Mononine 1000, 100 Ui/Ml, Polvo Y Disolvente Para Solucion Inyectable O Perfusion
"I
an
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
MONONINE® 1000, 100 Ul/ml, polvo y disolvente para solución inyectable o perfusión.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un vial con polvo contiene 1.000 UI de factor IX de la coagulación humano.
El producto reconstituido con 10 ml de agua para preparaciones inyectables contiene por 1 ml: 100 UI de factor IX de la coagulación humano/ml.
La potencia (UI) se determina según el método de coagulación de una fase de la Farmacopea Europea. La actividad específica media de MONONINE® 1000 es superior a 190 UI/mg de proteína.
Excipiente con un efecto conocido
Sodio (como cloruro): hasta 20,4 mg
Para ver lista completa de excipientes, ver Sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución para inyección o perfusión.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de hemorragias en pacientes con hemofilia B (deficiencia congénita de factor IX).
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia.
Posología
La dosis y la duración de la terapia de sustitución dependen de la gravedad de la deficiencia de factor IX, de la localización e importancia de la hemorragia y del cuadro clínico del paciente.
El número de unidades de factor IX administradas se expresa en Unidades Internacionales (UI), en relación con el estándar de la Organización mundial de la Salud (OMS) vigente para concentrados de factor IX. La actividad plasmática de factor IX se expresa como un porcentaje (en relación con el plasma humano normal) o en Unidades Internacionales (en relación con un estándar internacional para el factor IX en plasma).
La actividad de una Unidad Internacional (UI) de factor IX es equivalente a la cantidad de factor IX contenido en un ml de plasma humano normal. El cálculo de la dosis necesaria de factor IX se basa en la observación empírica de que 1 UI de factor IX por kilogramo de peso corporal incrementa la actividad plasmática de factor IX aproximadamente en un 1% sobre la actividad normal. La dosis requerida se determina mediante la fórmula siguiente:
Unidades requeridas = peso corporal (kg) x aumento deseado de factor IX [% o UI/dL] x 1.0* *Recíproco de la recuperación observada
ÍTTI
La posología, el método así como la frecuencia de administración se establecerán siempre en función de la eficacia clínica observada en cada caso. Los preparados de factor IX raramente requieren que se administren más de una vez al día cuando se administran en bolo.
En los siguientes episodios hemorrágicos, la actividad de factor IX no deberá ser inferior al nivel plasmático de actividad que se indica (en % del valor normal o UI/dl), durante el período correspondiente.
La siguiente tabla puede emplearse como guía de dosificación en episodios hemorrágicos y cirugía:
|
TABLA 1: INYECCIÓN INTRAVENOSA EN BOLO | ||
|
Tipo de hemorragia / tipo de cirugía |
Nivel de factor IX requerido (% o UI/dL) |
Frecuencia de dosificación (horas)/Duración de la terapia (días) |
|
HEMORRAGIAS | ||
|
Hemartrosis precoz y sangrado muscular u oral |
20-40 |
Repetir cada 24 horas. Al menos 1 día, hasta que el episodio hemorrágico manifestado por dolor se detenga o hasta cicatrización de la herida |
|
Hemartrosis más extensa y hemorragia muscular o hematoma |
30-60 |
Repetir la perfusión cada 24 horas durante 3-4 días o más hasta que el dolor y la discapacidad aguda se hayan resuelto |
|
Hemorragias con riesgo vital |
60-100 |
Repetir la perfusión cada 8-24 horas hasta que el riesgo desaparezca |
|
CIRUGÍA | ||
|
Menor Incluyendo extracciones dentales |
30-60 |
Cada 24 horas, al menos 1 día, hasta la cicatrización |
|
Mayor |
80-100 (pre y postoperatorio) |
Repetir la perfusión cada 8-24 horas, hasta la adecuada cicatrización de la herida, y luego tratamiento durante un mínimo de 7 días más para mantener un nivel de actividad de factor IX de 30 a 60% (UI/dL) |
|
TABLA 2: PERFUSIÓN CONTINUA EN CIRUGÍA | |
|
Niveles de factor IX necesarios para alcanzar la hemostasia |
40-100% (o UI/dL) |
|
Dosis de carga inicial para conseguir el nivel deseado |
Dosis única en bolo de 90 UI por kg p.c. (Rango 75100 UI kg p.c.), o guía de dosificación según farmacocinética |
|
Frecuencia de dosificación |
Perfusión intravenosa continua, dependiendo del aclaramiento y de los niveles de factor IX medidos |
|
Duración del tratamiento |
Durante 5 días. La continuación del tratamiento puede ser necesaria, dependiendo del tipo de cirugía |
Durante el tratamiento, se aconseja realizar un seguimiento apropiado de los niveles plasmáticos de factor IX a fin de determinar la dosis y la frecuencia de las perfusiones. En el caso concreto de la cirugía mayor, es imprescindible un control exacto de la terapia de sustitución mediante pruebas de la coagulación (actividad plasmática de factor IX). La respuesta individual de los pacientes al tratamiento con factor IX puede variar, tanto en los niveles de recuperación in vivo, como en la vida media.
En la profilaxis a largo plazo de las hemorragias en pacientes con hemofilia B grave, las dosis usuales son 20 a 40 UI de factor IX por kilo de peso corporal cada 3 ó 4 días. En algunos casos, especialmente en los pacientes más jóvenes, pueden ser necesarias dosis mayores a las calculadas o acortar los intervalos entre administraciones.
En los pacientes deberá controlarse, el desarrollo de inhibidores del factor IX. Si no se alcanzan los niveles de actividad plasmática de factor IX esperados, o si el sangrado no se controla con la dosis adecuada, deberá realizarse una prueba para determinar la presencia de inhibidores del factor IX. En pacientes con títulos altos de inhibidores, la terapia con factor IX puede ser no efectiva, por lo que deberán considerarse otras opciones terapéuticas.
El manejo de estos pacientes deberá ser dirigida por médicos con experiencia en el tratamiento de la hemofilia.
Ver también Sección 4.4.
Forma de administración
Reconstituir la preparación tal como se describe en el apartado 6.6. La preparación debe llevarse a temperatura ambiente o corporal antes de la administración. MONONINE® 1000 debe ser administrado por vía intravenosa lenta, para observar al paciente por si se presenta una reacción inmediata. Si se produce una reacción de la que se piense que pueda estar relacionada con la administración de MONONINE® 1000 debe disminuirse la velocidad de perfusión o bien interrumpirse la perfusión, según requiera la condición clínica del paciente (ver también Sección 4.4).
Inyección intravenosa en bolo
Realizar la venopunción con el equipo correspondiente que se acompaña con el producto. Conectar la jeringa al final luer del equipo de venopunción.
Inyectar lentamente por vía intravenosa a una velocidad confortable para el paciente (máx. 2 mL/minuto). Perfusión continua
MONONINE® 1000 debe reconstituirse con Agua para preparaciones inyectables tal como se describe en el Apartado 6.6. Después de la reconstitución, MONONINE® 1000 puede administrarse sin diluir, como perfusión continua, mediante una bomba de perfusión.
La potencia de la solución reconstituida de MONONINE® 1000 sin diluir es de aproximadamente 100 UI/mL.
Una solución diluida se obtiene de la siguiente forma:
-Usando una técnica aséptica, diluir la solución reconstituida, filtrar y transferir la cantidad apropiada de MONONINE® al volumen deseado de solución salina fisiológica.
-En diluciones de una titulación de hasta 1:10 (concentración de 10 UI de factor IX/mL), la actividad de factor IX permanece estable durante 24 horas.
-En titulaciones superiores puede producirse una disminución de la actividad de factor IX, ésta debe controlarse para mantener los niveles hemáticos deseados.
Ejemplo para diluir 1.000 UI de MONONINE® reconstituido:
|
Potencia deseada de la dilución |
10 UI/mL |
20 UI/mL |
|
Volumen de MONONINE® reconstituido |
10,0 mL |
10,0 mL |
|
Volumen necesario de solución salina fisiológica |
90,0 mL |
40,0 mL |
|
Dilución conseguida |
1:10 |
1:5 |
-Se recomienda el uso de bolsas IV y tubos de cloruro de polivinilo (PVC).
-Mezclar mediante agitación vigorosa y comprobar que la bolsa no tiene fugas.
-Se recomienda reemplazar las bolsas con MONONINE® recién diluido cada 12-24 horas.
Cuando se administra MONONINE® 1000 como perfusión continua, la velocidad de administración recomendada es de 4 UI/kg p.c./h para, mantener un estado de equilibrio de los niveles de factor IX de aproximadamente un 80%. Esto depende del perfil farmacocinético del paciente y del nivel de factor IX que se desee alcanzar. En pacientes, en los que se conoce el aclaramiento de factor IX, la velocidad de administración se puede calcular individualmente.
Velocidad de administración (UI/kg p.c./h) = aclaramiento (mL/h/kg p.c.) x incremento deseado de factor IX (UI/mL).
La seguridad y eficacia de la perfusión continua en niños no han sido estudiadas (Ver Sección 4.4). Por lo tanto, en niños y adolescentes, la perfusión continua de MONONINE® 1000 sólo debe considerarse, si los datos farmacocinéticos prequirúrgicos (p. ej.: incremento de la recuperación y aclaramiento) se obtuvieron a partir de la dosis calculada,y los niveles perioperativos deben ser controlados cuidadosamente.
4.3 Contraindicaciones
Hipersensibilidad al principio activo y a cualquier excipiente del preparado.
Hipersensibilidad conocida a la proteína murina.
Alto riesgo de trombosis o de coagulación intravascular diseminada (ver también 4.4).
4.4 Advertencias y precauciones especiales de empleo
Al igual que sucede con cualquier producto proteico para administración intravenosa, es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico. MONONINE® 1000 contiene trazas de proteínas murinas (los anticuerpos monoclonales murinos usados en su proceso de purificación). Aunque los niveles de proteína murina son extremadamente bajos (igual o inferior a 50 ng de proteína murina/ 100 UI), la perfusión de dichas proteínas podrían, teóricamente, provocar reacciones de hipersensibilidad.
Los pacientes deben ser informados sobre la aparición de síntomas precoces de reacciones de hipersensibilidad incluyendo, habones, urticaria generalizada, opresión torácica, respiración dificultosa, hipotensión y anafilaxia. Si se presentan estos síntomas, debe informárseles que debe interrumpirse inmediatamente la administración del producto y comunicarlo a su médico.
En caso de shock, se seguirán las pautas médicas vigentes para su tratamiento.
MONONINE 1000 contiene hasta 20,4 mg de sodio por 1000 UI, esto debe ser tenido en cuenta por los pacientes que observan una dieta controlada respecto al contenido de sodio.
4 de 1 1 MINISTWODE
SANIDAD, POLITICA SOCIAL E IGUALDAD Agencia es pañosa de medicamentos y productos sanéanos
ÍTTI
Tras tratamiento repetido con preparados de factor IX de la coagulación humano, los pacientes deben ser controlados respecto al desarrollo de anticuerpos contra el factor IX (inhibidores), que deben cuantificarse en Unidades Bethesda, usando un método biológico apropiado.
En la bibliografía existen informes que muestran una correlación entre la aparición de inhibidores del factor IX y reacciones alérgicas. Por lo tanto, los pacientes que experimenten reacciones alérgicas deberán ser controlados acerca de la presencia de inhibidores. Debe tenerse en cuenta que pacientes con inhibidores del factor IX, pueden presentar un riesgo aumentado de anafilaxia, con la consiguiente respuesta anamnésica para el factor IX.
Debido al riesgo de reacciones alérgicas con concentrados de factor IX, la administración inicial de factor IX debe realizarse bajo observación médica, de acuerdo con la opinión del médico responsable del tratamiento, en un lugar donde pueda proporcionarse asistencia médica adecuada para el tratamiento de reacciones alérgicas.
Ya que el uso de concentrados de complejos del factor IX ha sido, históricamente, asociado con el desarrollo de complicaciones tromboembólicas, y aún siendo el riesgo más elevado en productos de baja pureza, el uso de productos que contienen factor IX puede ser potencialmente arriesgado en pacientes con signos de fibrinolisis y en pacientes con coagulación intravascular diseminada (CID). Debido al riesgo potencial de complicaciones tromboembólicas, cuando se administra este producto a pacientes con enfermedad hepática, a pacientes en postoperatorio, a neonatos o a pacientes con riesgo de accidentes tromboembólicos o CID, el seguimiento clínico para la detección de signos precoces de trombosis y coagulopatía de consumo debe iniciarse usando un método de análisis biológico apropiado. En cada una de estas situaciones, deberá valorarse la relación entre el beneficio del tratamiento con MONONINE®
1000 y el riesgo de estas complicaciones.
No existen datos de eficacia y seguridad para la perfusión continua en niños, en particular se desconoce el potencial para desarrollar inhibidores (Ver, también Sección 4.2).
Seguridad vírica
Para prevenir la transmisión de enfermedades infecciosas a los pacientes, cuando se administran medicamentos derivados de la sangre o plasma humanos, se toman medidas estándar como la selección de donantes de sangre y plasma, análisis de las donaciones individuales y de las mezclas de plasma se determinan marcadores específicos de enfermedades infecciosas. Los fabricantes de estos productos incluirán en el proceso de producción, etapas para eliminar / inactivar virus. A pesar de estas medidas, cuando se administran medicamentos derivados de la sangre o plasma humanos, la posibilidad de transmisión de agentes infecciosos no se puede excluir totalmente. Esto también se refiere a virus desconocidos o emergentes y otro tipo de agentes infecciosos.
Estos procedimientos se consideran efectivos frente al virus capsulados como el VIH, VHB y VHC), así como para los virus no recubiertos de la hepatitis A y parvovirus B19.
Si Vd. es tratado regular o repetidamente con derivados plasmáticos que contengan factor IX, debe, en general, considerarse la vacunación contra la hepatitis A y B.
Se recomienda encarecidamente que cada vez que se administre MONONINE® 1000 que se anote el nombre del producto y el número del lote del producto, a fin de mantener la trazabilidad entre el paciente y el lote del producto administrado.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se conocen interacciones del factor IX de la coagulación humano con otros medicamentos.
Hay pocos datos disponibles en lo que se refiere a la utilización de ácido s-aminocaproico después de una perfusión inicial de MONONINE® 1000 para la prevención o tratamiento de hemorragias en la cavidad
ÍTTI
bucal, que se presenten como consecuencia de un traumatismo o de intervenciones dentales, tales como extracciones.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción animal con factor IX. Ya que la hemofilia B es excepcional en mujeres, no se dispone de experiencia clínica sobre el uso de factor IX durante el embarazo y lactancia. Por lo tanto, el factor IX debe usarse durante el embarazo y la lactancia sólo en el caso de que esté claramente indicado.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han observado efectos sobre la capacidad de conducir vehículos o utilizar maquinaria.
4.8 Reacciones adversas
Las siguientes reacciones adversas están basadas, tanto en los estudios de farmacovigilancia tras la comercialización del producto, como en la literatura científica.
Se usan las siguientes categorías estándares según su frecuencia:
-Muy habitual > 1/10 -Habitual > 1/100 y < 1/10 -No habitual 1/1.000 y < 1/100 -Rara 1/10.000 y < 1/1.000
-Muy rara < 1/10.0000 (incluyendo informes de casos aislados)
Trastornos renales y del aparato urinario
Se han descrito casos muy raros de síndrome nefrótico, seguido al intento de inducir inmunotolerancia en pacientes con hemofilia B con inhibidores contra el factor IX e historia de reacciones alérgicas.
Trastornos vasculares
Tras la administración de productos que contengan factor IX existe un riesgo potencial de episodios tromboembólicos, siendo el riesgo más elevado en productos de baja pureza. El uso de preparados de factor IX de baja pureza ha sido asociado con casos de infarto de miocardio, coagulación intravascular diseminada, trombosis venosa y embolismo pulmonar. El uso de factor IX de alta pureza raramente se asocia con estas reacciones adversas.
Trastornos generales y en el sitio de administración En raras ocasiones se ha observado fiebre.
Trastornos del sistema inmunitario
En pacientes tratados con preparados que contienen factor IX en raras ocasiones se han observado reacciones alérgicas o de hipersensibilidad (que pueden incluir angioedema, sensación de ardor (irritación) o flebitis en el lugar de la inyección/perfusión, escalofríos, enrojecimiento, urticaria generalizada, cefalea,habones, hipotensión, somnolencia, náuseas, desasosiego, taquicardia, opresión torácica, hormigueo,vómitos, disnea). En ciertos casos, estas reacciones han progresado hasta anafilaxia grave y en estos casos se ha descrito una asociación temporal con el desarrollo de inhibidores de factor IX (ver también Apartado4.4).
Los estudios de farmacovigilancia tras la comercialización del producto han informado que pacientes con hemofilia B pueden desarrollar anticuerpos neutralizantes del factor IX (inhibidores). La aparición de estos inhibidores se manifiesta como una respuesta clínica insuficiente. En estos casos se recomienda contactar con un centro especializado en hemofilia. En un ensayo clínico, 2 de 51(4%) pacientes previamente no tratados (PUPs) desarrollaron anticuerpos y en uno de estos pacientes, se asoció en dos ocasiones, con una reacción anafilactoide.
"I
an
Para información sobre “Seguridad vírica”, véase sección 4.4.
4.9 Sobredosis
No se ha informado sobre síntomas de sobredosificación con factor IX de la coagulación humano.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antihemorrágicos: factor IX de la coagulación. Código ATC: B02B D04.
El factor IX es una glicoproteína de cadena única, con un peso molecular aproximado de 68.000 Dalton.
Es un factor de la coagulación dependiente de la vitamina K y se sintetiza en el hígado. El factor IX es activado por el factor XIa en la vía intrínseca de la coagulación y en la vía extrínseca por el factor VII/complejo de factor tisular.
El factor IX activado, en combinación con el factor VIII activado, activa el factor X. El factor X activado convierte la protrombina en trombina. La trombina, a su vez, convierte el fibrinógeno en fibrina, con lo que se forma el coágulo.
La hemofilia B es una alteración de la coagulación sanguínea hereditaria ligada al sexo y se debe a una disminución de los niveles de factor IX que da lugar a un sangrado profuso en las articulaciones, músculos y órganos internos, ya sea de forma espontánea o a causa de un traumatismo accidental o quirúrgico. La terapia de sustitución aumenta los niveles plasmáticos de factor IX, obteniéndose una restauración temporal de la deficiencia de este factor y una corrección de la tendencia al sangrado.
Cuando se reconstituye el preparado, tal como se indica (ver 6.6), el resultado es una solución clara, incolora, isotónica de pH neutro, que contiene aproximadamente, 100 veces la actividad de factor IX hallada en un volumen equivalente de plasma.
Para dosificar a niños menores de 6 años, ver sección 4.2 (Método de administración).
5.2 Propiedades farmacocinéticas
La perfusión de MONONINE® 1000 en un breve período de tiempo en 38 pacientes con hemofilia B (estudio sobre recuperación) demostró un incremento medio de la recuperación de 1,71 UI/dL por UI/kg p.c. (Rango: 0,85-4,66). La vida media terminal determinada en un subgrupo de 28 pacientes fue de 14,9 horas (Rango: 7,2-22,7).
La farmacocinética de MONONINE® 1000 se determinó también en 12 pacientes (cirugía programada) antes del tratamiento con una perfusión continua de MONONINE® 1000 .
|
Parámetro |
Estudio sobre recuperación (n=38). Valores medios (rango) |
Cirugía programada (n=12) Valores medios (rango) |
|
Incremento de la recuperación (UI/dL por UI/kg) |
1,71 (0,85-4,66) |
1,21 (0,83-1,60) |
|
Vida media terminal (h) |
14,9 (7,2-22,7)** |
16,4 (8,7-36,6) |
|
Vida media inicial*** (h) |
n.d. |
2,46 (0,34-6,2) |
|
Área bajo la curva* (hxkg/mL) |
n.d. |
0,254 (0,147-0,408) |
|
Volumen en estado de equilibrio (mL/kg) |
n.d. |
111(77-146) |
|
Aclaramiento (mL/h/kg) |
n.d. |
4,27 (2,45-6,78) |
|
Tiempo medio de permanencia (h) |
n.d. |
27,4 (17,7-42,6) |
* Estandarizado a 1 UI de dosis /kg ** Basado en un subgrupo de 28 pacientes
*** Datos de sólo 4 pacientes de 12. Los restantes 8 pacientes siguieron un modelo simple monocompartimental. El proceso de distribución de MONONINE® sólo se observó ocasionalmente n.d. (no disponible) .
5.3 Datos preclínicos sobre seguridad
El factor IX de la coagulación plasmático humano es un constituyente normal del plasma humano y actúa como el factor IX endógeno. Carece de interés realizar pruebas de toxicidad con dosis única ya que las dosis altas pueden provocar sobrecarga.
El ensayo de toxicidad a dosis repetidas en animales es impracticable debido al desarrollo de anticuerpos frente a proteínas heterólogas (humanas).
Dado que la experiencia clínica no proporciona indicios de efecto carcinogénicos o mutagénicos del factor IX de la coagulación plasmático humano, no se considera necesario realizar estudios experimentales, particularmente en especies heterólogas..
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Histidina, manitol y cloruro sódico.
Ácido clorhídrico o hidróxido sódico (en pequeñas cantidades para ajustar el pH).
Disolvente suministrado:
Agua para preparaciones inyectables.
6.2 Incompatibilidades
Este producto no debe mezclarse con otros medicamentos, excepto con solución salina fisiológica.
6.3 Periodo de validez 2 años.
Después de la reconstitución, la estabilidad físico-química se ha demostrado para un período de 24 horas a temperatura ambiente (hasta un máximo de +25° C). Desde un punto de vista microbiológico, MONONINE® 1000 no contiene conservantes, por lo que desde un punto de vista microbiológico el producto reconstituido debe usarse inmediatamente.
Después de diluir (hasta 1:10) la estabilidad de la solución reconstituida de MONONINE® 1000 ha sido demostrada para un período de 24 horas..
6.4 Precauciones especiales de conservación
Debe conservarse entre +2° y +8° C (en refrigerador). No congelar, Mantener el vial en su estuche.
Durante el periodo de validez el producto (guardado en su embalaje original) puede ser sacado del refrigerador y almacenado a temperatura ambiente (hasta +25° C) durante un período único de 1 mes como máximo; el producto no debe refrigerarse durante este periodo. El inicio del almacenamiento a
.Uí1.
"I ■"
¡m
temperatura ambiente y el final de 1 mes a temperatura ambiente deben ser anotados en la caja. Al finalizar este período el producto no debe volver a guardarse en el refrigerador, por lo que debe usarse o descartarse.
6.5 Naturaleza y contenido del envase
Envases primarios:
Viales de vidrio de Tipo I con tapones contienen respectivamente: polvo con 1.000 UI y 10 ml de Agua para preparaciones inyectables.
Presentación Caja con 1000 UI 1 vial con polvo
1 vial con 10 mL de Agua para preparaciones inyectables 1 trasvasador con filtro 20/13 1 jeringa de 10 ml de un solo uso
1 equipo para venopunción
2 toallitas con alcohol 1 apósito adhesivo
6.6 Precauciones especiales de eliminación y otras manipulaciones
El producto no utilizado y los materiales desechados deberán eliminarse adecuadamente según los requerimientos locales.
Método de administración
Instrucciones generales
-La reconstitución y extracción de la solución para su administración se debe realizar bajo condiciones asépticas.
-Normalmente la solución es clara o ligeramente opalescente. El producto reconstituido debe ser revisado ópticamente para detectar partículas extrañas o decoloraciones después de la filtración / extracción, antes de la administración (véase “Reconstitución”). No utilizar soluciones turbias o que contengan depósitos (sedimentos o partículas).
Reconstitución
Atemperar el disolvente a temperatura ambiente. Retirar las cápsulas de los viales del polvo y del disolvente, desinfectar la superficie de los tapones de goma, dejándolos secar antes de proceder a abrir el envase que contiene el Mix2Vial.
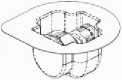
1
1. Abrir el envase que contiene el Mix2Vial, desgarrando el precinto
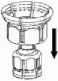
2
2. Retirar el Mix2Vial del contenedor, teniendo cuidado de no tocar ambos extremos del mismo. Coloque el vial del disolvente sobre una superficie plana y limpia. Sujetándolo firmemente, encaje el terminal azul del Mix2Vial en el tapón del disolvente.

3
3. Manteniendo el vial con el producto apoyado sobre una superficie firme, invertir el vial del disolvente con el Mix2Vial acoplado, y encajar el adaptador transparente en el tapón del vial del producto. El disolvente pasará automáticamente al vial con el polvo

^ 4

5
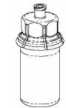
4. Con los dos viales aún acoplados, agitar suavemente mediante movimientos de rotación el vial con el polvo para garantizar la disolución total del polvo. No agitar el vial.
5. Sujetar con una mano el vial con el polvo con el Mix2Vial, sujetar con la otra mano el otro extremo que corresponde al vial del disolvente. Desenroscar el conjunto en 2 piezas. Llenar con aire una jeringa vacía y estéril.
Manteniendo el vial con el polvo en posición vertical, conectar la jeringa al Mix2Vial. Inyectar el aire en el vial con el polvo.
Trasvase de la solución y administración:
|
1 |
16 |
6. Manteniendo el émbolo de la jeringa presionado, invertir el sistema, aspirar la solución obtenida en la jeringa tirando despacio del émbolo. | ||
|
I |
7 |
7. Una vez que la solución obtenida ha sido transferida a la jeringa, sujetar firmemente el cuerpo de la jeringa (manteniendo el émbolo de la jeringa mirando hacia abajo) y desenroscar el Mix2Vial de la jeringa. |
Prefundir o inyectar inmediatamente por vía intravenosa lenta (ver Sección 4.2. Método de administración).
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
CSL Behring GmbH 35041 - Marburg. Alemania
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
N° de registro: 60.312
ílMt
ÍTTI
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN Fecha autorización: 23.12.94. Fecha última renovación: 18.07.2008
10. FECHA DE LA REVISIÓN DEL TEXTO
Julio 2011.