento de sustitución aumenta los niveles plasmáticos de factor VIII, lo cual permite corregir temporalmente la deficiencia del factor VIII y la tendencia al sangrado.
Se ha evaluado la inmunogenicidad de Nuwiq en ensayos clínicos en 135 pacientes con hemofilia A grave previamente tratados (74 pacientes adultos y 61 pacientes pediátricos). Ninguno de los pacientes desarrollaron inhibidores.
En un estudio clínico en 32 pacientes adultos con hemofilia A grave, la mediana de consumo de Nuwiq para profilaxis fue de 468,7 UI/kg/mes. La mediana de la dosis para tratar episodios hemorrágicos intercurrentes fue de 33,0 UI/kg en aquellos pacientes en profilaxis. En otro estudio clínico, se trató a demanda a 22 pacientes adultos. En total, se trataron 986 episodios hemorrágicos con una mediana de dosis de 30,9 UI/kg. En general, los sangrados menores requirieron una dosis ligeramente menor, mientras que los sangrados más graves requirieron hasta tres veces la mediana de dosis.
Población pediátrica
Se han obtenido los datos de 29 niños previamente tratados con edades entre 2 y 5 años, 31 niños entre 6 y 12 años y un adolescente de 14 años. La mediana de la dosis por inyección profiláctica fue de 37,8 UI/kg. Veinte pacientes utilizaron una mediana de dosis mayor de 45 UI/kg. La mediana del consumo de Nuwiq para profilaxis por mes fue de 521,9 UI/kg. Se requirió una mediana de dosis de Nuwiq más alta para tratar las hemorragias en niños (43,9 Ul/kg) que en adultos (33,0 Ul/kg), y una mediana de dosis más alta para tratar las hemorragias de moderadas a importantes que para las menores (78,2 UI/kg frente a 41,7 UI/kg). Los niños de menor edad requirieron en general medianas de dosis más altas (de 6 a 12 años: 43,9 UI/kg; de 2 a 5 años: 52,6 UI/kg).
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Nuwiq en uno o más grupos de la población pediátrica en tratamiento de la Hemofilia A (deficiencia congénita del Factor VIII) (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas (PK)
Tabla 2. Parámetros PK para Nuwiq (dosis: 50 UI/kg) en pacientes adultos previamente tratados (de 18 a 65 años de edad) con hemofilia A grave (n = 20)_
|
Parámetro PK |
Ensayo cromogénico | |
|
Valor medio ± SD |
Mediana (rango) | |
|
AUC (h*UI/ml) |
22,6 ± 8,0 |
22,3 (8,4 - 38,1) |
|
Tm (h) |
14,7 ± 10,4 |
12,5 (5,4 -55,6) |
|
IVR (%/UI/kg) |
2,5 ± 0,4 |
2,5 (1,7 -3,2) |
|
Cl (ml/h/kg) |
3,0 ± 1,2 |
2,7 (1,5-6,4) |
AUC = Área bajo la curva (FVIII:C), Tm = Semivida terminal,
IVR = Recuperación incremental in vivo , Cl = Aclaramiento, SD = Desviación estándar
Tabla 3. Parámetros PK para Nuwiq (dosis: 50 UI/kg) en niños previamente tratados de entre 6 y 12 años de edad con hemofilia A grave (n = 12)_
|
Parámetro PK |
Ensayo cromogénico | |
|
Valor medio ± SD |
Mediana (rango) | |
|
AUC (h*UI/ml) |
13,2 ± 3,4 |
12,8 (7,8 - 19,1) |
|
Tm (h) |
10,0 ± 1,9 |
9,9 (7,6 -14,1) |
|
IVR (%/UI/kg) |
1,9 ± 0,4 |
1,9 (1,2 -2,6) |
|
Cl (ml/h/kg) |
4,3 ± 1,2 |
4,2 (2,8 - 6,9) |
AUC = Área bajo la curva (FVIII:C), Tm = Semivida terminal,
IVR = Recuperación incremental in vivo , Cl = Aclaramiento, SD = Desviación estándar
Tabla 4. Parámetros PK para Nuwiq (dosis: 50 UI/kg) en niños previamente tratados de entre 2 y 5 años de edad con hemofilia A grave (n = 13)_
|
Parámetro PK |
Ensayo cromogénico | |
|
Valor medio ± SD |
Mediana (rango) | |
|
AUC (h*UI/ml) |
11,7 ± 5,3 |
10,5 (4,9 -23,8) |
|
Tm (h) |
9,5 ± 3,3 |
8,2 (4,3 - 17,3) |
|
IVR (%/UI/kg) |
1,9 ± 0,3 |
1,8 (1,5 -2,4) |
|
Cl (ml/h/kg) |
5,4 ± 2,4 |
5,1 ( 2,3 -10,9) |
AUC = Área bajo la curva (FVIII:C), Tm = Semivida terminal,
IVR = Recuperación incremental in vivo , Cl = Aclaramiento, SD = Desviación estándar Población pediátrica
Según lo extraído de publicaciones médicas, la recuperación y semivida fueron menores en los niños más pequeños que en los adultos y el aclaramiento más alto, lo que se puede deber en parte al mayor volumen de plasma conocido por kilogramo de peso corporal en los pacientes más jóvenes.
Subgrupos de peso ajustado
Tabla 5. Parámetros PK de peso ajustado para Nuwiq (dosis: 50 Ul/kg) en pacientes adultos
|
previamente tratados (de 18 a 65 años ( |
e edad) con hemofilia A grave (n = 20) | |||
|
Parámetro PK |
Todos (n=20) |
Peso normal (n=14) |
Pre-adiposo (n=4) |
Adiposo (n=2) |
|
Ensayo cromogénico valor medio ± SD | ||||
|
AUC (h*UI/ml) |
22,6 ± 8,0 |
20,4 ± 6,9 |
24,9 ± 8,9 |
33,5 ± 6,5 |
|
T1/2 (h) |
14,7 ± 10,4 |
14,7 ± 12,1 |
13,4 ± 5,9 |
17,2 ± 4,8 |
|
IVR (%/UI/kg) |
2,5 ± 0,4 |
2,4 ± 0,4 |
2,7 ± 0,4 |
2,8 ± 0,3 |
|
Cl (ml/h/kg) |
3,0 ± 1,2 |
3,2 ± 1,3 |
2,6 ± 1,0 |
1,8 ± 0,4 |
|
Mediana ensayo cromogénico (rango) | ||||
|
AUC (h*UI/ml) |
22,3 (8,4 -38,1) |
21,2 (8,4 -32,6) |
23,3 (17,4 - 35,5) |
33,5 (28,9 -38,1) |
|
T1/2 (h) |
12,5 (5,4 -55,6) |
12,3 (5,4 -55,6) |
11,2 (9,3 -22,0) |
17,2 (13,8 -20,6) |
|
IVR (%/UI/kg) |
2,5 (1,7 -3,2) |
2,4 (1,7 -3,1) |
2,8 (2,3 - 3,2) |
2,8 (2,6 -3,0) |
|
Cl (ml/h/kg) |
2,7 (1,5 -6,4) |
2,8 (1,7 -6,4) |
2,5 (1,6 -3,7) |
1,8 (1,5 -2,0) |
Peso normal: IMC 18,5-25 kg/m2, pre-adiposo: IMC 25-30 kg/m2, adiposo: IMC > 30 kg/m2,
SD = Desviación estándar
5.3 Datos preclínicos sobre seguridad
En los estudios preclínicos, Nuwiq se utilizó para restablecer la hemostasia de manera segura y eficaz en perros con hemofilia. Los estudios toxicológicos demostraron que los animales de laboratorio (ratas y monos cynomolgus) toleran bien la administración vía intravenosa local y la exposición sistémica.
No se realizaron estudios específicos con Nuwiq con administración repetida a largo plazo como los de toxicidad para la reproducción, toxicidad crónica y carcinogenia debido a la respuesta inmune a proteínas heterólogas en todas las especies mamíferas no humanas.
No se realizaron estudios sobre el potencial mutagénico de Nuwiq.
Las evaluaciones ex vivo utilizando un kit comercial de ensayo para cuantificar la respuesta de la célula T a las proteínas terapéuticas, ponen de manifiesto un bajo riesgo de inmunogenicidad.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo:
Sacarosa Cloruro de sodio Cloruro de calcio dihidratado Clorhidrato de arginina Citrato de sodio dihidratado Poloxamer 188
Disolvente:
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
Solo deben utilizarse los kits de inyección suministrados, ya que el tratamiento puede fallar a consecuencia de la adsorción del factor VIII humano de coagulación a las superficies internas de otros productos de inyección.
6.3 Periodo de validez
2 años
Durante el periodo de validez, el medicamento se puede almacenar a temperatura ambiente (hasta 25°C) durante un único periodo no superior a 1 mes. Una vez que el medicamento se saca de la nevera, no se debe volver a refrigerar. Registrar la fecha de inicio del almacenamiento a temperatura ambiente en el embalaje del producto. Conservar el vial en el embalaje exterior para protegerlo de la luz.
Tras la reconstitución, se ha comprobado la estabilidad química y física del producto conservado a temperatura ambiente durante 24 horas.
Desde un punto de vista microbiológico, el producto se debe utilizar inmediatamente después de la reconstitución. Si no se usa de forma inmediata, los tiempos y condiciones de conservación previas a su uso son responsabilidad del usuario.
Mantener la solución reconstituida a temperatura ambiente. No refrigerar una vez reconstituida.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior de cartón para protegerlo de la luz.
Para consultar la información sobre almacenamiento a temperatura ambiente y condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Cada envase de Nuwiq 1000 UI contiene:
- Polvo: 1000 UI de polvo en vial de vidrio tipo 1 de 8 ml, cerrado con tapón de bromobutilo recubierto y sellado con cápsula de cierre de aluminio flip-off
- Disolvente: 2,5 ml de agua para preparaciones inyectables en una jeringa precargada de vidrio de borosilicato
- 1 adaptador de vial estéril para reconstitución con 1 aguja de mariposa y 2 toallitas con alcohol Tamaño de envase de 1 unidad.
Puede que solamente se comercialicen algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
El polvo se debe reconstituir únicamente con el disolvente incluido (2,5 ml de agua para preparaciones inyectables) utilizando el kit de inyección suministrado. Mover suavemente el vial en círculos hasta que todo el polvo se haya disuelto. Tras la reconstitución, transferir la solución a la jeringa.
Inspeccionar visualmente el medicamento para comprobar si existen partículas o cambio de coloración antes de la administración. El medicamento reconstituido es una solución transparente e incolora libre de partículas extrañas y tiene un pH de entre 6,5 y 7,5. No use soluciones turbias o con sedimentos.
Instrucciones para la preparación y administración
1. Deje que la jeringa de disolvente (agua para preparaciones inyectables) y el polvo alcancen la temperatura ambiente en el vial cerrado. Puede hacerlo sujetándolos con las manos hasta que tengan la misma temperatura que las manos. No caliente de ninguna otra manera el vial y la jeringa precargada. Esta temperatura debe mantenerse durante la reconstitución.
2. Retire la cápsula de cierre de plástico del vial de polvo para dejar al descubierto las partes centrales del tapón de goma. No retire el tapón gris ni la anilla metálica que rodea la parte superior del vial.
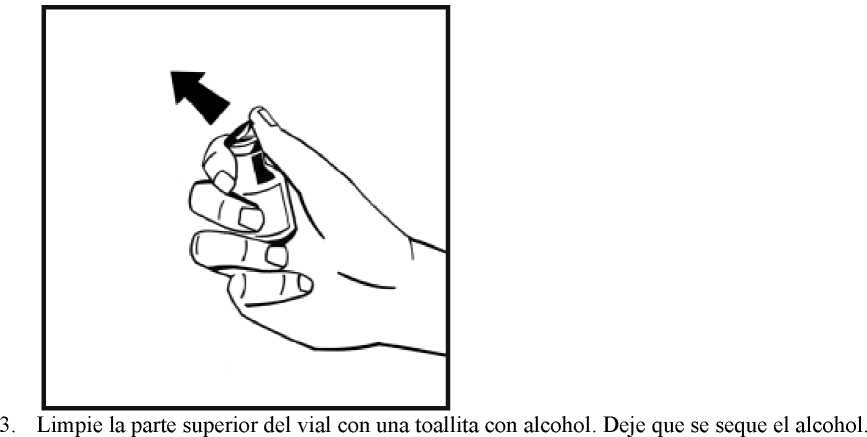
4. Retire la cubierta de papel del envase del adaptador del vial. No saque el adaptador de su envase.
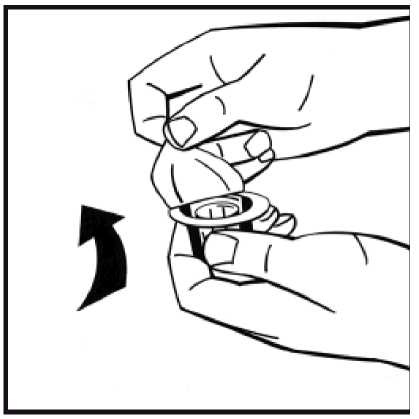
5. Coloque el vial de polvo en una superficie plana y sujételo. Tome el envase del adaptador y coloque el adaptador del vial sobre el centro del tapón de goma del vial de polvo. Presione el envase del adaptador firmemente hacia abajo hasta que la punta del adaptador atraviese el tapón de goma. El adaptador se acoplará al vial cuando esté hecho.
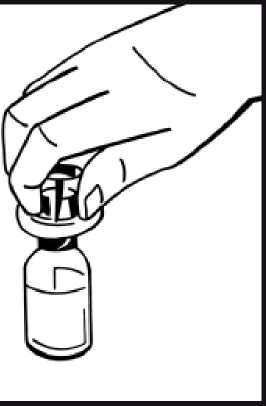
6. Retire la cubierta de papel del envase de la jeringa precargada. Sujete la varilla del émbolo de la jeringa por el extremo y no toque el eje. Fije el extremo con rosca de la varilla del émbolo al émbolo de la jeringa de disolvente. Gire la varilla del émbolo en el sentido de las agujas del reloj hasta que note una ligera resistencia.
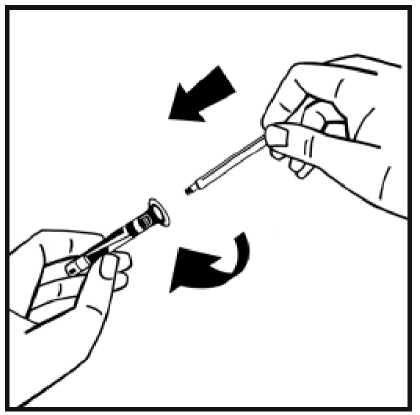
7. Rompa el precinto de la punta de plástico de protección de la jeringa de disolvente partiendo la perforación de la cápsula de cierre. No toque el interior de la cápsula de cierre ni la punta de la jeringa. En caso de no usar la solución inmediatamente, cierre la jeringa llena con la punta
de protección de plástico para almacenarla.
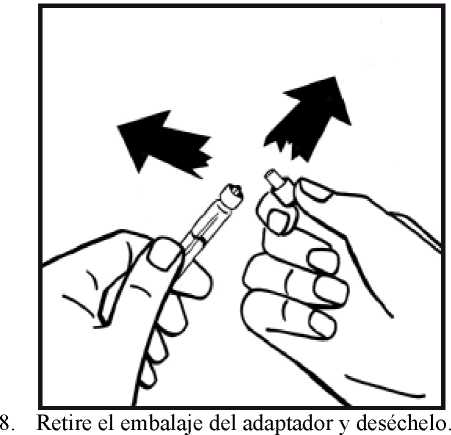
9. Acople firmemente la jeringa de disolvente al adaptador del vial girando en el sentido de las
agujas del reloj hasta que note una ligera resistencia.
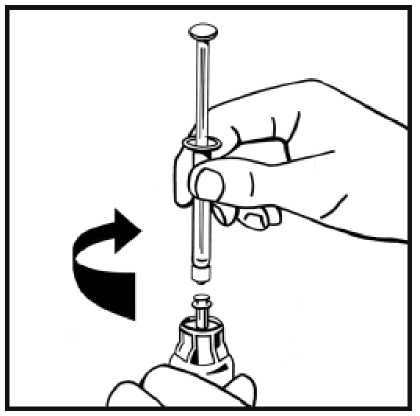
hacia abajo.
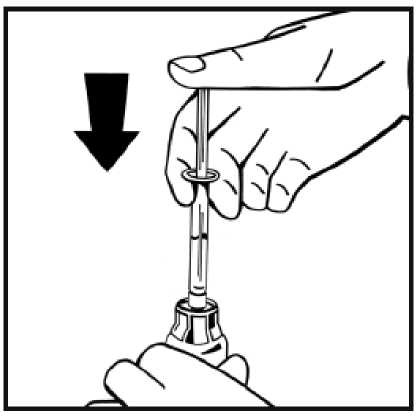
11. Sin retirar la jeringa, mueva suavemente o en círculos el vial unas cuantas veces para disolver el polvo. No agitar. Espere hasta que todo el polvo se disuelva completamente.
12. Fíjese en si la solución final tiene partículas antes de administrarla. La solución debe ser transparente e incolora, prácticamente libre de partículas visibles. No use soluciones turbias o con sedimentos.
13. Dé la vuelta al vial acoplado a la jeringa, y lentamente extraiga la solución a la jeringa. Asegúrese de transferir todo el contenido del vial a la jeringa.
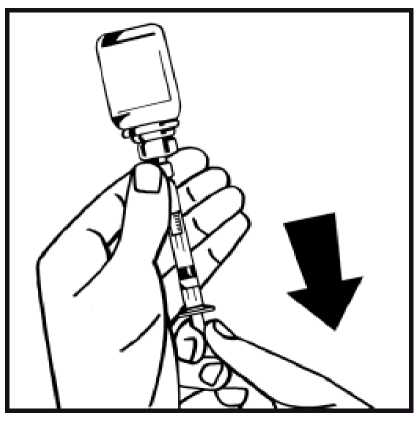
14. Separe la jeringa llena del adaptador del vial girando en sentido contrario a las agujas del reloj y deseche el vial vacío.
15. La solución estará preparada para su uso inmediato. No refrigerar.
16. Limpie la parte elegida para la inyección con una de las toallitas con alcohol suministradas.
17. Acople el kit de inyección suministrado a la jeringa.
Introduzca la aguja del kit de inyección en la vena elegida. Si ha utilizado un torniquete para hacer la vena más visible, deberá estar aflojado antes de empezar a inyectar la solución.
No deberá entrar sangre en la jeringa debido al riesgo de formación de coágulos de fibrina.
18. Inyecte la solución en la vena despacio, no más rápido de 4 ml por minuto.
Si utiliza más de un vial de polvo para un tratamiento, podrá usar la misma aguja de nuevo. El adaptador del vial y la jeringa son de un solo uso.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Octapharma AB Lars Forssells gata 23 112 75 Estocolmo Suecia
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/936/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 22 de julio de 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Nuwiq 2000 UI polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 2000 UI de Factor VIII de coagulación humano (rDNA), simoctocog alfa.
Nuwiq contiene aproximadamente 800 UI/ml de Factor VIII de coagulación humano (rDNA), simoctocog alfa tras la reconstitución.
La potencia (UI) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de Nuwiq es de aproximadamente 9500 UI/mg de proteína.
El simoctocog alfa (factor VIII de coagulación humano (rDNA)) es una proteína purificada que contiene 1440 aminoácidos. La secuencia de aminoácidos es comparable a la forma de 90 + 80 kDa del factor VIII humano plasmático (esto es, con el dominio B suprimido). Nuwiq se ha producido a partir de tecnología de ADN recombinante de células embrionarias de riñón humano (HEK) modificadas genéticamente 293F. No se ha añadido ningún material derivado de seres humanos o animales durante el proceso de fabricación ni al medicamento final.
Excipiente(s) con efecto conocido:
7,35 mg de sodio por ml de solución reconstituida (18,4 mg de sodio por vial).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: polvo friable de color entre blanco y blanquecino.
Disolvente: agua para preparaciones inyectables, líquido transparente e incoloro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de hemorragias en pacientes con hemofilia A (deficiencia congénita del factor VIII).
Nuwiq se puede usar en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico experimentado en el tratamiento de la hemofilia.
Pacientes no tratados anteriormente
La seguridad y eficacia de Nuwiq en pacientes no tratados previamente no se ha establecido todavía. Posología
La dosis y duración del tratamiento de sustitución dependen de la gravedad de la deficiencia del factor VIII, así como de la localización y alcance de las hemorragias y del estado clínico del paciente.
El número de unidades administradas del factor VIII se expresa en Unidades Internacionales (UI) las cuales están relacionadas con el estándar actual de la OMS para medicamentos con factor VIII. La actividad del factor VIII en plasma se expresa como un porcentaje (respecto al plasma humano normal) o en Unidades Internacionales (respecto a un Estándar Internacional para factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII equivale a la cantidad de factor VIII en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis requerida de factor VIII se basa en el dato empírico de que 1 Unidad Internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad del factor VIII del plasma en aproximadamente un 2% de la actividad normal o 2 UI/dl. La dosis necesaria se determina utilizando la siguiente fórmula:
I. Unidades requeridas (UI) = peso corporal (kg) x aumento deseado de factor VIII (%)(UI/dl) x 0,5 (UI/kg por UI/dl)
II. Aumento previsto del factor VIII (% de lo normal) = 2 x UI administradas
peso corporal (kg)
La dosis a administrar y la frecuencia de administración debe estar siempre orientada a la eficacia clínica en cada caso individual.
En el caso de los episodios hemorrágicos siguientes, la actividad del factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % de lo normal o UI/dl) en el periodo correspondiente. La siguiente tabla se puede utilizar como guía de dosificación en cirugía y en episodios hemorrágicos.
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (%) (UI/dL) |
Frecuencia de las dosis (horas) / duración del tratamiento (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente, hemorragia muscular u oral |
20-40 |
Repetir cada 12 a 24 horas. Al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma |
30-60 |
Repetir la perfusión cada 12 a 24 horas, entre 3 y 4 días o más, hasta que cese el dolor y la discapacidad aguda. |
|
Hemorragia potencialmente mortal |
60-100 |
Repetir la perfusión cada 8 a 24 horas hasta que se supere el peligro |
|
Cirugía | ||
|
Cirugía menor Incluyendo extracción dental |
30-60 |
Cada 24 horas, al menos 1 día, hasta lograr la curación. |
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (%) (UI/dL) |
Frecuencia de las dosis (horas) / duración del tratamiento (días) |
|
Cirugía mayor |
80-100 (pre y postoperatorio) |
Repetir la perfusión cada 8-24 horas hasta que se consiga una cicatrización adecuada de la herida, y después al menos durante otros 7 días de tratamiento para mantener una actividad de factor VIII del 30% al 60% (UI/dL). |
Profilaxis
En la profilaxis a largo plazo de episodios hemorrágicos en pacientes con hemofilia A grave, las dosis habituales son de 20 a 40 UI de factor VIII por kg de peso corporal, en intervalos de 2 a 3 días. En algunos casos, especialmente en los pacientes más jóvenes, puede ser necesario un intervalo de dosis más corto o dosis mayores.
Durante el transcurso del tratamiento, se aconseja la determinación adecuada de los niveles de factor VIII como guía de la dosis a administrar y la frecuencia de repetición de las inyecciones. En el caso de intervenciones, cirugía mayor en particular, es indispensable la monitorización precisa del tratamiento de sustitución mediante el análisis de la coagulación (actividad del factor VIII del plasma). La respuesta al factor VIII puede variar en cada paciente, revelándose unos valores diferentes de semivida y recuperación.
Población pediátrica
La posología es la misma en adultos y niños. Sin embargo, hay que tener en cuenta que para los niños pueden ser necesarios intervalos de dosis más cortos o dosis mayores. Los datos actualmente disponibles se describen en las secciones 4.8, 5.1 y 5.2.
No se dispone de datos en niños menores de 2 años de edad.
Forma de administración
Vía intravenosa.
Se recomienda no administrar más de 4 ml por minuto.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo Hipersensibilidad
Como con cualquier producto proteínico vía intravenosa, es posible que se den reacciones de hipersensibilidad de tipo alérgico. Nuwiq contiene trazas de proteínas de células huésped humanas distintas al factor VIII. Si aparecen síntomas de hipersensibilidad, se debe aconsejar a los pacientes que interrumpan inmediatamente el uso del medicamento y contacten con su médico. Los pacientes deben ser informados acerca de los signos iniciales de las reacciones de hipersensibilidad, que incluyen sarpullido, urticaria generalizada, opresión en el pecho, sibilancias, hipotensión y anafilaxia.
En caso de shock, se debe aplicar el tratamiento médico habitual para el shock. Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) del factor VIII es una complicación conocida en el tratamiento de individuos con hemofilia A. Estos inhibidores son normalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en unidades Bethesda (UB) por ml de plasma usando el ensayo modificado. El riesgo de desarrollar inhibidores se relaciona con la exposición al factor VIII, siendo este riesgo más alto en los 20 primeros días de exposición. Rara vez se pueden desarrollar inhibidores transcurridos los primeros 100 días de exposición.
Se han observado casos de inhibidor recurrente (de bajo título) tras cambiar de un producto de factor VIII a otro en pacientes previamente tratados con un periodo de exposición de más de 100 días y con antecedentes de desarrollo de inhibidores. Por ello, se recomienda realizar un seguimiento meticuloso del desarrollo de inhibidores en todos los pacientes tras cualquier cambio de producto.
En general, todos los pacientes tratados con productos del factor de coagulación VIII deben ser vigilados cuidadosamente, de cara al desarrollo de inhibidores, mediante las observaciones clínicas y pruebas de laboratorio adecuadas. Si no se alcanzan los niveles plasmáticos de actividad del factor VIII esperados o si no se controla la hemorragia mediante una dosis adecuada, se debe realizar una prueba de presencia de inhibidor del factor VIII. En pacientes con altos niveles de inhibidor, es posible que el tratamiento con factor VIII no resulte eficaz y se deban considerar otras opciones terapéuticas, como la inducción de inmunotolerancia (ITI). El tratamiento de esos pacientes debe ser dirigido por médicos con experiencia en el tratamiento de la hemofilia y de los inhibidores del factor VIII.
Complicaciones asociadas a los catéteres
Si se requiere un dispositivo de acceso venoso central (CVAD (por sus siglas en inglés)), hay que tener en cuenta el riesgo de complicaciones asociadas al CVAD, incluidas las infecciones localizadas, la bacteremia y la trombosis en el lugar de implantación del catéter.
Se recomienda encarecidamente que cada vez que se administre Nuwiq a un paciente, se registre el nombre y número de lote del producto para mantener un vínculo entre el paciente y el lote del medicamento.
Población pediátrica
Las advertencias y precauciones enumeradas se refieren tanto a adultos como a niños.
Consideraciones relativas al excipiente (contenido de sodio)
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por vial.
Sin embargo, dependiendo del peso corporal y la posología, podría administrársele al paciente más de un vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones con Nuwiq.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción animal con Nuwiq.
Debido a la escasa frecuencia de la hemofilia A en mujeres, no se tiene experiencia sobre el uso del factor VIII durante el embarazo o periodo de lactancia. Por lo tanto, Nuwiq solo se debe usar durante el embarazo y la lactancia si está claramente indicado. No hay datos disponibles de fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Nuwiq no influye sobre la capacidad para conducir y utilizar máquinas.
4.8 Reacciones adversas Resumen del perfil de seguridad
Se han observado con poca frecuencia reacciones alérgicas o de hipersensibilidad con preparados de FVIII (que pueden incluir angioedema, quemazón y punzadas en el lugar de la inyección, escalofríos, rubefacción, urticaria generalizada, cefalea, sarpullido, hipotensión, letargia, náuseas, inquietud, taquicardia, opresión en el pecho, cosquilleo, vómitos, sibilancias) y en algunos casos pueden empeorar hasta convertirse en anafilaxia grave, (incluido el shock anafiláctico).
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) del factor VIII. Si aparecen dichos inhibidores, puede manifestarse en forma de una respuesta clínica insuficiente. En estos casos, se recomienda ponerse en contacto con un centro especializado en hemofilia.
Tabla de reacciones adversas
Durante los estudios clínicos con Nuwiq en pacientes pediátricos previamente tratados (2 a 11 años, n = 58), adolescentes (12 a 17 años, n = 3) y adultos (n = 74) con hemofilia A grave, se notificaron un total de 8 reacciones adversas a medicamentos (RAM) (6 en adultos, 2 en niños) en 5 pacientes (3 adultos, 2 niños).
La Tabla 1 que se presenta a continuación sigue la Clasificación de Órganos del y Sistemas MedDRA (COS y nivel de términos preferentes).
Las frecuencias se han evaluado conforme a la convención siguiente: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
En cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 1. Frecuencia de las reacciones adversas a medicamentos (RAM) en ensayos clínicos en 135 pacientes con hemofilia A grave previamente tratados__
|
Clasificación estándar de órganos del sistema MedDRA |
Reacciones adversas |
Frecuencia* |
|
Trastornos del sistema nervioso |
Parestesia Dolor de cabeza |
Poco frecuentes |
|
Trastornos del oído y del laberinto |
Vértigo |
Poco frecuentes |
|
Trastornos gastrointestinales |
Sequedad de boca |
Poco frecuentes |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor de espalda |
Poco frecuentes |
|
Trastornos generales y alteraciones en el lugar de administración |
Inflamación en la zona de inyección Dolor en la zona de inyección |
Poco frecuentes |
|
Exploraciones complementarias |
Positivo por anticuerpos no neutralizantes anti-factor VIII |
Poco frecuentes |
* Todas estas RAM solo se produjeron una vez. Como el número total de pacientes analizados es de 135, la frecuencia no puede ser menor que "poco frecuente" si se produce una RAM.
Descripción de las reacciones adversas seleccionadas
Se detectó un anticuerpo no neutralizante anti-Factor VIII en un paciente adulto (ver Tabla 1). Se analizó la muestra en el laboratorio central en ocho diluciones. El resultado fue positivo solo en la dilución factor 1 y el título de anticuerpos fue muy bajo. No se detectó en este paciente ninguna actividad inhibidora, según las mediciones del ensayo de Bethesda modificado. La eficacia clínica y la recuperación in vivo de Nuwiq no se vieron afectadas en este paciente.
Población pediátrica
Se asume que la frecuencia, tipo y gravedad de las reacciones adversas son las mismas en niños que en adultos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se ha notificado ningún caso de sobredosis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos, factor VIII de la coagulación sanguínea, código ATC: B02BD02.
El complejo factor VIII/factor von Willebrand consiste en dos moléculas (factor VIII y factor de von Willebrand) con distintas funciones fisiológicas. Cuando se inyecta en un paciente hemofílico, el factor VIII se une con el factor von Willebrand circulante del paciente. El factor VIII activado actúa como cofactor del factor IX activado, acelerando la conversión del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Seguidamente, la trombina convierte el fibrinógeno en fibrina y se puede formar un coágulo. La hemofilia A es una alteración hereditaria de la coagulación de la sangre vinculada al sexo, debida a niveles reducidos de factor VIII:C y da como resultado hemorragias profusas en articulaciones, músculos u órganos internos, ya sea de forma espontánea, o tras un accidente o un traumatismo quirúrgico. El tratamiento de sustitución aumenta los niveles plasmáticos de factor VIII, lo cual permite corregir temporalmente la deficiencia del factor VIII y la tendencia al sangrado.
Se ha evaluado la inmunogenicidad de Nuwiq en ensayos clínicos en 135 pacientes con hemofilia A grave previamente tratados (74 pacientes adultos y 61 pacientes pediátricos). Ninguno de los pacientes desarrollaron inhibidores.
En un estudio clínico en 32 pacientes adultos con hemofilia A grave, la mediana de consumo de Nuwiq para profilaxis fue de 468,7 UI/kg/mes. La mediana de la dosis para tratar episodios hemorrágicos intercurrentes fue de 33,0 UI/kg en aquellos pacientes en profilaxis. En otro estudio clínico, se trató a demanda a 22 pacientes adultos. En total, se trataron 986 episodios hemorrágicos con una mediana de dosis de 30,9 UI/kg. En general, los sangrados menores requirieron una dosis ligeramente menor, mientras que los sangrados más graves requirieron hasta tres veces la mediana de dosis.
Población pediátrica
Se han obtenido los datos de 29 niños previamente tratados con edades entre 2 y 5 años, 31 niños entre 6 y 12 años y un adolescente de 14 años. La mediana de la dosis por inyección profiláctica fue de 37,8 UI/kg. Veinte pacientes utilizaron una mediana de dosis mayor de 45 UI/kg. La mediana del consumo de Nuwiq para profilaxis por mes fue de 521,9 UI/kg. Se requirió una mediana de dosis de Nuwiq más alta para tratar las hemorragias en niños (43,9 Ul/kg) que en adultos (33,0 Ul/kg), y una mediana de dosis más alta para tratar las hemorragias de moderadas a importantes que para las menores (78,2 UI/kg frente a 41,7 UI/kg). Los niños de menor edad requirieron en general medianas de dosis más altas (de 6 a 12 años: 43,9 UI/kg; de 2 a 5 años: 52,6 UI/kg).
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Nuwiq en uno o más grupos de la población pediátrica en tratamiento de la Hemofilia A (deficiencia congénita del Factor VIII) (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas (PK)
Tabla 2. Parámetros PK para Nuwiq (dosis: 50 UI/kg) en pacientes adultos previamente tratados (de 18 a 65 años de edad) con hemofilia A grave (n = 20)_
|
Parámetro PK |
Ensayo cromogénico | |
|
Valor medio ± SD |
Mediana (rango) | |
|
AUC (h*UI/ml) |
22,6 ± 8,0 |
22,3 (8,4 - 38,1) |
|
Tm (h) |
14,7 ± 10,4 |
12,5 (5,4 -55,6) |
|
IVR (%/UI/kg) |
2,5 ± 0,4 |
2,5 (1,7 -3,2) |
|
Cl (ml/h/kg) |
3,0 ± 1,2 |
2,7 (1,5-6,4) |
AUC = Área bajo la curva (FVIII:C), Tm = Semivida terminal,
IVR = Recuperación incremental in vivo , Cl = Aclaramiento, SD = Desviación estándar
Tabla 3. Parámetros PK para Nuwiq (dosis: 50 UI/kg) en niños previamente tratados de entre 6 y 12 años de edad con hemofilia A grave (n = 12)_
|
Parámetro PK |
Ensayo cromogénico | |
|
Valor medio ± SD |
Mediana (rango) | |
|
AUC (h*UI/ml) |
13,2 ± 3,4 |
12,8 (7,8 - 19,1) |
|
Tm (h) |
10,0 ± 1,9 |
9,9 (7,6 -14,1) |
|
IVR (%/UI/kg) |
1,9 ± 0,4 |
1,9 (1,2 -2,6) |
|
Cl (ml/h/kg) |
4,3 ± 1,2 |
4,2 (2,8 - 6,9) |
AUC = Área bajo la curva (FVIII:C), Tm = Semivida terminal,
IVR = Recuperación incremental in vivo , Cl = Aclaramiento, SD = Desviación estándar
Tabla 4. Parámetros PK para Nuwiq (dosis: 50 UI/kg) en niños previamente tratados de entre 2 y 5 años de edad con hemofilia A grave (n = 13)_
|
Parámetro PK |
Ensayo cromogénico | |
|
Valor medio ± SD |
Mediana (rango) | |
|
AUC (h*UI/ml) |
11,7 ± 5,3 |
10,5 (4,9 -23,8) |
|
Tm (h) |
9,5 ± 3,3 |
8,2 (4,3 - 17,3) |
|
IVR (%/UI/kg) |
1,9 ± 0,3 |
1,8 (1,5 -2,4) |
|
Cl (ml/h/kg) |
5,4 ± 2,4 |
5,1 ( 2,3 -10,9) |
AUC = Área bajo la curva (FVIII:C), Tm = Semivida terminal,
IVR = Recuperación incremental in vivo , Cl = Aclaramiento, SD = Desviación estándar Población pediátrica
Según lo extraído de publicaciones médicas, la recuperación y semivida fueron menores en los niños más pequeños que en los adultos y el aclaramiento más alto, lo que se puede deber en parte al mayor volumen de plasma conocido por kilogramo de peso corporal en los pacientes más jóvenes.
Subgrupos de peso ajustado
Tabla 5. Parámetros PK de peso ajustado para Nuwiq (dosis: 50 Ul/kg) en pacientes adultos
|
previamente tratados (de 18 a 65 años ( |
e edad) con hemofilia A grave (n = 20) | |||
|
Parámetro PK |
Todos (n=20) |
Peso normal (n=14) |
Pre-adiposo (n=4) |
Adiposo (n=2) |
|
Ensayo cromogénico valor medio ± SD | ||||
|
AUC (h*UI/ml) |
22,6 ± 8,0 |
20,4 ± 6,9 |
24,9 ± 8,9 |
33,5 ± 6,5 |
|
T1/2 (h) |
14,7 ± 10,4 |
14,7 ± 12,1 |
13,4 ± 5,9 |
17,2 ± 4,8 |
|
IVR (%/UI/kg) |
2,5 ± 0,4 |
2,4 ± 0,4 |
2,7 ± 0,4 |
2,8 ± 0,3 |
|
Cl (ml/h/kg) |
3,0 ± 1,2 |
3,2 ± 1,3 |
2,6 ± 1,0 |
1,8 ± 0,4 |
|
Mediana ensayo cromogénico (rango) | ||||
|
AUC (h*UI/ml) |
22,3 (8,4 -38,1) |
21,2 (8,4 -32,6) |
23,3 (17,4 - 35,5) |
33,5 (28,9 -38,1) |
|
T1/2 (h) |
12,5 (5,4 -55,6) |
12,3 (5,4 -55,6) |
11,2 (9,3 -22,0) |
17,2 (13,8 -20,6) |
|
IVR (%/UI/kg) |
2,5 (1,7 -3,2) |
2,4 (1,7 -3,1) |
2,8 (2,3 - 3,2) |
2,8 (2,6 -3,0) |
|
Cl (ml/h/kg) |
2,7 (1,5 -6,4) |
2,8 (1,7 -6,4) |
2,5 (1,6 -3,7) |
1,8 (1,5 -2,0) |
Peso normal: IMC 18,5-25 kg/m2, pre-adiposo: IMC 25-30 kg/m2, adiposo: IMC > 30 kg/m2,
SD = Desviación estándar
5.3 Datos preclínicos sobre seguridad
En los estudios preclínicos, Nuwiq se utilizó para restablecer la hemostasia de manera segura y eficaz en perros con hemofilia. Los estudios toxicológicos demostraron que los animales de laboratorio (ratas y monos cynomolgus) toleran bien la administración vía intravenosa local y la exposición sistémica.
No se realizaron estudios específicos con Nuwiq con administración repetida a largo plazo como los de toxicidad para la reproducción, toxicidad crónica y carcinogenia debido a la respuesta inmune a proteínas heterólogas en todas las especies mamíferas no humanas.
No se realizaron estudios sobre el potencial mutagénico de Nuwiq.
Las evaluaciones ex vivo utilizando un kit comercial de ensayo para cuantificar la respuesta de la célula T a las proteínas terapéuticas, ponen de manifiesto un bajo riesgo de inmunogenicidad.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo:
Sacarosa Cloruro de sodio Cloruro de calcio dihidratado Clorhidrato de arginina Citrato de sodio dihidratado Poloxamer 188
Disolvente:
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
Solo deben utilizarse los kits de inyección suministrados, ya que el tratamiento puede fallar a consecuencia de la adsorción del factor VIII humano de coagulación a las superficies internas de otros productos de inyección.
6.3 Periodo de validez
2 años
Durante el periodo de validez, el medicamento se puede almacenar a temperatura ambiente (hasta 25°C) durante un único periodo no superior a 1 mes. Una vez que el medicamento se saca de la nevera, no se debe volver a refrigerar. Registrar la fecha de inicio del almacenamiento a temperatura ambiente en el embalaje del producto. Conservar el vial en el embalaje exterior para protegerlo de la luz.
Tras la reconstitución, se ha comprobado la estabilidad química y física del producto conservado a temperatura ambiente durante 24 horas.
Desde un punto de vista microbiológico, el producto se debe utilizar inmediatamente después de la reconstitución. Si no se usa de forma inmediata, los tiempos y condiciones de conservación previas a su uso son responsabilidad del usuario.
Mantener la solución reconstituida a temperatura ambiente. No refrigerar una vez reconstituida.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior de cartón para protegerlo de la luz.
Para consultar la información sobre almacenamiento a temperatura ambiente y condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Cada envase de Nuwiq 2000 UI contiene:
- Polvo: 2000 UI de polvo en vial de vidrio tipo 1 de 8 ml, cerrado con tapón de bromobutilo recubierto y sellado con cápsula de cierre de aluminio flip-off
- Disolvente: 2,5 ml de agua para preparaciones inyectables en una jeringa precargada de vidrio de borosilicato
- 1 adaptador de vial estéril para reconstitución con 1 aguja de mariposa y 2 toallitas con alcohol Tamaño de envase de 1 unidad.
Puede que solamente se comercialicen algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
El polvo se debe reconstituir únicamente con el disolvente incluido (2,5 ml de agua para preparaciones inyectables) utilizando el kit de inyección suministrado. Mover suavemente el vial en círculos hasta que todo el polvo se haya disuelto. Tras la reconstitución, transferir la solución a la jeringa.
Inspeccionar visualmente el medicamento para comprobar si existen partículas o cambio de coloración antes de la administración. El medicamento reconstituido es una solución transparente e incolora libre de partículas extrañas y tiene un pH de entre 6,5 y 7,5. No use soluciones turbias o con sedimentos.
Instrucciones para la preparación y administración
1. Deje que la jeringa de disolvente (agua para preparaciones inyectables) y el polvo alcancen la temperatura ambiente en el vial cerrado. Puede hacerlo sujetándolos con las manos hasta que tengan la misma temperatura que las manos. No caliente de ninguna otra manera el vial y la jeringa precargada. Esta temperatura debe mantenerse durante la reconstitución.
2. Retire la cápsula de cierre de plástico del vial de polvo para dejar al descubierto las partes centrales del tapón de goma. No retire el tapón gris ni la anilla metálica que rodea la parte superior del vial.
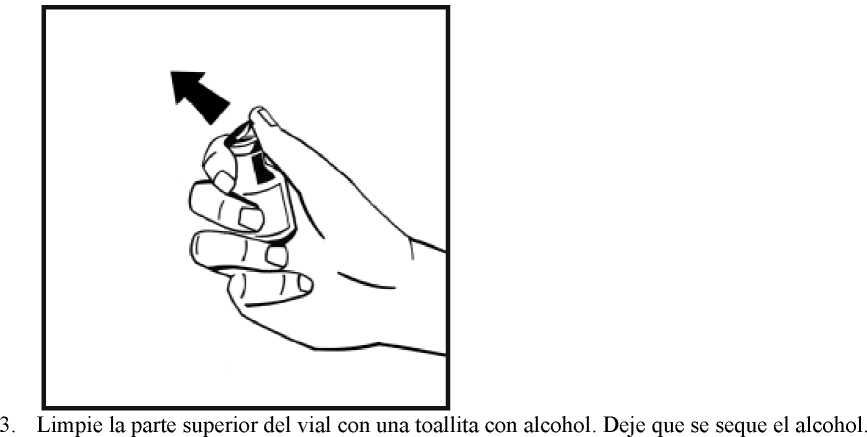
4. Retire la cubierta de papel del envase del adaptador del vial. No saque el adaptador de su envase.
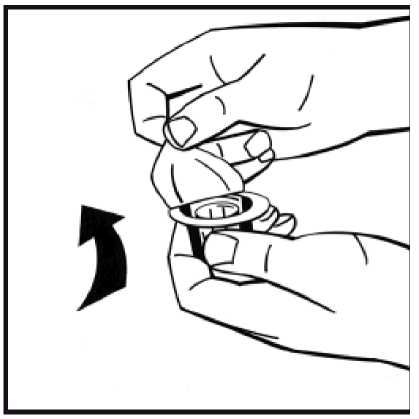
5. Coloque el vial de polvo en una superficie plana y sujételo. Tome el envase del adaptador y coloque el adaptador del vial sobre el centro del tapón de goma del vial de polvo. Presione el envase del adaptador firmemente hacia abajo hasta que la punta del adaptador atraviese el tapón de goma. El adaptador se acoplará al vial cuando esté hecho.
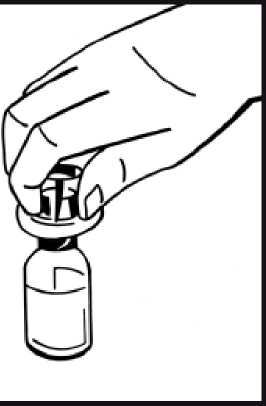
6. Retire la cubierta de papel del envase de la jeringa precargada. Sujete la varilla del émbolo de la jeringa por el extremo y no toque el eje. Fije el extremo con rosca de la varilla del émbolo al émbolo de la jeringa de disolvente. Gire la varilla del émbolo en el sentido de las agujas del reloj hasta que note una ligera resistencia.
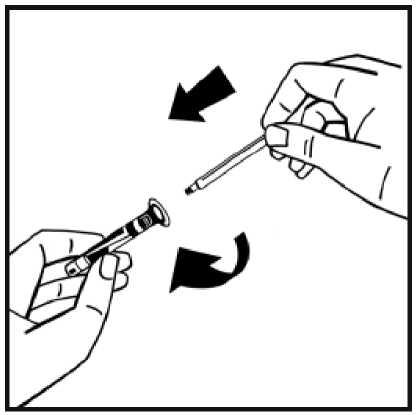
7. Rompa el precinto de la punta de plástico de protección de la jeringa de disolvente partiendo la perforación de la cápsula de cierre. No toque el interior de la cápsula de cierre ni la punta de la jeringa. En caso de no usar la solución inmediatamente, cierre la jeringa llena con la punta
de protección de plástico para almacenarla.
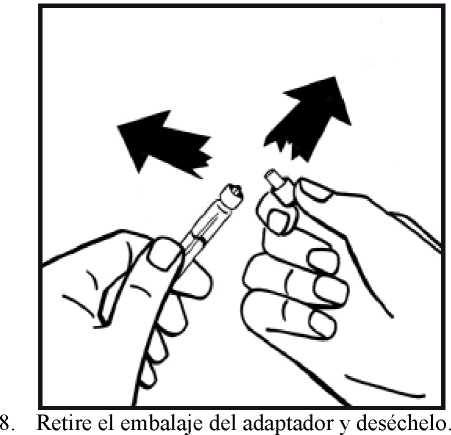
9. Acople firmemente la jeringa de disolvente al adaptador del vial girando en el sentido de las
agujas del reloj hasta que note una ligera resistencia.
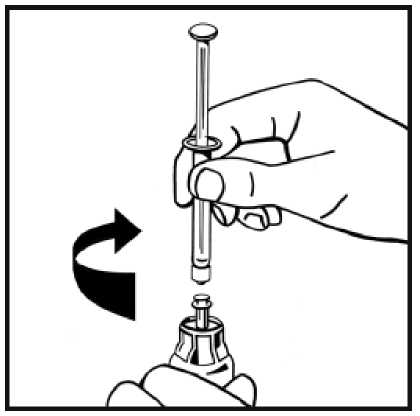
hacia abajo.
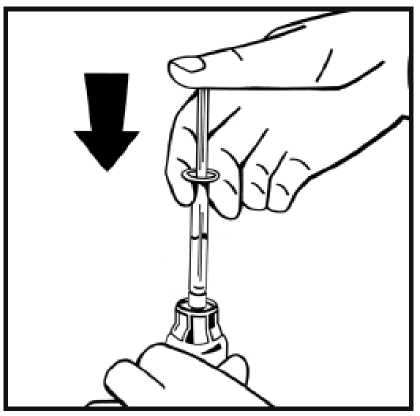
11. Sin retirar la jeringa, mueva suavemente o en círculos el vial unas cuantas veces para disolver el polvo. No agitar. Espere hasta que todo el polvo se disuelva completamente.
12. Fíjese en si la solución final tiene partículas antes de administrarla. La solución debe ser transparente e incolora, prácticamente libre de partículas visibles. No use soluciones turbias o con sedimentos.
13. Dé la vuelta al vial acoplado a la jeringa, y lentamente extraiga la solución a la jeringa. Asegúrese de transferir todo el contenido del vial a la jeringa.
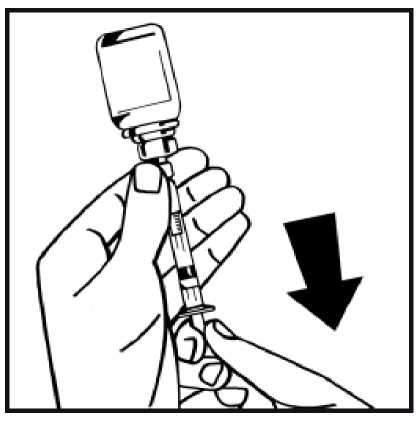
14. Separe la jeringa llena del adaptador del vial girando en sentido contrario a las agujas del reloj y deseche el vial vacío.
15. La solución estará preparada para su uso inmediato. No refrigerar.
16. Limpie la parte elegida para la inyección con una de las toallitas con alcohol suministradas.
17. Acople el kit de inyección suministrado a la jeringa.
Introduzca la aguja del kit de inyección en la vena elegida. Si ha utilizado un torniquete para hacer la vena más visible, deberá estar aflojado antes de empezar a inyectar la solución.
No deberá entrar sangre en la jeringa debido al riesgo de formación de coágulos de fibrina.
18. Inyecte la solución en la vena despacio, no más rápido de 4 ml por minuto.
Si utiliza más de un vial de polvo para un tratamiento, podrá usar la misma aguja de nuevo. El adaptador del vial y la jeringa son de un solo uso.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Octapharma AB Lars Forssells gata 23 112 75 Estocolmo Suecia
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/936/004
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 22 de julio de 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTE(S) DEL (DE LOS) PRINCIPIO(S) ACTIVO(S) BIOLÓGICO(S) Y FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE(S) DEL (DE LOS) PRINCIPIO(S) ACTIVO(S) BIOLÓGICO(S) Y FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del (de los) fabricante(s) del (de los) principio(s) activo(s) biológico(s).
Octapharma AB Lars Forssells gata 23 112 75 Estocolmo Suecia
Nombre y dirección del (de los) fabricante(s) responsable(s) de la liberación de los lotes
Octapharma AB Lars Forssells gata 23 112 75 Estocolmo Suecia
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará el primer informe periódico de seguridad para este medicamento en un plazo de 6 meses después de la autorización. Posteriormente, el titular de la autorización de comercialización presentará informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 ter, (párrafo 7), de la Directiva 2001/83/CE y publicados en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva
información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
Nuwiq 250 Unidades Internacionales
Polvo y disolvente para solución inyectable
simoctocog alfa (factor VIII de coagulación humano recombinante)
2. PRINCIPIO(S) ACTIVO(S)
250 Unidades Internacionales/vial simoctocog alfa (100 UI/ml tras la reconstitución)
3. LISTA DE EXCIPIENTES
Excipientes: Sacarosa, cloruro de sodio, cloruro de calcio dihidratado, clorhidrato de arginina, citrato de sodio dihidratado, poloxamer 188 Disolvente: Agua para preparaciones inyectables Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable
1 vial de polvo, 1 jeringa precargada, 1 adaptador de vial, 1 aguja de mariposa, 2 toallitas con alcohol 1 vial de polvo contiene 250 Unidades Internacionales simoctocog alfa.
1 jeringa precargada contiene 2,5 ml de agua para preparaciones inyectables.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía intravenosa tras la reconstitución.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Conservar en nevera. No congelar. Conservar en el embalaje original para protegerlo de la luz.
Se puede conservar a temperatura ambiente (hasta 25 °C) durante un único periodo no superior a 1 mes.
Fecha de salida de la nevera:
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Octapharma AB Lars Forssells gata 23 112 75 Estocolmo Suecia
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/936/001
13. NÚMERO DE LOTE
Lot
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Nuwiq 250
VIAL CON POLVO PARA SOLUCIÓN INYECTABLE
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Nuwiq 250 Unidades Internacionales, polvo para solución inyectable, simoctocog alfa (factor VIII de coagulación humano recombinante). Vía intravenosa tras la reconstitución.
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
250 UI/vial
6. OTROS
Logotipo de Octapharma
1. NOMBRE DEL MEDICAMENTO
Nuwiq 500 Unidades Internacionales
Polvo y disolvente para solución inyectable
simoctocog alfa (factor VIII de coagulación humano recombinante)
2. PRINCIPIO(S) ACTIVO(S)
500 Unidades Internacionales/vial simoctocog alfa (200 UI/ml tras la reconstitución)
3. LISTA DE EXCIPIENTES
Excipientes: Sacarosa, cloruro de sodio, cloruro de calcio dihidratado, clorhidrato de arginina, citrato de sodio dihidratado, poloxamer 188 Disolvente: Agua para preparaciones inyectables Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable
1 vial de polvo, 1 jeringa precargada, 1 adaptador de vial, 1 aguja de mariposa, 2 toallitas con alcohol 1 vial de polvo contiene 500 Unidades Internacionales simoctocog alfa.
1 jeringa precargada contiene 2,5 ml de agua para preparaciones inyectables.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía intravenosa tras la reconstitución.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Conservar en nevera. No congelar. Conservar en el embalaje original para protegerlo de la luz.
Se puede conservar a temperatura ambiente (hasta 25 °C) durante un único periodo no superior a 1 mes.
Fecha de salida de la nevera:
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Octapharma AB Lars Forssells gata 23 112 75 Estocolmo Suecia
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/936/002
13. NÚMERO DE LOTE
Lot
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Nuwiq 500
VIAL CON POLVO PARA SOLUCIÓN INYECTABLE
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Nuwiq 500 Unidades Internacionales, polvo para solución inyectable, simoctocog alfa (factor VIII de coagulación humano recombinante). Vía intravenosa tras la reconstitución.
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
500 UI/vial
6. OTROS
Logotipo de Octapharma
1. NOMBRE DEL MEDICAMENTO
Nuwiq 1000 Unidades Internacionales
Polvo y disolvente para solución inyectable
simoctocog alfa (factor VIII de coagulación humano recombinante)
2. PRINCIPIO(S) ACTIVO(S)
1000 Unidades Internacionales/vial simoctocog alfa (400 UI/ml tras la reconstitución)
3. LISTA DE EXCIPIENTES
Excipientes: Sacarosa, cloruro de sodio, cloruro de calcio dihidratado, clorhidrato de arginina, citrato de sodio dihidratado, poloxamer 188 Disolvente: Agua para preparaciones inyectables Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable
1 vial de polvo, 1 jeringa precargada, 1 adaptador de vial, 1 aguja de mariposa, 2 toallitas con alcohol 1 vial de polvo contiene 1000 Unidades Internacionales simoctocog alfa.
1 jeringa precargada contiene 2,5 ml de agua para preparaciones inyectables.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía intravenosa tras la reconstitución.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Conservar en nevera. No congelar. Conservar en el embalaje original para protegerlo de la luz.
Se puede conservar a temperatura ambiente (hasta 25 °C) durante un único periodo no superior a 1 mes.
Fecha de salida de la nevera:
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Octapharma AB Lars Forssells gata 23 112 75 Estocolmo Suecia
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/936/003
13. NÚMERO DE LOTE
Lot
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Nuwiq 1000
VIAL CON POLVO PARA SOLUCIÓN INYECTABLE
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Nuwiq 1000 Unidades Internacionales, polvo para solución inyectable, simoctocog alfa (factor VIII de coagulación humano recombinante). Vía intravenosa tras la reconstitución.
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
1000 UI/vial
6. OTROS
Logotipo de Octapharma
1. NOMBRE DEL MEDICAMENTO
Nuwiq 2000 Unidades Internacionales
Polvo y disolvente para solución inyectable
simoctocog alfa (factor VIII de coagulación humano recombinante)
2. PRINCIPIO(S) ACTIVO(S)
2000 Unidades Internacionales/vial simoctocog alfa (800 UI/ml tras la reconstitución)
3. LISTA DE EXCIPIENTES
Excipientes: Sacarosa, cloruro de sodio, cloruro de calcio dihidratado, clorhidrato de arginina, citrato de sodio dihidratado, poloxamer 188 Disolvente: Agua para preparaciones inyectables Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable
1 vial de polvo, 1 jeringa precargada, 1 adaptador de vial, 1 aguja de mariposa, 2 toallitas con alcohol 1 vial de polvo contiene 2000 Unidades Internacionales simoctocog alfa.
1 jeringa precargada contiene 2,5 ml de agua para preparaciones inyectables.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía intravenosa tras la reconstitución.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Conservar en nevera. No congelar. Conservar en el embalaje original para protegerlo de la luz.
Se puede conservar a temperatura ambiente (hasta 25 °C) durante un único periodo no superior a 1 mes.
Fecha de salida de la nevera:
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Octapharma AB Lars Forssells gata 23 112 75 Estocolmo Suecia
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/936/004
13. NÚMERO DE LOTE
Lot
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Nuwiq 2000
VIAL CON POLVO PARA SOLUCIÓN INYECTABLE
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Nuwiq 2000 Unidades Internacionales, polvo para solución inyectable, simoctocog alfa (factor VIII de coagulación humano recombinante). Vía intravenosa tras la reconstitución.
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
2000 UI/vial
6. OTROS
Logotipo de Octapharma
JERINGA PRECARGADA CON 2,5 ML DE AGUA PARA INYECTABLES
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Disolvente para Nuwiq
Agua para preparaciones inyectables
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
2,5 ml
6. OTROS
B. PROSPECTO
Prospecto: información para el usuario
Nuwiq 250 UI polvo y disolvente para solución inyectable Nuwiq 500 UI polvo y disolvente para solución inyectable Nuwiq 1000 UI polvo y disolvente para solución inyectable Nuwiq 2000 UI polvo y disolvente para solución inyectable
Simoctocog alfa (factor VIII de coagulación humano recombinante)
V Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico o enfermero, incluso si se tratara de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Nuwiq y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Nuwiq
3. Cómo usar Nuwiq
4. Posibles efectos secundarios
5. Conservación de Nuwiq
6. Contenido del envase e información adicional
1. Qué es Nuwiq y para qué se utiliza
Nuwiq contiene el principio activo factor VIII de coagulación humano recombinante (simoctocog alfa). El factor VIII es necesario para que la sangre forme coágulos y detenga la hemorragia. En pacientes con hemofilia A (deficiencia congénita del factor VIII), el factor VIII falta o no actúa correctamente.
Nuwiq reemplaza el factor VIII que falta y se utiliza para el tratamiento y la prevención de hemorragias en pacientes con hemofilia A y se puede usar en todos los grupos de edad.
2. Qué necesita saber antes de empezar a usar Nuwiq No use Nuwiq:
• si es alérgico al principio activo simoctocog alfa o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Si no está seguro de ello, pregunte a su médico.
Advertencias y precauciones
Consulte a su médico o farmacéutico o enfermero antes de empezar a usar Nuwiq.
Existe una rara posibilidad de que experimente una reacción anafiláctica (una súbita reacción alérgica grave) a Nuwiq. Debe poder reconocer los síntomas tempranos de las reacciones alérgicas, que se incluyen en la sección 4 "Reacciones alérgicas".
Si se produce cualquiera de estos síntomas, detenga la inyección de inmediato y póngase en contacto con su médico.
Si Nuwiq no logra controlar sus hemorragias, informe inmediatamente a su médico. Es posible que haya desarrollado ‘inhibidores del factor VIII’ y puede que su médico quiera realizar algunas pruebas para confirmarlo. Los inhibidores del factor VIII son anticuerpos que se encuentran en la sangre y que bloquean el factor VIII que está usando. Esto hace que el factor VIII resulte menos efectivo en el control de las hemorragias. Debe informar a su médico si le han tratado previamente con medicamentos con factor VIII, especialmente si desarrolló inhibidores, puesto que podría existir un riesgo mayor de que volviera a ocurrir.
Complicaciones asociadas a los catéteres
Si usted requiere un dispositivo de acceso venoso central (CVAD), hay que tener en cuenta el riesgo de complicaciones asociadas al CVAD, incluidas las infecciones localizadas, la presencia de bacterias en la sangre y la trombosis en el lugar de implantación del catéter.
Se recomienda encarecidamente que cada vez que se administre Nuwiq, se registre el nombre y número de lote del producto para mantener un vínculo entre usted y el lote del medicamento.
Uso de Nuwiq con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento.
Conducción y uso de máquinas
Nuwiq no influye sobre la capacidad para conducir y utilizar máquinas.
Nuwiq contiene sodio
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por vial.
Sin embargo, dependiendo de su peso corporal y de la posología, se le podría administrar más de un vial, lo que se debe tener en cuenta si sigue usted una dieta pobre en sodio.
3. Cómo usar Nuwiq
El tratamiento con Nuwiq será iniciado por un médico con experiencia en el cuidado de pacientes con hemofilia A. Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o enfermero. En caso de duda pregunte a su médico o enfermero.
Nuwiq se inyecta normalmente en una vena (intravenosamente) por su médico o un enfermero con experiencia en el cuidado de pacientes con hemofilia A. Usted mismo u otra persona también podrían inyectarle Nuwiq pero solo después de haber recibido la formación adecuada.
Su médico calculará su dosis de Nuwiq (en Unidades Internacionales = UI) dependiendo de su estado y peso corporal y de si se está utilizando como prevención o como tratamiento de las hemorragias. La frecuencia con que necesitará una inyección dependerá de lo bien que actúe Nuwiq en usted. Normalmente, el tratamiento de la hemofilia A es un tratamiento de por vida.
Prevención de las hemorragias
La dosis habitual de Nuwiq es de 20 a 40 UI por kg de peso corporal, suministradas cada 2 a 3 días. Sin embargo, en algunos casos, especialmente en los pacientes más jóvenes, podrían ser necesarias inyecciones más frecuentes o dosis más altas.
Tratamiento de las hemorragias
La dosis de Nuwiq se calcula dependiendo de su peso corporal y de los niveles de factor VIII que se deben alcanzar. Los niveles objetivos de factor VIII dependerán de la gravedad y localización de las hemorragias.
Si tiene la impresión de que el efecto de Nuwiq es insuficiente, consulte a su médico. Su médico le hará los análisis de laboratorio pertinentes para asegurarse de que tenga los niveles adecuados de factor VIII. Esto es de especial importancia si le van a someter a una cirugía mayor.
Pacientes que desarrollan inhibidores del factor VIII
Si su factor VIII plasmático no consigue alcanzar los niveles esperados con Nuwiq, o si no se consiguen controlar las hemorragias adecuadamente, se podría deber al desarrollo de inhibidores del factor VIII. Su médico lo comprobará. Puede que necesite una dosis más alta de Nuwiq o un producto diferente para controlar las hemorragias. No aumente la dosis total de Nuwiq para controlar sus hemorragias sin consultar a su médico.
Uso en niños
La forma en la que se utiliza Nuwiq en niños no difiere de la forma en la que se utiliza en los adultos. Puesto que es posible tener que administrar los medicamentos de factor VIII con más frecuencia en los niños, puede que sea necesario acoplar un dispositivo de acceso venoso central (CVAD, un conector externo que permite el acceso al flujo sanguíneo mediante un catéter sin inyección a través de la piel).
Si usa más Nuwiq del que debe
No se ha notificado ningún síntoma de sobredosis. Si ha inyectado más Nuwiq del que debe, informe a su médico.
Si olvidó usar Nuwiq
No tome una dosis doble para compensar una dosis olvidada. Proceda a administrar la siguiente dosis inmediatamente y prosiga con las recomendaciones de su médico o farmacéutico.
Si interrumpe el tratamiento con Nuwiq
No interrumpa el tratamiento con Nuwiq sin consultar a su médico o farmacéutico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Reacciones alérgicas
Debe poder reconocer los síntomas tempranos de las reacciones alérgicas. Si se producen graves reacciones alérgicas (anafilácticas) súbitas (muy raras, hasta 1 de cada 10.000), deberá detenerse la inyección inmediatamente. Contacte inmediatamente con su médico si nota alguno de los siguientes síntomas:
- erupción, sarpullido, ronchas, picor generalizado,
- hinchazón de labios y lengua,
- dificultad para respirar, sibilancias, opresión en el pecho,
- sensación de malestar general,
- mareo y pérdida de la consciencia.
Estos síntomas pueden ser síntomas tempranos de un shock anafiláctico. Si se produce cualquiera de estos síntomas, detenga la inyección de inmediato y póngase en contacto con su médico. Los síntomas graves requieren un tratamiento de urgencia inmediato.
Efectos adversos poco frecuentes que pueden afectar hasta a 1 de cada 100 pacientes
Cosquilleo o entumecimiento (parestesia), dolor de cabeza, inflamación y/o dolor en la zona de la inyección, dolor de espalda, vértigo, sequedad de boca.
Efectos adversos relacionados con los dispositivos de acceso venoso central (CVAD):
infección relacionada con el catéter, infección general (sistémica) y coágulo de sangre local en la zona
de inserción del catéter.
Comunicación de efectos adversos
Si experimenta efectos adversos, consulte a su médico o farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Nuwiq
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta después de abreviatura EXP. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2°C y 8°C). No congelar. Conservar el vial en el embalaje exterior de cartón para protegerlo de la luz.
Antes de que Nuwiq polvo se reconstituya, se puede conservar a temperatura ambiente (hasta 25°C) durante un único periodo no superior a 1 mes. Anote la fecha en la que empiece a conservar Nuwiq a temperatura ambiente en el embalaje del medicamento. No almacene de nuevo Nuwiq en la nevera, tras haber sido conservado a temperatura ambiente.
Usar la solución reconstituida inmediatamente después de la reconstitución.
Advertencias con respecto a ciertos signos visibles de deterioro
No utilice este medicamento si observa indicios visibles de deterioro del precinto del envase, especialmente de la jeringa y/o del vial.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de Nuwiq
Polvo:
- El principio activo es Factor VIII de coagulación humano recombinante (simoctocog alfa).
Cada vial de polvo contiene 250, 500, 1000 o 2000 UI de simoctocog alfa.
- Los demás componentes son sacarosa, cloruro de sodio, cloruro de calcio dihidratado, clorhidrato de arginina, citrato de sodio dihidratado y poloxamer 188
Disolvente:
Agua para preparaciones inyectables Aspecto de Nuwiq y contenido del envase
Nuwiq se suministra como polvo y disolvente para solución inyectable y es un polvo friable de color entre blanco y blanquecino en vial de vidrio. El disolvente es agua para preparaciones inyectables y se suministra en una jeringa de vidrio precargada.
Una vez reconstituida, la solución es transparente, incolora y libre de partículas extrañas.
Cada envase de Nuwiq contiene:
- 1 vial de polvo con 250, 500, 1000 o 2000 UI de simoctocog alfa
- 1 jeringa precargada con 2,5 ml de agua para inyectables
- 1 adaptador de vial
- 1 aguja de mariposa
- 2 toallitas con alcohol
Titular de la autorización de comercialización y responsable de la fabricación
Octapharma AB, Lars Forssells gata 23, 112 75 Estocolmo, Suecia
Para cualquier información acerca de este medicamento, contacte con el representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Octapharma Benelux (Belgium) Tél/Tel: +32 2 3730890 |
Ireland Octapharma Limited (UK) Tel: +44 161 8373770 |
Norge Octapharma AS Tlf: +47 63988860 |
|
Etarapna Octapharma Nordic AB (Sweden) Tea.: +46 8 56643000 |
Ísland Octapharma AS (Norway) Sími: +47 63988860 |
Osterreich Octapharma Handelsgesellschaft m.b.H Tel: +43 1 610321222 |
|
Ceská republika Octapharma CZ s.r.o. Tel: +420 266 793 510 |
Italia Kedrion S.p.A. Tel: +39 0583 1969603 |
Polska Octapharma Poland Sp. z o.o. Tel: +48 22 2082734 |
|
Danmark Octapharma Nordic AB (Sweden) Tlf: +46 8 56643000 |
Kúnpoq Octapharma Nordic AB (Sweden) Tn^: +46 8 56643000 |
Portugal Octapharma Produtos Farmacéuticos Tel: +351 21 8160820 |
|
Deutschland Octapharma GmbH Tel: +49 2173 9170 |
Latvija Octapharma Nordic AB (Sweden) Tel: +46 8 56643000 |
Romania Octapharma Nordic AB (Sweden) Tel: +46 8 56643000 |
|
Eesti Octapharma Nordic AB (Sweden) Tel: +46 8 56643000 |
Lietuva Octapharma Nordic AB (Sweden) Tel: +46 8 56643000 |
Slovenija Octapharma Nordic AB (Sweden) Tel: +46 8 56643000 |
|
EXXáSa Octapharma Hellas SA Tn^: +30 210 8986500 |
Luxembourg/Luxemburg Octapharma Benelux (Belgium) Tél/Tel: +32 2 3730890 |
Slovenská republika Octapharma AG, o.z.z.o. Tel: +421 2 54646701 |
|
España Octapharma S.A. Tel: +34 91 6487298 |
Magyarország Octapharma Nordic AB (Sweden) Tel: +46 8 56643000 |
Suomi/Finland Octapharma Nordic AB Puh/Tel: +358 9 85202710 |
|
France Octapharma France Tél: +33 1 41318000 |
Malta Octapharma Nordic AB (Sweden) |
Sverige Octapharma Nordic AB Tel: +46 8 56643000 |
Tel: +46 8 56643000
Fecha de la última revisión de este prospecto
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Esta información está destinada únicamente a profesionales del sector sanitario:
Tratamiento a demanda
La dosis a administrar y la frecuencia de administración debe siempre estar orientada a la eficacia clínica en cada caso individual.
En el caso de los episodios hemorrágicos siguientes, la actividad del factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % de lo normal o UI/dl) en el periodo correspondiente. La siguiente tabla se puede utilizar como guía de dosificación en cirugía y en episodios hemorrágicos.
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (%) (UI/dL) |
Frecuencia de las dosis (horas) / duración del tratamiento (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente, hemorragia muscular u oral |
20-40 |
Repetir cada 12 a 24 horas. Al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma |
30-60 |
Repetir la perfusión cada 12 a 24 horas, entre 3 y 4 días o más, hasta que cese el dolor y la discapacidad aguda. |
|
Hemorragia potencialmente mortal |
60-100 |
Repetir la perfusión cada 8 a 24 horas hasta que se supere el peligro |
|
Cirugía | ||
|
Cirugía menor Incluyendo extracción dental |
30-60 |
Cada 24 horas, al menos 1 día, hasta lograr la curación. |
|
Cirugía mayor |
80-100 (pre y postoperatorio) |
Repetir la perfusión cada 8-24 horas hasta que se consiga una cicatrización adecuada de la herida, y después al menos durante otros 7 días de tratamiento para mantener una actividad de factor VIII del 30% al 60% (UI/dL). |
INSTRUCCIONES PARA LA PREPARACIÓN Y ADMINISTRACIÓN
1. Deje que la jeringa de disolvente (agua para preparaciones inyectables) y el polvo alcancen la temperatura ambiente en el vial cerrado. Puede hacerlo sujetándolos con las manos hasta que tengan la misma temperatura que las manos. No caliente de ninguna otra manera el vial y la jeringa precargada. Esta temperatura debe mantenerse durante la reconstitución.
envase.

2. Retire la cápsula de cierre de plástico del vial de polvo para dejar al descubierto las partes centrales del tapón de goma. No retire el tapón gris ni la anilla metálica que rodea la parte superior del vial.

5. Coloque el vial de polvo en una superficie plana y sujételo. Tome el envase del adaptador y coloque el adaptador del vial sobre el centro del tapón de goma del vial de polvo. Presione el envase del adaptador firmemente hacia abajo hasta que la punta del adaptador atraviese el tapón de goma. El adaptador se acoplará al vial cuando esté hecho.

6. Retire la cubierta de papel del envase de la jeringa precargada. Sujete la varilla del émbolo de la jeringa por el extremo y no toque el eje. Fije el extremo con rosca de la varilla del émbolo al émbolo de la jeringa de disolvente. Gire la varilla del émbolo en el sentido de las agujas del reloj hasta que note una ligera resistencia.

7. Rompa el precinto de la punta de plástico de protección de la jeringa de disolvente partiendo la perforación de la cápsula de cierre. No toque el interior de la cápsula de cierre ni la punta de la jeringa. En caso de no usar la solución inmediatamente, cierre la jeringa llena con la punta
de protección de plástico para almacenarla.

9. Acople firmemente la jeringa de disolvente al adaptador del vial girando en el sentido de las
agujas del reloj hasta que note una ligera resistencia.

10. Inyecte lentamente todo el disolvente en el vial de polvo presionando la varilla del émbolo
hacia abajo.

11. Sin retirar la jeringa, mueva suavemente o en círculos el vial unas cuantas veces para disolver el polvo. No agitar. Espere hasta que todo el polvo se disuelva completamente.
12. Fíjese en si la solución final tiene partículas antes de administrarla. La solución debe ser transparente e incolora, prácticamente libre de partículas visibles. No use soluciones turbias o con sedimentos.
13. Dé la vuelta al vial acoplado a la jeringa, y lentamente extraiga la solución a la jeringa. Asegúrese de transferir todo el contenido del vial a la jeringa.

14. Separe la jeringa llena del adaptador del vial girando en sentido contrario a las agujas del reloj y deseche el vial vacío.
15. La solución estará preparada para su uso inmediato. No refrigerar.
16. Limpie la parte elegida para la inyección con una de las toallitas con alcohol suministradas.
17. Acople el kit de inyección suministrado a la jeringa.
Introduzca la aguja del kit de inyección en la vena elegida. Si ha utilizado un torniquete para hacer la vena más visible, deberá estar aflojado antes de empezar a inyectar la solución.
No deberá entrar sangre en la jeringa debido al riesgo de formación de coágulos de fibrina.
18. Inyecte la solución en la vena despacio, no más rápido de 4 ml por minuto.
Si utiliza más de un vial de polvo para un tratamiento, podrá usar la misma aguja de nuevo. El adaptador del vial y la jeringa son de un solo uso.
82