Nplate 500 Microgramos Polvo Y Disolvente Para Solucion Inyectable
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Nplate 250 microgramos polvo para solución inyectable Nplate 500 microgramos polvo para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Nplate 250 microgramos polvo para solución inyectable
Cada vial contiene 250 qg de romiplostim. Tras la reconstitución, un volumen liberado de 0,5 ml de solución contiene 250 qg de romiplostim (500 qg/ml). En cada vial se incluye una cantidad adicional para garantizar que se puedan administrar 250 qg de romiplostim.
Nplate 500 microgramos polvo para solución inyectable
Cada vial contiene 500 qg de romiplostim. Tras la reconstitución, un volumen liberado de 1 ml de solución contiene 500 qg de romiplostim (500 qg/ml). En cada vial se incluye una cantidad adicional para garantizar que se puedan administrar 500 qg de romiplostim.
Romiplostim se produce mediante tecnología del ADN recombinante en Escherichia coli (E. coli). Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo para solución inyectable (polvo para inyectable) El polvo es blanco.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Nplate está indicado para pacientes adultos con Púrpura Trombocitopénica Inmune (Idiopática) (PTI) crónica que son refractarios a otros tratamientos (por ejemplo, corticosteroides, inmunoglobulinas) (ver las secciones 4.2 y 5.1).
4.2 Posología y forma de administración
El tratamiento debe realizarse bajo la supervisión de un médico con experiencia en el tratamiento de patologías hematológicas.
Posología
Nplate debe administrarse una vez a la semana en forma de inyección subcutánea.
Dosis inicial
Cálculo de la dosis
|
Dosis inicial o posteriores una vez a la semana: |
Peso* en kg x dosis en pg/kg = dosis individual del paciente en pg. |
|
Volumen de administración: |
Dosis en pg x 1 ml 500 pg = cantidad que hay que inyectar en ml |
|
Ejemplo: |
La dosis inicial de un paciente de 75 kg es de 1 pg/kg de romiplostim. Dosis individual del paciente = 75 kg x 1 pg/kg = 75 pg La cantidad correspondiente de solución de Nplate que hay que inyectar = 75 pa x 1 ml = 0,15 ml 500 pg |
|
*Al calcular la dosis de romiplostim debe utilizarse siempre el peso corporal real del paciente en el momento de iniciar el tratamiento. Los futuros ajustes de la dosis se basarán únicamente en los cambios en los recuentos plaquetarios y se realizarán en incrementos de 1 pg/kg (consultar la siguiente tabla). | |
Ajustes de la dosis
Para calcular la dosis debe utilizarse siempre el peso corporal real del paciente en el momento de iniciar el tratamiento. La dosis semanal de romiplostim debe aumentarse en incrementos de 1 pg/kg hasta que el paciente alcance un recuento plaquetario > 50 x 109/l. Hay que evaluar los recuentos plaquetarios semanalmente hasta alcanzar un recuento estable (> 50 x 109/l durante al menos 4 semanas sin ajuste de la dosis). Posteriormente hay que evaluar los recuentos plaquetarios mensualmente. No se debe sobrepasar la dosis máxima semanal de 10 pg/kg.
Ajustar la dosis de la siguiente manera:
|
Recuento de plaquetas (x 109/l) |
Acción |
|
< 50 |
Aumentar la dosis semanal en 1 pg/kg |
|
> 150 durante dos semanas consecutivas |
Disminuir la dosis semanal en 1 pg/kg |
|
> 250 |
No administrar; continuar evaluando el recuento plaquetario semanalmente. Después de que el recuento plaquetario haya descendido a < 150 x 109/l, reiniciar la administración con una dosis semanal reducida en 1 pg/kg. |
Debido a la variabilidad interindividual de la respuesta plaquetaria, en algunos pacientes el recuento plaquetario puede caer bruscamente por debajo de 50 x 109/l tras reducir la dosis o la interrupción del tratamiento. En estos casos, si es clínicamente adecuado, pueden considerarse niveles de corte del recuento plaquetario mas elevados para la reducción de dosis (200 x 109/l) y la interrupción del tratamiento (400 x 109/l) de acuerdo con el criterio médico.
La pérdida de respuesta o la incapacidad de mantener una respuesta plaquetaria con romiplostim dentro del intervalo de dosis recomendado debe motivar la búsqueda de los factores causales (ver sección 4.4, pérdida de respuesta a romiplostim).
Interrupción del tratamiento
Debe interrumpirse la administración de romiplostim si, tras cuatro semanas de tratamiento a la mayor dosis semanal de 10 pg/kg, el recuento plaquetario no aumenta hasta un nivel suficiente que evite hemorragias clínicamente relevantes.
Los pacientes deben ser evaluados clínicamente de forma periódica y el médico que administra el tratamiento debe decidir la continuación del mismo de manera individual, y en pacientes no esplenectomizados, se debe incluir la evaluación relativa a la esplenectomía. Es posible que reaparezca la trombocitopenia tras la interrupción del tratamiento (ver sección 4.4).
Pacientes de edad avanzada (> 65 años)
No se han observado diferencias globales de seguridad o eficacia entre pacientes < 65 y > 65 años de edad (ver sección 5.1). Aunque según estos datos no se requiere un ajuste de la pauta posológica en pacientes de edad avanzada, se aconseja precaución, teniendo en cuenta el reducido número de pacientes de edad avanzada incluidos en los ensayos clínicos realizados hasta la fecha.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de romiplostim en niños menores de 18 años. No se dispone de datos.
Pacientes con insuficiencia hepática
No se recomienda utilizar romiplostim en pacientes con insuficiencia hepática de moderada a grave (escala Child-Pugh > 7) a menos que el beneficio esperado sea mayor que el riesgo identificado de trombosis venosa portal en pacientes con trombocitopenia asociada a insuficiencia hepática tratada con agonistas de la trombopoyetina (TPO) (ver sección 4.4).
Si el uso de romiplostim se considera necesario, el recuento de plaquetas debe vigilarse estrechamente para minimizar el riesgo de complicaciones tromboembólicas.
Pacientes con insuficiencia renal
No se han realizado ensayos clínicos formales en estas poblaciones de pacientes. Nplate debe administrarse con precaución a estos pacientes.
Forma de administración
Vía subcutánea.
Una vez reconstituido el polvo, la solución inyectable de Nplate se administra por vía subcutánea. El volumen de inyección puede ser muy pequeño. Durante la preparación de Nplate, se debe tener cuidado en el cálculo de la dosis y de la reconstitución con el volumen correcto de agua estéril para preparaciones inyectables. Debe prestarse una atención especial para asegurar que el volumen apropiado de Nplate se extrae del vial para la administración subcutánea. Debe emplearse una jeringa con graduaciones de 0,01 ml.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1 o a las proteínas derivadas de E. coli.
4.4 Advertencias y precauciones especiales de empleo
Reaparición de trombocitopenia v hemorragia tras la finalización del tratamiento
Es probable que la trombocitopenia reaparezca tras interrumpir el tratamiento con romiplostim. Si se suspende el tratamiento con romiplostim mientras están siendo administrados medicamentos anticoagulantes o antiplaquetarios, aumenta el riesgo de hemorragias. Los pacientes deben ser sometidos a un control estricto ante un posible descenso del recuento plaquetario v tratados médicamente para evitar las hemorragias tras la suspensión del tratamiento con romiplostim. Se recomienda que, si se interrumpe el tratamiento con romiplostim, se reinicie el tratamiento para la PTI conforme a las recomendaciones de tratamiento actuales. El tratamiento médico adicional puede incluir la interrupción del tratamiento con anticoagulantes, antiplaquetarios v/o ambos, la reversión de la anticoagulación o el tratamiento complementario con plaquetas.
Aumento de la reticulina en la médula ósea
Se cree que el aumento de la reticulina en la médula ósea es el resultado de la estimulación del receptor de la TPO, que conlleva un aumento de la cantidad de megacariocitos en la médula ósea, que en consecuencia liberarán citocinas. Los cambios morfológicos de las células de la sangre periférica pueden sugerir un aumento de la reticulina, que puede detectarse mediante una biopsia de la médula ósea. Por tanto, se recomienda realizar análisis para detectar anomalías morfológicas celulares utilizando un frotis sanguíneo periférico v un hemograma completo antes v durante el tratamiento con romiplostim. Ver la sección 4.8 para obtener información relativa a los aumentos de reticulina observados durante los ensavos clínicos con romiplostim.
Si se observa una pérdida de eficacia v un frotis sanguíneo periférico anormal en los pacientes, deberá interrumpirse la administración de romiplostim, se realizará una exploración física v se valorará la necesidad de realizar una biopsia de médula ósea con la tinción adecuada para detectar la presencia de reticulina. Si se encuentra disponible, deberá compararse con una biopsia de médula ósea anterior. Si se mantiene la eficacia pero se observa en los pacientes un frotis sanguíneo periférico anormal, el médico deberá seguir el criterio clínico adecuado, que incluve valorar la realización de una biopsia de médula ósea v deberá evaluar de nuevo los riesgos v beneficios de romiplostim v las opciones alternativas para el tratamiento de la PTI.
Complicaciones trombóticas/tromboembólicas
Los recuentos plaquetarios por encima del intervalo normal suponen un riesgo de complicaciones trombóticas/tromboembólicas. La incidencia de acontecimientos trombóticos/tromboembólicos observada en los ensavos clínicos fue 6,0% con romiplostim v 3,6% con placebo. Se debe tener precaución cuando se administre romiplostim a pacientes con factores de riesgo conocidos de tromboembolismo incluvendo, pero no limitados a, factores hereditarios (por ej. Factor V Leiden) o factores de riesgo adquiridos (por ej. deficiencia ATIII, síndrome antifosfolipídico), edad avanzada, pacientes con periodos prolongados de inmovilización, neoplasias, anticonceptivos v terapia hormonal sustitutiva, cirugía/traumatismo, obesidad v fumadores.
Se han notificado casos de acontecimientos tromboembólicos (ATEs), incluvendo trombosis venosa portal, en pacientes con enfermedad hepática crónica que reciben romiplostim. Romiplostim debe utilizarse con precaución en estas poblaciones. Deben seguirse las recomendaciones para el ajuste de la dosis (ver sección 4.2).
Errores de medicación
Los errores de medicación, incluvendo sobredosis e infradosis, se han notificado en pacientes que reciben Nplate, se deben seguir las guías de cálculo de dosis v ajuste de dosis (ver sección 4.2).
La sobredosis puede dar lugar a un aumento excesivo de los recuentos plaquetarios asociados con las complicaciones trombóticas/tromboembólicas. Si los recuentos plaquetarios aumentan excesivamente,
discontinuar Nplate y supervisar los recuentos plaquetarios. Reiniciar el tratamiento con Nplate de acuerdo con las recomendaciones de dosificación y administración. Una dosis insuficiente puede dar lugar a unos recuentos plaquetarios menores de lo esperado y posible sangrado. Los recuentos plaquetarios deben controlarse en los pacientes que reciben Nplate (ver secciones 4.2, 4.4 y 4.9).
Progresión de Síndromes Mielodisplásicos (SMD) existentes
El balance beneficio/riesgo para romiplostim se ha establecido favorable sólo para el tratamiento de la trombocitopenia asociada a PTI crónica y no se debe utilizar romiplostim en otras condiciones clínicas asociadas a trombocitopenia.
El diagnóstico de la PTI en adultos y en pacientes de edad avanzada debería haber sido confirmado por la exclusión de otras entidades clínicas que presentan trombocitopenia, en particular, se debe excluir el diagnóstico de SMD. Se debe realizar un aspirado y biopsia de médula ósea en el curso de la enfermedad y tratamiento, sobre todo en pacientes mayores de 60 años de edad, que presenten síntomas sistémicos o signos anormales, tales como las células blásticas periféricas aumentadas.
En estudios clínicos con romiplostim para el tratamiento de pacientes con SMD, se notificaron casos de un incremento transitorio del recuento de células blásticas y casos de progresión de la enfermedad de SMD a LMA. Un estudio aleatorizado, controlado con placebo, en pacientes con SMD tratados con romiplostim se interrumpió prematuramente debido a un exceso en el número de progresiones de la enfermedad a LMA y un aumento en los blastos circulantes de más del 10% en pacientes que recibían romiplostim. De los casos de progresión de la enfermedad de SMD a LMA que se observaron, los pacientes con clasificación RAEB-1 de SMD al inicio fueron más propensos a presentar progresión de la enfermedad a LMA en comparación con los de SMD de riesgo más bajo.
Romiplostim no se debe utilizar para el tratamiento de la trombocitopenia debida a SMD ni a ninguna otra causa de trombocitopenia que no sea la PTI, fuera de ensayos clínicos.
Pérdida de respuesta a romiplostim
Una pérdida de respuesta o la incapacidad de mantener una respuesta plaquetaria con el tratamiento con romiplostim dentro del intervalo de dosis recomendado, debe motivar la búsqueda de los factores causales incluyendo la inmunogenicidad (ver sección 4.8) y el aumento de reticulina en la médula ósea (ver más arriba).
Efectos de romiplostim sobre los glóbulos rojos y blancos
Se han observado alteraciones en parámetros relacionados con los glóbulos rojos (disminución) y blancos (incremento) en ensayos toxicológicos no-clínicos (ratas y monos), así como en pacientes con PTI. Pueden ocurrir anemia concomitante y leucocitosis (dentro de un intervalo de 4 semanas) en pacientes de forma independiente de la esplenectomía. Sin embargo, se han observado con más frecuencia en pacientes que han sido sometidos previamente a una esplenectomía. Debería considerarse la monitorización de dichos parámetros en los pacientes tratados con romiplostim.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones. Se desconocen las interacciones potenciales de romiplostim con medicamentos administrados conjuntamente a consecuencia de la unión a las proteínas plasmáticas.
Los medicamentos empleados en el tratamiento de la PTI en combinación con romiplostim en ensayos clínicos fueron corticosteroides, danazol y/o azatioprina, inmunoglobulina intravenosa (IGIV) e inmunoglobulina anti-D. Cuando se combine romiplostim con otros medicamentos para el tratamiento de la PTI deben controlarse los recuentos plaquetarios a fin de evitar recuentos plaquetarios fuera del intervalo recomendado (ver sección 4.2).
Debe reducirse o interrumpirse la administración de corticosteroides, danazol y azatioprina cuando se administran en combinación con romiplostim (ver sección 5.1). Cuando se reduzcan o interrumpan otros tratamientos para la PTI deben controlarse los recuentos plaquetarios a fin de evitar que se sitúen fuera del intervalo recomendado (ver sección 4.2).
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos o éstos son limitados relativos al uso de romiplostim en mujeres embarazadas.
Los estudios realizados en animales han mostrado que romiplostim traspasa la barrera placentaria y aumenta los recuentos plaquetarios fetales. En estudios con animales, también ocurrieron pérdida posimplementación y un ligero aumento en la mortalidad perinatal de las crías (ver sección 5.3).
Romiplostim está contraindicado durante el embarazo y en mujeres en edad fértil que no utilicen métodos anticonceptivos.
Lactancia
Se desconoce si romiplostim/metabolitos se excretan en la leche materna. No se puede excluir el riesgo en recién nacidos/lactantes. Se debe decidir si es necesario interrumpir la lactancia o interrumpir/abstenerse el tratamiento con romiplostim tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre.
Fertilidad
No hay datos disponibles sobre fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Nplate sobre la capacidad para conducir y utilizar máquinas es moderada. En los ensayos clínicos, algunos pacientes experimentaron episodios de mareos transitorios de leves a moderados.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Basándose en un análisis de todos los pacientes adultos con PTI que recibían romiplostim en 4 ensayos clínicos controlados y 5 no controlados, la incidencia global de todas las reacciones adversas en pacientes tratados con romiplostim fue de 91,5% (248/271). La duración media de exposición a romiplostim en esta población en estudio fue de 50 semanas.
Las reacciones adversas de mayor gravedad que pueden ocurrir durante el tratamiento con Nplate incluyen: recurrencia de trombocitopenia y sangrado después del cese del tratamiento, aumento de la reticulina en la médula ósea, complicaciones trombóticas/tromboembólicas, errores de medicación y progresión de SMD existente a LMA. Las reacciones adversas más frecuentes observadas incluyen reacciones de hipersensibilidad (incluyendo casos de erupción, urticaria y angioedema) y cefalea.
Listado tabulado de acontecimientos adversos
Las frecuencias se definen como: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10) poco frecuentes (> 1/1.000 a < 1/100), raras (> 1/10.000 a < 1/1.000); muy raras (< 1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Dentro de cada intervalo de frecuencia y según el sistema de clasificación de órganos de MedDRA, las reacciones adversas se enumeran en orden decreciente de incidencia.
|
Clasificación de órganos del sistema MedDRA |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Infecciones e infestaciones |
Infección del tracto respiratorio superior |
Gastroenteritis |
Gripe Infección localizada Nasofaringitis |
|
Neoplasias benignas, malignas y no especificadas (incluyendo quistes y pólipos) |
Mieloma múltiple Mielofibrosis | ||
|
Trastornos de la sangre y del sistema linfático |
Trastornos de la médula ósea* T rombocitopenia* Anemia |
Anemia aplásica Insuficiencia de la médula ósea Leucocitosis Esplenomegalia T rombocitanemia Recuento plaquetario aumentado Recuento plaquetario anormal | |
|
Trastornos del sistema inmunológico |
Hipersensibilidad* * |
Angioedema | |
|
Trastornos del metabolismo y de la nutrición |
Intolerancia al alcohol Anorexia Pérdida de apetito Deshidratación Gota | ||
|
Trastornos psiquiátricos |
Insomnio |
Depresión Sueños anormales | |
|
Trastornos del sistema nervioso |
Cefalea |
Mareos Migraña Parestesia |
Clonus Disgeusia Hipoestesia Hipogeusia Neuropatía periférica Trombosis del seno transverso |
|
Trastornos oculares |
Hemorragia conjuntival Alteración de la acomodación visual Ceguera Alteración ocular Prurito ocular Aumento del lagrimeo Papiloedema Alteración visual | ||
|
Trastornos del oído y del laberinto |
Vértigo | ||
|
Trastornos cardiacos |
Palpitaciones |
Infarto de miocardio Frecuencia cardiaca aumentada |
|
Clasificación de órganos del sistema MedDRA |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Trastornos vasculares |
Enrojecimiento |
Trombosis venosa profunda Hipotensión Embolismo periférico Isquemia periférica Flebitis Tromboflebitis superficial Trombosis Eritromelalgia | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Embolia pulmonar1 |
Tos Rinorrea Garganta seca Disnea Congestión nasal Dolor al respirar | |
|
Trastornos gastrointestinales |
Náuseas Diarrea Dolor abdominal Estreñimiento Dispepsia |
Vómitos Hemorragia rectal Mal aliento Disfagia Trastorno del reflujo gastroesofágico Hematoquecia Hemorragia bucal Malestar estomacal Estomatitis Decoloración dental | |
|
Trastornos hepatobiliares |
Trombosis venosa portal Aumento de transaminasas | ||
|
Trastornos de la piel y del tejido subcutáneo |
Prurito Equimosis Erupción |
Alopecia Reacción de fotosensibilidad Acné Dermatitis de contacto Sequedad de piel Eczema Eritema Erupción exfoliativa Crecimiento de pelo anormal Prurigo Púrpura Erupción papular Erupción pruriginosa Nódulos en la piel Olor anormal de la piel Urticaria |
|
Clasificación de órganos del sistema MedDRA |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia Mialgia Espasmo muscular Dolor en las extremidades Dolor en la espalda Dolor de huesos |
Tensión muscular Debilidad muscular Dolor en el hombro Espasmo muscular | |
|
Trastornos renales y urinarios |
Presencia de proteínas en orina | ||
|
Trastornos del aparato reproductor y de la mama |
Hemorragia vaginal | ||
|
Trastornos generales y alteraciones en el lugar de administración |
Fatiga Edema periférico Enfermedad semejante a la gripe Dolor Astenia Pirexia Escalofríos Irritación en el lugar de la inyección |
Hemorragia en el lugar de la inyección Dolor en el pecho Irritabilidad Malestar Edema facial Sensación de calor Sensación de inquietud | |
|
Exploraciones complementarias |
Aumento de presión arterial Aumento de lactato deshidrogenasa en sangre Aumento de la temperatura corporal Pérdida de peso Aumento de peso | ||
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Contusión |
Trombocitopenia después de la interrupción del tratamiento
Basándose en un análisis de todos los pacientes adultos con PTI que recibían romiplostim en 4 ensayos clínicos controlados y 5 no controlados, se notificaron 4 acontecimientos de trombocitopenia tras la interrupción del tratamiento, n = 271 (ver sección 4.4).
Progresión de Síndromes Mielodisplásicos (SMD) existentes
En un ensayo clínico aleatorizado, controlado con placebo, en pacientes con SMD, se interrumpió prematuramente el tratamiento con romiplostim debido a un incremento numérico de casos de progresión de la enfermedad de SMD a LMA y aumento transitorio en el recuento de células blásticas en pacientes tratados con romiplostim comparado con placebo. De los casos de progresión de la enfermedad de SMD a LMA que se observaron, los pacientes con clasificación RAEB-1 de SMD al inicio fueron más propensos a presentar progresión de la enfermedad a LMA (ver sección 4.4). La supervivencia global fue similar al placebo.
Aumento de la reticulina en la médula ósea
En los ensayos clínicos, se interrumpió la administración de romiplostim en 4 de 271 pacientes debido a la aparición de depósitos de reticulina en la médula ósea. En 6 pacientes adicionales, se observó reticulina tras biopsia de la médula ósea (ver sección 4.4).
Inmunogenicidad
En ensayos clínicos, pacientes adultos con PTI presentaron anticuerpos anti romiplostim.
Mientras que el 5,8% y 3,9% de los sujetos fueron positivos al desarrollo de anticuerpos de unión anti romiplostim y TPO respectivamente, sólo 2 sujetos (0,4%) dieron positivo a los anticuerpos neutralizantes anti romiplostim. Sin embargo, estos anticuerpos no generaron una reacción cruzada con la TPO endógena. Cuatro meses después del final de la administración, ambos sujetos dieron negativo a los anticuerpos neutralizantes anti romiplostim. La incidencia de anticuerpos pre-existentes anti romiplostim y TPO fue del 8,0% y 5,4%, respectivamente.
Al igual que ocurre con todas las proteínas terapéuticas, existe la posibilidad de que se origine inmunogenicidad. Si existe sospecha de formación de anticuerpos neutralizantes, ha de ponerse en contacto con el representante local del titular de la Autorización de Comercialización (ver sección 6 del prospecto) para realizar una prueba de anticuerpos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
No se observaron acontecimientos adversos en ratas a las que se administró una dosis única de
1.000 pg/kg o en monos tras la administración repetida de romiplostim a 500 pg/kg (100 ó 50 veces la dosis clínica máxima de 10 pg/kg, respectivamente).
En caso de sobredosis, los recuentos plaquetarios pueden aumentar excesivamente y dar lugar a complicaciones trombóticas/tromboembólicas. Si los recuentos plaquetarios son excesivamente elevados, interrumpir Nplate y monitorizar el recuento plaquetario. Reiniciar el tratamiento con Nplate de acuerdo con las recomendaciones de dosis y administración (ver secciones 4.2 y 4.4).
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antihemorrágicos, otros hemostáticos sistémicos. Código ATC: B02BX04 Mecanismo de acción
Romiplostim es una proteína de fusión Fc-péptido (cuerpo peptídico) que señala y activa las rutas de transcripción intracelular a través del receptor de la TPO (también denominado cMpl) para aumentar la producción de plaquetas. La molécula del cuerpo peptídico está formada por un dominio Fc de la inmunoglobulina humana IgGl, con cada subunidad de cadena simple unida mediante enlace covalente en el extremo C a una cadena peptídica que contiene dos dominios de unión del receptor de la TPO.
No existe homología secuencial de aminoácidos entre romiplostim y la TPO endógena. En ensayos clínicos y preclínicos ningún anticuerpo anti-romiplostim reaccionó de forma cruzada con la TPO endógena.
Eficacia clínica y seguridad
La seguridad y la eficacia de romiplostim han sido evaluadas durante tres años de tratamiento continuado. En los ensayos clínicos, el tratamiento con romiplostim produjo aumentos del recuento plaquetario dependientes de la dosis. El tiempo hasta alcanzar el efecto máximo sobre el recuento plaquetario es de aproximadamente 10-14 días y es independiente de la dosis. Tras una dosis única subcutánea de entre 1 y 10 qg/kg de romiplostim en pacientes con PTI, el recuento máximo de plaquetas era entre 1,3 y 14,9 veces superior al recuento de plaquetas de la situación basal tras un periodo de dos a tres semanas y la respuesta variaba entre pacientes. En la mayoría de los pacientes con PTI que recibieron 6 dosis semanales de 1 ó 3 qg/kg de romiplostim los recuentos plaquetarios estaban en el intervalo de 50 a 450 x 109/l. De los 271 pacientes que recibieron romiplostim en los ensayos clínicos sobre PTI, 55 (20%) tenían 65 o más años y 27 (10%) tenían 75 años o más. No se han observado diferencias de seguridad o eficacia entre los pacientes de edad avanzada o más jóvenes en los ensayos controlados con placebo.
Resultados de los ensayos pivotales controlados con placebo
Se evaluó la seguridad y eficacia de romiplostim en dos ensayos controlados con placebo, doble ciego, en adultos con PTI que habían finalizado como mínimo un tratamiento antes de su entrada en el ensayo y que son representativos de la totalidad del espectro de dichos pacientes con PTI.
En el ensayo SI (212) se evaluaron pacientes no esplenectomizados y que habían presentado una respuesta insuficiente o intolerancia a los tratamientos previos. Los pacientes habían sido diagnosticados de PTI aproximadamente 2 años antes del momento de su inclusión en el ensayo. Los pacientes tenían una mediana de 3 (intervalo, de 1 a 7) tratamientos para la PTI antes de su inclusión en el ensayo. Los tratamientos previos eran corticosteroides (el 90% de los pacientes), inmunoglobulinas (76%), rituximab (29%), terapias citotóxicas (21%), danazol (11%) y azatioprina (5%). Los pacientes tenían una mediana de recuento plaquetario de 19 x 109/l en el momento de su inclusión en el ensayo.
En el ensayo S2 (105) se evaluaron pacientes esplenectomizados y que seguían teniendo trombocitopenia. Los pacientes habían sido diagnosticados de PTI aproximadamente 8 años antes del momento de su inclusión en el ensayo. Además de esplenectomía, los pacientes habían recibido una mediana de seis (intervalo, de 3 a 10) tratamientos para la PTI antes de su inclusión en el ensayo. Los tratamientos previos fueron corticosteroides (el 98% de los pacientes), inmunoglobulinas (97%), rituximab (71%), danazol (37%), terapias citotóxicas (68%) y azatioprina (24%). Los pacientes tenían una mediana de recuento plaquetario de 14 x 109/l en el momento de su inclusión en el ensayo.
Ambos ensayos se diseñaron de manera similar. Los pacientes (> 18 años) fueron asignados aleatoriamente en una proporción 2:1 para recibir una dosis de inicio de 1 qg/kg de romiplostim o placebo. Los pacientes recibieron una única inyección subcutánea semanal durante 24 semanas. Las dosis se ajustaron para mantener (de 50 a 200 x 109/l) los recuentos plaquetarios. En ambos ensayos, se determinó la eficacia como un aumento de la proporción de pacientes que conseguía una respuesta plaquetaria duradera. La mediana de la dosis promedio semanal para los pacientes esplenectomizados era de 3 qg/kg y para los pacientes no esplenectomizados era de 2 qg/kg.
En ambos ensayos, una proporción significativamente superior de pacientes que recibían romiplostim alcanzó una respuesta plaquetaria duradera en comparación con los pacientes que recibían placebo. Después de las cuatro primeras semanas del ensayo, romiplostim mantuvo los recuentos plaquetarios > 50 x 109/l en entre un 50% y un 70% de los pacientes durante el periodo de seis meses de tratamiento en los ensayos controlados con placebo. En el grupo de placebo, entre el 0% y el 7% de los pacientes fueron capaces de mantener una respuesta del recuento plaquetario durante los seis meses de tratamiento. A continuación se presenta un resumen de las variables principales de eficacia.
Resumen de los resultados principales de eficacia de los ensayos controlados con placebo
|
Ensayo 1 pacientes no esplenectomizados |
Ensayo 2 pacientes esplenectomizados |
Ensayo 1 y 2 combinados | ||||
|
romiplostim (n = 41) |
Placebo (n = 21) |
romiplostim (n = 42) |
Placebo (n = 21) |
romiplostim (n = 83) |
Placebo (n = 42) | |
|
N° (%) de pacientes con respuesta plaquetaria duradera3 |
25 (61%) |
1 (5%) |
16 (38%) |
0 (0%) |
41 (50%) |
1 (2%) |
|
(IC del 95%) |
(45%, 76%) |
(0%, 24%) |
(24%, 54%) |
(0%, 16%) |
(38%, 61%) |
(0%, 13%) |
|
valor p |
< 0,0001 |
0,0013 |
< 0,0001 | |||
|
N° (%) de pacientes con respuesta plaquetaria globalb |
36 (88%) |
3 (14%) |
33 (79%) |
0 (0%) |
69 (83%) |
3 (7%) |
|
(IC del 95%) |
(74%, 96%) |
(3%, 36%) |
(63%, 90%) |
(0%, 16%) |
(73%, 91%) |
(2%, 20%) |
|
valor p |
< 0,0001 |
< 0,0001 |
< 0,0001 | |||
|
Media del n° de semanas con respuesta plaquetariac |
15 |
1 |
12 |
0 |
14 |
1 |
|
(de) |
3,5 |
7,5 |
7,9 |
0,5 |
7,8 |
2,5 |
|
valor p |
< 0,0001 |
< 0,0001 |
< 0,0001 | |||
|
N° (%) de pacientes que requieren tratamientos de rescated |
8(20%) |
13 (62%) |
11 (26%) |
12 (57%) |
19 (23%) |
25 (60%) |
|
(IC del 95%) |
(9%, 35%) |
(38%, 82%) |
(14%, 42%) |
(34%, 78%) |
(14%, 33%) |
(43%, 74%) |
|
valor p |
0,001 |
0,0 |
175 |
< 0,0001 | ||
|
Ensayo 1 pacientes no esplenectomizados |
Ensayo 2 pacientes esplenectomizados |
Ensayo 1 y 2 combinados | ||||
|
romiplostim (n = 41) |
Placebo (n = 21) |
romiplostim (n = 42) |
Placebo (n = 21) |
romiplostim (n = 83) |
Placebo (n = 42) | |
|
N° (%) de pacientes con respuesta plaquetaria duradera con dosis establee |
21 (51%) |
0 (0%) |
13 (31%) |
0 (0%) |
34 (41%) |
0 (0%) |
|
(IC del 95%) |
(35%, 67%) |
(0%, 16%) |
(18%, 47%) |
(0%, 16%) |
(30%, 52%) |
(0%, 8%) |
|
valor p |
0,0001 |
0,0046 |
< 0,0001 | |||
|
a La respuesta plaquetaria duradera se definió como un recuento plaquetario semanal > 50 x 109/l presente seis o más veces durante las semanas de estudio 18-25 en ausencia de tratamientos de rescate en cualquier momento durante el periodo de tratamiento. b La respuesta plaquetaria global se define como la consecución de respuestas plaquetarias duraderas o transitorias. La respuesta plaquetaria transitoria se definió como un recuento plaquetario semanal > 50 x 109/l presente cuatro o más veces durante las semanas de estudio 2-25, pero sin respuesta plaquetaria duradera. El paciente puede no presentar una respuesta semanal en las 8 semanas posteriores a la administración de cualquier medicamento de rescate. c El número de semanas con respuesta plaquetaria se define como el número de semanas con recuentos plaquetarios > 50 x 109/l durante las semanas 2-25 del estudio. El paciente puede no presentar una respuesta semanal en las 8 semanas posteriores a la administración de cualquier medicamento de rescate. d Los tratamientos de rescate se definen como cualquier tratamiento administrado para aumentar el recuento de plaquetas. Los pacientes que requirieron medicación de rescate no fueron considerados para la respuesta plaquetaria duradera. Los tratamientos de rescate permitidos en el ensayo fueron IGIV, transfusiones de plaquetas, inmunoglobulina anti-D y corticosteroides. e La dosis estable se definió como la dosis mantenida en ± 1 qg/kg durante las últimas ocho semanas de tratamiento. | ||||||
Resultados de los estudios comparados con el estándar de tratamiento (SOC- Standard of Care, por sus siglas en inglés) en pacientes no esplenectomizados
El estudio S3 (131) fue un ensayo abierto, aleatorizado de 52 semanas, en pacientes que recibieron romiplostim o medicación estándar de tratamiento (SOC). Este estudio evaluó pacientes no esplenectomizados con PTI y recuento plaquetario < 50 x 109/l. Romiplostim fue administrado a 157 pacientes por vía subcutánea (SC), con una inyección semanal, comenzando a una dosis de 3 qg/kg, y ajustada durante todo el estudio dentro de un rango de 1-10 qg/kg, para mantener el recuento plaquetario entre 50 y 200 x 109/l, 77 pacientes recibieron SOC, de acuerdo con el estándar de la práctica habitual o las guías terapéuticas.
La tasa de incidencia global de esplenectomia en pacientes fue del 8,9% (14 de 157 pacientes) en el grupo de romiplostim comparado con 36,4% (28 de 77 pacientes) en el grupo de SOC, con un Odds Ratio (romiplostim frente a SOC) de 0,17 (IC del 95%: 0,08; 0,35).
La incidencia global de pacientes con fracaso de tratamiento fue del 11,5% (18 de 157 pacientes) en el grupo de romiplostim comprado con 29,9% (23 de 77 pacientes) en el grupo de SOC, con un Odds Ratio (romiplostim frente a SOC) de 0,31 (IC del 95%: 0,15; 0,61).
De los 157 pacientes aleatorizados en el grupo de romiplostim, tres pacientes no recibieron romiplostim. Entre los 154 pacientes que recibieron romiplostim, la mediana total de exposición a romiplostim fue 52,0 semanas y en un rango de 2 a 53 semanas. La dosis semanal más frecuentemente utilizada fue entre 3-5 qg/kg (percentil 25-75 repectivamente; mediana 3 qg/kg).
De Ios 77 pacientes aleatorizados en el grupo de SOC, dos pacientes no recibieron ningún SOC. Entre Ios 75 pacientes que recibieron al menos una dosis de SOC, la mediana total de exposición a SOC fue 51 semanas y en un rango de 0,4 a 52 semanas.
Reducción de los tratamientos médicos concomitantes permitidos en la PTI
En ambos ensayos controlados con placebo, doble ciego, se permitió que, los pacientes que ya recibían tratamientos médicos para la PTI con una pauta constante de dosificación, continuaran recibiendo dichos tratamientos durante todo el ensayo (corticosteroides, danazol y/o azatioprina). Veintiún pacientes no esplenectomizados y 18 pacientes esplenectomizados recibieron tratamientos médicos para la PTI durante el ensayo (principalmente corticosteroides) al inicio del ensayo. Todos (100%) los pacientes esplenectomizados que recibieron romiplostim pudieron reducir la dosis en más de un 25% o interrumpir los tratamientos médicos concomitantes para la PTI al final del periodo del tratamiento, en comparación con el 17% de los pacientes tratados con placebo. El 73% de los pacientes no esplenectomizados que recibieron romiplostim pudieron reducir la dosis en más de un 25% o interrumpir los tratamientos médicos concomitantes para la PTI al final del periodo del tratamiento, en comparación con el 50% de los pacientes tratados con placebo (ver sección 4.5).
Acontecimientos hemorrágicos
Durante todo el desarrollo clínico en PTI se observó una relación inversa entre los acontecimientos hemorrágicos y los recuentos plaquetarios. Todos los acontecimientos hemorrágicos clínicamente significativos (grado > 3) se produjeron con recuentos plaquetarios < 30 x 109/l. Los acontecimientos hemorrágicos de grado > 2 se produjeron con recuentos plaquetarios < 50 x 109/l. No se observaron diferencias estadísticamente significativas en la incidencia global de acontecimientos hemorrágicos entre los pacientes tratados con Nplate y los tratados con placebo.
En los dos ensayos controlados con placebo, 9 pacientes presentaron un acontecimiento hemorrágico que fue considerado grave (5 [6,0%] romiplostim, 4 [9,8%] placebo; Odds Ratio [romiplostim/placebo] = 0,59; IC del 95% = (0,15; 2,31)). El 15% de los pacientes tratados con romiplostim y el 34% de los pacientes tratados con placebo presentaron acontecimientos hemorrágicos de grado 2 o superior (Odds Ratio; [romiplostim/placebo] = 0,35; IC del 95% = (0,14; 0,85)).
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Nplate en uno o más grupos de la población pediátrica en el tratamiento de trombocitopenia inmune (púrpura trombocitopénica idiopática) (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
La farmacocinética de romiplostim implica una distribución mediada por las células diana, que es presumiblemente mediada por receptores de la TPO sobre las plaquetas y otras células del linaje trombopoyético como los megacariocitos.
Absorción
Tras la administración subcutánea de entre 3 y 15 pg/kg de romiplostim, se obtuvieron las concentraciones séricas máximas de romiplostim en los pacientes con PTI tras 7-50 horas (mediana de 14 horas). Las concentraciones plasmáticas variaron de un paciente a otro y no se correlacionaron con la dosis administrada. Los niveles plasmáticos de romiplostim presentan una relación inversa con los recuentos plaquetarios.
Distribución
El volumen de distribución de romiplostim tras la administración intravenosa de romiplostim descendió de manera no lineal desde 122; 78,8 a 48,2 ml/kg para dosis intravenosas de 0,3; 1,0 y 10 Mg/kg, respectivamente administradas a sujetos sanos. Este descenso no lineal del volumen de distribución está en línea con la fijación mediada por células diana (megacariocitos y plaquetas) de romiplostim, que puede saturarse cuando se administran las dosis más altas.
Eliminación
La semivida de eliminación de romiplostim en pacientes con PTI oscila entre 1 y 34 días (mediana,
3,5 días). La eliminación de romiplostim plasmático depende en parte del receptor de la TPO en las plaquetas. En consecuencia, para una dosis dada los pacientes con recuentos plaquetarios elevados se asocian a bajas concentraciones plasmáticas y viceversa. En otro ensayo clínico sobre la PTI, no se observó acumulación en las concentraciones plasmáticas tras seis dosis semanales de romiplostim
(3 Mg/kg).
Poblaciones especiales
No se ha investigado la farmacocinética de romiplostim en pacientes con insuficiencia renal o hepática. La farmacocinética de romiplostim no parece estar afectada en un grado clínicamente significativo por la edad, el peso y el sexo.
5.3 Datos preclínicos sobre seguridad
Se realizaron ensayos toxicológicos a dosis múltiples de romiplostim en ratas durante cuatro semanas y en monos durante seis meses. En general, los efectos observados durante estos ensayos estaban relacionados con la actividad trombopoyética de romiplostim y fueron similares independientemente de la duración del ensayo. Las reacciones en el lugar de inyección también estaban relacionadas con la administración de romiplostim. Se ha observado mielofibrosis en la médula ósea de las ratas a todos los niveles de dosis evaluados. En estos ensayos, no se observó mielofibrosis en animales tras un periodo de recuperación de cuatro semanas después del tratamiento, lo que indicaba reversibilidad.
En un ensayo de toxicología de un mes de duración en ratas y monos, se observó una disminución leve del recuento de glóbulos rojos, hematocrito y hemoglobina. También se detectó un efecto estimulante en la producción de leucocitos, con un ligero aumento de los recuentos sanguíneos periféricos de neutrófilos, linfocitos, monocitos y eosinófilos. En el ensayo crónico de larga duración con monos, no se observaron efectos en los linajes eritroides y leucocitarios al administrar romiplostim durante seis meses rebajando su administración de tres veces a la semana a una. Además, en los ensayos pivotales de fase 3 romiplostim no afectó a los linajes de glóbulos rojos y blancos en comparación con los sujetos que recibieron placebo.
Debido a la formación de anticuerpos neutralizantes, los efectos farmacodinámicos de romiplostim en ratas descendían con frecuencia con una duración prolongada de la administración. Los ensayos toxicocinéticos no mostraron interacción de los anticuerpos en las concentraciones medidas. Aunque en los ensayos con animales se probaron dosis elevadas, debido a las diferencias entre las especies de laboratorio y los humanos en relación con la sensibilidad ante los efectos farmacodinámicos de romiplostim y el efecto de los anticuerpos neutralizantes, no se pueden calcular de forma fiable los márgenes de seguridad.
Carcinogénesis
No se ha investigado el potencial carcinogénico de romiplostim. Por tanto, el riesgo de carcinogénesis potencial en humanos sigue sin conocerse.
Toxicidad reproductora
En todos los ensayos de desarrollo se formaron anticuerpos neutralizantes, que pueden tener efectos de inhibición sobre romiplostim. En los ensayos de desarrollo embriofetal en ratones y ratas, sólo se observaron reducciones del peso corporal de la madre en ratones. En los ratones había signos de un aumento de pérdidas postimplantación. En los ensayos de desarrollo pre y postnatal en ratas se observó un aumento de la duración de la gestación y un ligero aumento en la incidencia de mortalidad perinatal de las crías. Se sabe que romiplostim atraviesa la barrera placentaria en las ratas y puede transmitirse de la madre al feto en desarrollo y estimular la producción plaquetaria del feto. No se han observado efectos de romiplostim sobre la fertilidad en ratas.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Manitol (E421)
Sacarosa
L-histidina
Ácido clorhídrico (para ajuste del pH)
Polisorbato 20
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección 6.6.
6.3 Periodo de validez 5 años.
Después de la reconstitución: Se ha demostrado estabilidad química y física en uso durante 24 horas a 25°C y durante 24 horas entre 2°C y 8°C, si se mantiene protegido de la luz y en el vial original.
Desde un punto de vista microbiológico, el medicamento debe usarse inmediatamente. Si no se usa inmediatamente, los tiempos y condiciones de conservación durante el uso antes de su utilización son responsabilidad del usuario y no deberían superar las 24 horas a 25 °C ó 24 horas en la nevera (entre 2°C y 8°C), protegido de la luz.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
Puede estar temporalmente fuera de la nevera durante un período máximo de 24 horas a temperatura ambiente (hasta 25 °C).
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Vial de 5 ml (vidrio transparente tipo 1) con tapón (caucho clorobutilo), precinto (aluminio) y una cápsula de cierre del tipo flip-off (polipropileno).
Envase con 1 ó 4 viales de romiplostim.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Nplate es un medicamento estéril pero sin conservantes y está indicado para un solo uso. Nplate debe reconstituirse siguiendo las recomendaciones de buena práctica aséptica.
Nplate 250 microgramos polvo para solución inyectable
Nplate 250 microgramos polvo para solución inyectable debe reconstituirse con 0,72 ml de agua estéril para preparaciones inyectables para producir un volumen liberado de 0,5 ml. En cada vial se incluye una cantidad adicional para garantizar que se puedan administrar 250 qg de romiplostim (ver abajo la tabla que incluye información sobre el contenido del vial).
Nplate 500 microgramos polvo para solución inyectable
Nplate 500 microgramos polvo para solución inyectable debe reconstituirse con 1,2 ml de agua estéril para preparaciones inyectables para producir un volumen liberado de 1 ml. En cada vial se incluye una cantidad adicional para garantizar que se puedan administrar 500 qg de romiplostim (ver abajo la tabla que incluye información sobre el contenido del vial).
Contenido del vial:
|
Vial de un solo uso de Nplate |
Contenido total de romiplostim en el vial |
Volumen de agua estéril para preparaciones inyectables |
Volumen y producto liberado |
Concentraci ón final | ||
|
250 qg |
375 qg |
añadir |
0,72 ml |
= |
250 qg en 0,5 ml |
500 qg/ml |
|
500 qg |
625 qg |
añadir |
1,2 ml |
= |
500 qg en 1 ml |
500 qg/ml |
No deben utilizarse soluciones de cloruro sódico o agua bacteriostática para reconstituir el medicamento.
El agua para preparaciones inyectables debe inyectarse en el vial. Durante la disolución, debe realizarse un movimiento circular suave e invertir el contenido del vial. No hay que sacudir ni agitar vigorosamente el vial. Por lo general, se tarda menos de 2 minutos en realizar la disolución de Nplate. Inspeccionar visualmente la solución en busca de partículas o decoloraciones antes de su administración. La solución reconstituida debe ser transparente e incolora y no debe administrarse si se observan partículas o decoloración.
Para las condiciones de conservación después de la reconstitución del medicamento, ver sección 6.3.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Amgen Europe B.V.
Minervum 7061 4817 ZK Breda Países Bajos
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/497/001
EU/1/08/497/003
EU/1/08/497/002
EU/1/08/497/004
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 4 febrero 2009 Fecha de la última renovación: 20 diciembre 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
1. NOMBRE DEL MEDICAMENTO
Nplate 250 microgramos polvo y disolvente para solución inyectable Nplate 500 microgramos polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Nplate 250 microgramos polvo y disolvente para solución inyectable
Cada vial contiene 250 qg de romiplostim. Tras la reconstitución, un volumen liberado de 0,5 ml de solución contiene 250 qg de romiplostim (500 qg/ml). En cada vial se incluye una cantidad adicional para garantizar que se puedan administrar 250 qg de romiplostim.
Nplate 500 microgramos polvo y disolvente para solución inyectable
Cada vial contiene 500 qg de romiplostim. Tras la reconstitución, un volumen liberado de 1 ml de solución contiene 500 qg de romiplostim (500 qg/ml). En cada vial se incluye una cantidad adicional para garantizar que se puedan administrar 500 qg de romiplostim.
Romiplostim se produce mediante tecnología del ADN recombinante en Escherichia coli (E. coli). Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable (polvo para inyectable) El polvo es blanco.
El disolvente es un líquido transparente incoloro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Nplate está indicado para pacientes adultos con Púrpura Trombocitopénica Inmune (Idiopática) (PTI) crónica que son refractarios a otros tratamientos (por ejemplo, corticosteroides, inmunoglobulinas) (ver las secciones 4.2 y 5.1).
4.2 Posología y forma de administración
El tratamiento debe realizarse bajo la supervisión de un médico con experiencia en el tratamiento de patologías hematológicas.
Posología
Nplate debe administrarse una vez a la semana en forma de inyección subcutánea.
Dosis inicial
|
Dosis inicial o posteriores una vez a la semana: |
Peso* en kg x dosis en pg/kg = dosis individual del paciente en pg. |
|
Volumen de administración: |
Dosis en pg x 1 ml 500 pg = cantidad que hay que inyectar en ml |
|
Ejemplo: |
La dosis inicial de un paciente de 75 kg es de 1 pg/kg de romiplostim. Dosis individual del paciente = 75 kg x 1 pg/kg = 75 pg La cantidad correspondiente de solución de Nplate que hay que inyectar = 75 pa x 1 ml = 0,15 ml 500 pg |
|
*Al calcular la dosis de romiplostim debe utilizarse siempre el peso corporal real del paciente en el momento de iniciar el tratamiento. Los futuros ajustes de la dosis se basarán únicamente en los cambios en los recuentos plaquetarios y se realizarán en incrementos de 1 pg/kg (consultar la siguiente tabla). | |
Ajustes de la dosis
Para calcular la dosis debe utilizarse siempre el peso corporal real del paciente en el momento de iniciar el tratamiento. La dosis semanal de romiplostim debe aumentarse en incrementos de 1 pg/kg hasta que el paciente alcance un recuento plaquetario > 50 x 109/l. Hay que evaluar los recuentos plaquetarios semanalmente hasta alcanzar un recuento estable (> 50 x 109/l durante al menos 4 semanas sin ajuste de la dosis). Posteriormente hay que evaluar los recuentos plaquetarios mensualmente. No se debe sobrepasar la dosis máxima semanal de 10 pg/kg.
Ajustar la dosis de la siguiente manera:
|
Recuento de plaquetas (x 109/l) |
Acción |
|
< 50 |
Aumentar la dosis semanal en 1 pg/kg |
|
> 150 durante dos semanas consecutivas |
Disminuir la dosis semanal en 1 pg/kg |
|
> 250 |
No administrar; continuar evaluando el recuento plaquetario semanalmente. Después de que el recuento plaquetario haya descendido a < 150 x 109/l, reiniciar la administración con una dosis semanal reducida en 1 pg/kg. |
Debido a la variabilidad interindividual de la respuesta plaquetaria, en algunos pacientes el recuento plaquetario puede caer bruscamente por debajo de 50 x 109/l tras reducir la dosis o la interrupción del tratamiento. En estos casos, si es clínicamente adecuado, pueden considerarse niveles de corte del recuento plaquetario mas elevados para la reducción de dosis (200 x 109/l) y la interrupción del tratamiento (400 x 109/l) de acuerdo con el criterio médico.
La pérdida de respuesta o la incapacidad de mantener una respuesta plaquetaria con romiplostim dentro del intervalo de dosis recomendado debe motivar la búsqueda de los factores causales (ver sección 4.4, pérdida de respuesta a romiplostim).
Interrupción del tratamiento
Debe interrumpirse la administración de romiplostim si, tras cuatro semanas de tratamiento a la mayor dosis semanal de 10 pg/kg, el recuento plaquetario no aumenta hasta un nivel suficiente que evite hemorragias clínicamente relevantes.
Los pacientes deben ser evaluados clínicamente de forma periódica y el médico que administra el tratamiento debe decidir la continuación del mismo de manera individual, y en pacientes no esplenectomizados, se debe incluir la evaluación relativa a la esplenectomía. Es posible que reaparezca la trombocitopenia tras la interrupción del tratamiento (ver sección 4.4).
Pacientes de edad avanzada (> 65 años)
No se han observado diferencias globales de seguridad o eficacia entre pacientes < 65 y > 65 años de edad (ver sección 5.1). Aunque según estos datos no se requiere un ajuste de la pauta posológica en pacientes de edad avanzada, se aconseja precaución, teniendo en cuenta el reducido número de pacientes de edad avanzada incluidos en los ensayos clínicos realizados hasta la fecha.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de romiplostim en niños menores de 18 años. No se dispone de datos.
Pacientes con insuficiencia hepática
No se recomienda utilizar romiplostim en pacientes con insuficiencia hepática de moderada a grave (escala Child-Pugh > 7) a menos que el beneficio esperado sea mayor que el riesgo identificado de trombosis venosa portal en pacientes con trombocitopenia asociada a insuficiencia hepática tratada con agonistas de la trombopoyetina (TPO) (ver sección 4.4).
Si el uso de romiplostim se considera necesario, el recuento de plaquetas debe vigilarse estrechamente para minimizar el riesgo de complicaciones tromboembólicas.
Pacientes con insuficiencia renal
No se han realizado ensayos clínicos formales en estas poblaciones de pacientes. Nplate debe administrarse con precaución a estos pacientes.
Forma de administración
Vía subcutánea.
Una vez reconstituido el polvo, la solución inyectable de Nplate se administra por vía subcutánea. El volumen de inyección puede ser muy pequeño. Durante la preparación de Nplate, se debe tener cuidado en el cálculo de la dosis y de la reconstitución con el volumen correcto de agua estéril para preparaciones inyectables. Debe prestarse una atención especial para asegurar que el volumen apropiado de Nplate se extrae del vial para la administración subcutánea. Debe emplearse una jeringa con graduaciones de 0,01 ml.
Los pacientes que tienen un recuento plaquetario estable > 50 x 109/l durante al menos 4 semanas sin ajuste de la dosis, según el criterio del médico supervisor, pueden autoadministrarse la solución inyectable de Nplate. Los pacientes elegibles para la autoadministración de Nplate deben estar formados en estos procedimientos.
Después de las primeras 4 semanas de autoadministración, el paciente debe ser supervisado de nuevo mientras realiza la reconstitución y administración de Nplate. Sólo los pacientes que demuestren capacidad de reconstituir y autoadministrarse Nplate, pueden seguir haciéndolo.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1 o a las proteínas derivadas de E. coli.
4.4 Advertencias y precauciones especiales de empleo
Reaparición de trombocitopenia v hemorragia tras la finalización del tratamiento
Es probable que la trombocitopenia reaparezca tras interrumpir el tratamiento con romiplostim. Si se suspende el tratamiento con romiplostim mientras están siendo administrados medicamentos anticoagulantes o antiplaquetarios, aumenta el riesgo de hemorragias. Los pacientes deben ser sometidos a un control estricto ante un posible descenso del recuento plaquetario v tratados médicamente para evitar las hemorragias tras la suspensión del tratamiento con romiplostim. Se recomienda que, si se interrumpe el tratamiento con romiplostim, se reinicie el tratamiento para la PTI conforme a las recomendaciones de tratamiento actuales. El tratamiento médico adicional puede incluir la interrupción del tratamiento con anticoagulantes, antiplaquetarios v/o ambos, la reversión de la anticoagulación o el tratamiento complementario con plaquetas.
Aumento de la reticulina en la médula ósea
Se cree que el aumento de la reticulina en la médula ósea es el resultado de la estimulación del receptor de la TPO, que conlleva un aumento de la cantidad de megacariocitos en la médula ósea, que en consecuencia liberarán citocinas. Los cambios morfológicos de las células de la sangre periférica pueden sugerir un aumento de la reticulina, que puede detectarse mediante una biopsia de la médula ósea. Por tanto, se recomienda realizar análisis para detectar anomalías morfológicas celulares utilizando un frotis sanguíneo periférico v un hemograma completo antes v durante el tratamiento con romiplostim. Ver la sección 4.8 para obtener información relativa a los aumentos de reticulina observados durante los ensavos clínicos con romiplostim.
Si se observa una pérdida de eficacia v un frotis sanguíneo periférico anormal en los pacientes, deberá interrumpirse la administración de romiplostim, se realizará una exploración física v se valorará la necesidad de realizar una biopsia de médula ósea con la tinción adecuada para detectar la presencia de reticulina. Si se encuentra disponible, deberá compararse con una biopsia de médula ósea anterior. Si se mantiene la eficacia pero se observa en los pacientes un frotis sanguíneo periférico anormal, el médico deberá seguir el criterio clínico adecuado, que incluve valorar la realización de una biopsia de médula ósea v deberá evaluar de nuevo los riesgos v beneficios de romiplostim v las opciones alternativas para el tratamiento de la PTI.
Complicaciones trombóticas/tromboembólicas
Los recuentos plaquetarios por encima del intervalo normal suponen un riesgo de complicaciones trombóticas/tromboembólicas. La incidencia de acontecimientos trombóticos/tromboembólicos observada en los ensavos clínicos fue 6,0% con romiplostim v 3,6% con placebo. Se debe tener precaución cuando se administre romiplostim a pacientes con factores de riesgo conocidos de tromboembolismo incluvendo, pero no limitados a, factores hereditarios (por ej. Factor V Leiden) o factores de riesgo adquiridos (por ej. deficiencia ATIII, síndrome antifosfolipídico), edad avanzada, pacientes con periodos prolongados de inmovilización, neoplasias, anticonceptivos v terapia hormonal sustitutiva, cirugía/traumatismo, obesidad v fumadores.
Se han notificado casos de acontecimientos tromboembólicos (ATEs), incluvendo trombosis venosa portal, en pacientes con enfermedad hepática crónica que reciben romiplostim. Romiplostim debe utilizarse con precaución en estas poblaciones. Deben seguirse las recomendaciones para el ajuste de la dosis (ver sección 4.2).
Los errores de medicación, incluyendo sobredosis e infradosis, se han notificado en pacientes que reciben Nplate, se deben seguir las guías de cálculo de dosis y ajuste de dosis (ver sección 4.2).
La sobredosis puede dar lugar a un aumento excesivo de los recuentos plaquetarios asociados con las complicaciones trombóticas/tromboembólicas. Si los recuentos plaquetarios aumentan excesivamente, discontinuar Nplate y supervisar los recuentos plaquetarios. Reiniciar el tratamiento con Nplate de acuerdo con las recomendaciones de dosificación y administración. Una dosis insuficiente puede dar lugar a unos recuentos plaquetarios menores de lo esperado y posible sangrado. Los recuentos plaquetarios deben controlarse en los pacientes que reciben Nplate (ver secciones 4.2, 4.4 y 4.9).
Progresión de Síndromes Mielodisplásicos (SMD) existentes
El balance beneficio/riesgo para romiplostim se ha establecido favorable sólo para el tratamiento de la trombocitopenia asociada a PTI crónica y no se debe utilizar romiplostim en otras condiciones clínicas asociadas a trombocitopenia
El diagnóstico de la PTI en adultos y en pacientes de edad avanzada debería haber sido confirmado por la exclusión de otras entidades clínicas que presentan trombocitopenia, en particular, se debe excluir el diagnóstico de SMD. Se debe realizar un aspirado y biopsia de médula ósea en el curso de la enfermedad y tratamiento, sobre todo en pacientes mayores de 60 años de edad, que presenten síntomas sistémicos o signos anormales, tales como las células blásticas periféricas aumentadas.
En estudios clínicos con romiplostim para el tratamiento de pacientes con SMD, se notificaron casos de un incremento transitorio del recuento de células blásticas y casos de progresión de la enfermedad de SMD a LMA. Un estudio aleatorizado, controlado con placebo en pacientes con SMD tratados con romiplostim, se interrumpió prematuramente debido a un exceso en el número de progresiones de la enfermedad a LMA y un aumento en los blastos circulantes de más del 10% en pacientes que recibían romiplostim. De los casos de progresión de la enfermedad de SMD a LMA que se observaron, los pacientes con clasificación RAEB-1 de SMD al inicio fueron más propensos a presentar progresión de la enfermedad a LMA en comparación con los de SMD de riesgo más bajo.
Romiplostim no se debe utilizar para el tratamiento de la trombocitopenia debida a SMD ni a ninguna otra causa de trombocitopenia que no sea la PTI, fuera de ensayos clínicos.
Pérdida de respuesta a romiplostim
Una pérdida de respuesta o la incapacidad de mantener una respuesta plaquetaria con el tratamiento con romiplostim dentro del intervalo de dosis recomendado, debe motivar la búsqueda de los factores causales incluyendo la inmunogenicidad (ver sección 4.8) y el aumento de reticulina en la médula ósea (ver más arriba).
Efectos de romiplostim sobre los glóbulos rojos y blancos
Se han observado alteraciones en parámetros relacionados con los glóbulos rojos (disminución) y blancos (incremento) en ensayos toxicológicos no-clínicos (ratas y monos) , así como en pacientes con PTI. Pueden ocurrir anemia concomitante y leucocitosis (dentro de un intervalo de 4 semanas) en pacientes de forma independiente de la esplenectomía. Sin embargo, se han observado con más frecuencia en pacientes que han sido sometidos previamente a una esplenectomía. Debería considerarse la monitorización de dichos parámetros en los pacientes tratados con romiplostim.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones. Se desconocen las interacciones potenciales de romiplostim con medicamentos administrados conjuntamente a consecuencia de la unión a las proteínas plasmáticas.
Los medicamentos empleados en el tratamiento de la PTI en combinación con romiplostim en ensayos clínicos fueron corticosteroides, danazol y/o azatioprina, inmunoglobulina intravenosa (IGIV) e inmunoglobulina anti-D. Cuando se combine romiplostim con otros medicamentos para el tratamiento de la PTI deben controlarse los recuentos plaquetarios a fin de evitar recuentos plaquetarios fuera del intervalo recomendado (ver sección 4.2).
Debe reducirse o interrumpirse la administración de corticosteroides, danazol y azatioprina cuando se administran en combinación con romiplostim (ver sección 5.1). Cuando se reduzcan o interrumpan otros tratamientos para la PTI deben controlarse los recuentos plaquetarios a fin de evitar que se sitúen fuera del intervalo recomendado (ver sección 4.2).
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos o éstos son limitados relativos al uso de romiplostim en mujeres embarazadas.
Los estudios realizados en animales han mostrado que romiplostim traspasa la barrera placentaria y aumenta los recuentos plaquetarios fetales. En estudios con animales, también ocurrieron pérdida posimplementación y un ligero aumento en la mortalidad perinatal de las crías (ver sección 5.3).
Romiplostim está contraindicado durante el embarazo y en mujeres en edad fértil que no utilicen métodos anticonceptivos.
Lactancia
Se desconoce si romiplostim/metabolitos se excretan en la leche materna. No se puede excluir el riesgo en recién nacidos/lactantes. Se debe decidir si es necesario interrumpir la lactancia o interrumpir/abstenerse el tratamiento con romiplostim tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre.
Fertilidad
No hay datos disponibles sobre fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Nplate sobre la capacidad para conducir y utilizar máquinas es moderada. En los ensayos clínicos, algunos pacientes experimentaron episodios de mareos transitorios de leves a moderados.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Basándose en un análisis de todos los pacientes adultos con PTI que recibían romiplostim en 4 ensayos clínicos controlados y 5 no controlados, la incidencia global de todas las reacciones adversas en pacientes tratados con romiplostim fue de 91,5% (248/271). La duración media de exposición a romiplostim en esta población en estudio fue de 50 semanas.
Las reacciones adversas de mayor gravedad que pueden ocurrir durante el tratamiento con Nplate incluyen recurrencia de trombocitopenia y sangrado después del cese del tratamiento, aumento de la reticulina en la médula ósea, complicaciones trombóticas/tromboembólicas, errores de medicación y progresión de SMD existente a LMA. Las reacciones adversas más frecuentes observadas incluyen reacciones de hipersensibilidad (incluyendo casos de erupción, urticaria y angioedema) y cefalea.
Listado tabulado de acontecimientos adversos
Las frecuencias se definen como: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100), raras (> 1/10.000 a < 1/1.000); muy raras (< 1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Dentro de cada intervalo de frecuencia y según el sistema de clasificación de órganos de MedDRA, las reacciones adversas se enumeran en orden decreciente de incidencia.
|
Clasificación de órganos del sistema MedDRA |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Infecciones e infestaciones |
Infección del tracto respiratorio superior |
Gastroenteritis |
Gripe Infección localizada Nasofaringitis |
|
Neoplasias benignas, malignas y no especificadas (incluyendo quistes y pólipos) |
Mieloma múltiple Mielofibrosis | ||
|
Trastornos de la sangre y del sistema linfático |
Trastornos de la médula ósea* T rombocitopenia* Anemia |
Anemia aplásica Insuficiencia de la médula ósea Leucocitosis Esplenomegalia T rombocitanemia Recuento plaquetario aumentado Recuento plaquetario anormal | |
|
Trastornos del sistema inmunológico |
Hipersensibilidad* * |
Angioedema | |
|
Trastornos del metabolismo y de la nutrición |
Intolerancia al alcohol Anorexia Pérdida de apetito Deshidratación Gota | ||
|
Trastornos psiquiátricos |
Insomnio |
Depresión Sueños anormales | |
|
Trastornos del sistema nervioso |
Cefalea |
Mareos Migraña Parestesia |
Clonus Disgeusia Hipoestesia Hipogeusia Neuropatía periférica Trombosis del seno transverso |
|
Clasificación de órganos del sistema MedDRA |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Trastornos oculares |
Hemorragia conjuntival Alteración de la acomodación visual Ceguera Alteración ocular Prurito ocular Aumento del lagrimeo Papiloedema Alteración visual | ||
|
Trastornos del oído y del laberinto |
Vértigo | ||
|
Trastornos cardiacos |
Palpitaciones |
Infarto de miocardio Frecuencia cardiaca aumentada | |
|
Trastornos vasculares |
Enrojecimiento |
Trombosis venosa profunda Hipotensión Embolismo periférico Isquemia periférica Flebitis Tromboflebitis superficial Trombosis Eritromelalgia | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Embolia pulmonar* |
Tos Rinorrea Garganta seca Disnea Congestión nasal Dolor al respirar | |
|
Trastornos gastrointestinales |
Náuseas Diarrea Dolor abdominal Estreñimiento Dispepsia |
Vómitos Hemorragia rectal Mal aliento Disfagia Trastorno del reflujo gastroesofágico Hematoquecia Hemorragia bucal Malestar estomacal Estomatitis Decoloración dental | |
|
Trastornos hepatobiliares |
Trombosis venosa portal Aumento de transaminasas |
|
Clasificación de órganos del sistema MedDRA |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Trastornos de la piel y del tejido subcutáneo |
Prurito Equimosis Erupción |
Alopecia Reacción de fotosensibilidad Acné Dermatitis de contacto Sequedad de piel Eczema Eritema Erupción exfoliativa Crecimiento de pelo anormal Prurigo Púrpura Erupción papular Erupción pruriginosa Nódulos en la piel Olor anormal de la piel Urticaria | |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia Mialgia Espasmo muscular Dolor en las extremidades Dolor en la espalda Dolor de huesos |
Tensión muscular Debilidad muscular Dolor en el hombro Espasmo muscular | |
|
Trastornos renales y urinarios |
Presencia de proteínas en orina | ||
|
Trastornos del aparato reproductor y de la mama |
Hemorragia vaginal | ||
|
Trastornos generales y alteraciones en el lugar de administración |
Fatiga Edema periférico Enfermedad semejante a la gripe Dolor Astenia Pirexia Escalofríos Irritación en el lugar de la inyección |
Hemorragia en el lugar de la inyección Dolor en el pecho Irritabilidad Malestar Edema facial Sensación de calor Sensación de inquietud | |
|
Exploraciones complementarias |
Aumento de presión arterial Aumento de lactato deshidrogenasa en sangre Aumento de la temperatura corporal Pérdida de peso Aumento de peso |
|
Clasificación de órganos del sistema MedDRA |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Contusión |
* ver sección 4.4
** Reacciones de hipersensibilidad incluyendo casos de erupción, urticaria y angioedema
Descripción de las reacciones adversas seleccionadas
Además, las reacciones listadas a continuación han sido consideradas relacionadas con el tratamiento con romiplostim.
Trombocitosis
Basándose en un análisis de todos los pacientes adultos con PTI que recibían romiplostim en 4 ensayos clínicos controlados y 5 no controlados, se notificaron 3 acontecimientos de trombocitosis, n = 271.
No se notificaron secuelas clínicas relacionadas con el recuento plaquetario elevado en ninguno de los 3 pacientes.
Trombocitopenia después de la interrupción del tratamiento
Basándose en un análisis de todos los pacientes adultos con PTI que recibían romiplostim en 4 ensayos clínicos controlados y 5 no controlados, se notificaron 4 acontecimientos de trombocitopenia tras la interrupción del tratamiento, n = 271 (ver sección 4.4).
Progresión de Síndromes Mielodisplásicos (SMD) existentes
En un ensayo clínico aleatorizado, controlado con placebo, en pacientes con SMD, se interrumpió prematuramente el tratamiento con romiplostim debido a un incremento numérico de casos de progresión de la enfermedad de SMD a LMA y aumento transitorio en el recuento de células blásticas en pacientes tratados con romiplostim comparado con placebo. De los casos de progresión de la enfermedad de SMD a LMA que se observaron, los pacientes con clasificación RAEB-1 de SMD al inicio fueron más propensos a presentar progresión de la enfermedad a LMA (ver sección 4.4). La supervivencia global fue similar al placebo.
Aumento de la reticulina en la médula ósea
En los ensayos clínicos, se interrumpió la administración de romiplostim en 4 de 271 pacientes debido a la aparición de depósitos de reticulina en la médula ósea. En 6 pacientes adicionales, se observó reticulina tras biopsia de la médula ósea (ver sección 4.4).
Inmunogenicidad
En ensayos clínicos, pacientes adultos con PTI presentaron anticuerpos anti romiplostim.
Mientras que el 5,8% y 3,9% de los sujetos fueron positivos al desarrollo de anticuerpos de unión anti romiplostim y TPO respectivamente, sólo 2 sujetos (0,4%) dieron positivo a los anticuerpos neutralizantes anti romiplostim. Sin embargo, estos anticuerpos no generaron una reacción cruzada con la TPO endógena. Cuatro meses después del final de la administración, ambos sujetos dieron negativo a los anticuerpos neutralizantes anti romiplostim. La incidencia de anticuerpos pre-existentes anti romiplostim y TPO fue del 8,0% y 5,4%, respectivamente.
Al igual que ocurre con todas las proteínas terapéuticas, existe la posibilidad de que se origine inmunogenicidad. Si existe sospecha de formación de anticuerpos neutralizantes, ha de ponerse en contacto con el representante local del titular de la Autorización de Comercialización (ver sección 6 del prospecto) para realizar una prueba de anticuerpos.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
No se observaron acontecimientos adversos en ratas a las que se administró una dosis única de 1.000 pg/kg o en monos tras la administración repetida de romiplostim a 500 pg/kg (100 ó 50 veces la dosis clínica máxima de 10 pg/kg, respectivamente).
En caso de sobredosis, los recuentos plaquetarios pueden aumentar excesivamente y dar lugar a complicaciones trombóticas/tromboembólicas. Si los recuentos plaquetarios son excesivamente elevados, interrumpir Nplate y monitorizar el recuento plaquetario. Reiniciar el tratamiento con Nplate de acuerdo con las recomendaciones de dosis y administración (ver secciones 4.2 y 4.4).
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antihemorrágicos, otros hemostáticos sistémicos. Código ATC: B02BX04 Mecanismo de acción
Romiplostim es una proteína de fusión Fc-péptido (cuerpo peptídico) que señala y activa las rutas de transcripción intracelular a través del receptor de la TPO (también denominado cMpl) para aumentar la producción de plaquetas. La molécula del cuerpo peptídico está formada por un dominio Fc de la inmunoglobulina humana IgGl, con cada subunidad de cadena simple unida mediante enlace covalente en el extremo C a una cadena peptídica que contiene dos dominios de unión del receptor de la TPO.
No existe homología secuencial de aminoácidos entre romiplostim y la TPO endógena. En ensayos clínicos y preclínicos ningún anticuerpo anti-romiplostim reaccionó de forma cruzada con la TPO endógena.
Eficacia clínica y seguridad
La seguridad y la eficacia de romiplostim han sido evaluadas durante tres años de tratamiento continuado. En los ensayos clínicos, el tratamiento con romiplostim produjo aumentos del recuento plaquetario dependientes de la dosis. El tiempo hasta alcanzar el efecto máximo sobre el recuento plaquetario es de aproximadamente 10-14 días y es independiente de la dosis. Tras una dosis única subcutánea de entre 1 y 10 pg/kg de romiplostim en pacientes con PTI, el recuento máximo de plaquetas era entre 1,3 y 14,9 veces superior al recuento de plaquetas de la situación basal tras un periodo de dos a tres semanas y la respuesta variaba entre pacientes. En la mayoría de los pacientes con PTI que recibieron 6 dosis semanales de 1 ó 3 pg/kg de romiplostim los recuentos plaquetarios estaban en el intervalo de 50 a 450 x 109/l. De los 271 pacientes que recibieron romiplostim en los ensayos clínicos sobre PTI, 55 (20%) tenían 65 o más años y 27 (10%) tenían 75 años o más. No se han observado diferencias de seguridad o eficacia entre los pacientes de edad avanzada o más jóvenes en los ensayos controlados con placebo.
Resultados de los ensayos pivotales controlados con placebo
Se evaluó la seguridad y eficacia de romiplostim en dos ensayos controlados con placebo, doble ciego, en adultos con PTI que habían finalizado como mínimo un tratamiento antes de su entrada en el ensayo y que son representativos de la totalidad del espectro de dichos pacientes con PTI.
En el ensayo SI (212) se evaluaron pacientes no esplenectomizados y que habían presentado una respuesta insuficiente o intolerancia a los tratamientos previos. Los pacientes habían sido diagnosticados de PTI aproximadamente 2 años antes del momento de su inclusión en el ensayo. Los pacientes tenían una mediana de 3 (intervalo, de 1 a 7) tratamientos para la PTI antes de su inclusión en el ensayo. Los tratamientos previos eran corticosteroides (el 90% de los pacientes), inmunoglobulinas (76%), rituximab (29%), terapias citotóxicas (21%), danazol (11%) y azatioprina (5%). Los pacientes tenían una mediana de recuento plaquetario de 19 x 109/l en el momento de su inclusión en el ensayo.
En el ensayo S2 (105) se evaluaron pacientes esplenectomizados y que seguían teniendo trombocitopenia. Los pacientes habían sido diagnosticados de PTI aproximadamente 8 años antes del momento de su inclusión en el ensayo. Además de esplenectomía, los pacientes habían recibido una mediana de seis (intervalo, de 3 a 10) tratamientos para la PTI antes de su inclusión en el ensayo. Los tratamientos previos fueron corticosteroides (el 98% de los pacientes), inmunoglobulinas (97%), rituximab (71%), danazol (37%), terapias citotóxicas (68%) y azatioprina (24%). Los pacientes tenían una mediana de recuento plaquetario de 14 x 109/l en el momento de su inclusión en el ensayo.
Ambos ensayos se diseñaron de manera similar. Los pacientes (> 18 años) fueron asignados aleatoriamente en una proporción 2:1 para recibir una dosis de inicio de 1 qg/kg de romiplostim o placebo. Los pacientes recibieron una única inyección subcutánea semanal durante 24 semanas. Las dosis se ajustaron para mantener (de 50 a 200 x 109/l) los recuentos plaquetarios. En ambos ensayos, se determinó la eficacia como un aumento de la proporción de pacientes que conseguía una respuesta plaquetaria duradera. La mediana de la dosis promedio semanal para los pacientes esplenectomizados era de 3 qg/kg y para los pacientes no esplenectomizados era de 2 qg/kg.
En ambos ensayos, una proporción significativamente superior de pacientes que recibían romiplostim alcanzó una respuesta plaquetaria duradera en comparación con los pacientes que recibían placebo. Después de las cuatro primeras semanas del ensayo, romiplostim mantuvo los recuentos plaquetarios > 50 x 109/l en entre un 50% y un 70% de los pacientes durante el periodo de seis meses de tratamiento en los ensayos controlados con placebo. En el grupo de placebo, entre el 0% y el 7% de los pacientes fueron capaces de mantener una respuesta del recuento plaquetario durante los seis meses de tratamiento. A continuación se presenta un resumen de las variables principales de eficacia.
Resumen de los resultados principales de eficacia de los ensayos controlados con placebo
|
Ensayo 1 pacientes no esplenectomizados |
Ensayo 2 pacientes esplenectomizados |
Ensayo 1 y 2 combinados | ||||
|
romiplostim (n = 41) |
Placebo (n = 21) |
romiplostim (n = 42) |
Placebo (n = 21) |
romiplostim (n = 83) |
Placebo (n = 42) | |
|
N° (%) de pacientes con respuesta plaquetaria duradera3 |
25 (61%) |
1 (5%) |
16 (38%) |
0 (0%) |
41 (50%) |
1 (2%) |
|
(IC del 95%) |
(45%, 76%) |
(0%, 24%) |
(24%, 54%) |
(0%, 16%) |
(38%, 61%) |
(0%, 13%) |
|
valor p |
< 0,0001 |
0,0013 |
< 0,0001 | |||
|
Ensayo 1 pacientes no esplenectomizados |
Ensayo 2 pacientes esplenectomizados |
Ensayo 1 y 2 combinados | ||||
|
romiplostim (n = 41) |
Placebo (n = 21) |
romiplostim (n = 42) |
Placebo (n = 21) |
romiplostim (n = 83) |
Placebo (n = 42) | |
|
N° (%) de pacientes con respuesta plaquetaria globalb |
36 (88%) |
3 (14%) |
33 (79%) |
0 (0%) |
69 (83%) |
3 (7%) |
|
(IC del 95%) |
(74%, 96%) |
(3%, 36%) |
(63%, 90%) |
(0%, 16%) |
(73%, 91%) |
(2%, 20%) |
|
valor p |
< 0,0001 |
< 0,0001 |
< 0,0001 | |||
|
Media del n° de semanas con respuesta plaquetariac |
15 |
1 |
12 |
0 |
14 |
1 |
|
(de) |
3,5 |
7,5 |
7,9 |
0,5 |
7,8 |
2,5 |
|
valor p |
< 0,0001 |
< 0,0001 |
< 0,0001 | |||
|
N° (%) de pacientes que requieren tratamientos de rescated |
8(20%) |
13 (62%) |
11 (26%) |
12 (57%) |
19 (23%) |
25 (60%) |
|
(IC del 95%) |
(9%, 35%) |
(38%, 82%) |
(14%, 42%) |
(34%, 78%) |
(14%, 33%) |
(43%, 74%) |
|
valor p |
0,001 |
0,0 |
175 |
< 0,0001 | ||
|
N° (%) de pacientes con respuesta plaquetaria duradera con dosis establee |
21 (51%) |
0 (0%) |
13 (31%) |
0 (0%) |
34 (41%) |
0 (0%) |
|
(IC del 95%) |
(35%, 67%) |
(0%, 16%) |
(18%, 47%) |
(0%, 16%) |
(30%, 52%) |
(0%, 8%) |
|
valor p |
0,0001 |
0,0046 |
< 0,0001 | |||
|
a La respuesta plaquetaria duradera se definió como un recuento plaquetario semanal > 50 x 109/l presente seis o más veces durante las semanas de estudio 18-25 en ausencia de tratamientos de rescate en cualquier momento durante el periodo de tratamiento. b La respuesta plaquetaria global se define como la consecución de respuestas plaquetarias duraderas o transitorias. La respuesta plaquetaria transitoria se definió como un recuento plaquetario semanal > 50 x 109/l presente cuatro o más veces durante las semanas de estudio 2-25, pero sin respuesta plaquetaria duradera. El paciente puede no presentar una respuesta semanal en las 8 semanas posteriores a la administración de cualquier medicamento de rescate. c El número de semanas con respuesta plaquetaria se define como el número de semanas con recuentos plaquetarios > 50 x 109/l durante las semanas 2-25 del estudio. El paciente puede no presentar una respuesta semanal en las 8 semanas posteriores a la administración de cualquier medicamento de rescate. d Los tratamientos de rescate se definen como cualquier tratamiento administrado para aumentar el recuento de plaquetas. Los pacientes que requirieron medicación de rescate no fueron considerados para la respuesta plaquetaria duradera. Los tratamientos de rescate permitidos en el ensayo fueron IGIV, transfusiones de plaquetas, inmunoglobulina anti-D y corticosteroides. e La dosis estable se definió como la dosis mantenida en ± 1 qg/kg durante las últimas ocho semanas de tratamiento. | ||||||
Resultados de los estudios comparados con el estándar de tratamiento (SOC- Standard of Care, por sus siglas en inglés) en pacientes no esplenectomizados
El estudio S3 (131) fue un ensayo abierto, aleatorizado de 52 semanas en pacientes que recibieron romiplostim o medicación estándar de tratamiento (SOC). Este estudio evaluó pacientes no
esplenectomizados con PTI y recuento plaquetario <50 x 109/1. Romiplostim fue administrado a 157 pacientes por vía subcutánea (SC), con una inyección semanal, comenzando a una dosis de 3 qg/kg, y ajustada durante todo el estudio dentro de un rango de 1-10 qg/kg, para mantener el recuento plaquetario entre 50 y 200 x 109/1, 77 pacientes recibieron SOC, de acuerdo con el estándar de la práctica habitual o las guías terapéuticas.
La tasa de incidencia global de esplenectomia en pacientes fue del 8,9% (14 de 157 pacientes) en el grupo de romiplostim comparado con 36,4% (28 de 77 pacientes) en el grupo de SOC, con un Odds Ratio (romiplostim frente a SOC) de 0,17 (IC del 95%: 0,08; 0,35).
La incidencia global de pacientes con fracaso de tratamiento fue del 11,5% (18 de 157 pacientes) en el grupo de romiplostim comprado con 29,9% (23 de 77 pacientes) en el grupo de SOC, con un Odds Ratio (romiplostim frente a SOC) de 0,31 (IC del 95%: 0,15; 0,61).
De los 157 pacientes aleatorizados en el grupo de romiplostim, tres pacientes no recibieron romiplostim. Entre los 154 pacientes que recibieron romiplostim, la mediana total de exposición a romiplostim fue 52,0 semanas y en un rango de 2 a 53 semanas. La dosis semanal más frecuentemente utilizada fue entre 3-5 qg/kg (percentil 25-75 repectivamente; mediana 3 qg/kg).
De los 77 pacientes aleatorizados en el grupo de SOC, dos pacientes no recibieron ningún SOC. Entre los 75 pacientes que recibieron al menos una dosis de SOC, la mediana total de exposición a SOC fue 51 semanas y en un rango de 0,4 a 52 semanas.
Reducción de los tratamientos médicos concomitantes permitidos en la PTI
En ambos ensayos controlados con placebo, doble ciego, se permitió que, los pacientes que ya recibían tratamientos médicos para la PTI con una pauta constante de dosificación, continuaran recibiendo dichos tratamientos durante todo el ensayo (corticosteroides, danazol y/o azatioprina). Veintiún pacientes no esplenectomizados y 18 pacientes esplenectomizados recibieron tratamientos médicos para la PTI durante el ensayo (principalmente corticosteroides) al inicio del ensayo. Todos (100%) los pacientes esplenectomizados que recibieron romiplostim pudieron reducir la dosis en más de un 25% o interrumpir los tratamientos médicos concomitantes para la PTI al final del periodo del tratamiento, en comparación con el 17% de los pacientes tratados con placebo. El 73% de los pacientes no esplenectomizados que recibieron romiplostim pudieron reducir la dosis en más de un 25% o interrumpir los tratamientos médicos concomitantes para la PTI al final del periodo del tratamiento, en comparación con el 50% de los pacientes tratados con placebo (ver sección 4.5).
Acontecimientos hemorrágicos
Durante todo el desarrollo clínico en PTI se observó una relación inversa entre los acontecimientos hemorrágicos y los recuentos plaquetarios. Todos los acontecimientos hemorrágicos clínicamente significativos (grado > 3) se produjeron con recuentos plaquetarios < 30 x 109/1. Los acontecimientos hemorrágicos de grado > 2 se produjeron con recuentos plaquetarios < 50 x 109/1. No se observaron diferencias estadísticamente significativas en la incidencia global de acontecimientos hemorrágicos entre los pacientes tratados con Nplate y los tratados con placebo.
En los dos ensayos controlados con placebo, 9 pacientes presentaron un acontecimiento hemorrágico que fue considerado grave (5 [6,0%] romiplostim, 4 [9,8%] placebo; Odds Ratio [romiplostim/placebo] = 0,59; IC del 95% = (0,15; 2,31)). El 15% de los pacientes tratados con romiplostim y el 34% de los pacientes tratados con placebo presentaron acontecimientos hemorrágicos de grado 2 o superior (Odds Ratio; [romiplostim/placebo] = 0,35; IC del 95% = (0,14; 0,85)).
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Nplate en uno o más grupos de la población pediátrica en el tratamiento de trombocitopenia inmune (púrpura trombocitopénica idiopática) (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
La farmacocinética de romiplostim implica una distribución mediada por las células diana, que es presumiblemente mediada por receptores de la TPO sobre las plaquetas y otras células del linaje trombopoyético como los megacariocitos.
Absorción
Tras la administración subcutánea de entre 3 y 15 pg/kg de romiplostim, se obtuvieron las concentraciones séricas máximas de romiplostim en los pacientes con PTI tras 7-50 horas (mediana de 14 horas). Las concentraciones plasmáticas variaron de un paciente a otro y no se correlacionaron con la dosis administrada. Los niveles plasmáticos de romiplostim presentan una relación inversa con los recuentos plaquetarios.
Distribución
El volumen de distribución de romiplostim tras la administración intravenosa de romiplostim descendió de manera no lineal desde 122; 78,8 a 48,2 ml/kg para dosis intravenosas de 0,3; 1,0 y 10 Mg/kg, respectivamente administradas a sujetos sanos. Este descenso no lineal del volumen de distribución está en línea con la fijación mediada por células diana (megacariocitos y plaquetas) de romiplostim, que puede saturarse cuando se administran las dosis más altas.
Eliminación
La semivida de eliminación de romiplostim en pacientes con PTI oscila entre 1 y 34 días (mediana,
3,5 días). La eliminación de romiplostim plasmático depende en parte del receptor de la TPO en las plaquetas. En consecuencia, para una dosis dada los pacientes con recuentos plaquetarios elevados se asocian a bajas concentraciones plasmáticas y viceversa. En otro ensayo clínico sobre la PTI, no se observó acumulación en las concentraciones plasmáticas tras seis dosis semanales de romiplostim
(3 Mg/kg).
Poblaciones especiales
No se ha investigado la farmacocinética de romiplostim en pacientes con insuficiencia renal o hepática. La farmacocinética de romiplostim no parece estar afectada en un grado clínicamente significativo por la edad, el peso y el sexo.
5.3 Datos preclínicos sobre seguridad
Se realizaron ensayos toxicológicos a dosis múltiples de romiplostim en ratas durante cuatro semanas y en monos durante seis meses. En general, los efectos observados durante estos ensayos estaban relacionados con la actividad trombopoyética de romiplostim y fueron similares independientemente de la duración del ensayo. Las reacciones en el lugar de inyección también estaban relacionadas con la administración de romiplostim. Se ha observado mielofibrosis en la médula ósea de las ratas a todos los niveles de dosis evaluados. En estos ensayos, no se observó mielofibrosis en animales tras un periodo de recuperación de cuatro semanas después del tratamiento, lo que indicaba reversibilidad.
En un ensayo de toxicología de un mes de duración en ratas y monos, se observó una disminución leve del recuento de glóbulos rojos, hematocrito y hemoglobina. También se detectó un efecto estimulante en la producción de leucocitos, con un ligero aumento de los recuentos sanguíneos periféricos de neutrófilos, Íinfocitos, monocitos y eosinófilos. En el ensayo crónico de larga duración con monos, no se observaron efectos en los linajes eritroides y leucocitarios al administrar romiplostim durante seis meses rebajando su administración de tres veces a la semana a una. Además, en los ensayos pivotales de fase 3 romiplostim no afectó a los linajes de glóbulos rojos y blancos en comparación con los sujetos que recibieron placebo.
Debido a la formación de anticuerpos neutralizantes, los efectos farmacodinámicos de romiplostim en ratas descendían con frecuencia con una duración prolongada de la administración. Los ensayos toxicocinéticos no mostraron interacción de los anticuerpos en las concentraciones medidas. Aunque en los ensayos con animales se probaron dosis elevadas, debido a las diferencias entre las especies de laboratorio y los humanos en relación con la sensibilidad ante los efectos farmacodinámicos de romiplostim y el efecto de los anticuerpos neutralizantes, no se pueden calcular de forma fiable los márgenes de seguridad.
Carcinogénesis
No se ha investigado el potencial carcinogénico de romiplostim. Por tanto, el riesgo de carcinogénesis potencial en humanos sigue sin conocerse.
Toxicidad reproductora
En todos los ensayos de desarrollo se formaron anticuerpos neutralizantes, que pueden tener efectos de inhibición sobre romiplostim. En los ensayos de desarrollo embriofetal en ratones y ratas, sólo se observaron reducciones del peso corporal de la madre en ratones. En los ratones había signos de un aumento de pérdidas postimplantación. En los ensayos de desarrollo pre y postnatal en ratas se observó un aumento de la duración de la gestación y un ligero aumento en la incidencia de mortalidad perinatal de las crías. Se sabe que romiplostim atraviesa la barrera placentaria en las ratas y puede transmitirse de la madre al feto en desarrollo y estimular la producción plaquetaria del feto. No se han observado efectos de romiplostim sobre la fertilidad en ratas.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Manitol (E421)
Sacarosa
L-histidina
Ácido clorhídrico (para ajuste del pH)
Polisorbato 20
Disolvente:
Agua para preparaciones inyectables
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección 6.6.
6.3 Periodo de validez 3 años.
Después de la reconstitución: Se ha demostrado estabilidad química y física en uso durante 24 horas a 25°C y durante 24 horas entre 2°C y 8°C, si se mantiene protegido de la luz y en el vial original.
Desde un punto de vista microbiológico, el medicamento debe usarse inmediatamente. Si no se usa inmediatamente, los tiempos y condiciones de conservación durante el uso antes de su utilización son responsabilidad del usuario y no deberían superar las 24 horas a 25°C ó 24 horas en la nevera (entre 2°C y 8°C), protegido de la luz.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
Puede estar temporalmente fuera de la nevera durante un período máximo de 24 horas a temperatura ambiente (hasta 25 °C).
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase Polvo:
Vial de 5 ml (vidrio transparente tipo 1) con tapón (caucho clorobutilo), precinto (aluminio) y una cápsula de cierre del tipo flip-off (polipropileno).
Disolvente:
Nplate 250 microgramos polvo y disolvente para solución inyectable: Jeringa precargada (vidrio transparente tipo 1 con émbolo de goma de bromobutilo) que contiene 0,72 ml de agua para preparaciones inyectables para reconstitución.
Nplate 500 microgramos polvo y disolvente para solución inyectable: Jeringa precargada (vidrio transparente tipo 1 con émbolo de goma de bromobutilo) que contiene 1,2 ml de agua para preparaciones inyectables para reconstitución.
Tamaño del envase:
Nplate 250 microgramos polvo y disolvente para solución inyectable:
Nplate se suministra en 1 envase o en envase múltiple con 4 envases. Cada envase contiene:
1 vial de 250 microgramos de romiplostim.
1 jeringa precargada que contiene 0,72 ml de agua para preparaciones inyectables para reconstitución. 1 émbolo para la jeringa precargada.
1 adaptador del vial estéril.
1 jeringa estéril de 1 ml con cierre Luer.
1 aguja de seguridad estéril.
4 toallitas de alcohol.
Nplate 500 microgramos polvo y disolvente para solución inyectable:
Nplate se suministra en 1 envase o en envase múltiple con 4 envases. Cada envase contiene:
1 vial de 500 microgramos de romiplostim.
1 jeringa precargada que contiene 1,2 ml de agua para preparaciones inyectables para reconstitución.
1 émbolo para la jeringa precargada.
1 adaptador del vial estéril.
1 jeringa estéril de 1 ml con cierre Luer.
1 aguja de seguridad estéril.
4 toallitas de alcohol.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Nplate es un medicamento estéril pero sin conservantes y está indicado para un solo uso. Nplate debe reconstituirse siguiendo las recomendaciones de buena práctica aséptica.
Nplate 250 microgramos polvo v disolvente para solución inyectable
Nplate 250 microgramos polvo para solución inyectable debe reconstituirse con 0,72 ml de agua estéril para preparaciones inyectables para producir un volumen liberado de 0,5 ml. En cada vial se incluye una cantidad adicional para garantizar que se puedan administrar 250 qg de romiplostim (ver abajo la tabla que incluye información sobre el contenido del vial).
Nplate 500 microgramos polvo v disolvente para solución inyectable
Nplate 500 microgramos polvo para solución inyectable debe reconstituirse con 1,2 ml de agua estéril para preparaciones inyectables para producir un volumen liberado de 1 ml. En cada vial se incluye una cantidad adicional para garantizar que se puedan administrar 500 qg de romiplostim (ver abajo la tabla que incluye información sobre el contenido del vial).
Contenido del vial:
|
Vial de un solo uso de Nplate |
Contenido total de romiplostim en el vial |
Volumen de agua estéril para preparaciones inyectables |
Volumen y producto liberado |
Concentra ción final | ||
|
250 qg |
375 qg |
añadir |
0,72 ml |
= |
250 qg en 0,5 ml |
500 qg/ml |
|
500 qg |
625 qg |
añadir |
1,2 ml |
= |
500 qg en 1 ml |
500 qg/ml |
Desde un punto de vista microbiológico, el producto debe usarse inmediatamente. Si no se usa inmediatamente, los tiempos y condiciones de conservación durante el uso antes de su utilización son responsabilidad del usuario y no deberían superar las 24 horas a 25°C ó 24 horas en la nevera (entre 2°C y 8°C), protegido de la luz.
1. Retire la cápsula de cierre de plástico del vial que
contiene el polvo de Nplate y limpie el tapón de goma con la toallita de alcohol suministrada.
2.
Acople el adaptador del vial al vial de Nplate despegando la tapa de papel del adaptador del vial, manteniendo el adaptador del vial en su envase. Manteniendo el vial sobre una mesa, presione hacia abajo el adaptador del vial hacia el centro del vial hasta que esté bien sujeto.
Nota: Para prevenir la contaminación del producto, no toque la punta del adaptador del vial o cierre Luer.
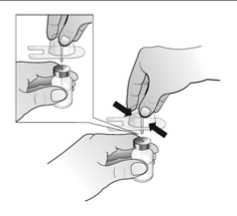
3.
4.
Una el émbolo a la jeringa precargada de agua para preparaciones inyectables, enroscando el émbolo en sentido de las agujas del reloj dentro de la jeringa, hasta que note una pequeña resistencia.
Sujetando la jeringa precargada de agua para preparaciones inyectables con una mano, doble la punta de la cubierta de plástico blanco hacia abajo con la otra mano. Esto romperá el precinto de la cubierta de plástico blanco. Una vez el precinto esté roto, tire de la cubierta para separar la tapa de goma gris de la punta de plástico transparente de la jeringa.
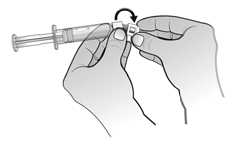
6. Manteniendo el vial sobre una mesa, acople la jeringa precargada con agua para preparaciones inyectables al adaptador del vial: sostenga la parte extema del adaptador del vial con una mano y enrosque la punta de la jeringa en sentido de las agujas del reloj en el adaptador con la otra mano, hasta que note una pequeña resistencia.
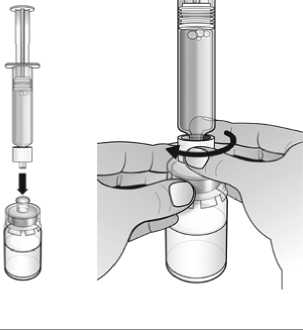
7. Inyecte muy lentamente y con cuidado toda el agua
dentro del vial con el polvo. El agua debe caer lentamente sobre el polvo. Remueva SUAVEMENTE el vial hasta que se disuelva todo el polvo y el líquido del vial sea transparente e incoloro.
No sacuda el vial.
Nota: Desde un punto de vista microbiológico, el producto debe utilizarse inmediatamente después de la reconstitución. Si el producto reconstituido no se usa inmediatamente, no retire la jeringa del adaptador del vial para conservar la integridad microbiológica.
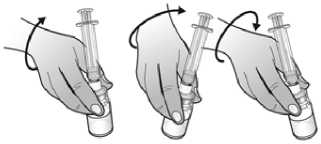
Nota: La disolución completa del polvo puede tardar hasta 2 minutos.
Antes de continuar:
Inspeccione visualmente la solución reconstituida de partículas y/o decoloración. La solución reconstituida debe ser transparente e incolora y no debe administrarse si se observan partículas y/o decoloración.
Asegúrese de que la solución está completamente disuelta antes de retirar la jeringa.
8. Retire la j eringa precargada vacía del adaptador del vial.
9. Saque la jeringa de administración de 1 ml del envase. Una la jeringa de 1 ml al adaptador del vial de la solución reconstituida girando la punta de la jeringa en el adaptador del vial hasta que note una pequeña resistencia.
10.
Ponga boca abajo el conjunto de jeringa y vial, de
manera que el vial del producto reconstituido quede por encima de la jeringa. Retire toda la solución del medicamento en la jeringa de administración.
Asegúrese que el émbolo permanece en la jeringa.
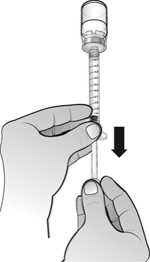
12.
13.
15.
16.
Asegúrese de que la jeringa de administración contiene la cantidad correcta de solución para la dosis del paciente, devolviendo cualquier exceso de solución al vial.
Nota: Saque todas las burbujas de aire de la jeringa para asegurar que la jeringa contiene la cantidad exacta de solución.
Desenrosque la jeringa de administración del adaptador del vial.
Acople la aguja de seguridad a la jeringa de administración cargada girando la aguja en sentido horario dentro de la punta del cierre Luer de la jeringa.
Prepare el lugar de inyección con una toallita nueva de alcohol. Tire de la cubierta de seguridad rosa hacia la jeringa alejándola de la aguja.
Retire el protector transparente de la aguja preparada
sosteniendo la jeringa en una mano y tirando del protector cuidadosamente con la otra mano.
Administre la inyección subcutánea siguiendo los protocolos locales y una buena técnica aséptica.
Tras la inyección, presione la tapa rosa de seguridad
empujándola hacia delante con la misma mano hasta que oiga un clic o note que se ha cerrado.
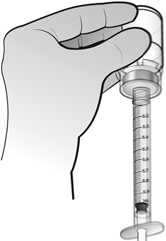
I
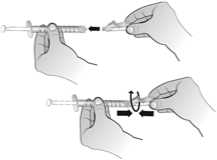
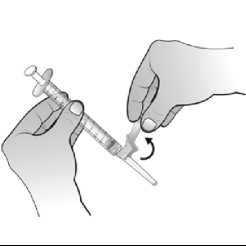
Para las condiciones de conservación del producto reconstituido, ver sección 6.3.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Amgen Europe B.V. Minervum 7061 4817 ZK Breda Países Bajos
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/497/005
EU/1/08/497/006
EU/1/08/497/007
EU/1/08/497/008
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 4 febrero 2009 Fecha de la última renovación: 20 diciembre 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
A. FABRICANTES DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTES DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
Nombre v dirección de los fabricantes del principio activo biológico
Amgen Inc
One Amgen Center Drive Thousand Oaks, CA 91320
usa
Amgen Manufacturing Limited State Road 31,
Km 24.6,
Juncos,
Puerto Rico 00777-4060
usa
Nombre v dirección de los fabricantes responsables de la liberación de los lotes
Amgen Europe BV Minervum 7061 NL-4817 ZK Breda Países Bajos
Amgen Technologv Ireland (ADL)
Potterv Road Dun Laoghaire Co Dublin Irlanda
El prospecto impreso del medicamento debe especificar el nombre v dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos;
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
• Medidas adicionales de minimización de riesgos
El TAC deberá acordar los detalles de las siguientes herramientas educacionales con la Autoridad Nacional Competente y deberá instaurar el programa a nivel nacional.
Calculadora de dosis
• A los médicos involucrados en la prescripción de romiplostim, se les facilitará una calculadora de dosis para simplificar el cálculo correcto de la dosis y guiar en los procedimientos correctos de reconstitución y de administración.
Kit de formación para la administración domiciliaria
• A los médicos que expresen un interés en iniciar la autoadministración en pacientes específicos, se les facilitará un kit de formación para la administración domiciliaria para esos pacientes. El kit de formación para la administración domiciliaria incluye materiales para los profesionales sanitarios sobre cómo seleccionar y formar a pacientes en la autoadministración de romiplostim; y para los pacientes, con el fin de ayudarles con el proceso de preparación y autoadministración de la dosis correcta de romiplostim.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
Nplate 250 microgramos polvo para solución inyectable romiplostim
2. PRINCIPIO ACTIVO
Vial que contiene 250 microgramos de romiplostim. Tras la reconstitución, un volumen liberado de 0,5 ml de solución contiene 250 microgramos de romiplostim (500 microgramos/ml).
3. LISTA DE EXCIPIENTES
Manitol (E421), sacarosa, l-histidina, ácido clorhídrico (para ajuste del pH) y polisorbato 20.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo para solución inyectable. 1 vial.
4 viales.
5. FORMA Y VÍA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD:
Después de la reconstitución: 24 horas si se almacena a 25°C o en nevera (entre 2°C y 8°C) si se mantiene en el vial original y protegido de la luz.
Conservar en nevera.
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Amgen Europe B.V. Minervum 7061 4817 ZK Breda Países Bajos
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/497/001
EU/1/08/497/003
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Nplate 250
ETIQUETA DEL VIAL
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Nplate 250 polvo para inyectable romiplostim
sc
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
exp
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
250 ^g
6. OTROS
Amgen Europe B.V.
1. NOMBRE DEL MEDICAMENTO
Nplate 500 microgramos polvo para solución inyectable romiplostim
2. PRINCIPIO ACTIVO
Vial que contiene 500 microgramos de romiplostim. Tras la reconstitución, un volumen liberado de 1 ml de solución contiene 500 microgramos de romiplostim (500 microgramos/ml).
3. LISTA DE EXCIPIENTES
Manitol (E421), sacarosa, l-histidina, ácido clorhídrico (para ajuste del pH) y polisorbato 20.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo para solución inyectable. 1 vial.
4 viales.
5. FORMA Y VÍA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD:
Después de la reconstitución: 24 horas si se almacena a 25°C o en nevera (entre 2°C y 8°C) si se mantiene en el vial original y protegido de la luz.
Conservar en nevera.
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Amgen Europe B.V. Minervum 7061 4817 ZK Breda Países Bajos
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/497/002
EU/1/08/497/004
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Nplate 500
ETIQUETA DEL VIAL
1. NOMBRE DEL MEDICAMENTO Y VÍA DE ADMINISTRACIÓN
Nplate 500 polvo para inyectable romiplostim
sc
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
exp
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
500 ^g
6. OTROS
Amgen Europe B.V.
1. NOMBRE DEL MEDICAMENTO
Nplate 250 microgramos polvo y disolvente para solución inyectable romiplostim
2. PRINCIPIO ACTIVO
Vial que contiene 250 microgramos de romiplostim. Tras la reconstitución, un volumen liberado de 0,5 ml de solución contiene 250 microgramos de romiplostim (500 microgramos/ml).
3. LISTA DE EXCIPIENTES
Polvo: Manitol (E421), sacarosa, l-histidina, ácido clorhídrico (para ajuste del pH) y polisorbato 20. Disolvente: agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Componente de un envase múltiple, no se vende por separado. 1 envase contiene:
1 vial de polvo para solución inyectable.
1 jeringa precargada que contiene 0,72 ml de disolvente.
1 émbolo para la jeringa precargada.
1 adaptador del vial estéril.
1 jeringa estéril de 1 ml con cierre Luer.
1 aguja de seguridad estéril.
4 toallitas de alcohol.
5. FORMA Y VÍA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
cad
Después de la reconstitución: 24 horas si se almacena a 25°C o en nevera (entre 2°C y 8°C) sí se mantiene en el vial original y protegido de la luz.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera.
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Cualquier medicamento no utilizado o material residual debe desecharse de acuerdo con los requerimientos locales.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Amgen Europe B.V. Minervum 7061 4817 ZK Breda Países Bajos
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/497/006 - 1 envase
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
1. NOMBRE DEL MEDICAMENTO
Nplate 250 microgramos polvo y disolvente para solución inyectable romiplostim
2. PRINCIPIO ACTIVO
Vial que contiene 250 microgramos de romiplostim. Tras la reconstitución, un volumen liberado de 0,5 ml de solución contiene 250 microgramos de romiplostim (500 microgramos/ml).
3. LISTA DE EXCIPIENTES
Polvo: Manitol (E421), sacarosa, l-histidina, ácido clorhídrico (para ajuste del pH) y polisorbato 20. Disolvente: agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Un envase contiene:
Envase múltiple: contiene 4 envases,
Cada envase contiene:
1 vial de polvo para solución inyectable.
1 jeringa precargada que contiene 0,72 ml de disolvente. 1 émbolo para la jeringa precargada.
1 adaptador del vial estéril.
1 jeringa estéril de 1 ml con cierre Luer.
1 aguja de seguridad estéril.
4 toallitas de alcohol.
5. FORMA Y VÍA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
cad
Después de la reconstitución: 24 horas si se almacena a 25°C o en nevera (entre 2°C y 8°C) sí se mantiene en el vial original y protegido de la luz.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera.
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Cualquier medicamento no utilizado o material residual debe desecharse de acuerdo con los requerimientos locales.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Amgen Europe B.V. Minervum 7061 4817 ZK Breda Países Bajos
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/497/005 - 1 envase EU/1/08/497/006 - 4 envases
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
1. NOMBRE DEL MEDICAMENTO
Nplate 500 microgramos polvo y disolvente para solución inyectable romiplostim
2. PRINCIPIO ACTIVO
Vial que contiene 500 microgramos de romiplostim. Tras la reconstitución, un volumen liberado de 1 ml de solución contiene 500 microgramos de romiplostim (500 microgramos/ml).
3. LISTA DE EXCIPIENTES
Polvo: Manitol (E421), sacarosa, l-histidina, ácido clorhídrico (para ajuste del pH) y polisorbato 20. Disolvente: agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Componente de un envase múltiple, no se vende por separado. 1 envase contiene:
1 vial de polvo para solución inyectable.
1 jeringa precargada que contiene 1,2 ml de disolvente.
1 émbolo para la jeringa precargada.
1 adaptador del vial estéril.
1 jeringa estéril de 1 ml con cierre Luer.
1 aguja de seguridad estéril.
4 toallitas de alcohol.
5. FORMA Y VÍA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
cad
Después de la reconstitución: 24 horas si se almacena a 25°C o en nevera (entre 2°C y 8°C) sí se mantiene en el vial original y protegido de la luz.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera.
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Cualquier medicamento no utilizado o material residual debe desecharse de acuerdo con los requerimientos locales.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Amgen Europe B.V. Minervum 7061 4817 ZK Breda Países Bajos
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/497/008 - 1 envase
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
1. NOMBRE DEL MEDICAMENTO
Nplate 500 microgramos polvo y disolvente para solución inyectable romiplostim
2. PRINCIPIO ACTIVO
Vial que contiene 500 microgramos de romiplostim. Tras la reconstitución, un volumen liberado de 1 ml de solución contiene 500 microgramos de romiplostim (500 microgramos/ml).
3. LISTA DE EXCIPIENTES
Polvo: Manitol (E421), sacarosa, l-histidina, ácido clorhídrico (para ajuste del pH) y polisorbato 20. Disolvente: agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Un envase contiene:
Envase múltiple: contiene 4 envases,
Cada envase contiene:
1 vial de polvo para solución inyectable.
1 jeringa precargada que contiene 1,2 ml de disolvente. 1 émbolo para la jeringa precargada.
1 adaptador del vial estéril.
1 jeringa estéril de 1 ml con cierre Luer.
1 aguja de seguridad estéril.
4 toallitas de alcohol.
5. FORMA Y VÍA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía subcutánea.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
cad
Después de la reconstitución: 24 horas si se almacena a 25°C o en nevera (entre 2°C y 8°C) sí se mantiene en el vial original y protegido de la luz.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera.
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Cualquier medicamento no utilizado o material residual debe desecharse de acuerdo con los requerimientos locales.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Amgen Europe B.V. Minervum 7061 4817 ZK Breda Países Bajos
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/08/497/007 - 1 envase EU/1/08/497/008 - 4 envases
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETA DEL AGUA PARA PREPARACIONES INYECTABLES
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Disolvente para Nplate
Agua para preparaciones inyectables
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
exp
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
0,72 ml 1,2 ml
6. OTROS
Para el KIT 250 ^g Para el KIT 500 ^g
B. PROSPECTO
Prospecto: información para el usuario
Nplate 250 microgramos polvo para solución inyectable Nplate 500 microgramos polvo para solución inyectable
Romiplostim
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Nplate y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Nplate
3. Cómo usar Nplate
4. Posibles efectos adversos
5. Conservación de Nplate
6. Contenido del envase e información adicional
1. Qué es Nplate y para qué se utiliza
El principio activo de Nplate es romiplostim, que es una proteína utilizada para tratar los recuentos bajos de plaquetas en pacientes con púrpura trombocitopénica inmune (idiopática) (siglas PTI). La PTI es una enfermedad en la que el sistema inmunitario de su cuerpo destruye sus propias plaquetas. Las plaquetas son las células de la sangre que ayudan a cicatrizar las heridas y forman coágulos de sangre. Los recuentos de plaquetas muy bajos pueden causar hematomas y hemorragias graves.
Nplate se utiliza en pacientes adultos (18 años o mayores) a los que se les podría o no extirpar el bazo debido a la PTI crónica y que han sido previamente tratados con corticosteroides o inmunoglobulinas, cuando estos tratamientos no funcionan.
Nplate funciona estimulando la médula ósea (parte del hueso que genera las células sanguíneas) para que produzca más plaquetas. Esto debería ayudar a evitar los hematomas y las hemorragias relacionadas con la PTI.
2. Qué necesita saber antes de empezar a usar Nplate
No use Nplate
• si es alérgico a romiplostim o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
• si es alérgico a otros medicamentos producidos mediante tecnología del ADN que utiliza el microorganismo Escherichia coli (E. coli).
Advertencias y precauciones
• Si deja de tomar Nplate es probable que vuelva a tener un recuento bajo de plaquetas (trombocitopenia). Si deja de tomar Nplate, su recuento de plaquetas tendrá que ser controlado, y su médico le comentará las precauciones adecuadas.
• Si tiene riesgo de aparición de coágulos en la sangre o sí Ios coágulos sanguíneos son frecuentes en su familia. El riesgo de coágulos en la sangre puede estar aumentado también sí:
- tiene problemas en el hígado;
- es usted una persona anciana (> 65 años);
- está postrado en la cama;
- tiene cáncer;
- está tomando píldoras anticonceptivas o terapia hormonal sustitutíva;
- se ha sometido recientemente a cirugía o ha padecido de una lesión;
- es obeso (sobrepeso);
- es fumador.
Consulte a su médico, farmacéutico o enfermero antes de empezar a usar Nplate.
Sí presenta un recuento de plaquetas muy alto, puede aumentar el riesgo de coágulos sanguíneos. Su médico ajustará su dosis de Nplate para asegurarse de que su recuento de plaquetas no sea demasiado alto.
Cambios en la médula ósea (aumento de reticulina y posible fíbrosis en la médula ósea)
El uso a largo plazo de Nplate puede causar cambios en su médula ósea. Estos cambios pueden llevar a que tenga las células de la sangre anormales o que su cuerpo produzca menor número de células sanguíneas. La forma leve de estos cambios en la médula ósea se llama “aumento de reticulina” y se ha observado en los ensayos clínicos de Nplate. Se desconoce si esto podría progresar a una forma más grave llamada “fíbrosis”. En sus análisis de sangre pueden aparecer señales de cambios en la médula ósea como anomalías. Su médico decidirá si un análisis anormal de sangre significa que se debería hacer un análisis de la médula ósea o sí debe interrumpir el tratamiento con Nplate.
Empeoramiento del cáncer de la sangre
Su médico puede decidir realizar una biopsia de la médula ósea si considera que es necesario asegurarse que tiene PTI y no otra enfermedad tal como Síndrome Mielodisplásico (SMD). Sí usted padece SMD y recibe Nplate, puede presentar un aumento del recuento de células blásticas y un empeoramiento de SMD hasta progresar a una leucemia mieloide aguda, que es un tipo de cáncer de la sangre.
Pérdida de respuesta a romiplostim
Sí presenta pérdida de respuesta a romiplostim o imposibilidad de mantener una respuesta a las plaquetas durante el tratamiento con romiplostim, su médico averiguará las razones del por qué, incluyendo si está presentando aumento en las fibras de médula ósea (reticulina) o sí ha desarrollado anticuerpos, que neutralizan la actividad de romiplostim.
Niños y adolescentes
No se recomienda el uso de Nplate en niños menores de 18 años.
Uso de Nplate con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Sí está también tomando otros medicamentos que evitan la formación de coágulos sanguíneos (tratamiento con anticoagulantes o antiplaquetarios) existe un mayor riesgo de hemorragias. Su médico le comentará este aspecto.
Sí está tomando corticosteroides, danazol y/o azatioprina, que los podría estar recibiendo para tratar su PTI, puede que haya que reducir o suprimir su administración al combinarlos con Nplate.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento. No se recomienda utilizar Nplate durante el embarazo a menos que su médico se lo indique.
Se desconoce si romiplostim se excreta en la leche humana. No se recomienda utilizar Nplate durante la lactancia. La decisión de interrumpir la lactancia o interrumpir el tratamiento con romiplostim debe tomarse teniendo en cuenta las ventajas de la lactancia para el bebé y las ventajas del tratamiento con romiplostim para la paciente.
Conducción y uso de máquinas
Consulte a su médico antes de conducir o utilizar máquinas, ya que algunos de los efectos adversos (ej: episodios temporales de mareos) pueden alterar su capacidad para llevar a cabo estas actividades de forma segura.
3. Cómo usar Nplate
Nplate debe administrarse bajo la supervisión directa de un médico que controle con precisión la cantidad de Nplate administrada.
Nplate se administra una vez a la semana mediante inyección debajo de la piel (subcutánea).
La dosis inicial es de 1 microgramo de Nplate por kilogramo de peso corporal una vez por semana. Su médico le indicará la cantidad de Nplate que debe utilizar. Nplate debe inyectarse una vez a la semana para el mantenimiento de los recuentos de plaquetas. Su médico le extraerá muestras de sangre regularmente para evaluar de qué manera están respondiendo sus plaquetas y ajustar la dosis si fuera necesario.
Una vez que su recuento de plaquetas esté controlado, su médico seguirá realizándole análisis de sangre de control. Su dosis puede ajustarse más adelante a fin de mantener un control a largo plazo de su recuento de plaquetas.
Si usa más Nplate del que debe
Su médico se asegurará de que usted reciba la cantidad adecuada de Nplate. Si ha recibido más Nplate del que debiera, puede que no presente ningún síntoma físico pero sus niveles de plaquetas en sangre pueden aumentar a niveles muy altos y esto puede aumentar el riesgo de coagulación de la sangre. Por tanto, si su médico sospecha que ha recibido más Nplate del que debiera, se recomienda que sea controlado para observar cualquier signo o síntoma de efectos secundarios y que se le administre el tratamiento adecuado de inmediato.
Si usa menos Nplate del que debe
Su médico se asegurará de que usted reciba la cantidad adecuada de Nplate. Si ha recibido menos Nplate del que debe, puede que no presente ningún síntoma físico pero sus niveles de plaquetas en sangre pueden disminuir a niveles bajos y esto puede producir riesgo de sangrado. Por tanto, si su médico sospecha que ha recibido menos Nplate del que debe, se recomienda que sea controlado para observar cualquier signo o síntoma de efectos secundarios y que se le administre el tratamiento adecuado de inmediato.
Si olvidó usar Nplate
Si olvida una dosis de Nplate, su médico le indicará cuándo debe recibir la siguiente dosis.
Si interrumpe el tratamiento con Nplate
Si deja de utilizar Nplate, es probable que vuelva a presentar un recuento de plaquetas bajo (trombocitopenia). Su médico decidirá si debe dejar de tomar Nplate.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Muy frecuentes: pueden afectar a más de 1 de cada 10 personas
• dolor de cabeza;
• reacción alérgica;
• infección de la vía respiratoria superior.
Frecuentes: pueden afectar hasta 1 de cada 10 personas
• alteración en la médula ósea, incluyendo aumento de las fibras de la médula ósea (reticulina);
• dificultad para conciliar el sueño (insomnio);
• mareos;
• hormigueo o entumecimiento de las manos o los pies (parestesia);
• migraña;
• enrojecimiento de la piel (rubor);
• coágulo de sangre en una arteria del pulmón (embolia pulmonar);
• náuseas;
• diarrea;
• dolor abdominal;
• indigestión (dispepsia);
• estreñimiento;
• picor en la piel (prurito);
• sangrado debajo de la piel (equimosis);
• hematomas (contusión);
• erupción cutánea;
• dolor en la articulación (artralgia);
• dolor muscular o debilidad (mialgia);
• dolor en manos y pies;
• espasmo muscular;
• dolor de espalda;
• dolor de huesos;
• cansancio (fatiga);
• reacción en el lugar de la inyección;
• hinchazón en las manos y los pies (edema periférico);
• síntomas similares a la gripe (enfermedad semejante a la gripe);
• dolor;
• debilidad (astenia);
• fiebre (pirexia);
• escalofríos;
• contusión;
• hinchazón de la cara, labios, boca, lengua o garganta que puede causar dificultad al tragar o respirar (angioedema);
• gastroenteritis;
• palpitaciones.
Frecuentes: pueden afectar hasta 1 de cada 10 personas (pueden ser observados en análisis de
sangre u orina)
• recuento bajo de plaquetas en sangre (trombocitopenia) y recuento bajo de plaquetas en sangre (trombocitopenia) tras interrumpir el tratamiento con Nplate;
• recuento de plaquetas más elevado de lo normal (trombocitosis);
• anemia.
Poco frecuentes: pueden afectar hasta 1 de cada 100 personas
• alteración de la medula ósea; trastorno de la medula ósea que provoca cicatrización (mielofibrosis); aumento del tamaño del bazo (esplenomegalia); sangrado de la vagina (hemorragia vaginal), sangrado en el recto (hemorragia rectal); sangrado de la boca (hemorragia bucal); sangrado en el lugar de la inyección (hemorragia en el lugar de la inyección);
• ataque al corazón (infarto de miocardio); aumento de la frecuencia cardíaca;
• mareo o sensación giratoria (vértigo);
• problemas con los ojos incluyendo: sangrado en los ojos (hemorragia conjuntival); dificultad para enfocar o visión borrosa (trastorno de acomodación visual, papiloedema o trastorno de los ojos); ceguera; picor en los ojos (prurito ocular); aumento de lágrimas (aumento del lagrimeo); o alteraciones visuales;
• problemas con el sistema digestivo incluyendo: vómitos; mal aliento (halitosis); dificultad para tragar (disfagia); indigestión o acidez gástrica (trastorno de reflujo gastroesofágico); sangre en las heces (hematoquecia); malestar estomacal; úlceras en la boca o ampollas en la boca (estomatitis); dientes decolorados (decoloración dental);
• pérdida de peso; aumento de peso; intolerancia al alcohol; pérdida de apetito (anorexia o disminución de apetito); deshidratación;
• sensación general de malestar (malestar); dolor en el pecho; irritabilidad; hinchazón de la cara (edema facial); sensación de calor; aumento de la temperatura corporal; sensación nerviosa;
• gripe; infección localizada; inflamación de las fosas de la nariz y la garganta (nasofaringitis);
• problemas en la nariz y la garganta incluyendo: tos; secreción nasal (rinorrea); garganta seca; falta de aliento o dificultad en respirar (disnea); congestión nasal; dolor al respirar (respiración dolorosa);
• articulaciones hinchadas doloridas, causadas por el ácido úrico (producto de degradación de los alimentos) (gota);
• rigidez muscular; debilidad muscular; dolor de hombros; contracciones musculares;
• problemas con el sistema nervioso incluyendo contracciones musculares involuntarias (clonus); sentido del gusto distorsionado (disgeusia); disminución del sentido del gusto (hipogeusia); sensación de disminución de la sensibilidad, especialmente en la piel (hipoaestesia); alteraciones en las funciones nerviosas en brazos y piernas (neuropatía periférica); coágulo sanguíneo en el seno transverso (trombosis de seno transverso);
• depresión; sueños anormales;
• pérdida de cabello (alopecia); sensibilidad a la luz (reacción de fotosensibilidad); acné; reacción alérgica en la piel por el contacto de alérgenos (dermatitis de contacto); manifestaciones de la piel con erupción y ampollas (eczema); piel seca; enrojecimiento de la piel (eritema); descamación grave o erupción descamativa (erupción exfoliativa); crecimiento anormal del pelo; engrasamiento y picor de la piel por rascarse repetidamente (prurigo); sangrado debajo de la superficie de la piel o moretones debajo de la piel (purpura); erupción de la piel con picor (erupción papular); erupción de la piel con picor (erupción pruriginosa); erupción con picor generalizada (urticaria); protuberancia en la piel (nódulo en la piel); olor anormal de la piel (olor anormal de la piel);
• problemas con la circulación incluyendo coágulos sanguíneos en la vena del hígado (trombosis de la vena porta); trombosis venosa profunda; presión sanguínea baja (hipotensión); aumento de la presión sanguínea; bloqueo de un vaso sanguíneo (embolismo periférico); flujo sanguíneo reducido en las manos, tobillos o pies (isquemia periférica); hinchazón y coagulación en una vena, que puede ser extremamente blanda al tacto (flebitis o tromboflebitis superficial); coagulación sanguínea (trombosis);
• una alteración rara caracterizada por períodos de dolor con quemazón, enrojecimiento y calor en los pies y manos (eritromelalgia).
Poco frecuentes: pueden afectar hasta 1 de cada 100 personas (pueden ser observados en análisis de sangre u orina)
• un tipo raro de anemia en la cual las células rojas de la sangre, las células blancas de la sangre y las plaquetas están reducidas en número (anemia aplásica);
• aumento del recuento de las células blancas de la sangre (leucocitosis);
• exceso en la producción de plaquetas (trombocitaemia); aumento en el recuento de plaquetas; recuento anormal de las células de la sangre que previenen las hemorragias (recuento de plaquetas anormal);
• alteraciones en algún análisis de la sangre (aumento de las transaminasas; aumento de la lactato deshidrogenasa de la sangre);
• o cáncer de las células blancas de la sangre (mieloma múltiple);
• proteínas en la orina.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Nplate
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase y en la etiqueta del vial después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
Este medicamento puede estar temporalmente fuera de la nevera durante un período máximo de 24 horas a temperatura ambiente (hasta 250C). Debe ser devuelto a la nevera para conservarlo durante más de 24 horas.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Nplate
- El principio activo es romiplostim.
Cada vial de Nplate 250 microgramos polvo para solución inyectable contiene un total de 375 microgramos de romiplostim. Se ha añadido un volumen adicional en cada vial para asegurar que se pueden administrar 250 microgramos de romiplostim Tras la reconstitución, un volumen liberado de 0,5 ml de solución contiene 250 microgramos de romiplostim (500 microgramos/ml).
Cada vial de Nplate 500 microgramos polvo para solución inyectable contiene un total de 625 microgramos de romiplostim. Se ha añadido un volumen adicional en cada vial para asegurar que se pueden administrar 500 microgramos de romiplostim Tras la reconstitución, un volumen liberado de 1 ml de solución contiene 500 microgramos de romiplostim (500 microgramos/ml).
- Los demás componentes son manitol (E421), sacarosa, l-histidina, ácido clorhídrico (para ajuste del pH) y polisorbato 20.
Aspecto del producto y contenido del envase
Nplate es un polvo blanco para solución inyectable que se suministra en un vial de vidrio de 5 ml. Cada envase contiene 1 ó 4 viales de 250 microgramos ó 500 microgramos de romiplostim.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación:
Amgen Europe B.V.
Minervum 7061 4817 ZK Breda Países Bajos
Titular de la autorización de comercialización:
Amgen Europe B.V.
Minervum 7061 4817 ZK Breda Países Bajos
Responsable de la fabricación:
Amgen Technology Ireland (ADL)
Pottery Road Dun Laoghaire Co Dublin Irlanda
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización.
Belgie/Belgique/Belgien
s.a. Amgen n.v.
Tel/Tél: +32 (0)2 7752711
Etarapun
Am^^h Etarapna EOOfl Tea.: +359 (0)2 424 7440
Ceská republika
Amgen s.r.o.
Tel: +420 221 773 500
Danmark
Amgen filial af Amgen AB, Sverige Tlf: +45 39617500
Lietuva
Amgen Switzerland AG Vilniaus filialas Tel: +370 5 219 7474
Luxembourg/Luxemburg
s.a. Amgen Belgique/Belgien Tel/Tél: +32 (0)2 7752711
Magyarország
Amgen Kft.
Tel.: +36 1 35 44 700
Malta
Amgen B.V.
The Netherlands Tel: +31 (0)76 5732500
|
Eesti Amgen Switzerland AG Vilniaus filíalas Tel: +372 586 09553 |
Norge Amgen AB Tel: +47 23308000 |
|
EXláda Amgen EAAág OapgaKswrKá E.n.E. T^A,: +30 210 344700o |
Osterreich Amgen GmbH Tel: +43 (0)1 50 217 |
|
España Amgen S.A. Tel: +34 93 600 18 60 |
Polska Amgen Biotechnologia Sp. z o.o. Tel.: +48 22 581 3000 |
|
France Amgen S.A.S. Tél: +33 (0)9 69 363 363 |
Portugal Amgen Biofarmaceutica, Lda. Tel: +351 21 422055o |
|
Hrvatska Amgen d.o.o. Tel: +385 (0)1 562 57 20 |
Romania Amgen Romanía SRL Tel: +4021 527 3000 |
|
Ireland Amgen Limited United Kingdom Tel: +44 (0)1223 420305 |
Slovenija AMGEN zdravila d.o.o. Tel: +386 (0)1 585 1767 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Amgen Slovakia s.r.o. Tel: +421 33 321 13 22 |
|
Italia Amgen S.r.l. Tel: +39 02 6241121 |
Suomi/Finland Amgen AB, sivuliike Suomessa/Amgen AB, filial i Finland Puh/Tel: +358 (0)9 5490050o |
|
Kúnpoq C.A. Papaellinas Ltd ThA.: +357 22741 741 |
Sverige Amgen AB Tel: +46 (0)8 6951100 |
|
Latvija Amgen Switzerland AG Rrgas filíale Tel: +371 257 25888 |
United Kingdom Amgen Limited Tel: +44 (0)1223 420305 |
Fecha de la última revisión de este prospecto en Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
Esta información está destinada únicamente a profesionales del sector sanitario:
Nplate es un producto estéril pero sin conservantes y está indicado para un solo uso. Nplate debe reconstituirse siguiendo las recomendaciones de buena práctica aséptica.
• Nplate 250 microgramos polvo para solución inyectable debe reconstituirse con 0,72 ml de agua estéril para preparaciones inyectables para producir un volumen liberado de 0,5 ml. En cada vial se incluye una cantidad adicional para garantizar que se puedan administrar 250 qg de romiplostim (ver abajo la tabla que incluye información sobre el contenido del vial).
o
• Nplate 500 microgramos polvo para solución inyectable debe reconstituirse con 1,2 ml de
agua estéril para preparaciones inyectables para producir un volumen liberado de 1 ml. En cada vial se incluye una cantidad adicional para garantizar que se puedan administrar 500 qg de romiplostim (ver abajo la tabla que incluye información sobre el contenido del vial).
Contenido del vial:
|
Vial de un solo uso de Nplate |
Contenido total de romiplostim en el vial |
Volumen de agua estéril para preparaciones inyectables |
Volumen y producto liberado |
Concentra ción final | ||
|
250 qg |
375 qg |
añadir |
0,72 ml |
= |
250 qg en 0,5 ml |
500 qg/ml |
|
500 qg |
625 qg |
añadir |
1,2 ml |
= |
500 qg en 1 ml |
500 qg/ml |
No deben utilizarse soluciones de cloruro sódico o agua bacteriostática para reconstituir el medicamento.
El agua para preparaciones inyectables debe inyectarse en el vial. Durante la disolución, debe realizarse un movimiento circular suave e invertir el contenido del vial. No hay que sacudir ni agitar vigorosamente el vial. Por lo general, se tarda menos de 2 minutos en realizar la disolución de Nplate. Inspeccionar visualmente la solución en busca de partículas o decoloraciones antes de su administración. La solución reconstituida debe ser transparente e incolora y no debe administrarse si se observan partículas y/o decoloración.
Desde un punto de vista microbiológico, el medicamento debe usarse inmediatamente. Si no se usa inmediatamente, los tiempos y condiciones de conservación durante el uso antes de su utilización son responsabilidad del usuario y no deberían superar las 24 horas a 25°C ó 24 horas en la nevera (entre 2°C y 8°C), protegido de la luz.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
Prospecto: información para el usuario
Nplate 250 microgramos polvo y disolvente para solución inyectable Nplate 500 microgramos polvo y disolvente para solución inyectable
Romiplostim
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Nplate y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Nplate
3. Cómo usar Nplate
4. Posibles efectos adversos
5. Conservación de Nplate
6. Contenido del envase e información adicional
7. Instrucciones para preparar y administrar una inyección de Nplate
1. Qué es Nplate y para qué se utiliza
El principio activo de Nplate es romiplostim, que es una proteína utilizada para tratar los recuentos bajos de plaquetas en pacientes con púrpura trombocitopénica inmune (idiopática) (siglas PTI). La PTI es una enfermedad en la que el sistema inmunitario de su cuerpo destruye sus propias plaquetas. Las plaquetas son las células de la sangre que ayudan a cicatrizar las heridas y forman coágulos de sangre. Los recuentos de plaquetas muy bajos pueden causar hematomas y hemorragias graves.
Nplate se utiliza en pacientes adultos (18 años o mayores) a los que se les podría o no extirpar el bazo debido a la PTI crónica y que han sido previamente tratados con corticosteroides o inmunoglobulinas, cuando estos tratamientos no funcionan.
Nplate funciona estimulando la médula ósea (parte del hueso que genera las células sanguíneas) para que produzca más plaquetas. Esto debería ayudar a evitar los hematomas y las hemorragias relacionadas con la PTI.
2. Qué necesita saber antes de empezar a usar Nplate No use Nplate
• si es alérgico a romiplostim o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
• si es alérgico a otros medicamentos producidos mediante tecnología del ADN que utiliza el microorganismo Escherichia coli (E. coli).
Advertencias y precauciones
• Si deja de tomar Nplate es probable que vuelva a tener un recuento bajo de plaquetas (trombocitopenia). Si deja de tomar Nplate, su recuento de plaquetas tendrá que ser controlado, y su médico le comentará las precauciones adecuadas.
• Si tiene riesgo de aparición de coágulos en la sangre o si los coágulos sanguíneos son frecuentes en su familia. El riesgo de coágulos en la sangre puede estar también aumentado si:
- tiene problemas en el hígado;
- es usted una persona anciana (> 65 años);
- está postrado en la cama;
- tiene cáncer;
- está tomando píldoras anticonceptivas o terapia hormonal sustitutiva;
- se ha sometido recientemente a cirugía o ha padecido de una lesión;
- es obeso (sobrepeso);
- es fumador.
Consulte a su médico, farmacéutico o enfermero antes de empezar a usar Nplate.
Si presenta un recuento de plaquetas muy alto, puede aumentar el riesgo de coágulos sanguíneos. Su médico ajustará su dosis de Nplate para asegurarse de que su recuento de plaquetas no sea demasiado alto.
Cambios en la médula ósea (aumento de reticulina y posible fibrosis en la médula ósea)
El uso a largo plazo de Nplate puede causar cambios en su médula ósea. Estos cambios pueden llevar a que tenga las células de la sangre anormales o que su cuerpo produzca menor número de células sanguíneas. La forma leve de estos cambios en la médula ósea se llama “aumento de reticulina” y se ha observado en los ensayos clínicos de Nplate. Se desconoce si esto podría progresar a una forma más grave llamada “fibrosis”. En sus análisis de sangre pueden aparecer señales de cambios en la médula ósea como anomalías. Su médico decidirá si un análisis anormal de sangre significa que se debería hacer un análisis de la médula ósea o si debe interrumpir el tratamiento con Nplate.
Empeoramiento del cáncer de la sangre
Su médico puede decidir realizar una biopsia de la médula ósea si considera que es necesario asegurarse que tiene PTI y no otra enfermedad tal como Síndrome Mielodisplásico (SMD). Si usted padece SMD y recibe Nplate, puede presentar un aumento del recuento de células blásticas y un empeoramiento de SMD hasta progresar a una leucemia mieloide aguda, que es un tipo de cáncer de la sangre.
Pérdida de respuesta a romiplostim
Si presenta pérdida de respuesta a romiplostim o imposibilidad de mantener una respuesta a las plaquetas durante el tratamiento con romiplostim, su médico averiguará las razones del por qué, incluyendo si está presentando aumento en las fibras de médula ósea (reticulina) o si ha desarrollado anticuerpos, que neutralizan la actividad de romiplostim.
Niños y adolescentes
No se recomienda el uso de Nplate en niños menores de 18 años.
Uso de Nplate con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Si está también tomando otros medicamentos que evitan la formación de coágulos sanguíneos (tratamiento con anticoagulantes o antiplaquetarios) existe un mayor riesgo de hemorragias. Su médico le comentará este aspecto.
Si está tomando corticosteroides, danazol y/o azatioprina, que los podría estar recibiendo para tratar su PTI, puede que haya que reducir o suprimir su administración al combinarlos con Nplate.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento. No se recomienda utilizar Nplate durante el embarazo a menos que su médico se lo indique.
Se desconoce si romiplostim se excreta en la leche humana. No se recomienda utilizar Nplate durante la lactancia. La decisión de interrumpir la lactancia o interrumpir el tratamiento con romiplostim debe tomarse teniendo en cuenta las ventajas de la lactancia para el bebé y las ventajas del tratamiento con romiplostim para la paciente.
Conducción y uso de máquinas
Consulte a su médico antes de conducir o utilizar máquinas, ya que algunos de los efectos adversos (ej: episodios temporales de mareos) pueden alterar su capacidad para llevar a cabo estas actividades de forma segura.
3. Cómo usar Nplate
Nplate debe administrarse bajo la supervisión directa de un médico que controle con precisión la cantidad de Nplate administrada.
Nplate se administra una vez a la semana mediante inyección debajo de la piel (subcutánea).
La dosis inicial es de 1 microgramo de Nplate por kilogramo de peso corporal una vez por semana. Su médico le indicará la cantidad de Nplate que debe utilizar. Nplate debe inyectarse una vez a la semana para el mantenimiento de los recuentos de plaquetas. Su médico le extraerá muestras de sangre regularmente para evaluar de qué manera están respondiendo sus plaquetas y ajustar la dosis si fuera necesario.
Una vez que su recuento de plaquetas esté controlado, su médico seguirá realizándole análisis de sangre de control. Su dosis puede ajustarse más adelante a fin de mantener un control a largo plazo de su recuento de plaquetas.
Siga siempre exactamente las instrucciones de administración de Nplate indicadas por su médico. En caso de duda, consulte de nuevo a su médico cómo usar Nplate.
Instrucciones para preparar y administrar una inyección de Nplate
Después de una formación adecuada, su médico también le puede permitir autoinyectarse Nplate. Por favor, lea las instrucciones al final de este prospecto sobre cómo inyectar Nplate, tal como ha comentado con su médico. Si su médico le ha permitido autoinyectarse, debe hacer un seguimiento con él todos los meses para que determine si Nplate está funcionando o si debe considerar otro tratamiento.
Tras el primer mes de autoinyectarse Nplate, necesitará demostrar que todavía puede preparar e inyectarse Nplate correctamente.
Si usa más Nplate del que debe
Su médico se asegurará de que usted reciba la cantidad adecuada de Nplate. Si ha recibido más Nplate del que debiera, puede que no presente ningún síntoma físico pero sus niveles de plaquetas en sangre pueden aumentar a niveles muy altos y esto puede aumentar el riesgo de coagulación de la sangre. Por tanto, si su médico sospecha que ha recibido más Nplate del que debiera, se recomienda que sea controlado para observar cualquier signo o síntoma de efectos secundarios y que se le administre el tratamiento adecuado de inmediato.
Si su médico ha permitido que se autoinyecte y utiliza más Nplate del que debería, informe a su médico inmediatamente.
Si usa menos Nplate del que debe
Su médico se asegurará de que usted reciba la cantidad adecuada de Nplate. Si ha recibido menos Nplate del que debe, puede que no presente ningún síntoma físico pero sus niveles de plaquetas en sangre pueden disminuir a niveles bajos y esto puede producir riesgo de sangrado. Por tanto, si su médico sospecha que ha recibido menos Nplate del que debe, se recomienda que sea controlado para observar cualquier signo o síntoma de efectos secundarios y que se le administre el tratamiento adecuado de inmediato.
Si su médico ha permitido que se autoinyecte y utiliza menos Nplate del que debería, informe a su médico inmediatamente.
Si olvidó usar Nplate
Si olvida una dosis de Nplate, su médico le indicará cuándo debe recibir la siguiente dosis.
Si su médico ha permitido que se autoinyecte y olvidó la administración de su inyección, informe a su médico inmediatamente.
Si interrumpe el tratamiento con Nplate
Si deja de utilizar Nplate, es probable que vuelva a presentar un recuento de plaquetas bajo (trombocitopenia). Su médico decidirá si debe dejar de tomar Nplate.
Autoinyectarse Nplate
Su médico puede decidir que es mejor que se inyecte Nplate. Su médico, enfermero o farmacéutico le mostrará cómo se inyecta Nplate. No intente inyectarse usted mismo si no se ha formado. Es muy importante que prepare Nplate correctamente y tome la dosis correcta (ver la sección 7. Instrucciones para la preparación y administración de una inyección de Nplate, al final de este prospecto).
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Muy frecuentes: pueden afectar a más de 1 de cada 10 personas
• dolor de cabeza;
• reacción alérgica;
• infección de la vía respiratoria superior.
Frecuentes: pueden afectar hasta 1 de cada 10 personas
• alteración en la médula ósea, incluyendo aumento de las fibras de la médula ósea (reticulina);
• dificultad para conciliar el sueño (insomnio);
• mareos;
• hormigueo o entumecimiento de las manos o Ios pies (parestesia);
• migraña;
• enrojecimiento de la piel (rubor);
• coágulo de sangre en una arteria del pulmón (embolia pulmonar);
• náuseas;
• diarrea;
• dolor abdominal;
• indigestión (dispepsia);
• estreñimiento;
• picor en la piel (prurito);
• sangrado debajo de la piel (equimosis);
• hematomas (contusión);
• erupción cutánea;
• dolor en la articulación (artralgia);
• dolor muscular o debilidad (mialgia);
• dolor en manos y pies;
• espasmo muscular;
• dolor de espalda;
• dolor de huesos;
• cansancio (fatiga);
• reacción en el lugar de la inyección;
• hinchazón en las manos y los pies (edema periférico);
• síntomas similares a la gripe (enfermedad semejante a la gripe);
• dolor;
• debilidad (astenia);
• fiebre (pirexia);
• escalofríos;
• contusión;
• hinchazón de la cara, labios, boca, lengua o garganta que puede causar dificultad al tragar o respirar (angioedema);
• gastroenteritis;
• palpitaciones.
Frecuentes: pueden afectar hasta 1 de cada 10 personas (pueden ser observados en análisis de sangre u orina)
• recuento bajo de plaquetas en sangre (trombocitopenia) y recuento bajo de plaquetas en sangre (trombocitopenia) tras interrumpir el tratamiento con Nplate;
• recuento de plaquetas más elevado de lo normal (trombocitosis);
• anemia.
Poco frecuentes: pueden afectar hasta 1 de cada 100 personas
• alteración de la medula ósea; trastorno de la medula ósea que provoca cicatrización (mielofibrosis); aumento del tamaño del bazo (esplenomegalia); sangrado de la vagina (hemorragia vaginal), sangrado en el recto (hemorragia rectal); sangrado de la boca (hemorragia bucal); sangrado en el lugar de la inyección (hemorragia en el lugar de la inyección);
• ataque al corazón (infarto de miocardio); aumento de la frecuencia cardíaca;
• mareo o sensación giratoria (vértigo);
• problemas con los ojos incluyendo: sangrado en los ojos (hemorragia conjuntival); dificultad para enfocar o visión borrosa (trastorno de acomodación visual, papiloedema o trastorno de los ojos); ceguera; picor en los ojos (prurito ocular); aumento de lágrimas (aumento del lagrimeo); o alteraciones visuales;
• problemas con el sistema digestivo incluyendo: vómitos; mal aliento (halitosis); dificultad para tragar (disfagia); indigestión o acidez gástrica (trastorno de reflujo gastroesofágico); sangre en las heces (hematoquecia); malestar estomacal; úlceras en la boca o ampollas en la boca (estomatitis); dientes decolorados (decoloración dental);
• pérdida de peso; aumento de peso; intolerancia al alcohol; pérdida de apetito (anorexia o disminución de apetito); deshidratación;
• sensación general de malestar (malestar); dolor en el pecho; irritabilidad; hinchazón de la cara (edema facial); sensación de calor; aumento de la temperatura corporal; sensación nerviosa;
• gripe; infección localizada; inflamación de las fosas de la nariz y la garganta (nasofaringitis);
• problemas en la nariz y la garganta incluyendo: tos; secreción nasal (rinorrea); garganta seca; falta de aliento o dificultad en respirar (disnea); congestión nasal; dolor al respirar (respiración dolorosa);
• articulaciones hinchadas doloridas, causadas por el ácido úrico (producto de degradación de los alimentos) (gota);
• rigidez muscular; debilidad muscular; dolor de hombros; contracciones musculares;
• problemas con el sistema nervioso incluyendo contracciones musculares involuntarias (clonus); sentido del gusto distorsionado (disgeusia); disminución del sentido del gusto (hipogeusia); sensación de disminución de la sensibilidad, especialmente en la piel (hipoaestesia); alteraciones en las funciones nerviosas en brazos y piernas (neuropatía periférica); coágulo sanguíneo en el seno transverso (trombosis de seno transverso);
• depresión; sueños anormales;
• pérdida de cabello (alopecia); sensibilidad a la luz (reacción de fotosensibilidad); acné; reacción alérgica en la piel por el contacto de alérgenos (dermatitis de contacto); manifestaciones de la piel con erupción y ampollas (eczema); piel seca; enrojecimiento de la piel (eritema); descamación grave o erupción descamativa (erupción exfoliativa); crecimiento anormal del pelo; engrasamiento y picor de la piel por rascarse repetidamente (prurigo); sangrado debajo de la superficie de la piel o moretones debajo de la piel (purpura); erupción de la piel con picor (erupción papular); erupción de la piel con picor (erupción pruriginosa); erupción con picor generalizada (urticaria); protuberancia en la piel (nódulo en la piel); olor anormal de la piel (olor anormal de la piel);
• problemas con la circulación incluyendo coágulos sanguíneos en la vena del hígado (trombosis de la vena porta); trombosis venosa profunda; presión sanguínea baja (hipotensión); aumento de la presión sanguínea; bloqueo de un vaso sanguíneo (embolismo periférico); flujo sanguíneo reducido en las manos, tobillos o pies (isquemia periférica); hinchazón y coagulación en una vena, que puede ser extremamente blanda al tacto (flebitis o tromboflebitis superficial); coagulación sanguínea (trombosis);
• una alteración rara caracterizada por períodos de dolor con quemazón, enrojecimiento y calor en los pies y manos (eritromelalgia).
Poco frecuentes: pueden afectar hasta 1 de cada 100 personas (pueden ser observados en análisis
de sangre u orina)
• un tipo raro de anemia en la cual las células rojas de la sangre, las células blancas de la sangre y las plaquetas están reducidas en número (anemia aplásica);
• aumento del recuento de las células blancas de la sangre (leucocitosis);
• exceso en la producción de plaquetas (trombocitaemia); aumento en el recuento de plaquetas; recuento anormal de las células de la sangre que previenen las hemorragias (recuento de plaquetas anormal);
• alteraciones en algunas análisis de la sangre (aumento de las transaminasas; aumento de la lactato deshidrogenasa de la sangre);
• o cáncer de las células blancas de la sangre (mieloma múltiple);
• proteínas en la orina.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero,
incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede
comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V.
Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Nplate
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase y en la etiqueta del vial después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
Este medicamento puede estar temporalmente fuera de la nevera durante un período máximo de 24 horas a temperatura ambiente (hasta 250C). Debe ser devuelto a la nevera para conservarlo durante más de 24 horas.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Nplate
- El principio activo es romiplostim.
Cada vial de Nplate 250 microgramos polvo para solución inyectable contiene un total de 375 microgramos de romiplostim. Se ha añadido un volumen adicional en cada vial para asegurar que se pueden administrar 250 microgramos de romiplostim Tras la disolución, una cantidad de volumen liberado de 0,5 ml de solución contiene 250 microgramos de romiplostim (500 microgramos/ml).
Cada vial de Nplate 500 microgramos polvo para solución inyectable contiene un total de 625 microgramos de romiplostim. Se ha añadido un volumen adicional en cada vial para asegurar que se pueden administrar 500 microgramos de romiplostim. Tras la disolución, una cantidad de volumen liberado de 1 ml de solución contiene 500 microgramos de romiplostim (500 microgramos/ml).
- Los demás componentes son:
Polvo: manitol (E421), sacarosa, l-histidina, ácido clorhídrico (para ajuste del pH) y polisorbato 20.
Disolvente: agua para preparaciones inyectables.
Aspecto del producto y contenido del envase
Nplate es un polvo blanco para solución inyectable que se suministra en un vial de vidrio de 5 ml.
Nplate se suministra en un envase o envase múltiple con 4 envases. Cada envase contiene:
1 vial de 250 microgramos ó 500 microgramos de romiplostim.
1 jeringa precargada que contiene 0,72 ó 1,2 ml de agua para preparaciones inyectables para reconstitución.
1 émbolo para la jeringa precargada.
1 adaptador del vial estéril.
1 jeringa estéril de 1 mi con cierre Luer.
1 aguja de seguridad estéril.
4 toallitas de alcohol.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación:
Amgen Europe B.V.
Minervum 7061 4817 ZK Breda Países Bajos
Titular de la autorización de comercialización:
Amgen Europe B.V.
Minervum 7061 4817 ZK Breda Países Bajos
Responsable de la fabricación:
Amgen Technology Ireland (ADL)
Pottery Road Dun Laoghaire Co Dublin Irlanda
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización.
Belgie/Belgique/Belgien
s.a. Amgen n.v.
Tel/Tél: +32 (0)2 7752711
Etarapun
Am^^h Etarapna EOOfl Tea.: +359 (0)2 424 7440
Ceská republika
Amgen s.r.o.
Tel: +420 221 773 500
Danmark
Amgen filial af Amgen AB, Sverige Tlf: +45 39617500
Deutschland
amgen GmbH
Tel.: +49 89 1490960 Eesti
Amgen Switzerland AG Vilniaus filialas Tel: +372 586 09553
Lietuva
Amgen Switzerland AG Vilniaus filialas Tel: +370 5 219 7474
Luxembourg/Luxemburg
s.a. Amgen Belgique/Belgien Tel/Tél: +32 (0)2 7752711
Magyarország
Amgen Kft.
Tel.: +36 1 35 44 700
Malta
Amgen B.V.
The Netherlands Tel: +31 (0)76 5732500
Nederland
Amgen B.V.
Tel: +31 (0)76 5732500
Norge
Amgen AB Tel: +47 23308000
Suomi/Finland
Italia
Amgen S.r.l.
Tel: +39 02 6241121
Tel: +4021 527 3000
Portugal
Amgen Biofarmaceutica, Lda.
Osterreich
Amgen GmbH Tel: +43 (0)1 50 217
Polska
Amgen Biotechnologia Sp. z o.o. Tel.: +48 22 581 3000
Tel: +351 21 4220550
Amgen AB, sivuliike Suomessa/Amgen AB, filial i Finland
Puh/Tel: +358 (0)9 54900500
Latvija United Kingdom
Amgen Switzerland AG Rlgas filíale Amgen Limited
Tel: +371 257 25888 Tel: +44 (0)1223 420305
Fecha de la última revisión de este prospecto en Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
7. Instrucciones para la preparación y administración de una inyección de Nplate
Esta sección contiene información sobre cómo autoinyectarse Nplate. Es importante que no intente autoadministrarse la inyección si no ha recibido formación de su médico, enfermero o farmacéutico. Si usted tiene preguntas acerca de cómo ponerse la inyección, consulte a su médico, enfermero o farmacéutico. Es muy importante que el producto esté preparado correctamente y que la dosis que se administra sea correcta.
Esta sección se divide en las siguiente subsecciones:
Antes de comenzar
Paso 1. Coloque los materiales para la inyección
Paso 2. Prepare el vial para su uso, coloque el adaptador del vial
Paso 3. Prepare la jeringa de agua estéril
Paso 4. Disuelva Nplate inyectando el agua en el vial
Paso 5. Prepare una nueva jeringa para la inyección
Paso 6. Prepare la aguja para la inyección
Paso 7. Elija y prepare la zona de inyección
Paso 8. Inyecte el líquido de Nplate
Paso 9. Eliminación de los suministros
Antes de comenzar
Lea todas las instrucciones de uso cuidadosamente. Estas instrucciones son para pacientes que ya están formados por su profesional sanitario, como su médico, enfermero o farmacéutico, en la autoinyección. Si no ha sido entrenado, por favor póngase en contacto con su profesional sanitario.
El kit de autoinyección de Nplate debe mantenerse en el envase original hasta su utilización con el fin de proteger de la luz el vial de Nplate. Mantener el kit de autoinyección de Nplate refrigerado entre 20C y 80C.
Una vez que se ha disuelto Nplate, inyecte inmediatamente.
Es posible que quede un exceso de Nplate después de la administración de la dosis prescrita. ¡No reutilice Nplate! Cualquier exceso de Nplate disuelto, debe ser desechado inmediatamente después de completar el proceso de la inyección. El sobrante de Nplate del vial NUNCA debe ser reutilizado para otra inyección.
Paso 1. Coloque los materiales para la inyección Realice lo siguiente:
• Seleccione una superficie de trabajo plana y bien iluminada, como una mesa.
• Coja el kit de autoinyección de Nplate de la nevera. No utilizar si está congelado. Si tiene alguna duda sobre la conservación, contacte con su profesional sanitario para más instrucciones. Verifique la fecha de caducidad incluida en el kit de autoinyección. Si la fecha de caducidad ha pasado, no lo utilice. No continúe y comuníqueselo a su profesional sanitario.
• Nota: Si su médico le ha indicado que la dosis de Nplate requiere más de una inyección, necesitará utilizar más de un kit de autoinyección. Siga los pasos que se describen en este prospecto y utilice tantos kits de autoinyección, como sean necesarios para completar la dosis prescrita de Nplate.
• Asegúrese que tiene los siguientes componentes:

1 vial de polvo de 250 microgramos ó 500 microgramos x1
Adaptador del vial de 13mm x1

Émbolo para jeringa precargada de agua estéril x1
Jeringa precargada de agua estéril x1

Jeringa de 1 ml con cierre Luer x1
Aguja de seguridad para la inyección x1

• No abra los elementos hasta que esté indicado en las instrucciones.
• No utilice componentes que hayan sido alterados o dañados.
• No reutilizar los componentes.
Paso 2. Prepare el vial para su uso, coloque el adaptador del vial Utilizar: 2 paquetes de las toallitas de alcohol, 1 vial y 1 paquete de adaptador del vial. Realice lo siguiente:

• Lávese las manos con jabón y agua caliente.
• Limpie la superficie de trabajo plana con una toallita de alcohol nueva.
• Retire el tapón de plástico rojo (250 microgramos) o azul (500 microgramos) del vial.
• Utilizando una nueva toallita de alcohol, limpie el tapón del vial.




• No toque el tapón del vial, después de limpiarlo.
Retire el papel hacia atrás lentamente del adaptador del vial, manteniendo el adaptador del vial en el envase de plástico.
No toque el tapón del vial o la punta del adaptador de vial
Manteniendo el vial en la mesa, y manteniendo el adaptador del vial en el embalaje de plástico, alinee la punta del adaptador del vial con el centro del tapón del vial.
Empuje el adaptador del vial hacia abajo hasta que el vial esté firmemente en su lugar y no pueda empujar más hacia abajo.
Retire el envase de plástico del adaptador del vial, dejando el adaptador del vial en el vial.

No toque la parte de arriba del adaptador del vial.
Paso 3. Prepare la jeringa de agua estéril
Utilizar: La jeringa precargada de agua estéril y el émbolo.
Antes de iniciar el Paso 3, por favor tenga en cuenta lo siguiente:
• El plástico transparente del émbolo siempre DEBE estar conectado antes de romper la punta blanca de la jeringa precargada de agua. Lleve a cabo el paso 3a antes del paso 3b.
Realice lo siguiente:




• Paso 3a: Una el émbolo de plástico
transparente a la jeringa precargada de agua estéril, colocando el extremo roscado del émbolo en la jeringa y gire con cuidado la varilla en sentido horario sobre la jeringa de pistón gris, hasta que sienta una ligera resistencia. No apriete demasiado.
Paso 3b: Sosteniendo la jeringa con una mano, doblar la punta de la cubierta de plástico blanco hacia abajo con la otra mano. Esto romperá el sello de la tapa de plástico blanco.
• Una vez que se rompe el sello, retire el plástico blanco de la cubierta. Verá la goma gris en la tapa.
Paso 4. Disuelva Nplate, inyectando el agua en el vial
Utilizar: Jeringa precargada de agua estéril y vial con adaptador de vial adjunto. Antes de iniciar el Paso 4, por favor tenga en cuenta lo siguiente:
• Disuelva lentamente y con cuidado. Es una proteína y como tal, puede ser fácilmente dañada por agitación de la mezcla inadecuada y excesiva.
Realice lo siguiente:


Presione lenta y suavemente
Manteniendo el vial en la mesa, coloque la jeringa precargada de agua en el adaptador del vial, sosteniendo un lado del adaptador del vial con una mano y con la otra mano girando la punta de la jeringa en sentido horario dentro del adaptador hasta que note una ligera resistencia.
Muy lenta y suavemente, presione el émbolo hacia abajo, para inyectar toda el agua de la jeringa en el vial. El agua debe fluir lentamente sobre el polvo.
No fuerce el agua dentro del vial.
Nota: Después de inyectar el agua en el vial, es normal que el émbolo vuelva hacia arriba. No tiene que mantener la presión sobre el émbolo para el resto del Paso 4.
Antes de continuar:
• Asegúrese que toda el agua se inyecta desde la jeringa al vial antes de disolverlo.

• Manteniendo entre sus dedos la zona donde se conectan el vial y su adaptador, remueva suavemente el vial con una rotación de su muñeca hasta que todo el polvo se haya disuelto y el líquido en el vial sea claro e incoloro.
• Remueva suavemente el vial
No agite el vial.

• No haga rodar el vial entre las palmas
• Nota: el polvo puede tardar hasta 2 minutos en disolverse completamente.
Antes de continuar:
• Inspeccionar visualmente el líquido disuelto en busca de partículas y/o decoloración. Debe ser claro e incoloro y completamente disuelto.
Nota: Si hay cualquier color o partículas en el líquido, contacte con su profesional sanitario.
• Asegúrese que el líquido está completamente disuelto antes de quitar la jeringa.

Cuando Nplate esté completamente disuelto, quite la jeringa vacía del adaptador del vial, girándola en sentido contrario a las agujas del reloj.
• Deseche la jeringa vacía en un contenedor de objetos punzantes o peligrosos. Mantenga el vial disuelto de Nplate. Inmediatamente prepare una nueva jeringa para la inyección.
• No retarde la inyección de Nplate.
Paso 5. Prepare una nueva jeringa para la inyección
Utilizar: Una nueva jeringa de 1 ml del envase y el vial con Nplate disuelto y trasparente.
Antes de continuar:
• Revise su dosis antes de iniciar este paso. Nota: Nplate líquido es muy potente, por lo que la exactitud y la medida de la dosis son importantes.
• Asegúrese que se han eliminado todas las burbujas de aire antes de la inyección.
Realice lo siguiente:

marca de 1 ml

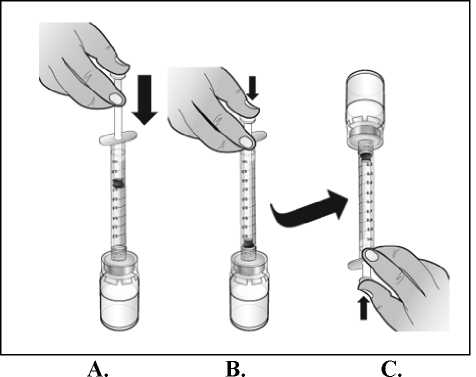
Gire hacia abajo
Extraiga una jeringa de 1 ml del envase.
Coja aire dentro de la jeringa hasta la marca de 1 ml.
No tire del émbolo más allá de la marca de 1 ml.
Coloque la jeringa de 1 ml al adaptador del vial del Nplate disuelto, girando la punta de la jeringa en sentido horario sobre el adaptador del vial hasta que note una ligera resistencia.
A.
B.
C.
Introduzca aire dentro del vial.
Mantenga la presión sobre el émbolo.
Gire el conjunto del vial y la jeringa hacia arriba, de manera que el vial esté directamente encima de la jeringa.
Succione todo el contenido del líquido dentro de la jeringa.



Burbujas de aire: Correcto
Incorrecto
- El volumen máximo liberado del vial de 250 microgramos es 0,5 ml y para el vial de 500 microgramos es 1 ml.
No tire del émbolo más allá del final de la
jeringa.
Asegúrese de que el émbolo permanece en la jeringa.
Compruebe y retire todas las burbujas de aire de la jeringa.
- Golpee suavemente la jeringa con sus dedos para separar las burbujas del líquido.
- Lentamente presione el émbolo hacia arriba para forzar las burbujas de aire fuera de la jeringa.
• Lentamente presione el émbolo para dejar únicamente la cantidad prescrita por su profesional sanitario.

Ajustar la cantidad a su dosis prescrita
• Asegúrese que la punta de la cabeza del émbolo se alinea con la marca de la jeringa correspondiente a su dosis prescrita. Si fuera necesario, devuelva líquido al vial para conseguir la dosis deseada.
• Haga una comprobación final para asegurar que la cantidad correcta de líquido para su dosis está en la jeringa y
que todas las burbujas de aire se han retirado.
Antes de continuar:
• Asegúrese que la cantidad correcta de líquido para su dosis se queda en la jeringa.
• Asegúrese que todas las burbujas de aire se retiran de la jeringa.

• Una vez que todas las burbujas se han
eliminado y la jeringa está llena con la dosis correcta, retire la jeringa del adaptador del vial.
Mantenga la jeringa llena en su mano y no toque la punta de la jeringa.

No ponga la jeringa boca abajo después de quitar el vial.
Paso 6. Prepare la aguja para la inyección
Utilizar: La jeringa llena con la dosis medida de Nplate y la aguja de seguridad. Realice lo siguiente:


Manteniendo la jeringa en la palma de su mano con la punta boca arriba, retire la aguja de seguridad del envase.
Coloque la aguja de seguridad en la jeringa llena. Aplique con fuerza la aguja de seguridad en la jeringa girando en sentido horario para cerrar la punta del cierre Luer.
El producto ya está preparado para la inyección. INMEDIATAMENTE continúe con el paso 7.
Paso 7. Elija y prepare la zona de inyección Utilizar: Nuevas toallitas de alcohol.
Realice lo siguiente:



Seleccione las zonas de inyección. Se
recomiendan 3 zonas para la inyección de
Nplate:
- Parte delantera del muslo medio
- Abdomen, excepto el área de 5 cm alrededor del ombligo
- Si otra persona le inyecta, también puede utilizar la parte exterior del brazo superior
- Rote las diferentes zonas para cada inyección.
No inyecte en zonas donde la piel está blanda, amoratada o dura.
No inyecte en zonas con cicatrices o estrías.
Limpie la zona donde se inyectará Nplate con una toallita de alcohol, usando un movimiento circular.
No toque el área otra vez, antes de inyectar.
Paso 8. Inyecte el líquido de Nplate
Utilizar: Conjunto con la jeringa llena y la aguja.
Realice lo siguiente:
• Tire de la cubierta rosa de seguridad
(hacia la jeringa y lejos de la aguja).
Retire el protector transparente de la aguja, manteniendo la jeringa en una mano y tirando cuidadosamente del protector directamente con la otra mano.




Retire el protector transparente de la aguja antes de inyectar.
Con una mano, pellizque suavemente la zona limpia de la piel y manténgala firmemente. Con la otra mano, sostenga la jeringa (como un lápiz) con un ángulo de
45° respecto a la piel.
Con un movimiento corto y rápido, introduzca la aguja dentro de la piel.
Inyecte la dosis prescrita subcutáneamente, tal y como le ha indicado su doctor, enfermero o farmacéutico.
Cuando la jeringa esté vacía, saque la aguja de la piel, manteniendo cuidadosamente el mismo ángulo en el que se insertó.
Puede sangrar un poco en la zona de inyección. Puede presionar con una algodón o una gasa en la zona de inyección durante 10 segundos.
No frote la zona de inyección. Si es
necesario, puede cubrir la zona de inyección con una tirita.
Después de la inyección, use su pulgar (o la punta de su dedo), para activar la cubierta rosa de seguridad, presionando la cubierta hacia delante, usando la misma mano hasta que oiga y/o note un clic y se cierre por encima de la aguja.
Confirme visualmente que la punta de la aguja está cubierta. Cubra siempre la aguja con la cubierta de seguridad rosa antes de deshacerse de ella.

Paso 9. Eliminación de los suministros
Realice lo siguiente:
• Inmediatamente deseche la jeringa con la aguja cubierta dentro de un contenedor de objetos punzantes.
• Inmediatamente deseche el vial de Nplate utilizado dentro de un contenedor de residuos adecuado.
• Asegúrese que todos los materiales restantes se desechan en los contenedores adecuados.
El dispositivo de inyección y el vial de Nplate NUNCA deben ser reutilizados.
• Deseche la aguja usada y la jeringa en un contenedor resistente a pinchazos.
• Deseche cualquier resto de Nplate en un contenedor de residuos adecuado. El sobrante de un vial de Nplate NUNCA puede ser reutilizado para otra inyección.
92
ver sección 4.4