Memantina Cinfa 5 Mg/Pulsacion Solucion Oral Efg
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Memantina cinfa 5 mg/pulsación, solución oral EFG.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada pulsación del dosificador (una pulsación) libera 0,5 ml de solución conteniendo 5 mg de hidrocloruro de memantina, equivalente a 4,16 mg de memantina.
Excipientes de efecto conocido:
Cada mililitro de solución contiene 100 mg de sorbitol líquido no cristalizable (E420) y 0,5 mg de potasio, ver sección 4.4.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA Solución oral.
La solución es transparente y de incolora a ligeramente amarillenta.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento de pacientes con enfermedad de Alzheimer de moderada a grave.
4.2 Posología y forma de administración
El tratamiento debe ser iniciado y supervisado por un médico con experiencia en el diagnóstico y tratamiento de la demencia de Alzheimer. El tratamiento se debe iniciar únicamente si se dispone de un cuidador que monitorice regularmente la toma del fármaco por parte del paciente. Se debe realizar el diagnóstico siguiendo las directrices actuales. La tolerabilidad y la dosis de memantina se deben reevaluar de forma regular, preferiblemente dentro de los 3 meses posteriores al inicio del tratamiento. Por lo tanto, el beneficio clínico de memantina y la tolerabilidad del paciente al tratamiento se deben reevaluar de forma regular de acuerdo a las directrices clínicas vigentes. El tratamiento de mantenimiento puede continuarse mientras el beneficio terapéutico sea favorable y el paciente tolere el tratamiento con memantina. La interrupción del tratamiento con memantina se debe considerar cuando ya no se evidencie su efecto terapéutico o si el paciente no tolera el tratamiento.
Este medicamento debe tomarse una vez al día, a la misma hora cada día. La solución no debe verterse, pulverizarse o administrarse dentro de la boca directamente desde el dosificador del frasco, debe colocarse en una cuchara o dentro de un vaso de agua usando el dosificador.
Para instrucciones detalladas sobre la preparación y el manejo del producto, ver sección 6.6.
La solución puede tomarse con o sin alimentos.
Adultos:
Ajuste de la dosis
La dosis máxima diaria es de 20 mg al día. Para reducir el riesgo de sufrir efectos adversos, la dosis de mantenimiento se alcanza incrementando la dosis 5 mg cada semana durante las primeras 3 semanas de la siguiente manera:
Semana 1 (día 1-7):
El paciente debe tomar 0,5 ml de solución (5 mg) equivalente a una pulsación al día hacia abajo durante 7 días.
Semana 2 (día 8-14):
El paciente debe tomar 1 ml de solución (10 mg) equivalente a dos pulsaciones al día hacia abajo durante 7 días.
Semana 3 (día 15-21):
El paciente debe tomar 1,5 ml de solución (15 mg) equivalente a tres pulsaciones al día hacia abajo durante 7 días.
A partir de la semana 4:
El paciente debe tomar 2 ml de solución (20 mg) equivalente a cuatro pulsaciones al día administradas hacia abajo de una sola vez.
Dosis de mantenimiento
La dosis recomendada de mantenimiento es de 20 mg al día.
Pacientes de edad avanzada: Basándose en estudios clínicos, la dosis recomendada para los pacientes mayores de 65 años es de 20 mg al día (2 ml solución, equivalente a cuatro pulsaciones hacia abajo) tal como se ha descrito anteriormente.
Niños y adolescentes menores de 18 años: No se recomienda el uso de este medicamento en niños menores de 18 años debido a una falta de datos de seguridad y eficacia.
Insuficiencia renal: En pacientes con función renal levemente afectada (aclaramiento de creatinina de 50 -80 ml/min), no es necesario ajustar la dosis. En pacientes con insuficiencia renal moderada (aclaramiento de creatinina de 30 - 49 ml/min), la dosis diaria debe ser de 10 mg al día (1 ml solución, equivalente a dos pulsaciones hacia abajo Si se tolera bien después de, al menos 7 días de tratamiento, la dosis podría aumentarse hasta 20 mg/día de acuerdo con el esquema de titulación estándar. En pacientes con insuficiencia renal grave (aclaramiento de creatinina de 5-29 ml/min.) la dosis diaria debe ser de 10 mg al día (1 ml solución, equivalente a dos pulsaciones hacia abajo).
Insuficiencia hepática: En pacientes con insuficiencia hepática leve o moderada (Child-Pugh A y Child-Pugh B) no es necesario ajustar la dosis. No existen datos disponibles sobre el uso de memantina en pacientes con insuficiencia hepática grave. No se recomienda la administración deeste medicamento a pacientes con insuficiencia hepática grave.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Se recomienda precaución en el tratamiento de pacientes con epilepsia, antecedentes de crisis convulsivas o en pacientes con factores de riesgo para padecer epilepsia.
Se debe evitar la administración concomitante de antagonistas del N-metil-D-aspartato (NMDA) como la amantadina, la ketamina y el dextrometorfano. Estos compuestos actúan sobre el mismo sistema receptor que memantina y, por lo tanto, las reacciones adversas (principalmente relacionadas con el sistema nervioso central [SNC]) pueden ser más frecuentes o más intensas (ver sección 4.5).
Todos aquellos factores que aumenten el pH urinario (ver sección 5.2 “Eliminación”) pueden requerir una monitorización rigurosa del paciente. Entre estos factores se incluyen cambios drásticos en la dieta, por ejemplo de carnívora a vegetariana, o una ingesta masiva de tampones gástricos alcalinizantes. Asimismo, el pH urinario puede estar elevado en estados de acidosis tubular renal(ATR) o infecciones graves del tracto urinario por bacterias del género Proteus.
En la mayoría de los ensayos clínicos, se excluyeron aquellos pacientes con antecedentes de infarto de miocardio reciente, enfermedad cardíaca congestiva (NYHA III-IV) o hipertensión no controlada. Como consecuencia, los datos en estos pacientes son limitados y los pacientes que presentan estas condiciones deben supervisarse cuidadosamente.
Excipientes: La solución oral contiene sorbitol. Los pacientes con intolerancia hereditaria a la fructosa, no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Debido a los efectos farmacológicos y al mecanismo de acción de la memantina, pueden producirse las siguientes interacciones:
• El mecanismo de acción sugiere que los efectos de la L-dopa, los agonistas dopaminérgicos y los anticolinérgicos pueden aumentar con el tratamiento concomitante de antagonistas del NMDA como memantina. Se pueden reducir los efectos de los barbitúricos y de los neurolépticos. La administración concomitante de memantina y agentes antiespasmódicos, como el dantroleno o el baclofeno, puede modificar sus efectos y hacer necesario un ajuste de la dosis.
• Se debe evitar el uso concomitante de memantina y amantadina, por el riesgo de psicosis farmacotóxica. Los dos compuestos están químicamente relacionados con los antagonistas del NMDA. Esto mismo podría aplicarse para la ketamina y el dextrometorfano (ver sección 4.4).
También hay un caso clínico publicado sobre el posible riesgo de la combinación de memantina y fenitoína.
• Otros principios activos, como cimetidina, ranitidina, procainamida, quinidina, quinina y nicotina, que utilizan el mismo sistema de transporte catiónico renal que la amantadina, posiblemente también interaccionen con la memantina lo que conlleva un riesgo potencial de aumento de los niveles plasmáticos.
• Cuando se co-administra memantina junto con hidroclorotiazida (HCTZ) o con cualquier combinación con HCTZ existe la posibilidad de que se produzca una disminución en los niveles séricos de la HCTZ.
• En la experiencia post-comercialización, se ha informado de casos aislados de incremento del cociente internacional normalizado (INR), en pacientes tratados concomitantemente con warfarina. Aunque no se ha establecido relación causal, es aconsejable realizar una monitorización estrecha del tiempo de protrombina o INR, en pacientes tratados concomitantemente con anticoagulantes orales.
En estudios farmacocinéticos (FC) a dosis únicas realizados en sujetos jóvenes sanos, no se han observado interacciones relevantes principio activo-principio activo entre memantina y gliburida /metformina o donepezilo.
En un ensayo clínico realizado en sujetos jóvenes sanos, no se han observado efectos relevantes de memantina sobre la farmacocinética de la galantamina.
Memantina no inhibió las isoformas CYP 1A2, 2A6, 2C9, 2D6, 2E1, 3A, la flavina monooxigenasa, la epóxido hidrolasa o la sulfonación in vitro.
4.6 Fertilidad, embarazo y lactancia
Embarazo:
No se dispone de datos clínicos sobre la utilización de memantina durante el embarazo. Estudios con animales indican un riesgo potencial de disminución del crecimiento intrauterino con niveles de exposición idénticos o ligeramente más altos que los niveles de exposición en humanos (ver sección 5.3). No se conoce el riesgo potencial para humanos. Memantina no debe utilizarse durante el embarazo excepto que sea considerado claramente necesario.
Lactancia:
.‘ítp.
untes
an
Se desconoce si memantina se excreta por la leche materna pero, teniendo en cuenta la lipofilia del principio activo, es probable que así sea. Las mujeres que tomen memantina deben suspender la lactancia materna.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La enfermedad de Alzheimer de moderada a grave afecta normalmente la capacidad de conducción y compromete la capacidad para utilizar máquinas. Además, este medicamento presenta una influencia de leve a moderada sobre la capacidad de para conducir y de utilizar máquinas, de forma que se debe advertir especialmente a los pacientes ambulatorios para que tomen precauciones especiales.
4.8 Reacciones adversas
En los ensayos clínicos en pacientes con demencia de leve a grave, en los que se incluyeron 1784 pacientes tratados con este medicamento y 1595 pacientes tratados con placebo, la incidencia global de reacciones adversas con este medicamento no difirió de la de aquellos tratados con placebo; las reacciones adversas fueron, por lo general de leves a moderadas en gravedad. Las reacciones adversas con mayor frecuencia de aparición que se observaron con una incidencia superior en el grupo de este medicamento respecto al grupo placebo fueron vértigo (6,3% frente a 5,6%, respectivamente), cefalea (5,2% frente a 3,9%), estreñimiento (4,6% frente a 2,6%), somnolencia (3,4% frente a 2,2%) e hipertensión (4,1% frente a 2,8%).
Las reacciones adversas al medicamento (RAM) enumeradas en la siguiente tabla proceden de los ensayos clínicos realizados con este medicamento y la experiencia post-comercialización. Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
Las reacciones adversas se categorizan conforme al sistema de clasificación por órganos, usando el siguiente convenio: muy frecuentes (> 1/10), frecuentes (de >1/100 a < 1/10), poco frecuentes (de > 1/1.000 a < 1/100), raras (de >1/10.000 a < 1/1.000), muy raras (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
|
Sistema de clasificación por órganos |
Frecuencia |
Reacción adversa |
|
Infecciones e infestaciones |
Poco frecuentes |
Infecciones fúngicas |
|
Trastornos del sistema inmunológico |
Frecuentes |
Hipersensibilidad al medicamento |
|
Trastornos psiquiátricos |
Frecuentes |
Somnolencia |
|
Poco frecuentes |
Confusión | |
|
Poco frecuentes |
Alucinaciones1 | |
|
Frecuencia no conocida |
Reacciones psicóticas2 | |
|
Trastornos del sistema nervioso |
Frecuentes |
Vértigo |
|
Frecuentes |
Alteraciones del equilibrio | |
|
Poco frecuentes |
Alteración de la marcha | |
|
Muy raras |
Convulsiones | |
|
Trastornos cardíacos |
Poco frecuentes |
Insuficiencia cardíaca |
|
Trastornos vasculares |
Frecuentes |
Hipertensión |
|
Poco frecuentes |
Trombosis venosa/tromboembolismo | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Frecuentes |
Disnea |
|
Trastornos gastrointestinales |
Frecuentes |
Estreñimiento |
|
Poco frecuentes |
Vómitos | |
|
Frecuencia no conocida |
Pancreatitis2 | |
|
Trastornos hepatobiliares |
Frecuentes |
Pruebas de función hepática elevadas |
|
Frecuencia no conocida |
Hepatitis |
Trastornos generales y alteraciones en el lugar de administración
|
Frecuentes |
Cefalea |
|
Poco frecuentes |
Fatiga |
1 Las alucinaciones se han observado principalmente en pacientes con enfermedad de Alzheimer grave.
2 Se han notificado casos aislados en la experiencia post-comercialización.
La enfermedad de Alzheimer ha sido asociada con depresión, ideación suicida y suicidio. En la experiencia post-comercialización, se ha notificado la aparición de estos acontecimientos en pacientes tratados con este medicamento.
Notificación de sospechas de reacciones adversas
Es importante notificar las sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: w ww.notificaRAM.es .
4.9 Sobredosis
Solo se dispone de experiencia limitada en casos de sobredosis de los ensayos clínicos y de la experiencia post-comercialización.
Síntomas: Sobredosis relativamente altas (200 mg y 105 mg/día durante 3 días respectivamente) se han asociado únicamente con síntomas como cansancio, debilidad y/o diarrea o han sido asintomáticas. En casos de sobredosis por debajo de 140 mg o dosis no conocida aparecieron en los pacientes síntomas a nivel del sistema nervioso central (confusión, adormecimiento, somnolencia, vértigo, agitación, agresividad, alucinaciones y alteraciones de la marcha) y/o de origen gastrointestinal (vómitos y diarreas). En el caso más extremo de sobredosis, el paciente sobrevivió a la ingesta oral de un total de 2000 mg de memantina con efectos a nivel del sistema nervioso central (coma durante 10 días, y posterior diplopía y agitación). El paciente recibió tratamiento sintomático y plasmaféresis, recuperándose sin secuelas permanentes.
En otro caso de sobredosis grave, el paciente también sobrevivió y se recuperó. Dicho paciente había recibido 400 mg de memantina por vía oral, y experimentó síntomas a nivel del sistema nervioso central tales como inquietud, psicosis, alucinaciones visuales, proconvulsividad, somnolencia, estupor e inconsciencia.
Tratamiento: En caso de sobredosis, el tratamiento debe ser sintomático. No existe antídoto específico para la intoxicación o sobredosis. Se deben utilizar procedimientos clínicos estándar para la eliminación del principio activo de forma apropiada, por ej.: lavado gástrico, carbón activado (interrupción de la recirculación enterohepática potencial), acidificación de la orina, diuresis forzada.
En caso de aparición de signos y síntomas de sobrestimulación general del sistema nervioso central (SNC), se debe considerar llevar a cabo un tratamiento clínico sintomático cuidadoso.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Otros fármacos anti-demencia, código ATC: N06DX01.
Existe una evidencia cada vez más clara de que el mal funcionamiento de la neurotransmisión glutamatérgica, en particular en los receptores NMDA, contribuye tanto a la expresión de los síntomas como a la progresión de la enfermedad hacia demencia neurodegenerativa.
Memantina es un antagonista no competitivo de los receptores NMDA, de afinidad moderada y voltaje dependiente. Modula los efectos de los niveles tónicos de glutamato elevados patológicamente que pueden ocasionar disfunción neuronal.
Ensayos clínicos
Un ensayo pivotal de monoterapia en pacientes que padecían enfermedad de Alzheimer de moderada a grave (puntuación total en el miniexamen cognoscitivo (MMSE, mini mental state examination) al inicio del estudio 3 - 14) incluyó un total de 252 pacientes. El estudio mostró los efectos beneficiosos del tratamiento con memantina respecto al tratamiento con placebo después de 6 meses (análisis de casos observados para el estudio CIBIC-plus (siglas en inglés que corresponden a la escala de impresión de cambio basada en la entrevista del médico, mas los comentarios del cuidador): p=0,025; ADCS-ADLsev (siglas en inglés que corresponden al estudio cooperativo de la enfermedad de Alzheimer - actividades de la vida diaria): p=0,003; SIB (siglas en inglés que corresponden a la batería de deterioro grave): p=0,002).
Un estudio pivotal de memantina en monoterapia en el tratamiento de la enfermedad de Alzheimer de leve a moderada (puntuación total MMSE al inicio del estudio de 10 a 22) incluyó a 403 pacientes. Los pacientes tratados con memantina mostraron un efecto superior a placebo estadísticamente significativo, en las variables principales: ADAS-cog (siglas en inglés que corresponden a la escala de valoración de la enfermedad de Alzheimer-subescala cognitiva) (p=0,003) y CIBIC-plus (p=0,004) en la semana 24 LOCF (siglas en inglés que corresponden al método de arrastre de la última observación realizada). En otro ensayo de monoterapia en enfermedad de Alzheimer de leve a moderada se aleatorizaron un total de 470 pacientes (puntuación total MMSE al inicio del estudio de 11 - 23). En el análisis primario definido prospectivamente no se encontró diferencia estadísiticamente significativa en las variables primarias de eficacia en la semana 24.
Un meta-análisis de los pacientes con enfermedad de Alzheimer de moderada a grave (puntuación total MMSE < 20) de seis estudios en fase III, controlados con placebo en estudios a 6 meses (incluyendo estudios de monoterapia y estudios con pacientes con una dosis estable de un inhibidor de la acetilcolinesterasa), mostró que había un efecto estadísticamente significativo a favor del tratamiento con memantina en las áreas cognitiva, global y funcional. Cuando los pacientes fueron identificados con un empeoramiento conjunto en las tres áreas, los resultados mostraron un efecto estadísticamente significativo de memantina en la prevención del empeoramiento, el doble de los pacientes tratados con placebo en comparación con los pacientes tratados con memantina mostraron empeoramiento en las tres áreas (21% frente 11%, p<0,0001).
5.2 Propiedades farmacocinéticas
Absorción: Memantina tiene una biodisponibilidad absoluta de aproximadamente el 100%. La tmáx está entre 3 y 8 horas. No hay indicios de la influencia de alimentos en la absorción de memantina.
Distribución: Las dosis diarias de 20 mg producen concentraciones plasmáticas constantes de memantina que oscilan entre 70 y 150 ng/ml (0,5 - 1 pmol) con importantes variaciones interindividuales. Cuando se administraron dosis diarias de 5 a 30 mg, se obtuvo un índice medio de líquido cefalorraquídeo (LCR)/suero de 0,52. El volumen de distribución es de aproximadamente 10l/kg. Alrededor del 45% de memantina se une a proteínas plasmáticas.
Biotransformación: En el hombre, aproximadamente el 80% del material circulante relacionado con memantina está presente como compuesto inalterado. Los principales metabolitos en humanos son N- 3,5-dimetil-gludantano, la mezcla isomérica de 4- y 6-hidroxi-memantina y 1-nitroso-3,5-dimetiladamantano. Ninguno de estos metabolitos muestra actividad antagonista NMDA. No se ha detectado in vitro metabolismo catalizado por citocromo P 450.
En un estudio con 14C-memantina administrado vía oral, se recuperó una media del 84% de la dosis dentro de los 20 días, excretándose más del 99% por vía renal.
Eliminación: Memantina se elimina de manera monoexponencial con una ti terminal de 60 a 100 horas. En voluntarios con función renal normal, el aclaramiento total (Cltot) asciende a 170 ml/min/1,73 m2 y parte del aclaramiento total renal se logra por secreción tubular.
La función renal también incluye la reabsorción tubular, probablemente mediada por proteínas transportadoras de cationes. La tasa de eliminación renal de la memantina en condiciones de orina alcalina puede reducirse en un factor entre 7 y 9 (ver sección 4.4). La alcalinización de la orina se puede producir por cambios drásticos en la dieta, por ejemplo de carnívora a vegetariana, o por una ingesta masiva de tampones gástricos alcalinizantes.
Linealidad: Los estudios en voluntarios han demostrado una farmacocinética lineal en el intervalo de dosis de 10 a 40 mg.
Relación farmacocinética/farmacodinámica: A una dosis de memantina de 20 mg al día los niveles en LCR concuerdan con el valor ki (ki=constante de inhibición) de memantina, que es de 0,5 pmol en la corteza frontal humana.
5.3 Datos preclínicos sobre seguridad
En estudios a corto plazo en ratas, memantina al igual que otros antagonistas del NMDA, indujo vacuolización neuronal y necrosis (lesiones de Olney) únicamente tras dosis que producían picos muy altos de concentraciones séricas. La ataxia y otros signos preclínicos precedieron a la vacuolización ya la necrosis. Como estos efectos no se observaron en roedores ni en no roedores en estudios a largo plazo, se desconoce la importancia clínica de estos hallazgos.
Se observaron cambios oculares en estudios de toxicidad de dosis repetidas en roedores y perros, pero no en monos. Los exámenes específicos oftalmoscópicos realizados en estudios clínicos conmemantina no revelaron cambios oculares.
En roedores se observó fosfolipidosis en macrófagos pulmonares producido por la acumulación de memantina en lisosomas. Este efecto se ha observado en otros principios activos con propiedades anfifílicas catiónicas. Existe una posible relación entre esta acumulación y la vacuolización observada en los pulmones. Este efecto se observó solamente en roedores a dosis altas. Se desconoce la importancia clínica de estos hallazgos.
No se observó genotoxicidad en los ensayos estándar realizados con memantina. No hubo evidencias de carcinogenicidad en los estudios en ratones y ratas hasta su muerte. Memantina no resultó teratogénica ni en ratas ni en conejos, incluso a dosis tóxicas para la madre y no se observó ningún efecto adverso de memantina sobre la fertilidad. En ratas, se observó una reducción del crecimiento fetal a niveles de exposición idénticos o ligeramente más altos que los niveles de exposición humana.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Sorbato potásico E202
Sorbitol líquido no cristalizable E420
Agua purificada
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez 30 meses.
Una vez abierto, el contenido del frasco debe ser utilizado en 12 semanas.
6.4 Precauciones especiales de conservación
Este medicamento no requiere condiciones especiales de conservación.
El frasco con el dosificador montado, debe guardarse y transportarse únicamente en posición vertical.
6.5 Naturaleza y contenido del envase
Frasco de vidrio de color ámbar (Tipo III) que contiene 100 ml de solución oral con tapón de rosca de polietileno y acompañado de un dosificador.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Ninguna especial.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
Instrucciones para el correcto uso del dosificador:
Antes del primer uso, el dosificador debe enroscarse en el frasco. Para sacar el tapón de rosca del frasco debe girarse en el sentido contrario a las agujas del reloj y desenroscarse completamente (fig.1).
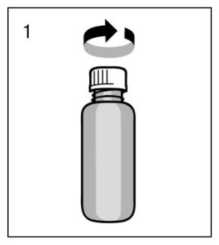
Montaje del dosificador en el frasco:
El dosificador debe sacarse de la bolsa de plástico (fig.2) y colocarse encima del frasco, introduciendo hacia abajo el tubo de plástico dentro del frasco con cuidado. El dosificador debe mantenerse en el cuello del frasco y girarse en el sentido de las agujas del reloj hasta que esté unido firmemente (fig.3). Para el uso
deseado, el dosisficador sólo debe enroscarse una vez al iniciar el uso y nunca debe desenroscarse.
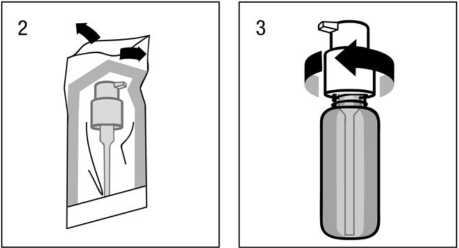
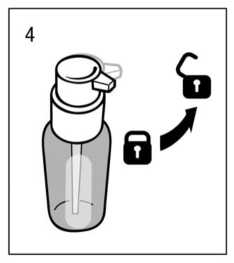
Uso del dosificador para dispensación:
El cabezal del dosificador tiene dos posiciones y se gira fácilmente - dirección contraria al sentido delas agujas del reloj (posición abierta) y dirección en el sentido de las agujas del reloj (posición cerrada). El
cabezal del dosificador no debe pulsarse mientras esté en la posición cerrada. La solución debe dispensarse por tanto en la posición abierta. Para ello, el cabezal del dosisficador debe girarse un octavo de giro en la dirección que indica la flecha, hasta encontrar resistencia (fig.4). El dosificador está entonces listo para su uso.
Preparación del dosificador:
Cuando se utiliza por primera vez, el dosificador no dispensa la cantidad correcta de solución oral. Por tanto debe prepararse (cebarse) pulsando el cabezal del dosificador hacia abajo completamente durante cinco veces sucesivas (fig.5).
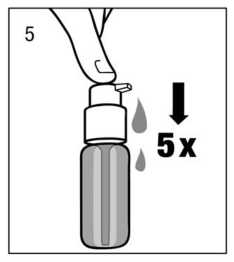
La solución así dispensada debe desecharse. La siguiente vez que el cabezal del dosificador se pulsa hacia abajo completamente (equivalente a una pulsación), ya dispensa la dosis correcta (1 pulsación es equivalente a 0,5 ml de solución oral y contiene 5 mg del principio activo clorhidrato de memantina; fig.6).
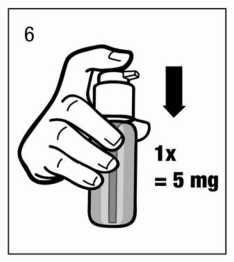
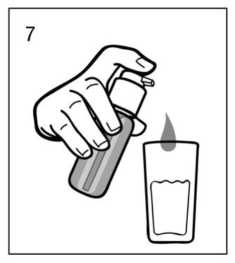
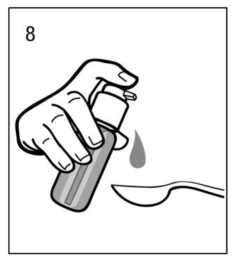
Uso correcto del dosificador:
Debe colocarse un vaso con un poco de agua o una cuchara debajo de la boquilla y el cabezal del dosificador debe pulsarse hacia abajo firmemente pero tranquilamente y de forma sostenida (no demasiado despacio) hasta el final (fig.7, fig.8).
ÜE
El cabezal puede entonces soltarse y está listo para la siguiente pulsación.
El dosificador debe usarse únicamente con memantina cinfa 10 mg/ml solución en el frasco proporcionado, no para otros productos o envases. Si el dosificador no funciona correctamente como se describe durante su uso y de acuerdo a las instrucciones, el paciente debería consultar a su médico o a un farmacéutico. El dosificador debe cerrarse después de su uso.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Laboratorios Cinfa, S.A.
C/ Olaz-Chipi, 10 - Polígono Industrial Areta.
31620 Huarte - Pamplona (Navarra)-España
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Diciembre 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
10 de 10