Neorecormon 3000 Ui Solucion Inyectable En Jeringa Precargada
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
NeoRecormon Multidosis 50.000 UI liofilizado y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un vial contiene 50.000 unidades internacionales (UI) correspondientes a 415 microgramos de epoetina beta* (eritropoyetina humana recombinante).
Una ampolla contiene 10 ml de disolvente (agua para preparaciones inyectables con alcohol bencílico y cloruro de benzalconio como conservantes).
Un ml de solución reconstituida contiene 5.000 UI de epoetina beta.
* producida en células de ovarios de hámster chino (CHO) mediante tecnología de ADN recombinante.
Excipientes con efecto conocido:
Fenilalanina (hasta 5,0 mg/vial)
Sodio (menos de 1 mmol por dosis)
Alcohol bencílico (hasta 40 mg por ampolla de disolvente multidosis)
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Liofilizado y disolvente para solución inyectable. Liofilizado blanco y disolvente incoloro y transparente.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
NeoRecormon está indicado para:
- Tratamiento de la anemia sintomática asociada a la insuficiencia renal crónica en pacientes adultos y pediátricos.
- Tratamiento de la anemia sintomática en pacientes adultos con neoplasias no mieloides tratados con quimioterapia.
- Aumentar el rendimiento de la sangre autóloga de pacientes incluidos en un programa de predonación.
Debe sopesarse su uso en esta indicación frente al riesgo aumentado de episodios tromboembólicos que han sido notificados. Sólo debe administrarse el tratamiento a pacientes con anemia moderada (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], sin deficiencia de hierro) si no se dispone de procedimientos para conservar la sangre o si éstos son insuficientes cuando la cirugía mayor electiva programada requiera un gran volumen de sangre (4 o más unidades de sangre en mujeres o 5 o más unidades en hombres). Ver sección 5.1.
4.2 Posología y forma de administración
La terapia con NeoRecormon debe iniciarse por médicos con experiencia en las indicaciones arriba
mencionadas. Como se han observado reacciones anafilácticas en algunos casos aislados, se
recomienda administrar la primera dosis bajo control médico.
Posología
Tratamiento de la anemia sintomática en pacientes adultos y pediátricos con insuficiencia renal crónica
Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente. NeoRecormon se puede administrar tanto por vía subcutánea como intravenosa con el fin de aumentar la concentración de hemoglobina hasta un nivel no superior a 12 g/dl (7,5 mmol/l). En pacientes que no están sometidos a hemodiálisis es preferible utilizar la vía subcutánea para evitar la punción de venas periféricas. En caso de administración intravenosa, la solución debe ser inyectada a lo largo de unos 2 minutos, p.ej. en pacientes en hemodiálisis por vía de la fístula arteriovenosa al final de la diálisis.
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones se pueden observar valores individuales de hemoglobina superiores o inferiores a los niveles deseados. La variabilidad en los niveles de hemoglobina se debe controlar mediante ajuste de la dosis con el objeto de mantener los valores de hemoglobina dentro del intervalo entre 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Se debe evitar un nivel de hemoglobina de forma continuada por encima de 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando los valores de hemoglobina sean superiores a 12 g/dl (7,5 mmol/l).
Debe evitarse un aumento de hemoglobina superior a 2 g/dl (1,25 mmol/l) durante un periodo de cuatro semanas. Si esto ocurre, se debe hacer un ajuste adecuado de la dosis según las instrucciones incluidas en esta misma sección. Si la tasa de aumento de hemoglobina es mayor de 2 g/dl (1,25 mmol/l) en un mes o si el nivel de hemoglobina está aumentando y se acerca a 12 g/dl (7,45 mmol/l), debe reducirse la dosis en aproximadamente un 25%. Si el nivel de hemoglobina sigue aumentando, se debe interrumpir el tratamiento hasta que el nivel de hemoglobina empiece a disminuir, momento en el que se podrá reiniciar el tratamiento con una dosis aproximadamente un 25 % inferior a la dosis administrada previamente.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis eficaz más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia al tiempo que se mantiene una concentración de hemoglobina inferior o igual a 12 g/dl (7,45 mmol/l).
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica. En los pacientes con una respuesta deficiente de la hemoglobina a NeoRecormon, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.4 y 5.1).
En presencia de hipertensión o de enfermedades cardiovasculares, cerebrovasculares o vasculoperiféricas, se debe determinar de modo individual el incremento semanal y el valor objetivo de la hemoglobina teniendo en cuenta el cuadro clínico.
El tratamiento con NeoRecormon se divide en dos fases:
1. Fase de corrección
- Administración subcutánea:
- La dosis inicial es 3 x 20 UI/kg por semana. La dosis puede incrementarse cada 4 semanas en 3 x 20 UI/kg y semana si el aumento de la hemoglobina no ha sido adecuado (< 0,25 g/dl por semana).
- La dosis semanal puede dividirse en dosis diarias.
- Administración intravenosa:
La dosis inicial es 3 x 40 UI/kg por semana, y puede aumentarse al cabo de 4 semanas a 80 UI/kg -tres veces por semana- y si son necesarios incrementos ulteriores serán de 20 UI/kg tres veces por semana, con intervalos mensuales.
Por ambas vías de administración, la dosis máxima no debe superar 720 UI/kg por semana.
2. Fase de mantenimiento
Para mantener el nivel de hemoglobina entre 10 y 12 g/dl, la dosis es inicialmente reducida a la mitad de la previamente administrada. Posteriormente, se ajustará la dosis individualmente para el paciente a intervalos de una o dos semanas (dosis de mantenimiento).
En caso de administración subcutánea, la dosis semanal puede administrarse en una inyección única o fraccionada en tres o siete inyecciones. Los pacientes que permanezcan estables en el régimen de una dosis única semanal pueden pasar a una administración única cada dos semanas. En este caso, puede ser necesario un aumento de la dosis.
Los resultados de los estudios clínicos en niños han revelado que, en general, a menor edad se necesita una dosis mayor NeoRecormon. No obstante, hay que seguir el programa posológico recomendado ya que no puede predecirse la respuesta individual.
El tratamiento con NeoRecormon es normalmente crónico. Sin embargo, en caso necesario puede interrumpirse en cualquier momento. Los datos sobre la pauta posológica de una vez a la semana se han obtenido de estudios clínicos con una duración de tratamiento de 24 semanas.
Tratamiento de anemia sintomática inducida por quimioterapia en pacientes con cáncer Se debe administrar NeoRecormon por vía subcutánea a pacientes con anemia (por ejemplo, si la concentración de hemoglobina es menor o igual a 10 g/dl [6,2 mmol/l]). Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente.
La dosis semanal puede administrarse como una inyección por semana o en dosis divididas entre 3 y 7 veces por semana.
La dosis inicial recomendada es de 30.000 UI por semana (que corresponde aproximadamente a 450 UI/kg de peso corporal por semana, en base al peso medio del paciente).
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones es posible llegar a observar en un paciente valores de hemoglobina superiores o inferiores a los esperados. La variabilidad de la hemoglobina se deberá manejar ajustando la dosis para mantener los valores de la hemoglobina dentro del intervalo de 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Deben evitarse concentraciones sostenidas de hemoglobina superiores a 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando se observen valores de hemoglobina superiores a los 12 g/dl (7,5 mmol/l).
Si a las 4 semanas de tratamiento el valor de hemoglobina aumenta hasta al menos 1 g/dl (0,62 mmol/l), se debe continuar con la dosis que en ese momento se esté administrando. Si los valores de hemoglobina no han aumentado al menos 1 g/dl (0,62 mmol/l), se debe considerar duplicar la dosis semanal. Si a las 8 semanas de tratamiento el valor de hemoglobina no aumenta hasta al menos 1 g/dl (0,62 mmol/l), es improbable que se produzca respuesta y, por consiguiente, el tratamiento debe ser interrumpido.
El tratamiento debe continuar durante las 4 semanas posteriores al final de la quimioterapia.
La dosis máxima no debe exceder de 60.000 UI por semana.
Una vez se ha alcanzado el objetivo terapéutico del paciente, la dosis debe reducirse del 25 al 50 % con el fin de mantener el valor de hemoglobina en ese nivel. Debe realizarse el ajuste posológico adecuado.
Si el nivel de hemoglobina excede los 12 g/dl (7,5 mmol/l) se debe reducir la dosis entre un 25 y un 50% aproximadamente. Si el nivel de hemoglobina excede de 13 g/dl (8,1 mmol), se debe interrumpir temporalmente el tratamiento con NeoRecormon. El tratamiento debe reiniciarse con una dosis aproximadamente un 25% inferior a la dosis previamente administrada después de que los niveles de hemoglobina desciendan hasta un valor menor o igual a 12 g/dl (7,5 mmol/l).
Si al cabo de 4 semanas el incremento de hemoglobina es superior a 2 g/dl (1,3 mmol/l), la dosis debe reducirse entre un 25 y un 50 %.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia.
Tratamiento para incrementar el rendimiento de la sangre autóloga donada
La solución reconstituida puede administrarse por vía intravenosa, en unos 2 minutos, o
subcutáneamente.
NeoRecormon se administra dos veces por semana durante 4 semanas. En aquellas ocasiones en que el hematocrito del paciente permite la donación de sangre, es decir el hematocrito > 33 %, NeoRecormon se administra al final de la donación de sangre.
Durante la totalidad del período de tratamiento no debe excederse un hematocrito del 48 %.
La dosis debe ser determinada por el equipo quirúrgico, individualmente para cada paciente, en función de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos:
1. La cantidad de sangre pre-donada necesaria dependerá de la pérdida prevista de sangre, de los medios de conservación empleados y del estado físico del paciente.
Este volumen debe ser equivalente a la cantidad de sangre que se considera necesaria para evitar transfusiones de sangre homóloga.
La cantidad de sangre pre-donada necesaria se expresa en unidades en las cuales una unidad de nomograma es equivalente a 180 ml de glóbulos rojos.
2. La capacidad de donar sangre depende predominantemente del volumen sanguíneo del paciente y del hematocrito basal. Ambas variables determinan la reserva endógena de eritrocitos, que puede ser calculada conforme a la fórmula siguiente:
Reserva endógena de eritrocitos = volumen sanguíneo (ml) x (hematocrito - 33)/ 100 mujeres: volumen sanguíneo (ml) = 41 (ml/kg) x peso corporal (kg) + 1200 (ml)
hombres: volumen sanguíneo (ml) = 44 (ml/kg) x peso corporal (kg) + 1600 (ml)
(peso corporal: > 45 kg)
La indicación para un tratamiento con NeoRecormon y, en su caso, la dosis individual debe ser determinada a partir de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos según las gráficas siguientes.
Paciente femenino Cantidad de sangre pre-donada necesaria [unidades]
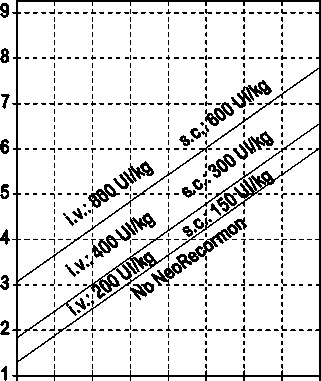
0 100 200 300 400 500 600 700 800
Reserva eritrocitaria endógena [ml]
Paciente masculino Cantidad de sangre pre-donada necesaria [unidades]
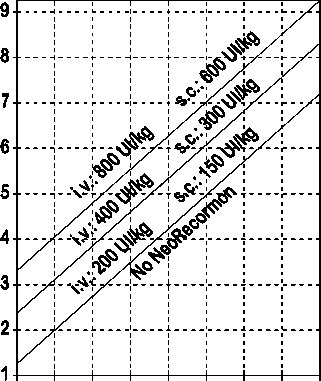
0 100 200 300 400 500 600 700 800
Reserva eritrocitaria endógena [ml]
La dosi s única así determinada debe administrarse dos veces por semana a lo largo de 4 semanas. La dosis máxima no debe sobrepasar 1.600 UI/kg peso corporal por semana en administración intravenosa ó 1.200 UI/kg por semana en administración subcutánea.
Forma de administración
Este preparado multidosis se puede utilizar para varios pacientes. Para evitar el riesgo de infección cruzada, se deben emplear siempre técnicas asépticas y utilizarse jeringas y agujas estériles desechables para cada administración. En cada ocasión solo debe haber un vial de NeoRecormon Multidosis en uso (es decir, reconstituido).
El producto reconstituido es una solución incolora, de transparente a ligeramente opalescente.
Para las instrucciones de reconstitución del medicamento antes de su administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Hipertensión mal controlada.
En la indicación para “Aumentar el rendimiento de sangre autóloga”: infarto de miocardio o accidente cerebrovascular en el mes anterior al tratamiento, angina de pecho inestable, riesgo aumentado de trombosis venosa profunda, como por ejemplo en aquellos con historial de enfermedad tromboembólica venosa.
NeoRecormon Multidosis contiene alcohol bencílico como conservante y por tanto no debe administrarse a lactantes o niños menores de 3 años.
4.4 Advertencias y precauciones especiales de empleo
NeoRecormon debe usarse con cautela en presencia de anemia refractaria con exceso de blastos en transformación, epilepsia, trombocitosis e insuficiencia hepática crónica. Y deben excluirse las deficiencias de ácido fólico y vitamina BJ2 pues reducen la eficacia de NeoRecormon.
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica, ya que las dosis acumuladas altas de epoetina pueden asociarse a un mayor riesgo de mortalidad, y de acontecimientos cardiovasculares y cerebrovasculares graves. En pacientes con una respuesta deficiente de la hemoglobina a epoetinas, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.2 y 5.1).
Para garantizar una eritropoyesis eficaz, se debe evaluar el nivel de hierro en todos los pacientes antes y durante el tratamiento, y puede ser necesario un tratamiento suplementario con hierro administrado de acuerdo con las guías terapéuticas.
La sobrecarga grave de aluminio debido al tratamiento de la insuficiencia renal puede comprometer la eficacia de NeoRecormon.
La indicación de tratamiento con NeoRecormon en enfermos nefroscleróticos aún no sometidos a diálisis debe definirse individualmente, ya que la posible aceleración de la insuficiencia renal no puede descartarse con certeza.
Aplasia pura de células rojas (APCR)
Se han notificado casos de APCR causada por anticuerpos neutralizantes antieritropoyetina, asociados a tratamientos con eritropoyetinas, incluido NeoRecormon. Estos anticuerpos han presentado reacción cruzada con todas las proteínas eritropoyéticas, por los que los pacientes en los que se sospeche o se haya confirmado la presencia de anticuerpos neutralizantes contra eritropoyetina no deben ser tratados con NeoRecormon (ver sección 4.8).
APCR en pacientes con Hepatitis C
Se debe interrumpir inmediatamente el tratamiento con epoetina, y realizar ensayos de anticuerpos antieritropoyetina si se produce una disminución paradójica de la hemoglobina y se desarrolla anemia severa asociada con bajos recuentos de reticulocitos. En pacientes con hepatitis C tratados con interferon y ribavirina se han notificados casos cuando las epoetinas se han utilizado concomitantemente. Las epoetinas no están aprobadas en el tratamiento de la anemia asociada a la hepatitis C.
Monitorización de la tensión arterial
Puede aparecer un aumento de la tensión arterial o un agravamiento de la hipertensión ya existente, especialmente en los casos en los que el hematocrito aumenta rápidamente. Estos incrementos en la tensión arterial pueden ser tratados con medicamentos. Si no pueden ser controlados con medicación se recomienda interrumpir transitoriamente la terapia con NeoRecormon. En particular al principio de la terapia se recomienda la monitorización regular de la tensión arterial; también durante la diálisis. Pueden producirse crisis hipertensivas con síntomas de encefalopatías y que requieren atención médica inmediata y cuidados médicos intensivos. Debe prestarse especial atención a las cefaleas súbitas lacerantes hemicraneales como posible signo de advertencia.
Insuficiencia renal crónica
En pacientes con insuficiencia renal crónica puede sobrevenir un aumento dosis-dependiente moderado en la cifra plaquetaria dentro del margen normal durante el tratamiento con NeoRecormon, especialmente después de la administración intravenosa. La regresión tiene lugar en el curso de la prosecución de la terapia. Se recomienda el control regular de la cifra de plaquetas durante las 8 primeras semanas de tratamiento.
Concentración de hemoglobina
En pacientes con insuficiencia renal crónica, la concentración de hemoglobina en la fase de mantenimiento no debe exceder el límite superior del rango recomendado en la sección 4.2. En ensayos clínicos se observó un aumento del riesgo de fallecimiento y de acontecimientos cardiovasculares graves o acontecimientos cerebrovasculares incluido el infarto, cuando se administraron agentes estimulantes de la eritropoyesis (AEEs) con el fin de alcanzar un nivel de hemoglobina superior a 12 g/dl (7,5 mmol/l).
En ensayos clínicos controlados no se han observado beneficios significativos atribuibles a la administración de epoetinas cuando se aumentaba la concentración de hemoglobina por encima del nivel necesario para controlar los síntomas de anemia y evitar las transfusiones sanguíneas.
Efectos sobre el crecimiento tumoral
Las epoetinas son factores de crecimiento que estimulan de forma primaria la producción de glóbulos rojos. Los receptores de epoetina pueden expresarse sobre la superficie de una variedad de células tumorales. Como sucede con todos los factores de crecimiento, existe la preocupación de que las epoetinas pudieran estimular el crecimiento de tumores. En varios ensayos controlados, no se ha observado que las epoetinas mejoren la supervivencia global o que disminuyan el riesgo de progresión del tumor en pacientes con anemia asociada con cáncer.
En ensayos clínicos controlados, se ha observado que el uso de NeoRecormon y otros agentes estimulantes de la eritropoyesis (AEEs):
- acortaba el tiempo de progresión del tumor en pacientes con cáncer avanzado de cabeza y cuello que recibían radioterapia cuando se administraba para conseguir una concentración de hemoglobina por encima de 14 g/dl (8,7 mmol/l),
- acortaba la supervivencia global y aumentaba el número de fallecimientos atribuidos a la progresión de la enfermedad a los 4 meses, en pacientes con cáncer de mama metastásico que recibían quimioterapia cuando se administraba para conseguir una concentración de hemoglobina entre los 12 y los 14 g/dl (7,5-8,7 mmol/l),
- aumentaba el riesgo de fallecimiento cuando se administraba para conseguir una concentración de hemoglobina de 12 g/dl (7,5 mmol/l) en pacientes con enfermedad maligna activa y que no recibían ni quimioterapia ni radioterapia. El uso de los AEEs no está indicado en esa población de pacientes.
En vista de lo anterior, en algunas situaciones clínicas la transfusión sanguínea debe ser el tratamiento de elección para la anemia en pacientes con cáncer. La decisión de administrar eritropoyetinas recomb inantes se tomará en base a la evaluación de la relación beneficio/riesgo junto con la aceptación individual del paciente, teniendo en cuenta el contexto clínico específico. Los factores que deben considerarse en esta evaluación son el tipo de tumor y su estadio, el grado de anemia, la esperanza de vida, el entorno en el que el paciente está siendo tratado y la preferencia del paciente (ver sección 5.1).
Puede haber un aumento de la tensión arterial que puede ser tratada con medicamentos. Se recomienda por tanto monitorizar la tensión arterial, en particular en la fase inicial de tratamiento en pacientes con cáncer.
En pacientes con cáncer también deberán monitorizarse regularmente el recuento plaquetario y el valor de hemoglobina.
En pacientes en programa de predonación de sangre autóloga, puede sobrevenir un aumento de la cifra de plaquetas, generalmente dentro de los límites normales. Por tanto, se recomienda la determinación de esta cifra al menos una vez por semana. Si se produce un aumento plaquetario de más de 150 x 109/l o si las plaquetas ascienden por encima del margen normal, se debe suspender el tratamiento con NeoRecormon.
En pacientes con insuficiencia renal crónica, a menudo se precisa aumentar la dosis de heparina durante la hemodiálisis en la terapia con NeoRecormon debido al aumento del hematocrito. Si la heparinización no es óptima es posible que se produzca la oclusión del sistema de diálisis.
En pacientes con alteración renal crónica con riesgo de trombosis de derivación, se debe considerar la revisión temprana de la derivación y profilaxis de la trombosis mediante la administración, por ejemplo, de ácido acetilsalicílico.
Los niveles séricos de potasio y fosfato deben ser controlados regularmente durante la terapia con NeoRecormon. Se ha informado de aumentos de potasio en unos pocos pacientes urémicos que recibieron NeoRecormon si bien no se ha establecido una relación causal. En caso de observar un nivel elevado o creciente de potasio se debe considerar la interrupción de la administración de NeoRecormon hasta que el nivel haya sido corregido.
Para el uso de NeoRecormon en un programa de predonación de sangre autóloga deben ser observadas las directrices oficiales sobre donación de sangre, en particular las siguientes:
- sólo deben donar los pacientes con hematocrito > 33 % (hemoglobina >11 g/dl [6,83 mmol/l]);
- debe procederse con especial cuidado con pacientes con un peso inferior a 50 kg;
- el volumen individual extraído no debe superar aproximadamente el 12 % del volumen sanguíneo estimado del paciente.
El tratamiento debe reservarse a pacientes en los que se considere particularmente importante evitar la transfusión de sangre homóloga teniendo en cuenta la evaluación riesgo/beneficio de las transfusiones homólogas.
Uso indebido
Un uso indebido del producto por personas sanas puede llevar a un aumento excesivo del volumen de hematocrito, lo cual puede asociarse con complicaciones del sistema cardiovascular con riesgo para la vida.
Excipientes
NeoRecormon Multidosis contiene, como máximo, 5,0 mg de fenilalanina por vial como excipiente. Este hecho se deberá tener en cuenta en los pacientes afectados por formas graves de fenilcetonuria.
La solución reconstituida de NeoRecormon Multidosis contiene alcohol bencílico que puede provocar reacciones tóxicas y reacciones anafilactoides en niños menores de 3 años.
Este medicamento contiene menos de 1 mmol (23 mg) de sodio por dosis, por lo que se considera esencialmente “exento de sodio”.
Trazabilidad de NeoRecormon
Para mejorar la trazabilidad de los agentes estimulantes de la eritropoyesis (AEEs), se debe registrar (o indicar) claramente el nombre comercial del AEE administrado en la historia clínica del paciente.
4.5 Interacción con otros medicamentos y otras formas de interacción
Los resultados clínicos obtenidos hasta el presente no ponen de manifiesto interacción alguna de NeoRecormon con otros medicamentos.
Los estudios llevados a cabo en animales mostraron que la epoetina beta no potencia la mielotoxicidad de los medicamentos citostáticos, tales como etopósido, cisplatino, ciclofosfamida y fluorouracilo.
4.6 Fertilidad, embarazo y lactancia
Fertilidad
Los estudios en animales no muestran efectos dañinos directos o indirectos sobre el embarazo, desarrollo embrional/fetal, parto o desarrollo postnatal (ver sección 5.3).
Embarazo
No se dispone de datos clínicos con epoetina beta en embarazos de riesgo.
Se debe tener precaución cuando se prescriba a mujeres embarazadas.
Lactancia
Se desconoce si la epoetina beta se excreta en la leche materna. La decisión de continuar/interrumpir la lactancia o de continuar/interrumpir el tratamiento con epoetina beta se debe hacer teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento con epoetina beta para la madre.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NeoRecormon sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas Resumen del perfil de seguridad
Basados en los resultados de los ensayos clínicos que incluyen 1.725 pacientes, se espera que aproximadamente el 8 % de los pacientes tratados con NeoRecormon sufran reacciones adversas.
Pacientes anémicos con insuficiencia renal crónica
La reacción adversa más frecuente durante el tratamiento con NeoRecormon es un aumento de la tensión arterial o agravamiento de la hipertensión existente, especialmente en casos de aumento rápido del hematocrito (ver sección 4.4). En algunos pacientes cuya tensión arterial es normal o baja también pueden darse crisis hipertensivas con síntomas de encefalopatías (p.ej. cefaleas y estados de confusión, trastornos sensitivomotores - como alteraciones del habla o de la ambulación - hasta convulsiones tónico-clónicas) (ver sección 4.4).
Puede darse trombosis de derivación, especialmente en pacientes que tienen tendencia a la hipotensión o cuyas fístulas arteriovenosas presentan complicaciones (p.ej. estenosis, aneurismas), ver sección 4.4. En la mayoría de los casos se observa una caída en los valores séricos de ferritina simultáneamente con el aumento del volumen de hematocrito (ver sección 4.4). Además, en casos aislados se han observado incrementos transitorios en los niveles séricos de potasio y fosfato (ver sección 4.4).
En casos aislados se ha notificado aplasia pura de células rojas (APCR) mediada por anticuerpos neutralizantes antieritropoyetina asociada al tratamiento con NeoRecormon. En caso de que se diagnostique APCR mediada por anticuerpos antieritropoyetina, el paciente deberá interrumpir el tratamiento con NeoRecormon, el cual no se deberá sustituir por otra proteína eritropoyética (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 1, que figura más adelante.
Pacientes con cáncer
El dolor de cabeza y la hipertensión relacionados con el tratamiento con epoetina beta son frecuentes y pueden tratarse con medicamentos (ver sección 4.4).
En algunos pacientes se observa un descenso de los valores de hierro sérico (ver sección 4.4).
Estudios clínicos han puesto de manifiesto que la frecuencia de acontecimientos tromboembólicos es más alta en pacientes con cáncer tratados con NeoRecormon que en los controles no tratados o tratados con placebo. En pacientes tratados con NeoRecormon, esta incidencia es del 7 % comparado con el 4 % en el grupo control; este hecho no está asociado con un incremento en la mortalidad tromboembólica comparado con los grupos control.
Estas reacciones adversas se enumeran en la Tabla 2, que figura más adelante.
Pacientes incluidos en un programa de predonación de sangre autóloga Los pacientes incluidos en un programa de predonación de sangre autóloga han demostrado una frecuencia ligeramente superior de accidentes tromboembólicos. Sin embargo, no se pudo establecer una relación causal con el tratamiento con NeoRecormon.
En ensayos clínicos controlados con placebo, la deficiencia transitoria de hierro fue más acusada en los pacientes tratados con NeoRecormon que en los pacientes control (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 3, que figura más adelante.
Tabla de reacciones adversas
Las reacciones adversas se enumeran de acuerdo con la clasificación de órganos del sistema MedDRA y por categoría de frecuencia.
Las categorías de frecuencia se definen utilizando la siguiente convención:
muy frecuentes (> 1/10), frecuentes (> 1/100 hasta < 1/10), poco frecuentes (> 1/1.000 hasta < 1/100), raras (> 1/10.000 hasta < 1/1.000); muy raras (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 1: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos clínicos
|
controlados en pacientes con I |
RC | |
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión Crisis hipertensivas |
Frecuente Poco frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
|
Trastornos de la sangre y del sistema linfático |
Trombosis de derivación Trombocitosis |
Raras Muy raras |
Tabla 2 : Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con cáncer__
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión |
Frecuente |
|
Trastornos de la sangre y del sistema linfático |
Acontecimientos tromboembólicos |
Frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Tabla 3: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes incluidos en un programa de predonación de sangre autóloga
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Descripción de reacciones adversas seleccionadas
Raramente pueden ocurrir reacciones cutáneas relacionadas con el tratamiento con epoetina beta como sarpullido, prurito, urticaria o reacciones en el lugar de la inyección. En casos muy raros se han notificado reacciones anafilácticas, relacionadas con el tratamiento con epoetina beta. Sin embargo, en los ensayos clínicos controlados no se encontró una mayor incidencia de reacciones de hipersensibilidad.
Se han comunicado casos muy raros de síntomas gripales relacionados con el tratamiento con epoetina beta, como fiebre, escalofríos, dolores de cabeza, dolor de las extremidades, malestar general, y/o dolor óseo, particularmente al inicio del tratamiento. Estas reacciones son de intensidad leve a moderada y desaparecen al cabo de un par de horas o días.
Los resultados obtenidos en un ensayo clínico controlado realizado con epoetina alfa o darbepoetina alfa, notificaron que la incidencia de infarto es frecuente .
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
El margen terapéutico de NeoRecormon es muy amplio. No se han observado síntomas de sobredosis incluso a niveles séricos muy elevados.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antianémico, código ATC: B03XA01 Mecanismo de acción
La eritropoyetina es una glucoproteína que estimula la formación de eritrocitos a partir de sus progenitores asignados. Actúa como factor estimulante de la mitosis y hormona de diferenciación.
La epoetina beta, el principio activo de NeoRecormon, es idéntica en su composición aminoácida e hidrocarbonada a la eritropoyetina aislada de la orina de pacientes anémicos.
La eficacia biológica de la epoetina beta ha sido demostrada tras administración intravenosa y subcutánea en varios modelos animales in vivo (ratas normales y urémicas, ratones policitémicos, perros). Con la administración de epoetina beta aumentan el número de eritrocitos, los valores de Hb y la cifra de reticulocitos, al igual que la velocidad de incorporación de 59Fe.
In vitro (cultivo citológico de bazo de ratón) ha sido hallada una mayor incorporación de 3H-timidina en las células eritroides nucleadas del bazo tras incubación con epoetina beta.
Las investigaciones en cultivos de células de la médula ósea humana han revelado que la epoetina beta estimula específicamente la eritropoyesis y no afecta a la leucopoyesis. No han sido detectadas acciones citotóxicas de la epoetina beta sobre la médula ósea o sobre células cutáneas humanas.
Tras una dosis única de epoetina beta no se ha observado ningún efecto en el comportamiento o la actividad locomotora del ratón ni en las funciones circulatoria o respiratoria del perro.
Eficacia clínica y seguridad
En un ensayo aleatorizado, doble ciego, controlado con placebo, realizado en 4.038 pacientes con enfermedad renal crónica no sometidos a diálisis, con una diabetes tipo 2 y los niveles de hemoglobina < 11 g/dl, los pacientes recibieron tratamiento bien con darbepoetina alfa para alcanzar niveles de hemoglobina de 13 g/dl o con placebo (ver sección 4.4). El ensayo no cumplió su objetivo principal de demostrar una disminución en el riesgo de todas las causas de mortalidad, la morbilidad cardiovascular o enfermedad renal terminal. El análisis de los componentes individuales de la variable combinada mostró los siguientes HR (IC 95%): fallecimiento 1,05 (0,92-1,21), infarto cerebrovascular 1,92 (1,38-2,68), insuficiencia cardíaca congestiva 0,89 (0,74-1,08), infarto de miocardio 0,96 (0,751,23), hospitalización por isquemia del miocardio 0,84 (0,55-1,27), enfermedad renal terminal 1,02 (0,87-1,18).
En pacientes con IRC (tratados con diálisis, no tratados con diálisis, pacientes diabéticos y pacientes no diabéticos) se han realizado análisis agrupados post hoc de los estudios clínicos con AEEs. Se observó una tendencia a un aumento de las estimaciones del riesgo para la mortalidad por todas las causas y para los acontecimientos cardiovasculares y cerebrovasculares asociados a dosis acumuladas más altas de AEE independientemente de que los pacientes padecieran o no diabetes y de que recibieran o no tratamiento con diálisis (ver las secciones 4.2 y 4.4).
La eritropoyetina es un factor de crecimiento que estimula de forma primaria la producción de glóbulos rojos. Los receptores de eritropoyetina se encuentran también presentes en la superficie de algunas líneas celulares malignas.
Se ha estudiado la supervivencia y la progresión tumoral en cinco grandes ensayos controlados que incluyeron a 2.833 pacientes, de los cuales cuatro fueron ensayos doble-ciego controlados con placebo y uno fue un ensayo abierto. Dos de los ensayos reclutaron pacientes que estaban siendo tratados con quimioterapia. El nivel de hemoglobina que se quería alcanzar fue >13 g/dl en dos de los ensayos y de entre 12 y 14 g/dl en los otros tres. En el ensayo abierto no se observaron diferencias en la supervivencia global, entre los pacientes tratados con eritropoyetina humana recombinante y el grupo control. En los cuatro ensayos controlados con placebo, el índice de riesgo (hazard ratio) para la supervivencia global osciló entre 1,25 y 2,47 en favor de los grupos control. En todos estos ensayos se ha observado un aumento de la mortalidad inexplicable y estadísticamente significativo en pacientes que presentaban anemia asociada con diversos tipos frecuentes de cáncer y que recibieron eritropoyetina humana recombinante, en comparación con los grupos control. Las diferencias observadas en la incidencia de trombosis y complicaciones relacionadas, entre los pacientes que recibieron eritropoyetina humana recombinante y los que formaban parte de los grupos control, no permiten explicar satisfactoriamente los resultados de supervivencia global observados en los ensayos.
Un meta-análisis basado en datos individuales de pacientes, que incluyó datos de los 12 ensayos clínicos controlados realizados con NeoRecormon en pacientes anémicos con cáncer (n=2301), mostró un valo r estimado del índice de riesgo (hazard ratio) de supervivencia global de 1,13 en favor de los grupos control (IC 95 %, CI 0,87, 1,46). En pacientes con un valor inicial de hemoglobina < 10 g/dl (n=899), el valor estimado del índice de riesgo (hazard ratio) para la supervivencia fue de 0,98 (IC 95% 0,68 al 1,40). Se observó un aumento del riesgo relativo de acontecimientos tromboembólicos en la población general (RR 1,62, IC 95 %: 1,13, 2,31).
Asimismo, se ha realizado un análisis de datos a nivel de paciente, en más de 13.900 pacientes con cáncer (tratados con quimioterapia, radioterapia, quimioradioterapia o sin tratamiento) que participaban en un total de 53 ensayos clínicos controlados en los que se administraban diversas epoetinas. En este meta-análisis se obtuvo un índice de riesgo (hazard ratio) para la supervivencia global de 1,06 a favor de los grupos control (IC 95%: 1,00, 1,12; 53 ensayos y 13.933 pacientes) y para los pacientes con cáncer que recibían quimioterapia, el índice de riesgo (hazard ratio) para la supervivencia global fue de 1,04 (IC 95%: 0,97, 1,11; 38 ensayos y 10.441 pacientes). Este meta-análisis además indica de forma consistente un aumento significativo del riesgo relativo de episodios tromboembólicos en pacientes con cáncer tratados con eritropoyetina recombinante humana (ver sección 4.4).
En casos muy raros, se han producido anticuerpos neutralizantes anti-eritropoyetina con o sin aplasia pura de células rojas (APCR) durante el tratamiento con rHuEPO.
5.2 Propiedades farmacocinéticas
Los estudios farmacocinéticos en voluntarios sanos y pacientes urémicos muestran que la semivida de la epoetina beta administrada por vía i.v. es de 4 a 12 horas y que el volumen de distribución corresponde de una a dos veces el volumen plasmático. Se han hallado resultados análogos en experimentos en animales con ratas normales y urémicas.
Tras la administración subcutánea de epoetina beta a pacientes urémicos la absorción retardada se traduce en una meseta de concentración sérica, cuya cota máxima se alcanza al cabo de 12 - 28 horas de media. El semiperíodo de vida terminal es mayor que después de la administración intravenosa, siendo la media de 13-28 horas.
La biodisponibilidad de la epoetina beta después de la administración subcutánea está comprendida entre el 23 y el 42 % en comparación con la administración intravenosa.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad de dosis repetidas, genotoxicidad y toxicidad para la reproducción.
Un estudio de carcinogenicidad con homólogos de eritropoyetina en ratones no reveló ningún signo de potencial proliferativo o carcinogénico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Liofilizado
Urea,
Cloruro sódico,
Polisorbato 20,
Fosfato monosódico,
Fosfato disódico,
Cloruro cálcico,
Glicina,
L-Leucina,
L-Isoleucina,
L-Treonina,
L-Ácido glutámico,
L-Fenilalanina.
Disolvente Alcohol bencílico,
Cloruro de benzalconio,
Agua para preparaciones inyectables.
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros medicamentos excepto con los mencionados en la sección 6.6 (contenido de la ampolla de disolvente acompañante).
6.3 Periodo de validez
3 años.
La solución reconstituida en uso ha demostrado estabilidad química y física durante un mes entre 2°C-8°C. Desde el punto de vista microbiológico, una vez abierta, la solución reconstituida puede almacenarse durante un máximo de un mes a 2°C-8°C. Si la duración y las condiciones de almacenamiento son distintas, el usuario asumirá la responsabilidad.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
Conservar el vial en el embalaje exterior, para protegerlo de la luz.
Con fines de uso ambulatorio, el producto sin reconstituir se puede mantener fuera de la nevera durante un único periodo de hasta 5 días a temperatura ambiente (no superior a 25°C).
La solución reconstituida debe permanecer fuera de la nevera sólo el tiempo necesario para preparar las inyecciones.
Para las condiciones de conservación del medicamento reconstituido, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Liofilizado: Vial (vidrio Tipo I) con tapón (goma teflonizada).
Disolvente: Ampolla de10 ml (vidrio Tipo I).
Envases de 1vial que contienen 50.000 UI de epoetina beta, 1 ampolla que contiene 10 ml de disolvente, un dispositivo de reconstitución y extracción, 1 aguja (21G2, de acero inoxidable) y 1 jeringa desechable (polipropileno y polietileno).
6.6 Precauciones especiales de eliminación y otras manipulaciones
NeoRecormon Multidosis se suministra en viales con liofilizado para solución inyectable, que se disuelve con el contenido de la ampolla de disolvente que le acompaña con ayuda de un dispositivo de reconstitución y extracción conforme a las instrucciones que a continuación se indican. Sólo pueden ser inyectadas soluciones claras o ligeramente opalescentes, incoloras y prácticamente libres de partículas visibles. No usar materiales de vidrio para la inyección, usar exclusivamente materiales de plástico.
Esto es un preparado multidosis del cual se pueden extraer diferentes dosis individuales durante un período de un mes después de la disolución. Para evitar el riesgo de contaminación del contenido es conveniente que siempre se observen las técnicas asépticas (por ejemplo, usar jeringas y agujas desechables, estériles, para administrar cada dosis) y seguir atentamente las instrucciones de uso de más abajo. Antes de extraer cada dosis, es conveniente desinfectar con alcohol el tapón de goma del dispositivo de extracción, para prevenir la contaminación del contenido debido a repetidas inserciones de agujas.
Preparación de la solución de NeoRecormon Multidosis
(1) Saque del envase el vial con la sustancia liofilizada. Escriba la fecha de reconstitución y la de caducidad en la etiqueta (la caducidad es 1 mes después de la reconstitución).
(2) Quite el protector de plástico del vial.
(3) Desinfecte la tapa de goma con alcohol.
(4) Extraiga el dispositivo de reconstitución y de extracción (que permite el intercambio de aire estéril) fuera del envoltorio y quite el protector del dispositivo.
(5) Coloque el dispositivo en el vial hasta que el cierre se coloque en su sitio
(6) Coloque la aguja verde en la jeringa que contiene el envase y quite el protector de la aguja.
(7) Mantenga la ampolla OPC (One-Point-Cut) con la punta azul hacia arriba. Agite o golpee suavemente la ampolla para que el líquido que esté en el cuello de la ampolla baje al cuerpo. Sujete el cuello de la ampolla y rómpalo en dirección contraria a usted. Transfiera todo el disolvente hacia la jeringa. Desinfecte con alcohol el tapón de goma del dispositivo a utilizar.
(8) Perfore el tapón con la aguja hasta una profundidad de 1 cm aproximadamente y lentamente inyecte el disolvente en el vial. Después retire la jeringa (con aguja) del dispositivo.
(9) Gire el vial suavemente hasta que el liofilizado se haya disuelto. No agite. Compruebe que la disolución es clara, incolora y prácticamente libre de partículas. Ponga el tapón protector encima del dispositivo.
(10) Antes y después de la reconstitución, NeoRecormon Multidosis debe ser conservado a 2°C - 8°C (nevera).
Preparación de una sola inyección
(1) Antes de extraer cada dosis desinfecte con alcohol el tapón de goma del dispositivo a utilizar.
(2) Coloque una aguja de 26G dentro de la jeringa apropiada desechable (máx. 1 ml).
(3) Saque el protector de la aguja e inserte la aguja a través del tapón de goma del dispositivo. Extraiga la solución de NeoRecormon hacia dentro de la jeringa, saque el aire de la jeringa hacia el vial y ajuste la cantidad de solución de NeoRecormon en la jeringa, a la dosis prescrita. Después retire la jeringa (con aguja) del dispositivo.
(4) Sustituya la aguja por una nueva (la nueva debería tener el tamaño que suele utilizarse para inyecciones).
(5) Saque el protector de la aguja y cuidadosamente saque el aire de la aguja, sujetando la jeringa verticalmente, y suavemente apriete el émbolo hacia arriba hasta que una gota de líquido aparezca en la punta de la aguja.
Para una inyección subcutánea, limpie la piel en el lugar donde se va a poner la inyección, utilizando un algodón con alcohol. Forme un pliegue de piel pellizcando la piel con el pulgar y el índice. Sujete la jeringa por la parte más próxima a la aguja e inserte la aguja en la piel con un movimiento rápido y firme. Inyecte la solución de NeoRecormon. Extraiga la aguja rápidamente y haga presión sobre el lugar de inyección con una gasa seca y estéril.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
8. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/019
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16 de julio de 1997 Fecha de la última revalidación: 16 de julio de 2007
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 500 UI solución inyectable en jeringa precargada
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Una jeringa precargada con 0,3 ml de solución para inyección contiene 500 unidades internacionales (UI) correspondientes a 4,15 microgramos de epoetina beta* (eritropoyetina recombinante humana).
Un ml de solución para inyección contiene 1.667 UI de epoetina beta.
* producida en células de ovarios de hámster chino (CHO) mediante tecnología de ADN recombinante.
Excipientes con efecto conocido:
Fenilalanina (hasta 0,3 mg/jeringa).
Sodio (menos de 1 mmol/jeringa)
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución incolora, de transparente a ligeramente opalescente.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
NeoRecormon está indicado para:
- Tratamiento de la anemia sintomática asociada a la insuficiencia renal crónica en pacientes adultos y pediátricos.
- Prevención de la anemia en prematuros con un peso corporal al nacer de 750 a 1.500 g y una edad gestacional de menos de 34 semanas.
- Tratamiento de la anemia sintomática en pacientes adultos con neoplasias no mieloides tratados con quimioterapia.
- Aumentar el rendimiento de la sangre autóloga de pacientes incluidos en un programa de predonación.
Debe sopesarse su uso en esta indicación frente al riesgo aumentado de episodios tromboembólicos que han sido notificados. Sólo debe administrarse el tratamiento a pacientes con anemia moderada (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], sin deficiencia de hierro) si no se dispone de procedimientos para conservar la sangre o si éstos son insuficientes cuando la cirugía mayor electiva programada requiera un gran volumen de sangre (4 o más unidades de sangre en mujeres o 5 o más unidades en hombres). Ver sección 5.1.
4.2 Posología y forma de administración
La terapia con NeoRecormon debe iniciarse por médicos con experiencia en las indicaciones arriba
mencionadas. Como se han observado reacciones anafilácticas en algunos casos aislados, se
recomienda administrar la primera dosis bajo control médico.
Posología
Tratamiento de la anemia sintomática en pacientes adultos y pediátricos con insuficiencia renal crónica
Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente. NeoRecormon se puede administrar tanto por vía subcutánea como intravenosa con el fin de aumentar la concentración de hemoglobina hasta un nivel no superior a 12 g/dl (7,5 mmol/l). En pacientes que no están sometidos a hemodiálisis es preferible utilizar la vía subcutánea para evitar la punción de venas periféricas. En caso de administración intravenosa, la solución debe ser inyectada a lo largo de unos 2 minutos, p.ej. en pacientes en hemodiálisis por vía de la fístula arteriovenosa al final de la diálisis.
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones se pueden observar valores individuales de hemoglobina superiores o inferiores a los niveles deseados. La variabilidad en los niveles de hemoglobina se debe controlar mediante ajuste de la dosis con el objeto de mantener los valores de hemoglobina dentro del intervalo entre 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Se debe evitar un nivel de hemoglobina de forma continuada por encima de 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando los valores de hemoglobina sean superiores a 12 g/dl (7,5 mmol/l).
Debe evitarse un aumento de hemoglobina superior a 2 g/dl (1,25 mmol/l) durante un periodo de cuatro semanas. Si esto ocurre, se debe hacer un ajuste adecuado de la dosis según las instrucciones incluidas en esta misma sección. Si la tasa de aumento de hemoglobina es mayor de 2 g/dl (1,25 mmol/l) en un mes o si el nivel de hemoglobina está aumentando y se acerca a 12 g/dl (7,45 mmol/l), debe reducirse la dosis en aproximadamente un 25%. Si el nivel de hemoglobina sigue aumentando, se debe interrumpir el tratamiento hasta que el nivel de hemoglobina empiece a disminuir, momento en el que se podrá reiniciar el tratamiento con una dosis aproximadamente un 25 % inferior a la dosis administrada previamente.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis eficaz más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia al tiempo que se mantiene una concentración de hemoglobina inferior o igual a 12 g/dl (7,45 mmol/l).
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica. En los pacientes con una respuesta deficiente de la hemoglobina a NeoRecormon, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.4 y 5.1).
En presencia de hipertensión o de enfermedades cardiovasculares, cerebrovasculares o vasculoperiféricas, se debe determinar de modo individual el incremento semanal y el valor objetivo de la hemoglobina teniendo en cuenta el cuadro clínico.
El tratamiento con NeoRecormon se divide en dos fases:
1. Fase de corrección
- Administración subcutánea:
La dosis inicial es 3 x 20 UI/kg por semana. La dosis puede incrementarse cada 4 semanas en 3 x 20 UI/kg y semana si el aumento de la hemoglobina no ha sido adecuado (< 0,25 g/dl por semana).
La dosis semanal puede dividirse en dosis diarias.
- Administración intravenosa:
La dosis inicial es 3 x 40 UI/kg por semana, y puede aumentarse al cabo de 4 semanas a 80 UI/kg -tres veces por semana- y si son necesarios incrementos ulteriores serán de 20 UI/kg tres veces por semana, con intervalos mensuales.
Por ambas vías de administración, la dosis máxima no debe superar 720 UI/kg por semana.
2. Fase de mantenimiento
Para mantener el nivel de hemoglobina entre 10 y 12 g/dl, la dosis es inicialmente reducida a la mitad de la cantidad previamente administrada. Posteriormente, se ajustará la dosis individualmente para el paciente a intervalos de una o dos semanas (dosis de mantenimiento).
En caso de administración subcutánea, la dosis semanal puede administrarse en una inyección única o fraccionada en tres o siete inyecciones. Los pacientes que permanezcan estables en el régimen de una dosis única semanal pueden pasar a una administración única cada dos semanas. En este caso, puede ser necesario un aumento de la dosis.
Los resultados de los estudios clínicos en niños han revelado que, en general, a menor edad, se necesita mayor dosis de NeoRecormon. No obstante, hay que seguir el programa posológico recomendado ya que no puede predecirse la respuesta individual.
El tratamiento con NeoRecormon es normalmente crónico. Sin embargo, en caso necesario puede interrumpirse en cualquier momento. Los datos sobre la pauta posológica de una vez a la semana se han obtenido de estudios clínicos con una duración de tratamiento de 24 semanas.
Prevención de anemia en prematuros
La solución se administra por vía subcutánea a una dosis de 3 x 250 UI/kg p.c. por semana. Es probable que los niños que ya hayan recibido una transfusión previa cuando se inicie el tratamiento con NeoRecormon no se beneficien tanto como los prematuros no transfundidos. La duración de tratamiento recomendada es de 6 semanas.
Tratamiento de anemia sintomática inducida por quimioterapia en pacientes con cáncer Se debe administrar NeoRecormon por vía subcutánea a pacientes con anemia (por ejemplo, si la concentración de hemoglobina es menor o igual a 10 g/dl [6,2 mmol/l]). Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente.
La dosis semanal puede administrarse como una inyección por semana o en dosis divididas entre 3 y 7 veces por semana.
La dosis inicial recomendada es de 30.000 UI por semana (que corresponde aproximadamente a 450 UI/kg de peso corporal por semana, en base al peso medio del paciente).
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones es posible llegar a observar en un paciente valores de hemoglobina superiores o inferiores a los esperados. La variabilidad de la hemoglobina se deberá manejar ajustando la dosis para mantener los valores de la hemoglobina dentro del intervalo de 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Deben evitarse concentraciones sostenidas de hemoglobina superiores a 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando se observen valores de hemoglobina superiores a los 12 g/dl (7,5 mmol/l).
Si a las 4 semanas de tratamiento el valor de hemoglobina aumenta hasta al menos 1 g/dl (0,62 mmol/l), se debe continuar con la dosis que en ese momento se esté administrando. Si los valores de hemoglobina no han aumentado al menos 1 g/dl (0,62 mmol/l), se debe considerar duplicar la dosis semanal. Si a las 8 semanas de tratamiento el valor de hemoglobina no aumenta hasta al menos 1 g/dl (0,62 mmol/l), es improbable que se produzca respuesta y, por consiguiente, el tratamiento debe ser interrumpido.
El tratamiento debe continuar durante las 4 semanas posteriores al final de la quimioterapia.
La dosis máxima no debe exceder de 60.000 UI por semana.
Una vez se ha alcanzado el objetivo terapéutico del paciente, la dosis debe reducirse del 25 al 50 % con el fin de mantener el valor de hemoglobina en ese nivel. Debe realizarse el ajuste posológico adecuado.
Si el nivel de hemoglobina excede los 12 g/dl (7,5 mmol/l) se debe reducir la dosis entre un 25 y un
50 % aproximadamente. Si el nivel de hemoglobina excede de 13 g/dl (8,1 mmol), se debe interrumpir temporalmente el tratamiento con NeoRecormon. El tratamiento debe reiniciarse con una dosis aproximadamente un 25 % inferior a la dosis previamente administrada después de que los niveles de hemoglobina desciendan hasta un valor menor o igual a 12 g/dl (7,5 mmol/l) o por debajo.
51 al cabo de 4 semanas el incremento de hemoglobina es superior a 2 g/dl (1,3 mmol/l), la dosis debe reducirse entre un 25 y un 50 %.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia.
Tratamiento para incrementar el rendimiento de la sangre autóloga donada La solución puede administrarse por vía intravenosa, en unos 2 minutos, o subcutáneamente. NeoRecormon se administra dos veces por semana durante 4 semanas. En aquellas ocasiones en que el hematocrito del paciente permite la donación de sangre, es decir el hematocrito es > 33 %, NeoRecormon se administra al final de la donación de sangre.
Durante la totalidad del período de tratamiento no debe excederse un hematocrito del 48 %.
La dosis debe ser determinada por el equipo quirúrgico, individualmente para cada paciente, en función de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos:
1. La cantidad de sangre pre-donada necesaria dependerá de la pérdida prevista de sangre, de los medios de conservación empleados y del estado físico del paciente.
Este volumen debe ser equivalente a la cantidad de sangre que se considera necesaria para evitar transfusiones de sangre homóloga.
La cantidad de sangre pre-donada necesaria se expresa en unidades en las cuales una unidad de nomograma es equivalente a 180 ml de glóbulos rojos.
2. La capacidad de donar sangre depende predominantemente del volumen sanguíneo del paciente y del hematocrito basal. Ambas variables determinan la reserva endógena de eritrocitos, que puede ser calculada conforme a la fórmula siguiente:
Reserva endógena de eritrocitos = volumen sanguíneo (ml) x (hematocrito - 33)/ 100 mujeres: volumen sanguíneo (ml) = 41 (ml/kg) x peso corporal (kg) + 1200 (ml)
hombres: volumen sanguíneo (ml) = 44 (ml/kg) x peso corporal (kg) + 1600 (ml)
(peso corporal: > 45 kg)
La indicación para un tratamiento con NeoRecormon y, en su caso, la dosis individual debe ser determinada a partir de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos según las gráficas siguientes:
Paciente femenino Paciente masculino
Cantidad de sangre pre-donada Cantidad de sangre pre-donada
necesaria [unidades] necesaria [unidades]
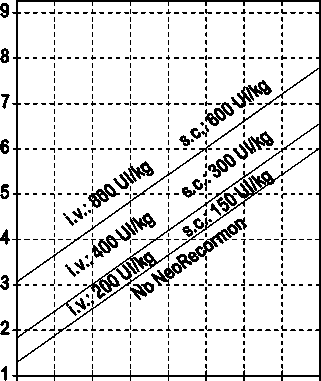
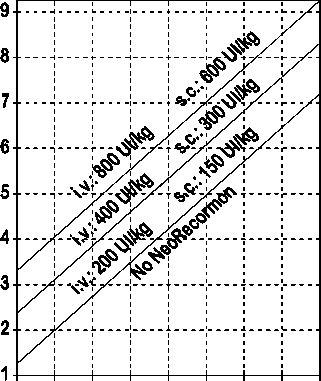
O 100 200 300 400 500 600 700 800 0 100 200 300 400 500 600 700 800
Reserva eritrocitaria endógena [ml] Reserva eritrocitaria endógena [ml]
La dosis única así determinada debe administrarse dos veces por semana a lo largo de 4 semanas. La dosis máxima no debe sobrepasar 1.600 UI/kg peso corporal por semana en administración intravenosa ó 1.200 UI/kg por semana en administración subcutánea.
Forma de administración
La jeringa precargada de NeoRecormon está lista para su uso. Sólo se pueden inyectar soluciones claras o ligeramente opalescentes, incoloras y prácticamente libres de partículas visibles. NeoRecormon en jeringa precargada es un producto estéril pero sin conservantes. Bajo ninguna circunstancia debe administrarse más de una dosis por jeringa; el medicamento es solo para uso individual.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Hipertensión mal controlada.
En la indicación para “Aumentar el rendimiento de sangre autóloga”: infarto de miocardio o accidente cerebrovascular en el mes anterior al tratamiento, angina de pecho inestable, riesgo aumentado de trombosis venosa profunda, como por ejemplo en aquellos con historial de enfermedad tromboembólica venosa.
4.4 Advertencias y precauciones especiales de empleo
NeoRecormon debe usarse con cautela en presencia de anemia refractaria con exceso de blastos en transformación, epilepsia, trombocitosis e insuficiencia hepática crónica. Y deben excluirse las deficiencias de ácido fólico y vitamina B12 pues reducen la eficacia de NeoRecormon.
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica, ya que las dosis acumuladas altas de epoetina pueden asociarse a un mayor riesgo de mortalidad, y de acontecimientos cardiovasculares y cerebrovasculares graves. En pacientes con una respuesta deficiente de la hemoglobina a epoetinas, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.2 y 5.1).
Para garantizar una eritropoyesis eficaz, se debe evaluar el nivel de hierro en todos los pacientes antes y durante el tratamiento, y puede ser necesario un tratamiento suplementario con hierro administrado de acuerdo con las guías terapéuticas.
La sobrecarga grave de aluminio debido al tratamiento de la insuficiencia renal puede comprometer la eficacia de NeoRecormon.
La indicación de tratamiento con NeoRecormon en enfermos nefroscleróticos aún no sometidos a diálisis debe definirse individualmente, ya que la posible aceleración de la insuficiencia renal no puede descartarse con certeza.
Aplasia pura de células rojas (APCR)
Se han notificado casos de APCR causada por anticuerpos neutralizantes antieritropoyetina, asociados a tratamientos con eritropoyetinas, incluido NeoRecormon. Estos anticuerpos han presentado reacción cruzada con todas las proteínas eritropoyéticas, por los que los pacientes en los que se sospeche o se haya confirmado la presencia de anticuerpos neutralizantes contra eritropoyetina no deben ser tratados con NeoRecormon (ver sección 4.8).
APCR en pacientes con hepatitis C
Se debe interrumpir inmediatamente el tratamiento con epoetina, y realizar ensayos de anticuerpos antieritropoyetina si se produce una disminución paradójica de la hemoglobina y se desarrolla anemia severa asociada con bajos recuentos de reticulocitos. En pacientes con hepatitis C tratados con interferon y ribavirina se han notificados casos cuando las epoetinas se han utilizado concomitantemente. Las epoetinas no están aprobadas en el tratamiento de la anemia asociada a la hepatitis C.
Monitorización de la tensión arterial
Puede aparecer un aumento de la tensión arterial o un agravamiento de la hipertensión ya existente, especialmente en los casos en los que el hematocrito aumenta rápidamente. Estos incrementos en la tensión arterial pueden ser tratados con medicamentos. Si no pueden ser controlados con medicación se recomienda interrumpir transitoriamente la terapia con NeoRecormon. En particular al principio de la terapia se recomienda la monitorización regular de la tensión arterial; también durante la diálisis. Pueden producirse crisis hipertensivas con síntomas de encefalopatías y que requieren atención médica inmediata y cuidados médicos intensivos. Debe prestarse especial atención a las cefaleas súbitas lacerantes hemicraneales como posible signo de advertencia.
Insuficiencia renal crónica
En pacientes coninsuficiencia renal crónica puede sobrevenir un aumento dosis-dependiente moderado en la cifra plaquetaria dentro del margen normal durante el tratamiento con NeoRecormon, especialmente después de la administración intravenosa. La regresión tiene lugar en el curso de la prosecución de la terapia. Se recomienda el control regular de la cifra de plaquetas durante las 8 primeras semanas de tratamiento.
Concentración de hemoglobina
En pacientes con insuficiencia renal crónica, la concentración de hemoglobina en la fase de mantenimiento no debe exceder el límite superior del rango recomendado en la sección 4.2. En ensayos clínicos se observó un aumento del riesgo de fallecimiento y de acontecimientos cardiovasculares graves o acontecimientos cerebrovasculares incluido el infarto, cuando se administraron agentes estimulantes de la eritropoyesis (AEEs) con el fin de alcanzar un nivel de hemoglobina superior a 12 g/dl (7,5 mmol/l).
En ensayos clínicos controlados no se han observado beneficios significativos atribuibles a la administración de epoetinas cuando se aumentaba la concentración de hemoglobina por encima del nivel necesario para controlar los síntomas de anemia y evitar las transfusiones sanguíneas.
En niños prematuros puede producirse un ligero aumento de la cifra plaquetaria, en especial hasta el día 12 -14 de vida, por lo tanto las plaquetas deben controlarse regularmente.
Efectos sobre el crecimiento tumoral
Las epoetinas son factores de crecimiento que estimulan de forma primaria la producción de glóbulos rojos. Los receptores de epoetina pueden expresarse sobre la superficie de una variedad de células tumorales. Como sucede con todos los factores de crecimiento, existe la preocupación de que las epoetinas pudieran estimular el crecimiento de tumores. En varios ensayos controlados, no se ha observado que las epoetinas mejoren la supervivencia global o que disminuyan el riesgo de progresión del tumor en pacientes con anemia asociada con cáncer.
En ensayos clínicos controlados, se ha observado que el uso de NeoRecormon y otros agentes estimulantes de la eritropoyesis (AEEs):
- acortaba el tiempo de progresión del tumor en pacientes con cáncer avanzado de cabeza y cuello que recibían radioterapia cuando se administraba para conseguir una concentración de hemoglobina por encima de 14 g/dl (8,7 mmol/l),
- acortaba la supervivencia global y aumentaba el número de fallecimientos atribuidos a la progresión de la enfermedad a los 4 meses, en pacientes con cáncer de mama metastásico que recibían quimioterapia cuando se administraba para conseguir una concentración de hemoglobina entre los 12 y los 14 g/dl (7,5-8,7 mmol/l),
- aumentaba el riesgo de fallecimiento cuando se administraba para conseguir una concentración de hemoglobina de 12 g/dl (7,5 mmol/l) en pacientes con enfermedad maligna activa y que no recibían ni quimioterapia ni radioterapia. El uso de los AEEs no está indicado en esa población de pacientes.
En vista de lo anterior, en algunas situaciones clínicas la transfusión sanguínea debe ser el tratamiento de elección para la anemia en pacientes con cáncer. La decisión de administrar eritropoyetinas recombinantes se tomará en base a la evaluación de la relación beneficio/riesgo junto con la aceptación individual del paciente, teniendo en cuenta el contexto clínico específico. Los factores que deben considerarse en esta evaluación son el tipo de tumor y su estadio, el grado de anemia, la esperanza de vida, el entorno en el que el paciente está siendo tratado y la preferencia del paciente (ver sección 5.1).
Puede haber un aumento de la tensión arterial que puede ser tratada con medicamentos. Se recomienda por tanto monitorizar la tensión arterial, en particular en la fase inicial de tratamiento en pacientes con cáncer.
En pacientes con cáncer también deberán monitorizarse regularmente el recuento plaquetario y el valor de hemoglobina.
En pacientes en programa de predonación de sangre autóloga, puede sobrevenir un aumento de la cifra de plaquetas, generalmente dentro de los límites normales. Por tanto, se recomienda la determinación de esta cifra al menos una vez por semana. Si se produce un aumento plaquetario de más de 150 x 109/l o si las plaquetas ascienden por encima del margen normal, se debe suspender el tratamiento con NeoRecormon.
En niños prematuros no se podría descartar el riesgo potencial de que la eritropoyetina provoque retinopatía, por lo tanto se debe tener precaución y la decisión de tratar a un niño prematuro se debe tomar teniendo en cuenta el beneficio potencial y el riesgo de este tratamiento, así como las opciones alternativas de tratamiento que estén disponibles.
En pacientes con insuficiencia renal crónica, a menudo se precisa aumentar la dosis de heparina durante la hemodiálisis en la terapia con NeoRecormon debido al aumento del volumen de hematocrito. Si la heparinización no es óptima es posible que se produzca la oclusión del sistema de diálisis.
En pacientes con alteración renal crónica con riesgo de trombosis de derivación, se debe considerar la revisión temprana de la derivación y profilaxis de la trombosis mediante la administración, por ejemplo, de ácido acetilsalicílico.
Los niveles séricos de potasio y fosfato deben ser controlados regularmente durante la terapia con NeoRecormon. Se ha informado de aumentos de potasio en unos pocos pacientes urémicos que recibieron NeoRecormon si bien no se ha establecido una relación causal. En caso de observar un nivel elevado o creciente de potasio se debe considerar la interrupción de la administración de NeoRecormon hasta que el nivel haya sido corregido.
Para el uso de NeoRecormon en un programa de predonación de sangre autóloga deben ser observadas las directrices oficiales sobre donación de sangre, en particular las siguientes:
- sólo deben donar los pacientes con hematocrito > 33 % (hemoglobina >11 g/dl [6,83 mmol/l]);
- debe procederse con especial cuidado con pacientes con un peso inferior a 50 kg;
- el volumen individual extraído no debe superar aproximadamente el 12 % del volumen sanguíneo estimado del paciente.
El tratamiento debe reservarse a pacientes en los que se considere particularmente importante evitar la transfusión de sangre homóloga teniendo en cuenta la evaluación riesgo/beneficio de las transfusiones homólogas.
Uso indebido
Un uso indebido del producto por personas sanas puede llevar a un aumento excesivo del volumen de hematocrito, lo cual puede asociarse con complicaciones del sistema cardiovascular con riesgo para la vida.
Excipientes
NeoRecormon en jeringa precargada contiene, como máximo, 0,3 mg de fenilalanina por jeringa como excipiente. Este hecho se deberá tener en cuenta en los pacientes afectados por formas graves de fenilcetonuria.
Este medicamento contiene menos de 1 mmol (23 mg) de sodio por jeringa, por lo que se considera esencialmente “exento de sodio”.
Trazabilidad de NeoRecormon
Para mejorar la trazabilidad de los agentes estimulantes de la eritropoyesis (AEEs), se debe registrar (o indicar) claramente el nombre comercial del AEE administrado en la historia clínica del paciente.
4.5 Interacción con otros medicamentos y otras formas de interacción
Los resultados clínicos obtenidos hasta el presente no ponen de manifiesto interacción alguna de NeoRecormon con otros medicamentos.
Los estudios llevados a cabo en animales mostraron que la epoetina beta no potencia la mielotoxicidad de los medicamentos citostáticos, tales como etopósido, cisplatino, ciclofosfamida y fluorouracilo.
4.6 Fertilidad, embarazo y lactancia
Fertilidad
Los estudios en animales no muestran efectos dañinos directos o indirectos sobre el embarazo, desarrollo embrional/fetal, parto o desarrollo postnatal (ver sección 5.3).
Embarazo
No hay datos relativos al uso de NeoRecormon en mujeres embarazadas.
Se debe tener precaución cuando se prescriba a mujeres embarazadas.
Lactancia
Se desconoce si la epoetina beta se excreta en la leche materna. La decisión de continuar/interrumpir la lactancia o de continuar/interrumpir el tratamiento con epoetina beta se debe hacer teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento con epoetina beta para la madre.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NeoRecormon sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Basados en los resultados de los ensayos clínicos que incluyen 1.725 pacientes, se espera que aproximadamente el 8 % de los pacientes tratados con NeoRecormon sufran reacciones adversas.
Pacientes anémicos con insuficiencia renal crónica
La reacción adversa más frecuente durante el tratamiento con NeoRecormon es un aumento de la tensión arterial o agravamiento de la hipertensión existente, especialmente en casos de aumento rápido del hematocrito (ver sección 4.4). En algunos pacientes cuya tensión arterial es normal o baja también pueden darse crisis hipertensivas con síntomas de encefalopatías (p.ej. cefaleas y estados de confusión, trastornos sensitivomotores - como alteraciones del habla o de la ambulación - hasta convulsiones tónico-clónicas) (ver sección 4.4).
Puede darse trombosis de derivación, especialmente en pacientes que tienen tendencia a la hipotensión o cuyas fístulas arteriovenosas presentan complicaciones (p.ej. estenosis, aneurismas), ver sección 4.4. En la mayoría de los casos se observa una caída en los valores séricos de ferritina simultáneamente con el aumento del volumen de hematocrito (ver sección 4.4). Además, en casos aislados se han observado incrementos transitorios en los niveles séricos de potasio y fosfato (ver sección 4.4).
En casos aislados se ha notificado aplasia pura de células rojas (APCR) mediada por anticuerpos neutralizantes antieritropoyetina asociada al tratamiento con NeoRecormon. En caso de que se diagnostique APCR mediada por anticuerpos antieritropoyetina, el paciente deberá interrumpir el tratamiento con NeoRecormon, el cual no se deberá sustituir por otra proteína eritropoyética (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 1, que figura más adelante.
Pacientes con cáncer
El dolor de cabeza y la hipertensión relacionados con el tratamiento con epoetina beta son frecuentes y pueden tratarse con medicamentos (ver sección 4.4).
En algunos pacientes se observa un descenso de los valores de hierro sérico (ver sección 4.4).
Estudios clínicos han puesto de manifiesto que la frecuencia de acontecimientos tromboembólicos es más alta en pacientes con cáncer tratados con NeoRecormon que en los controles no tratados o tratados con placebo. En pacientes tratados con NeoRecormon, esta incidencia es del 7 % comparado con el 4 % en el grupo control; este hecho no está asociado con un incremento en la mortalidad tromboembólica comparado con los grupos control.
Estas reacciones adversas se enumeran en la Tabla 2, que figura más adelante.
Pacientes incluidos en un programa de predonación de sangre autóloga Los pacientes incluidos en un programa de predonación de sangre autóloga han demostrado una frecuencia ligeramente superior de accidentes tromboembólicos. Sin embargo, no se pudo establecer una relación causal con el tratamiento con NeoRecormon.
En ensayos clínicos controlados con placebo, la deficiencia transitoria de hierro fue más acusada en los pacientes tratados con NeoRecormon que en los pacientes control (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 3, que figura más adelante.
Tabla de reacciones adversas
Las reacciones adversas se enumeran de acuerdo con la clasificación de órganos del sistema MedDRA y por categoría de frecuencia.
Las categorías de frecuencia se definen utilizando la siguiente convención:
muy frecuentes (> 1/10), frecuentes (> 1/100 hasta < 1/10), poco frecuentes (> 1/1.000 hasta <1 /100), raras (> 1/10.000 hasta < 1/1.000); muy raras (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 1: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con IRC_
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión Crisis hipertensivas |
Frecuente Poco frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
|
Trastornos de la sangre y |
Trombosis de derivación |
Raras |
|
del sistema linfático |
Trombocitosis |
Muy raras |
Tabla 2: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con cáncer__
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión |
Frecuente |
|
Trastornos de la sangre y del sistema linfático |
Acontecimientos tromboembólicos |
Frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Tabla 3: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes incluidos en un programa de predonación de sangre autóloga
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Prematuros
Es muy frecuente un descenso de los valores de ferritina sérica (ver sección 4.4).
Descripción de reacciones adversas seleccionadas
Raramente pueden ocurrir reacciones cutáneas relacionadas con el tratamiento con epoetina beta como sarpullido, prurito, urticaria o reacciones en el lugar de la inyección. En casos muy raros se han notificado reacciones anafilácticas relacionadas con el tratamiento con epoetina beta. Sin embargo, en los ensayos clínicos controlados no se encontró una mayor incidencia de reacciones de hipersensibilidad.
Se han comunicado casos muy raros de síntomas gripales relacionados con el tratamiento con epoetina beta como fiebre, escalofríos, dolores de cabeza, dolor de las extremidades, malestar general, y/o dolor óseo, particularmente al inicio del tratamiento. Estas reacciones son de intensidad leve a moderada y desaparecen al cabo de un par de horas o días.
Los resultados obtenidos en un ensayo clínico controlado realizado con epoetina alfa o darbepoetina alfa, notificaron que la incidencia de infarto es frecuente.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
El margen terapéutico de NeoRecormon es muy amplio. No se han observado síntomas de sobredosis incluso a niveles séricos muy elevados.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antianémico, código ATC: B03XA01 Mecanismo de acción
La eritropoyetina es una glucoproteína que estimula la formación de eritrocitos a partir de sus progenitores asignados. Actúa como factor estimulante de la mitosis y hormona de diferenciación.
La epoetina beta, el principio activo de NeoRecormon, es idéntica en su composición aminoácida e hidrocarbonada a la eritropoyetina aislada de la orina de pacientes anémicos.
La eficacia biológica de la epoetina beta ha sido demostrada tras administración intravenosa y subcutánea en varios modelos animales in vivo (ratas normales y urémicas, ratones policitémicos, perros). Con la administración de epoetina beta aumentan el número de eritrocitos, los valores de Hb y la cifra de reticulocitos, al igual que la velocidad de incorporación de 59Fe.
In vitro (cultivo citológico de bazo de ratón) ha sido hallada una mayor incorporación de 3H-timidina en las células eritroides nucleadas del bazo tras incubación con epoetina beta.
Las investigaciones en cultivos de células de la médula ósea humana han revelado que la epoetina beta estimula específicamente la eritropoyesis y no afecta a la leucopoyesis. No han sido detectadas acciones citotóxicas de la epoetina beta sobre la médula ósea o sobre células cutáneas humanas.
Tras una dosis única de epoetina beta no se ha observado ningún efecto en el comportamiento o la actividad locomotora del ratón ni en las funciones circulatoria o respiratoria del perro.
Eficacia clínica y seguridad
En un ensayo aleatorizado, doble ciego, controlado con placebo, realizado en 4.038 pacientes con enfermedad renal crónica no sometidos a diálisis, con una diabetes tipo 2 y los niveles de hemoglobina < 11 g/dl, los pacientes recibieron tratamiento bien con darbepoetina alfa para alcanzar niveles de hemoglobina de 13 g/dl o con placebo (ver sección 4.4). El ensayo no cumplió su objetivo principal de demostrar una disminución en el riesgo de todas las causas de mortalidad, la morbilidad cardiovascular o enfermedad renal terminal. El análisis de los componentes individuales de la variable combinada mostró los siguientes HR (IC 95%): fallecimiento 1,05 (0,92-1,21), infarto cerebrovascular 1,92 (1,38-2,68), insuficiencia cardíaca congestiva 0,89 (0,74-1,08), infarto de miocardio 0,96 (0,751,23), hospitalización por isquemia del miocardio 0,84 (0,55-1,27), enfermedad renal terminal 1,02 (0,87-1,18).
En pacientes con IRC (tratados con diálisis, no tratados con diálisis, pacientes diabéticos y pacientes no diabéticos) se han realizado análisis agrupados post hoc de los estudios clínicos con AEEs. Se observó una tendencia a un aumento de las estimaciones del riesgo para la mortalidad por todas las causas y para los acontecimientos cardiovasculares y cerebrovasculares asociados a dosis acumuladas más altas de AEE independientemente de que los pacientes padecieran o no diabetes y de que recibieran o no tratamiento con diálisis (ver las secciones 4.2 y 4.4).
La eritropoyetina es un factor de crecimiento que estimula de forma primaria la producción de glóbulos rojos. Los receptores de eritropoyetina se encuentran también presentes en la superficie de algunas líneas celulares malignas.
Se ha estudiado la supervivencia y la progresión tumoral en cinco grandes ensayos controlados que incluyeron a 2.833 pacientes, de los cuales cuatro fueron ensayos doble-ciego controlados con placebo y uno fue un ensayo abierto. Dos de los ensayos reclutaron pacientes que estaban siendo tratados con quimioterapia. El nivel de hemoglobina que se quería alcanzar fue >13 g/dl en dos de los ensayos y de entre 12 y 14 g/dl en los otros tres. En el ensayo abierto no se observaron diferencias en la supervivencia global, entre los pacientes tratados con eritropoyetina humana recombinante y el grupo control. En los cuatro ensayos controlados con placebo, el índice de riesgo (hazard ratio) para la supervivencia global osciló entre 1,25 y 2,47 en favor de los grupos control. En todos estos ensayos se ha observado un aumento de la mortalidad inexplicable y estadísticamente significativo en pacientes que presentaban anemia asociada con diversos tipos frecuentes de cáncer y que recibieron eritropoyetina humana recombinante, en comparación con los grupos control. Las diferencias observadas en la incidencia de trombosis y complicaciones relacionadas, entre los pacientes que recibieron eritropoyetina humana recombinante y los que formaban parte de los grupos control, no permiten explicar satisfactoriamente los resultados de supervivencia global observados en los ensayos.
Un meta-análisis basado en datos individuales de pacientes, que incluyó datos de los 12 ensayos clínicos controlados realizados con NeoRecormon en pacientes anémicos con cáncer (n=2301), mostró un valor estimado del índice de riesgo (hazard ratio) de supervivencia global de 1,13 en favor de los grupos control (IC 95 %, CI 0,87, 1,46). En pacientes con un valor inicial de hemoglobina < 10 g/dl (n=899), el valor estimado del índice de riesgo (hazard ratio) para la supervivencia fue de 0,98 (IC 95% 0,68 al 1,40). Se observó un aumento del riesgo relativo de acontecimientos tromboembólicos en la población general (RR 1,62, IC 95 %: 1,13, 2,31).
Asimismo, se ha realizado un análisis de datos a nivel de paciente, en más de 13.900 pacientes con cáncer (tratados con quimioterapia, radioterapia, quimioradioterapia o sin tratamiento) que participaban en un total de 53 ensayos clínicos controlados en los que se administraban diversas epoetinas. En este meta-análisis se obtuvo un índice de riesgo (hazard ratio) para la supervivencia global de 1,06 a favor de los grupos control (IC 95%: 1,00, 1,12; 53 ensayos y 13.933 pacientes) y para los pacientes con cáncer que recibían quimioterapia, el índice de riesgo (hazard ratio) para la supervivencia global fue de 1,04 (IC 95%: 0,97, 1,11; 38 ensayos y 10.441 pacientes). Este meta-análisis además indica de forma consistente un aumento significativo del riesgo relativo de episodios tromboembólicos en pacientes con cáncer tratados con eritropoyetina recombinante humana (ver sección 4.4).
En casos muy raros, se han producido anticuerpos neutralizantes anti-eritropoyetina con o sin aplasia pura de células rojas (APCR) durante el tratamiento con rHuEPO.
5.2 Propiedades farmacocinéticas
Los estudios farmacocinéticos en voluntarios sanos y pacientes urémicos muestran que la semivida de la epoetina beta administrada por vía i.v. es de 4 a 12 horas y que el volumen de distribución corresponde de una a dos veces el volumen plasmático. Se han hallado resultados análogos en experimentos en animales con ratas normales y urémicas.
Tras la administración subcutánea de epoetina beta a pacientes urémicos la absorción retardada se traduce en una meseta de concentración sérica, cuya cota máxima se alcanza al cabo de 12 - 28 horas de media. El semiperíodo de vida terminal es mayor que después de la administración intravenosa, siendo la media de 13-28 horas.
La biodisponibilidad de la epoetina beta después de la administración subcutánea está comprendida entre el 23 y el 42 % en comparación con la administración intravenosa.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad de dosis repetidas, genotoxicidad y toxicidad para la reproducción.
Un estudio de carcinogenicidad con homólogos de eritropoyetina en ratones no reveló ningún signo de potencial proliferativo o carcinogénico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Urea,
Cloruro sódico,
Polisorbato 20,
Fosfato monosódico dihidratado,
Fosfato disódico dodecahidratado,
Cloruro cálcico dihidratado,
Glicina,
L-Leucina,
L-Isoleucina,
L-Treonina,
L-Ácido glutámico,
L-Fenilalanina,
Agua para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
Conservar la jeringa precargada en el embalaje exterior, para protegerlo de la luz.
Con fines de uso ambulatorio, se puede mantener el producto fuera de la nevera durante un único periodo de hasta 3 días a temperatura ambiente (no superior a 25°C).
6.5 Naturaleza y contenido del envase
Jeringa precargada (vidrio tipo I), con un protector y un émbolo (goma teflonizada) con una aguja de inyección (30G1/2). Cada jeringa contiene 0,3 ml de solución.
Tamaños de envase de 1 jeringa precargada y 1 aguja o de 6 jeringas precargadas y 6 agujas.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
En primer lugar, ¡lávese las manos!
1. Saque una jeringa del envase y asegúrese que la solución es clara, incolora y prácticamente libre de partículas visibles. Retire el protector de la jeringa.
2. Saque una aguja del envase, fíjela sobre la jeringa y retire la tapa protectora de la aguja.
3. Expulse el aire de la jeringa y aguja manteniendo la jeringa vertical y presionando suavemente el émbolo hacia arriba. Continúe presionando el émbolo hasta que la cantidad de solución de NeoRecormon en la jeringa sea la prescrita.
4. Limpie la piel en el lugar de inyección con un algodón con alcohol. Haga un pliegue tomando la piel entre el índice y el pulgar. Sujete el cuerpo de la jeringa por la parte más próxima a la aguja e inserte la aguja en el pliegue cutáneo con acción firme y rápida. Inyecte la solución de NeoRecormon. Retire la aguja rápidamente y aplique presión sobre el lugar de inyección con un apósito seco y estéril.
Este medicamento es solo para uso individual. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/025 -026
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16 de julio de 1997 Fecha de la última revalidación: 16 de julio de 2007
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 2.000 UI solución inyectable en jeringa precargada
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Una jeringa precargada con 0,3 ml de solución para inyección contiene 2.000 unidades internacionales (UI) correspondientes a 16,6 microgramos de epoetina beta* (eritropoyetina recombinante humana).
Un ml de solución para inyección contiene 6.667 UI de epoetina beta.
* producida en células de ovarios de hámster chino (CHO) mediante tecnología de ADN recombinante.
Excipientes con efecto conocido:
Fenilalanina (hasta 0,3 mg/jeringa)
Sodio (menos de 1 mmol/jeringa)
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución incolora, de transparente a ligeramente opalescente.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
NeoRecormon está indicado para:
- Tratamiento de la anemia sintomática asociada a la insuficiencia renal crónica en pacientes adultos y pediátricos.
- Prevención de la anemia en prematuros con un peso corporal al nacer de 750 a 1.500 g y una edad gestacional de menos de 34 semanas.
- Tratamiento de la anemia sintomática en pacientes adultos con neoplasias no mieloides tratados con quimioterapia.
- Aumentar el rendimiento de la sangre autóloga de pacientes incluidos en un programa de predonación.
Debe sopesarse su uso en esta indicación frente al riesgo aumentado de episodios tromboembólicos que han sido notificados. Sólo debe administrarse el tratamiento a pacientes con anemia moderada (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], sin deficiencia de hierro) si no se dispone de procedimientos para conservar la sangre o si éstos son insuficientes cuando la cirugía mayor electiva programada requiera un gran volumen de sangre (4 o más unidades de sangre en mujeres o 5 o más unidades en hombres). Ver sección 5.1.
4.2 Posología y forma de administración
La terapia con NeoRecormon debe iniciarse por médicos con experiencia en las indicaciones arriba
mencionadas. Como se han observado reacciones anafilácticas en algunos casos aislados, se
recomienda administrar la primera dosis bajo control médico.
Posología
Tratamiento de la anemia sintomática en pacientes adultos y pediátricos con insuficiencia renal crónica
Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente. NeoRecormon se puede administrar tanto por vía subcutánea como intravenosa con el fin de aumentar la concentración de hemoglobina hasta un nivel no superior a 12 g/dl (7,5 mmol/l). En pacientes que no están sometidos a hemodiálisis es preferible utilizar la vía subcutánea para evitar la punción de venas periféricas. En caso de administración intravenosa, la solución debe ser inyectada a lo largo de unos 2 minutos, p.ej. en pacientes en hemodiálisis por vía de la fístula arteriovenosa al final de la diálisis.
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones se pueden observar valores individuales de hemoglobina superiores o inferiores a los niveles deseados. La variabilidad en los niveles de hemoglobina se debe controlar mediante ajuste de la dosis con el objeto de mantener los valores de hemoglobina dentro del intervalo entre 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Se debe evitar un nivel de hemoglobina de forma continuada por encima de 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando los valores de hemoglobina sean superiores a 12 g/dl (7,5 mmol/l).
Debe evitarse un aumento de hemoglobina superior a 2 g/dl (1,25 mmol/l) durante un periodo de cuatro semanas. Si esto ocurre, se debe hacer un ajuste adecuado de la dosis según las instrucciones incluidas en esta misma sección. Si la tasa de aumento de hemoglobina es mayor de 2 g/dl (1,25 mmol/l) en un mes o si el nivel de hemoglobina está aumentando y se acerca a 12 g/dl (7,45 mmol/l), debe reducirse la dosis en aproximadamente un 25%. Si el nivel de hemoglobina sigue aumentando, se debe interrumpir el tratamiento hasta que el nivel de hemoglobina empiece a disminuir, momento en el que se podrá reiniciar el tratamiento con una dosis aproximadamente un 25 % inferior a la dosis administrada previamente.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis eficaz más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia al tiempo que se mantiene una concentración de hemoglobina inferior o igual a 12 g/dl (7,45 mmol/l).
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica. En los pacientes con una respuesta deficiente de la hemoglobina a NeoRecormon, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.4 y 5.1).
En presencia de hipertensión o de enfermedades cardiovasculares, cerebrovasculares o vasculoperiféricas, se debe determinar de modo individual el incremento semanal y el valor objetivo de la hemoglobina teniendo en cuenta el cuadro clínico.
El tratamiento con NeoRecormon se divide en dos fases:
1. Fase de corrección
- Administración subcutánea:
La dosis inicial es 3 x 20 UI/kg por semana. La dosis puede incrementarse cada 4 semanas en 3 x 20 UI/kg y semana si el aumento de la hemoglobina no ha sido adecuado (< 0,25 g/dl por semana).
La dosis semanal puede dividirse en dosis diarias.
- Administración intravenosa:
La dosis inicial es 3 x 40 UI/kg por semana, y puede aumentarse al cabo de 4 semanas a 80 UI/kg -tres veces por semana- y si son necesarios incrementos ulteriores serán de 20 UI/kg tres veces por semana, con intervalos mensuales.
Por ambas vías de administración, la dosis máxima no debe superar 720 UI/kg por semana.
2. Fase de mantenimiento
Para mantener el nivel de hemoglobina entre 10 y 12 g/dl, la dosis es inicialmente reducida a la mitad de la cantidad previamente administrada. Posteriormente, se ajustará la dosis individualmente para el paciente a intervalos de una o dos semanas (dosis de mantenimiento).
En caso de administración subcutánea, la dosis semanal puede administrarse en una inyección única o fraccionada en tres o siete inyecciones. Los pacientes que permanezcan estables en el régimen de una dosis única semanal pueden pasar a una administración única cada dos semanas. En este caso, puede ser necesario un aumento de la dosis.
Los resultados de los estudios clínicos en niños han revelado que, en general, a menor edad, se necesita mayor dosis de NeoRecormon. No obstante, hay que seguir el programa posológico recomendado ya que no puede predecirse la respuesta individual.
El tratamiento con NeoRecormon es normalmente crónico. Sin embargo, en caso necesario puede interrumpirse en cualquier momento. Los datos sobre la pauta posológica de una vez a la semana se han obtenido de estudios clínicos con una duración de tratamiento de 24 semanas.
Prevención de anemia en prematuros
La solución se administra por vía subcutánea a una dosis de 3 x 250 UI/kg p.c. por semana. Es probable que los niños que ya hayan recibido una transfusión previa cuando se inicie el tratamiento con NeoRecormon no se beneficien tanto como los prematuros no transfundidos. La duración de tratamiento recomendada es de 6 semanas.
Tratamiento de anemia sintomática inducida por quimioterapia en pacientes con cáncer Se debe administrar NeoRecormon por vía subcutánea a pacientes con anemia (por ejemplo, si la concentración de hemoglobina es menor o igual a 10 g/dl [6,2 mmol/l]). Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente.
La dosis semanal puede administrarse como una inyección por semana o en dosis divididas entre 3 y 7 veces por semana.
La dosis inicial recomendada es de 30.000 UI por semana (que corresponde aproximadamente a 450 UI/kg de peso corporal por semana, en base al peso medio del paciente).
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones es posible llegar a observar en un paciente valores de hemoglobina superiores o inferiores a los esperados. La variabilidad de la hemoglobina se deberá manejar ajustando la dosis para mantener los valores de la hemoglobina dentro del intervalo de 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Deben evitarse concentraciones sostenidas de hemoglobina superiores a 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando se observen valores de hemoglobina superiores a los 12 g/dl (7,5 mmol/l).
Si a las 4 semanas de tratamiento el valor de hemoglobina aumenta hasta al menos 1 g/dl (0,62 mmol/l), se debe continuar con la dosis que en ese momento se esté administrando. Si los valores de hemoglobina no han aumentado al menos 1 g/dl (0,62 mmol/l), se debe considerar duplicar la dosis semanal. Si a las 8 semanas de tratamiento el valor de hemoglobina no aumenta hasta al menos 1 g/dl (0,62 mmol/l), es improbable que se produzca respuesta y, por consiguiente, el tratamiento debe ser interrumpido.
El tratamiento debe continuar durante las 4 semanas posteriores al final de la quimioterapia.
La dosis máxima no debe exceder de 60.000 UI por semana.
Una vez se ha alcanzado el objetivo terapéutico del paciente, la dosis debe reducirse del 25 al 50 % con el fin de mantener el valor de hemoglobina en ese nivel. Debe realizarse el ajuste posológico adecuado.
Si el nivel de hemoglobina excede los 12 g/dl (7,5 mmol/l) se debe reducir la dosis entre un 25 y un
50 % aproximadamente. Si el nivel de hemoglobina excede de 13 g/dl (8,1 mmol), se debe interrumpir temporalmente el tratamiento con NeoRecormon. El tratamiento debe reiniciarse con una dosis aproximadamente un 25 % inferior a la dosis previamente administrada después de que los niveles de hemoglobina desciendan hasta un valor menor o igual a 12 g/dl (7,5 mmol/l) o por debajo.
51 al cabo de 4 semanas el incremento de hemoglobina es superior a 2 g/dl (1,3 mmol/l), la dosis debe reducirse entre un 25 y un 50 %.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia.
Tratamiento para incrementar el rendimiento de la sangre autóloga donada La solución puede administrarse por vía intravenosa, en unos 2 minutos, o subcutáneamente. NeoRecormon se administra dos veces por semana durante 4 semanas. En aquellas ocasiones en que el hematocrito del paciente permite la donación de sangre, es decir el hematocrito es > 33 %, NeoRecormon se administra al final de la donación de sangre.
Durante la totalidad del período de tratamiento no debe excederse un hematocrito del 48 %.
La dosis debe ser determinada por el equipo quirúrgico, individualmente para cada paciente, en función de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos:
1. La cantidad de sangre pre-donada necesaria dependerá de la pérdida prevista de sangre, de los medios de conservación empleados y del estado físico del paciente.
Este volumen debe ser equivalente a la cantidad de sangre que se considera necesaria para evitar transfusiones de sangre homóloga.
La cantidad de sangre pre-donada necesaria se expresa en unidades en las cuales una unidad de nomograma es equivalente a 180 ml de glóbulos rojos.
2. La capacidad de donar sangre depende predominantemente del volumen sanguíneo del paciente y del hematocrito basal. Ambas variables determinan la reserva endógena de eritrocitos, que puede ser calculada conforme a la fórmula siguiente:
Reserva endógena de eritrocitos = volumen sanguíneo (ml) x (hematocrito - 33)/ 100 mujeres: volumen sanguíneo (ml) = 41 (ml/kg) x peso corporal (kg) + 1200 (ml)
hombres: volumen sanguíneo (ml) = 44 (ml/kg) x peso corporal (kg) + 1600 (ml)
(peso corporal: > 45 kg)
La indicación para un tratamiento con NeoRecormon y, en su caso, la dosis individual debe ser determinada a partir de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos según las gráficas siguientes:
Paciente femenino Paciente masculino
Cantidad de sangre pre-donada Cantidad de sangre pre-donada
necesaria [unidades] necesaria [unidades]
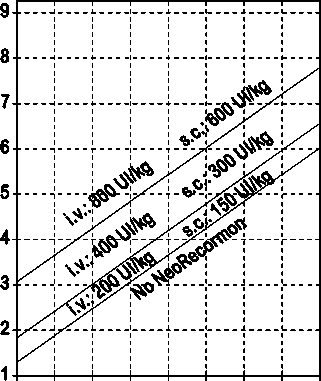
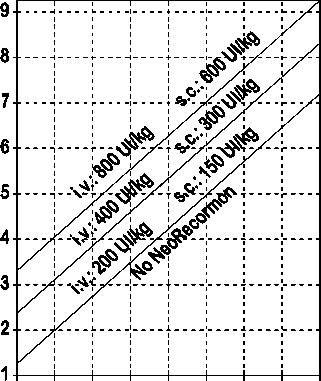
O 100 200 300 400 500 600 700 800 0 100 200 300 400 500 600 700 800
Reserva eritrocitaria endógena [ml] Reserva eritrocitaria endógena [ml]
La dosis única así determinada debe administrarse dos veces por semana a lo largo de 4 semanas. La dosis máxima no debe sobrepasar 1.600 UI/kg peso corporal por semana en administración intravenosa ó 1.200 UI/kg por semana en administración subcutánea.
Forma de administración
La jeringa precargada de NeoRecormon está lista para su uso. Sólo se pueden inyectar soluciones claras o ligeramente opalescentes, incoloras y prácticamente libres de partículas visibles. NeoRecormon en jeringa precargada es un producto estéril pero sin conservantes. Bajo ninguna circunstancia debe administrarse más de una dosis por jeringa; el medicamento es solo para uso individual.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Hipertensión mal controlada.
En la indicación para “Aumentar el rendimiento de sangre autóloga”: infarto de miocardio o accidente cerebrovascular en el mes anterior al tratamiento, angina de pecho inestable, riesgo aumentado de trombosis venosa profunda, como por ejemplo en aquellos con historial de enfermedad tromboembólica venosa.
4.4 Advertencias y precauciones especiales de empleo
NeoRecormon debe usarse con cautela en presencia de anemia refractaria con exceso de blastos en transformación, epilepsia, trombocitosis e insuficiencia hepática crónica. Y deben excluirse las deficiencias de ácido fólico y vitamina B12 pues reducen la eficacia de NeoRecormon.
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica, ya que las dosis acumuladas altas de epoetina pueden asociarse a un mayor riesgo de mortalidad, y de acontecimientos cardiovasculares y cerebrovasculares graves. En pacientes con una respuesta deficiente de la hemoglobina a epoetinas, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.2 y 5.1).
Para garantizar una eritropoyesis eficaz, se debe evaluar el nivel de hierro en todos los pacientes antes y durante el tratamiento, y puede ser necesario un tratamiento suplementario con hierro administrado de acuerdo con las guías terapéuticas.
La sobrecarga grave de aluminio debido al tratamiento de la insuficiencia renal puede comprometer la eficacia de NeoRecormon.
La indicación de tratamiento con NeoRecormon en enfermos nefroscleróticos aún no sometidos a diálisis debe definirse individualmente, ya que la posible aceleración de la insuficiencia renal no puede descartarse con certeza.
Aplasia pura de células rojas (APCR)
Se han notificado casos de APCRcausada por anticuerpos neutralizantes antieritropoyetina, asociados a tratamientos con eritropoyetinas, incluido NeoRecormon. Estos anticuerpos han presentado reacción cruzada con todas las proteínas eritropoyéticas, por los que los pacientes en los que se sospeche o se haya confirmado la presencia de anticuerpos neutralizantes contra eritropoyetina no deben ser tratados con NeoRecormon (ver sección 4.8).
APCR en pacientes con hepatitis C
Se debe interrumpir inmediatamente el tratamiento con epoetina, y realizar ensayos de anticuerpos antieritropoyetina si se produce una disminución paradójica de la hemoglobina y se desarrolla anemia severa asociada con bajos recuentos de reticulocitos. En pacientes con hepatitis C tratados con interferon y ribavirina se han notificados casos cuando las epoetinas se han utilizado concomitantemente. Las epoetinas no están aprobadas en el tratamiento de la anemia asociada a la hepatitis C.
Monitorización de la tensión arterial
Puede aparecer un aumento de la tensión arterial o un agravamiento de la hipertensión ya existente, especialmente en los casos en los que el hematocrito aumenta rápidamente. Estos incrementos en la tensión arterial pueden ser tratados con medicamentos. Si no pueden ser controlados con medicación se recomienda interrumpir transitoriamente la terapia con NeoRecormon. En particular al principio de la terapia se recomienda la monitorización regular de la tensión arterial; también durante la diálisis. Pueden producirse crisis hipertensivas con síntomas de encefalopatías y que requieren atención médica inmediata y cuidados médicos intensivos. Debe prestarse especial atención a las cefaleas súbitas lacerantes hemicraneales como posible signo de advertencia.
Insuficiencia renal crónica
En pacientes con insuficiencia renal crónica puede sobrevenir un aumento dosis-dependiente moderado en la cifra plaquetaria dentro del margen normal durante el tratamiento con NeoRecormon, especialmente después de la administración intravenosa. La regresión tiene lugar en el curso de la prosecución de la terapia. Se recomienda el control regular de la cifra de plaquetas durante las 8 primeras semanas de tratamiento.
Concentración de hemoglobina
En pacientes con insuficiencia renal crónica, la concentración de hemoglobina en la fase de mantenimiento no debe exceder el límite superior del rango recomendado en la sección 4.2. En ensayos clínicos se observó un aumento del riesgo de fallecimiento y de acontecimientos cardiovasculares graves o acontecimientos cerebrovasculares incluido el infarto, cuando se administraron agentes estimulantes de la eritropoyesis (AEEs) con el fin de alcanzar un nivel de hemoglobina superior a 12 g/dl (7,5 mmol/l).
En ensayos clínicos controlados no se han observado beneficios significativos atribuibles a la administración de epoetinas cuando se aumentaba la concentración de hemoglobina por encima del nivel necesario para controlar los síntomas de anemia y evitar las transfusiones sanguíneas.
En niños prematuros puede producirse un ligero aumento de la cifra plaquetaria, en especial hasta el día 12 -14 de vida, por lo tanto las plaquetas deben controlarse regularmente.
Efectos sobre el crecimiento tumoral
Las epoetinas son factores de crecimiento que estimulan de forma primaria la producción de glóbulos rojos. Los receptores de epoetina pueden expresarse sobre la superficie de una variedad de células tumorales. Como sucede con todos los factores de crecimiento, existe la preocupación de que las epoetinas pudieran estimular el crecimiento de tumores. En varios ensayos controlados, no se ha observado que las epoetinas mejoren la supervivencia global o que disminuyan el riesgo de progresión del tumor en pacientes con anemia asociada con cáncer.
En ensayos clínicos controlados, se ha observado que el uso de NeoRecormon y otros agentes estimulantes de la eritropoyesis (AEEs):
- acortaba el tiempo de progresión del tumor en pacientes con cáncer avanzado de cabeza y cuello que recibían radioterapia cuando se administraba para conseguir una concentración de hemoglobina por encima de 14 g/dl (8,7 mmol/l),
- acortaba la supervivencia global y aumentaba el número de fallecimientos atribuidos a la progresión de la enfermedad a los 4 meses, en pacientes con cáncer de mama metastásico que recibían quimioterapia cuando se administraba para conseguir una concentración de hemoglobina entre los 12 y los 14 g/dl (7,5-8,7 mmol/l),
- aumentaba el riesgo de fallecimiento cuando se administraba para conseguir una concentración de hemoglobina de 12 g/dl (7,5 mmol/l) en pacientes con enfermedad maligna activa y que no recibían ni quimioterapia ni radioterapia. El uso de los AEEs no está indicado en esa población de pacientes.
En vista de lo anterior, en algunas situaciones clínicas la transfusión sanguínea debe ser el tratamiento de elección para la anemia en pacientes con cáncer. La decisión de administrar eritropoyetinas recombinantes se tomará en base a la evaluación de la relación beneficio/riesgo junto con la aceptación individual del paciente, teniendo en cuenta el contexto clínico específico. Los factores que deben considerarse en esta evaluación son el tipo de tumor y su estadio, el grado de anemia, la esperanza de vida, el entorno en el que el paciente está siendo tratado y la preferencia del paciente (ver sección 5.1).
Puede haber un aumento de la tensión arterial que puede ser tratada con medicamentos. Se recomienda por tanto monitorizar la tensión arterial, en particular en la fase inicial de tratamiento en pacientes con cáncer.
En pacientes con cáncer también deberán monitorizarse regularmente el recuento plaquetario y el valor de hemoglobina.
En pacientes en programa de predonación de sangre autóloga, puede sobrevenir un aumento de la cifra de plaquetas, generalmente dentro de los límites normales. Por tanto, se recomienda la determinación de esta cifra al menos una vez por semana. Si se produce un aumento plaquetario de más de 150 x 109/l o si las plaquetas ascienden por encima del margen normal, se debe suspender el tratamiento con NeoRecormon.
En niños prematuros no se podría descartar el riesgo potencial de que la eritropoyetina provoque retinopatía, por lo tanto se debe tener precaución y la decisión de tratar a un niño prematuro se debe tomar teniendo en cuenta el beneficio potencial y el riesgo de este tratamiento, así como las opciones alternativas de tratamiento que estén disponibles.
En pacientes con insuficiencia renal crónica, a menudo se precisa aumentar la dosis de heparina durante la hemodiálisis en la terapia con NeoRecormon debido al aumento del volumen de hematocrito. Si la heparinización no es óptima es posible que se produzca la oclusión del sistema de diálisis.
En pacientes con alteración renal crónica con riesgo de trombosis de derivación, se debe considerar la revisión temprana de la derivación y profilaxis de la trombosis mediante la administración, por ejemplo, de ácido acetilsalicílico.
Los niveles séricos de potasio y fosfato deben ser controlados regularmente durante la terapia con NeoRecormon. Se ha informado de aumentos de potasio en unos pocos pacientes urémicos que recibieron NeoRecormon si bien no se ha establecido una relación causal. En caso de observar un nivel elevado o creciente de potasio se debe considerar la interrupción de la administración de NeoRecormon hasta que el nivel haya sido corregido.
Para el uso de NeoRecormon en un programa de predonación de sangre autóloga deben ser observadas las directrices oficiales sobre donación de sangre, en particular las siguientes:
- sólo deben donar los pacientes con hematocrito > 33 % (hemoglobina >11 g/dl [6,83 mmol/l]);
- debe procederse con especial cuidado con pacientes con un peso inferior a 50 kg;
- el volumen individual extraído no debe superar aproximadamente el 12 % del volumen sanguíneo estimado del paciente.
El tratamiento debe reservarse a pacientes en los que se considere particularmente importante evitar la transfusión de sangre homóloga teniendo en cuenta la evaluación riesgo/beneficio de las transfusiones homólogas.
Uso indebido
Un uso indebido del producto por personas sanas puede llevar a un aumento excesivo del volumen de hematocrito, lo cual puede asociarse con complicaciones del sistema cardiovascular con riesgo para la vida.
Excipientes
NeoRecormon en jeringa precargada contiene, como máximo, 0,3 mg de fenilalanina por jeringa como excipiente. Este hecho se deberá tener en cuenta en los pacientes afectados por formas graves de fenilcetonuria.
Este medicamento contiene menos de 1 mmol (23 mg) de sodio por jeringa, por lo que se considera esencialmente “exento de sodio”.
Trazabilidad de NeoRecormon
Para mejorar la trazabilidad de los agentes estimulantes de la eritropoyesis (AEEs), se debe registrar (o indicar) claramente el nombre comercial del AEE administrado en la historia clínica del paciente.
4.5 Interacción con otros medicamentos y otras formas de interacción
Los resultados clínicos obtenidos hasta el presente no ponen de manifiesto interacción alguna de NeoRecormon con otros medicamentos.
Los estudios llevados a cabo en animales mostraron que la epoetina beta no potencia la mielotoxicidad de los medicamentos citostáticos, tales como etopósido, cisplatino, ciclofosfamida y fluorouracilo.
4.6 Fertilidad, embarazo y lactancia
Fertilidad
Los estudios en animales no muestran efectos dañinos directos o indirectos sobre el embarazo, desarrollo embrional/fetal, parto o desarrollo postnatal (ver sección 5.3).
Embarazo
No hay datos relativos al uso de NeoRecormon en mujeres embarazadas.
Se debe tener precaución cuando se prescriba a mujeres embarazadas.
Lactancia
Se desconoce si la epoetina beta se excreta en la leche materna. La decisión de continuar/interrumpir la lactancia o de continuar/interrumpir el tratamiento con epoetina beta se debe hacer teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento con epoetina beta para la madre.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NeoRecormon sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Basados en los resultados de los ensayos clínicos que incluyen 1.725 pacientes, se espera que aproximadamente el 8 % de los pacientes tratados con NeoRecormon sufran reacciones adversas.
Pacientes anémicos con insuficiencia renal crónica
La reacción adversa más frecuente durante el tratamiento con NeoRecormon es un aumento de la tensión arterial o agravamiento de la hipertensión existente, especialmente en casos de aumento rápido del hematocrito (ver sección 4.4). En algunos pacientes cuya tensión arterial es normal o baja también pueden darse crisis hipertensivas con síntomas de encefalopatías (p.ej. cefaleas y estados de confusión, trastornos sensitivomotores - como alteraciones del habla o de la ambulación - hasta convulsiones tónico-clónicas) (ver sección 4.4).
Puede darse trombosis de derivación, especialmente en pacientes que tienen tendencia a la hipotensión o cuyas fístulas arteriovenosas presentan complicaciones (p.ej. estenosis, aneurismas), ver sección 4.4. En la mayoría de los casos se observa una caída en los valores séricos de ferritina simultáneamente con el aumento del volumen de hematocrito (ver sección 4.4). Además, en casos aislados se han observado incrementos transitorios en los niveles séricos de potasio y fosfato (ver sección 4.4).
En casos aislados se ha notificado aplasia pura de células rojas (APCR) mediada por anticuerpos neutralizantes antieritropoyetina asociada al tratamiento con NeoRecormon. En caso de que se diagnostique APCR mediada por anticuerpos antieritropoyetina, el paciente deberá interrumpir el tratamiento con NeoRecormon, el cual no se deberá sustituir por otra proteína eritropoyética (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 1, que figura más adelante.
Pacientes con cáncer
El dolor de cabeza y la hipertensión relacionados con el tratamiento con epoetina beta son frecuentes y pueden tratarse con medicamentos (ver sección 4.4).
En algunos pacientes se observa un descenso de los valores de hierro sérico (ver sección 4.4).
Estudios clínicos han puesto de manifiesto que la frecuencia de acontecimientos tromboembólicos es más alta en pacientes con cáncer tratados con NeoRecormon que en los controles no tratados o tratados con placebo. En pacientes tratados con NeoRecormon, esta incidencia es del 7 % comparado con el 4 % en el grupo control; este hecho no está asociado con un incremento en la mortalidad tromboembólica comparado con los grupos control.
Estas reacciones adversas se enumeran en la Tabla 2, que figura más adelante.
Pacientes incluidos en un programa de predonación de sangre autóloga Los pacientes incluidos en un programa de predonación de sangre autóloga han demostrado una frecuencia ligeramente superior de accidentes tromboembólicos. Sin embargo, no se pudo establecer una relación causal con el tratamiento con NeoRecormon.
En ensayos clínicos controlados con placebo, la deficiencia transitoria de hierro fue más acusada en los pacientes tratados con NeoRecormon que en los pacientes control (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 3, que figura más adelante.
Tabla de reacciones adversas
Las reacciones adversas se enumeran de acuerdo con la clasificación de órganos del sistema MedDRA y por categoría de frecuencia.
Las categorías de frecuencia se definen utilizando la siguiente convención:
muy frecuentes (> 1/10), frecuentes (> 1/100 hasta <1/10), poco frecuentes (>1/1.000 hasta <1/100), raras (>1/10.000 hasta <1/1.000); muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 1: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con IRC_
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión Crisis hipertensivas |
Frecuente Poco frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
|
Trastornos de la sangre y |
Trombosis de derivación |
Raras |
|
del sistema linfático |
Trombocitosis |
Muy raras |
Tabla 2: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con cáncer__
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión |
Frecuente |
|
Trastornos de la sangre y del sistema linfático |
Acontecimientos tromboembólicos |
Frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Tabla 3: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes incluidos en un programa de predonación de sangre autóloga
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
-Prematuros
Es muy frecuente un descenso de los valores de ferritina sérica (ver sección 4.4).
Descripción de reacciones adversas seleccionadas
Raramente pueden ocurrir reacciones cutáneas relacionadas con el tratamiento con epoetina beta como sarpullido, prurito, urticaria o reacciones en el lugar de la inyección. En casos muy raros se han notificado reacciones anafilácticas relacionadas con el tratamiento con epoetina beta. Sin embargo, en los ensayos clínicos controlados no se encontró una mayor incidencia de reacciones de hipersensibilidad.
Se han comunicado casos muy raros de síntomas gripales relacionados con el tratamiento con epoetina beta como fiebre, escalofríos, dolores de cabeza, dolor de las extremidades, malestar general, y/o dolor óseo, particularmente al inicio del tratamiento. Estas reacciones son de intensidad leve a moderada y desaparecen al cabo de un par de horas o días.
Los resultados obtenidos en un ensayo clínico controlado realizado con epoetina alfa o darbepoetina alfa, notificaron que la incidencia de infarto es frecuente.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
El marg en terapéutico de NeoRecormon es muy amplio. No se han observado síntomas de sobredosis incluso a niveles séricos muy elevados.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antianémico, código ATC: B03XA01 Mecanismo de acción
La eritropoyetina es una glucoproteína que estimula la formación de eritrocitos a partir de sus progenitores asignados. Actúa como factor estimulante de la mitosis y hormona de diferenciación.
La epoetina beta, el principio activo de NeoRecormon, es idéntica en su composición aminoácida e hidrocarbonada a la eritropoyetina aislada de la orina de pacientes anémicos.
La eficacia biológica de la epoetina beta ha sido demostrada tras administración intravenosa y subcutánea en varios modelos animales in vivo (ratas normales y urémicas, ratones policitémicos, perros). Con la administración de epoetina beta aumentan el número de eritrocitos, los valores de Hb y la cifra de reticulocitos, al igual que la velocidad de incorporación de 59Fe.
In vitro (cultivo citológico de bazo de ratón) ha sido hallada una mayor incorporación de 3H-timidina en las células eritroides nucleadas del bazo tras incubación con epoetina beta.
Las investigaciones en cultivos de células de la médula ósea humana han revelado que la epoetina beta estimula específicamente la eritropoyesis y no afecta a la leucopoyesis. No han sido detectadas acciones citotóxicas de la epoetina beta sobre la médula ósea o sobre células cutáneas humanas.
Tras una dosis única de epoetina beta no se ha observado ningún efecto en el comportamiento o la actividad locomotora del ratón ni en las funciones circulatoria o respiratoria del perro.
Eficacia clínica y seguridad
En un ensayo aleatorizado, doble ciego, controlado con placebo, realizado en 4.038 pacientes con enfermedad renal crónica no sometidos a diálisis, con una diabetes tipo 2 y los niveles de hemoglobina < 11 g/dl, los pacientes recibieron tratamiento bien con darbepoetina alfa para alcanzar niveles de hemoglobina de 13 g/dl o con placebo (ver sección 4.4). El ensayo no cumplió su objetivo principal de demostrar una disminución en el riesgo de todas las causas de mortalidad, la morbilidad cardiovascular o enfermedad renal terminal. El análisis de los componentes individuales de la variable combinada mostró los siguientes HR (IC 95%): fallecimiento 1,05 (0,92-1,21), infarto cerebrovascular 1,92 (1,38-2,68), insuficiencia cardíaca congestiva 0,89 (0,74-1,08), infarto de miocardio 0,96 (0,751,23), hospitalización por isquemia del miocardio 0,84 (0,55-1,27), enfermedad renal terminal 1,02 (0,87-1,18).
En pacientes con IRC (tratados con diálisis, no tratados con diálisis, pacientes diabéticos y pacientes no diabéticos) se han realizado análisis agrupados post hoc de los estudios clínicos con AEEs. Se observó una tendencia a un aumento de las estimaciones del riesgo para la mortalidad por todas las causas y para los acontecimientos cardiovasculares y cerebrovasculares asociados a dosis acumuladas más altas de AEE independientemente de que los pacientes padecieran o no diabetes y de que recibieran o no tratamiento con diálisis (ver las secciones 4.2 y 4.4).
La eritropoyetina es un factor de crecimiento que estimula de forma primaria la producción de glóbulos rojos. Los receptores de eritropoyetina se encuentran también presentes en la superficie de algunas líneas celulares malignas.
Se ha estudiado la supervivencia y la progresión tumoral en cinco grandes ensayos controlados que incluyeron a 2.833 pacientes, de los cuales cuatro fueron ensayos doble-ciego controlados con placebo y uno fue un ensayo abierto. Dos de los ensayos reclutaron pacientes que estaban siendo tratados con quimioterapia. El nivel de hemoglobina que se quería alcanzar fue >13 g/dl en dos de los ensayos y de entre 12 y 14 g/dl en los otros tres. En el ensayo abierto no se observaron diferencias en la supervivencia global, entre los pacientes tratados con eritropoyetina humana recombinante y el grupo control. En los cuatro ensayos controlados con placebo, el índice de riesgo (hazard ratio) para la supervivencia global osciló entre 1,25 y 2,47 en favor de los grupos control. En todos estos ensayos se ha observado un aumento de la mortalidad inexplicable y estadísticamente significativo en pacientes que presentaban anemia asociada con diversos tipos frecuentes de cáncer y que recibieron eritropoyetina humana recombinante, en comparación con los grupos control. Las diferencias observadas en la incidencia de trombosis y complicaciones relacionadas, entre los pacientes que recibieron eritropoyetina humana recombinante y los que formaban parte de los grupos control, no permiten explicar satisfactoriamente los resultados de supervivencia global observados en los ensayos.
Un meta-análisis basado en datos individuales de pacientes, que incluyó datos de los 12 ensayos clínicos controlados realizados con NeoRecormon en pacientes anémicos con cáncer (n=2301), mostró un valor estimado del índice de riesgo (hazard ratio) de supervivencia global de 1,13 en favor de los grupos control (IC 95 %, CI 0,87, 1,46). En pacientes con un valor inicial de hemoglobina < 10 g/dl (n=899), el valor estimado del índice de riesgo (hazard ratio) para la supervivencia fue de 0,98 (IC 95% 0,68 al 1,40). Se observó un aumento del riesgo relativo de acontecimientos tromboembólicos en la población general (RR 1,62, IC 95 %: 1,13, 2,31).
Asimismo, se ha realizado un análisis de datos a nivel de paciente, en más de 13.900 pacientes con cáncer (tratados con quimioterapia, radioterapia, quimioradioterapia o sin tratamiento) que participaban en un total de 53 ensayos clínicos controlados en los que se administraban diversas epoetinas. En este meta-análisis se obtuvo un índice de riesgo (hazard ratio) para la supervivencia global de 1,06 a favor de los grupos control (IC 95%: 1,00, 1,12; 53 ensayos y 13.933 pacientes) y para los pacientes con cáncer que recibían quimioterapia, el índice de riesgo (hazard ratio) para la supervivencia global fue de 1,04 (IC 95%: 0,97, 1,11; 38 ensayos y 10.441 pacientes). Este meta-análisis además indica de forma consistente un aumento significativo del riesgo relativo de episodios tromboembólicos en pacientes con cáncer tratados con eritropoyetina recombinante humana (ver sección 4.4).
En casos muy raros, se han producido anticuerpos neutralizantes anti-eritropoyetina con o sin aplasia pura de células rojas (APCR) durante el tratamiento con rHuEPO.
5.2 Propiedades farmacocinéticas
Los estudios farmacocinéticos en voluntarios sanos y pacientes urémicos muestran que la semivida de la epoetina beta administrada por vía i.v. es de 4 a 12 horas y que el volumen de distribución corresponde de una a dos veces el volumen plasmático. Se han hallado resultados análogos en experimentos en animales con ratas normales y urémicas.
Tras la administración subcutánea de epoetina beta a pacientes urémicos la absorción retardada se traduce en una meseta de concentración sérica, cuya cota máxima se alcanza al cabo de 12 - 28 horas de media. El semiperíodo de vida terminal es mayor que después de la administración intravenosa, siendo la media de 13-28 horas.
La biodisponibilidad de la epoetina beta después de la administración subcutánea está comprendida entre el 23 y el 42 % en comparación con la administración intravenosa.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad de dosis repetidas, genotoxicidad y toxicidad para la reproducción.
Un estudio de carcinogenicidad con homólogos de eritropoyetina en ratones no reveló ningún signo de potencial proliferativo o carcinogénico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Urea,
Cloruro sódico,
Polisorbato 20,
Fosfato monosódico dihidratado,
Fosfato disódico dodecahidratado,
Cloruro cálcico dihidratado,
Glicina,
L-Leucina,
L-Isoleucina,
L-Treonina,
L-Ácido glutámico,
L-Fenilalanina,
Agua para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
Conservar la jeringa precargada en el embalaje exterior, para protegerlo de la luz.
Con fines de uso ambulatorio, se puede mantener el producto fuera de la nevera durante un único periodo de hasta 3 días a temperatura ambiente (no superior a 25°C).
6.5 Naturaleza y contenido del envase
Jeringa precargada (vidrio tipo I), con un protector y un émbolo (goma teflonizada) con una aguja de inyección (27G1/2). Cada jeringa contiene 0,3 ml de solución.
Tamaños de envase de 1 jeringa precargada y 1 aguja o de 6 jeringas precargadas y 6 agujas.
Puede que solamente estén comercializados algunos tamaños de envase.
6.6 Precauciones especiales de eliminación y otras manipulaciones
En primer lugar, ¡lávese las manos!
1. Saque una jeringa del envase y asegúrese que la solución es clara, incolora y prácticamente libre de partículas visibles. Retire el protector de la jeringa.
2. Saque una aguja del envase, fíjela sobre la jeringa y retire la tapa protectora de la aguja.
3. Expulse el aire de la jeringa y aguja manteniendo la jeringa vertical y presionando suavemente el émbolo hacia arriba. Continúe presionando el émbolo hasta que la cantidad de solución de NeoRecormon en la jeringa sea la prescrita.
4. Limpie la piel en el lugar de inyección con un algodón con alcohol. Haga un pliegue tomando la piel entre el índice y el pulgar. Sujete el cuerpo de la jeringa por la parte más próxima a la aguja e inserte la aguja en el pliegue cutáneo con acción firme y rápida. Inyecte la solución de NeoRecormon. Retire la aguja rápidamente y aplique presión sobre el lugar de inyección con un apósito seco y estéril.
Este medicamento es solo para uso individual. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/029 -030
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16 de julio de 1997 Fecha de la última revalidación: 16 de julio de 2007.
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 3.000 UI solución inyectable en jeringa precargada
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Una jeringa precargada con 0,3 ml de solución para inyección contiene 3.000 unidades internacionales (UI) correspondientes a 24,9 microgramos de epoetina beta* (eritropoyetina recombinante humana).
Un ml de solución para inyección contiene 10.000 UI de epoetina beta.
* producida en células de ovarios de hámster chino (CHO) mediante tecnología de ADN recombinante.
Excipientes con efecto conocido:
Fenilalanina (hasta 0,3 mg/jeringa)
Sodio (menos de 1 mmol/jeringa)
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución incolora, de transparente a ligeramente opalescente.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
NeoRecormon está indicado para:
- Tratamiento de la anemia sintomática asociada a la insuficiencia renal crónica en pacientes adultos y pediátricos.
- Prevención de la anemia en prematuros con un peso corporal al nacer de 750 a 1.500 g y una edad gestacional de menos de 34 semanas.
- Tratamiento de la anemia sintomática en pacientes adultos con neoplasias no mieloides tratados con quimioterapia.
- Aumentar el rendimiento de la sangre autóloga de pacientes incluidos en un programa de predonación.
Debe sopesarse su uso en esta indicación frente al riesgo aumentado de episodios tromboembólicos que han sido notificados. Sólo debe administrarse el tratamiento a pacientes con anemia moderada (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], sin deficiencia de hierro) si no se dispone de procedimientos para conservar la sangre o si éstos son insuficientes cuando la cirugía mayor electiva programada requiera un gran volumen de sangre (4 o más unidades de sangre en mujeres o 5 o más unidades en hombres). Ver sección 5.1.
4.2 Posología y forma de administración
La terapia con NeoRecormon debe iniciarse por médicos con experiencia en las indicaciones arriba
mencionadas. Como se han observado reacciones anafilácticas en algunos casos aislados, se
recomienda administrar la primera dosis bajo control médico.
Posología
Tratamiento de la anemia sintomática en pacientes adultos y pediátricos con insuficiencia renal crónica
Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente. NeoRecormon se puede administrar tanto por vía subcutánea como intravenosa con el fin de aumentar la concentración de hemoglobina hasta un nivel no superior a 12 g/dl (7,5 mmol/l). En pacientes que no están sometidos a hemodiálisis es preferible utilizar la vía subcutánea para evitar la punción de venas periféricas. En caso de administración intravenosa, la solución debe ser inyectada a lo largo de unos 2 minutos, p.ej. en pacientes en hemodiálisis por vía de la fístula arteriovenosa al final de la diálisis.
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones se pueden observar valores individuales de hemoglobina superiores o inferiores a los niveles deseados. La variabilidad en los niveles de hemoglobina se debe controlar mediante ajuste de la dosis con el objeto de mantener los valores de hemoglobina dentro del intervalo entre 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Se debe evitar un nivel de hemoglobina de forma continuada por encima de 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando los valores de hemoglobina sean superiores a 12 g/dl (7,5 mmol/l).
Debe evitarse un aumento de hemoglobina superior a 2 g/dl (1,25 mmol/l) durante un periodo de cuatro semanas. Si esto ocurre, se debe hacer un ajuste adecuado de la dosis según las instrucciones incluidas en esta misma sección. Si la tasa de aumento de hemoglobina es mayor de 2 g/dl (1,25 mmol/l) en un mes o si el nivel de hemoglobina está aumentando y se acerca a 12 g/dl (7,45 mmol/l), debe reducirse la dosis en aproximadamente un 25%. Si el nivel de hemoglobina sigue aumentando, se debe interrumpir el tratamiento hasta que el nivel de hemoglobina empiece a disminuir, momento en el que se podrá reiniciar el tratamiento con una dosis aproximadamente un 25 % inferior a la dosis administrada previamente.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis eficaz más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia al tiempo que se mantiene una concentración de hemoglobina inferior o igual a 12 g/dl (7,45 mmol/l).
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica. En los pacientes con una respuesta deficiente de la hemoglobina a NeoRecormon, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.4 y 5.1).
En presencia de hipertensión o de enfermedades cardiovasculares, cerebrovasculares o vasculoperiféricas, se debe determinar de modo individual el incremento semanal y el valor objetivo de la hemoglobina teniendo en cuenta el cuadro clínico.
El tratamiento con NeoRecormon se divide en dos fases:
1. Fase de corrección
- Administración subcutánea:
La dosis inicial es 3 x 20 UI/kg por semana. La dosis puede incrementarse cada 4 semanas en 3 x 20 UI/kg y semana si el aumento de la hemoglobina no ha sido adecuado (< 0,25 g/dl por semana).
La dosis semanal puede dividirse en dosis diarias.
- Administración intravenosa:
La dosis inicial es 3 x 40 UI/kg por semana, y puede aumentarse al cabo de 4 semanas a 80 UI/kg -tres veces por semana- y si son necesarios incrementos ulteriores serán de 20 UI/kg tres veces por semana, con intervalos mensuales.
Por ambas vías de administración, la dosis máxima no debe superar 720 UI/kg por semana.
2. Fase de mantenimiento
Para mantener el nivel de hemoglobina entre 10 y 12 g/dl, la dosis es inicialmente reducida a la mitad de la cantidad previamente administrada. Posteriormente, se ajustará la dosis individualmente para el paciente a intervalos de una o dos semanas (dosis de mantenimiento).
En caso de administración subcutánea, la dosis semanal puede administrarse en una inyección única o fraccionada en tres o siete inyecciones. Los pacientes que permanezcan estables en el régimen de una dosis única semanal pueden pasar a una administración única cada dos semanas. En este caso, puede ser necesario un aumento de la dosis.
Los resultados de los estudios clínicos en niños han revelado que, en general, a menor edad, se necesita mayor dosis de NeoRecormon. No obstante, hay que seguir el programa posológico recomendado ya que no puede predecirse la respuesta individual.
El tratamiento con NeoRecormon es normalmente crónico. Sin embargo, en caso necesario puede interrumpirse en cualquier momento. Los datos sobre la pauta posológica de una vez a la semana se han obtenido de estudios clínicos con una duración de tratamiento de 24 semanas.
Prevención de anemia en prematuros
La solución se administra por vía subcutánea a una dosis de 3 x 250 UI/kg p.c. por semana. Es probable que los niños que ya hayan recibido una transfusión previa cuando se inicie el tratamiento con NeoRecormon no se beneficien tanto como los prematuros no transfundidos. La duración de tratamiento recomendada es de 6 semanas.
Tratamiento de anemia sintomática inducida por quimioterapia en pacientes con cáncer Se debe administrar NeoRecormon por vía subcutánea a pacientes con anemia (por ejemplo, si la concentración de hemoglobina es menor o igual a 10 g/dl [6,2 mmol/l]). Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente.
La dosis semanal puede administrarse como una inyección por semana o en dosis divididas entre 3 y 7 veces por semana.
La dosis inicial recomendada es de 30.000 UI por semana (que corresponde aproximadamente a 450 UI/kg de peso corporal por semana, en base al peso medio del paciente).
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones es posible llegar a observar en un paciente valores de hemoglobina superiores o inferiores a los esperados. La variabilidad de la hemoglobina se deberá manejar ajustando la dosis para mantener los valores de la hemoglobina dentro del intervalo de 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Deben evitarse concentraciones sostenidas de hemoglobina superiores a 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando se observen valores de hemoglobina superiores a los 12 g/dl (7,5 mmol/l).
Si a las 4 semanas de tratamiento el valor de hemoglobina aumenta hasta al menos 1 g/dl (0,62 mmol/l), se debe continuar con la dosis que en ese momento se esté administrando. Si los valores de hemoglobina no han aumentado al menos 1 g/dl (0,62 mmol/l), se debe considerar duplicar la dosis semanal. Si a las 8 semanas de tratamiento el valor de hemoglobina no aumenta hasta al menos 1 g/dl (0,62 mmol/l), es improbable que se produzca respuesta y, por consiguiente, el tratamiento debe ser interrumpido.
El tratamiento debe continuar durante las 4 semanas posteriores al final de la quimioterapia.
La dosis máxima no debe exceder de 60.000 UI por semana.
Una vez se ha alcanzado el objetivo terapéutico del paciente, la dosis debe reducirse del 25 al 50 % con el fin de mantener el valor de hemoglobina en ese nivel. Debe realizarse el ajuste posológico adecuado.
Si el nivel de hemoglobina excede los 12 g/dl (7,5 mmol/l) se debe reducir la dosis entre un 25 y un
50 % aproximadamente. Si el nivel de hemoglobina excede de 13 g/dl (8,1 mmol), se debe interrumpir temporalmente el tratamiento con NeoRecormon. El tratamiento debe reiniciarse con una dosis aproximadamente un 25 % inferior a la dosis previamente administrada después de que los niveles de hemoglobina desciendan hasta un valor menor o igual a 12 g/dl (7,5 mmol/l) o por debajo.
51 al cabo de 4 semanas el incremento de hemoglobina es superior a 2 g/dl (1,3 mmol/l), la dosis debe reducirse entre un 25 y un 50 %.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia.
Tratamiento para incrementar el rendimiento de la sangre autóloga donada La solución puede administrarse por vía intravenosa, en unos 2 minutos, o subcutáneamente. NeoRecormon se administra dos veces por semana durante 4 semanas. En aquellas ocasiones en que el hematocrito del paciente permite la donación de sangre, es decir el hematocrito es > 33 %, NeoRecormon se administra al final de la donación de sangre.
Durante la totalidad del período de tratamiento no debe excederse un hematocrito del 48 %.
La dosis debe ser determinada por el equipo quirúrgico, individualmente para cada paciente, en función de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos:
1. La cantidad de sangre pre-donada necesaria dependerá de la pérdida prevista de sangre, de los medios de conservación empleados y del estado físico del paciente.
Este volumen debe ser equivalente a la cantidad de sangre que se considera necesaria para evitar transfusiones de sangre homóloga.
La cantidad de sangre pre-donada necesaria se expresa en unidades en las cuales una unidad de nomograma es equivalente a 180 ml de glóbulos rojos.
2. La capacidad de donar sangre depende predominantemente del volumen sanguíneo del paciente y del hematocrito basal. Ambas variables determinan la reserva endógena de eritrocitos, que puede ser calculada conforme a la fórmula siguiente:
Reserva endógena de eritrocitos = volumen sanguíneo (ml) x (hematocrito - 33)/ 100 mujeres: volumen sanguíneo (ml) = 41 (ml/kg) x peso corporal (kg) + 1200 (ml)
hombres: volumen sanguíneo (ml) = 44 (ml/kg) x peso corporal (kg) + 1600 (ml)
(peso corporal: > 45 kg)
La indicación para un tratamiento con NeoRecormon y, en su caso, la dosis individual debe ser determinada a partir de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos según las gráficas siguientes:
Paciente femenino Paciente masculino
Cantidad de sangre pre-donada Cantidad de sangre pre-donada
necesaria [unidades] necesaria [unidades]
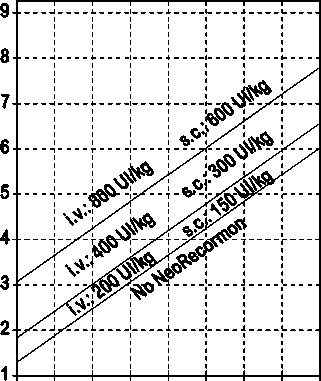
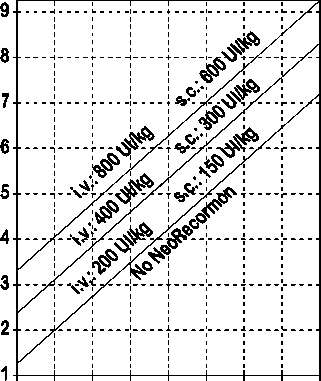
O 100 200 300 400 500 600 700 800 0 100 200 300 400 500 600 700 800
Reserva eritrocitaria endógena [ml] Reserva eritrocitaria endógena [ml]
La dosis única así determinada debe administrarse dos veces por semana a lo largo de 4 semanas. La dosis máxima no debe sobrepasar 1.600 UI/kg peso corporal por semana en administración intravenosa ó 1.200 UI/kg por semana en administración subcutánea.
Forma de administración
La jeringa precargada de NeoRecormon está lista para su uso. Sólo se pueden inyectar soluciones claras o ligeramente opalescentes, incoloras y prácticamente libres de partículas visibles. NeoRecormon en jeringa precargada es un producto estéril pero sin conservantes. Bajo ninguna circunstancia debe administrarse más de una dosis por jeringa; el medicamento es solo para uso individual.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Hipertensión mal controlada.
En la indicación para “Aumentar el rendimiento de sangre autóloga”: infarto de miocardio o accidente cerebrovascular en el mes anterior al tratamiento, angina de pecho inestable, riesgo aumentado de trombosis venosa profunda, como por ejemplo en aquellos con historial de enfermedad tromboembólica venosa.
4.4 Advertencias y precauciones especiales de empleo
NeoRecormon debe usarse con cautela en presencia de anemia refractaria con exceso de blastos en transformación, epilepsia, trombocitosis e insuficiencia hepática crónica. Y deben excluirse las deficiencias de ácido fólico y vitamina B12 pues reducen la eficacia de NeoRecormon.
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica, ya que las dosis acumuladas altas de epoetina pueden asociarse a un mayor riesgo de mortalidad, y de acontecimientos cardiovasculares y cerebrovasculares graves. En pacientes con una respuesta deficiente de la hemoglobina a epoetinas, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.2 y 5.1).
Para garantizar una eritropoyesis eficaz, se debe evaluar el nivel de hierro en todos los pacientes antes y durante el tratamiento, y puede ser necesario un tratamiento suplementario con hierro administrado de acuerdo con las guías terapéuticas.
La sobrecarga grave de aluminio debido al tratamiento de la insuficiencia renal puede comprometer la eficacia de NeoRecormon.
La indicación de tratamiento con NeoRecormon en enfermos nefroscleróticos aún no sometidos a diálisis debe definirse individualmente, ya que la posible aceleración de la insuficiencia renal no puede descartarse con certeza.
Aplasia pura de células rojas (APCR)
Se han notificado casos de APCR causada por anticuerpos neutralizantes antieritropoyetina, asociados a tratamientos con eritropoyetinas, incluido NeoRecormon. Estos anticuerpos han presentado reacción cruzada con todas las proteínas eritropoyéticas, por los que los pacientes en los que se sospeche o se haya confirmado la presencia de anticuerpos neutralizantes contra eritropoyetina no deben ser tratados con NeoRecormon (ver sección 4.8).
APCR en pacientes con hepatitis C
Se debe interrumpir inmediatamente el tratamiento con epoetina, y realizar ensayos de anticuerpos antieritropoyetina si se produce una disminución paradójica de la hemoglobina y se desarrolla anemia severa asociada con bajos recuentos de reticulocitos. En pacientes con hepatitis C tratados con interferon y ribavirina se han notificados casos cuando las epoetinas se han utilizado concomitantemente. Las epoetinas no están aprobadas en el tratamiento de la anemia asociada a la hepatitis C.
Monitorización de la tensión arterial
Puede aparecer un aumento de la tensión arterial o un agravamiento de la hipertensión ya existente, especialmente en los casos en los que el hematocrito aumenta rápidamente. Estos incrementos en la tensión arterial pueden ser tratados con medicamentos. Si no pueden ser controlados con medicación se recomienda interrumpir transitoriamente la terapia con NeoRecormon. En particular al principio de la terapia se recomienda la monitorización regular de la tensión arterial; también durante la diálisis. Pueden producirse crisis hipertensivas con síntomas de encefalopatías y que requieren atención médica inmediata y cuidados médicos intensivos. Debe prestarse especial atención a las cefaleas súbitas lacerantes hemicraneales como posible signo de advertencia.
Insuficiencia renal crónica
En pacientes_con insuficiencia renal crónica puede sobrevenir un aumento dosis-dependiente moderado en la cifra plaquetaria dentro del margen normal durante el tratamiento con NeoRecormon, especialmente después de la administración intravenosa. La regresión tiene lugar en el curso de la prosecución de la terapia. Se recomienda el control regular de la cifra de plaquetas durante las 8 primeras semanas de tratamiento.
Concentración de hemoglobina
En pacientes con insuficiencia renal crónica, la concentración de hemoglobina en la fase de mantenimiento no debe exceder el límite superior del rango recomendado en la sección 4.2. En ensayos clínicos se observó un aumento del riesgo de fallecimiento y de acontecimientos cardiovasculares graves o acontecimientos cerebrovasculares incluido el infarto, cuando se administraron agentes estimulantes de la eritropoyesis (AEEs) con el fin de alcanzar un nivel de hemoglobina superior a 12 g/dl (7,5 mmol/l).
En ensayos clínicos controlados no se han observado beneficios significativos atribuibles a la administración de epoetinas cuando se aumentaba la concentración de hemoglobina por encima del nivel necesario para controlar los síntomas de anemia y evitar las transfusiones sanguíneas.
En niños prematuros puede producirse un ligero aumento de la cifra plaquetaria, en especial hasta el día 12 -14 de vida, por lo tanto las plaquetas deben controlarse regularmente.
Efectos sobre el crecimiento tumoral
Las epoetinas son factores de crecimiento que estimulan de forma primaria la producción de glóbulos rojos. Los receptores de epoetina pueden expresarse sobre la superficie de una variedad de células tumorales. Como sucede con todos los factores de crecimiento, existe la preocupación de que las epoetinas pudieran estimular el crecimiento de tumores. En varios ensayos controlados, no se ha observado que las epoetinas mejoren la supervivencia global o que disminuyan el riesgo de progresión del tumor en pacientes con anemia asociada con cáncer.
En ensayos clínicos controlados, se ha observado que el uso de NeoRecormon y otros agentes estimulantes de la eritropoyesis (AEEs):
- acortaba el tiempo de progresión del tumor en pacientes con cáncer avanzado de cabeza y cuello que recibían radioterapia cuando se administraba para conseguir una concentración de hemoglobina por encima de 14 g/dl (8,7 mmol/l),
- acortaba la supervivencia global y aumentaba el número de fallecimientos atribuidos a la progresión de la enfermedad a los 4 meses, en pacientes con cáncer de mama metastásico que recibían quimioterapia cuando se administraba para conseguir una concentración de hemoglobina entre los 12 y los 14 g/dl (7,5-8,7 mmol/l),
- aumentaba el riesgo de fallecimiento cuando se administraba para conseguir una concentración de hemoglobina de 12 g/dl (7,5 mmol/l) en pacientes con enfermedad maligna activa y que no recibían ni quimioterapia ni radioterapia. El uso de los AEEs no está indicado en esa población de pacientes.
En vista de lo anterior, en algunas situaciones clínicas la transfusión sanguínea debe ser el tratamiento de elección para la anemia en pacientes con cáncer. La decisión de administrar eritropoyetinas recombinantes se tomará en base a la evaluación de la relación beneficio/riesgo junto con la aceptación individual del paciente, teniendo en cuenta el contexto clínico específico. Los factores que deben considerarse en esta evaluación son el tipo de tumor y su estadio, el grado de anemia, la esperanza de vida, el entorno en el que el paciente está siendo tratado y la preferencia del paciente (ver sección 5.1).
Puede haber un aumento de la tensión arterial que puede ser tratada con medicamentos. Se recomienda por tanto monitorizar la tensión arterial, en particular en la fase inicial de tratamiento en pacientes con cáncer.
En pacientes con cáncer también deberán monitorizarse regularmente el recuento plaquetario y el valor de hemoglobina.
En pacientes en^programa de predonación de sangre autóloga, puede sobrevenir un aumento de la cifra de plaquetas, generalmente dentro de los límites normales. Por tanto, se recomienda la determinación de esta cifra al menos una vez por semana. Si se produce un aumento plaquetario de más de 150 x 109/l o si las plaquetas ascienden por encima del margen normal, se debe suspender el tratamiento con NeoRecormon.
En niños prematuros no se podría descartar el riesgo potencial de que la eritropoyetina provoque retinopatía, por lo tanto se debe tener precaución y la decisión de tratar a un niño prematuro se debe tomar teniendo en cuenta el beneficio potencial y el riesgo de este tratamiento, así como las opciones alternativas de tratamiento que estén disponibles.
En pacientes con msuficiencia renal crónica, a menudo se precisa aumentar la dosis de heparina durante la hemodiálisis en la terapia con NeoRecormon debido al aumento del volumen de hematocrito. Si la heparinización no es óptima es posible que se produzca la oclusión del sistema de diálisis.
En pacientes con alteración renal crónica con riesgo de trombosis de derivación, se debe considerar la revisión temprana de la derivación y profilaxis de la trombosis mediante la administración, por ejemplo, de ácido acetilsalicílico.
Los niveles séricos de potasio y fosfato deben ser controlados regularmente durante la terapia con NeoRecormon. Se ha informado de aumentos de potasio en unos pocos pacientes urémicos que recibieron NeoRecormon si bien no se ha establecido una relación causal. En caso de observar un nivel elevado o creciente de potasio se debe considerar la interrupción de la administración de NeoRecormon hasta que el nivel haya sido corregido.
Para el uso de NeoRecormon en un programa de predonación de sangre autóloga deben ser observadas las directrices oficiales sobre donación de sangre, en particular las siguientes:
- sólo deben donar los pacientes con hematocrito > 33 % (hemoglobina >11 g/dl [6,83 mmol/l]);
- debe procederse con especial cuidado con pacientes con un peso inferior a 50 kg;
- el volumen individual extraído no debe superar aproximadamente el 12 % del volumen sanguíneo estimado del paciente.
El tratamiento debe reservarse a pacientes en los que se considere particularmente importante evitar la transfusión de sangre homóloga teniendo en cuenta la evaluación riesgo/beneficio de las transfusiones homólogas.
Uso indebido
Un uso indebido del producto por personas sanas puede llevar a un aumento excesivo del volumen de hematocrito, lo cual puede asociarse con complicaciones del sistema cardiovascular con riesgo para la vida.
Excipientes
NeoRecormon en jeringa precargada contiene, como máximo, 0,3 mg de fenilalanina por jeringa como excipiente. Este hecho se deberá tener en cuenta en los pacientes afectados por formas graves de fenilcetonuria.
Este medicamento contiene menos de 1 mmol (23 mg) de sodio por jeringa, por lo que se considera esencialmente “exento de sodio”.
Trazabilidad de NeoRecormon
Para mejorar la trazabilidad de los agentes estimulantes de la eritropoyesis (AEEs), se debe registrar (o indicar) claramente el nombre comercial del AEE administrado en la historia clínica del paciente.
4.5 Interacción con otros medicamentos y otras formas de interacción
Los resultados clínicos obtenidos hasta el presente no ponen de manifiesto interacción alguna de NeoRecormon con otros medicamentos.
Los estudios llevados a cabo en animales mostraron que la epoetina beta no potencia la mielotoxicidad de los medicamentos citostáticos, tales como etopósido, cisplatino, ciclofosfamida y fluorouracilo.
4.6 Fertilidad, embarazo y lactancia
Fertilidad
Los estudios en animales no muestran efectos dañinos directos o indirectos sobre el embarazo, desarrollo embrional/fetal, parto o desarrollo postnatal (ver sección 5.3).
Embarazo
No hay datos relativos al uso de NeoRecormon en mujeres embarazadas.
Se debe tener precaución cuando se prescriba a mujeres embarazadas.
Lactancia
Se desconoce si la epoetina beta se excreta en la leche materna. La decisión de continuar/interrumpir la lactancia o de continuar/interrumpir el tratamiento con epoetina beta se debe hacer teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento con epoetina beta para la madre.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NeoRecormon sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas Resumen del perfil de seguridad
Basados en los resultados de los ensayos clínicos que incluyen 1.725 pacientes, se espera que aproximadamente el 8 % de los pacientes tratados con NeoRecormon sufran reacciones adversas.
- Pacientes anémicos con insuficiencia renal crónica
La reacción adversa más frecuente durante el tratamiento con NeoRecormon es un aumento de la tensión arterial o agravamiento de la hipertensión existente, especialmente en casos de aumento rápido del hematocrito (ver sección 4.4). En algunos pacientes cuya tensión arterial es normal o baja también pueden darse crisis hipertensivas con síntomas de encefalopatías (p.ej. cefaleas y estados de confusión, trastornos sensitivomotores - como alteraciones del habla o de la ambulación - hasta convulsiones tónico-clónicas) (ver sección 4.4).
Puede darse trombosis de derivación, especialmente en pacientes que tienen tendencia a la hipotensión o cuyas fístulas arteriovenosas presentan complicaciones (p.ej. estenosis, aneurismas), ver sección 4.4. En la mayoría de los casos se observa una caída en los valores séricos de ferritina simultáneamente con el aumento del volumen de hematocrito (ver sección 4.4). Además, en casos aislados se han observado incrementos transitorios en los niveles séricos de potasio y fosfato (ver sección 4.4).
En casos aislados se ha notificado aplasia pura de células rojas (APCR) mediada por anticuerpos neutralizantes antieritropoyetina asociada al tratamiento con NeoRecormon. En caso de que se diagnostique APCR mediada por anticuerpos antieritropoyetina, el paciente deberá interrumpir el tratamiento con NeoRecormon, el cual no se deberá sustituir por otra proteína eritropoyética (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 1, que figura más adelante.
Pacientes con cáncer
El dolor de cabeza y la hipertensión relacionados con el tratamiento con epoetina beta son frecuentes y pueden tratarse con medicamentos (ver sección 4.4).
En algunos pacientes se observa un descenso de los valores de hierro sérico (ver sección 4.4).
Estudios clínicos han puesto de manifiesto que la frecuencia de acontecimientos tromboembólicos es más alta en pacientes con cáncer tratados con NeoRecormon que en los controles no tratados o tratados con placebo. En pacientes tratados con NeoRecormon, esta incidencia es del 7 % comparado con el 4 % en el grupo control; este hecho no está asociado con un incremento en la mortalidad tromboembólica comparado con los grupos control.
Estas reacciones adversas se enumeran en la Tabla 2, que figura más adelante.
Pacientes incluidos en un programa de predonación de sangre autóloga Los pacientes incluidos en un programa de predonación de sangre autóloga han demostrado una frecuencia ligeramente superior de accidentes tromboembólicos. Sin embargo, no se pudo establecer una relación causal con el tratamiento con NeoRecormon.
En ensayos clínicos controlados con placebo, la deficiencia transitoria de hierro fue más acusada en los pacientes tratados con NeoRecormon que en los pacientes control (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 3, que figura más adelante.
Tabla de reacciones adversas
Las reacciones adversas se enumeran de acuerdo con la clasificación de órganos del sistema MedDRA y por categoría de frecuencia.
Las categorías de frecuencia se definen utilizando la siguiente convención:
muy frecuentes (> 1/10), frecuentes (> 1/100 hasta <1/10), poco frecuentes (>1/1.000 hasta <1/100), raras (>1/10.000 hasta <1/1.000); muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 1: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con IRC_
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión Crisis hipertensivas |
Frecuente Poco frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
|
Trastornos de la sangre y |
Trombosis de derivación |
Raras |
|
del sistema linfático |
Trombocitosis |
Muy raras |
Tabla 2: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con cáncer__
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión |
Frecuente |
|
Trastornos de la sangre y del sistema linfático |
Acontecimientos tromboembólicos |
Frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Tabla 3: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes incluidos en un programa de predonación de sangre autóloga
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Prematuros
Es muy frecuente un descenso de los valores de ferritina sérica (ver sección 4.4).
Descripción de reacciones adversas seleccionadas
Raramente pueden ocurrir reacciones cutáneas relacionadas con el tratamiento con epoetina beta como sarpullido, prurito, urticaria o reacciones en el lugar de la inyección. En casos muy raros se han notificado reacciones anafilácticas relacionadas con el tratamiento con epoetina beta. Sin embargo, en los ensayos clínicos controlados no se encontró una mayor incidencia de reacciones de hipersensibilidad.
Se han comunicado casos muy raros de síntomas gripales relacionados con el tratamiento con epoetina beta como fiebre, escalofríos, dolores de cabeza, dolor de las extremidades, malestar general, y/o dolor óseo, particularmente al inicio del tratamiento. Estas reacciones son de intensidad leve a moderada y desaparecen al cabo de un par de horas o días.
Los resultados obtenidos en un ensayo clínico controlado realizado con epoetina alfa o darbepoetina alfa, notificaron que la incidencia de infarto es frecuente.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
El margen terapéutico de NeoRecormon es muy amplio. No se han observado síntomas de sobredosis incluso a niveles séricos muy elevados.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 P ropiedades farmacodinámicas
Grupo farmacoterapéutico: antianémico, código ATC: B03XA01 Mecanismo de acción
La eritropoyetina es una glucoproteína que estimula la formación de eritrocitos a partir de sus progenitores asignados. Actúa como factor estimulante de la mitosis y hormona de diferenciación.
La epoetina beta, el principio activo de NeoRecormon, es idéntica en su composición aminoácida e hidrocarbonada a la eritropoyetina aislada de la orina de pacientes anémicos.
La eficacia biológica de la epoetina beta ha sido demostrada tras administración intravenosa y subcutánea en varios modelos animales in vivo (ratas normales y urémicas, ratones policitémicos, perros). Con la administración de epoetina beta aumentan el número de eritrocitos, los valores de Hb y la cifra de reticulocitos, al igual que la velocidad de incorporación de 59Fe.
In vitro (cultivo citológico de bazo de ratón) ha sido hallada una mayor incorporación de 3H-timidina en las células eritroides nucleadas del bazo tras incubación con epoetina beta.
Las investigaciones en cultivos de células de la médula ósea humana han revelado que la epoetina beta estimula específicamente la eritropoyesis y no afecta a la leucopoyesis. No han sido detectadas acciones citotóxicas de la epoetina beta sobre la médula ósea o sobre células cutáneas humanas.
Tras una dosis única de epoetina beta no se ha observado ningún efecto en el comportamiento o la actividad locomotora del ratón ni en las funciones circulatoria o respiratoria del perro.
Eficacia clínica y seguridad
En un ensayo aleatorizado, doble ciego, controlado con placebo, realizado en 4.038 pacientes con enfermedad renal crónica no sometidos a diálisis, con una diabetes tipo 2 y los niveles de hemoglobina < 11 g/dl, los pacientes recibieron tratamiento bien con darbepoetina alfa para alcanzar niveles de hemoglobina de 13 g/dl o con placebo (ver sección 4.4). El ensayo no cumplió su objetivo principal de demostrar una disminución en el riesgo de todas las causas de mortalidad, la morbilidad cardiovascular o enfermedad renal terminal. El análisis de los componentes individuales de la variable combinada mostró los siguientes HR (IC 95%): fallecimiento 1,05 (0,92-1,21), infarto cerebrovascular 1,92 (1,38-2,68), insuficiencia cardíaca congestiva 0,89 (0,74-1,08), infarto de miocardio 0,96 (0,751,23), hospitalización por isquemia del miocardio 0,84 (0,55-1,27), enfermedad renal terminal 1,02 (0,87-1,18).
En pacientes con IRC (tratados con diálisis, no tratados con diálisis, pacientes diabéticos y pacientes no diabéticos) se han realizado análisis agrupados post hoc de los estudios clínicos con AEEs. Se observó una tendencia a un aumento de las estimaciones del riesgo para la mortalidad por todas las causas y para los acontecimientos cardiovasculares y cerebrovasculares asociados a dosis acumuladas más altas de AEE independientemente de que los pacientes padecieran o no diabetes y de que recibieran o no tratamiento con diálisis (ver las secciones 4.2 y 4.4).
La eritropoyetina es un factor de crecimiento que estimula de forma primaria la producción de glóbulos rojos. Los receptores de eritropoyetina se encuentran también presentes en la superficie de algunas líneas celulares malignas.
Se ha estudiado la supervivencia y la progresión tumoral en cinco grandes ensayos controlados que incluyeron a 2.833 pacientes, de los cuales cuatro fueron ensayos doble-ciego controlados con placebo y uno fue un ensayo abierto. Dos de los ensayos reclutaron pacientes que estaban siendo tratados con quimioterapia. El nivel de hemoglobina que se quería alcanzar fue >13 g/dl en dos de los ensayos y de entre 12 y 14 g/dl en los otros tres. En el ensayo abierto no se observaron diferencias en la supervivencia global, entre los pacientes tratados con eritropoyetina humana recombinante y el grupo control. En los cuatro ensayos controlados con placebo, el índice de riesgo (hazard ratio) para la supervivencia global osciló entre 1,25 y 2,47 en favor de los grupos control. En todos estos ensayos se ha observado un aumento de la mortalidad inexplicable y estadísticamente significativo en pacientes que presentaban anemia asociada con diversos tipos frecuentes de cáncer y que recibieron eritropoyetina humana recombinante, en comparación con los grupos control. Las diferencias observadas en la incidencia de trombosis y complicaciones relacionadas, entre los pacientes que recibieron eritropoyetina humana recombinante y los que formaban parte de los grupos control, no permiten explicar satisfactoriamente los resultados de supervivencia global observados en los ensayos.
Un meta-análisis basado en datos individuales de pacientes, que incluyó datos de los 12 ensayos clínicos controlados realizados con NeoRecormon en pacientes anémicos con cáncer (n=2301), mostró un valor estimado del índice de riesgo (hazard ratio) de supervivencia global de 1,13 en favor de los grupos control (IC 95 %, CI 0,87, 1,46). En pacientes con un valor inicial de hemoglobina < 10 g/dl (n=899), el valor estimado del índice de riesgo (hazard ratio) para la supervivencia fue de 0,98 (IC 95% 0,68 al 1,40). Se observó un aumento del riesgo relativo de acontecimientos tromboembólicos en la población general (RR 1,62, IC 95 %: 1,13, 2,31).
Asimismo, se ha realizado un análisis de datos a nivel de paciente, en más de 13.900 pacientes con cáncer (tratados con quimioterapia, radioterapia, quimioradioterapia o sin tratamiento) que participaban en un total de 53 ensayos clínicos controlados en los que se administraban diversas epoetinas. En este meta-análisis se obtuvo un índice de riesgo (hazard ratio) para la supervivencia global de 1,06 a favor de los grupos control (IC 95%: 1,00, 1,12; 53 ensayos y 13.933 pacientes) y para los pacientes con cáncer que recibían quimioterapia, el índice de riesgo (hazard ratio) para la supervivencia global fue de 1,04 (IC 95%: 0,97, 1,11; 38 ensayos y 10.441 pacientes). Este meta-análisis además indica de forma consistente un aumento significativo del riesgo relativo de episodios tromboembólicos en pacientes con cáncer tratados con eritropoyetina recombinante humana (ver sección 4.4).
En casos muy raros, se han producido anticuerpos neutralizantes anti-eritropoyetina con o sin aplasia pura de células rojas (APCR) durante el tratamiento con rHuEPO.
5.2 Propiedades farmacocinéticas
Los estudios farmacocinéticos en voluntarios sanos y pacientes urémicos muestran que la semivida de la epoetina beta administrada por vía i.v. es de 4 a 12 horas y que el volumen de distribución corresponde de una a dos veces el volumen plasmático. Se han hallado resultados análogos en experimentos en animales con ratas normales y urémicas.
Tras la administración subcutánea de epoetina beta a pacientes urémicos la absorción retardada se traduce en una meseta de concentración sérica, cuya cota máxima se alcanza al cabo de 12 - 28 horas de media. El semiperíodo de vida terminal es mayor que después de la administración intravenosa, siendo la media de 13-28 horas.
La biodisponibilidad de la epoetina beta después de la administración subcutánea está comprendida entre el 23 y el 42 % en comparación con la administración intravenosa.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad de dosis repetidas, genotoxicidad y toxicidad para la reproducción.
Un estudio de carcinogenicidad con homólogos de eritropoyetina en ratones no reveló ningún signo de potencial proliferativo o carcinogénico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Urea,
Cloruro sódico,
Polisorbato 20,
Fosfato monosódico dihidratado,
Fosfato disódico dodecahidratado,
Cloruro cálcico dihidratado,
Glicina,
L-Leucina,
L-Isoleucina,
L-Treonina,
L-Ácido glutámico,
L-Fenilalanina,
Agua para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
Conservar la jeringa precargada en el embalaje exterior, para protegerlo de la luz.
Con fines de uso ambulatorio, se puede mantener el producto fuera de la nevera durante un único periodo de hasta 3 días a temperatura ambiente (no superior a 25°C).
6.5 Naturaleza y contenido del envase
Jeringa precargada (vidrio tipo I), con un protector y un émbolo (goma teflonizada) con una aguja de inyección (27G1/2). Cada jeringa contiene 0,3 ml de solución.
Tamaños de envase de 1 jeringa precargada y 1 aguja o de 6 jeringas precargadas y 6 agujas.
Puede que solamente estén comercializados algunos tamaños de envase.
6.6 Precauciones especiales de eliminación y otras manipulaciones
En primer lugar, ¡lávese las manos!
1. Saque una jeringa del envase y asegúrese que la solución es clara, incolora y prácticamente libre de partículas visibles. Retire el protector de la jeringa.
2. Saque una aguja del envase, fíjela sobre la jeringa y retire la tapa protectora de la aguja.
3. Expulse el aire de la jeringa y aguja manteniendo la jeringa vertical y presionando suavemente el émbolo hacia arriba. Continúe presionando el émbolo hasta que la cantidad de solución de NeoRecormon en la jeringa sea la prescrita.
4. Limpie la piel en el lugar de inyección con un algodón con alcohol. Haga un pliegue tomando la piel entre el índice y el pulgar. Sujete el cuerpo de la jeringa por la parte más próxima a la aguja e inserte la aguja en el pliegue cutáneo con acción firme y rápida. Inyecte la solución de NeoRecormon. Retire la aguja rápidamente y aplique presión sobre el lugar de inyección con un apósito seco y estéril.
Este medicamento es solo para uso individual. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/031 -032
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16 de julio de 1997 Fecha de la última revalidación: 16 de julio de 2007
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 4.000 UI solución inyectable en jeringa precargada
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Una jeringa precargada con 0,3 ml de solución para inyección contiene 4.000 unidades internacionales (UI) correspondientes a 33,2 microgramos de epoetina beta* (eritropoyetina recombinante humana).
Un ml de solución para inyección contiene 13.333 UI de epoetina beta.
* producida en células de ovarios de hámster chino (CHO) mediante tecnología de ADN recombinante.
Excipientes con efecto conocido:
Fenilalanina (hasta 0,3 mg/jeringa).
Sodio (menos de 1 mmol/jeringa)
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución incolora, de transparente a ligeramente opalescente.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
NeoRecormon está indicado para:
- Tratamiento de la anemia sintomática asociada a la insuficiencia renal crónica en pacientes adultos y pediátricos.
- Prevención de la anemia en prematuros con un peso corporal al nacer de 750 a 1.500 g y una edad gestacional de menos de 34 semanas.
- Tratamiento de la anemia sintomática en pacientes adultos con neoplasias no mieloides tratados con quimioterapia.
- Aumentar el rendimiento de la sangre autóloga de pacientes incluidos en un programa de predonación.
Debe sopesarse su uso en esta indicación frente al riesgo aumentado de episodios tromboembólicos que han sido notificados. Sólo debe administrarse el tratamiento a pacientes con anemia moderada (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], sin deficiencia de hierro) si no se dispone de procedimientos para conservar la sangre o si éstos son insuficientes cuando la cirugía mayor electiva programada requiera un gran volumen de sangre (4 o más unidades de sangre en mujeres o 5 o más unidades en hombres). Ver sección 5.1.
4.2 Posología y forma de administración
La terapia con NeoRecormon debe iniciarse por médicos con experiencia en las indicaciones arriba
mencionadas. Como se han observado reacciones anafilácticas en algunos casos aislados, se
recomienda administrar la primera dosis bajo control médico.
Posología
Tratamiento de la anemia sintomática en pacientes adultos y pediátricos con insuficiencia renal crónica
Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente. NeoRecormon se puede administrar tanto por vía subcutánea como intravenosa con el fin de aumentar la concentración de hemoglobina hasta un nivel no superior a 12 g/dl (7,5 mmol/l). En pacientes que no están sometidos a hemodiálisis es preferible utilizar la vía subcutánea para evitar la punción de venas periféricas. En caso de administración intravenosa, la solución debe ser inyectada a lo largo de unos 2 minutos, p.ej. en pacientes en hemodiálisis por vía de la fístula arteriovenosa al final de la diálisis.
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones se pueden observar valores individuales de hemoglobina superiores o inferiores a los niveles deseados. La variabilidad en los niveles de hemoglobina se debe controlar mediante ajuste de la dosis con el objeto de mantener los valores de hemoglobina dentro del intervalo entre 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Se debe evitar un nivel de hemoglobina de forma continuada por encima de 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando los valores de hemoglobina sean superiores a 12 g/dl (7,5 mmol/l).
Debe evitarse un aumento de hemoglobina superior a 2 g/dl (1,25 mmol/l) durante un periodo de cuatro semanas. Si esto ocurre, se debe hacer un ajuste adecuado de la dosis según las instrucciones incluidas en esta misma sección. Si la tasa de aumento de hemoglobina es mayor de 2 g/dl (1,25 mmol/l) en un mes o si el nivel de hemoglobina está aumentando y se acerca a 12 g/dl (7,45 mmol/l), debe reducirse la dosis en aproximadamente un 25%. Si el nivel de hemoglobina sigue aumentando, se debe interrumpir el tratamiento hasta que el nivel de hemoglobina empiece a disminuir, momento en el que se podrá reiniciar el tratamiento con una dosis aproximadamente un 25 % inferior a la dosis administrada previamente.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis eficaz más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia al tiempo que se mantiene una concentración de hemoglobina inferior o igual a 12 g/dl (7,45 mmol/l).
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica. En los pacientes con una respuesta deficiente de la hemoglobina a NeoRecormon, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.4 y 5.1).
En presencia de hipertensión o de enfermedades cardiovasculares, cerebrovasculares o vasculoperiféricas, se debe determinar de modo individual el incremento semanal y el valor objetivo de la hemoglobina teniendo en cuenta el cuadro clínico.
El tratamiento con NeoRecormon se divide en dos fases:
1. Fase de corrección
- Administración subcutánea:
La dosis inicial es 3 x 20 UI/kg por semana. La dosis puede incrementarse cada 4 semanas en 3 x 20 UI/kg y semana si el aumento de la hemoglobina no ha sido adecuado (< 0,25 g/dl por semana).
La dosis semanal puede dividirse en dosis diarias.
- Administración intravenosa:
La dosis inicial es 3 x 40 UI/kg por semana, y puede aumentarse al cabo de 4 semanas a 80 UI/kg -tres veces por semana- y si son necesarios incrementos ulteriores serán de 20 UI/kg tres veces por semana, con intervalos mensuales.
Por ambas vías de administración, la dosis máxima no debe superar 720 UI/kg por semana.
2. Fase de mantenimiento
Para mantener el nivel de hemoglobina entre 10 y 12 g/dl, la dosis es inicialmente reducida a la mitad de la cantidad previamente administrada. Posteriormente, se ajustará la dosis individualmente para el paciente a intervalos de una o dos semanas (dosis de mantenimiento).
En caso de administración subcutánea, la dosis semanal puede administrarse en una inyección única o fraccionada en tres o siete inyecciones. Los pacientes que permanezcan estables en el régimen de una dosis única semanal pueden pasar a una administración única cada dos semanas. En este caso, puede ser necesario un aumento de la dosis.
Los resultados de los estudios clínicos en niños han revelado que, en general, a menor edad, se necesita mayor dosis de NeoRecormon. No obstante, hay que seguir el programa posológico recomendado ya que no puede predecirse la respuesta individual.
El tratamiento con NeoRecormon es normalmente crónico. Sin embargo, en caso necesario puede interrumpirse en cualquier momento. Los datos sobre la pauta posológica de una vez a la semana se han obtenido de estudios clínicos con una duración de tratamiento de 24 semanas.
Prevención de anemia en prematuros
La solución se administra por vía subcutánea a una dosis de 3 x 250 UI/kg p.c. por semana. Es probable que los niños que ya hayan recibido una transfusión previa cuando se inicie el tratamiento con NeoRecormon no se beneficien tanto como los prematuros no transfundidos. La duración de tratamiento recomendada es de 6 semanas.
Tratamiento de anemia sintomática inducida por quimioterapia en pacientes con cáncer Se debe administrar NeoRecormon por vía subcutánea a pacientes con anemia (por ejemplo, si la concentración de hemoglobina es menor o igual a 10 g/dl [6,2 mmol/l]). Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente.
La dosis semanal puede administrarse como una inyección por semana o en dosis divididas entre 3 y 7 veces por semana.
La dosis inicial recomendada es de 30.000 UI por semana (que corresponde aproximadamente a 450 UI/kg de peso corporal por semana, en base al peso medio del paciente).
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones es posible llegar a observar en un paciente valores de hemoglobina superiores o inferiores a los esperados. La variabilidad de la hemoglobina se deberá manejar ajustando la dosis para mantener los valores de la hemoglobina dentro del intervalo de 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Deben evitarse concentraciones sostenidas de hemoglobina superiores a 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando se observen valores de hemoglobina superiores a los 12 g/dl (7,5 mmol/l).
Si a las 4 semanas de tratamiento el valor de hemoglobina aumenta hasta al menos 1 g/dl (0,62 mmol/l), se debe continuar con la dosis que en ese momento se esté administrando. Si los valores de hemoglobina no han aumentado al menos 1 g/dl (0,62 mmol/l), se debe considerar duplicar la dosis semanal. Si a las 8 semanas de tratamiento el valor de hemoglobina no aumenta hasta al menos 1 g/dl (0,62 mmol/l), es improbable que se produzca respuesta y, por consiguiente, el tratamiento debe ser interrumpido.
El tratamiento debe continuar durante las 4 semanas posteriores al final de la quimioterapia.
La dosis máxima no debe exceder de 60.000 UI por semana.
Una vez se ha alcanzado el objetivo terapéutico del paciente, la dosis debe reducirse del 25 al 50 % con el fin de mantener el valor de hemoglobina en ese nivel. Debe realizarse el ajuste posológico adecuado.
Si el nivel de hemoglobina excede los 12 g/dl (7,5 mmol/l) se debe reducir la dosis entre un 25 y un
50 % aproximadamente. Si el nivel de hemoglobina excede de 13 g/dl (8,1 mmol), se debe interrumpir temporalmente el tratamiento con NeoRecormon. El tratamiento debe reiniciarse con una dosis aproximadamente un 25 % inferior a la dosis previamente administrada después de que los niveles de hemoglobina desciendan hasta un valor menor o igual a 12 g/dl (7,5 mmol/l) o por debajo.
51 al cabo de 4 semanas el incremento de hemoglobina es superior a 2 g/dl (1,3 mmol/l), la dosis debe reducirse entre un 25 y un 50 %.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia.
Tratamiento para incrementar el rendimiento de la sangre autóloga donada La solución puede administrarse por vía intravenosa, en unos 2 minutos, o subcutáneamente. NeoRecormon se administra dos veces por semana durante 4 semanas. En aquellas ocasiones en que el hematocrito del paciente permite la donación de sangre, es decir el hematocrito es > 33 %, NeoRecormon se administra al final de la donación de sangre.
Durante la totalidad del período de tratamiento no debe excederse un hematocrito del 48 %.
La dosis debe ser determinada por el equipo quirúrgico, individualmente para cada paciente, en función de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos:
1. La cantidad de sangre pre-donada necesaria dependerá de la pérdida prevista de sangre, de los medios de conservación empleados y del estado físico del paciente.
Este volumen debe ser equivalente a la cantidad de sangre que se considera necesaria para evitar transfusiones de sangre homóloga.
La cantidad de sangre pre-donada necesaria se expresa en unidades en las cuales una unidad de nomograma es equivalente a 180 ml de glóbulos rojos.
2. La capacidad de donar sangre depende predominantemente del volumen sanguíneo del paciente y del hematocrito basal. Ambas variables determinan la reserva endógena de eritrocitos, que puede ser calculada conforme a la fórmula siguiente:
Reserva endógena de eritrocitos = volumen sanguíneo (ml) x (hematocrito - 33)/ 100 mujeres: volumen sanguíneo (ml) = 41 (ml/kg) x peso corporal (kg) + 1200 (ml)
hombres: volumen sanguíneo (ml) = 44 (ml/kg) x peso corporal (kg) + 1600 (ml)
(peso corporal: > 45 kg)
La indicación para un tratamiento con NeoRecormon y, en su caso, la dosis individual debe ser determinada a partir de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos según las gráficas siguientes:
Paciente femenino Paciente masculino
Cantidad de sangre pre-donada Cantidad de sangre pre-donada
necesaria [unidades] necesaria [unidades]
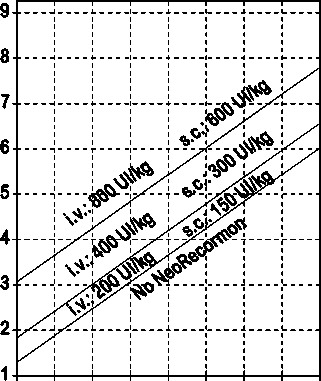
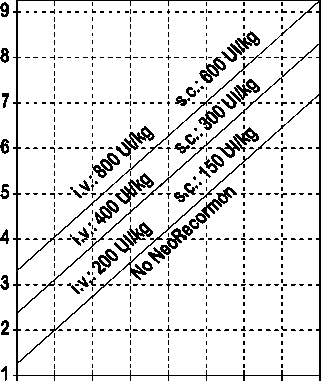
O 100 200 300 400 500 600 700 800 0 100 200 300 400 500 600 700 800
Reserva eritrocitaria endógena [ml] Reserva eritrocitaria endógena [ml]
La dosis única así determinada debe administrarse dos veces por semana a lo largo de 4 semanas. La dosis máxima no debe sobrepasar 1.600 UI/kg peso corporal por semana en administración intravenosa ó 1.200 UI/kg por semana en administración subcutánea.
Forma de administración
La jeringa precargada de NeoRecormon está lista para su uso. Sólo se pueden inyectar soluciones claras o ligeramente opalescentes, incoloras y prácticamente libres de partículas visibles. NeoRecormon en jeringa precargada es un producto estéril pero sin conservantes. Bajo ninguna circunstancia debe administrarse más de una dosis por jeringa; el medicamento es solo para uso individual.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Hipertensión mal controlada.
En la indicación para “Aumentar el rendimiento de sangre autóloga”: infarto de miocardio o accidente cerebrovascular en el mes anterior al tratamiento, angina de pecho inestable, riesgo aumentado de trombosis venosa profunda, como por ejemplo en aquellos con historial de enfermedad tromboembólica venosa.
4.4 Advertencias y precauciones especiales de empleo
NeoRecormon debe usarse con cautela en presencia de anemia refractaria con exceso de blastos en transformación, epilepsia, trombocitosis e insuficiencia hepática crónica. Y deben excluirse las deficiencias de ácido fólico y vitamina B12 pues reducen la eficacia de NeoRecormon.
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica, ya que las dosis acumuladas altas de epoetina pueden asociarse a un mayor riesgo de mortalidad, y de acontecimientos cardiovasculares y cerebrovasculares graves. En pacientes con una respuesta deficiente de la hemoglobina a epoetinas, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.2 y 5.1).
Para garantizar una eritropoyesis eficaz, se debe evaluar el nivel de hierro en todos los pacientes antes y durante el tratamiento, y puede ser necesario un tratamiento suplementario con hierro administrado de acuerdo con las guías terapéuticas.
La sobrecarga grave de aluminio debido al tratamiento de la insuficiencia renal puede comprometer la eficacia de NeoRecormon.
La indicación de tratamiento con NeoRecormon en enfermos nefroscleróticos aún no sometidos a diálisis debe definirse individualmente, ya que la posible aceleración de la insuficiencia renal no puede descartarse con certeza.
Aplasia pura de células rojas (APCR)
Se han notificado casos de APCR causada por anticuerpos neutralizantes antieritropoyetina, asociados a tratamientos con eritropoyetinas, incluido NeoRecormon. Estos anticuerpos han presentado reacción cruzada con todas las proteínas eritropoyéticas, por los que los pacientes en los que se sospeche o se haya confirmado la presencia de anticuerpos neutralizantes contra eritropoyetina no deben ser tratados con NeoRecormon (ver sección 4.8).
APCR en pacientes con hepatitis C
Se debe interrumpir inmediatamente el tratamiento con epoetina, y realizar ensayos de anticuerpos antieritropoyetina si se produce una disminución paradójica de la hemoglobina y se desarrolla anemia severa asociada con bajos recuentos de reticulocitos. En pacientes con hepatitis C tratados con interferon y ribavirina se han notificados casos cuando las epoetinas se han utilizado concomitantemente. Las epoetinas no están aprobadas en el tratamiento de la anemia asociada a la hepatitis C.
Monitorización de la tensión arterial
Puede aparecer un aumento de la tensión arterial o un agravamiento de la hipertensión ya existente, especialmente en los casos en los que el hematocrito aumenta rápidamente. Estos incrementos en la tensión arterial pueden ser tratados con medicamentos. Si no pueden ser controlados con medicación se recomienda interrumpir transitoriamente la terapia con NeoRecormon. En particular al principio de la terapia se recomienda la monitorización regular de la tensión arterial; también durante la diálisis. Pueden producirse crisis hipertensivas con síntomas de encefalopatías y que requieren atención médica inmediata y cuidados médicos intensivos. Debe prestarse especial atención a las cefaleas súbitas lacerantes hemicraneales como posible signo de advertencia.
Insuficiencia renal crónica
En pacientes con insuficiencia renal crónica puede sobrevenir un aumento dosis-dependiente moderado en la cifra plaquetaria dentro del margen normal durante el tratamiento con NeoRecormon, especialmente después de la administración intravenosa. La regresión tiene lugar en el curso de la prosecución de la terapia. Se recomienda el control regular de la cifra de plaquetas durante las 8 primeras semanas de tratamiento.
Concentración de hemoglobina
En pacientes con insuficiencia renal crónica, la concentración de hemoglobina en la fase de mantenimiento no debe exceder el límite superior del rango recomendado en la sección 4.2. En ensayos clínicos se observó un aumento del riesgo de fallecimiento y de acontecimientos cardiovasculares graves o acontecimientos cerebrovasculares incluido el infarto, cuando se administraron agentes estimulantes de la eritropoyesis (AEEs) con el fin de alcanzar un nivel de hemoglobina superior a 12 g/dl (7,5 mmol/l).
En ensayos clínicos controlados no se han observado beneficios significativos atribuibles a la administración de epoetinas cuando se aumentaba la concentración de hemoglobina por encima del nivel necesario para controlar los síntomas de anemia y evitar las transfusiones sanguíneas.
En niños prematuros puede producirse un ligero aumento de la cifra plaquetaria, en especial hasta el día 12 -14 de vida, por lo tanto las plaquetas deben controlarse regularmente.
Efectos sobre el crecimiento tumoral
Las epoetinas son factores de crecimiento que estimulan de forma primaria la producción de glóbulos rojos. Los receptores de epoetina pueden expresarse sobre la superficie de una variedad de células tumorales. Como sucede con todos los factores de crecimiento, existe la preocupación de que las epoetinas pudieran estimular el crecimiento de tumores. En varios ensayos controlados, no se ha
observado que las epoetinas mejoren la supervivencia global o que disminuyan el riesgo de progresión del tumor en pacientes con anemia asociada con cáncer.
En ensayos clínicos controlados, se ha observado que el uso de NeoRecormon y otros agentes estimulantes de la eritropoyesis (AEEs):
- acortaba el tiempo de progresión del tumor en pacientes con cáncer avanzado de cabeza y cuello que recibían radioterapia cuando se administraba para conseguir una concentración de hemoglobina por encima de 14 g/dl (8,7 mmol/l),
- acortaba la supervivencia global y aumentaba el número de fallecimientos atribuidos a la progresión de la enfermedad a los 4 meses, en pacientes con cáncer de mama metastásico que recibían quimioterapia cuando se administraba para conseguir una concentración de hemoglobina entre los 12 y los 14 g/dl (7,5-8,7 mmol/l),
- aumentaba el riesgo de fallecimiento cuando se administraba para conseguir una concentración de hemoglobina de 12 g/dl (7,5 mmol/l) en pacientes con enfermedad maligna activa y que no recibían ni quimioterapia ni radioterapia. El uso de los AEEs no está indicado en esa población de pacientes.
En vista de lo anterior, en algunas situaciones clínicas la transfusión sanguínea debe ser el tratamiento de elección para la anemia en pacientes con cáncer. La decisión de administrar eritropoyetinas recombinantes se tomará en base a la evaluación de la relación beneficio/riesgo junto con la aceptación individual del paciente, teniendo en cuenta el contexto clínico específico. Los factores que deben considerarse en esta evaluación son el tipo de tumor y su estadio, el grado de anemia, la esperanza de vida, el entorno en el que el paciente está siendo tratado y la preferencia del paciente (ver sección 5.1).
Puede haber un aumento de la tensión arterial que puede ser tratada con medicamentos. Se recomienda por tanto monitorizar la tensión arterial, en particular en la fase inicial de tratamiento en pacientes con cáncer.
En pacientes con cáncer también deberán monitorizarse regularmente el recuento plaquetario y el valor de hemoglobina.
En pacientes en programa de predonación de sangre autóloga, puede sobrevenir un aumento de la cifra de plaquetas, generalmente dentro de los límites normales. Por tanto, se recomienda la determinación de esta cifra al menos una vez por semana. Si se produce un aumento plaquetario de más de 150 x 109/l o si las plaquetas ascienden por encima del margen normal, se debe suspender el tratamiento con NeoRecormon.
En niños prematuros no se podría descartar el riesgo potencial de que la eritropoyetina provoque retinopatía, por lo tanto se debe tener precaución y la decisión de tratar a un niño prematuro se debe tomar teniendo en cuenta el beneficio potencial y el riesgo de este tratamiento, así como las opciones alternativas de tratamiento que estén disponibles.
En pacientes con insuficiencia renal crónica, a menudo se precisa aumentar la dosis de heparina durante la hemodiálisis en la terapia con NeoRecormon debido al aumento del volumen de hematocrito. Si la heparinización no es óptima es posible que se produzca la oclusión del sistema de diálisis.
En pacientes con alteración renal crónica con riesgo de trombosis de derivación, se debe considerar la revisión temprana de la derivación y profilaxis de la trombosis mediante la administración, por ejemplo, de ácido acetilsalicílico.
Los niveles séricos de potasio y fosfato deben ser controlados regularmente durante la terapia con NeoRecormon. Se ha informado de aumentos de potasio en unos pocos pacientes urémicos que recibieron NeoRecormon si bien no se ha establecido una relación causal. En caso de observar un nivel elevado o creciente de potasio se debe considerar la interrupción de la administración de NeoRecormon hasta que el nivel haya sido corregido.
Para el uso de NeoRecormon en un programa de predonación de sangre autóloga deben ser observadas las directrices oficiales sobre donación de sangre, en particular las siguientes:
- sólo deben donar los pacientes con hematocrito > 33 % (hemoglobina >11 g/dl [6,83 mmol/l]);
- debe procederse con especial cuidado con pacientes con un peso inferior a 50 kg;
- el volumen individual extraído no debe superar aproximadamente el 12 % del volumen sanguíneo estimado del paciente.
El tratamiento debe reservarse a pacientes en los que se considere particularmente importante evitar la transfusión de sangre homóloga teniendo en cuenta la evaluación riesgo/beneficio de las transfusiones homólogas.
Uso indebido
Un uso indebido del producto por personas sanas puede llevar a un aumento excesivo del volumen de hematocrito, lo cual puede asociarse con complicaciones del sistema cardiovascular con riesgo para la vida.
Excipientes
NeoRecormon en jeringa precargada contiene, como máximo, 0,3 mg de fenilalanina por jeringa como excipiente. Este hecho se deberá tener en cuenta en los pacientes afectados por formas graves de fenilcetonuria.
Este medicamento contiene menos de 1 mmol (23 mg) de sodio por jeringa, por lo que se considera esencialmente “exento de sodio”.
Trazabilidad de NeoRecormon
Para mejorar la trazabilidad de los agentes estimulantes de la eritropoyesis (AEEs), se debe registrar (o indicar) claramente el nombre comercial del AEE administrado en la historia clínica del paciente..
4.5 Interacción con otros medicamentos y otras formas de interacción
Los resultados clínicos obtenidos hasta el presente no ponen de manifiesto interacción alguna de NeoRecormon con otros medicamentos.
Los estudios llevados a cabo en animales mostraron que la epoetina beta no potencia la mielotoxicidad de los medicamentos citostáticos, tales como etopósido, cisplatino, ciclofosfamida y fluorouracilo.
4.6 Fertilidad, embarazo y lactancia
Fertilidad
Los estudios en animales no muestran efectos dañinos directos o indirectos sobre el embarazo, desarrollo embrional/fetal, parto o desarrollo postnatal (ver sección5.3).
Embarazo
No hay datos relativos al uso de NeoRecormon en mujeres embarazadas.
Se debe tener precaución cuando se prescriba a mujeres embarazadas.
Lactancia
Se desconoce si la epoetina beta se excreta en la leche materna. La decisión de continuar/interrumpir la lactancia o de continuar/interrumpir el tratamiento con epoetina beta se debe hacer teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento con epoetina beta para la madre
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NeoRecormon sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Basados en los resultados de los ensayos clínicos que incluyen 1.725 pacientes, se espera que aproximadamente el 8 % de los pacientes tratados con NeoRecormon sufran reacciones adversas.
Pacientes anémicos con insuficiencia renal crónica
La reacción adversa más frecuente durante el tratamiento con NeoRecormon es un aumento de la tensión arterial o agravamiento de la hipertensión existente, especialmente en casos de aumento rápido del hematocrito (ver sección 4.4). En algunos pacientes cuya tensión arterial es normal o baja también pueden darse crisis hipertensivas con síntomas de encefalopatías (p.ej. cefaleas y estados de confusión, trastornos sensitivomotores - como alteraciones del habla o de la ambulación - hasta convulsiones tónico-clónicas) (ver sección 4.4).
Puede darse trombosis de derivación, especialmente en pacientes que tienen tendencia a la hipotensión o cuyas fístulas arteriovenosas presentan complicaciones (p.ej. estenosis, aneurismas), ver sección 4.4. En la mayoría de los casos se observa una caída en los valores séricos de ferritina simultáneamente con el aumento del volumen de hematocrito (ver sección 4.4). Además, en casos aislados se han observado incrementos transitorios en los niveles séricos de potasio y fosfato (ver sección 4.4).
En casos aislados se ha notificado aplasia pura de células rojas (APCR) mediada por anticuerpos neutralizantes antieritropoyetina asociada al tratamiento con NeoRecormon. En caso de que se diagnostique APCR mediada por anticuerpos antieritropoyetina, el paciente deberá interrumpir el tratamiento con NeoRecormon, el cual no se deberá sustituir por otra proteína eritropoyética (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 1, que figura más adelante.
Pacientes con cáncer
El dolor de cabeza y la hipertensión relacionados con el tratamiento con epoetina beta son frecuentes y pueden tratarse con medicamentos (ver sección 4.4).
En algunos pacientes se observa un descenso de los valores de hierro sérico (ver sección 4.4).
Estudios clínicos han puesto de manifiesto que la frecuencia de acontecimientos tromboembólicos es más alta en pacientes con cáncer tratados con NeoRecormon que en los controles no tratados o tratados con placebo. En pacientes tratados con NeoRecormon, esta incidencia es del 7 % comparado con el 4 % en el grupo control; este hecho no está asociado con un incremento en la mortalidad tromboembólica comparado con los grupos control.
Estas reacciones adversas se enumeran en la Tabla 2, que figura más adelante.
Pacientes incluidos en un programa de predonación de sangre autóloga Los pacientes incluidos en un programa de predonación de sangre autóloga han demostrado una frecuencia ligeramente superior de accidentes tromboembólicos. Sin embargo, no se pudo establecer una relación causal con el tratamiento con NeoRecormon.
En ensayos clínicos controlados con placebo, la deficiencia transitoria de hierro fue más acusada en los pacientes tratados con NeoRecormon que en los pacientes control (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 3, que figura más adelante.
Tabla de reacciones adversas
Las reacciones adversas se enumeran de acuerdo con la clasificación de órganos del sistema MedDRA y por categoría de frecuencia.
Las categorías de frecuencia se definen utilizando la siguiente convención:
muy frecuentes (> 1/10), frecuentes (> 1/100 hasta <1/10), poco frecuentes (>1/1.000 hasta <1/100), raras (>1/10.000 hasta <1/1.000); muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 1: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con IRC_
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión Crisis hipertensivas |
Frecuente Poco frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
|
Trastornos de la sangre y |
Trombosis de derivación |
Raras |
|
del sistema linfático |
Trombocitosis |
Muy raras |
Tabla 2: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con cáncer__
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión |
Frecuente |
|
Trastornos de la sangre y del sistema linfático |
Acontecimientos tromboembólicos |
Frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Tabla 3: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes incluidos en un programa de predonación de sangre autóloga
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Prematuros
Es muy frecuente un descenso de los valores de ferritina sérica (ver sección 4.4).
Descripción de reacciones adversas seleccionadas
Raramente pueden ocurrir reacciones cutáneas relacionadas con el tratamiento con epoetina beta como sarpullido, prurito, urticaria o reacciones en el lugar de la inyección. En casos muy raros se han notificado reacciones anafilácticas relacionadas con el tratamiento con epoetina beta. Sin embargo, en los ensayos clínicos controlados no se encontró una mayor incidencia de reacciones de hipersensibilidad.
Se han comunicado casos muy raros de síntomas gripales relacionados con el tratamiento con epoetina beta como fiebre, escalofríos, dolores de cabeza, dolor de las extremidades, malestar general, y/o dolor óseo, particularmente al inicio del tratamiento. Estas reacciones son de intensidad leve a moderada y desaparecen al cabo de un par de horas o días.
Los resultados obtenidos en un ensayo clínico controlado realizado con epoetina alfa o darbepoetina alfa, notificaron que la incidencia de infarto es frecuente.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
El margen terapéutico de NeoRecormon es muy amplio. No se han observado síntomas de sobredosis incluso a niveles séricos muy elevados.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antianémico, código ATC: B03XA01 Mecanismo de acción
La eritropoyetina es una glucoproteína que estimula la formación de eritrocitos a partir de sus progenitores asignados. Actúa como factor estimulante de la mitosis y hormona de diferenciación.
La epoetina beta, el principio activo de NeoRecormon, es idéntica en su composición aminoácida e hidrocarbonada a la eritropoyetina aislada de la orina de pacientes anémicos.
La eficacia biológica de la epoetina beta ha sido demostrada tras administración intravenosa y subcutánea en varios modelos animales in vivo (ratas normales y urémicas, ratones policitémicos, perros). Con la administración de epoetina beta aumentan el número de eritrocitos, los valores de Hb y la cifra de reticulocitos, al igual que la velocidad de incorporación de 59Fe.
In vitro (cultivo citológico de bazo de ratón) ha sido hallada una mayor incorporación de 3H-timidina en las células eritroides nucleadas del bazo tras incubación con epoetina beta.
Las investigaciones en cultivos de células de la médula ósea humana han revelado que la epoetina beta estimula específicamente la eritropoyesis y no afecta a la leucopoyesis. No han sido detectadas acciones citotóxicas de la epoetina beta sobre la médula ósea o sobre células cutáneas humanas.
Tras una dosis única de epoetina beta no se ha observado ningún efecto en el comportamiento o la actividad locomotora del ratón ni en las funciones circulatoria o respiratoria del perro.
Eficacia clínica y seguridad
En un ensayo aleatorizado, doble ciego, controlado con placebo, realizado en 4.038 pacientes con enfermedad renal crónica no sometidos a diálisis, con una diabetes tipo 2 y los niveles de hemoglobina < 11 g/dl, los pacientes recibieron tratamiento bien con darbepoetina alfa para alcanzar niveles de hemoglobina de 13 g/dl o con placebo (ver sección 4.4). El ensayo no cumplió su objetivo principal de demostrar una disminución en el riesgo de todas las causas de mortalidad, la morbilidad cardiovascular o enfermedad renal terminal. El análisis de los componentes individuales de la variable combinada mostró los siguientes HR (IC 95%): fallecimiento 1,05 (0,92-1,21), infarto cerebrovascular 1,92 (1,38-2,68), insuficiencia cardíaca congestiva 0,89 (0,74-1,08), infarto de miocardio 0,96 (0,751,23), hospitalización por isquemia del miocardio 0,84 (0,55-1,27), enfermedad renal terminal 1,02 (0,87-1,18).
En pacientes con IRC (tratados con diálisis, no tratados con diálisis, pacientes diabéticos y pacientes no diabéticos) se han realizado análisis agrupados post hoc de los estudios clínicos con AEEs. Se observó una tendencia a un aumento de las estimaciones del riesgo para la mortalidad por todas las causas y para los acontecimientos cardiovasculares y cerebrovasculares asociados a dosis acumuladas más altas de AEE independientemente de que los pacientes padecieran o no diabetes y de que recibieran o no tratamiento con diálisis (ver las secciones 4.2 y 4.4).
La eritropoyetina es un factor de crecimiento que estimula de forma primaria la producción de glóbulos rojos. Los receptores de eritropoyetina se encuentran también presentes en la superficie de algunas líneas celulares malignas.
Se ha estudiado la supervivencia y la progresión tumoral en cinco grandes ensayos controlados que incluyeron a 2.833 pacientes, de los cuales cuatro fueron ensayos doble-ciego controlados con placebo y uno fue un ensayo abierto. Dos de los ensayos reclutaron pacientes que estaban siendo tratados con quimioterapia. El nivel de hemoglobina que se quería alcanzar fue >13 g/dl en dos de los ensayos y de entre 12 y 14 g/dl en los otros tres. En el ensayo abierto no se observaron diferencias en la supervivencia global, entre los pacientes tratados con eritropoyetina humana recombinante y el grupo control. En los cuatro ensayos controlados con placebo, el índice de riesgo (hazard ratio) para la supervivencia global osciló entre 1,25 y 2,47 en favor de los grupos control. En todos estos ensayos se ha observado un aumento de la mortalidad inexplicable y estadísticamente significativo en pacientes que presentaban anemia asociada con diversos tipos frecuentes de cáncer y que recibieron eritropoyetina humana recombinante, en comparación con los grupos control. Las diferencias observadas en la incidencia de trombosis y complicaciones relacionadas, entre los pacientes que recibieron eritropoyetina humana recombinante y los que formaban parte de los grupos control, no permiten explicar satisfactoriamente los resultados de supervivencia global observados en los ensayos.
Un meta-análisis basado en datos individuales de pacientes, que incluyó datos de los 12 ensayos clínicos controlados realizados con NeoRecormon en pacientes anémicos con cáncer (n=2301), mostró un valor estimado del índice de riesgo (hazard ratio) de supervivencia global de 1,13 en favor de los grupos control (IC 95 %, CI 0,87, 1,46). En pacientes con un valor inicial de hemoglobina < 10 g/dl (n=899), el valor estimado del índice de riesgo (hazard ratio) para la supervivencia fue de 0,98 (IC 95% 0,68 al 1,40). Se observó un aumento del riesgo relativo de acontecimientos tromboembólicos en la población general (RR 1,62, IC 95 %: 1,13, 2,31).
Asimismo, se ha realizado un análisis de datos a nivel de paciente, en más de 13.900 pacientes con cáncer (tratados con quimioterapia, radioterapia, quimioradioterapia o sin tratamiento) que participaban en un total de 53 ensayos clínicos controlados en los que se administraban diversas epoetinas. En este meta-análisis se obtuvo un índice de riesgo (hazard ratio) para la supervivencia global de 1,06 a favor de los grupos control (IC 95%: 1,00, 1,12; 53 ensayos y 13.933 pacientes) y para los pacientes con cáncer que recibían quimioterapia, el índice de riesgo (hazard ratio) para la supervivencia global fue de 1,04 (IC 95%: 0,97, 1,11; 38 ensayos y 10.441 pacientes). Este meta-análisis además indica de forma consistente un aumento significativo del riesgo relativo de episodios tromboembólicos en pacientes con cáncer tratados con eritropoyetina recombinante humana (ver sección 4.4).
En casos muy raros, se han producido anticuerpos neutralizantes anti-eritropoyetina con o sin aplasia pura de células rojas (APCR) durante el tratamiento con rHuEPO.
5.2 Propiedades farmacocinéticas
Los estudios farmacocinéticos en voluntarios sanos y pacientes urémicos muestran que la semivida de la epoetina beta administrada por vía i.v. es de 4 a 12 horas y que el volumen de distribución corresponde de una a dos veces el volumen plasmático. Se han hallado resultados análogos en experimentos en animales con ratas normales y urémicas.
Tras la administración subcutánea de epoetina beta a pacientes urémicos la absorción retardada se traduce en una meseta de concentración sérica, cuya cota máxima se alcanza al cabo de 12 - 28 horas de media. El semiperíodo de vida terminal es mayor que después de la administración intravenosa, siendo la media de 13-28 horas.
La biodisponibilidad de la epoetina beta después de la administración subcutánea está comprendida entre el 23 y el 42 % en comparación con la administración intravenosa.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad de dosis repetidas, genotoxicidad y toxicidad para la reproducción.
Un estudio de carcinogenicidad con homólogos de eritropoyetina en ratones no reveló ningún signo de potencial proliferativo o carcinogénico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Urea,
Cloruro sódico,
Polisorbato 20,
Fosfato monosódico dihidratado,
Fosfato disódico dodecahidratado,
Cloruro cálcico dihidratado,
Glicina,
L-Leucina,
L-Isoleucina,
L-Treonina,
L-Ácido glutámico,
L-Fenilalanina,
Agua para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
Conservar la jeringa precargada en el embalaje exterior, para protegerlo de la luz.
Con fines de uso ambulatorio, se puede mantener el producto fuera de la nevera durante un único periodo de hasta 3 días a temperatura ambiente (no superior a 25°C).
6.5 Naturaleza y contenido del envase
Jeringa precargada (vidrio tipo I), con un protector y un émbolo (goma teflonizada) con una aguja de inyección (27G1/2). Cada jeringa contiene 0,3 ml de solución.
Tamaños de envase de 1 jeringa precargada y 1 aguja o de 6 jeringas precargadas y 6 agujas.
Puede que solamente estén comercializados algunos tamaños de envase.
6.6 Precauciones especiales de eliminación y otras manipulaciones
En primer lugar, ¡lávese las manos!
1. Saque una jeringa del envase y asegúrese que la solución es clara, incolora y prácticamente libre de partículas visibles. Retire el protector de la jeringa.
2. Saque una aguja del envase, fíjela sobre la jeringa y retire la tapa protectora de la aguja.
3. Expulse el aire de la jeringa y aguja manteniendo la jeringa vertical y presionando suavemente el émbolo hacia arriba. Continúe presionando el émbolo hasta que la cantidad de solución de NeoRecormon en la jeringa sea la prescrita.
4. Limpie la piel en el lugar de inyección con un algodón con alcohol. Haga un pliegue tomando la piel entre el índice y el pulgar. Sujete el cuerpo de la jeringa por la parte más próxima a la aguja e inserte la aguja en el pliegue cutáneo con acción firme y rápida. Inyecte la solución de NeoRecormon. Retire la aguja rápidamente y aplique presión sobre el lugar de inyección con un apósito seco y estéril.
Este medicamento es solo para uso individual. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/041 -042
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16 de julio de 1997 Fecha de la última revalidación: 16 de julio de 2007
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 5.000 UI solución inyectable en jeringa precargada
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Una jeringa precargada con 0,3 ml de solución para inyección contiene 5.000 unidades internacionales (UI) correspondientes a 41,5 microgramos de epoetina beta* (eritropoyetina recombinante humana).
Un ml de solución para inyección contiene 16.667 UI de epoetina beta.
* producida en células de ovarios de hámster chino (CHO) mediante tecnología de ADN recombinante.
Excipientes con efecto conocido:
Fenilalanina (hasta 0,3 mg/jeringa).
Sodio (menos de 1 mmol/jeringa)
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución incolora, de transparente a ligeramente opalescente.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
NeoRecormon está indicado para:
- Tratamiento de la anemia sintomática asociada a la insuficiencia renal crónica en pacientes adultos y pediátricos.
- Prevención de la anemia en prematuros con un peso corporal al nacer de 750 a 1.500 g y una edad gestacional de menos de 34 semanas.
- Tratamiento de la anemia sintomática en pacientes adultos con neoplasias no mieloides tratados con quimioterapia.
- Aumentar el rendimiento de la sangre autóloga de pacientes incluidos en un programa de predonación.
Debe sopesarse su uso en esta indicación frente al riesgo aumentado de episodios tromboembólicos que han sido notificados. Sólo debe administrarse el tratamiento a pacientes con anemia moderada (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], sin deficiencia de hierro) si no se dispone de procedimientos para conservar la sangre o si éstos son insuficientes cuando la cirugía mayor electiva programada requiera un gran volumen de sangre (4 o más unidades de sangre en mujeres o 5 o más unidades en hombres). Ver sección 5.1.
4.2 Posología y forma de administración
La terapia con NeoRecormon debe iniciarse por médicos con experiencia en las indicaciones arriba
mencionadas. Como se han observado reacciones anafilácticas en algunos casos aislados, se
recomienda administrar la primera dosis bajo control médico.
Posología
Tratamiento de la anemia sintomática en pacientes adultos y pediátricos con insuficiencia renal crónica
Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente. NeoRecormon se puede administrar tanto por vía subcutánea como intravenosa con el fin de aumentar la concentración de hemoglobina hasta un nivel no superior a 12 g/dl (7,5 mmol/l). En pacientes que no están sometidos a hemodiálisis es preferible utilizar la vía subcutánea para evitar la punción de venas periféricas. En caso de administración intravenosa, la solución debe ser inyectada a lo largo de unos 2 minutos, p.ej. en pacientes en hemodiálisis por vía de la fístula arteriovenosa al final de la diálisis.
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones se pueden observar valores individuales de hemoglobina superiores o inferiores a los niveles deseados. La variabilidad en los niveles de hemoglobina se debe controlar mediante ajuste de la dosis con el objeto de mantener los valores de hemoglobina dentro del intervalo entre 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Se debe evitar un nivel de hemoglobina de forma continuada por encima de 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando los valores de hemoglobina sean superiores a 12 g/dl (7,5 mmol/l).
Debe evitarse un aumento de hemoglobina superior a 2 g/dl (1,25 mmol/l) durante un periodo de cuatro semanas. Si esto ocurre, se debe hacer un ajuste adecuado de la dosis según las instrucciones incluidas en esta misma sección. Si la tasa de aumento de hemoglobina es mayor de 2 g/dl (1,25 mmol/l) en un mes o si el nivel de hemoglobina está aumentando y se acerca a 12 g/dl (7,45 mmol/l), debe reducirse la dosis en aproximadamente un 25%. Si el nivel de hemoglobina sigue aumentando, se debe interrumpir el tratamiento hasta que el nivel de hemoglobina empiece a disminuir, momento en el que se podrá reiniciar el tratamiento con una dosis aproximadamente un 25 % inferior a la dosis administrada previamente.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis eficaz más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia al tiempo que se mantiene una concentración de hemoglobina inferior o igual a 12 g/dl (7,45 mmol/l).
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica. En los pacientes con una respuesta deficiente de la hemoglobina a NeoRecormon, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.4 y 5.1).
En presencia de hipertensión o de enfermedades cardiovasculares, cerebrovasculares o vasculoperiféricas, se debe determinar de modo individual el incremento semanal y el valor objetivo de la hemoglobina teniendo en cuenta el cuadro clínico.
El tratamiento con NeoRecormon se divide en dos fases:
1. Fase de corrección
- Administración subcutánea:
La dosis inicial es 3 x 20 UI/kg por semana. La dosis puede incrementarse cada 4 semanas en 3 x 20 UI/kg y semana si el aumento de la hemoglobina no ha sido adecuado (< 0,25 g/dl por semana).
La dosis semanal puede dividirse en dosis diarias.
- Administración intravenosa:
La dosis inicial es 3 x 40 UI/kg por semana, y puede aumentarse al cabo de 4 semanas a 80 UI/kg -tres veces por semana- y si son necesarios incrementos ulteriores serán de 20 UI/kg tres veces por semana, con intervalos mensuales.
Por ambas vías de administración, la dosis máxima no debe superar 720 UI/kg por semana.
2. Fase de mantenimiento
Para mantener el nivel de hemoglobina entre 10 y 12 g/dl, la dosis es inicialmente reducida a la mitad de la cantidad previamente administrada. Posteriormente, se ajustará la dosis individualmente para el paciente a intervalos de una o dos semanas (dosis de mantenimiento).
En caso de administración subcutánea, la dosis semanal puede administrarse en una inyección única o fraccionada en tres o siete inyecciones. Los pacientes que permanezcan estables en el régimen de una dosis única semanal pueden pasar a una administración única cada dos semanas. En este caso, puede ser necesario un aumento de la dosis.
Los resultados de los estudios clínicos en niños han revelado que, en general, a menor edad, se necesita mayor dosis de NeoRecormon. No obstante, hay que seguir el programa posológico recomendado ya que no puede predecirse la respuesta individual.
El tratamiento con NeoRecormon es normalmente crónico. Sin embargo, en caso necesario puede interrumpirse en cualquier momento. Los datos sobre la pauta posológica de una vez a la semana se han obtenido de estudios clínicos con una duración de tratamiento de 24 semanas.
Prevención de anemia en prematuros
La solución se administra por vía subcutánea a una dosis de 3 x 250 UI/kg p.c. por semana. Es probable que los niños que ya hayan recibido una transfusión previa cuando se inicie el tratamiento con NeoRecormon no se beneficien tanto como los prematuros no transfundidos. La duración de tratamiento recomendada es de 6 semanas.
Tratamiento de anemia sintomática inducida por quimioterapia en pacientes con cáncer Se debe administrar NeoRecormon por vía subcutánea a pacientes con anemia (por ejemplo, si la concentración de hemoglobina es menor o igual a 10 g/dl [6,2 mmol/l]). Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente.
La dosis semanal puede administrarse como una inyección por semana o en dosis divididas entre 3 y 7 veces por semana.
La dosis inicial recomendada es de 30.000 UI por semana (que corresponde aproximadamente a 450 UI/kg de peso corporal por semana, en base al peso medio del paciente).
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones es posible llegar a observar en un paciente valores de hemoglobina superiores o inferiores a los esperados. La variabilidad de la hemoglobina se deberá manejar ajustando la dosis para mantener los valores de la hemoglobina dentro del intervalo de 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Deben evitarse concentraciones sostenidas de hemoglobina superiores a 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando se observen valores de hemoglobina superiores a los 12 g/dl (7,5 mmol/l).
Si a las 4 semanas de tratamiento el valor de hemoglobina aumenta hasta al menos 1 g/dl (0,62 mmol/l), se debe continuar con la dosis que en ese momento se esté administrando. Si los valores de hemoglobina no han aumentado al menos 1 g/dl (0,62 mmol/l), se debe considerar duplicar la dosis semanal. Si a las 8 semanas de tratamiento el valor de hemoglobina no aumenta hasta al menos 1 g/dl (0,62 mmol/l), es improbable que se produzca respuesta y, por consiguiente, el tratamiento debe ser interrumpido.
El tratamiento debe continuar durante las 4 semanas posteriores al final de la quimioterapia.
La dosis máxima no debe exceder de 60.000 UI por semana.
Una vez se ha alcanzado el objetivo terapéutico del paciente, la dosis debe reducirse del 25 al 50 % con el fin de mantener el valor de hemoglobina en ese nivel. Debe realizarse el ajuste posológico adecuado.
Si el nivel de hemoglobina excede los 12 g/dl (7,5 mmol/l) se debe reducir la dosis entre un 25 y un
50 % aproximadamente. Si el nivel de hemoglobina excede de 13 g/dl (8,1 mmol), se debe interrumpir temporalmente el tratamiento con NeoRecormon. El tratamiento debe reiniciarse con una dosis aproximadamente un 25 % inferior a la dosis previamente administrada después de que los niveles de hemoglobina desciendan hasta un valor menor o igual a 12 g/dl (7,5 mmol/l) o por debajo.
51 al cabo de 4 semanas el incremento de hemoglobina es superior a 2 g/dl (1,3 mmol/l), la dosis debe reducirse entre un 25 y un 50 %.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia.
Tratamiento para incrementar el rendimiento de la sangre autóloga donada La solución puede administrarse por vía intravenosa, en unos 2 minutos, o subcutáneamente. NeoRecormon se administra dos veces por semana durante 4 semanas. En aquellas ocasiones en que el hematocrito del paciente permite la donación de sangre, es decir el hematocrito es > 33 %, NeoRecormon se administra al final de la donación de sangre.
Durante la totalidad del período de tratamiento no debe excederse un hematocrito del 48 %.
La dosis debe ser determinada por el equipo quirúrgico, individualmente para cada paciente, en función de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos:
1. La cantidad de sangre pre-donada necesaria dependerá de la pérdida prevista de sangre, de los medios de conservación empleados y del estado físico del paciente.
Este volumen debe ser equivalente a la cantidad de sangre que se considera necesaria para evitar transfusiones de sangre homóloga.
La cantidad de sangre pre-donada necesaria se expresa en unidades en las cuales una unidad de nomograma es equivalente a 180 ml de glóbulos rojos.
2. La capacidad de donar sangre depende predominantemente del volumen sanguíneo del paciente y del hematocrito basal. Ambas variables determinan la reserva endógena de eritrocitos, que puede ser calculada conforme a la fórmula siguiente:
Reserva endógena de eritrocitos = volumen sanguíneo (ml) x (hematocrito - 33)/ 100 mujeres: volumen sanguíneo (ml) = 41 (ml/kg) x peso corporal (kg) + 1200 (ml)
hombres: volumen sanguíneo (ml) = 44 (ml/kg) x peso corporal (kg) + 1600 (ml)
(peso corporal: > 45 kg)
La indicación para un tratamiento con NeoRecormon y, en su caso, la dosis individual debe ser determinada a partir de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos según las gráficas siguientes:
Paciente femenino Paciente masculino
Cantidad de sangre pre-donada Cantidad de sangre pre-donada
necesaria [unidades] necesaria [unidades]
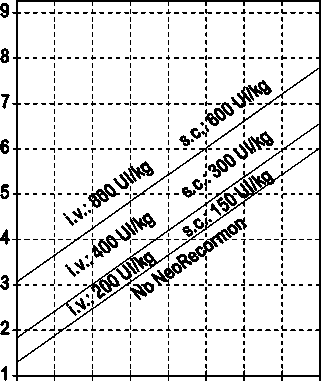
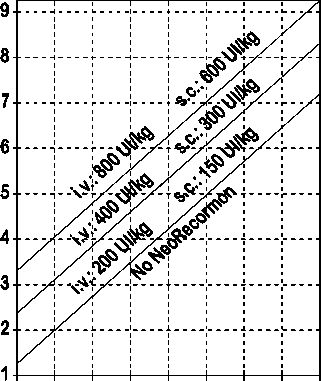
O 100 200 300 400 500 600 700 800 0 100 200 300 400 500 600 700 800
Reserva eritrocitaria endógena [ml] Reserva eritrocitaria endógena [ml]
La dosis única así determinada debe administrarse dos veces por semana a lo largo de 4 semanas. La dosis máxima no debe sobrepasar 1.600 UI/kg peso corporal por semana en administración intravenosa ó 1.200 UI/kg por semana en administración subcutánea.
Forma de administración
La jeringa precargada de NeoRecormon está lista para su uso. Sólo se pueden inyectar soluciones claras o ligeramente opalescentes, incoloras y prácticamente libres de partículas visibles. NeoRecormon en jeringa precargada es un producto estéril pero sin conservantes. Bajo ninguna circunstancia debe administrarse más de una dosis por jeringa; el medicamento es solo para uso individual.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Hipertensión mal controlada.
En la indicación para “Aumentar el rendimiento de sangre autóloga”: infarto de miocardio o accidente cerebrovascular en el mes anterior al tratamiento, angina de pecho inestable, riesgo aumentado de trombosis venosa profunda, como por ejemplo en aquellos con historial de enfermedad tromboembólica venosa.
4.4 Advertencias y precauciones especiales de empleo
NeoRecormon debe usarse con cautela en presencia de anemia refractaria con exceso de blastos en transformación, epilepsia, trombocitosis e insuficiencia hepática crónica. Y deben excluirse las deficiencias de ácido fólico y vitamina B12 pues reducen la eficacia de NeoRecormon.
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica, ya que las dosis acumuladas altas de epoetina pueden asociarse a un mayor riesgo de mortalidad, y de acontecimientos cardiovasculares y cerebrovasculares graves. En pacientes con una respuesta deficiente de la hemoglobina a epoetinas, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.2 y 5.1).
Para garantizar una eritropoyesis eficaz, se debe evaluar el nivel de hierro en todos los pacientes antes y durante el tratamiento, y puede ser necesario un tratamiento suplementario con hierro administrado de acuerdo con las guías terapéuticas.
La sobrecarga grave de aluminio debido al tratamiento de la insuficiencia renal puede comprometer la eficacia de NeoRecormon.
La indicación de tratamiento con NeoRecormon en enfermos nefroscleróticos aún no sometidos a diálisis debe definirse individualmente, ya que la posible aceleración de la insuficiencia renal no puede descartarse con certeza.
Aplasia pura de células rojas (APCR)
Se han notificado casos de APCR causada por anticuerpos neutralizantes antieritropoyetina, asociados a tratamientos con eritropoyetinas, incluido NeoRecormon. Estos anticuerpos han presentado reacción cruzada con todas las proteínas eritropoyéticas, por los que los pacientes en los que se sospeche o se haya confirmado la presencia de anticuerpos neutralizantes contra eritropoyetina no deben ser tratados con NeoRecormon (ver sección 4.8).
APCR en pacientes con hepatitis C
Se debe interrumpir inmediatamente el tratamiento con epoetina, y realizar ensayos de anticuerpos antieritropoyetina si se produce una disminución paradójica de la hemoglobina y se desarrolla anemia severa asociada con bajos recuentos de reticulocitos. En pacientes con hepatitis C tratados con interferon y ribavirina se han notificados casos cuando las epoetinas se han utilizado concomitantemente. Las epoetinas no están aprobadas en el tratamiento de la anemia asociada a la hepatitis C.
Monitorización de la tensión arterial
Puede aparecer un aumento de la tensión arterial o un agravamiento de la hipertensión ya existente, especialmente en los casos en los que el hematocrito aumenta rápidamente. Estos incrementos en la tensión arterial pueden ser tratados con medicamentos. Si no pueden ser controlados con medicación se recomienda interrumpir transitoriamente la terapia con NeoRecormon. En particular al principio de la terapia se recomienda la monitorización regular de la tensión arterial; también durante la diálisis. Pueden producirse crisis hipertensivas con síntomas de encefalopatías y que requieren atención médica inmediata y cuidados médicos intensivos. Debe prestarse especial atención a las cefaleas súbitas lacerantes hemicraneales como posible signo de advertencia.
Insuficiencia renal crónica
En pacientes con insuficiencia renal crónica puede sobrevenir un aumento dosis-dependiente moderado en la cifra plaquetaria dentro del margen normal durante el tratamiento con NeoRecormon, especialmente después de la administración intravenosa. La regresión tiene lugar en el curso de la prosecución de la terapia. Se recomienda el control regular de la cifra de plaquetas durante las 8 primeras semanas de tratamiento.
Concentración de hemoglobina
En pacientes con insuficiencia renal crónica, la concentración de hemoglobina en la fase de mantenimiento no debe exceder el límite superior del rango recomendado en la sección 4.2. En ensayos clínicos se observó un aumento del riesgo de fallecimiento y de acontecimientos cardiovasculares graves o acontecimientos cerebrovasculares incluido el infarto, cuando se administraron agentes estimulantes de la eritropoyesis (AEEs) con el fin de alcanzar un nivel de hemoglobina superior a 12 g/dl (7,5 mmol/l).
En ensayos clínicos controlados no se han observado beneficios significativos atribuibles a la administración de epoetinas cuando se aumentaba la concentración de hemoglobina por encima del nivel necesario para controlar los síntomas de anemia y evitar las transfusiones sanguíneas.
En niños prematuros puede producirse un ligero aumento de la cifra plaquetaria, en especial hasta el día 12 -14 de vida, por lo tanto las plaquetas deben controlarse regularmente.
Efectos sobre el crecimiento tumoral
Las epoetinas son factores de crecimiento que estimulan de forma primaria la producción de glóbulos rojos. Los receptores de epoetina pueden expresarse sobre la superficie de una variedad de células tumorales. Como sucede con todos los factores de crecimiento, existe la preocupación de que las epoetinas pudieran estimular el crecimiento de tumores. En varios ensayos controlados, no se ha observado que las epoetinas mejoren la supervivencia global o que disminuyan el riesgo de progresión del tumor en pacientes con anemia asociada con cáncer.
En ensayos clínicos controlados, se ha observado que el uso de NeoRecormon y otros agentes estimulantes de la eritropoyesis (AEEs):
- acortaba el tiempo de progresión del tumor en pacientes con cáncer avanzado de cabeza y cuello que recibían radioterapia cuando se administraba para conseguir una concentración de hemoglobina por encima de 14 g/dl (8,7 mmol/l),
- acortaba la supervivencia global y aumentaba el número de fallecimientos atribuidos a la progresión de la enfermedad a los 4 meses, en pacientes con cáncer de mama metastásico que recibían quimioterapia cuando se administraba para conseguir una concentración de hemoglobina entre los 12 y los 14 g/dl (7,5-8,7 mmol/l),
- aumentaba el riesgo de fallecimiento cuando se administraba para conseguir una concentración de hemoglobina de 12 g/dl (7,5 mmol/l) en pacientes con enfermedad maligna activa y que no recibían ni quimioterapia ni radioterapia. El uso de los AEEs no está indicado en esa población de pacientes.
En vista de lo anterior, en algunas situaciones clínicas la transfusión sanguínea debe ser el tratamiento de elección para la anemia en pacientes con cáncer. La decisión de administrar eritropoyetinas recombinantes se tomará en base a la evaluación de la relación beneficio/riesgo junto con la aceptación individual del paciente, teniendo en cuenta el contexto clínico específico. Los factores que deben considerarse en esta evaluación son el tipo de tumor y su estadio, el grado de anemia, la esperanza de vida, el entorno en el que el paciente está siendo tratado y la preferencia del paciente (ver sección 5.1).
Puede haber un aumento de la tensión arterial que puede ser tratada con medicamentos. Se recomienda por tanto monitorizar la tensión arterial, en particular en la fase inicial de tratamiento en pacientes con cáncer.
En pacientes con cáncer también deberán monitorizarse regularmente el recuento plaquetario y el valor de hemoglobina.
En pacientes en programa de predonación de sangre autóloga, puede sobrevenir un aumento de la cifra de plaquetas, generalmente dentro de los límites normales. Por tanto, se recomienda la determinación de esta cifra al menos una vez por semana. Si se produce un aumento plaquetario de más de 150 x 109/l o si las plaquetas ascienden por encima del margen normal, se debe suspender el tratamiento con NeoRecormon.
En niños prematuros no se podría descartar el riesgo potencial de que la eritropoyetina provoque retinopatía, por lo tanto se debe tener precaución y la decisión de tratar a un niño prematuro se debe tomar teniendo en cuenta el beneficio potencial y el riesgo de este tratamiento, así como las opciones alternativas de tratamiento que estén disponibles.
En pacientes con insuficiencia renal crónica, a menudo se precisa aumentar la dosis de heparina durante la hemodiálisis en la terapia con NeoRecormon debido al aumento del volumen de hematocrito. Si la heparinización no es óptima es posible que se produzca la oclusión del sistema de diálisis.
En pacientes con alteración renal crónica con riesgo de trombosis de derivación, se debe considerar la revisión temprana de la derivación y profilaxis de la trombosis mediante la administración, por ejemplo, de ácido acetilsalicílico.
Los niveles séricos de potasio y fosfato deben ser controlados regularmente durante la terapia con NeoRecormon. Se ha informado de aumentos de potasio en unos pocos pacientes urémicos que recibieron NeoRecormon si bien no se ha establecido una relación causal. En caso de observar un nivel elevado o creciente de potasio se debe considerar la interrupción de la administración de NeoRecormon hasta que el nivel haya sido corregido.
Para el uso de NeoRecormon en un programa de predonación de sangre autóloga deben ser observadas las directrices oficiales sobre donación de sangre, en particular las siguientes:
- sólo deben donar los pacientes con hematocrito > 33 % (hemoglobina >11 g/dl [6,83 mmol/l]);
- debe procederse con especial cuidado con pacientes con un peso inferior a 50 kg;
- el volumen individual extraído no debe superar aproximadamente el 12 % del volumen sanguíneo estimado del paciente.
El tratamiento debe reservarse a pacientes en los que se considere particularmente importante evitar la transfusión de sangre homóloga teniendo en cuenta la evaluación riesgo/beneficio de las transfusiones homólogas.
Uso indebido
Un uso indebido del producto por personas sanas puede llevar a un aumento excesivo del volumen de hematocrito, lo cual puede asociarse con complicaciones del sistema cardiovascular con riesgo para la vida.
Excipientes
NeoRecormon en jeringa precargada contiene, como máximo, 0,3 mg de fenilalanina por jeringa como excipiente. Este hecho se deberá tener en cuenta en los pacientes afectados por formas graves de fenilcetonuria.
Este medicamento contiene menos de 1 mmol (23 mg) de sodio por jeringa, por lo que se considera esencialmente “exento de sodio”.
Trazabilidad de NeoRecormon
Para mejorar la trazabilidad de los agentes estimulantes de la eritropoyesis (AEEs), se debe registrar (o indicar) claramente el nombre comercial del AEE administrado en la historia clínica del paciente.
4.5 Interacción con otros medicamentos y otras formas de interacción
Los resultados clínicos obtenidos hasta el presente no ponen de manifiesto interacción alguna de NeoRecormon con otros medicamentos.
Los estudios llevados a cabo en animales mostraron que la epoetina beta no potencia la mielotoxicidad de los medicamentos citostáticos, tales como etopósido, cisplatino, ciclofosfamida y fluorouracilo.
4.6 Fertilidad, embarazo y lactancia
Fertilidad
Los estudios en animales no muestran efectos dañinos directos o indirectos sobre el embarazo, desarrollo embrional/fetal, parto o desarrollo postnatal (ver sección 5.3).
Embarazo
No hay datos relativos al uso de NeoRecormon en mujeres embarazadas.
Se debe tener precaución cuando se prescriba a mujeres embarazadas.
Lactancia
Se desconoce si la epoetina beta se excreta en la leche materna. La decisión de continuar/interrumpir la lactancia o de continuar/interrumpir el tratamiento con epoetina beta se debe hacer teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento con epoetina beta para la madre.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NeoRecormon sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Basados en los resultados de los ensayos clínicos que incluyen 1.725 pacientes, se espera que aproximadamente el 8 % de los pacientes tratados con NeoRecormon sufran reacciones adversas.
- Pacientes anémicos con insuficiencia renal crónica
La reacción adversa más frecuente durante el tratamiento con NeoRecormon es un aumento de la tensión arterial o agravamiento de la hipertensión existente, especialmente en casos de aumento rápido del hematocrito (ver sección 4.4). En algunos pacientes cuya tensión arterial es normal o baja también pueden darse crisis hipertensivas con síntomas de encefalopatías (p.ej. cefaleas y estados de confusión, trastornos sensitivomotores - como alteraciones del habla o de la ambulación - hasta convulsiones tónico-clónicas) (ver sección 4.4).
Puede darse trombosis de derivación, especialmente en pacientes que tienen tendencia a la hipotensión o cuyas fístulas arteriovenosas presentan complicaciones (p.ej. estenosis, aneurismas), ver sección 4.4. En la mayoría de los casos se observa una caída en los valores séricos de ferritina simultáneamente con el aumento del volumen de hematocrito (ver sección 4.4). Además, en casos aislados se han observado incrementos transitorios en los niveles séricos de potasio y fosfato (ver sección 4.4).
En casos aislados se ha notificado aplasia pura de células rojas (APCR) mediada por anticuerpos neutralizantes antieritropoyetina asociada al tratamiento con NeoRecormon. En caso de que se diagnostique APCR mediada por anticuerpos antieritropoyetina, el paciente deberá interrumpir el tratamiento con NeoRecormon, el cual no se deberá sustituir por otra proteína eritropoyética (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 1, que figura más adelante.
- Pacientes con cáncer
El dolor de cabeza y la hipertensión relacionados con el tratamiento con epoetina beta son frecuentes y pueden tratarse con medicamentos (ver sección 4.4).
En algunos pacientes se observa un descenso de los valores de hierro sérico (ver sección 4.4).
Estudios clínicos han puesto de manifiesto que la frecuencia de acontecimientos tromboembólicos es más alta en pacientes con cáncer tratados con NeoRecormon que en los controles no tratados o tratados con placebo. En pacientes tratados con NeoRecormon, esta incidencia es del 7 % comparado con el 4 % en el grupo control; este hecho no está asociado con un incremento en la mortalidad tromboembólica comparado con los grupos control.
Estas reacciones adversas se enumeran en la Tabla 2, que figura más adelante.
- Pacientes incluidos en un programa de predonación de sangre autóloga
Los pacientes incluidos en un programa de predonación de sangre autóloga han demostrado una frecuencia ligeramente superior de accidentes tromboembólicos. Sin embargo, no se pudo establecer una relación causal con el tratamiento con NeoRecormon.
En ensayos clínicos controlados con placebo, la deficiencia transitoria de hierro fue más acusada en los pacientes tratados con NeoRecormon que en los pacientes control (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 3, que figura más adelante.
Tabla de reacciones adversas
Las reacciones adversas se enumeran de acuerdo con la clasificación de órganos del sistema MedDRA y por categoría de frecuencia.
Las categorías de frecuencia se definen utilizando la siguiente convención:
muy frecuentes (> 1/10), frecuentes (> 1/100 hasta <1/10), poco frecuentes (>1/1.000 hasta <1/100), raras (>1/10.000 hasta <1/1.000); muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 1: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con IRC_
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión Crisis hipertensivas |
Frecuente Poco frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
|
Trastornos de la sangre y |
Trombosis de derivación |
Raras |
|
del sistema linfático |
Trombocitosis |
Muy raras |
Tabla 2: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con cáncer__
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión |
Frecuente |
|
Trastornos de la sangre y del sistema linfático |
Acontecimientos tromboembólicos |
Frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Tabla 3: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes incluidos en un programa de predonación de sangre autóloga
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
- Prematuros
Es muy frecuente un descenso de los valores de ferritina sérica (ver sección 4.4).
Descripción de reacciones adversas seleccionadas
Raramente pueden ocurrir reacciones cutáneas relacionadas con el tratamiento con epoetina beta como sarpullido, prurito, urticaria o reacciones en el lugar de la inyección. En casos muy raros se han notificado reacciones anafilácticas relacionadas con el tratamiento con epoetina beta. Sin embargo, en los ensayos clínicos controlados no se encontró una mayor incidencia de reacciones de hipersensibilidad.
Se han comunicado casos muy raros de síntomas gripales relacionados con el tratamiento con epoetina beta como fiebre, escalofríos, dolores de cabeza, dolor de las extremidades, malestar general, y/o dolor óseo, particularmente al inicio del tratamiento. Estas reacciones son de intensidad leve a moderada y desaparecen al cabo de un par de horas o días.
Los resultados obtenidos en un ensayo clínico controlado realizado con epoetina alfa o darbepoetina alfa, notificaron que la incidencia de infarto es frecuente.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
El margen terapéutico de NeoRecormon es muy amplio. No se han observado síntomas de sobredosis incluso a niveles séricos muy elevados.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antianémico, código ATC: B03XA01 Mecanismo de acción
La eritropoyetina es una glucoproteína que estimula la formación de eritrocitos a partir de sus progenitores asignados. Actúa como factor estimulante de la mitosis y hormona de diferenciación.
La epoetina beta, el principio activo de NeoRecormon, es idéntica en su composición aminoácida e hidrocarbonada a la eritropoyetina aislada de la orina de pacientes anémicos.
La eficacia biológica de la epoetina beta ha sido demostrada tras administración intravenosa y subcutánea en varios modelos animales in vivo (ratas normales y urémicas, ratones policitémicos, perros). Con la administración de epoetina beta aumentan el número de eritrocitos, los valores de Hb y la cifra de reticulocitos, al igual que la velocidad de incorporación de 59Fe.
In vitro (cultivo citológico de bazo de ratón) ha sido hallada una mayor incorporación de 3H-timidina en las células eritroides nucleadas del bazo tras incubación con epoetina beta.
Las investigaciones en cultivos de células de la médula ósea humana han revelado que la epoetina beta estimula específicamente la eritropoyesis y no afecta a la leucopoyesis. No han sido detectadas acciones citotóxicas de la epoetina beta sobre la médula ósea o sobre células cutáneas humanas.
Tras una dosis única de epoetina beta no se ha observado ningún efecto en el comportamiento o la actividad locomotora del ratón ni en las funciones circulatoria o respiratoria del perro.
Eficacia clínica y seguridad
En un ensayo aleatorizado, doble ciego, controlado con placebo, realizado en 4.038 pacientes con enfermedad renal crónica no sometidos a diálisis, con una diabetes tipo 2 y los niveles de hemoglobina < 11 g/dl, los pacientes recibieron tratamiento bien con darbepoetina alfa para alcanzar niveles de hemoglobina de 13 g/dl o con placebo (ver sección 4.4). El ensayo no cumplió su objetivo principal de demostrar una disminución en el riesgo de todas las causas de mortalidad, la morbilidad cardiovascular o enfermedad renal terminal. El análisis de los componentes individuales de la variable combinada mostró los siguientes HR (IC 95%): fallecimiento 1,05 (0,92-1,21), infarto cerebrovascular 1,92 (1,38-2,68), insuficiencia cardíaca congestiva 0,89 (0,74-1,08), infarto de miocardio 0,96 (0,751,23), hospitalización por isquemia del miocardio 0,84 (0,55-1,27), enfermedad renal terminal 1,02 (0,87-1,18).
En pacientes con IRC (tratados con diálisis, no tratados con diálisis, pacientes diabéticos y pacientes no diabéticos) se han realizado análisis agrupados post hoc de los estudios clínicos con AEEs. Se observó una tendencia a un aumento de las estimaciones del riesgo para la mortalidad por todas las causas y para los acontecimientos cardiovasculares y cerebrovasculares asociados a dosis acumuladas más altas de AEE independientemente de que los pacientes padecieran o no diabetes y de que recibieran o no tratamiento con diálisis (ver las secciones 4.2 y 4.4).
La eritropoyetina es un factor de crecimiento que estimula de forma primaria la producción de glóbulos rojos. Los receptores de eritropoyetina se encuentran también presentes en la superficie de algunas líneas celulares malignas.
Se ha estudiado la supervivencia y la progresión tumoral en cinco grandes ensayos controlados que incluyeron a 2.833 pacientes, de los cuales cuatro fueron ensayos doble-ciego controlados con placebo y uno fue un ensayo abierto. Dos de los ensayos reclutaron pacientes que estaban siendo tratados con quimioterapia. El nivel de hemoglobina que se quería alcanzar fue >13 g/dl en dos de los ensayos y de entre 12 y 14 g/dl en los otros tres. En el ensayo abierto no se observaron diferencias en la supervivencia global, entre los pacientes tratados con eritropoyetina humana recombinante y el grupo control. En los cuatro ensayos controlados con placebo, el índice de riesgo (hazard ratio) para la supervivencia global osciló entre 1,25 y 2,47 en favor de los grupos control. En todos estos ensayos se ha observado un aumento de la mortalidad inexplicable y estadísticamente significativo en pacientes que presentaban anemia asociada con diversos tipos frecuentes de cáncer y que recibieron eritropoyetina humana recombinante, en comparación con los grupos control. Las diferencias observadas en la incidencia de trombosis y complicaciones relacionadas, entre los pacientes que recibieron eritropoyetina humana recombinante y los que formaban parte de los grupos control, no permiten explicar satisfactoriamente los resultados de supervivencia global observados en los ensayos.
Un meta-análisis basado en datos individuales de pacientes, que incluyó datos de los 12 ensayos clínicos controlados realizados con NeoRecormon en pacientes anémicos con cáncer (n=2301), mostró un valor estimado del índice de riesgo (hazard ratio) de supervivencia global de 1,13 en favor de los grupos control (IC 95 %, CI 0,87, 1,46). En pacientes con un valor inicial de hemoglobina < 10 g/dl (n=899), el valor estimado del índice de riesgo (hazard ratio) para la supervivencia fue de 0,98 (IC 95% 0,68 al 1,40). Se observó un aumento del riesgo relativo de acontecimientos tromboembólicos en la población general (RR 1,62, IC 95 %: 1,13, 2,31).
Asimismo, se ha realizado un análisis de datos a nivel de paciente, en más de 13.900 pacientes con cáncer (tratados con quimioterapia, radioterapia, quimioradioterapia o sin tratamiento) que participaban en un total de 53 ensayos clínicos controlados en los que se administraban diversas epoetinas. En este meta-análisis se obtuvo un índice de riesgo (hazard ratio) para la supervivencia global de 1,06 a favor de los grupos control (IC 95%: 1,00, 1,12; 53 ensayos y 13.933 pacientes) y para los pacientes con cáncer que recibían quimioterapia, el índice de riesgo (hazard ratio) para la supervivencia global fue de 1,04 (IC 95%: 0,97, 1,11; 38 ensayos y 10.441 pacientes). Este meta-análisis además indica de forma consistente un aumento significativo del riesgo relativo de episodios tromboembólicos en pacientes con cáncer tratados con eritropoyetina recombinante humana (ver sección 4.4).
En casos muy raros, se han producido anticuerpos neutralizantes anti-eritropoyetina con o sin aplasia pura de células rojas (APCR) durante el tratamiento con rHuEPO.
5.2 Propiedades farmacocinéticas
Los estudios farmacocinéticos en voluntarios sanos y pacientes urémicos muestran que la semivida de la epoetina beta administrada por vía i.v. es de 4 a 12 horas y que el volumen de distribución corresponde de una a dos veces el volumen plasmático. Se han hallado resultados análogos en experimentos en animales con ratas normales y urémicas.
Tras la administración subcutánea de epoetina beta a pacientes urémicos la absorción retardada se traduce en una meseta de concentración sérica, cuya cota máxima se alcanza al cabo de 12 - 28 horas de media. El semiperíodo de vida terminal es mayor que después de la administración intravenosa, siendo la media de 13-28 horas.
La biodisponibilidad de la epoetina beta después de la administración subcutánea está comprendida entre el 23 y el 42 % en comparación con la administración intravenosa.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad de dosis repetidas, genotoxicidad y toxicidad para la reproducción.
Un estudio de carcinogenicidad con homólogos de eritropoyetina en ratones no reveló ningún signo de potencial proliferativo o carcinogénico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Urea,
Cloruro sódico,
Polisorbato 20,
Fosfato monosódico dihidratado,
Fosfato disódico dodecahidratado,
Cloruro cálcico dihidratado,
Glicina,
L-Leucina,
L-Isoleucina,
L-Treonina,
L-Ácido glutámico,
L-Fenilalanina,
Agua para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
Conservar la jeringa precargada en el embalaje exterior, para protegerlo de la luz.
Con fines de uso ambulatorio, se puede mantener el producto fuera de la nevera durante un único periodo de hasta 3 días a temperatura ambiente (no superior a 25°C).
6.5 Naturaleza y contenido del envase
Jeringa precargada (vidrio tipo I), con un protector y un émbolo (goma teflonizada) con una aguja de inyección (27G1/2). Cada jeringa contiene 0,3 ml de solución.
Tamaños de envase de 1 jeringa precargada y 1 aguja o de 6 jeringas precargadas y 6 agujas.
Puede que solamente estén comercializados algunos tamaños de envase.
6.6 Precauciones especiales de eliminación y otras manipulaciones
En primer lugar, ¡lávese las manos!
1. Saque una jeringa del envase y asegúrese que la solución es clara, incolora y prácticamente libre de partículas visibles. Retire el protector de la jeringa.
2. Saque una aguja del envase, fíjela sobre la jeringa y retire la tapa protectora de la aguja.
3. Expulse el aire de la jeringa y aguja manteniendo la jeringa vertical y presionando suavemente el émbolo hacia arriba. Continúe presionando el émbolo hasta que la cantidad de solución de NeoRecormon en la jeringa sea la prescrita.
4. Limpie la piel en el lugar de inyección con un algodón con alcohol. Haga un pliegue tomando la piel entre el índice y el pulgar. Sujete el cuerpo de la jeringa por la parte más próxima a la aguja e inserte la aguja en el pliegue cutáneo con acción firme y rápida. Inyecte la solución de NeoRecormon. Retire la aguja rápidamente y aplique presión sobre el lugar de inyección con un apósito seco y estéril.
Este medicamento es solo para uso individual. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/033 -034
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16 de julio de 1997 Fecha de la última revalidación: 16 de julio de 2007
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 6.000 UI solución inyectable en jeringa precargada
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Una jeringa precargada con 0,3 ml de solución para inyección contiene 6.000 unidades internacionales (UI) correspondientes a 49,8 microgramos de epoetina beta* (eritropoyetina recombinante humana).
Un ml de solución para inyección contiene 20.000 UI de epoetina beta.
* producida en células de ovarios de hámster chino (CHO) mediante tecnología de ADN recombinante.
Excipientes con efecto conocido:
Fenilalanina (hasta 0,3 mg/jeringa).
Sodio (menos de 1 mmol/jeringa)
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución incolora, de transparente a ligeramente opalescente.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
NeoRecormon está indicado para:
- Tratamiento de la anemia sintomática asociada a la insuficiencia renal crónica en pacientes adultos y pediátricos.
- Prevención de la anemia en prematuros con un peso corporal al nacer de 750 a 1.500 g y una edad gestacional de menos de 34 semanas.
- Tratamiento de la anemia sintomática en pacientes adultos con neoplasias no mieloides tratados con quimioterapia.
- Aumentar el rendimiento de la sangre autóloga de pacientes incluidos en un programa de predonación.
Debe sopesarse su uso en esta indicación frente al riesgo aumentado de episodios tromboembólicos que han sido notificados. Sólo debe administrarse el tratamiento a pacientes con anemia moderada (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], sin deficiencia de hierro) si no se dispone de procedimientos para conservar la sangre o si éstos son insuficientes cuando la cirugía mayor electiva programada requiera un gran volumen de sangre (4 o más unidades de sangre en mujeres o 5 o más unidades en hombres). Ver sección 5.1.
4.2 Posología y forma de administración
La terapia con NeoRecormon debe iniciarse por médicos con experiencia en las indicaciones arriba
mencionadas. Como se han observado reacciones anafilácticas en algunos casos aislados, se
recomienda administrar la primera dosis bajo control médico.
Posología
Tratamiento de la anemia sintomática en pacientes adultos y pediátricos con insuficiencia renal crónica
Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente. NeoRecormon se puede administrar tanto por vía subcutánea como intravenosa con el fin de aumentar la concentración de hemoglobina hasta un nivel no superior a 12 g/dl (7,5 mmol/l). En pacientes que no están sometidos a hemodiálisis es preferible utilizar la vía subcutánea para evitar la punción de venas periféricas._En caso de administración intravenosa, la solución debe ser inyectada a lo largo de unos 2 minutos, p.ej. en pacientes en hemodiálisis por vía de la fístula arteriovenosa al final de la diálisis.
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones se pueden observar valores individuales de hemoglobina superiores o inferiores a los niveles deseados. La variabilidad en los niveles de hemoglobina se debe controlar mediante ajuste de la dosis con el objeto de mantener los valores de hemoglobina dentro del intervalo entre 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Se debe evitar un nivel de hemoglobina de forma continuada por encima de 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando los valores de hemoglobina sean superiores a 12 g/dl (7,5 mmol/l).
Debe evitarse un aumento de hemoglobina superior a 2 g/dl (1,25 mmol/l) durante un periodo de cuatro semanas. Si esto ocurre, se debe hacer un ajuste adecuado de la dosis según las instrucciones incluidas en esta misma sección. Si la tasa de aumento de hemoglobina es mayor de 2 g/dl (1,25 mmol/l) en un mes o si el nivel de hemoglobina está aumentando y se acerca a 12 g/dl (7,45 mmol/l), debe reducirse la dosis en aproximadamente un 25%. Si el nivel de hemoglobina sigue aumentando, se debe interrumpir el tratamiento hasta que el nivel de hemoglobina empiece a disminuir, momento en el que se podrá reiniciar el tratamiento con una dosis aproximadamente un 25 % inferior a la dosis administrada previamente.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis eficaz más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia al tiempo que se mantiene una concentración de hemoglobina inferior o igual a 12 g/dl (7,45 mmol/l).
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica. En los pacientes con una respuesta deficiente de la hemoglobina a NeoRecormon, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.4 y 5.1).
En presencia de hipertensión o de enfermedades cardiovasculares, cerebrovasculares o vasculoperiféricas, se debe determinar de modo individual el incremento semanal y el valor objetivo de la hemoglobina teniendo en cuenta el cuadro clínico.
El tratamiento con NeoRecormon se divide en dos fases:
1. Fase de corrección
- Administración subcutánea:
La dosis inicial es 3 x 20 UI/kg por semana. La dosis puede incrementarse cada 4 semanas en 3 x 20 UI/kg y semana si el aumento de la hemoglobina no ha sido adecuado (< 0,25 g/dl por semana).
La dosis semanal puede dividirse en dosis diarias.
- Administración intravenosa:
La dosis inicial es 3 x 40 UI/kg por semana, y puede aumentarse al cabo de 4 semanas a 80 UI/kg -tres veces por semana- y si son necesarios incrementos ulteriores serán de 20 UI/kg tres veces por semana, con intervalos mensuales.
Por ambas vías de administración, la dosis máxima no debe superar 720 UI/kg por semana.
2. Fase de mantenimiento
Para mantener el nivel de hemoglobina entre 10 y 12 g/dl, la dosis es inicialmente reducida a la mitad de la cantidad previamente administrada. Posteriormente, se ajustará la dosis individualmente para el paciente a intervalos de una o dos semanas (dosis de mantenimiento).
En caso de administración subcutánea, la dosis semanal puede administrarse en una inyección única o fraccionada en tres o siete inyecciones. Los pacientes que permanezcan estables en el régimen de una dosis única semanal pueden pasar a una administración única cada dos semanas. En este caso, puede ser necesario un aumento de la dosis.
Los resultados de los estudios clínicos en niños han revelado que, en general, a menor edad, se necesita mayor dosis de NeoRecormon. No obstante, hay que seguir el programa posológico recomendado ya que no puede predecirse la respuesta individual.
El tratamiento con NeoRecormon es normalmente crónico. Sin embargo, en caso necesario puede interrumpirse en cualquier momento. Los datos sobre la pauta posológica de una vez a la semana se han obtenido de estudios clínicos con una duración de tratamiento de 24 semanas.
Prevención de anemia en prematuros
La solución se administra por vía subcutánea a una dosis de 3 x 250 UI/kg p.c. por semana. Es probable que los niños que ya hayan recibido una transfusión previa cuando se inicie el tratamiento con NeoRecormon no se beneficien tanto como los prematuros no transfundidos. La duración de tratamiento recomendada es de 6 semanas.
Tratamiento de anemia sintomática inducida por quimioterapia en pacientes con cáncer Se debe administrar NeoRecormon por vía subcutánea a pacientes con anemia (por ejemplo, si la concentración de hemoglobina es menor o igual a 10 g/dl [6,2 mmol/l]). Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente.
La dosis semanal puede administrarse como una inyección por semana o en dosis divididas entre 3 y 7 veces por semana.
La dosis inicial recomendada es de 30.000 UI por semana (que corresponde aproximadamente a 450 UI/kg de peso corporal por semana, en base al peso medio del paciente).
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones es posible llegar a observar en un paciente valores de hemoglobina superiores o inferiores a los esperados. La variabilidad de la hemoglobina se deberá manejar ajustando la dosis para mantener los valores de la hemoglobina dentro del intervalo de 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Deben evitarse concentraciones sostenidas de hemoglobina superiores a 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando se observen valores de hemoglobina superiores a los 12 g/dl (7,5 mmol/l).
Si a las 4 semanas de tratamiento el valor de hemoglobina aumenta hasta al menos 1 g/dl (0,62 mmol/l), se debe continuar con la dosis que en ese momento se esté administrando. Si los valores de hemoglobina no han aumentado al menos 1 g/dl (0,62 mmol/l), se debe considerar duplicar la dosis semanal. Si a las 8 semanas de tratamiento el valor de hemoglobina no aumenta hasta al menos 1 g/dl (0,62 mmol/l), es improbable que se produzca respuesta y, por consiguiente, el tratamiento debe ser interrumpido.
El tratamiento debe continuar durante las 4 semanas posteriores al final de la quimioterapia.
La dosis máxima no debe exceder de 60.000 UI por semana.
Una vez se ha alcanzado el objetivo terapéutico del paciente, la dosis debe reducirse del 25 al 50 % con el fin de mantener el valor de hemoglobina en ese nivel. Debe realizarse el ajuste posológico adecuado.
Si el nivel de hemoglobina excede los 12 g/dl (7,5 mmol/l) se debe reducir la dosis entre un 25 y un
50 % aproximadamente. Si el nivel de hemoglobina excede de 13 g/dl (8,1 mmol), se debe interrumpir temporalmente el tratamiento con NeoRecormon. El tratamiento debe reiniciarse con una dosis aproximadamente un 25 % inferior a la dosis previamente administrada después de que los niveles de hemoglobina desciendan hasta un valor menor o igual a 12 g/dl (7,5 mmol/l) o por debajo.
51 al cabo de 4 semanas el incremento de hemoglobina es superior a 2 g/dl (1,3 mmol/l), la dosis debe reducirse entre un 25 y un 50 %.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia.
Tratamiento para incrementar el rendimiento de la sangre autóloga donada La solución puede administrarse por vía intravenosa, en unos 2 minutos, o subcutáneamente. NeoRecormon se administra dos veces por semana durante 4 semanas. En aquellas ocasiones en que el hematocrito del paciente permite la donación de sangre, es decir el hematocrito es > 33 %, NeoRecormon se administra al final de la donación de sangre.
Durante la totalidad del período de tratamiento no debe excederse un hematocrito del 48 %.
La dosis debe ser determinada por el equipo quirúrgico, individualmente para cada paciente, en función de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos:
1. La cantidad de sangre pre-donada necesaria dependerá de la pérdida prevista de sangre, de los medios de conservación empleados y del estado físico del paciente.
Este volumen debe ser equivalente a la cantidad de sangre que se considera necesaria para evitar transfusiones de sangre homóloga.
La cantidad de sangre pre-donada necesaria se expresa en unidades en las cuales una unidad de nomograma es equivalente a 180 ml de glóbulos rojos.
2. La capacidad de donar sangre depende predominantemente del volumen sanguíneo del paciente y del hematocrito basal. Ambas variables determinan la reserva endógena de eritrocitos, que puede ser calculada conforme a la fórmula siguiente:
Reserva endógena de eritrocitos = volumen sanguíneo (ml) x (hematocrito - 33)/ 100 mujeres: volumen sanguíneo (ml) = 41 (ml/kg) x peso corporal (kg) + 1200 (ml)
hombres: volumen sanguíneo (ml) = 44 (ml/kg) x peso corporal (kg) + 1600 (ml)
(peso corporal: > 45 kg)
La indicación para un tratamiento con NeoRecormon y, en su caso, la dosis individual debe ser determinada a partir de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos según las gráficas siguientes:
Paciente femenino Paciente masculino
Cantidad de sangre pre-donada Cantidad de sangre pre-donada
necesaria [unidades] necesaria [unidades]


O 100 200 300 400 500 600 700 800 0 100 200 300 400 500 600 700 800
Reserva eritrocitaria endógena [ml] Reserva eritrocitaria endógena [ml]
La dosis única así determinada debe administrarse dos veces por semana a lo largo de 4 semanas. La dosis máxima no debe sobrepasar 1.600 UI/kg peso corporal por semana en administración intravenosa ó 1.200 UI/kg por semana en administración subcutánea.
Forma de administración
La jeringa precargada de NeoRecormon está lista para su uso. Sólo se pueden inyectar soluciones claras o ligeramente opalescentes, incoloras y prácticamente libres de partículas visibles. NeoRecormon en jeringa precargada es un producto estéril pero sin conservantes. Bajo ninguna circunstancia debe administrarse más de una dosis por jeringa; el medicamento es solo para uso individual.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Hipertensión mal controlada.
En la indicación para “Aumentar el rendimiento de sangre autóloga”: infarto de miocardio o accidente cerebrovascular en el mes anterior al tratamiento, angina de pecho inestable, riesgo aumentado de trombosis venosa profunda, como por ejemplo en aquellos con historial de enfermedad tromboembólica venosa.
4.4 Advertencias y precauciones especiales de empleo
NeoRecormon debe usarse con cautela en presencia de anemia refractaria con exceso de blastos en transformación, epilepsia, trombocitosis e insuficiencia hepática crónica. Y deben excluirse las deficiencias de ácido fólico y vitamina B12 pues reducen la eficacia de NeoRecormon.
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica, ya que las dosis acumuladas altas de epoetina pueden asociarse a un mayor riesgo de mortalidad, y de acontecimientos cardiovasculares y cerebrovasculares graves. En pacientes con una respuesta deficiente de la hemoglobina a epoetinas, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.2 y 5.1).
Para garantizar una eritropoyesis eficaz, se debe evaluar el nivel de hierro en todos los pacientes antes y durante el tratamiento, y puede ser necesario un tratamiento suplementario con hierro administrado de acuerdo con las guías terapéuticas.
La sobrecarga grave de aluminio debido al tratamiento de la insuficiencia renal puede comprometer la eficacia de NeoRecormon.
La indicación de tratamiento con NeoRecormon en enfermos nefroscleróticos aún no sometidos a diálisis debe definirse individualmente, ya que la posible aceleración de la insuficiencia renal no puede descartarse con certeza.
Aplasia pura de células rojas (APCR)
Se han notificado casos de APCR causada por anticuerpos neutralizantes antieritropoyetina, asociados a tratamientos con eritropoyetinas, incluido NeoRecormon. Estos anticuerpos han presentado reacción cruzada con todas las proteínas eritropoyéticas, por los que los pacientes en los que se sospeche o se haya confirmado la presencia de anticuerpos neutralizantes contra eritropoyetina no deben ser tratados con NeoRecormon (ver sección 4.8).
APCR en pacientes con hepatitis C
Se debe interrumpir inmediatamente el tratamiento con epoetina, y realizar ensayos de anticuerpos antieritropoyetina si se produce una disminución paradójica de la hemoglobina y se desarrolla anemia severa asociada con bajos recuentos de reticulocitos. En pacientes con hepatitis C tratados con interferon y ribavirina se han notificados casos cuando las epoetinas se han utilizado concomitantemente. Las epoetinas no están aprobadas en el tratamiento de la anemia asociada a la hepatitis C.
Monitorización de la tensión arterial
Puede aparecer un aumento de la tensión arterial o un agravamiento de la hipertensión ya existente, especialmente en los casos en los que el hematocrito aumenta rápidamente. Estos incrementos en la tensión arterial pueden ser tratados con medicamentos. Si no pueden ser controlados con medicación se recomienda interrumpir transitoriamente la terapia con NeoRecormon. En particular al principio de la terapia se recomienda la monitorización regular de la tensión arterial; también durante la diálisis. Pueden producirse crisis hipertensivas con síntomas de encefalopatías y que requieren atención médica inmediata y cuidados médicos intensivos. Debe prestarse especial atención a las cefaleas súbitas lacerantes hemicraneales como posible signo de advertencia.
Insuficiencia renal crónica
En pacientes con insuficiencia renal crónica puede sobrevenir un aumento dosis-dependiente moderado en la cifra plaquetaria dentro del margen normal durante el tratamiento con NeoRecormon, especialmente después de la administración intravenosa. La regresión tiene lugar en el curso de la prosecución de la terapia. Se recomienda el control regular de la cifra de plaquetas durante las 8 primeras semanas de tratamiento.
Concentración de hemoglobina
En pacientes con insuficiencia renal crónica, la concentración de hemoglobina en la fase de mantenimiento no debe exceder el límite superior del rango recomendado en la sección 4.2. En ensayos clínicos se observó un aumento del riesgo de fallecimiento y de acontecimientos cardiovasculares graves o acontecimientos cerebrovasculares incluido el infarto, cuando se administraron agentes estimulantes de la eritropoyesis (AEEs) con el fin de alcanzar un nivel de hemoglobina superior a 12 g/dl (7,5 mmol/l).
En ensayos clínicos controlados no se han observado beneficios significativos atribuibles a la administración de epoetinas cuando se aumentaba la concentración de hemoglobina por encima del nivel necesario para controlar los síntomas de anemia y evitar las transfusiones sanguíneas.
En niños prematuros puede producirse un ligero aumento de la cifra plaquetaria, en especial hasta el día 12 -14 de vida, por lo tanto las plaquetas deben controlarse regularmente.
Efectos sobre el crecimiento tumoral
Las epoetinas son factores de crecimiento que estimulan de forma primaria la producción de glóbulos rojos. Los receptores de epoetina pueden expresarse sobre la superficie de una variedad de células tumorales. Como sucede con todos los factores de crecimiento, existe la preocupación de que las epoetinas pudieran estimular el crecimiento de tumores. En varios ensayos controlados, no se ha observado que las epoetinas mejoren la supervivencia global o que disminuyan el riesgo de progresión del tumor en pacientes con anemia asociada con cáncer.
En ensayos clínicos controlados, se ha observado que el uso de NeoRecormon y otros agentes estimulantes de la eritropoyesis (AEEs):
- acortaba el tiempo de progresión del tumor en pacientes con cáncer avanzado de cabeza y cuello que recibían radioterapia cuando se administraba para conseguir una concentración de hemoglobina por encima de 14 g/dl (8,7 mmol/l),
- acortaba la supervivencia global y aumentaba el número de fallecimientos atribuidos a la progresión de la enfermedad a los 4 meses, en pacientes con cáncer de mama metastásico que recibían quimioterapia cuando se administraba para conseguir una concentración de hemoglobina entre los 12 y los 14 g/dl (7,5-8,7 mmol/l),
- aumentaba el riesgo de fallecimiento cuando se administraba para conseguir una concentración de hemoglobina de 12 g/dl (7,5 mmol/l) en pacientes con enfermedad maligna activa y que no recibían ni quimioterapia ni radioterapia. El uso de los AEEs no está indicado en esa población de pacientes.
En vista de lo anterior, en algunas situaciones clínicas la transfusión sanguínea debe ser el tratamiento de elección para la anemia en pacientes con cáncer. La decisión de administrar eritropoyetinas recombinantes se tomará en base a la evaluación de la relación beneficio/riesgo junto con la aceptación individual del paciente, teniendo en cuenta el contexto clínico específico. Los factores que deben considerarse en esta evaluación son el tipo de tumor y su estadio, el grado de anemia, la esperanza de vida, el entorno en el que el paciente está siendo tratado y la preferencia del paciente (ver sección 5.1).
Puede haber un aumento de la tensión arterial que puede ser tratada con medicamentos. Se recomienda por tanto monitorizar la tensión arterial, en particular en la fase inicial de tratamiento en pacientes con cáncer.
En pacientes con cáncer también deberán monitorizarse regularmente el recuento plaquetario y el valor de hemoglobina.
En pacientes en programa de predonación de sangre autóloga, puede sobrevenir un aumento de la cifra de plaquetas, generalmente dentro de los límites normales. Por tanto, se recomienda la determinación de esta cifra al menos una vez por semana. Si se produce un aumento plaquetario de más de 150 x 109/l o si las plaquetas ascienden por encima del margen normal, se debe suspender el tratamiento con NeoRecormon.
En niños prematuros no se podría descartar el riesgo potencial de que la eritropoyetina provoque retinopatía, por lo tanto se debe tener precaución y la decisión de tratar a un niño prematuro se debe tomar teniendo en cuenta el beneficio potencial y el riesgo de este tratamiento, así como las opciones alternativas de tratamiento que estén disponibles.
En pacientes con insuficiencia renal crónica, a menudo se precisa aumentar la dosis de heparina durante la hemodiálisis en la terapia con NeoRecormon debido al aumento del volumen de hematocrito. Si la heparinización no es óptima es posible que se produzca la oclusión del sistema de diálisis.
En pacientes con alteración renal crónica con riesgo de trombosis de derivación, se debe considerar la revisión temprana de la derivación y profilaxis de la trombosis mediante la administración, por ejemplo, de ácido acetilsalicílico.
Los niveles séricos de potasio y fosfato deben ser controlados regularmente durante la terapia con NeoRecormon. Se ha informado de aumentos de potasio en unos pocos pacientes urémicos que recibieron NeoRecormon si bien no se ha establecido una relación causal. En caso de observar un nivel elevado o creciente de potasio se debe considerar la interrupción de la administración de NeoRecormon hasta que el nivel haya sido corregido.
Para el uso de NeoRecormon en un programa de predonación de sangre autóloga deben ser observadas las directrices oficiales sobre donación de sangre, en particular las siguientes:
- sólo deben donar los pacientes con hematocrito > 33 % (hemoglobina >11 g/dl [6,83 mmol/l]);
- debe procederse con especial cuidado con pacientes con un peso inferior a 50 kg;
- el volumen individual extraído no debe superar aproximadamente el 12 % del volumen sanguíneo estimado del paciente.
El tratamiento debe reservarse a pacientes en los que se considere particularmente importante evitar la transfusión de sangre homóloga teniendo en cuenta la evaluación riesgo/beneficio de las transfusiones homólogas.
Uso indebido
Un uso indebido del producto por personas sanas puede llevar a un aumento excesivo del volumen de hematocrito, lo cual puede asociarse con complicaciones del sistema cardiovascular con riesgo para la vida.
Excipientes
NeoRecormon en jeringa precargada contiene, como máximo, 0,3 mg de fenilalanina por jeringa como excipiente. Este hecho se deberá tener en cuenta en los pacientes afectados por formas graves de fenilcetonuria.
Este medicamento contiene menos de 1 mmol (23 mg) de sodio por jeringa, por lo que se considera esencialmente “exento de sodio”.
Trazabilidad de NeoRecormon
Para mejorar la trazabilidad de los agentes estimulantes de la eritropoyesis (AEEs), se debe registrar (o indicar) claramente el nombre comercial del AEE administrado en la historia clínica del paciente.
4.5 Interacción con otros medicamentos y otras formas de interacción
Los resultados clínicos obtenidos hasta el presente no ponen de manifiesto interacción alguna de NeoRecormon con otros medicamentos.
Los estudios llevados a cabo en animales mostraron que la epoetina beta no potencia la mielotoxicidad de los medicamentos citostáticos, tales como etopósido, cisplatino, ciclofosfamida y fluorouracilo.
4.6 Fertilidad, embarazo y lactancia
Fertilidad
Los estudios en animales no muestran efectos dañinos directos o indirectos sobre el embarazo, desarrollo embrional/fetal, parto o desarrollo postnatal (ver sección 5.3).
Embarazo
No hay datos relativos al uso de NeoRecormon en mujeres embarazadas.
Se debe tener precaución cuando se prescriba a mujeres embarazadas
Lactancia
Se desconoce si la epoetina beta se excreta en la leche materna. La decisión de continuar/interrumpir la lactancia o de continuar/interrumpir el tratamiento con epoetina beta se debe hacer teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento con epoetina beta para la madre.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NeoRecormon sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Basados en los resultados de los ensayos clínicos que incluyen 1.725 pacientes, se espera que aproximadamente el 8 % de los pacientes tratados con NeoRecormon sufran reacciones adversas.
Pacientes anémicos con insuficiencia renal crónica
La reacción adversa más frecuente durante el tratamiento con NeoRecormon es un aumento de la tensión arterial o agravamiento de la hipertensión existente, especialmente en casos de aumento rápido del hematocrito (ver sección 4.4). En algunos pacientes cuya tensión arterial es normal o baja también pueden darse crisis hipertensivas con síntomas de encefalopatías (p.ej. cefaleas y estados de confusión, trastornos sensitivomotores - como alteraciones del habla o de la ambulación - hasta convulsiones tónico-clónicas) (ver sección 4.4).
Puede darse trombosis de derivación, especialmente en pacientes que tienen tendencia a la hipotensión o cuyas fístulas arteriovenosas presentan complicaciones (p.ej. estenosis, aneurismas), ver sección 4.4. En la mayoría de los casos se observa una caída en los valores séricos de ferritina simultáneamente con el aumento del volumen de hematocrito (ver sección 4.4). Además, en casos aislados se han observado incrementos transitorios en los niveles séricos de potasio y fosfato (ver sección 4.4).
En casos aislados se ha notificado aplasia pura de células rojas (APCR) mediada por anticuerpos neutralizantes antieritropoyetina asociada al tratamiento con NeoRecormon. En caso de que se diagnostique APCR mediada por anticuerpos antieritropoyetina, el paciente deberá interrumpir el tratamiento con NeoRecormon, el cual no se deberá sustituir por otra proteína eritropoyética (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 1, que figura más adelante.
Pacientes con cáncer
El dolor de cabeza y la hipertensión relacionados con el tratamiento con epoetina beta son frecuentes y pueden tratarse con medicamentos (ver sección 4.4).
En algunos pacientes se observa un descenso de los valores de hierro sérico (ver sección 4.4).
Estudios clínicos han puesto de manifiesto que la frecuencia de acontecimientos tromboembólicos es más alta en pacientes con cáncer tratados con NeoRecormon que en los controles no tratados o tratados con placebo. En pacientes tratados con NeoRecormon, esta incidencia es del 7 % comparado con el 4 % en el grupo control; este hecho no está asociado con un incremento en la mortalidad tromboembólica comparado con los grupos control.
Estas reacciones adversas se enumeran en la Tabla 2, que figura más adelante.
Pacientes incluidos en un programa de predonación de sangre autóloga Los pacientes incluidos en un programa de predonación de sangre autóloga han demostrado una frecuencia ligeramente superior de accidentes tromboembólicos. Sin embargo, no se pudo establecer una relación causal con el tratamiento con NeoRecormon.
En ensayos clínicos controlados con placebo, la deficiencia transitoria de hierro fue más acusada en los pacientes tratados con NeoRecormon que en los pacientes control (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 3, que figura más adelante.
Tabla de reacciones adversas
Las reacciones adversas se enumeran de acuerdo con la clasificación de órganos del sistema MedDRA y por categoría de frecuencia.
Las categorías de frecuencia se definen utilizando la siguiente convención:
muy frecuentes (> 1/10), frecuentes (> 1/100 hasta <1/10), poco frecuentes (>1/1.000 hasta <1/100), raras (>1/10.000 hasta <1/1.000); muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 1: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con IRC_
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión Crisis hipertensivas |
Frecuente Poco frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
|
Trastornos de la sangre y |
Trombosis de derivación |
Raras |
|
del sistema linfático |
Trombocitosis |
Muy raras |
Tabla 2: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con cáncer__
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión |
Frecuente |
|
Trastornos de la sangre y del sistema linfático |
Acontecimientos tromboembólicos |
Frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Tabla 3: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes incluidos en un programa de predonación de sangre autóloga
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Prematuros
Es muy frecuente un descenso de los valores de ferritina sérica (ver sección 4.4).
Descripción de reacciones adversas seleccionadas
Raramente pueden ocurrir reacciones cutáneas relacionadas con el tratamiento con epoetina beta como sarpullido, prurito, urticaria o reacciones en el lugar de la inyección. En casos muy raros se han notificado reacciones anafilácticas relacionadas con el tratamiento con epoetina beta. Sin embargo, en los ensayos clínicos controlados no se encontró una mayor incidencia de reacciones de hipersensibilidad.
Se han comunicado casos muy raros de síntomas gripales relacionados con el tratamiento con epoetina beta como fiebre, escalofríos, dolores de cabeza, dolor de las extremidades, malestar general, y/o dolor óseo, particularmente al inicio del tratamiento. Estas reacciones son de intensidad leve a moderada y desaparecen al cabo de un par de horas o días.
Los resultados obtenidos en un ensayo clínico controlado realizado con epoetina alfa o darbepoetina alfa, notificaron que la incidencia de infarto es frecuente.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
El margen terapéutico de NeoRecormon es muy amplio. No se han observado síntomas de sobredosis incluso a niveles séricos muy elevados.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antianémico, código ATC: B03XA01 Mecanismo de acción
La eritropoyetina es una glucoproteína que estimula la formación de eritrocitos a partir de sus progenitores asignados. Actúa como factor estimulante de la mitosis y hormona de diferenciación.
La epoetina beta, el principio activo de NeoRecormon, es idéntica en su composición aminoácida e hidrocarbonada a la eritropoyetina aislada de la orina de pacientes anémicos.
La eficacia biológica de la epoetina beta ha sido demostrada tras administración intravenosa y subcutánea en varios modelos animales in vivo (ratas normales y urémicas, ratones policitémicos, perros). Con la administración de epoetina beta aumentan el número de eritrocitos, los valores de Hb y la cifra de reticulocitos, al igual que la velocidad de incorporación de 59Fe.
In vitro (cultivo citológico de bazo de ratón) ha sido hallada una mayor incorporación de 3H-timidina en las células eritroides nucleadas del bazo tras incubación con epoetina beta.
Las investigaciones en cultivos de células de la médula ósea humana han revelado que la epoetina beta estimula específicamente la eritropoyesis y no afecta a la leucopoyesis. No han sido detectadas acciones citotóxicas de la epoetina beta sobre la médula ósea o sobre células cutáneas humanas.
Tras una dosis única de epoetina beta no se ha observado ningún efecto en el comportamiento o la actividad locomotora del ratón ni en las funciones circulatoria o respiratoria del perro.
Eficacia clínica y seguridad
En un ensayo aleatorizado, doble ciego, controlado con placebo, realizado en 4.038 pacientes con enfermedad renal crónica no sometidos a diálisis, con una diabetes tipo 2 y los niveles de hemoglobina < 11 g/dl, los pacientes recibieron tratamiento bien con darbepoetina alfa para alcanzar niveles de hemoglobina de 13 g/dl o con placebo (ver sección 4.4). El ensayo no cumplió su objetivo principal de demostrar una disminución en el riesgo de todas las causas de mortalidad, la morbilidad cardiovascular o enfermedad renal terminal. El análisis de los componentes individuales de la variable combinada mostró los siguientes HR (IC 95%): fallecimiento 1,05 (0,92-1,21), infarto cerebrovascular 1,92 (1,38-2,68), insuficiencia cardíaca congestiva 0,89 (0,74-1,08), infarto de miocardio 0,96 (0,751,23), hospitalización por isquemia del miocardio 0,84 (0,55-1,27), enfermedad renal terminal 1,02 (0,87-1,18).
En pacientes con IRC (tratados con diálisis, no tratados con diálisis, pacientes diabéticos y pacientes no diabéticos) se han realizado análisis agrupados post hoc de los estudios clínicos con AEEs. Se observó una tendencia a un aumento de las estimaciones del riesgo para la mortalidad por todas las causas y para los acontecimientos cardiovasculares y cerebrovasculares asociados a dosis acumuladas más altas de AEE independientemente de que los pacientes padecieran o no diabetes y de que recibieran o no tratamiento con diálisis (ver las secciones 4.2 y 4.4).
La eritropoyetina es un factor de crecimiento que estimula de forma primaria la producción de glóbulos rojos. Los receptores de eritropoyetina se encuentran también presentes en la superficie de algunas líneas celulares malignas.
Se ha estudiado la supervivencia y la progresión tumoral en cinco grandes ensayos controlados que incluyeron a 2.833 pacientes, de los cuales cuatro fueron ensayos doble-ciego controlados con placebo y uno fue un ensayo abierto. Dos de los ensayos reclutaron pacientes que estaban siendo tratados con quimioterapia. El nivel de hemoglobina que se quería alcanzar fue >13 g/dl en dos de los ensayos y de entre 12 y 14 g/dl en los otros tres. En el ensayo abierto no se observaron diferencias en la supervivencia global, entre los pacientes tratados con eritropoyetina humana recombinante y el grupo control. En los cuatro ensayos controlados con placebo, el índice de riesgo (hazard ratio) para la supervivencia global osciló entre 1,25 y 2,47 en favor de los grupos control. En todos estos ensayos se ha observado un aumento de la mortalidad inexplicable y estadísticamente significativo en pacientes que presentaban anemia asociada con diversos tipos frecuentes de cáncer y que recibieron eritropoyetina humana recombinante, en comparación con los grupos control. Las diferencias observadas en la incidencia de trombosis y complicaciones relacionadas, entre los pacientes que recibieron eritropoyetina humana recombinante y los que formaban parte de los grupos control, no permiten explicar satisfactoriamente los resultados de supervivencia global observados en los ensayos.
Un meta-análisis basado en datos individuales de pacientes, que incluyó datos de los 12 ensayos clínicos controlados realizados con NeoRecormon en pacientes anémicos con cáncer (n=2301), mostró un valor estimado del índice de riesgo (hazard ratio) de supervivencia global de 1,13 en favor de los grupos control (IC 95 %, CI 0,87, 1,46). En pacientes con un valor inicial de hemoglobina < 10 g/dl (n=899), el valor estimado del índice de riesgo (hazard ratio) para la supervivencia fue de 0,98 (IC 95% 0,68 al 1,40). Se observó un aumento del riesgo relativo de acontecimientos tromboembólicos en la población general (RR 1,62, IC 95 %: 1,13, 2,31).
Asimismo, se ha realizado un análisis de datos a nivel de paciente, en más de 13.900 pacientes con cáncer (tratados con quimioterapia, radioterapia, quimioradioterapia o sin tratamiento) que participaban en un total de 53 ensayos clínicos controlados en los que se administraban diversas epoetinas. En este meta-análisis se obtuvo un índice de riesgo (hazard ratio) para la supervivencia global de 1,06 a favor de los grupos control (IC 95%: 1,00, 1,12; 53 ensayos y 13.933 pacientes) y para los pacientes con cáncer que recibían quimioterapia, el índice de riesgo (hazard ratio) para la supervivencia global fue de 1,04 (IC 95%: 0,97, 1,11; 38 ensayos y 10.441 pacientes). Este meta-análisis además indica de forma consistente un aumento significativo del riesgo relativo de episodios tromboembólicos en pacientes con cáncer tratados con eritropoyetina recombinante humana (ver sección 4.4).
En casos muy raros, se han producido anticuerpos neutralizantes anti-eritropoyetina con o sin aplasia pura de células rojas (APCR) durante el tratamiento con rHuEPO.
5.2 Propiedades farmacocinéticas
Los estudios farmacocinéticos en voluntarios sanos y pacientes urémicos muestran que la semivida de la epoetina beta administrada por vía i.v. es de 4 a 12 horas y que el volumen de distribución corresponde de una a dos veces el volumen plasmático. Se han hallado resultados análogos en experimentos en animales con ratas normales y urémicas.
Tras la administración subcutánea de epoetina beta a pacientes urémicos la absorción retardada se traduce en una meseta de concentración sérica, cuya cota máxima se alcanza al cabo de 12 - 28 horas de media. El semiperíodo de vida terminal es mayor que después de la administración intravenosa, siendo la media de 13-28 horas.
La biodisponibilidad de la epoetina beta después de la administración subcutánea está comprendida entre el 23 y el 42 % en comparación con la administración intravenosa.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad de dosis repetidas, genotoxicidad y toxicidad para la reproducción.
Un estudio de carcinogenicidad con homólogos de eritropoyetina en ratones no reveló ningún signo de potencial proliferativo o carcinogénico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Urea,
Cloruro sódico,
Polisorbato 20,
Fosfato monosódico dihidratado,
Fosfato disódico dodecahidratado,
Cloruro cálcico dihidratado,
Glicina,
L-Leucina,
L-Isoleucina,
L-Treonina,
L-Ácido glutámico,
L-Fenilalanina,
Agua para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
Conservar la jeringa precargada en el embalaje exterior, para protegerlo de la luz.
Con fines de uso ambulatorio, se puede mantener el producto fuera de la nevera durante un único periodo de hasta 3 días a temperatura ambiente (no superior a 25°C).
6.5 Naturaleza y contenido del envase
Jeringa precargada (vidrio tipo I), con un protector y un émbolo (goma teflonizada) con una aguja de inyección (27G1/2). Cada jeringa contiene 0,3 ml de solución.
Tamaños de envase de 1 jeringa precargada y 1 aguja o de 6 jeringas precargadas y 6 agujas.
Puede que solamente estén comercializados algunos tamaños de envase.
6.6 Precauciones especiales de eliminación y otras manipulaciones
En primer lugar, ¡lávese las manos!
1. S aque una jeringa del envase y asegúrese que la solución es clara, incolora y prácticamente libre de partículas visibles. Retire el protector de la jeringa.
2. Saque una aguja del envase, fíjela sobre la jeringa y retire la tapa protectora de la aguja.
3. Expulse el aire de la jeringa y aguja manteniendo la jeringa vertical y presionando suavemente el émbolo hacia arriba. Continúe presionando el émbolo hasta que la cantidad de solución de NeoRecormon en la jeringa sea la prescrita.
4. Limpie la piel en el lugar de inyección con un algodón con alcohol. Haga un pliegue tomando la piel entre el índice y el pulgar. Sujete el cuerpo de la jeringa por la parte más próxima a la aguja e inserte la aguja en el pliegue cutáneo con acción firme y rápida. Inyecte la solución de NeoRecormon. Retire la aguja rápidamente y aplique presión sobre el lugar de inyección con un apósito seco y estéril.
Este medicamento es solo para uso individual. La eliminación del medicamento no utilizado y de
todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa
local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/043 -044
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16 de julio de 1997 Fecha de la última revalidación: 16 de julio de 2007
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 10.000 UI solución inyectable en jeringa precargada
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Una jeringa precargada con 0,6 ml de solución para inyección contiene 10.000 unidades internacionales (UI) correspondientes a 83 microgramos de epoetina beta* (eritropoyetina recombinante humana).
Un ml de solución para inyección contiene 16.667 UI de epoetina beta
* producida en células de ovarios de hámster chino (CHO) mediante tecnología de ADN recombinante.
Excipientes con efecto conocido:
Fenilalanina (hasta 0,3 mg/jeringa).
Sodio (menos de 1 mmol/jeringa)
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución incolora, de transparente a ligeramente opalescente.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
NeoRecormon está indicado para:
- Tratamiento de la anemia sintomática asociada a la insuficiencia renal crónica en pacientes adultos y pediátricos.
- Prevención de la anemia en prematuros con un peso corporal al nacer de 750 a 1.500 g y una edad gestacional de menos de 34 semanas.
- Tratamiento de la anemia sintomática en pacientes adultos con neoplasias no mieloides tratados con quimioterapia.
- Aumentar el rendimiento de la sangre autóloga de pacientes incluidos en un programa de predonación.
Debe sopesarse su uso en esta indicación frente al riesgo aumentado de episodios tromboembólicos que han sido notificados. Sólo debe administrarse el tratamiento a pacientes con anemia moderada (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], sin deficiencia de hierro) si no se dispone de procedimientos para conservar la sangre o si éstos son insuficientes cuando la cirugía mayor electiva programada requiera un gran volumen de sangre (4 o más unidades de sangre en mujeres o 5 o más unidades en hombres). Ver sección 5.1.
4.2 Posología y forma de administración
La terapia con NeoRecormon debe iniciarse por médicos con experiencia en las indicaciones arriba
mencionadas. Como se han observado reacciones anafilácticas en algunos casos aislados, se
recomienda administrar la primera dosis bajo control médico.
Posología
Tratamiento de la anemia sintomática en pacientes adultos y pediátricos con insuficiencia renal crónica
Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente. NeoRecormon se puede administrar tanto por vía subcutánea como intravenosa con el fin de aumentar la concentración de hemoglobina hasta un nivel no superior a 12 g/dl (7,5 mmol/l). En pacientes que no están sometidos a hemodiálisis es preferible utilizar la vía subcutánea para evitar la punción de venas periféricas. En caso de administración intravenosa, la solución debe ser inyectada a lo largo de unos 2 minutos, p.ej. en pacientes en hemodiálisis por vía de la fístula arteriovenosa al final de la diálisis.
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones se pueden observar valores individuales de hemoglobina superiores o inferiores a los niveles deseados. La variabilidad en los niveles de hemoglobina se debe controlar mediante ajuste de la dosis con el objeto de mantener los valores de hemoglobina dentro del intervalo entre 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Se debe evitar un nivel de hemoglobina de forma continuada por encima de 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando los valores de hemoglobina sean superiores a 12 g/dl (7,5 mmol/l).
Debe evitarse un aumento de hemoglobina superior a 2 g/dl (1,25 mmol/l) durante un periodo de cuatro semanas. Si esto ocurre, se debe hacer un ajuste adecuado de la dosis según las instrucciones incluidas en esta misma sección. Si la tasa de aumento de hemoglobina es mayor de 2 g/dl (1,25 mmol/l) en un mes o si el nivel de hemoglobina está aumentando y se acerca a 12 g/dl (7,45 mmol/l), debe reducirse la dosis en aproximadamente un 25%. Si el nivel de hemoglobina sigue aumentando, se debe interrumpir el tratamiento hasta que el nivel de hemoglobina empiece a disminuir, momento en el que se podrá reiniciar el tratamiento con una dosis aproximadamente un 25 % inferior a la dosis administrada previamente.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis eficaz más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia al tiempo que se mantiene una concentración de hemoglobina inferior o igual a 12 g/dl (7,45 mmol/l).
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica. En los pacientes con una respuesta deficiente de la hemoglobina a NeoRecormon, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.4 y 5.1).
En presencia de hipertensión o de enfermedades cardiovasculares, cerebrovasculares o vasculoperiféricas, se debe determinar de modo individual el incremento semanal y el valor objetivo de la hemoglobina teniendo en cuenta el cuadro clínico.
El tratamiento con NeoRecormon se divide en dos fases:
1. Fase de corrección
- Administración subcutánea:
La dosis inicial es 3 x 20 UI/kg por semana. La dosis puede incrementarse cada 4 semanas en 3 x 20 UI/kg y semana si el aumento de la hemoglobina no ha sido adecuado (< 0,25 g/dl por semana).
La dosis semanal puede dividirse en dosis diarias.
- Administración intravenosa:
La dosis inicial es 3 x 40 UI/kg por semana, y puede aumentarse al cabo de 4 semanas a 80 UI/kg -tres veces por semana- y si son necesarios incrementos ulteriores serán de 20 UI/kg tres veces por semana, con intervalos mensuales.
Por ambas vías de administración, la dosis máxima no debe superar 720 UI/kg por semana.
2. Fase de mantenimiento
Para mantener el nivel de hemoglobina entre 10 y 12 g/dl, la dosis es inicialmente reducida a la mitad de la cantidad previamente administrada. Posteriormente, se ajustará la dosis individualmente para el paciente a intervalos de una o dos semanas (dosis de mantenimiento).
En caso de administración subcutánea, la dosis semanal puede administrarse en una inyección única o fraccionada en tres o siete inyecciones. Los pacientes que permanezcan estables en el régimen de una dosis única semanal pueden pasar a una administración única cada dos semanas. En este caso, puede ser necesario un aumento de la dosis.
Los resultados de los estudios clínicos en niños han revelado que, en general, a menor edad, se necesita mayor dosis de NeoRecormon. No obstante, hay que seguir el programa posológico recomendado ya que no puede predecirse la respuesta individual.
El tratamiento con NeoRecormon es normalmente crónico. Sin embargo, en caso necesario puede interrumpirse en cualquier momento. Los datos sobre la pauta posológica de una vez a la semana se han obtenido de estudios clínicos con una duración de tratamiento de 24 semanas.
Prevención de anemia en prematuros
La solución se administra por vía subcutánea a una dosis de 3 x 250 UI/kg p.c. por semana. Es probable que los niños que ya hayan recibido una transfusión previa cuando se inicie el tratamiento con NeoRecormon no se beneficien tanto como los prematuros no transfundidos. La duración de tratamiento recomendada es de 6 semanas.
Tratamiento de anemia sintomática inducida por quimioterapia en pacientes con cáncer Se debe administrar NeoRecormon por vía subcutánea a pacientes con anemia (por ejemplo, si la concentración de hemoglobina es menor o igual a 10 g/dl [6,2 mmol/l]). Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente.
La dosis semanal puede administrarse como una inyección por semana o en dosis divididas entre 3 y 7 veces por semana.
La dosis inicial recomendada es de 30.000 UI por semana (que corresponde aproximadamente a 450 UI/kg de peso corporal por semana, en base al peso medio del paciente).
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones es posible llegar a observar en un paciente valores de hemoglobina superiores o inferiores a los esperados. La variabilidad de la hemoglobina se deberá manejar ajustando la dosis para mantener los valores de la hemoglobina dentro del intervalo de 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Deben evitarse concentraciones sostenidas de hemoglobina superiores a 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando se observen valores de hemoglobina superiores a los 12 g/dl (7,5 mmol/l).
Si a las 4 semanas de tratamiento el valor de hemoglobina aumenta hasta al menos 1 g/dl (0,62 mmol/l), se debe continuar con la dosis que en ese momento se esté administrando. Si los valores de hemoglobina no han aumentado al menos 1 g/dl (0,62 mmol/l), se debe considerar duplicar la dosis semanal. Si a las 8 semanas de tratamiento el valor de hemoglobina no aumenta hasta al menos 1 g/dl (0,62 mmol/l), es improbable que se produzca respuesta y, por consiguiente, el tratamiento debe ser interrumpido.
El tratamiento debe continuar durante las 4 semanas posteriores al final de la quimioterapia.
La dosis máxima no debe exceder de 60.000 UI por semana.
Una vez se ha alcanzado el objetivo terapéutico del paciente, la dosis debe reducirse del 25 al 50 % con el fin de mantener el valor de hemoglobina en ese nivel. Debe realizarse el ajuste posológico adecuado.
Si el nivel de hemoglobina excede los 12 g/dl (7,5 mmol/l) se debe reducir la dosis entre un 25 y un
50 % aproximadamente. Si el nivel de hemoglobina excede de 13 g/dl (8,1 mmol), se debe interrumpir temporalmente el tratamiento con NeoRecormon. El tratamiento debe reiniciarse con una dosis aproximadamente un 25 % inferior a la dosis previamente administrada después de que los niveles de hemoglobina desciendan hasta un valor menor o igual a 12 g/dl (7,5 mmol/l) o por debajo.
51 al cabo de 4 semanas el incremento de hemoglobina es superior a 2 g/dl (1,3 mmol/l), la dosis debe reducirse entre un 25 y un 50 %.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia.
Tratamiento para incrementar el rendimiento de la sangre autóloga donada La solución puede administrarse por vía intravenosa, en unos 2 minutos, o subcutáneamente. NeoRecormon se administra dos veces por semana durante 4 semanas. En aquellas ocasiones en que el hematocrito del paciente permite la donación de sangre, es decir el hematocrito es > 33 %, NeoRecormon se administra al final de la donación de sangre.
Durante la totalidad del período de tratamiento no debe excederse un hematocrito del 48 %.
La dosis debe ser determinada por el equipo quirúrgico, individualmente para cada paciente, en función de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos:
1. La cantidad de sangre pre-donada necesaria dependerá de la pérdida prevista de sangre, de los medios de conservación empleados y del estado físico del paciente.
Este volumen debe ser equivalente a la cantidad de sangre que se considera necesaria para evitar transfusiones de sangre homóloga.
La cantidad de sangre pre-donada necesaria se expresa en unidades en las cuales una unidad de nomograma es equivalente a 180 ml de glóbulos rojos.
2. La capacidad de donar sangre depende predominantemente del volumen sanguíneo del paciente y del hematocrito basal. Ambas variables determinan la reserva endógena de eritrocitos, que puede ser calculada conforme a la fórmula siguiente:
Reserva endógena de eritrocitos = volumen sanguíneo (ml) x (hematocrito - 33)/ 100 mujeres: volumen sanguíneo (ml) = 41 (ml/kg) x peso corporal (kg) + 1200 (ml)
hombres: volumen sanguíneo (ml) = 44 (ml/kg) x peso corporal (kg) + 1600 (ml)
(peso corporal: > 45 kg)
La indicación para un tratamiento con NeoRecormon y, en su caso, la dosis individual debe ser determinada a partir de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos según las gráficas siguientes:
Paciente femenino Paciente masculino
Cantidad de sangre pre-donada Cantidad de sangre pre-donada
necesaria [unidades] necesaria [unidades]


O 100 200 300 400 500 600 700 800 0 100 200 300 400 500 600 700 800
Reserva eritrocitaria endógena [ml] Reserva eritrocitaria endógena [ml]
La dosis única así determinada debe administrarse dos veces por semana a lo largo de 4 semanas. La dosis máxima no debe sobrepasar 1.600 UI/kg peso corporal por semana en administración intravenosa ó 1.200 UI/kg por semana en administración subcutánea.
Forma de administración
La jeringa precargada de NeoRecormon está lista para su uso. Sólo se pueden inyectar soluciones claras o ligeramente opalescentes, incoloras y prácticamente libres de partículas visibles. NeoRecormon en jeringa precargada es un producto estéril pero sin conservantes. Bajo ninguna circunstancia debe administrarse más de una dosis por jeringa; el medicamento es solo para uso individual.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Hipertensión mal controlada.
En la indicación para “Aumentar el rendimiento de sangre autóloga”: infarto de miocardio o accidente cerebrovascular en el mes anterior al tratamiento, angina de pecho inestable, riesgo aumentado de trombosis venosa profunda, como por ejemplo en aquellos con historial de enfermedad tromboembólica venosa.
4.4 Advertencias y precauciones especiales de empleo
NeoRecormon debe usarse con cautela en presencia de anemia refractaria con exceso de blastos en transformación, epilepsia, trombocitosis e insuficiencia hepática crónica. Y deben excluirse las deficiencias de ácido fólico y vitamina B12 pues reducen la eficacia de NeoRecormon.
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica, ya que las dosis acumuladas altas de epoetina pueden asociarse a un mayor riesgo de mortalidad, y de acontecimientos cardiovasculares y cerebrovasculares graves. En pacientes con una respuesta deficiente de la hemoglobina a epoetinas, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.2 y 5.1).
Para garantizar una eritropoyesis eficaz, se debe evaluar el nivel de hierro en todos los pacientes antes y durante el tratamiento, y puede ser necesario un tratamiento suplementario con hierro administrado de acuerdo con las guías terapéuticas.
La sobrecarga grave de aluminio debido al tratamiento de la insuficiencia renal puede comprometer la eficacia de NeoRecormon.
La indicación de tratamiento con NeoRecormon en enfermos nefroscleróticos aún no sometidos a diálisis debe definirse individualmente, ya que la posible aceleración de la insuficiencia renal no puede descartarse con certeza.
Aplasia pura de células rojas (APCR)
Se han notificado casos de APCR causada por anticuerpos neutralizantes antieritropoyetina, asociados a tratamientos con eritropoyetinas, incluido NeoRecormon. Estos anticuerpos han presentado reacción cruzada con todas las proteínas eritropoyéticas, por los que los pacientes en los que se sospeche o se haya confirmado la presencia de anticuerpos neutralizantes contra eritropoyetina no deben ser tratados con NeoRecormon (ver sección 4.8).
APCR en pacientes con hepatitis C
Se debe interrumpir inmediatamente el tratamiento con epoetina, y realizar ensayos de anticuerpos antieritropoyetina si se produce una disminución paradójica de la hemoglobina y se desarrolla anemia severa asociada con bajos recuentos de reticulocitos. En pacientes con hepatitis C tratados con interferon y ribavirina se han notificados casos cuando las epoetinas se han utilizado concomitantemente. Las epoetinas no están aprobadas en el tratamiento de la anemia asociada a la hepatitis C.
Monitorización de la tensión arterial
Puede aparecer un aumento de la tensión arterial o un agravamiento de la hipertensión ya existente, especialmente en los casos en los que el hematocrito aumenta rápidamente. Estos incrementos en la tensión arterial pueden ser tratados con medicamentos. Si no pueden ser controlados con medicación se recomienda interrumpir transitoriamente la terapia con NeoRecormon. En particular al principio de la terapia se recomienda la monitorización regular de la tensión arterial; también durante la diálisis. Pueden producirse crisis hipertensivas con síntomas de encefalopatías y que requieren atención médica inmediata y cuidados médicos intensivos. Debe prestarse especial atención a las cefaleas súbitas lacerantes hemicraneales como posible signo de advertencia.
Insuficiencia renal crónica
En pacientes con insuficiencia renal crónica puede sobrevenir un aumento dosis-dependiente moderado en la cifra plaquetaria dentro del margen normal durante el tratamiento con NeoRecormon, especialmente después de la administración intravenosa. La regresión tiene lugar en el curso de la prosecución de la terapia. Se recomienda el control regular de la cifra de plaquetas durante las 8 primeras semanas de tratamiento.
Concentración de hemoglobina
En pacientes con insuficiencia renal crónica, la concentración de hemoglobina en la fase de mantenimiento no debe exceder el límite superior del rango recomendado en la sección 4.2. En ensayos clínicos se observó un aumento del riesgo de fallecimiento y de acontecimientos cardiovasculares graves o acontecimientos cerebrovasculares incluido el infarto, cuando se administraron agentes estimulantes de la eritropoyesis (AEEs) con el fin de alcanzar un nivel de hemoglobina superior a 12 g/dl (7,5 mmol/l).
En ensayos clínicos controlados no se han observado beneficios significativos atribuibles a la administración de epoetinas cuando se aumentaba la concentración de hemoglobina por encima del nivel necesario para controlar los síntomas de anemia y evitar las transfusiones sanguíneas.
En niños prematuros puede producirse un ligero aumento de la cifra plaquetaria, en especial hasta el día 12 -14 de vida, por lo tanto las plaquetas deben controlarse regularmente.
Efectos sobre el crecimiento tumoral
Las epoetinas son factores de crecimiento que estimulan de forma primaria la producción de glóbulos rojos. Los receptores de epoetina pueden expresarse sobre la superficie de una variedad de células tumorales. Como sucede con todos los factores de crecimiento, existe la preocupación de que las epoetinas pudieran estimular el crecimiento de tumores. En varios ensayos controlados, no se ha observado que las epoetinas mejoren la supervivencia global o que disminuyan el riesgo de progresión del tumor en pacientes con anemia asociada con cáncer.
En ensayos clínicos controlados, se ha observado que el uso de NeoRecormon y otros agentes estimulantes de la eritropoyesis (AEEs):
- acortaba el tiempo de progresión del tumor en pacientes con cáncer avanzado de cabeza y cuello que recibían radioterapia cuando se administraba para conseguir una concentración de hemoglobina por encima de 14 g/dl (8,7 mmol/l),
- acortaba la supervivencia global y aumentaba el número de fallecimientos atribuidos a la progresión de la enfermedad a los 4 meses, en pacientes con cáncer de mama metastásico que recibían quimioterapia cuando se administraba para conseguir una concentración de hemoglobina entre los 12 y los 14 g/dl (7,5-8,7 mmol/l),
- aumentaba el riesgo de fallecimiento cuando se administraba para conseguir una concentración de hemoglobina de 12 g/dl (7,5 mmol/l) en pacientes con enfermedad maligna activa y que no recibían ni quimioterapia ni radioterapia. El uso de los AEEs no está indicado en esa población de pacientes.
En vista de lo anterior, en algunas situaciones clínicas la transfusión sanguínea debe ser el tratamiento de elección para la anemia en pacientes con cáncer. La decisión de administrar eritropoyetinas recombinantes se tomará en base a la evaluación de la relación beneficio/riesgo junto con la aceptación individual del paciente, teniendo en cuenta el contexto clínico específico. Los factores que deben considerarse en esta evaluación son el tipo de tumor y su estadio, el grado de anemia, la esperanza de vida, el entorno en el que el paciente está siendo tratado y la preferencia del paciente (ver sección 5.1).
Puede haber un aumento de la tensión arterial que puede ser tratada con medicamentos. Se recomienda por tanto monitorizar la tensión arterial, en particular en la fase inicial de tratamiento en pacientes con cáncer.
En pacientes con cáncer también deberán monitorizarse regularmente el recuento plaquetario y el valor de hemoglobina.
En pacientes en programa de predonación de sangre autóloga, puede sobrevenir un aumento de la cifra de plaquetas, generalmente dentro de los límites normales. Por tanto, se recomienda la determinación de esta cifra al menos una vez por semana. Si se produce un aumento plaquetario de más de 150 x 109/l o si las plaquetas ascienden por encima del margen normal, se debe suspender el tratamiento con NeoRecormon.
En niños prematuros no se podría descartar el riesgo potencial de que la eritropoyetina provoque retinopatía, por lo tanto se debe tener precaución y la decisión de tratar a un niño prematuro se debe tomar teniendo en cuenta el beneficio potencial y el riesgo de este tratamiento, así como las opciones alternativas de tratamiento que estén disponibles.
En pacientes con insuficiencia renal crónica, a menudo se precisa aumentar la dosis de heparina durante la hemodiálisis en la terapia con NeoRecormon debido al aumento del volumen de hematocrito. Si la heparinización no es óptima es posible que se produzca la oclusión del sistema de diálisis.
En pacientes con alteración renal crónica con riesgo de trombosis de derivación, se debe considerar la revisión temprana de la derivación y profilaxis de la trombosis mediante la administración, por ejemplo, de ácido acetilsalicílico.
Los niveles séricos de potasio y fosfato deben ser controlados regularmente durante la terapia con NeoRecormon. Se ha informado de aumentos de potasio en unos pocos pacientes urémicos que recibieron NeoRecormon si bien no se ha establecido una relación causal. En caso de observar un nivel elevado o creciente de potasio se debe considerar la interrupción de la administración de NeoRecormon hasta que el nivel haya sido corregido.
Para el uso de NeoRecormon en un programa de predonación de sangre autóloga deben ser observadas las directrices oficiales sobre donación de sangre, en particular las siguientes:
- sólo deben donar los pacientes con hematocrito > 33 % (hemoglobina >11 g/dl [6,83 mmol/l]);
- debe procederse con especial cuidado con pacientes con un peso inferior a 50 kg;
- el volumen individual extraído no debe superar aproximadamente el 12 % del volumen sanguíneo estimado del paciente.
El tratamiento debe reservarse a pacientes en los que se considere particularmente importante evitar la transfusión de sangre homóloga teniendo en cuenta la evaluación riesgo/beneficio de las transfusiones homólogas.
Uso indebido
Un uso indebido del producto por personas sanas puede llevar a un aumento excesivo del volumen de hematocrito, lo cual puede asociarse con complicaciones del sistema cardiovascular con riesgo para la vida.
Excipientes
NeoRecormon en jeringa precargada contiene, como máximo, 0,3 mg de fenilalanina por jeringa como excipiente. Este hecho se deberá tener en cuenta en los pacientes afectados por formas graves de fenilcetonuria.
Este medicamento contiene menos de 1 mmol (23 mg) de sodio por jeringa, por lo que se considera esencialmente “exento de sodio”.
Trazabilidad de NeoRecormon
Para mejorar la trazabilidad de los agentes estimulantes de la eritropoyesis (AEEs), se debe registrar (o indicar) claramente el nombre comercial del AEE administrado en la historia clínica del paciente.
4.5 Interacción con otros medicamentos y otras formas de interacción
Los resultados clínicos obtenidos hasta el presente no ponen de manifiesto interacción alguna de NeoRecormon con otros medicamentos.
Los estudios llevados a cabo en animales mostraron que la epoetina beta no potencia la mielotoxicidad de los medicamentos citostáticos, tales como etopósido, cisplatino, ciclofosfamida y fluorouracilo.
4.6 Fertilidad, embarazo y lactancia
Fertilidad
Los estudios en animales no muestran efectos dañinos directos o indirectos sobre el embarazo, desarrollo embrional/fetal, parto o desarrollo postnatal (ver sección 5.3).
Embarazo
No hay datos relativos al uso de NeoRecormon en mujeres embarazadas.
Se debe tener precaución cuando se prescriba a mujeres embarazadas
Lactancia
Se desconoce si la epoetina beta se excreta en la leche materna. La decisión de continuar/interrumpir la lactancia o de continuar/interrumpir el tratamiento con epoetina beta se debe hacer teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento con epoetina beta para la madre.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NeoRecormon sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Basados en los resultados de los ensayos clínicos que incluyen 1.725 pacientes, se espera que aproximadamente el 8 % de los pacientes tratados con NeoRecormon sufran reacciones adversas.
Pacientes anémicos con insuficiencia renal crónica
La reacción adversa más frecuente durante el tratamiento con NeoRecormon es un aumento de la tensión arterial o agravamiento de la hipertensión existente, especialmente en casos de aumento rápido del hematocrito (ver sección 4.4). En algunos pacientes cuya tensión arterial es normal o baja también pueden darse crisis hipertensivas con síntomas de encefalopatías (p.ej. cefaleas y estados de confusión, trastornos sensitivomotores - como alteraciones del habla o de la ambulación - hasta convulsiones tónico-clónicas) (ver sección 4.4).
Puede darse trombosis de derivación, especialmente en pacientes que tienen tendencia a la hipotensión o cuyas fístulas arteriovenosas presentan complicaciones (p.ej. estenosis, aneurismas), ver sección 4.4. En la mayoría de los casos se observa una caída en los valores séricos de ferritina simultáneamente con el aumento del volumen de hematocrito (ver sección 4.4). Además, en casos aislados se han observado incrementos transitorios en los niveles séricos de potasio y fosfato (ver sección 4.4).
En casos aislados se ha notificado aplasia pura de células rojas (APCR) mediada por anticuerpos neutralizantes antieritropoyetina asociada al tratamiento con NeoRecormon. En caso de que se diagnostique APCR mediada por anticuerpos antieritropoyetina, el paciente deberá interrumpir el tratamiento con NeoRecormon, el cual no se deberá sustituir por otra proteína eritropoyética (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 1, que figura más adelante.
Pacientes con cáncer
El dolor de cabeza y la hipertensión relacionados con el tratamiento con epoetina beta son frecuentes y pueden tratarse con medicamentos (ver sección 4.4).
En algunos pacientes se observa un descenso de los valores de hierro sérico (ver sección 4.4).
Estudios clínicos han puesto de manifiesto que la frecuencia de acontecimientos tromboembólicos es más alta en pacientes con cáncer tratados con NeoRecormon que en los controles no tratados o tratados con placebo. En pacientes tratados con NeoRecormon, esta incidencia es del 7 % comparado con el 4 % en el grupo control; este hecho no está asociado con un incremento en la mortalidad tromboembólica comparado con los grupos control.
Estas reacciones adversas se enumeran en la Tabla 2, que figura más adelante.
Pacientes incluidos en un programa de predonación de sangre autóloga Los pacientes incluidos en un programa de predonación de sangre autóloga han demostrado una frecuencia ligeramente superior de accidentes tromboembólicos. Sin embargo, no se pudo establecer una relación causal con el tratamiento con NeoRecormon.
En ensayos clínicos controlados con placebo, la deficiencia transitoria de hierro fue más acusada en los pacientes tratados con NeoRecormon que en los pacientes control (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 3, que figura más adelante.
Tabla de reacciones adversas
Las reacciones adversas se enumeran de acuerdo con la clasificación de órganos del sistema MedDRA y por categoría de frecuencia.
Las categorías de frecuencia se definen utilizando la siguiente convención:
muy frecuentes (> 1/10), frecuentes (> 1/100 hasta <1/10), poco frecuentes (>1/1.000 hasta <1/100), raras (>1/10.000 hasta <1/1.000); muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 1: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con IRC_
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión Crisis hipertensivas |
Frecuente Poco frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
|
Trastornos de la sangre y |
Trombosis de derivación |
Raras |
|
del sistema linfático |
Trombocitosis |
Muy raras |
Tabla 2: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con cáncer__
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión |
Frecuente |
|
Trastornos de la sangre y del sistema linfático |
Acontecimientos tromboembólicos |
Frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Tabla 3: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes incluidos en un programa de predonación de sangre autóloga
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Prematuros
Es muy frecuente un descenso de los valores de ferritina sérica (ver sección 4.4).
Descripción de reacciones adversas seleccionadas
Raramente pueden ocurrir reacciones cutáneas relacionadas con el tratamiento con epoetina beta como sarpullido, prurito, urticaria o reacciones en el lugar de la inyección. En casos muy raros se han notificado reacciones anafilácticas relacionadas con el tratamiento con epoetina beta. Sin embargo, en los ensayos clínicos controlados no se encontró una mayor incidencia de reacciones de hipersensibilidad.
Se han comunicado casos muy raros de síntomas gripales relacionados con el tratamiento con epoetina beta como fiebre, escalofríos, dolores de cabeza, dolor de las extremidades, malestar general, y/o dolor óseo, particularmente al inicio del tratamiento. Estas reacciones son de intensidad leve a moderada y desaparecen al cabo de un par de horas o días.
Los resultados obtenidos en un ensayo clínico controlado realizado con epoetina alfa o darbepoetina alfa, notificaron que la incidencia de infarto es frecuente.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
El margen terapéutico de NeoRecormon es muy amplio. No se han observado síntomas de sobredosis incluso a niveles séricos muy elevados.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antianémico, código ATC: B03XA01 Mecanismo de acción
La eritropoyetina es una glucoproteína que estimula la formación de eritrocitos a partir de sus progenitores asignados. Actúa como factor estimulante de la mitosis y hormona de diferenciación.
La epoetina beta, el principio activo de NeoRecormon, es idéntica en su composición aminoácida e hidrocarbonada a la eritropoyetina aislada de la orina de pacientes anémicos.
La eficacia biológica de la epoetina beta ha sido demostrada tras administración intravenosa y subcutánea en varios modelos animales in vivo (ratas normales y urémicas, ratones policitémicos, perros). Con la administración de epoetina beta aumentan el número de eritrocitos, los valores de Hb y la cifra de reticulocitos, al igual que la velocidad de incorporación de 59Fe.
In vitro (cultivo citológico de bazo de ratón) ha sido hallada una mayor incorporación de 3H-timidina en las células eritroides nucleadas del bazo tras incubación con epoetina beta.
Las investigaciones en cultivos de células de la médula ósea humana han revelado que la epoetina beta estimula específicamente la eritropoyesis y no afecta a la leucopoyesis. No han sido detectadas acciones citotóxicas de la epoetina beta sobre la médula ósea o sobre células cutáneas humanas.
Tras una dosis única de epoetina beta no se ha observado ningún efecto en el comportamiento o la actividad locomotora del ratón ni en las funciones circulatoria o respiratoria del perro.
Eficacia clínica y seguridad
En un ensayo aleatorizado, doble ciego, controlado con placebo, realizado en 4.038 pacientes con enfermedad renal crónica no sometidos a diálisis, con una diabetes tipo 2 y los niveles de hemoglobina < 11 g/dl, los pacientes recibieron tratamiento bien con darbepoetina alfa para alcanzar niveles de hemoglobina de 13 g/dl o con placebo (ver sección 4.4). El ensayo no cumplió su objetivo principal de demostrar una disminución en el riesgo de todas las causas de mortalidad, la morbilidad cardiovascular o enfermedad renal terminal. El análisis de los componentes individuales de la variable combinada mostró los siguientes HR (IC 95%): fallecimiento 1,05 (0,92-1,21), infarto cerebrovascular 1,92 (1,38-2,68), insuficiencia cardíaca congestiva 0,89 (0,74-1,08), infarto de miocardio 0,96 (0,751,23), hospitalización por isquemia del miocardio 0,84 (0,55-1,27), enfermedad renal terminal 1,02 (0,87-1,18).
En pacientes con IRC (tratados con diálisis, no tratados con diálisis, pacientes diabéticos y pacientes no diabéticos) se han realizado análisis agrupados post hoc de los estudios clínicos con AEEs. Se observó una tendencia a un aumento de las estimaciones del riesgo para la mortalidad por todas las causas y para los acontecimientos cardiovasculares y cerebrovasculares asociados a dosis acumuladas más altas de AEE independientemente de que los pacientes padecieran o no diabetes y de que recibieran o no tratamiento con diálisis (ver las secciones 4.2 y 4.4).
La eritropoyetina es un factor de crecimiento que estimula de forma primaria la producción de glóbulos rojos. Los receptores de eritropoyetina se encuentran también presentes en la superficie de algunas líneas celulares malignas.
Se ha estudiado la supervivencia y la progresión tumoral en cinco grandes ensayos controlados que incluyeron a 2.833 pacientes, de los cuales cuatro fueron ensayos doble-ciego controlados con placebo y uno fue un ensayo abierto. Dos de los ensayos reclutaron pacientes que estaban siendo tratados con quimioterapia. El nivel de hemoglobina que se quería alcanzar fue >13 g/dl en dos de los ensayos y de entre 12 y 14 g/dl en los otros tres. En el ensayo abierto no se observaron diferencias en la supervivencia global, entre los pacientes tratados con eritropoyetina humana recombinante y el grupo control. En los cuatro ensayos controlados con placebo, el índice de riesgo (hazard ratio) para la supervivencia global osciló entre 1,25 y 2,47 en favor de los grupos control. En todos estos ensayos se ha observado un aumento de la mortalidad inexplicable y estadísticamente significativo en pacientes que presentaban anemia asociada con diversos tipos frecuentes de cáncer y que recibieron eritropoyetina humana recombinante, en comparación con los grupos control. Las diferencias observadas en la incidencia de trombosis y complicaciones relacionadas, entre los pacientes que recibieron eritropoyetina humana recombinante y los que formaban parte de los grupos control, no permiten explicar satisfactoriamente los resultados de supervivencia global observados en los ensayos.
Un meta-análisis basado en datos individuales de pacientes, que incluyó datos de los 12 ensayos clínicos controlados realizados con NeoRecormon en pacientes anémicos con cáncer (n=2301), mostró un valor estimado del índice de riesgo (hazard ratio) de supervivencia global de 1,13 en favor de los grupos control (IC 95 %, CI 0,87, 1,46). En pacientes con un valor inicial de hemoglobina < 10 g/dl (n=899), el valor estimado del índice de riesgo (hazard ratio) para la supervivencia fue de 0,98 (IC 95% 0,68 al 1,40). Se observó un aumento del riesgo relativo de acontecimientos tromboembólicos en la población general (RR 1,62, IC 95 %: 1,13, 2,31).
Asimismo, se ha realizado un análisis de datos a nivel de paciente, en más de 13.900 pacientes con cáncer (tratados con quimioterapia, radioterapia, quimioradioterapia o sin tratamiento) que participaban en un total de 53 ensayos clínicos controlados en los que se administraban diversas epoetinas. En este meta-análisis se obtuvo un índice de riesgo (hazard ratio) para la supervivencia global de 1,06 a favor de los grupos control (IC 95%: 1,00, 1,12; 53 ensayos y 13.933 pacientes) y para los pacientes con cáncer que recibían quimioterapia, el índice de riesgo (hazard ratio) para la supervivencia global fue de 1,04 (IC 95%: 0,97, 1,11; 38 ensayos y 10.441 pacientes). Este meta-análisis además indica de forma consistente un aumento significativo del riesgo relativo de episodios tromboembólicos en pacientes con cáncer tratados con eritropoyetina recombinante humana (ver sección 4.4).
En casos muy raros, se han producido anticuerpos neutralizantes anti-eritropoyetina con o sin aplasia pura de células rojas (APCR) durante el tratamiento con rHuEPO.
5.2 Propiedades farmacocinéticas
Los estudios farmacocinéticos en voluntarios sanos y pacientes urémicos muestran que la semivida de la epoetina beta administrada por vía i.v. es de 4 a 12 horas y que el volumen de distribución corresponde de una a dos veces el volumen plasmático. Se han hallado resultados análogos en experimentos en animales con ratas normales y urémicas.
Tras la administración subcutánea de epoetina beta a pacientes urémicos la absorción retardada se traduce en una meseta de concentración sérica, cuya cota máxima se alcanza al cabo de 12 - 28 horas de media. El semiperíodo de vida terminal es mayor que después de la administración intravenosa, siendo la media de 13-28 horas.
La biodisponibilidad de la epoetina beta después de la administración subcutánea está comprendida entre el 23 y el 42 % en comparación con la administración intravenosa.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad de dosis repetidas, genotoxicidad y toxicidad para la reproducción.
Un estudio de carcinogenicidad con homólogos de eritropoyetina en ratones no reveló ningún signo de potencial proliferativo o carcinogénico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Urea,
Cloruro sódico,
Polisorbato 20,
Fosfato monosódico dihidratado,
Fosfato disódico dodecahidratado,
Cloruro cálcico dihidratado,
Glicina,
L-Leucina,
L-Isoleucina,
L-Treonina,
L-Ácido glutámico,
L-Fenilalanina,
Agua para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
Conservar la jeringa precargada en el embalaje exterior, para protegerlo de la luz.
Con fines de uso ambulatorio, se puede mantener el producto fuera de la nevera durante un único periodo de hasta 3 días a temperatura ambiente (no superior a 25°C).
6.5 Naturaleza y contenido del envase
Jeringa precargada (vidrio tipo I), con un protector y un émbolo (goma teflonizada) con una aguja de inyección (27G1/2). Cada jeringa contiene 0,6 ml de solución.
Tamaños de envase de 1 jeringa precargada y 1 aguja o de 6 jeringas precargadas y 6 agujas.
Puede que solamente estén comercializados algunos tamaños de envase.
6.6 Precauciones especiales de eliminación y otras manipulaciones
En primer lugar, ¡lávese las manos!
1. Saque una jeringa del envase y asegúrese que la solución es clara, incolora y prácticamente libre de partículas visibles. Retire el protector de la jeringa.
2. Saque una aguja del envase, fíjela sobre la jeringa y retire la tapa protectora de la aguja.
3. Expulse el aire de la jeringa y aguja manteniendo la jeringa vertical y presionando suavemente el émbolo hacia arriba. Continúe presionando el émbolo hasta que la cantidad de solución de NeoRecormon en la jeringa sea la prescrita.
4. Limpie la piel en el lugar de inyección con un algodón con alcohol. Haga un pliegue tomando la piel entre el índice y el pulgar. Sujete el cuerpo de la jeringa por la parte más próxima a la aguja e inserte la aguja en el pliegue cutáneo con acción firme y rápida. Inyecte la solución de NeoRecormon. Retire la aguja rápidamente y aplique presión sobre el lugar de inyección con un apósito seco y estéril.
Este medicamento es solo para uso individual. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/035 -036
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16 de julio de 1997 Fecha de la última renovación: 16 de julio de 2007
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 20.000 UI solución inyectable en jeringa precargada
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Una jeringa precargada con 0,6 ml de solución para inyección contiene 20.000 unidades internacionales (UI) correspondientes a 166 microgramos de epoetina beta* (eritropoyetina recombinante humana).
Un ml de solución para inyección contiene 33.333 UI de epoetina beta.
* producida en células de ovarios de hámster chino (CHO) mediante tecnología de ADN recombinante.
Excipientes con efecto conocido:
Fenilalanina (hasta 0,3 mg/jeringa).
Sodio (menos de 1 mmol/jeringa)
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución incolora, de transparente a ligeramente opalescente.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
NeoRecormon está indicado para:
- Tratamiento de la anemia sintomática asociada a la insuficiencia renal crónica en pacientes adultos y pediátricos.
- Prevención de la anemia en prematuros con un peso corporal al nacer de 750 a 1.500g y una edad gestacional de menos de 34 semanas.
- Tratamiento de la anemia sintomática en pacientes adultos con neoplasias no mieloides tratados con quimioterapia.
- Aumentar el rendimiento de la sangre autóloga de pacientes incluidos en un programa de predonación.
Debe sopesarse su uso en esta indicación frente al riesgo aumentado de episodios tromboembólicos que han sido notificados. Sólo debe administrarse el tratamiento a pacientes con anemia moderada (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], sin deficiencia de hierro) si no se dispone de procedimientos para conservar la sangre o si éstos son insuficientes cuando la cirugía mayor electiva programada requiera un gran volumen de sangre (4 o más unidades de sangre en mujeres o 5 o más unidades en hombres). Ver sección 5.1.
4.2 Posología y forma de administración
La terapia con NeoRecormon debe iniciarse por médicos con experiencia en las indicaciones arriba
mencionadas. Como se han observado reacciones anafilácticas en algunos casos aislados, se
recomienda administrar la primera dosis bajo control médico.
Posología
Tratamiento de la anemia sintomática en pacientes adultos y pediátricos con insuficiencia renal crónica
Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente. NeoRecormon se puede administrar tanto por vía subcutánea como intravenosa con el fin de aumentar la concentración de hemoglobina hasta un nivel no superior a 12 g/dl (7,5 mmol/l). En pacientes que no están sometidos a hemodiálisis es preferible utilizar la vía subcutánea para evitar la punción de venas periféricas._En caso de administración intravenosa, la solución debe ser inyectada a lo largo de unos 2 minutos, p.ej. en pacientes en hemodiálisis por vía de la fístula arteriovenosa al final de la diálisis.
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones se pueden observar valores individuales de hemoglobina superiores o inferiores a los niveles deseados. La variabilidad en los niveles de hemoglobina se debe controlar mediante ajuste de la dosis con el objeto de mantener los valores de hemoglobina dentro del intervalo entre 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Se debe evitar un nivel de hemoglobina de forma continuada por encima de 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando los valores de hemoglobina sean superiores a 12 g/dl (7,5 mmol/l).
Debe evitarse un aumento de hemoglobina superior a 2 g/dl (1,25 mmol/l) durante un periodo de cuatro semanas. Si esto ocurre, se debe hacer un ajuste adecuado de la dosis según las instrucciones incluidas en esta misma sección. Si la tasa de aumento de hemoglobina es mayor de 2 g/dl (1,25 mmol/l) en un mes o si el nivel de hemoglobina está aumentando y se acerca a 12 g/dl (7,45 mmol/l), debe reducirse la dosis en aproximadamente un 25%. Si el nivel de hemoglobina sigue aumentando, se debe interrumpir el tratamiento hasta que el nivel de hemoglobina empiece a disminuir, momento en el que se podrá reiniciar el tratamiento con una dosis aproximadamente un 25 % inferior a la dosis administrada previamente.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis eficaz más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia al tiempo que se mantiene una concentración de hemoglobina inferior o igual a 12 g/dl (7,45 mmol/l).
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica. En los pacientes con una respuesta deficiente de la hemoglobina a NeoRecormon, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.4 y 5.1).
En presencia de hipertensión o de enfermedades cardiovasculares, cerebrovasculares o vasculoperiféricas, se debe determinar de modo individual el incremento semanal y el valor objetivo de la hemoglobina teniendo en cuenta el cuadro clínico.
El tratamiento con NeoRecormon se divide en dos fases:
1. Fase de corrección
- Administración subcutánea:
La dosis inicial es 3 x 20 UI/kg por semana. La dosis puede incrementarse cada 4 semanas en 3 x 20 UI/kg y semana si el aumento de la hemoglobina no ha sido adecuado (< 0,25 g/dl por semana).
La dosis semanal puede dividirse en dosis diarias.
- Administración intravenosa:
La dosis inicial es 3 x 40 UI/kg por semana, y puede aumentarse al cabo de 4 semanas a 80 UI/kg -tres veces por semana- y si son necesarios incrementos ulteriores serán de 20 UI/kg tres veces por semana, con intervalos mensuales.
Por ambas vías de administración, la dosis máxima no debe superar 720 UI/kg por semana.
2. Fase de mantenimiento
Para mantener el nivel de hemoglobina entre 10 y 12 g/dl, la dosis es inicialmente reducida a la mitad de la cantidad previamente administrada. Posteriormente, se ajustará la dosis individualmente para el paciente a intervalos de una o dos semanas (dosis de mantenimiento).
En caso de administración subcutánea, la dosis semanal puede administrarse en una inyección única o fraccionada en tres o siete inyecciones. Los pacientes que permanezcan estables en el régimen de una dosis única semanal pueden pasar a una administración única cada dos semanas. En este caso, puede ser necesario un aumento de la dosis.
Los resultados de los estudios clínicos en niños han revelado que, en general, a menor edad, se necesita mayor dosis de NeoRecormon. No obstante, hay que seguir el programa posológico recomendado ya que no puede predecirse la respuesta individual.
El tratamiento con NeoRecormon es normalmente crónico. Sin embargo, en caso necesario puede interrumpirse en cualquier momento. Los datos sobre la pauta posológica de una vez a la semana se han obtenido de estudios clínicos con una duración de tratamiento de 24 semanas.
Prevención de anemia en prematuros
La solución se administra por vía subcutánea a una dosis de 3 x 250 UI/kg p.c. por semana. Es probable que los niños que ya hayan recibido una transfusión previa cuando se inicie el tratamiento con NeoRecormon no se beneficien tanto como los prematuros no transfundidos. La duración de tratamiento recomendada es de 6 semanas.
Tratamiento de anemia sintomática inducida por quimioterapia en pacientes con cáncer Se debe administrar NeoRecormon por vía subcutánea a pacientes con anemia (por ejemplo, si la concentración de hemoglobina es menor o igual a 10 g/dl [6,2 mmol/l]). Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente.
La dosis semanal puede administrarse como una inyección por semana o en dosis divididas entre 3 y 7 veces por semana.
La dosis inicial recomendada es de 30.000 UI por semana (que corresponde aproximadamente a 450 UI/kg de peso corporal por semana, en base al peso medio del paciente).
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones es posible llegar a observar en un paciente valores de hemoglobina superiores o inferiores a los esperados. La variabilidad de la hemoglobina se deberá manejar ajustando la dosis para mantener los valores de la hemoglobina dentro del intervalo de 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Deben evitarse concentraciones sostenidas de hemoglobina superiores a 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando se observen valores de hemoglobina superiores a los 12 g/dl (7,5 mmol/l).
Si a las 4 semanas de tratamiento el valor de hemoglobina aumenta hasta al menos 1 g/dl (0,62 mmol/l), se debe continuar con la dosis que en ese momento se esté administrando. Si los valores de hemoglobina no han aumentado al menos 1 g/dl (0,62 mmol/l), se debe considerar duplicar la dosis semanal. Si a las 8 semanas de tratamiento el valor de hemoglobina no aumenta hasta al menos 1 g/dl (0,62 mmol/l), es improbable que se produzca respuesta y, por consiguiente, el tratamiento debe ser interrumpido.
El tratamiento debe continuar durante las 4 semanas posteriores al final de la quimioterapia.
La dosis máxima no debe exceder de 60.000 UI por semana.
Una vez se ha alcanzado el objetivo terapéutico del paciente, la dosis debe reducirse del 25 al 50 % con el fin de mantener el valor de hemoglobina en ese nivel. Debe realizarse el ajuste posológico adecuado.
Si el nivel de hemoglobina excede los 12 g/dl (7,5 mmol/l) se debe reducir la dosis entre un 25 y un
50 % aproximadamente. Si el nivel de hemoglobina excede de 13 g/dl (8,1 mmol), se debe interrumpir temporalmente el tratamiento con NeoRecormon. El tratamiento debe reiniciarse con una dosis aproximadamente un 25 % inferior a la dosis previamente administrada después de que los niveles de hemoglobina desciendan hasta un valor menor o igual a 12 g/dl (7,5 mmol/l) o por debajo.
51 al cabo de 4 semanas el incremento de hemoglobina es superior a 2 g/dl (1,3 mmol/l), la dosis debe reducirse entre un 25 y un 50 %.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia.
Tratamiento para incrementar el rendimiento de la sangre autóloga donada La solución puede administrarse por vía intravenosa, en unos 2 minutos, o subcutáneamente. NeoRecormon se administra dos veces por semana durante 4 semanas. En aquellas ocasiones en que el hematocrito del paciente permite la donación de sangre, es decir el hematocrito es > 33 %, NeoRecormon se administra al final de la donación de sangre.
Durante la totalidad del período de tratamiento no debe excederse un hematocrito del 48 %.
La dosis debe ser determinada por el equipo quirúrgico, individualmente para cada paciente, en función de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos:
1. La cantidad de sangre pre-donada necesaria dependerá de la pérdida prevista de sangre, de los medios de conservación empleados y del estado físico del paciente.
Este volumen debe ser equivalente a la cantidad de sangre que se considera necesaria para evitar transfusiones de sangre homóloga.
La cantidad de sangre pre-donada necesaria se expresa en unidades en las cuales una unidad de nomograma es equivalente a 180 ml de glóbulos rojos.
2. La capacidad de donar sangre depende predominantemente del volumen sanguíneo del paciente y del hematocrito basal. Ambas variables determinan la reserva endógena de eritrocitos, que puede ser calculada conforme a la fórmula siguiente:
Reserva endógena de eritrocitos = volumen sanguíneo (ml) x (hematocrito - 33)/ 100 mujeres: volumen sanguíneo (ml) = 41 (ml/kg) x peso corporal (kg) + 1200 (ml)
hombres: volumen sanguíneo (ml) = 44 (ml/kg) x peso corporal (kg) + 1600 (ml)
(peso corporal: > 45 kg)
La indicación para un tratamiento con NeoRecormon y, en su caso, la dosis individual debe ser determinada a partir de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos según las gráficas siguientes:
Paciente femenino Paciente masculino
Cantidad de sangre pre-donada Cantidad de sangre pre-donada
necesaria [unidades] necesaria [unidades]


O 100 200 300 400 500 600 700 800 0 100 200 300 400 500 600 700 800
Reserva eritrocitaria endógena [ml] Reserva eritrocitaria endógena [ml]
La dosis única así determinada debe administrarse dos veces por semana a lo largo de 4 semanas. La dosis máxima no debe sobrepasar 1.600 UI/kg peso corporal por semana en administración intravenosa ó 1.200 UI/kg por semana en administración subcutánea.
Forma de administración
La jeringa precargada de NeoRecormon está lista para su uso. Sólo se pueden inyectar soluciones claras o ligeramente opalescentes, incoloras y prácticamente libres de partículas visibles. NeoRecormon en jeringa precargada es un producto estéril pero sin conservantes. Bajo ninguna circunstancia debe administrarse más de una dosis por jeringa; el medicamento es solo para uso individual.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Hipertensión mal controlada.
En la indicación para “Aumentar el rendimiento de sangre autóloga”: infarto de miocardio o accidente cerebrovascular en el mes anterior al tratamiento, angina de pecho inestable, riesgo aumentado de trombosis venosa profunda, como por ejemplo en aquellos con historial de enfermedad tromboembólica venosa.
4.4 Advertencias y precauciones especiales de empleo
NeoRecormon debe usarse con cautela en presencia de anemia refractaria con exceso de blastos en transformación, epilepsia, trombocitosis e insuficiencia hepática crónica. Y deben excluirse las deficiencias de ácido fólico y vitamina B12 pues reducen la eficacia de NeoRecormon.
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica, ya que las dosis acumuladas altas de epoetina pueden asociarse a un mayor riesgo de mortalidad, y de acontecimientos cardiovasculares y cerebrovasculares graves. En pacientes con una respuesta deficiente de la hemoglobina a epoetinas, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.2 y 5.1).
Para garantizar una eritropoyesis eficaz, se debe evaluar el nivel de hierro en todos los pacientes antes y durante el tratamiento, y puede ser necesario un tratamiento suplementario con hierro administrado de acuerdo con las guías terapéuticas.
La sobrecarga grave de aluminio debido al tratamiento de la insuficiencia renal puede comprometer la eficacia de NeoRecormon.
La indicación de tratamiento con NeoRecormon en enfermos nefroscleróticos aún no sometidos a diálisis debe definirse individualmente, ya que la posible aceleración de la insuficiencia renal no puede descartarse con certeza.
Aplasia pura de células rojas (APCR)
Se han notificado casos de APCR causada por anticuerpos neutralizantes antieritropoyetina, asociados a tratamientos con eritropoyetinas, incluido NeoRecormon. Estos anticuerpos han presentado reacción cruzada con todas las proteínas eritropoyéticas, por los que los pacientes en los que se sospeche o se haya confirmado la presencia de anticuerpos neutralizantes contra eritropoyetina no deben ser tratados con NeoRecormon (ver sección 4.8).
APCR en pacientes con hepatitis C
Se debe interrumpir inmediatamente el tratamiento con epoetina, y realizar ensayos de anticuerpos antieritropoyetina si se produce una disminución paradójica de la hemoglobina y se desarrolla anemia severa asociada con bajos recuentos de reticulocitos. En pacientes con hepatitis C tratados con interferon y ribavirina se han notificados casos cuando las epoetinas se han utilizado concomitantemente. Las epoetinas no están aprobadas en el tratamiento de la anemia asociada a la hepatitis C.
Monitorización de la tensión arterial
Puede aparecer un aumento de la tensión arterial o un agravamiento de la hipertensión ya existente, especialmente en los casos en los que el hematocrito aumenta rápidamente. Estos incrementos en la tensión arterial pueden ser tratados con medicamentos. Si no pueden ser controlados con medicación se recomienda interrumpir transitoriamente la terapia con NeoRecormon. En particular al principio de la terapia se recomienda la monitorización regular de la tensión arterial; también durante la diálisis. Pueden producirse crisis hipertensivas con síntomas de encefalopatías y que requieren atención médica inmediata y cuidados médicos intensivos. Debe prestarse especial atención a las cefaleas súbitas lacerantes hemicraneales como posible signo de advertencia.
Insuficiencia renal crónica
En pacientes con insuficiencia renal crónica puede sobrevenir un aumento dosis-dependiente moderado en la cifra plaquetaria dentro del margen normal durante el tratamiento con NeoRecormon, especialmente después de la administración intravenosa. La regresión tiene lugar en el curso de la prosecución de la terapia. Se recomienda el control regular de la cifra de plaquetas durante las 8 primeras semanas de tratamiento.
Concentración de hemoglobina
En pacientes con insuficiencia renal crónica, la concentración de hemoglobina en la fase de mantenimiento no debe exceder el límite superior del rango recomendado en la sección 4.2. En ensayos clínicos se observó un aumento del riesgo de fallecimiento y de acontecimientos cardiovasculares graves o acontecimientos cerebrovasculares incluido el infarto, cuando se administraron agentes estimulantes de la eritropoyesis (AEEs) con el fin de alcanzar un nivel de hemoglobina superior a 12 g/dl (7,5 mmol/l).
En ensayos clínicos controlados no se han observado beneficios significativos atribuibles a la administración de epoetinas cuando se aumentaba la concentración de hemoglobina por encima del nivel necesario para controlar los síntomas de anemia y evitar las transfusiones sanguíneas.
En niños prematuros puede producirse un ligero aumento de la cifra plaquetaria, en especial hasta el día 12 -14 de vida, por lo tanto las plaquetas deben controlarse regularmente.
Efectos sobre el crecimiento tumoral
Las epoetinas son factores de crecimiento que estimulan de forma primaria la producción de glóbulos rojos. Los receptores de epoetina pueden expresarse sobre la superficie de una variedad de células tumorales. Como sucede con todos los factores de crecimiento, existe la preocupación de que las epoetinas pudieran estimular el crecimiento de tumores. En varios ensayos controlados, no se ha observado que las epoetinas mejoren la supervivencia global o que disminuyan el riesgo de progresión del tumor en pacientes con anemia asociada con cáncer.
En ensayos clínicos controlados, se ha observado que el uso de NeoRecormon y otros agentes estimulantes de la eritropoyesis (AEEs):
- acortaba el tiempo de progresión del tumor en pacientes con cáncer avanzado de cabeza y cuello que recibían radioterapia cuando se administraba para conseguir una concentración de hemoglobina por encima de 14 g/dl (8,7 mmol/l),
- acortaba la supervivencia global y aumentaba el número de fallecimientos atribuidos a la progresión de la enfermedad a los 4 meses, en pacientes con cáncer de mama metastásico que recibían quimioterapia cuando se administraba para conseguir una concentración de hemoglobina entre los 12 y los 14 g/dl (7,5-8,7 mmol/l),
- aumentaba el riesgo de fallecimiento cuando se administraba para conseguir una concentración de hemoglobina de 12 g/dl (7,5 mmol/l) en pacientes con enfermedad maligna activa y que no recibían ni quimioterapia ni radioterapia. El uso de los AEEs no está indicado en esa población de pacientes.
En vista de lo anterior, en algunas situaciones clínicas la transfusión sanguínea debe ser el tratamiento de elección para la anemia en pacientes con cáncer. La decisión de administrar eritropoyetinas recombinantes se tomará en base a la evaluación de la relación beneficio/riesgo junto con la aceptación individual del paciente, teniendo en cuenta el contexto clínico específico. Los factores que deben considerarse en esta evaluación son el tipo de tumor y su estadio, el grado de anemia, la esperanza de vida, el entorno en el que el paciente está siendo tratado y la preferencia del paciente (ver sección 5.1).
Puede haber un aumento de la tensión arterial que puede ser tratada con medicamentos. Se recomienda por tanto monitorizar la tensión arterial, en particular en la fase inicial de tratamiento en pacientes con cáncer.
En pacientes con cáncer también deberán monitorizarse regularmente el recuento plaquetario y el valor de hemoglobina.
En pacientes en programa de predonación de sangre autóloga, puede sobrevenir un aumento de la cifra de plaquetas, generalmente dentro de los límites normales. Por tanto, se recomienda la determinación de esta cifra al menos una vez por semana. Si se produce un aumento plaquetario de más de 150 x 109/l o si las plaquetas ascienden por encima del margen normal, se debe suspender el tratamiento con NeoRecormon.
En niños prematuros no se podría descartar el riesgo potencial de que la eritropoyetina provoque retinopatía, por lo tanto se debe tener precaución y la decisión de tratar a un niño prematuro se debe tomar teniendo en cuenta el beneficio potencial y el riesgo de este tratamiento, así como las opciones alternativas de tratamiento que estén disponibles.
En pacientes con insuficiencia renal crónica, a menudo se precisa aumentar la dosis de heparina durante la hemodiálisis en la terapia con NeoRecormon debido al aumento del volumen de hematocrito. Si la heparinización no es óptima es posible que se produzca la oclusión del sistema de diálisis.
En pacientes con alteración renal crónica con riesgo de trombosis de derivación, se debe considerar la revisión temprana de la derivación y profilaxis de la trombosis mediante la administración, por ejemplo, de ácido acetilsalicílico.
Los niveles séricos de potasio y fosfato deben ser controlados regularmente durante la terapia con NeoRecormon. Se ha informado de aumentos de potasio en unos pocos pacientes urémicos que recibieron NeoRecormon si bien no se ha establecido una relación causal. En caso de observar un nivel elevado o creciente de potasio se debe considerar la interrupción de la administración de NeoRecormon hasta que el nivel haya sido corregido.
Para el uso de NeoRecormon en un programa de predonación de sangre autóloga deben ser observadas las directrices oficiales sobre donación de sangre, en particular las siguientes:
- sólo deben donar los pacientes con hematocrito > 33 % (hemoglobina >11 g/dl [6,83 mmol/l]);
- debe procederse con especial cuidado con pacientes con un peso inferior a 50 kg;
- el volumen individual extraído no debe superar aproximadamente el 12 % del volumen sanguíneo estimado del paciente.
El tratamiento debe reservarse a pacientes en los que se considere particularmente importante evitar la transfusión de sangre homóloga teniendo en cuenta la evaluación riesgo/beneficio de las transfusiones homólogas.
Uso indebido
Un uso indebido del producto por personas sanas puede llevar a un aumento excesivo del volumen de hematocrito, lo cual puede asociarse con complicaciones del sistema cardiovascular con riesgo para la vida.
Excipientes
NeoRecormon en jeringa precargada contiene, como máximo, 0,3 mg de fenilalanina por jeringa como excipiente. Este hecho se deberá tener en cuenta en los pacientes afectados por formas graves de fenilcetonuria.
Este medicamento contiene menos de 1 mmol (23 mg) de sodio por jeringa, por lo que se considera esencialmente “exento de sodio”.
Trazabilidad de NeoRecormon
Para mejorar la trazabilidad de los agentes estimulantes de la eritropoyesis (AEEs), se debe registrar (o indicar) claramente el nombre comercial del AEE administrado en la historia clínica del paciente.
4.5 Interacción con otros medicamentos y otras formas de interacción
Los resultados clínicos obtenidos hasta el presente no ponen de manifiesto interacción alguna de NeoRecormon con otros medicamentos.
Los estudios llevados a cabo en animales mostraron que la epoetina beta no potencia la mielotoxicidad de los medicamentos citostáticos, tales como etopósido, cisplatino, ciclofosfamida y fluorouracilo.
4.6 Fertilidad, embarazo y lactancia
Fertilidad
Los estudios en animales no muestran efectos dañinos directos o indirectos sobre el embarazo, desarrollo embrional/fetal, parto o desarrollo postnatal (ver sección 5.3).
Embarazo
No hay datos relativos al uso de NeoRecormon en mujeres embarazadas.
Se debe tener precaución cuando se prescriba a mujeres embarazadas
Lactancia
Se desconoce si la epoetina beta se excreta en la leche materna. La decisión de continuar/interrumpir la lactancia o de continuar/interrumpir el tratamiento con epoetina beta se debe hacer teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento con epoetina beta para la madre.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NeoRecormon sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Basados en los resultados de los ensayos clínicos que incluyen 1.725 pacientes, se espera que aproximadamente el 8 % de los pacientes tratados con NeoRecormon sufran reacciones adversas.
Pacientes anémicos con insuficiencia renal crónica
La reacción adversa más frecuente durante el tratamiento con NeoRecormon es un aumento de la tensión arterial o agravamiento de la hipertensión existente, especialmente en casos de aumento rápido del hematocrito (ver sección 4.4). En algunos pacientes cuya tensión arterial es normal o baja también pueden darse crisis hipertensivas con síntomas de encefalopatías (p.ej. cefaleas y estados de confusión, trastornos sensitivomotores - como alteraciones del habla o de la ambulación - hasta convulsiones tónico-clónicas) (ver sección 4.4).
Puede darse trombosis de derivación, especialmente en pacientes que tienen tendencia a la hipotensión o cuyas fístulas arteriovenosas presentan complicaciones (p.ej. estenosis, aneurismas), ver sección 4.4. En la mayoría de los casos se observa una caída en los valores séricos de ferritina simultáneamente con el aumento del volumen de hematocrito (ver sección 4.4). Además, en casos aislados se han observado incrementos transitorios en los niveles séricos de potasio y fosfato (ver sección 4.4).
En casos aislados se ha notificado aplasia pura de células rojas (APCR) mediada por anticuerpos neutralizantes antieritropoyetina asociada al tratamiento con NeoRecormon. En caso de que se diagnostique APCR mediada por anticuerpos antieritropoyetina, el paciente deberá interrumpir el tratamiento con NeoRecormon, el cual no se deberá sustituir por otra proteína eritropoyética (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 1, que figura más adelante.
Pacientes con cáncer
El dolor de cabeza y la hipertensión relacionados con el tratamiento con epoetina beta son frecuentes y pueden tratarse con medicamentos (ver sección 4.4).
En algunos pacientes se observa un descenso de los valores de hierro sérico (ver sección 4.4).
Estudios clínicos han puesto de manifiesto que la frecuencia de acontecimientos tromboembólicos es más alta en pacientes con cáncer tratados con NeoRecormon que en los controles no tratados o tratados con placebo. En pacientes tratados con NeoRecormon, esta incidencia es del 7 % comparado con el 4 % en el grupo control; este hecho no está asociado con un incremento en la mortalidad tromboembólica comparado con los grupos control.
Estas reacciones adversas se enumeran en la Tabla 2, que figura más adelante.
Pacientes incluidos en un programa de predonación de sangre autóloga Los pacientes incluidos en un programa de predonación de sangre autóloga han demostrado una frecuencia ligeramente superior de accidentes tromboembólicos. Sin embargo, no se pudo establecer una relación causal con el tratamiento con NeoRecormon.
En ensayos clínicos controlados con placebo, la deficiencia transitoria de hierro fue más acusada en los pacientes tratados con NeoRecormon que en los pacientes control (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 3, que figura más adelante.
Tabla de reacciones adversas
Las reacciones adversas se enumeran de acuerdo con la clasificación de órganos del sistema MedDRA y por categoría de frecuencia.
Las categorías de frecuencia se definen utilizando la siguiente convención:
muy frecuentes (> 1/10), frecuentes (> 1/100 hasta <1/10), poco frecuentes (>1/1.000 hasta <1/100), raras (>1/10.000 hasta <1/1.000); muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 1: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con IRC_
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión Crisis hipertensivas |
Frecuente Poco frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
|
Trastornos de la sangre y |
Trombosis de derivación |
Raras |
|
del sistema linfático |
Trombocitosis |
Muy raras |
Tabla 2: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con cáncer__
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión |
Frecuente |
|
Trastornos de la sangre y del sistema linfático |
Acontecimientos tromboembólicos |
Frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Tabla 3: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes incluidos en un programa de predonación de sangre autóloga
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Prematuros
Es muy frecuente un descenso de los valores de ferritina sérica (ver sección 4.4).
Descripción de reacciones adversas seleccionadas
Raramente pueden ocurrir reacciones cutáneas relacionadas con el tratamiento con epoetina beta como sarpullido, prurito, urticaria o reacciones en el lugar de la inyección. En casos muy raros se han notificado reacciones anafilácticas relacionadas con el tratamiento con epoetina beta. Sin embargo, en los ensayos clínicos controlados no se encontró una mayor incidencia de reacciones de hipersensibilidad.
Se han comunicado casos muy raros de síntomas gripales relacionados con el tratamiento con epoetina beta como fiebre, escalofríos, dolores de cabeza, dolor de las extremidades, malestar general, y/o dolor óseo, particularmente al inicio del tratamiento. Estas reacciones son de intensidad leve a moderada y desaparecen al cabo de un par de horas o días.
Los resultados obtenidos en un ensayo clínico controlado realizado con epoetina alfa o darbepoetina alfa, notificaron que la incidencia de infarto es frecuente.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
El margen terapéutico de NeoRecormon es muy amplio. No se han observado síntomas de sobredosis incluso a niveles séricos muy elevados.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antianémico, código ATC: B03XA01 Mecanismo de acción
La eritropoyetina es una glucoproteína que estimula la formación de eritrocitos a partir de sus progenitores asignados. Actúa como factor estimulante de la mitosis y hormona de diferenciación.
La epoetina beta, el principio activo de NeoRecormon, es idéntica en su composición aminoácida e hidrocarbonada a la eritropoyetina aislada de la orina de pacientes anémicos.
La eficacia biológica de la epoetina beta ha sido demostrada tras administración intravenosa y subcutánea en varios modelos animales in vivo (ratas normales y urémicas, ratones policitémicos, perros). Con la administración de epoetina beta aumentan el número de eritrocitos, los valores de Hb y la cifra de reticulocitos, al igual que la velocidad de incorporación de 59Fe.
In vitro (cultivo citológico de bazo de ratón) ha sido hallada una mayor incorporación de 3H-timidina en las células eritroides nucleadas del bazo tras incubación con epoetina beta.
Las investigaciones en cultivos de células de la médula ósea humana han revelado que la epoetina beta estimula específicamente la eritropoyesis y no afecta a la leucopoyesis. No han sido detectadas acciones citotóxicas de la epoetina beta sobre la médula ósea o sobre células cutáneas humanas.
Tras una dosis única de epoetina beta no se ha observado ningún efecto en el comportamiento o la actividad locomotora del ratón ni en las funciones circulatoria o respiratoria del perro.
Eficacia clínica y seguridad
En un ensayo aleatorizado, doble ciego, controlado con placebo, realizado en 4.038 pacientes con enfermedad renal crónica no sometidos a diálisis, con una diabetes tipo 2 y los niveles de hemoglobina < 11 g/dl, los pacientes recibieron tratamiento bien con darbepoetina alfa para alcanzar niveles de hemoglobina de 13 g/dl o con placebo (ver sección 4.4). El ensayo no cumplió su objetivo principal de demostrar una disminución en el riesgo de todas las causas de mortalidad, la morbilidad cardiovascular o enfermedad renal terminal. El análisis de los componentes individuales de la variable combinada mostró los siguientes HR (IC 95%): fallecimiento 1,05 (0,92-1,21), infarto cerebrovascular 1,92 (1,38-2,68), insuficiencia cardíaca congestiva 0,89 (0,74-1,08), infarto de miocardio 0,96 (0,751,23), hospitalización por isquemia del miocardio 0,84 (0,55-1,27), enfermedad renal terminal 1,02 (0,87-1,18).
En pacientes con IRC (tratados con diálisis, no tratados con diálisis, pacientes diabéticos y pacientes no diabéticos) se han realizado análisis agrupados post hoc de los estudios clínicos con AEEs. Se observó una tendencia a un aumento de las estimaciones del riesgo para la mortalidad por todas las causas y para los acontecimientos cardiovasculares y cerebrovasculares asociados a dosis acumuladas más altas de AEE independientemente de que los pacientes padecieran o no diabetes y de que recibieran o no tratamiento con diálisis (ver las secciones 4.2 y 4.4).
La eritropoyetina es un factor de crecimiento que estimula de forma primaria la producción de glóbulos rojos. Los receptores de eritropoyetina se encuentran también presentes en la superficie de algunas líneas celulares malignas.
Se ha estudiado la supervivencia y la progresión tumoral en cinco grandes ensayos controlados que incluyeron a 2.833 pacientes, de los cuales cuatro fueron ensayos doble-ciego controlados con placebo y uno fue un ensayo abierto. Dos de los ensayos reclutaron pacientes que estaban siendo tratados con quimioterapia. El nivel de hemoglobina que se quería alcanzar fue >13 g/dl en dos de los ensayos y de entre 12 y 14 g/dl en los otros tres. En el ensayo abierto no se observaron diferencias en la supervivencia global, entre los pacientes tratados con eritropoyetina humana recombinante y el grupo control. En los cuatro ensayos controlados con placebo, el índice de riesgo (hazard ratio) para la supervivencia global osciló entre 1,25 y 2,47 en favor de los grupos control. En todos estos ensayos se ha observado un aumento de la mortalidad inexplicable y estadísticamente significativo en pacientes que presentaban anemia asociada con diversos tipos frecuentes de cáncer y que recibieron eritropoyetina humana recombinante, en comparación con los grupos control. Las diferencias observadas en la incidencia de trombosis y complicaciones relacionadas, entre los pacientes que recibieron eritropoyetina humana recombinante y los que formaban parte de los grupos control, no permiten explicar satisfactoriamente los resultados de supervivencia global observados en los ensayos.
Un meta-análisis basado en datos individuales de pacientes, que incluyó datos de los 12 ensayos clínicos controlados realizados con NeoRecormon en pacientes anémicos con cáncer (n=2301), mostró un valor estimado del índice de riesgo (hazard ratio) de supervivencia global de 1,13 en favor de los grupos control (IC 95 %, CI 0,87, 1,46). En pacientes con un valor inicial de hemoglobina < 10 g/dl (n=899), el valor estimado del índice de riesgo (hazard ratio) para la supervivencia fue de 0,98 (IC 95% 0,68 al 1,40). Se observó un aumento del riesgo relativo de acontecimientos tromboembólicos en la población general (RR 1,62, IC 95 %: 1,13, 2,31).
Asimismo, se ha realizado un análisis de datos a nivel de paciente, en más de 13.900 pacientes con cáncer (tratados con quimioterapia, radioterapia, quimioradioterapia o sin tratamiento) que participaban en un total de 53 ensayos clínicos controlados en los que se administraban diversas epoetinas. En este meta-análisis se obtuvo un índice de riesgo (hazard ratio) para la supervivencia global de 1,06 a favor de los grupos control (IC 95%: 1,00, 1,12; 53 ensayos y 13.933 pacientes) y para los pacientes con cáncer que recibían quimioterapia, el índice de riesgo (hazard ratio) para la supervivencia global fue de 1,04 (IC 95%: 0,97, 1,11; 38 ensayos y 10.441 pacientes). Este meta-análisis además indica de forma consistente un aumento significativo del riesgo relativo de episodios tromboembólicos en pacientes con cáncer tratados con eritropoyetina recombinante humana (ver sección 4.4).
En casos muy raros, se han producido anticuerpos neutralizantes anti-eritropoyetina con o sin aplasia pura de células rojas (APCR) durante el tratamiento con rHuEPO.
5.2 Propiedades farmacocinéticas
Los estudios farmacocinéticos en voluntarios sanos y pacientes urémicos muestran que la semivida de la epoetina beta administrada por vía i.v. es de 4 a 12 horas y que el volumen de distribución corresponde de una a dos veces el volumen plasmático. Se han hallado resultados análogos en experimentos en animales con ratas normales y urémicas.
Tras la administración subcutánea de epoetina beta a pacientes urémicos la absorción retardada se traduce en una meseta de concentración sérica, cuya cota máxima se alcanza al cabo de 12 - 28 horas de media. El semiperíodo de vida terminal es mayor que después de la administración intravenosa, siendo la media de 13-28 horas.
La biodisponibilidad de la epoetina beta después de la administración subcutánea está comprendida entre el 23 y el 42 % en comparación con la administración intravenosa.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad de dosis repetidas, genotoxicidad y toxicidad para la reproducción.
Un estudio de carcinogenicidad con homólogos de eritropoyetina en ratones no reveló ningún signo de potencial proliferativo o carcinogénico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Urea,
Cloruro sódico,
Polisorbato 20,
Fosfato monosódico dihidratado,
Fosfato disódico dodecahidratado,
Cloruro cálcico dihidratado,
Glicina,
L-Leucina,
L-Isoleucina,
L-Treonina,
L-Ácido glutámico,
L-Fenilalanina,
Agua para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
Conservar la jeringa precargada en el embalaje exterior, para protegerlo de la luz.
Con fines de uso ambulatorio, se puede mantener el producto fuera de la nevera durante un único periodo de hasta 3 días a temperatura ambiente (no superior a 25°C).
6.5 Naturaleza y contenido del envase
Jeringa precargada (vidrio tipo I), con un protector y un émbolo (goma teflonizada) con una aguja de inyección (27G1/2). Cada jeringa contiene 0,6 ml de solución.
Tamaños de envase de 1 jeringa precargada y 1 aguja o de 6 jeringas precargadas y 6 agujas.
Puede que solamente estén comercializados algunos tamaños de envase.
6.6 Precauciones especiales de eliminación y otras manipulaciones
En primer lugar, ¡lávese las manos!
1. Saque una jeringa del envase y asegúrese que la solución es clara, incolora y prácticamente libre de partículas visibles. Retire el protector de la jeringa.
2. Saque una aguja del envase, fíjela sobre la jeringa y retire la tapa protectora de la aguja.
3. Expulse el aire de la jeringa y aguja manteniendo la jeringa vertical y presionando suavemente el émbolo hacia arriba. Continúe presionando el émbolo hasta que la cantidad de solución de NeoRecormon en la jeringa sea la prescrita.
4. Limpie la piel en el lugar de inyección con un algodón con alcohol. Haga un pliegue tomando la piel entre el índice y el pulgar. Sujete el cuerpo de la jeringa por la parte más próxima a la aguja e inserte la aguja en el pliegue cutáneo con acción firme y rápida. Inyecte la solución de NeoRecormon. Retire la aguja rápidamente y aplique presión sobre el lugar de inyección con un apósito seco y estéril.
Este medicamento es solo para uso individual. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/037 -038
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16 de julio de 1997 Fecha de la última revalidación: 16 de julio de 2007.
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 30.000 UI solución inyectable en jeringa precargada
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Una jeringa precargada con 0,6 ml de solución para inyección contiene 30.000 unidades internacionales (UI) correspondientes a 250 microgramos de epoetina beta* (eritropoyetina recombinante humana).
Un ml de solución para inyección contiene 50.000 UI de epoetina beta.
* producida en células de ovarios de hámster chino (CHO) mediante tecnología de ADN recombinante.
Excipientes con efecto conocido:
Fenilalanina (hasta 0,3 mg/jeringa).
Sodio (menos de 1 mmol/jeringa)
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución incolora, de transparente a ligeramente opalescente.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
NeoRecormon está indicado para:
- Tratamiento de la anemia sintomática asociada a la insuficiencia renal crónica en pacientes adultos y pediátricos.
- Prevención de la anemia en prematuros con un peso corporal al nacer de 750 a 1.500 g y una edad gestacional de menos de 34 semanas.
- Tratamiento de la anemia sintomática en pacientes adultos con neoplasias no mieloides tratados con quimioterapia.
- Aumentar el rendimiento de la sangre autóloga de pacientes incluidos en un programa de predonación.
Debe sopesarse su uso en esta indicación frente al riesgo aumentado de episodios tromboembólicos que han sido notificados. Sólo debe administrarse el tratamiento a pacientes con anemia moderada (Hb 10 - 13 g/dl [6,21 - 8,07 mmol/l], sin deficiencia de hierro) si no se dispone de procedimientos para conservar la sangre o si éstos son insuficientes cuando la cirugía mayor electiva programada requiera un gran volumen de sangre (4 o más unidades de sangre en mujeres o 5 o más unidades en hombres). Ver sección 5.1.
4.2 Posología y forma de administración
La terapia con NeoRecormon debe iniciarse por médicos con experiencia en las indicaciones arriba
mencionadas. Como se han observado reacciones anafilácticas en algunos casos aislados, se
recomienda administrar la primera dosis bajo control médico.
Posología
Tratamiento de la anemia sintomática en pacientes adultos y pediátricos con insuficiencia renal crónica
Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente. NeoRecormon se puede administrar tanto por vía subcutánea como intravenosa con el fin de aumentar la concentración de hemoglobina hasta un nivel no superior a 12 g/dl (7,5 mmol/l). En pacientes que no están sometidos a hemodiálisis es preferible utilizar la vía subcutánea para evitar la punción de venas periféricas. En caso de administración intravenosa, la solución debe ser inyectada a lo largo de unos 2 minutos, p.ej. en pacientes en hemodiálisis por vía de la fístula arteriovenosa al final de la diálisis.
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones se pueden observar valores individuales de hemoglobina superiores o inferiores a los niveles deseados. La variabilidad en los niveles de hemoglobina se debe controlar mediante ajuste de la dosis con el objeto de mantener los valores de hemoglobina dentro del intervalo entre 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Se debe evitar un nivel de hemoglobina de forma continuada por encima de 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando los valores de hemoglobina sean superiores a 12 g/dl (7,5 mmol/l).
Debe evitarse un aumento de hemoglobina superior a 2 g/dl (1,25 mmol/l) durante un periodo de cuatro semanas. Si esto ocurre, se debe hacer un ajuste adecuado de la dosis según las instrucciones incluidas en esta misma sección. Si la tasa de aumento de hemoglobina es mayor de 2 g/dl (1,25 mmol/l) en un mes o si el nivel de hemoglobina está aumentando y se acerca a 12 g/dl (7,45 mmol/l), debe reducirse la dosis en aproximadamente un 25%. Si el nivel de hemoglobina sigue aumentando, se debe interrumpir el tratamiento hasta que el nivel de hemoglobina empiece a disminuir, momento en el que se podrá reiniciar el tratamiento con una dosis aproximadamente un 25 % inferior a la dosis administrada previamente.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis eficaz más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia al tiempo que se mantiene una concentración de hemoglobina inferior o igual a 12 g/dl (7,45 mmol/l).
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica. En los pacientes con una respuesta deficiente de la hemoglobina a NeoRecormon, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.4 y 5.1).
En presencia de hipertensión o de enfermedades cardiovasculares, cerebrovasculares o vasculoperiféricas, se debe determinar de modo individual el incremento semanal y el valor objetivo de la hemoglobina teniendo en cuenta el cuadro clínico.
El tratamiento con NeoRecormon se divide en dos fases:
1. Fase de corrección
- Administración subcutánea:
La dosis inicial es 3 x 20 UI/kg por semana. La dosis puede incrementarse cada 4 semanas en 3 x 20 UI/kg y semana si el aumento de la hemoglobina no ha sido adecuado (< 0,25 g/dl por semana).
La dosis semanal puede dividirse en dosis diarias.
- Administración intravenosa:
La dosis inicial es 3 x 40 UI/kg por semana, y puede aumentarse al cabo de 4 semanas a 80 UI/kg -tres veces por semana- y si son necesarios incrementos ulteriores serán de 20 UI/kg tres veces por semana, con intervalos mensuales.
Por ambas vías de administración, la dosis máxima no debe superar 720 UI/kg por semana.
2. Fase de mantenimiento
Para mantener el nivel de hemoglobina entre 10 y 12 g/dl, la dosis es inicialmente reducida a la mitad de la cantidad previamente administrada. Posteriormente, se ajustará la dosis individualmente para el paciente a intervalos de una o dos semanas (dosis de mantenimiento).
En caso de administración subcutánea, la dosis semanal puede administrarse en una inyección única o fraccionada en tres o siete inyecciones. Los pacientes que permanezcan estables en el régimen de una dosis única semanal pueden pasar a una administración única cada dos semanas. En este caso, puede ser necesario un aumento de la dosis.
Los resultados de los estudios clínicos en niños han revelado que, en general, a menor edad, se necesita mayor dosis de NeoRecormon. No obstante, hay que seguir el programa posológico recomendado ya que no puede predecirse la respuesta individual.
El tratamiento con NeoRecormon es normalmente crónico. Sin embargo, en caso necesario puede interrumpirse en cualquier momento. Los datos sobre la pauta posológica de una vez a la semana se han obtenido de estudios clínicos con una duración de tratamiento de 24 semanas.
Prevención de anemia en prematuros
La solución se administra por vía subcutánea a una dosis de 3 x 250 UI/kg p.c. por semana. Es probable que los niños que ya hayan recibido una transfusión previa cuando se inicie el tratamiento con NeoRecormon no se beneficien tanto como los prematuros no transfundidos. La duración de tratamiento recomendada es de 6 semanas.
Tratamiento de anemia sintomática inducida por quimioterapia en pacientes con cáncer Se debe administrar NeoRecormon por vía subcutánea a pacientes con anemia (por ejemplo, si la concentración de hemoglobina es menor o igual a 10 g/dl[6,2 mmol/l]). Los síntomas de la anemia y sus secuelas pueden variar en función de la edad, el sexo, y el grado de anemia. Por ello es necesario que el médico realice un seguimiento de la evolución clínica y el estado de cada paciente.
La dosis semanal puede administrarse como una inyección por semana o en dosis divididas entre 3 y 7 veces por semana.
La dosis inicial recomendada es de 30.000 UI por semana (que corresponde aproximadamente a 450 UI/kg de peso corporal por semana, en base al peso medio del paciente).
Debido a la variabilidad intraindividual de los pacientes, en ciertas ocasiones es posible llegar a observar en un paciente valores de hemoglobina superiores o inferiores a los esperados. La variabilidad de la hemoglobina se deberá manejar ajustando la dosis para mantener los valores de la hemoglobina dentro del intervalo de 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l). Deben evitarse concentraciones sostenidas de hemoglobina superiores a 12 g/dl (7,5 mmol/l); más adelante se proporcionan instrucciones para ajustar adecuadamente la dosis cuando se observen valores de hemoglobina superiores a los 12 g/dl (7,5 mmol/l).
Si a las 4 semanas de tratamiento el valor de hemoglobina aumenta hasta al menos 1 g/dl (0,62 mmol/l), se debe continuar con la dosis que en ese momento se esté administrando. Si los valores de hemoglobina no han aumentado al menos 1 g/dl (0,62 mmol/l), se debe considerar duplicar la dosis semanal. Si a las 8 semanas de tratamiento el valor de hemoglobina no aumenta hasta al menos 1 g/dl (0,62 mmol/l), es improbable que se produzca respuesta y, por consiguiente, el tratamiento debe ser interrumpido.
El tratamiento debe continuar durante las 4 semanas posteriores al final de la quimioterapia.
La dosis máxima no debe exceder de 60.000 UI por semana.
Una vez se ha alcanzado el objetivo terapéutico del paciente, la dosis debe reducirse del 25 al 50 % con el fin de mantener el valor de hemoglobina en ese nivel. Debe realizarse el ajuste posológico adecuado.
Si el nivel de hemoglobina excede los 12 g/dl (7,5 mmol/l) se debe reducir la dosis entre un 25 y un
50 % aproximadamente. Si el nivel de hemoglobina excede de 13 g/dl (8,1 mmol), se debe interrumpir temporalmente el tratamiento con NeoRecormon. El tratamiento debe reiniciarse con una dosis aproximadamente un 25 % inferior a la dosis previamente administrada después de que los niveles de hemoglobina desciendan hasta un valor menor o igual a 12 g/dl (7,5 mmol/l) o por debajo.
51 al cabo de 4 semanas el incremento de hemoglobina es superior a 2 g/dl (1,3 mmol/l), la dosis debe reducirse entre un 25 y un 50 %.
Se debe monitorizar adecuadamente a los pacientes para garantizar que se utiliza la dosis más baja autorizada de NeoRecormon que permita un control adecuado de los síntomas de la anemia.
Tratamiento para incrementar el rendimiento de la sangre autóloga donada La solución puede administrarse por vía intravenosa, en unos 2 minutos, o subcutáneamente. NeoRecormon se administra dos veces por semana durante 4 semanas. En aquellas ocasiones en que el hematocrito del paciente permite la donación de sangre, es decir el hematocrito es > 33 %, NeoRecormon se administra al final de la donación de sangre.
Durante la totalidad del período de tratamiento no debe excederse un hematocrito del 48 %.
La dosis debe ser determinada por el equipo quirúrgico, individualmente para cada paciente, en función de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos:
1. La cantidad de sangre pre-donada necesaria dependerá de la pérdida prevista de sangre, de los medios de conservación empleados y del estado físico del paciente.
Este volumen debe ser equivalente a la cantidad de sangre que se considera necesaria para evitar transfusiones de sangre homóloga.
La cantidad de sangre pre-donada necesaria se expresa en unidades en las cuales una unidad de nomograma es equivalente a 180 ml de glóbulos rojos.
2. La capacidad de donar sangre depende predominantemente del volumen sanguíneo del paciente y del hematocrito basal. Ambas variables determinan la reserva endógena de eritrocitos, que puede ser calculada conforme a la fórmula siguiente:
Reserva endógena de eritrocitos = volumen sanguíneo (ml) x (hematocrito - 33)/ 100 mujeres: volumen sanguíneo (ml) = 41 (ml/kg) x peso corporal (kg) + 1200 (ml)
hombres: volumen sanguíneo (ml) = 44 (ml/kg) x peso corporal (kg) + 1600 (ml)
(peso corporal: > 45 kg)
La indicación para un tratamiento con NeoRecormon y, en su caso, la dosis individual debe ser determinada a partir de la cantidad de sangre pre-donada necesaria y de la reserva endógena de eritrocitos según las gráficas siguientes:
Paciente femenino Paciente masculino
Cantidad de sangre pre-donada Cantidad de sangre pre-donada
necesaria [unidades] necesaria [unidades]


O 100 200 300 400 500 600 700 800 0 100 200 300 400 500 600 700 800
Reserva eritrocitaria endógena [ml] Reserva eritrocitaria endógena [ml]
La dosis única así determinada debe administrarse dos veces por semana a lo largo de 4 semanas. La dosis máxima no debe sobrepasar 1.600 UI/kg peso corporal por semana en administración intravenosa ó 1.200 UI/kg por semana en administración subcutánea.
Forma de administración
La jeringa precargada de NeoRecormon está lista para su uso. Sólo se pueden inyectar soluciones claras o ligeramente opalescentes, incoloras y prácticamente libres de partículas visibles. NeoRecormon en jeringa precargada es un producto estéril pero sin conservantes. Bajo ninguna circunstancia debe administrarse más de una dosis por jeringa; el medicamento es solo para uso individual.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Hipertensión mal controlada.
En la indicación para “Aumentar el rendimiento de sangre autóloga”: infarto de miocardio o accidente cerebrovascular en el mes anterior al tratamiento, angina de pecho inestable, riesgo aumentado de trombosis venosa profunda, como por ejemplo en aquellos con historial de enfermedad tromboembólica venosa.
4.4 Advertencias y precauciones especiales de empleo
NeoRecormon debe usarse con cautela en presencia de anemia refractaria con exceso de blastos en transformación, epilepsia, trombocitosis e insuficiencia hepática crónica. Y deben excluirse las deficiencias de ácido fólico y vitamina B12 pues reducen la eficacia de NeoRecormon.
Se debe tener precaución al aumentar de forma escalonada las dosis de NeoRecormon en pacientes con insuficiencia renal crónica, ya que las dosis acumuladas altas de epoetina pueden asociarse a un mayor riesgo de mortalidad, y de acontecimientos cardiovasculares y cerebrovasculares graves. En pacientes con una respuesta deficiente de la hemoglobina a epoetinas, se deben considerar explicaciones alternativas para la respuesta deficiente (ver las secciones 4.2 y 5.1).
Para garantizar una eritropoyesis eficaz, se debe evaluar el nivel de hierro en todos los pacientes antes y durante el tratamiento, y puede ser necesario un tratamiento suplementario con hierro administrado de acuerdo con las guías terapéuticas.
La sobrecarga grave de aluminio debido al tratamiento de la insuficiencia renal puede comprometer la eficacia de NeoRecormon.
La indicación de tratamiento con NeoRecormon en enfermos nefroscleróticos aún no sometidos a diálisis debe definirse individualmente, ya que la posible aceleración de la insuficiencia renal no puede descartarse con certeza.
Aplasia pura de células rojas (APCR)
Se han notificado casos de APCR causada por anticuerpos neutralizantes antieritropoyetina, asociados a tratamientos con eritropoyetinas, incluido NeoRecormon. Estos anticuerpos han presentado reacción cruzada con todas las proteínas eritropoyéticas, por los que los pacientes en los que se sospeche o se haya confirmado la presencia de anticuerpos neutralizantes contra eritropoyetina no deben ser tratados con NeoRecormon (ver sección 4.8).
APCR en pacientes con hepatitis C
Se debe interrumpir inmediatamente el tratamiento con epoetina, y realizar ensayos de anticuerpos antieritropoyetina si se produce una disminución paradójica de la hemoglobina y se desarrolla anemia severa asociada con bajos recuentos de reticulocitos. En pacientes con hepatitis C tratados con interferon y ribavirina se han notificados casos cuando las epoetinas se han utilizado concomitantemente. Las epoetinas no están aprobadas en el tratamiento de la anemia asociada a la hepatitis C.
Monitorización de la tensión arterial
Puede aparecer un aumento de la tensión arterial o un agravamiento de la hipertensión ya existente, especialmente en los casos en los que el hematocrito aumenta rápidamente. Estos incrementos en la tensión arterial pueden ser tratados con medicamentos. Si no pueden ser controlados con medicación se recomienda interrumpir transitoriamente la terapia con NeoRecormon. En particular al principio de la terapia se recomienda la monitorización regular de la tensión arterial; también durante la diálisis. Pueden producirse crisis hipertensivas con síntomas de encefalopatías y que requieren atención médica inmediata y cuidados médicos intensivos. Debe prestarse especial atención a las cefaleas súbitas lacerantes hemicraneales como posible signo de advertencia.
Insuficiencia renal crónica
En pacientes coninsuficiencia renal crónica puede sobrevenir un aumento dosis-dependiente moderado en la cifra plaquetaria dentro del margen normal durante el tratamiento con NeoRecormon, especialmente después de la administración intravenosa. La regresión tiene lugar en el curso de la prosecución de la terapia. Se recomienda el control regular de la cifra de plaquetas durante las 8 primeras semanas de tratamiento.
Concentración de hemoglobina
En pacientes con insuficiencia renal crónica, la concentración de hemoglobina en la fase de mantenimiento no debe exceder el límite superior del rango recomendado en la sección 4.2. En ensayos clínicos se observó un aumento del riesgo de fallecimiento y de acontecimientos cardiovasculares graves o acontecimientos cerebrovasculares incluido el infarto, cuando se administraron agentes estimulantes de la eritropoyesis (AEEs) con el fin de alcanzar un nivel de hemoglobina superior a 12 g/dl (7,5 mmol/l).
En ensayos clínicos controlados no se han observado beneficios significativos atribuibles a la administración de epoetinas cuando se aumentaba la concentración de hemoglobina por encima del nivel necesario para controlar los síntomas de anemia y evitar las transfusiones sanguíneas.
En niños prematuros puede producirse un ligero aumento de la cifra plaquetaria, en especial hasta el día 12 -14 de vida, por lo tanto las plaquetas deben controlarse regularmente.
Efectos sobre el crecimiento tumoral
Las epoetinas son factores de crecimiento que estimulan de forma primaria la producción de glóbulos rojos. Los receptores de epoetina pueden expresarse sobre la superficie de una variedad de células tumorales. Como sucede con todos los factores de crecimiento, existe la preocupación de que las epoetinas pudieran estimular el crecimiento de tumores. En varios ensayos controlados, no se ha observado que las epoetinas mejoren la supervivencia global o que disminuyan el riesgo de progresión del tumor en pacientes con anemia asociada con cáncer.
En ensayos clínicos controlados, se ha observado que el uso de NeoRecormon y otros agentes estimulantes de la eritropoyesis (AEEs):
- acortaba el tiempo de progresión del tumor en pacientes con cáncer avanzado de cabeza y cuello que recibían radioterapia cuando se administraba para conseguir una concentración de hemoglobina por encima de 14 g/dl (8,7 mmol/l),
- acortaba la supervivencia global y aumentaba el número de fallecimientos atribuidos a la progresión de la enfermedad a los 4 meses, en pacientes con cáncer de mama metastásico que recibían quimioterapia cuando se administraba para conseguir una concentración de hemoglobina entre los 12 y los 14 g/dl (7,5-8,7 mmol/l),
- aumentaba el riesgo de fallecimiento cuando se administraba para conseguir una concentración de hemoglobina de 12 g/dl (7,5 mmol/l) en pacientes con enfermedad maligna activa y que no recibían ni quimioterapia ni radioterapia. El uso de los AEEs no está indicado en esa población de pacientes.
En vista de lo anterior, en algunas situaciones clínicas la transfusión sanguínea debe ser el tratamiento de elección para la anemia en pacientes con cáncer. La decisión de administrar eritropoyetinas recombinantes se tomará en base a la evaluación de la relación beneficio/riesgo junto con la aceptación individual del paciente, teniendo en cuenta el contexto clínico específico. Los factores que deben considerarse en esta evaluación son el tipo de tumor y su estadio, el grado de anemia, la esperanza de vida, el entorno en el que el paciente está siendo tratado y la preferencia del paciente (ver sección 5.1).
Puede haber un aumento de la tensión arterial que puede ser tratada con medicamentos. Se recomienda por tanto monitorizar la tensión arterial, en particular en la fase inicial de tratamiento en pacientes con cáncer.
En pacientes con cáncer también deberán monitorizarse regularmente el recuento plaquetario y el valor de hemoglobina.
En pacientes en programa de predonación de sangre autóloga, puede sobrevenir un aumento de la cifra de plaquetas, generalmente dentro de los límites normales. Por tanto, se recomienda la determinación de esta cifra al menos una vez por semana. Si se produce un aumento plaquetario de más de 150 x 109/l o si las plaquetas ascienden por encima del margen normal, se debe suspender el tratamiento con NeoRecormon.
En niños prematuros no se podría descartar el riesgo potencial de que la eritropoyetina provoque retinopatía, por lo tanto se debe tener precaución y la decisión de tratar a un niño prematuro se debe tomar teniendo en cuenta el beneficio potencial y el riesgo de este tratamiento, así como las opciones alternativas de tratamiento que estén disponibles.
En pacientes con insuficiencia renal crónica, a menudo se precisa aumentar la dosis de heparina durante la hemodiálisis en la terapia con NeoRecormon debido al aumento del volumen de hematocrito. Si la heparinización no es óptima es posible que se produzca la oclusión del sistema de diálisis.
En pacientes con alteración renal crónica con riesgo de trombosis de derivación, se debe considerar la revisión temprana de la derivación y profilaxis de la trombosis mediante la administración, por ejemplo, de ácido acetilsalicílico.
Los niveles séricos de potasio y fosfato deben ser controlados regularmente durante la terapia con NeoRecormon. Se ha informado de aumentos de potasio en unos pocos pacientes urémicos que recibieron NeoRecormon si bien no se ha establecido una relación causal. En caso de observar un nivel elevado o creciente de potasio se debe considerar la interrupción de la administración de NeoRecormon hasta que el nivel haya sido corregido.
Para el uso de NeoRecormon en un programa de predonación de sangre autóloga deben ser observadas las directrices oficiales sobre donación de sangre, en particular las siguientes:
- sólo deben donar los pacientes con hematocrito > 33 % (hemoglobina >11 g/dl [6,83 mmol/l]);
- debe procederse con especial cuidado con pacientes con un peso inferior a 50 kg;
- el volumen individual extraído no debe superar aproximadamente el 12 % del volumen sanguíneo estimado del paciente.
El tratamiento debe reservarse a pacientes en los que se considere particularmente importante evitar la transfusión de sangre homóloga teniendo en cuenta la evaluación riesgo/beneficio de las transfusiones homólogas.
Uso indebido
Un uso indebido del producto por personas sanas puede llevar a un aumento excesivo del volumen de hematocrito, lo cual puede asociarse con complicaciones del sistema cardiovascular con riesgo para la vida.
Excipientes
NeoRecormon en jeringa precargada contiene, como máximo, 0,3 mg de fenilalanina por jeringa como excipiente. Este hecho se deberá tener en cuenta en los pacientes afectados por formas graves de fenilcetonuria.
Este medicamento contiene menos de 1 mmol (23 mg) de sodio por jeringa, por lo que se considera esencialmente “exento de sodio”.
Trazabilidad de NeoRecormon
Para mejorar la trazabilidad de los agentes estimulantes de la eritropoyesis (AEEs), se debe registrar (o indicar) claramente el nombre comercial del AEE administrado en la historia clínica del paciente.
4.5 Interacción con otros medicamentos y otras formas de interacción
Los resultados clínicos obtenidos hasta el presente no ponen de manifiesto interacción alguna de NeoRecormon con otros medicamentos.
Los estudios llevados a cabo en animales mostraron que la epoetina beta no potencia la mielotoxicidad de los medicamentos citostáticos, tales como etopósido, cisplatino, ciclofosfamida y fluorouracilo.
4.6 Fertilidad, embarazo y lactancia
Fertilidad
Los estudios en animales no muestran efectos dañinos directos o indirectos sobre el embarazo, desarrollo embrional/fetal, parto o desarrollo postnatal (ver sección 5.3).
Embarazo
No hay datos relativos al uso de NeoRecormon en mujeres embarazadas.
Se debe tener precaución cuando se prescriba a mujeres embarazadas
Lactancia
Se desconoce si la epoetina beta se excreta en la leche materna. La decisión de continuar/interrumpir la lactancia o de continuar/interrumpir el tratamiento con epoetina beta se debe hacer teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento con epoetina beta para la madre.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de NeoRecormon sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Basados en los resultados de los ensayos clínicos que incluyen 1.725 pacientes, se espera que aproximadamente el 8 % de los pacientes tratados con NeoRecormon sufran reacciones adversas.
Pacientes anémicos con insuficiencia renal crónica
La reacción adversa más frecuente durante el tratamiento con NeoRecormon es un aumento de la tensión arterial o agravamiento de la hipertensión existente, especialmente en casos de aumento rápido del hematocrito (ver sección 4.4). En algunos pacientes cuya tensión arterial es normal o baja también pueden darse crisis hipertensivas con síntomas de encefalopatías (p.ej. cefaleas y estados de confusión, trastornos sensitivomotores - como alteraciones del habla o de la ambulación - hasta convulsiones tónico-clónicas) (ver sección 4.4).
Puede darse trombosis de derivación, especialmente en pacientes que tienen tendencia a la hipotensión o cuyas fístulas arteriovenosas presentan complicaciones (p.ej. estenosis, aneurismas), ver sección 4.4. En la mayoría de los casos se observa una caída en los valores séricos de ferritina simultáneamente con el aumento del volumen de hematocrito (ver sección 4.4). Además, en casos aislados se han observado incrementos transitorios en los niveles séricos de potasio y fosfato (ver sección 4.4).
En casos aislados se ha notificado aplasia pura de células rojas (APCR) mediada por anticuerpos neutralizantes antieritropoyetina asociada al tratamiento con NeoRecormon. En caso de que se diagnostique APCR mediada por anticuerpos antieritropoyetina, el paciente deberá interrumpir el tratamiento con NeoRecormon, el cual no se deberá sustituir por otra proteína eritropoyética (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 1, que figura más adelante.
Pacientes con cáncer
El dolor de cabeza y la hipertensión relacionados con el tratamiento con epoetina beta son frecuentes y pueden tratarse con medicamentos (ver sección 4.4).
En algunos pacientes se observa un descenso de los valores de hierro sérico (ver sección 4.4).
Estudios clínicos han puesto de manifiesto que la frecuencia de acontecimientos tromboembólicos es más alta en pacientes con cáncer tratados con NeoRecormon que en los controles no tratados o tratados con placebo. En pacientes tratados con NeoRecormon, esta incidencia es del 7 % comparado con el 4 % en el grupo control; este hecho no está asociado con un incremento en la mortalidad tromboembólica comparado con los grupos control.
Estas reacciones adversas se enumeran en la Tabla 2, que figura más adelante.
Pacientes incluidos en un programa de predonación de sangre autóloga Los pacientes incluidos en un programa de predonación de sangre autóloga han demostrado una frecuencia ligeramente superior de accidentes tromboembólicos. Sin embargo, no se pudo establecer una relación causal con el tratamiento con NeoRecormon.
En ensayos clínicos controlados con placebo, la deficiencia transitoria de hierro fue más acusada en los pacientes tratados con NeoRecormon que en los pacientes control (ver sección 4.4).
Estas reacciones adversas se enumeran en la Tabla 3, que figura más adelante.
Tabla de reacciones adversas
Las reacciones adversas se enumeran de acuerdo con la clasificación de órganos del sistema MedDRA y por categoría de frecuencia.
Las categorías de frecuencia se definen utilizando la siguiente convención:
muy frecuentes (> 1/10), frecuentes (> 1/100 hasta <1/10), poco frecuentes (>1/1.000 hasta <1/100), raras (>1/10.000 hasta <1/1.000); muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 1: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con IRC_
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión Crisis hipertensivas |
Frecuente Poco frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
|
Trastornos de la sangre y |
Trombosis de derivación |
Raras |
|
del sistema linfático |
Trombocitosis |
Muy raras |
Tabla 2: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes con cáncer__
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos vasculares |
Hipertensión |
Frecuente |
|
Trastornos de la sangre y del sistema linfático |
Acontecimientos tromboembólicos |
Frecuente |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
Tabla 3: Reacciones adversas que se atribuyen al tratamiento con NeoRecormon en ensayos
clínicos controlados en pacientes incluidos en un programa de predonación de sangre autóloga
|
Sistema de Clasificación de Órganos |
Reacción adversa |
Frecuencia |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuente |
- Prematuros
Es muy frecuente un descenso de los valores de ferritina sérica (ver sección 4.4).
Descripción de reacciones adversas seleccionadas
Raramente pueden ocurrir reacciones cutáneas relacionadas con el tratamiento con epoetina beta como sarpullido, prurito, urticaria o reacciones en el lugar de la inyección. En casos muy raros se han notificado reacciones anafilácticas relacionadas con el tratamiento con epoetina beta. Sin embargo, en los ensayos clínicos controlados no se encontró una mayor incidencia de reacciones de hipersensibilidad.
Se han comunicado casos muy raros de síntomas gripales relacionados con el tratamiento con epoetina beta como fiebre, escalofríos, dolores de cabeza, dolor de las extremidades, malestar general, y/o dolor óseo, particularmente al inicio del tratamiento. Estas reacciones son de intensidad leve a moderada y desaparecen al cabo de un par de horas o días.
Los resultados obtenidos en un ensayo clínico controlado realizado con epoetina alfa o darbepoetina alfa, notificaron que la incidencia de infarto es frecuente.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
El margen terapéutico de NeoRecormon es muy amplio. No se han observado síntomas de sobredosis incluso a niveles séricos muy elevados.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antianémico, código ATC: B03XA01 Mecanismo de acción
La eritropoyetina es una glucoproteína que estimula la formación de eritrocitos a partir de sus progenitores asignados. Actúa como factor estimulante de la mitosis y hormona de diferenciación.
La epoetina beta, el principio activo de NeoRecormon, es idéntica en su composición aminoácida e hidrocarbonada a la eritropoyetina aislada de la orina de pacientes anémicos.
La eficacia biológica de la epoetina beta ha sido demostrada tras administración intravenosa y subcutánea en varios modelos animales in vivo (ratas normales y urémicas, ratones policitémicos, perros). Con la administración de epoetina beta aumentan el número de eritrocitos, los valores de Hb y la cifra de reticulocitos, al igual que la velocidad de incorporación de 59Fe.
In vitro (cultivo citológico de bazo de ratón) ha sido hallada una mayor incorporación de 3H-timidina en las células eritroides nucleadas del bazo tras incubación con epoetina beta.
Las investigaciones en cultivos de células de la médula ósea humana han revelado que la epoetina beta estimula específicamente la eritropoyesis y no afecta a la leucopoyesis. No han sido detectadas acciones citotóxicas de la epoetina beta sobre la médula ósea o sobre células cutáneas humanas.
Tras una dosis única de epoetina beta no se ha observado ningún efecto en el comportamiento o la actividad locomotora del ratón ni en las funciones circulatoria o respiratoria del perro.
Eficacia clínica y seguridad
En un ensayo aleatorizado, doble ciego, controlado con placebo, realizado en 4.038 pacientes con enfermedad renal crónica no sometidos a diálisis, con una diabetes tipo 2 y los niveles de hemoglobina < 11 g/dl, los pacientes recibieron tratamiento bien con darbepoetina alfa para alcanzar niveles de hemoglobina de 13 g/dl o con placebo (ver sección 4.4). El ensayo no cumplió su objetivo principal de demostrar una disminución en el riesgo de todas las causas de mortalidad, la morbilidad cardiovascular o enfermedad renal terminal. El análisis de los componentes individuales de la variable combinada mostró los siguientes HR (IC 95%): fallecimiento 1,05 (0,92-1,21), infarto cerebrovascular 1,92 (1,38-2,68), insuficiencia cardíaca congestiva 0,89 (0,74-1,08), infarto de miocardio 0,96 (0,751,23), hospitalización por isquemia del miocardio 0,84 (0,55-1,27), enfermedad renal terminal 1,02 (0,87-1,18).
En pacientes con IRC (tratados con diálisis, no tratados con diálisis, pacientes diabéticos y pacientes no diabéticos) se han realizado análisis agrupados post hoc de los estudios clínicos con AEEs. Se observó una tendencia a un aumento de las estimaciones del riesgo para la mortalidad por todas las causas y para los acontecimientos cardiovasculares y cerebrovasculares asociados a dosis acumuladas más altas de AEE independientemente de que los pacientes padecieran o no diabetes y de que recibieran o no tratamiento con diálisis (ver las secciones 4.2 y 4.4).
La eritropoyetina es un factor de crecimiento que estimula de forma primaria la producción de glóbulos rojos. Los receptores de eritropoyetina se encuentran también presentes en la superficie de algunas líneas celulares malignas.
Se ha estudiado la supervivencia y la progresión tumoral en cinco grandes ensayos controlados que incluyeron a 2.833 pacientes, de los cuales cuatro fueron ensayos doble-ciego controlados con placebo y uno fue un ensayo abierto. Dos de los ensayos reclutaron pacientes que estaban siendo tratados con quimioterapia. El nivel de hemoglobina que se quería alcanzar fue >13 g/dl en dos de los ensayos y de entre 12 y 14 g/dl en los otros tres. En el ensayo abierto no se observaron diferencias en la supervivencia global, entre los pacientes tratados con eritropoyetina humana recombinante y el grupo control. En los cuatro ensayos controlados con placebo, el índice de riesgo (hazard ratio) para la supervivencia global osciló entre 1,25 y 2,47 en favor de los grupos control. En todos estos ensayos se ha observado un aumento de la mortalidad inexplicable y estadísticamente significativo en pacientes que presentaban anemia asociada con diversos tipos frecuentes de cáncer y que recibieron eritropoyetina humana recombinante, en comparación con los grupos control. Las diferencias observadas en la incidencia de trombosis y complicaciones relacionadas, entre los pacientes que recibieron eritropoyetina humana recombinante y los que formaban parte de los grupos control, no permiten explicar satisfactoriamente los resultados de supervivencia global observados en los ensayos.
Un meta-análisis basado en datos individuales de pacientes, que incluyó datos de los 12 ensayos clínicos controlados realizados con NeoRecormon en pacientes anémicos con cáncer (n=2301), mostró un valor estimado del índice de riesgo (hazard ratio) de supervivencia global de 1,13 en favor de los grupos control (IC 95 %, CI 0,87, 1,46). En pacientes con un valor inicial de hemoglobina < 10 g/dl (n=899), el valor estimado del índice de riesgo (hazard ratio) para la supervivencia fue de 0,98 (IC 95% 0,68 al 1,40). Se observó un aumento del riesgo relativo de acontecimientos tromboembólicos en la población general (RR 1,62, IC 95 %: 1,13, 2,31).
Asimismo, se ha realizado un análisis de datos a nivel de paciente, en más de 13.900 pacientes con cáncer (tratados con quimioterapia, radioterapia, quimioradioterapia o sin tratamiento) que participaban en un total de 53 ensayos clínicos controlados en los que se administraban diversas epoetinas. En este meta-análisis se obtuvo un índice de riesgo (hazard ratio) para la supervivencia global de 1,06 a favor de los grupos control (IC 95%: 1,00, 1,12; 53 ensayos y 13.933 pacientes) y para los pacientes con cáncer que recibían quimioterapia, el índice de riesgo (hazard ratio) para la supervivencia global fue de 1,04 (IC 95%: 0,97, 1,11; 38 ensayos y 10.441 pacientes). Este meta-análisis además indica de forma consistente un aumento significativo del riesgo relativo de episodios tromboembólicos en pacientes con cáncer tratados con eritropoyetina recombinante humana (ver sección 4.4).
En casos muy raros, se han producido anticuerpos neutralizantes anti-eritropoyetina con o sin aplasia pura de células rojas (APCR) durante el tratamiento con rHuEPO.
5.2 Propiedades farmacocinéticas
Los estudios farmacocinéticos en voluntarios sanos y pacientes urémicos muestran que la semivida de la epoetina beta administrada por vía i.v. es de 4 a 12 horas y que el volumen de distribución corresponde de una a dos veces el volumen plasmático. Se han hallado resultados análogos en experimentos en animales con ratas normales y urémicas.
Tras la administración subcutánea de epoetina beta a pacientes urémicos la absorción retardada se traduce en una meseta de concentración sérica, cuya cota máxima se alcanza al cabo de 12 - 28 horas de media. El semiperíodo de vida terminal es mayor que después de la administración intravenosa, siendo la media de 13-28 horas.
La biodisponibilidad de la epoetina beta después de la administración subcutánea está comprendida entre el 23 y el 42 % en comparación con la administración intravenosa.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales sobre los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad de dosis repetidas, genotoxicidad y toxicidad para la reproducción.
Un estudio de carcinogenicidad con homólogos de eritropoyetina en ratones no reveló ningún signo de potencial proliferativo o carcinogénico.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Urea,
Cloruro sódico,
Polisorbato 20,
Fosfato monosódico dihidratado,
Fosfato disódico dodecahidratado,
Cloruro cálcico dihidratado,
Glicina,
L-Leucina,
L-Isoleucina,
L-Treonina,
L-Ácido glutámico,
L-Fenilalanina,
Agua para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
Conservar la jeringa precargada en el embalaje exterior, para protegerlo de la luz.
Con fines de uso ambulatorio, se puede mantener el producto fuera de la nevera durante un único periodo de hasta 3 días a temperatura ambiente (no superior a 25°C).
6.5 Naturaleza y contenido del envase
Jeringa precargada (vidrio tipo I), con un protector y un émbolo (goma teflonizada) con una aguja de inyección (27G1/2). Cada jeringa contiene 0,6 ml de solución.
Tamaños de envase de 1 jeringa precargada y 1 aguja o de 4 jeringas precargadas y 4 agujas.
Puede que solamente estén comercializados algunos tamaños de envase.
6.6 Precauciones especiales de eliminación y otras manipulaciones
En primer lugar, ¡lávese las manos!
1. Saque una jeringa del envase y asegúrese que la solución es clara, incolora y prácticamente libre de partículas visibles. Retire el protector de la jeringa.
2. Saque una aguja del envase, fíjela sobre la jeringa y retire la tapa protectora de la aguja.
3. Expulse el aire de la jeringa y aguja manteniendo la jeringa vertical y presionando suavemente el émbolo hacia arriba. Continúe presionando el émbolo hasta que la cantidad de solución de NeoRecormon en la jeringa sea la prescrita.
4. Limpie la piel en el lugar de inyección con un algodón con alcohol. Haga un pliegue tomando la piel entre el índice y el pulgar. Sujete el cuerpo de la jeringa por la parte más próxima a la aguja e inserte la aguja en el pliegue cutáneo con acción firme y rápida. Inyecte la solución de NeoRecormon. Retire la aguja rápidamente y aplique presión sobre el lugar de inyección con un apósito seco y estéril.
Este medicamento es solo para uso individual. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
8. NÚMEROS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/045-046
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16 de julio de 1997 Fecha de la última revalidación: 16 de julio de 2007.
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/
A. FABRICANTE(S) DEL (DE LOS) PRINCIPIO(S) ACTIVO(S) BIOLÓGICO(S) Y FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE(S) DEL (DE LOS) PRINCIPIO(S) ACTIVO(S) BIOLÓGICO(S) Y FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante del principio activo biológico
Roche Diagnostics GMBH Nonnenwald 2 D-82377 Penzberg Alemania
Nombre y dirección del fabricante responsables de la liberación de los lotes
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (Ver anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2)
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva
información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
NeoRecormon Multidosis 50.000 UI liofilizado y disolvente para solución inyectable Epoetina beta
1 vial contiene 50.000 UI de epoetina beta
1 vial contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico, fosfato disódico, cloruro cálcico, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico y L-Fenilalanina 1 ampolla contiene disolvente (alcohol bencílico y cloruro de benzalconio en agua para preparaciones inyectables)
Este medicamento contiene fenilalanina y sodio y el disolvente contiene alcohol bencílico. Para mayor información consultar el prospecto
Liofilizado y disolvente para solución inyectable
1 vial con liofilizado (50.000 UI) para solución para inyección. 1 ampolla con disolvente conservante (10 ml), 1 dispositivo de reconstitución y extracción, 1 jeringa desechable (10 ml), 1 aguja (21G2)
Uso subcutáneo e intravenoso después de reconstitución como se indica. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
CAD
La solución reconstituida es estable durante 1 mes si se conserva entre 2°C - 8 °C (en nevera)
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera
Conservar en el embalaje exterior, para protegerlo de la luz
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/019
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
(Texto de la tapa interior) Instrucciones para el manejo/empleo Ver prospecto
Texto del soporte
NeoRecormon Multidosis 50.000 UI 5.000 Ul/ml
Conservar entre 2°C - 8°C
Seguir siempre técnicas asépticas
Utilizar jeringuillas y agujas estériles y desechables para cada dosis
|
5.000 Ul/ml | |||||||||
|
500 UI |
1.000 UI |
1.500 UI |
2.000 UI |
2.500 UI |
3.000 UI |
3.500 UI |
4.000 UI |
4.500 UI |
5.000 UI |
|
0,1 ml |
0,2 ml |
0,3 ml |
0,4 ml |
0,5 ml |
0,6 ml |
0,7 ml |
0,8 ml |
0,9 ml |
1,0 ml |
neorecormon 50.000 UI
Incluido el código de barras 2D que lleva el identificador único
PC:
SN:
NN:
ETIQUETAS/VIALES
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
NeoRecormon Multidosis liofilizado para inyectable 50.000 UI Uso IV/SC
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento
3. FECHA DE CADUCIDAD
EXP
Reconst. lili CAD i l l l l l i
Una vez abierta, la solución reconstituida debe almacenarse durante un máximo de un mes a 2°C - 8°C
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
5.000 UI/1 ml
6. OTROS
ETIQUETAS/AMPOLLAS
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
10 ml de disolvente para NeoRecormon Multidosis 50.000 UI
(agua para preparaciones inyectables, alcohol bencílico y cloruro de benzalconio)
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
NeoRecormon 500 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 500 UI de epoetina beta
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
1 jeringa precargada (0,3 ml) y 1 aguja (30G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/025
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 500 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
NeoRecormon 500 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 500 UI de epoetina beta
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
6 jeringas precargadas (0,3 ml) y 6 agujas (30G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/026
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 500 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLISTER
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 500 UI solución inyectable en jeringa precargada Epoetina beta
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Ltd.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETAS
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
NeoRecormon 500 UI inyectable Uso IV/SC
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
0,3 ml
6. OTROS
NeoRecormon 2.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 2.000 UI de epoetina beta
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
1 jeringa precargada (0,3 ml) y 1 aguja (27G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/029
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 2.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
NeoRecormon 2.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 2.000 UI de epoetina beta
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
6 jeringas precargadas (0,3 ml) y 6 agujas (27G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/030
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 2.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLISTER
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 2.000 UI solución inyectable en jeringa precargada Epoetina beta
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Ltd.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETAS
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
NeoRecormon 2.000 UI inyectable Uso IV/SC
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
0,3 ml
6. OTROS
NeoRecormon 3.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 3.000 UI de epoetina beta.
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
1 jeringa precargada (0,3 ml) y 1 aguja (27G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/031
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 3.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
NeoRecormon 3.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 3.000 UI de epoetina beta
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
6 jeringas precargadas (0,3 ml) y 6 agujas (27G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/032
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 3.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLISTER
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 3.000 UI solución inyectable en jeringa precargada Epoetina beta
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Ltd.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
ETIQUETAS
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
NeoRecormon 3.000 UI inyectable Uso IV/SC
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
0,3 ml
6. OTROS
NeoRecormon 4.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 4.000 UI de epoetina beta
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
1 jeringa precargada (0,3 ml) y 1 aguja (27G1/2)
Uso subcutáneo e intravenosa. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/041
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 4.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
NeoRecormon 4.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 4.000 UI de epoetina beta.
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
6 jeringas precargadas (0,3 ml) y 6 agujas (27G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/042
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 4.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLISTER
1. NOMBRE DEL MEDICAMENTO
NeoRecorrmon 4.000 UI solución inyectable en jeringa precargada Epoetina beta
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Ltd.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
ETIQUETAS
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
NeoRecormon 4.000 UI inyectable Uso IV/SC
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
0,3 ml
6. OTROS
NeoRecormon 5.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 5.000 UI de epoetina beta
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
1 jeringa precargada (0,3 ml) y 1 aguja (27G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/033
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 5.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
NeoRecormon 5.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 5.000 UI de epoetina beta
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
6 jeringas precargadas (0,3 ml) y 6 agujas (27G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/034
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 5.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLISTER
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 5.000 UI solución inyectable en jeringa precargada Epoetina beta
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Ltd.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETAS
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
NeoRecormon 5.000 UI inyectable Uso IV/SC
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
0,3 ml
6. OTROS
NeoRecormon 6.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 6.000 UI de epoetina beta
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
1 jeringa precargada (0,3 ml) y 1 aguja (27G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/043
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 6.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
NeoRecormon 6.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 6.000 UI de epoetina beta
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
6 jeringas precargadas (0,3 ml) y 6 agujas (27G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/044
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 6.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLISTER
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 6.000 UI solución inyectable en jeringa precargada Epoetina beta
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Ltd.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETAS
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
NeoRecormon 6.000 UI inyectable Uso IV/SC
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
0,3 ml
6. OTROS
NeoRecormon 10.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 10.000 UI de epoetina beta
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
1 jeringa precargada (0,6 ml) y 1 aguja (27G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/035
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 10.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
NeoRecormon 10.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 10.000 UI de epoetina beta
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
6 jeringas precargadas (0,6 ml) y 6 agujas (27G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/036
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 10.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLISTER
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 10.000 UI solución inyectable en jeringa precargada Epoetina beta
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Ltd.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETAS
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
NeoRecormon 10.000 UI inyectable Uso IV/SC
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
0,6 ml
6. OTROS
NeoRecormon 20.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 20.000 UI de epoetina beta
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
1 jeringa precargada (0,6 ml) y 1 aguja (27G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/037
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 20.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
NeoRecormon 20.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 20.000 UI de epoetina beta
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
6 jeringas precargadas (0,6 ml) y 6 agujas (27G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/038
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 20.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLISTER
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 20.000 UI solución inyectable en jeringa precargada Epoetina beta
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Ltd.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETAS
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
NeoRecormon 20.000 UI inyectable Uso IV/SC
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
0,6 ml
6. OTROS
NeoRecormon 30.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 30.000 UI de epoetina beta
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
1 jeringa precargada (0,6 ml) y 1 aguja (27G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/045
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 30.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
NeoRecormon 30.000 UI solución inyectable en jeringa precargada Epoetina beta
1 jeringa precargada contiene 30.000 UI de epoetina beta.
1 jeringa contiene: Urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico, L-Fenilalanina y agua para preparaciones inyectables Este medicamento contiene fenilalanina y sodio. Para mayor información consultar el prospecto
Solución inyectable
4 jeringas precargadas (0,6 ml) y 4 agujas (27G1/2)
Uso subcutáneo e intravenoso. Leer el prospecto antes de utilizar este medicamento
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/031/046
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
neorecormon 30.000 UI
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLISTER
1. NOMBRE DEL MEDICAMENTO
NeoRecormon 30.000 UI solución inyectable en jeringa precargada Epoetina beta
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Ltd.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETAS
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
NeoRecormon 30.000 UI inyectable Uso IV/SC
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
0,6 ml
6. OTROS
B. PROSPECTO
Prospecto: información para el usuario
NeoRecormon multidosis 50.000 UI liofilizado y disolvente para solución inyectable
Epoetina beta
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
• Conserve este prospecto, ya que puede tener que volver a leerlo.
• Si tiene alguna duda, consulte a su médico o farmacéutico.
• Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
• Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto.Ver sección 4.
Contenido del prospecto:
1. Qué es NeoRecormon y para qué se utiliza
2. Qué necesita saber antes de empezar a usar NeoRecormon
3. Cómo usar NeoRecormon
4. Posibles efectos adversos
5. Conservación de NeoRecormon
6. Contenido del envase e información adicional
1. Qué es NeoRecormon y para qué se utiliza
Esta presentación de NeoRecormon contiene liofilizado blanco y disolvente. Una vez disuelto, NeoRecormon se inyecta debajo de la piel (subcutáneamente) o en la vena (intravenosamente).
Contiene epoetina beta, una hormona que estimula la producción de glóbulos rojos. Epoetina beta se produce por tecnología genética especializada y funciona exactamente de la misma forma que la hormona eritropoyetina natural humana.
Debe consultar a un médico si empeora o si no mejora.
NeoRecormon está indicado para:
• Tratamiento de la anemia sintomática provocada por enfermedad renal crónica (anemia renal) en pacientes sometidos a diálisis o que aún no están sometidos a diálisis.
• Tratamiento de la anemia con síntomas relacionados en pacientes adultos con cáncer tratados con quimioterapia.
• Tratamiento de personas donantes de su propia sangre antes de ser sometidas a una intervención quirúrgica. Las inyecciones de epoetina beta aumentarán la cantidad de sangre que puede extraerse de su cuerpo antes de la operación, para ponérsela durante la intervención o después de la misma (esto es una transfusión autóloga).
2. Qué necesita saber antes de empezar a usarNeoRecormon No use NeoRecormon
• si es alérgico a la epoetina beta o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6) o al ácido benzoico, metabolito del alcohol bencílico
• si tiene problemas de presión sanguínea que no pueden ser controlados
• si va a donar su propia sangre antes de someterse a una operación y:
• ha sufrido un infarto de miocardio o accidente cerebrovascular en el mes anterior al tratamiento
• si presenta angina de pecho inestable - nuevo dolor de pecho o dolor en aumento
• si tiene riesgo de que se le formen coágulos de sangre en las venas (trombosis de las venas profundas) - p. ej. si ha sufrido anteriormente coágulos
• en bebés o niños menores de 3 años, ya que el disolvente de NeoRecormon Multidosis contiene alcohol bencílico como conservante
Si sufriera cualquiera de estas situaciones o pudiera llegar a padecerlas, dígaselo a su médico. Advertencias y precauciones
Consulte a su médico antes de empezar a usar NeoRecormon
• si su anemia no mejora con el tratamiento con epoetina
• si tiene bajos niveles de algunas vitaminas del complejo B (ácido fólico o vitamina B12)
• si tiene niveles altos de aluminio en sangre
• si el recuento de plaquetas es alto
• si tiene enfermedad hepática crónica
• si tiene epilepsia
• si ha desarrollado anticuerpos anti-eritropoyetina y aplasia pura de células rojas (disminución o anulación de la producción de glóbulos rojos) durante una exposición previa a cualquier sustancia eritropoyética. En este caso, no debe cambiar a NeoRecormon.
Tenga especial cuidado con otros medicamentos que estimulan la producción de glóbulos rojos:
NeoRecormon es uno de los agentes estimuladores de la producción de glóbulos rojos como lo hace la proteína humana eritropoyetina. Su médico deberá siempre registrar el producto exacto que usted está utilizando.
Advertencia especial:
Durante el tratamiento con Neorecormon
Si usted es un paciente con enfermedad renal crónica, y en particular si no responde adecuadamente a NeoRecormon, su médico controlará su dosis de NeoRecormon ya que el aumento repetido de la dosis de NeoRecormon si no está respondiendo al tratamiento puede aumentar el riesgo de tener un problema de corazón o de los vasos sanguíneos, y podría aumentar el riesgo de infarto de miocardio, accidente cerebrovascular y muerte.
Si usted es un paciente con cáncer debe saber que NeoRecormon puede actuar como factor de crecimiento de las células de la sangre y que en algunas circunstancias puede tener un efecto negativo sobre el cáncer. Dependiendo de su situación individual puede ser preferible una transfusión de sangre. Por favor, háblelo con su médico.
Si usted es un enfermo nefrosclerótico aún no sometido a diálisis, su médico decidirá si el tratamiento es conveniente para usted. Esto es debido a que no se puede descartar con certeza la posible aceleración de la insuficiencia renal.
Su médico puede realizar análisis de sangre de forma regular para controlar:
• sus niveles de potasio. Si usted presenta niveles de potasio altos o en aumento su doctor debe reconsiderar su tratamiento
• su recuento de plaquetas. Durante el tratamiento con epoetina el número de plaquetas puede aumentar de forma leve a moderada y esto puede producir cambios en la coagulación sanguínea.
Si usted es un paciente con alteración renal sometido a hemodiálisis, su médico deberá ajustar su dosis de heparina. Esto evitará la obstrucción de los tubos del sistema de diálisis.
Si usted es un paciente con alteración renal sometido a hemodiálisis y con riesgo de trombosis de derivación podrían formarse coágulos (trombosis) en su derivación (vaso utilizado para conectar al sistema de diálisis). Su médico podría recetarle ácido acetil salicílico o modificar la derivación.
Si usted está donando su propia sangre antes de ser operado, su médico necesitará:
• comprobar que usted puede donar sangre, especialmente si su peso es inferior a 50 kg
• comprobar que usted tiene nivel suficiente de glóbulos rojos (hemoglobina de al menos 11 g/dl)
• asegurarse que solo puede extraerle de una vez el 12 % de su volumen sanguíneo.
No haga un uso inapropiado de NeoRecormon:
El uso inapropiado de NeoRecormon por personas sanas puede llevar a un aumento de las células sanguíneas y, en consecuencia, espesamiento de la sangre que puede asociarse con complicaciones cardíacas o vasculares con riesgo para la vida.
Uso de NeoRecormon con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento, incluso los adquiridos sin receta.
Embarazo, lactancia y fertilidad
No se ha obtenido experiencia adecuada con NeoRecormon en mujeres durante el embarazo y la lactancia. Pida consejo a su médico o farmacéutico antes de tomar cualquier medicamento. NeoRecormon no ha mostrado evidencia de alteración de la fertilidad en animales. Se desconoce el riesgo potencial en humanos.
Conducción y uso de máquinas
No se han descrito efectos sobre la capacidad para conducir y utilizar máquinas.
NeoRecormon contiene fenilalanina, alcohol bencílico y sodio
Este medicamento contiene fenilalanina. Puede ser perjudicial para personas con fenilcetonuria.
Si tiene fenilcetonuria, consulte con su médico acerca del tratamiento con NeoRecormon.
NeoRecormon contiene hasta 40 mg de alcohol bencílico como conservante por ampolla de disolvente, y por lo tanto, no debe administrarse a lactantes o niños menores de tres años.
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis, esto es, esencialmente “exento de sodio”.
3. Cómo usar NeoRecormon
La terapia con NeoRecormon debe iniciarse por un médico con experiencia en su enfermedad. La primera dosis se administra normalmente bajo control médico ante la posibilidad de que se produzca una reacción alérgica.
Las inyecciones de NeoRecormon deben pues ser administradas por personal sanitario entrenado (enfermero), médico u otro profesional. (Ver instrucciones de uso al final de este prospecto).
Dosis de NeoRecormon
La dosis de NeoRecormon depende del estado de su enfermedad, la forma en que se administra la inyección (bajo la piel o en una vena) y su peso corporal. Su médico calculará la dosis apropiada para usted.
Su médico utilizará la dosis eficaz más baja para controlar los síntomas de su anemia.
Si no responde adecuadamente a NeoRecormon, su médico controlará su dosis y le informará si necesita cambiar la dosis de NeoRecormon.
• Anemia sintomática provocada por enfermedad renal crónica
Las inyecciones se administran bajo la piel o en una vena. Si la solución se le administra en vena, debe ser inyectada a lo largo de unos 2 minutos, p.ej. los pacientes en hemodiálisis recibirán la inyección por vía de la fístula arteriovenosa al final de la diálisis.
Los pacientes no hemodializados recibirán normalmente la administración bajo la piel.
El tratamiento con NeoRecormon se divide en dos fases:
a) Corrección de la anemia
La dosis inicial para la administración bajo la piel es de 20 UI por inyección por cada kg de peso, administrada tres veces por semana.
Después de 4 semanas, el médico le hará pruebas y si la respuesta al tratamiento no es suficiente, puede incrementarle la dosis a 40 UI/kg por inyección, administrada tres veces por semana. El médico puede seguir aumentando su dosis a intervalos mensuales si fuera necesario.
La dosis semanal puede dividirse en dosis diarias.
La dosis inicial para la administración en vena es de 40 UI por inyección por cada kg de peso corporal administradas tres veces por semana.
Después de 4 semanas, su médico le hará pruebas y, si la respuesta al tratamiento no es suficiente, puede incrementarle la dosis a 80 UI/kg por inyección, administradas tres veces por semana. El médico puede seguir aumentando la dosis a intervalos mensuales si fuera necesario.
Para ambos tipos de inyección, la dosis máxima no debe superar 720 UI por cada kg de su peso corporal por semana.
b) Mantenimiento de los niveles de glóbulos rojos
Dosis de mantenimiento: Una vez que sus glóbulos rojos alcanzan un nivel aceptable, la dosis se reduce a la mitad de la dosis utilizada para corregir la anemia. La dosis semanal puede administrarse una vez por semana o dividirse en tres o siete dosis por semana. Si sus glóbulos rojos permanecen estables en un régimen de una dosis única semanal, puede pasar a una administración única cada dos semanas. En este caso, puede ser necesario un aumento de la dosis.
El médico puede ajustar su dosis cada una o dos semanas hasta encontrar su dosis individual de mantenimiento. Los niños iniciarán el tratamiento siguiendo las mismas normas. En los ensayos clínicos, los niños normalmente necesitaron dosis más altas de NeoRecormon (cuanto más pequeño es el niño, mayor es la dosis).
El tratamiento con NeoRecormon normalmente es un tratamiento a largo plazo. No obstante, puede interrumpirse en cualquier momento en caso necesario.
• Adultos con anemia sintomática con cáncer tratados con quimioterapia Las inyecciones se administran bajo la piel.
Su médico puede iniciar el tratamiento con NeoRecormon si su nivel de hemoglobina es menor o igual de 10 g/dl. Después de comenzar el tratamiento, su médico mantendrá su nivel de hemoglobina entre 10 y 12 g/dl.
La dosis semanal inicial es de 30.000 UI. Esta dosis puede administrarse en una inyección por semana, o en dosis divididas de 3 a 7 inyecciones por semana. Su médico tomará muestras de sangre de forma regular. En función de los resultados de los análisis, podrá aumentarle o reducirle la dosis o interrumpir el tratamiento. Los valores de hemoglobina no excederán de 12 g/dl.
El tratamiento debe continuar hasta 4 semanas después de terminar la quimioterapia.
La dosis máxima no debe exceder de 60.000 UI por semana.
• Pacientes donantes de su propia sangre antes de someterse a cirugía
Las inyecciones se administran en vena en unos 2 minutos, o debajo de la piel.
La dosis de NeoRecormon dependerá de su condición, de los niveles de glóbulos rojos y de la cantidad de sangre que donará antes de la intervención.
La dosis calculada por el médico se administra dos veces por semana durante 4 semanas. Cuando usted done la sangre, recibirá NeoRecormon al final de la donación.
La dosis máxima no debe exceder
• para inyección en vena: 1.600 UI por kg de peso por semana
• para inyecciones bajo la piel: 1.200 UI por kg de peso por semana
Si le inyectan demasiado NeoRecormon
Si cree que le han inyectado más NeoRecormon del que debiera, contacte con su médico. Es poco probable que sea grave. Incluso en presencia de altos niveles en sangre no han sido observados síntomas de intoxicación.
Si olvidó usar NeoRecormon
Si cree que ha olvidado una inyección o le han inyectado muy poco, dígaselo a su médico.
No se administre una dosis doble para compensar las dosis olvidadas.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede tener efectos adversos, aunque no todas
las personas los sufran.
Efectos adversos que pueden afectar a cualquier paciente
• La mayor parte de los pacientes (muy frecuentes pueden afectar a más de 1 de cada 10 personas) presenta niveles bajos de hierro en la sangre. Casi todos los pacientes deben ser tratados con suplementos de hierro durante el tratamiento con NeoRecormon.
• Rara vez (pueden afectar hasta 1 de cada 1.000 personas) han aparecido reacciones alérgicas o de la piel como sarpullido o urticaria, picor o reacciones en el lugar de la inyección.
• Muy rara vez (pueden afectar hasta 1 de cada 10.000 personas) han aparecido formas severas de reacción alérgica, especialmente después de la inyección. Estas han de tratarse de inmediato. Si usted presenta sibilancias o dificultad para respirar; inflamación en la lengua, cara o garganta o alrededor del lugar de la inyección; si siente mareo o desmayos o si se cae, llame al médico de inmediato.
• Muy rara vez (pueden afectar hasta 1 de cada 10.000 personas) la gente experimenta síntomas gripales, especialmente cuando inician el tratamiento. Estos síntomas incluyen fiebre, escalofríos, dolores de cabeza, dolor de las extremidades, dolor de huesos y/o malestar general. Estas reacciones son normalmente leves o moderadas y desaparecen en unas horas o días.
Efectos adversos adicionales en pacientes con enfermedad renal crónica (anemia renal)
• Los efectos adversos más comunes (muy frecuentes pueden afectar a más de 1 de cada 10 personas) son aumentos de la presión sanguínea, empeoramiento de la presión sanguínea existente y dolor de cabeza. Su médico comprobará regularmente su presión sanguínea, especialmente al inicio del tratamiento y podrá tratar la presión sanguínea elevada con medicamentos apropiados o interrumpir temporalmente el tratamiento con NeoRecormon.
• Si tiene dolores de cabeza, especialmente súbitos, punzantes, tipo migraña, confusión, alteración del habla, inestabilidad al andar, ataques o convulsiones llame enseguida al médico. Pueden ser síntomas de presión sanguínea extremadamente alta (crisis hipertensiva), aunque la presión sanguínea sea habitualmente normal o baja. Esta sintomatología debe tratarse de inmediato.
• Si usted tiene presión sanguínea baja o complicaciones de derivación puede padecer una trombosis de derivación (un coágulo de sangre en el vaso utilizado para conectar al sistema de diálisis).
• Muy rara vez (pueden afectar hasta 1 de cada 10.000 personas), los pacientes presentan niveles elevados de potasio o fosfatos en sangre. Estos pueden ser tratados por su médico.
• Se ha observado aplasia pura de células rojas (APCR) causada por anticuerpos neutralizantes durante el tratamiento con eritropoyetina, incluidos algunos casos aislados durante el tratamiento con NeoRecormon. APCR quiere decir que el organismo para o reduce la producción de glóbulos rojos. Esto causa una anemia grave, entre cuyos síntomas se incluye cansancio inusual y carencia de energía. Si su organismo produce anticuerpos neutralizantes, su médico interrumpirá la terapia con NeoRecormon, y determinará la mejor forma de actuar para tratar su anemia.
Efectos adversos adicionales en adultos con cáncer tratados con quimioterapia
• Ocasionalmente puede producirse un aumento de la presión sanguínea y dolor de cabeza.
Su médico puede tratar la presión sanguínea elevada con fármacos.
• Se ha observado un aumento de la incidencia de coágulos de sangre.
Efectos adversos adicionales en pacientes donantes de su propia sangre antes de someterse a cirugía
• Se ha observado un ligero aumento de la incidencia de coágulos de sangre.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de NeoRecormon
• Mantener este medicamento fuera de la vista y del alcance de los niños.
• No utilice NeoRecormon después de la fecha de caducidad que aparece en la caja y en la etiqueta.
• Conservar en nevera (entre 2°C - 8°C).
• El vial puede mantenerse fuera de la nevera durante un único periodo máximo de hasta 5 días a temperatura ambiente (no superior a 25°C).
• La solución reconstituida es estable durante un mes si se conserva entre 2°C - 8°C en nevera.
• Conservar el envase en el embalaje exterior para protegerlo de la luz.
• Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico como deshacerse de los envases y de los medicamentos que no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de NeoRecormon
• El principio activo es la epoetina beta. Un vial contiene 50.000 UI (unidades internacionales) de epoetina beta.
• Los otros componentes son:
En el liofilizado: urea, cloruro sódico, polisorbato 20, fosfato monosódico, fosfato disódico, cloruro cálcico, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico y L-Fenilalanina.
• En el disolvente: alcohol bencílico y cloruro de benzalconio como conservantes y agua para preparaciones inyectables.
Aspecto de NeoRecormon y contenido del envase
NeoRecormon Multidosis es un liofilizado y un disolvente para solución inyectable. El liofilizado es blanco y el disolvente es incoloro y transparente.
Cada envase contiene un vial con epoetina beta 50.000 UI, 1 ampolla con 10 ml de disolvente, un dispositivo de reconstitución y extracción, una aguja (21G2) y una jeringa desechable.
Titular de la autorización de comercialización
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
Responsable de la fabricación
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien
N.V. Roche S.A.
Tél/Tel: +32 (0) 2 525 82 11
Etarapna
Pom Etarapua EOOfl Tea: +359 2 818 44 44
Ceská republika
Roche s. r. o.
Tel: +420 -2 20382111
Danmark
Roche a/s
Tlf: +45 - 36 39 99 99
Deutschland
Roche Pharma AG Tel: +49 (0) 7624 140
Eesti
Roche Eesti OÜ Tel: + 372 -6 177 380
Lietuva
UAB “Roche Lietuva”
Tel: +370 5 2546799
Luxembourg/Luxemburg
(Voir/siehe Belgique/Belgien)
Magyarország
Roche (Magyarország) Kft. Tel: +36 - 23 446 800
Malta
(See United Kingdom)
Nederland
Roche Nederland B.V. Tel: +31 (0) 348 438050
Norge
Roche Norge AS Tlf: +47 - 22 78 90 00
|
EXXáSa Roche (Hellas) A.E. Tn^: +3 0 210 61 66 100 |
Osterreich Roche Austria GmbH Tel: +43 (0) 1 27739 |
|
España Roche Farma S.A. Tel: +34 -91 324 81 00 |
Polska Roche Polska Sp.z o.o. Tel: +48 -22 345 18 88 |
|
France Roche Tél: +33 (0)1 47 61 40 00 |
Portugal Roche Farmacéutica Química, Lda Tel: +351 -21 425 70 00 |
|
Hrvatska Roche d.o.o Tel: +385 1 4722 333 |
Romania Roche Romania S.R.L. Tel: +40 21 206 47 01 |
|
Ireland Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700 |
Slovenija Roche farmacevtska druzba d.o.o. Tel: +386 - 1 360 26 00 |
|
Ísland Roche a/s c/o Icepharma hf Sími: +354 540 8000 |
Slovenská republika Roche Slovensko, s.r.o. Tel: +421 -2 52638201 |
|
Italia Roche S.p.A. Tel: +39 -039 2471 |
Suomi/Finland Roche Oy Puh/Tel: +358 (0) 10 554 500 |
|
Kúnpoq r.A.Zxa^áxn? & £ra AxS. Tn^: +357 - 22 76 62 76 |
Sverige Roche AB Tel: +46 (0) 8 726 1200 |
|
Latvija Roche Latvija SIA Tel: +371 -6 7039831 |
United Kingdom Roche Products Ltd. Tel: +44 (0) 1707 366000 |
Fecha de la última revisión de este prospecto: <{MM/AAAA}> <{mes AAAA}>
La información detallada de este medicamento está disponible en al página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/
Esta información está destinada únicamente a profesionales del sector sanitario:
Esta preparación multidosis se puede utilizar para varios pacientes durante un período de un mes después de su reconstitución. Para evitar el riesgo de contaminación del contenido es conveniente que siempre se sigan las técnicas asépticas y se usen jeringas y agujas desechables, estériles, para administrar cada dosis. Por favor, compruebe que solo hay un vial de NeoRecormon Multidosis en uso (es decir, reconstituido) en cada ocasión.
No mezcle NeoRecormon con otras inyecciones o soluciones para perfusión.
Use exclusivamente materiales para inyección de plástico.
Instrucciones de uso
En primer lugar, ¡lávese las manos!
Preparación de la solución de NeoRecormon Multidosis
(1) Saque del envase el vial liofilizado. Escriba la fecha de reconstitución y la de caducidad en la etiqueta (la caducidad es 1 mes después de la reconstitución).
(2) Quite el protector de plástico del vial.
(3) Desinfecte la tapa de goma con alcohol.
(4) Extraiga el dispositivo de reconstitución y de extracción (que permite el intercambio de aire estéril) fuera del envoltorio y quite el protector del dispositivo.
(5) Coloque el dispositivo en el vial hasta que el cierre se coloque en su sitio.
(6) Coloque la aguja verde en la jeringa que contiene el envase y quite el protector de la aguja.
(7) Mantenga la ampolla OPC (One-Point-Cut) con la punta azul hacia arriba. Agite o golpee suavemente la ampolla para que el líquido que esté en el cuello de la ampolla baje al cuerpo. Sujete el cuello de la ampolla y rómpalo en dirección contraria a usted. Transfiera el disolvente hacia la jeringa. Desinfecte con alcohol el tapón de goma del dispositivo.
(8) Perfore el tapón con la aguja hasta una profundidad de 1 cm aproximadamente y lentamente inyecte el disolvente en el vial. Después retire la jeringa (con aguja) del dispositivo.
(9) Gire el vial suavemente hasta que el liofilizado se haya disuelto. No agite. Compruebe que la disolución es transparente, incolora y exenta de partículas. De no ser así, no administre la inyección. Ponga el tapón protector encima del dispositivo.
(10) Antes y después de la reconstitución, NeoRecormon Multidosis debe ser conservado en nevera entre 2°C - 8°C.
Preparación de una sola inyección
(1) Antes de extraer cada dosis desinfecte con alcohol el tapón de goma del dispositivo.
(2) Coloque una aguja de 26G dentro de la jeringa apropiada desechable (máx. 1 ml).
(3) Saque el protector de la aguja e inserte la aguja a través del tapón de goma del dispositivo. Extraiga la solución de NeoRecormon hacia dentro de la jeringa, saque el aire de la jeringa hacia el vial y ajuste la cantidad de solución de NeoRecormon en la jeringa, a la dosis prescrita. Después retire la jeringa (con aguja) del dispositivo.
(4) Sustituya la aguja por una nueva (la nueva debería tener el tamaño que suele utilizarse para inyecciones).
(5) Saque el protector de la aguja y cuidadosamente saque el aire de la aguja, sujetando la jeringa verticalmente y apriete suavemente el émbolo hacia arriba.
Para una inyección subcutánea, limpie la piel en el lugar donde se va a poner la inyección, utilizando un algodón con alcohol. Forme un pliegue de piel pellizcando la piel con el pulgar y el índice. Sujete la jeringa por la parte más próxima a la aguja e inserte la aguja en la piel con un movimiento rápido y firme. Inyecte la solución de NeoRecormon. Extraiga la aguja rápidamente y haga presión sobre el lugar de inyección con una gasa seca y estéril.
Prospecto: información para el usuario
NeoRecormon 500 UI NeoRecormon 2.000 UI NeoRecormon 3.000 UI NeoRecormon 4.000 UI NeoRecormon 5.000 UI NeoRecormon 6.000 UI NeoRecormon 10.000 UI NeoRecormon 20.000 UI NeoRecormon 30.000 UI solución inyectable en jeringa precargada Epoetina beta
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque
contiene información importante para usted.
• Conserve este prospecto, ya que puede tener que volver a leerlo.
• Si tiene alguna duda, consulte a su médico o farmacéutico.
• Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
• Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto.Versección 4.
Contenido del prospecto
1. Qué es NeoRecormon y para qué se utiliza
2. Qué necesita saber antes de empezar a usar NeoRecormon
3. Cómo usar NeoRecormon
4. Posibles efectos adversos
5. Conservación de NeoRecormon
6. Contenido del envase e información adicional
1. Qué es NeoRecormon y para qué se utiliza
NeoRecormon es una solución clara, incolora para inyectar debajo de la piel (subcutáneamente) o en la vena (intravenosamente). Contiene epoetina beta, una hormona que estimula la producción de glóbulos rojos. Epoetina beta se produce por tecnología genética especializada y funciona exactamente de la misma forma que la hormona eritropoyetina natural humana.
Debe consultar a su médico si empeora o si no mejora.
NeoRecormon está indicado para:
• Tratamiento de la anemia sintomática provocada por enfermedad renal crónica (anemia renal) en pacientes sometidos a diálisis o que aún no están sometidos a diálisis.
• Prevención de la anemia en niños prematuros (con un peso corporal al nacer de 750 a 1.500 g y una edad gestacional de menos de 34 semanas).
• Tratamiento de la anemia con síntomas relacionados en pacientes adultos con cáncer tratados con quimioterapia.
• Tratamiento de personas donantes de su propia sangre antes de ser sometidas a una intervención quirúrgica. Las inyecciones de epoetina beta aumentarán la cantidad de sangre que puede extraerse de su cuerpo antes de la operación, para ponérsela durante la intervención o después de la misma (esto es una transfusión autóloga).
2. Qué necesita saber antes de empezar a usarNeoRecormon No use NeoRecormon:
• si es alérgico a la epoetina beta o a cualquiera de los demás componentes de este medicamento
(incluidos en la sección 6).
• si tiene problemas de presión sanguínea que no pueden ser controlados
• si va a donar su propia sangre antes de someterse a una operación y:
• ha sufrido un infarto de miocardio o accidente cerebrovascular en el mes anterior al tratamiento
• si presenta angina de pecho inestable - nuevo dolor de pecho o dolor en aumento
• si tiene riesgo de que se le formen coágulos de sangre en las venas (trombosis de las venas profundas) - p. ej. si ha sufrido anteriormente coágulos.
Si sufriera cualquiera de estas situaciones o pudiera llegar a padecerlas, dígaselo a su médico de inmediato.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar NeoRecormon
• Si su bebé necesita tratamiento con NeoRecormon, su bebé será vigilado atentamente por los posibles efectos sobre el ojo.
• si su anemia no mejora con el tratamiento con epoetina
• si tiene bajos niveles de algunas vitaminas del complejo B (ácido fólico o vitamina B12)
• si tiene niveles altos de aluminio en sangre
• si el recuento de plaquetas es alto
• si tiene enfermedad hepática crónica
• si tiene epilepsia
• si ha desarrollado anticuerpos anti-eritropoyetina y aplasia pura de células rojas
(disminución o anulación de la producción de glóbulos rojos) durante una exposición previa a cualquier sustancia eritropoyética. En este caso, no debe cambiar a NeoRecormon.
Tenga especial cuidado con otros medicamentos que estimulan la producción de glóbulos rojos:
NeoRecormon es uno de los agentes estimuladores de la producción de glóbulos rojos como lo hace la proteína humana eritropoyetina. Su médico deberá siempre registrar el producto exacto que usted está utilizando.
Advertencia especial
Durante el tratamiento con Neorecormon
Si usted es un paciente con enfermedad renal crónica, y en particular si no responde adecuadamente a NeoRecormon, su médico controlará su dosis de NeoRecormon ya que el aumento repetido de la dosis de NeoRecormon si no está respondiendo al tratamiento puede aumentar el riesgo de tener un problema de corazón o de los vasos sanguíneos, y podría aumentar el riesgo de infarto de miocardio, accidente cerebrovascular y muerte.
Si usted es un paciente con cáncer debe saber que NeoRecormon puede actuar como factor de crecimiento de las células de la sangre y que en algunas circunstancias puede tener un efecto negativo sobre el cáncer. Dependiendo de su situación individual puede ser preferible una transfusión de sangre. Por favor, háblelo con su médico.
Si usted es un enfermo nefrosclerótico aún no sometido a diálisis, su médico decidirá si el tratamiento es conveniente para usted. Esto es debido a que no se puede descartar con certeza la posible aceleración de la insuficiencia renal.
Su médico puede realizar análisis de sangre de forma regular para controlar:
• sus niveles de potasio. Si usted presenta niveles de potasio altos o en aumento su doctor debe reconsiderar su tratamiento
• su recuento de plaquetas. Durante el tratamiento con epoetina el número de plaquetas puede aumentar de forma leve a moderada y esto puede producir cambios en la coagulación sanguínea.
Si usted es un paciente con alteración renal sometido a hemodiálisis, su médico deberá ajustar su dosis de heparina. Esto evitará la obstrucción de los tubos del sistema de diálisis.
Si usted es un paciente con alteración renal sometido a hemodiálisis y con riesgo de trombosis de derivación podrían formarse coágulos (trombosis) en su derivación (vaso utilizado para conectar al sistema de diálisis). Su médico podría recetarle ácido acetil salicílico o modificar la derivación.
Si usted está donando su propia sangre antes de ser operado, su médico necesitará:
• comprobar que usted puede donar sangre, especialmente si su peso es inferior a 50 kg
• comprobar que usted tiene nivel suficiente de glóbulos rojos (hemoglobina de al menos 11 g/dl)
• asegurarse que solo puede extraerle de una vez el 12 % de su volumen sanguíneo.
No haga un uso inapropiado de NeoRecormon:
El uso inapropiado de NeoRecormon por personas sanas puede llevar a un aumento de las células sanguíneas y, en consecuencia, un espesamiento de la sangre que puede asociarse con complicaciones cardíacas o vasculares con riesgo para la vida.
Uso de NeoRecormon con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento, incluso los adquiridos sin receta.
Embarazo, lactancia y fertilidad
No se ha obtenido experiencia adecuada con NeoRecormon en mujeres durante el embarazo y la lactancia. Pida consejo a su médico o farmacéutico antes de tomar cualquier medicamento. NeoRecormon no ha mostrado evidencia de alteración de la fertilidad en animales. Se desconoce el riesgo potencial en humanos.
Conducción y uso de máquinas
No se han descrito efectos sobre la capacidad para conducir y utilizar máquinas.
NeoRecormon contiene fenilalanina y sodio
Este medicamento contiene fenilalanina. Puede ser perjudicial para personas con fenilcetonuria.
Si tiene fenilcetonuria, consulte con su médico acerca del tratamiento con NeoRecormon.
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis, esto es, esencialmente “exento de sodio”.
3. Cómo usar NeoRecormon
La terapia con NeoRecormon debe iniciarse por un médico con experiencia en su enfermedad. La primera dosis se administra normalmente bajo control médico ante la posibilidad de que se produzca una reacción alérgica.
Las inyecciones de NeoRecormon deben pues ser administradas por personal sanitario entrenado (enfermero), médico u otro profesional. Una vez que le hayan enseñado, puede inyectarse la solución usted mismo.
NeoRecormon jeringa precargada está lista para su uso. Cada jeringa debe utilizarse para administrar una dosis única. No mezcle NeoRecormon con otras inyecciones o soluciones para perfusión.
Instrucciones de uso
En primer lugar, ¡lávese las manos!
1 Saque una jeringa de la caja Compruebe si el líquido de la jeringa:
• ¿está claro?
• ¿es incoloro?
• ¿está libre de partículas?
Si la respuesta a cualquiera de la preguntas es negativa, no se administre la inyección.
Deshágase de la jeringa y coja otra distinta.
Si la respuesta a todas las cuestiones es afirmativa, retire el tapón de la jeringa y continúe por el paso 2.
2 Saque la aguja de la caja, fíjela bien sobre la jeringa y retire la tapa protectora de la aguja.
3 Elimine el aire de la jeringa y aguja. Hágalo dando unos golpecitos suaves en la mitad superior de la jeringa. Esto provocará que cualquier burbuja de aire salga al exterior. Sujete la jeringa vertical y la aguja hacia arriba y presione suavemente el émbolo hacia arriba. Mantenga presionado el émbolo hasta que la cantidad de NeoRecormon en la jeringa se corresponda con la dosis prescrita.
4 Limpie la piel en el lugar de inyección con un algodón con alcohol. Haga un pliegue tomando la piel entre el índice y el pulgar.
5 Sujetando el cuerpo de la jeringa por la parte más próxima a la aguja, inserte la aguja en el
pliegue cutáneo con acción firme y rápida. Inyecte la solución de NeoRecormon. Retire la aguja rápidamente y aplique presión sobre el lugar de inyección con un apósito seco y estéril.
Dosis de NeoRecormon
La dosis de NeoRecormon depende del estado de su enfermedad, la forma en que se administra la inyección (bajo la piel o en una vena) y su peso corporal. Su médico calculará la dosis apropiada para usted.
Su médico utilizará la dosis eficaz más baja para controlar los síntomas de su anemia.
Si no responde adecuadamente a NeoRecormon, su médico controlará su dosis y le informará si necesita cambiar la dosis de NeoRecormon.
• Anemia sintomática provocada por enfermedad renal crónica
Las inyecciones se administran bajo la piel o en una vena. Si la solución se le administra en vena, debe ser inyectada a lo largo de unos 2 minutos, p.ej. en pacientes en hemodiálisis por vía de la fístula arteriovenosa al final de la diálisis.
Los pacientes no hemodializados recibirán normalmente la administración bajo la piel.
El tratamiento con NeoRecormon se divide en dos fases: a) Corrección de la anemia
La dosis inicial para la administración bajo la piel es de 20 UI por inyección por cada kg de peso, administrada tres veces por semana.
Después de 4 semanas, el médico le hará pruebas y puede incrementarle la dosis a 40 UI/kg por inyección, administrada tres veces por semana. El médico puede seguir aumentando su dosis a intervalos mensuales si fuera necesario.
La dosis semanal puede dividirse en dosis diarias.
La dosis inicial para la administración en vena es de 40 UI por inyección por cada kg de peso corporal administradas tres veces por semana.
Después de 4 semanas, su médico le hará pruebas y si la respuesta al tratamiento no es suficiente, puede incrementarle la dosis a 80 UI/kg por inyección, administradas tres veces por semana. El médico puede seguir aumentando la dosis a intervalos mensuales si fuera necesario.
Para ambos tipos de inyección, la dosis máxima no debe superar 720 UI por cada kg de su peso corporal por semana.
b) Mantenimiento de los niveles de glóbulos rojos
Dosis de mantenimiento: Una vez que sus glóbulos rojos alcancen un nivel aceptable, la dosis se reduce a la mitad de la dosis utilizada para corregir la anemia. La dosis semanal puede administrarse una vez por semana, o dividirse en tres o siete dosis por semana. Si sus glóbulos rojos permanecen estables en un régimen de una dosis única semanal pueden pasar a una administración única cada dos semanas. En este caso, puede ser necesario un aumento de la dosis.
El médico puede ajustar su dosis cada una o dos semanas hasta encontrar su dosis individual de mantenimiento.
Los niños iniciarán el tratamiento siguiendo las mismas normas. En los ensayos clínicos, los niños normalmente necesitaron dosis más altas de NeoRecormon (cuanto más pequeño es el niño, mayor es la dosis).
El tratamiento con NeoRecormon normalmente es un tratamiento a largo plazo. No obstante, puede interrumpirse en cualquier momento en caso necesario.
• Anemia en niños prematuros
Las inyecciones se administran bajo la piel.
La dosis inicial es de 250 UI inyectadas por cada kg de peso, tres veces por semana.
Es probable que los bebés prematuros que ya hayan recibido una transfusión previa cuando se inicie el tratamiento con NeoRecormon no se beneficien tanto como los prematuros no transfundidos.
La duración de tratamiento recomendada es de 6 semanas.
• Adultos con anemia sintomática con cáncer tratados con quimioterapia Las inyecciones se administran bajo la piel.
Su médico puede iniciar el tratamiento con NeoRecormon si su nivel de hemoglobina es menor o igual a 10 g/dl. Después de comenzar el tratamiento, su médico mantendrá su nivel de hemoglobina entre 10 y 12 g/dl.
La dosis semanal inicial es de 30.000 UI. Esta dosis se puede administrar en una inyección semanal, o puede dividirse en 3 a 7 inyecciones por semana. Su médico tomará muestras de sangre de forma regular. En función de los resultados de los análisis, podrá aumentarle o reducirle la dosis o interrumpirle el tratamiento. Los valores de hemoglobina no excederán de 12 g/dl.
El tratamiento debe continuar hasta 4 semanas después de terminar la quimioterapia.
La dosis máxima no debe exceder de 60.000 UI por semana.
• Pacientes donantes de su propia sangre antes de someterse a cirugía
Las inyecciones se administran en vena en unos 2 minutos, o debajo de la piel.
La dosis de NeoRecormon dependerá de su condición, de los niveles de glóbulos rojos y de la cantidad de sangre que donará antes de la intervención.
La dosis calculada por el médico se administra dos veces por semana durante 4 semanas. Cuando usted done sangre, recibirá NeoRecormon al final de la donación.
La dosis máxima no debe exceder
• para inyección en vena: 1.600 UI por kg de peso por semana
• para inyecciones bajo la piel: 1.200 UI por kg de peso por semana
Si inyecta demasiado NeoRecormon
No aumente la dosis que le ha dado su médico. Si cree que se ha inyectado más NeoRecormon del que debiera, contacte con su médico. Es poco probable que sea grave. Incluso en presencia de altos niveles en sangre no han sido observados síntomas de intoxicación.
Si olvidó usar NeoRecormon
Si ha olvidado una inyección o se ha inyectado muy poco, dígaselo a su médico.
No se administre una dosis doble para compensar las dosis olvidadas.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede tener efectos adversos, aunque no todas
las personas los sufran.
Efectos adversos que pueden afectar a cualquier paciente
• La mayor parte de los pacientes (muy frecuentes pueden afectar a más de 1 de cada 10 personas) presenta niveles bajos de hierro en la sangre. Casi todos los pacientes deben ser tratados con suplementos de hierro durante el tratamiento con NeoRecormon.
• Rara vez (pueden afectar hasta 1 de cada 1.000 personas) han aparecido reacciones alérgicas o de la piel como sarpullido o urticaria o reacciones en el lugar de la inyección.
• Muy rara vez (pueden afectar hasta 1 de cada 10.000 personas) han aparecido formas severas de reacción alérgica, especialmente después de la inyección. Estas han de tratarse de inmediato. Si usted presenta sibilancias o dificultad para respirar; inflamación en la lengua, cara, garganta o alrededor del lugar de la inyección; si siente mareo o desmayos o si se cae llame al médico de inmediato.
• Muy rara vez (pueden afectar hasta 1 de cada 10.000 personas) la gente experimenta síntomas gripales, especialmente cuando inician el tratamiento. Estos síntomas incluyen
fiebre, escalofríos, dolores de cabeza, dolor de las extremidades, dolor de huesos y/o malestar general. Estas reacciones fueron normalmente leves o moderadas y desaparecieron en unas horas o días.
Efectos adversos en pacientes con enfermedad renal crónica (anemia renal)
• Los efectos adversos más comunes (muy frecuentes pueden afectar a más de 1 de cada 10 personas) son aumentos de la presión sanguínea, empeoramiento de la presión sanguínea existente y dolor de cabeza. Su médico deberá tratar la presión sanguínea elevada con medicamentos apropiados o interrumpir temporalmente el tratamiento con NeoRecormon.
• Si tiene dolores de cabeza, especialmente súbitos, punzantes, tipo migraña, confusión, alteración del habla, inestabilidad al andar, ataques o convulsiones llame enseguida al médico. Pueden ser síntomas de presión sanguínea extremadamente alta (crisis hipertensiva), aunque la presión sanguínea sea habitualmente normal o baja. Esta sintomatología debe tratarse de inmediato.
• Si usted tiene presión sanguínea baja o complicaciones de derivación puede padecer una trombosis de derivación (un coágulo de sangre en el vaso utilizado para conectar al sistema de diálisis).
• Muy rara vez (pueden afectar hasta 1 de cada 10.000 personas), los pacientes presentan niveles elevados de potasio o fosfatos en sangre. Estos pueden ser tratados por su médico.
• Se ha observado aplasia pura de células rojas (APCR) causada por anticuerpos neutralizantes durante el tratamiento con eritropoyetina, incluidos algunos casos aislados durante el tratamiento con NeoRecormon. APCR quiere decir que el organismo para o reduce la producción de glóbulos rojos. Esto causa una anemia grave, entre cuyos síntomas se incluye cansancio inusual y carencia de energía. Si su organismo produce anticuerpos neutralizantes, su médico interrumpirá la terapia con NeoRecormon, y determinará la mejor forma de actuar para tratar su anemia.
Efectos adversos adicionales en adultos con cáncer tratados con quimioterapia
• Ocasionalmente puede producirse un aumento de la presión sanguínea y dolor de cabeza. Su médico puede tratar la presión sanguínea elevada con fármacos.
• Se ha observado un aumento de la incidencia de coágulos de sangre.
Efectos adversos adicionales en pacientes donantes de su propia sangre antes de someterse a cirugía
• Se ha observado un ligero aumento de la incidencia de coágulos de sangre.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de NeoRecormon
• Mantener este medicamento fuera de la vista y del alcance de los niños.
• No utilice NeoRecormon después de la fecha de caducidad que aparece en la caja y en la etiqueta.
• Conservar en nevera (entre 2°C - 8°C).
• La jeringa puede mantenerse fuera de la nevera durante un único periodo de hasta 3 días a temperatura ambiente (no superior a 25°C).
• Mantener la jeringa precargada en el embalaje exterior, para protegerla de la luz.
• Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico como deshacerse de los envases y de los medicamentos que no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de NeoRecormon
• El principio activo es la epoetina beta. 1 jeringa precargada contiene 500, 2.000, 3.000, 4.000, 5.000, 6.000, 10.000, 20.000 ó 30.000 UI (unidades internacionales) de epoetina beta en 0,3 ml ó 0,6 ml de solución.
• Los otros componentes son:
urea, cloruro sódico, polisorbato 20, fosfato monosódico dihidratado, fosfato disódico dodecahidratado, cloruro cálcico dihidratado, glicina, L-Leucina, L-Isoleucina, L-Treonina, L-Ácido glutámico y L-Fenilalanina y agua para preparaciones inyectables.
Aspecto de NeoRecormon y contenido del envase
NeoRecormon es una solución en jeringa precargada para inyección.
La Solución es incolora, de transparente a ligeramente opalescente.
NeoRecormon 500 UI, 2.000 UI, 3.000 UI, 4.000 UI, 5.000 UI y 6.000 UI: cada jeringa precargada contiene 0,3 ml de solución.
NeoRecormon 10.000 UI, 20.000 UI y 30.000 UI: cada jeringa precargada contiene 0,6 ml de solución.
NeoRecormon se presenta en los siguientes tamaños de envase:
NeoRecormon 500 UI 1 jeringa precargada con 1 aguja (30G1/2) o 6 jeringas precargadas con 6 agujas (30G1/2).
NeoRecormon 2.000 UI, 3.000 UI, 4.000 UI, 5.000 UI, 6.000 UI, 10.000 UI y 20.000 UI 1 jeringa precargada con 1 aguja (27G1/2) o 6 jeringas precargadas con 6 agujas (27G1/2).
NeoRecormon 30.000 UI 1 jeringa precargada con 1 aguja (27G1/2) o 4 jeringas precargadas con 4 agujas (27G1/2).
Puede que solamente algunos tamaños de envase estén comercializados.
Titular de la autorización de comercialización
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
Responsable de la fabricación
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien N.V. Roche S.A. Tél/Tel: +32 (0) 2 525 82 11 |
Lietuva UAB “Roche Lietuva” Tel: +370 5 2546799 |
|
Etarapna Pom Etarapua EOOfl Tea: +359 2 818 44 44 |
Luxembourg/Luxemburg (Voir/siehe Belgique/Belgien) |
|
Ceská republika Roche s. r. o. Tel: +420 -2 20382111 |
Magyarország Roche (Magyarország) Kft. Tel: +36 - 23 446 800 |
|
Danmark Roche a/s Tlf: +45 - 36 39 99 99 |
Malta (ara Renju Unit) |
|
Deutschland Roche Pharma AG Tel: +49 (0) 7624 140 |
Nederland Roche Nederland B.V. Tel: +31 (0) 348 438050 |
|
Eesti Roche Eesti OÜ Tel: + 372 -6 177 380 |
Norge Roche Norge AS Tlf: +47 - 22 78 90 00 |
|
EXXáSa Roche (Hellas) A.E. T^: +30 210 61 66 100 |
Osterreich Roche Austria GmbH Tel: +43 (0) 1 27739 |
|
España Roche Farma S.A. Tel: +34 -91 324 81 00 |
Polska Roche Polska Sp.z o.o. Tel: +48 -22 345 18 88 |
|
France Roche Tél: +33 (0) 1 47 61 40 00 |
Portugal Roche Farmacéutica Química, Lda Tel: +351 -21 425 70 00 |
|
Hrvatska Roche d.o.o Tel: +385 1 4722 333 |
Romania Roche Romania S.R.L. Tel: +40 21 206 47 01 |
|
Ireland Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700 |
Slovenija Roche farmacevtska druzba d.o.o. Tel: +386 - 1 360 26 00 |
|
Ísland Roche a/s c/o Icepharma hf Sími: +354 540 8000 |
Slovenská republika Roche Slovensko, s.r.o. Tel: +421 -2 52638201 |
|
Italia Roche S.p.A. Tel: +39 -039 2471 |
Suomi/Finland Roche Oy Puh/Tel: +358 (0) 10 554 500 |
Kúrcpog
r.A.Zxa^áxn? & Sia AxS. Tqk +3 57 - 22 76 62 76
Sverige
Roche AB
Tel: +46 (0) 8 726 1200
Latvija
Roche Latvija SIA
Tel: +371 -6 7039831
United Kingdom
Roche Products Ltd.
Tel: +44 (0) 1707 366000
Fecha de la última revisión de este prospecto: <{MM/AAAA}> <{mes AAAA}>
La información detallada de este medicamento está disponible en al página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/
225