Lynparza 50Mg Capsulas Duras
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Lynparza 50 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula dura contiene 50 mg de olaparib.
Para consultar la lista completa de excipientes, ver sección 6.1
3. FORMA FARMACÉUTICA
Cápsula dura.
Cápsula dura de tamaño 0, blanca, opaca, marcada con “OLAPARIB 50 mg” y el logotipo de AstraZeneca en tinta negra.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Lynparza está indicado como monoterapia para el tratamiento de mantenimiento de pacientes adultas con cáncer de ovario epitelial seroso de alto grado, trompa de Falopio, o peritoneal primario, con mutación BRCA (germinal y/o somática), sensible a platino, en recaída, que están en respuesta (respuesta completa o parcial) a quimioterapia basada en platino.
4.2 Posología y forma de administración
El tratamiento con Lynparza se debe iniciar y supervisar por un médico con experiencia en el empleo de medicamentos antineoplásicos.
Antes de que se inicie el tratamiento con Lynparza, las pacientes deben tener confirmación de una mutación del gen sensible al cáncer de mama (BRCA) (bien germinal o somática). El estado de la mutación BRCA debe estar determinado por un laboratorio con experiencia mediante un método de análisis validado (ver sección 5.1).
Existen datos limitados en pacientes con tumores y mutación somática BRCA (ver sección 5.1).
Para pacientes con mutaciones BRCA se debe realizar consejo genético de acuerdo a las regulaciones locales.
Posología
La dosis recomendada de Lynparza es 400 mg (ocho cápsulas) dos veces al día, equivalente a una dosis total diaria de 800 mg.
Las pacientes deben iniciar el tratamiento con Lynparza, no más tarde de transcurridas 8 semanas después de la finalización de su última dosis del régimen que contiene platino.
Se recomienda continuar el tratamiento hasta progresión de la enfermedad subyacente. No existen datos de retratamiento con Lynparza tras recaída posterior (ver sección 5.1).
Dosis olvidada
Si una paciente olvida una dosis de Lynparza, debe tomar su siguiente dosis normal en el momento programado.
Ajustes de dosis para reacciones adversas
El tratamiento puede ser interrumpido para tratar reacciones adversas tales como náuseas, vómitos, diarrea y anemia y se puede considerar una reducción de la dosis (ver sección 4.8).
La reducción de dosis recomendada es a 200 mg dos veces al día (equivalente a una dosis total diaria de 400 mg).
Si se requiere una reducción final de la dosis de forma adicional, se podría considerar una reducción a 100 mg dos veces al día (equivalente a una dosis total diaria de 200 mg).
Ajustes de dosis para la administración concomitante con inhibidores del CYP3A No se recomienda el uso concomitante con inhibidores potentes ni moderados del CYP3A y se deben considerar agentes alternativos. Si es necesario administrar de forma concomitante un inhibidor potente o moderado del CYP3A, se recomienda reducir la dosis de olaparib a 150 mg tomado dos veces al día (equivalente a una dosis total diaria de 300 mg) con un inhibidor potente del CYP3A o a 200 mg tomado dos veces al día (equivalente a una dosis total diaria de 400 mg) con un inhibidor moderado de CYP3A (ver secciones 4.4 y 4.5).
Pacientes de edad avanzada
No se requiere ajuste en la dosis inicial para pacientes de edad avanzada. Se dispone de datos clínicos limitados en pacientes de 75 años o mayores.
Insuficiencia renal
La dosis recomendada de Lynparza en pacientes con insuficiencia renal moderada (aclaramiento de creatinina de 31 a 50 ml/min) es 300 mg dos veces al día (equivalente a una dosis total de 600 mg al día) (ver sección 5.2).
Lynparza se puede administrar a pacientes con insuficiencia renal leve (aclaramiento de creatinina de 51 a 80 ml/min) sin ajuste de dosis.
En pacientes con insuficiencia renal grave o con enfermedad renal en estadio terminal (aclaramiento de creatinina <30 ml/min) no se recomienda Lynparza, ya que no se dispone de datos en estos pacientes. Lynparza sólo se puede usar en pacientes con insuficiencia renal grave, si el beneficio supera el posible riesgo, debiéndose monitorizar cuidadosamente estas pacientes en cuanto a la función renal y acontecimientos adversos.
Insuficiencia hepática
No se ha estudiado el efecto de la insuficiencia hepática en la exposición a Lynparza. Por tanto, no se recomienda el uso de Lynparza en pacientes con insuficiencia hepática (bilirrubina sérica superior a 1,5 veces el límite superior normal), ya que no se ha establecido la seguridad y eficacia.
Pacientes no caucásicas
Existen datos clínicos limitados en pacientes no caucásicas; no obstante, no es necesario ajuste de dosis en función de la raza (ver sección 5.2).
Pacientes con estado funcional de 2 a 4
Existen datos clínicos muy limitados en pacientes con estado funcional de 2 a 4.
Población pediátrica
No se ha establecido la seguridad y eficacia de Lynparza en niños y adolescentes.
No se dispone de datos.
Forma de administración
Lynparza se administra por vía oral.
Debido al efecto de la comida en la absorción de olaparib, las pacientes deben tomar Lynparza al menos una hora después de las comidas, y abstenerse de comer preferiblemente hasta dos horas más tarde.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1 Lactancia durante el tratamiento y 1 mes después de la última dosis (ver sección 4.6).
4.4 Advertencias y precauciones especiales de empleo
Toxicidad hematológica
Se ha notificado toxicidad hematológica en pacientes tratadas con olaparib, incluyendo el diagnóstico clínico y/o los hallazgos de laboratorio de anemia generalmente leve o moderada (CTCAE grado 1 ó 2), neutropenia, trombocitopenia y linfopenia. Las pacientes no deben iniciar el tratamiento con Lynparza hasta que se hayan recuperado de la toxicidad hematológica causada por la terapia antineoplásica previa (los niveles de hemoglobina, plaquetas y neutrófilos deben estar dentro del rango normal o CTCAE grado 1). Se recomienda realizar pruebas al inicio, seguidas de una monitorización mensual, del recuento sanguíneo completo durante los primeros 12 meses de tratamiento y de forma periódica a partir de este momento, para monitorizar los cambios clínicamente significativos en cualquier parámetro durante el tratamiento.
Si una paciente desarrolla toxicidad hematológica grave o dependencia de transfusión sanguínea, se debe interrumpir el tratamiento con Lynparza y se debe iniciar un análisis hematológico adecuado. Si los parámetros sanguíneos permanecen siendo clínicamente anormales tras 4 semanas de la interrupción de la dosis de Lynparza, es recomendable un análisis de la médula ósea y/o un análisis citogenético de sangre.
Síndrome Mielodisplásico/Leucemia Mieloide Aguda
En un pequeño número de pacientes que recibieron Lynparza, solo o en combinación con otro medicamento antineoplásico, se han notificado casos de Síndrome Mielodisplásico/Leucemia Mieloide Aguda (SMD/LMA); siendo la mayoría de los casos mortales. La duración de la terapia con olaparib en pacientes que desarrollaron SMD/LMA varió de < 6 meses a > 2años. Los casos fueron típicos de SDM secundario/LMA relacionada con la terapia para el cáncer. Todas las pacientes presentaban factores potenciales que contribuían al desarrollo de SDM/LMA; la mayoría de los casos fueron en portadoras de la mutación gBRCA y algunas de las pacientes tenían antecedentes previos de cáncer o displasia de médula ósea. Todas habían recibido regímenes previos de quimioterapia basada en platino y muchas habían recibido también otros agentes que dañan el ADN y radioterapia. Si durante el tratamiento con Lynparza, se confirma SDM y/o LMA, es recomendable que se trate a la paciente adecuadamente. Si se recomienda una terapia antineoplásica adicional, se debe interrumpir el tratamiento con Lynparza y no administrarse en combinación con dicha terapia antineoplásica.
Neumonitis
En un pequeño número de pacientes que estaban recibiendo olaparib se ha notificado neumonitis y algunas de ellas han sido mortales. Los informes de neumonitis carecían de un patrón clínico coherente y se confundían con varios factores de predisposición (cáncer y/o metástasis en pulmones, enfermedad pulmonar subyacente, antecedentes de tabaquismo, y/o quimioterapia y radioterapia previas). Si las pacientes presentan síntomas respiratorios nuevos o empeoramiento de los mismos tales como disnea, tos y fiebre, o se produce una anomalía radiológica, se debe interrumpir el tratamiento con Lynparza e iniciar una rápida investigación. Si se confirma neumonitis, se debe interrumpir el tratamiento con Lynparza y tratar a la paciente adecuadamente.
Toxicidad embriofetal
Según su mecanismo de acción (inhibición de PARP), olaparib podría causar daño fetal cuando se administra a una mujer embarazada. Estudios no clínicos en ratas han mostrado que olaparib causa efectos adversos en la supervivencia embriofetal e induce malformaciones fetales importantes, a exposiciones por debajo de las esperadas a la dosis recomendada para humanos de 400 mg dos veces al día.
Embarazo/anticoncepción
Lynparza no se debe usar durante el embarazo, ni en mujeres en edad fértil que no utilicen métodos anticonceptivos fiables durante la terapia y durante 1 mes después de recibir la última dosis de Lynparza (ver sección 4.6).
Interacciones
No se recomienda la administración concomitante con inhibidores potentes o moderados del CYP3A (ver sección 4.5). Si es necesario administrar de forma concomitante un inhibidor potente o moderado del CYP3A, se debe reducir la dosis de olaparib (ver secciones 4.2 y 4.5).
No se recomienda la administración concomitante con inductores potentes del CYP3A (ver sección 4.5). En caso de que una paciente que ya esté recibiendo olaparib precise tratamiento con un inductor potente del CYP3A, el médico prescriptor debe tener en cuenta que la eficacia de olaparib puede reducirse substancialmente (ver sección 4.5).
En el caso de que una paciente que ya esté recibiendo olaparib precise tratamiento con un inhibidor de la Pm se recomienda una monitorización cuidadosa de los acontecimientos adversos asociados a olaparib y el tratamiento de dichos acontecimientos mediante una reducción de la dosis (ver sección 4.2).
4.5 Interacción con otros medicamentos y otras formas de interacción
Interacciones farmacodinámicas
Estudios clínicos de olaparib en combinación con otros medicamentos antineoplásicos, incluyendo fármacos que dañan el ADN, indican una potenciación y prolongación de toxicidad mielosupresora. La dosis recomendada de Lynparza en monoterapia no es adecuada para la combinación con otros medicamentos antineoplásicos.
No se ha estudiado la combinación de olaparib con vacunas o agentes inmunosupresores. Por consiguiente, se debe tomar precaución si estos fármacos se administran de forma concomitante con olaparib y las pacientes deben ser monitorizadas minuciosamente.
Interacciones farmacocinéticas
Efecto de otros fármacos sobre olaparib
CYP3A4/5 son los isoenzimas predominantemente responsables de la eliminación metabólica de olaparib. Un estudio clínico para evaluar el impacto de rifampicina, un inductor conocido del CYP3A, ha demostrado que la administración concomitante con olaparib disminuye la Cmax media de olaparib un 71% (ratio de tratamiento: 0,29; 90% IC: 0,24-0,33) y el AUC medio un 87% (ratio de tratamiento: 0,13; 90% IC: 0,11-0,16). Por lo tanto, se recomienda evitar la administración de inductores potentes conocidos de este isoenzima (por ej., fenitoína, rifampicina, rifapentina, carbamazepina, nevirapina, fenobarbital e hierba de San Juan) con olaparib, ya que es posible que pueda reducirse sustancialmente la eficacia de olaparib (ver sección 4.4). La magnitud del efecto de los inductores moderados a potentes (por ej. efavirenz, rifabutina) sobre la exposición a olaparib no ha sido establecida, por lo que no se recomienda la administración concomitante de olaparib con estos fármacos.
Un estudio clínico para evaluar el impacto de itraconazol, un inhibidor conocido del CYP3A, ha demostrado que la administración concomitante con olaparib aumenta la Cmax media de olaparib del orden de 1,42 veces (90% IC: 1,33-1,52) y el AUC medio del orden de 2,70-veces (90% CI: 2,44-2,97). Por lo tanto, se recomienda evitar la administración de inhibidores conocidos potentes (por ej. itraconazol, telitromicina, claritromicina, inhibidores de la proteasa potenciados con ritonavir o cobicistat, boceprevir, telaprevir) o moderados de este isoenzima (por ej. eritromicina, diltiazem, fluconazol, verapamilo) con olaparib (ver sección 4.4). Si es necesario administrar de forma concomitante inhibidores potentes o moderados del CYP3A, se debe reducir la dosis de olaparib. Se recomienda reducir la dosis de olaparib a 150 mg tomado dos veces al día (equivalente a una dosis total diaria de 300 mg) con un inhibidor potente del CYP3A o a 200 mg tomado dos veces al día (equivalente a una dosis total diaria de 400 mg) con un inhibidor moderado del CYP3A (ver secciones 4.2 y 4.4). Tampoco se recomienda el consumo de zumo de pomelo durante el tratamiento con olaparib.
Olaparib in vitro es un sustrato para el transportador de eflujo P-gp y, por lo tanto, los inhibidores de la P-gp podrían aumentar la exposición a olaparib (ver sección 4.4).
Efecto de olaparib sobre otros fármacos
Olaparib in vitro es un inhibidor del CYP3A4 y se espera que in vivo sea un inhibidor leve.. Por tanto, se debe tener precaución cuando se combinan sustratos sensibles del CYP3A4 o sustancias con un estrecho margen terapéutico (por ej. simvastatina, cisaprida, ciclosporina, alacaloides ergotamínicos, fentanilo, pimozida, sirolimus, tacrolimus y quetiapina) con olaparib. Se recomienda realizar una monitorización clínica apropiada a las pacientes que reciben sustratos del CYP3A con un margen terapéutico estrecho de forma concomitante con olaparib.
Se ha observado in vitro inducción del CYP1A2, 2B6 y 3A4, siendo más probable que la inducción del CYP2B6 alcance un grado clínicamente relevante. Tampoco puede excluirse el potencial de olaparib para inducir CYP2C9, CYP2C19 y P-gp. Por lo tanto, la administración concomitante de olaparib puede reducir la exposición a sustratos de estos enzimas metabólicos y de la proteína transportadora. La eficacia de los anticonceptivos hormonales puede reducirse si se administran junto con olaparib (ver también secciones 4.4 y 4.6).
Olaparib in vitro es un inhibidor del transportador de eflujo P-gp (IC50=76 ^M), por lo tanto, no puede excluirse que olaparib pueda causar interacciones farmacológicas relevantes con sustratos de P-gp (por ej. simvastatina, pravastatina, dabigatran, digoxina, colchicina). Se recomienda realizar una monitorización clínica adecuada a las pacientes que reciben de forma concomitante este tipo de medicación.
Se ha observado in vitro que olaparib es un inhibidor de OATP1B1, OCT1, OCT2, OAT3, MATE1 y MATE2K. No se puede excluir que olaparib pueda aumentar la exposición a los sustratos de OATP1B1 (por ej., bosentán, glibenclamida, repaglinida, estatinas y valsartán), OCT1 (por ej., metformina) OCT2 (por ej., creatinina sérica), OAT3 (por ej. furosemida y metotrexato), MATe1 (por ej. metformina) y MATE2K (por ej. metformina). En particular, se debe tener precaución si se administra olaparib en combinación con cualquier estatina.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil/anticoncepción en mujeres
Las mujeres en edad fértil no se deben quedar embarazadas mientras estén tomando Lynparza y no deben estar embarazadas al inicio del tratamiento. Antes del tratamiento se debe realizar un test de embarazo en todas las mujeres premenopáusicas. Las mujeres en edad fértil deben utilizar métodos anticonceptivos eficaces durante la terapia y durante 1 mes después de recibir la última dosis de Lynparza. La eficacia de los anticonceptivos hormonales puede reducirse si se administran de forma concomitante con olaparib, ya que no se puede excluir que olaparib pueda reducir la exposición a sustratos del CYP3A mediante la inducción del enzima. Por lo tanto, se debe considerar un método anticonceptivo no hormonal adicional y tests de embarazo regulares durante el tratamiento (ver sección 4.5).
Embarazo
Los estudios en animales han mostrado toxicidad reproductiva, incluyendo efectos teratogénicos graves y efectos en la supervivencia embriofetal en la rata en exposiciones sistémicas de la madre inferiores a aquellas en humanos a dosis terapéuticas (ver sección 5.3). No se dispone de datos del uso de olaparib en mujeres embarazadas, sin embargo, teniendo en cuenta el mecanismo de acción de olaparib, Lynparza no se debe usar durante el embarazo ni en mujeres en edad fértil que no utilicen un método anticonceptivo fiable durante el tratamiento y durante 1 mes después de recibir la última dosis de Lynparza. (Ver párrafo anterior: “Mujeres en edad fértil/anticoncepción en mujeres”, para información adicional sobre los métodos anticonceptivos y las pruebas de embarazo.)
Lactancia
No se dispone de estudios en animales de la excreción de olaparib en la leche materna. Se desconoce si olaparib/o sus metabolitos se excretan en la leche humana. Lynparza está contraindicado durante la lactancia y durante 1 mes después de haber recibido la última dosis, dadas las propiedades farmacológicas del medicamento (ver sección 4.3).
Fertilidad
No existen datos clínicos sobre fertilidad. En estudios en animales, no se observó ningún efecto sobre la concepción, aunque sí existen efectos adversos sobre la supervivencia embriofetal (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Durante el tratamiento con Lynparza se han notificado astenia, fatiga y mareo, y aquellas pacientes que experimenten estos síntomas deben tener precaución cuando conduzcan o utilicen máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Olaparib en monoterapia se ha asociado con reacciones adversas generalmente leves o moderadas (CTCAE grado 1 ó 2) y por lo general no requieren interrupción del tratamiento. En los ensayos clínicos en pacientes que recibieron olaparib en monoterapia las reacciones adversas observadas con mayor frecuencia (> 10%) fueron náuseas, vómitos, diarrea, dispepsia, fatiga, cefalea, disgeusia, disminución del apetito, mareo, anemia, neutropenia, linfopenia, elevación del volumen corpuscular medio y aumento en la creatinina.
Tabla de reacciones adversas
En estudios clínicos con pacientes que recibieron Lynparza en monoterapia se han identificado las siguientes reacciones adversas. Su frecuencia se presenta utilizando la clasificación de frecuencias CIOMS III, enumeradas por la Clasificación de Órganos y del Sistema (COS) MedDRA y en el nivel de término preferido. Las frecuencias de aparición de las reacciones adversas se definen como: muy frecuentes (> 1/10); frecuentes (> 1/100 a < 1/10); poco frecuentes (> 1/1.000 a < 1/100); raras (> 1/10.000 a < 1/1000); muy raras (< 1/10.000). Esta sección incluye únicamente datos derivados de estudios completados donde se conoce la exposición de la paciente.
Representa la incidencia de los hallazgos de laboratorio, no de las reacciones adversas notificadas. Las disminuciones para hemoglobina, recuento absoluto de neutrófilos, plaquetas y linfocitos fueron CTAE grado 2 o superior.
Tabla 1 Tabla de reacciones adversas
|
Reacciones Adversas | ||
|
Clasificación de Órganos y del Sistema MedDRA |
Frecuencia de CTCAE todos los grados |
Frecuencia de CTCAE grado 3 y superior |
|
Trastornos del metabolismo y de la nutrición |
Muy frecuentes Disminución del apetito |
Poco frecuentes Disminución del apetito |
|
Trastornos del sistema nervioso |
Muy frecuentes Cefalea, Mareo, Disgeusia |
Poco frecuentes Cefalea, Mareo |
|
Trastornos gastrointestinales |
Muy frecuentes Náuseas, Vómitos, Diarrea, Dispepsia Frecuentes Dolor en la parte superior del abdomen, Estomatitis |
Frecuentes Náuseas, Vómitos, Diarrea Poco frecuentes Dolor en la parte superior del abdomen, Estomatitis |
|
Trastornos generales y alteraciones en el lugar de administración |
Muy frecuentes Fatiga (incluyendo astenia) |
Frecuentes Fatiga (incluyendo astenia) |
|
Exploraciones complementarias |
Muy frecuentes Anemia (disminución de hemoglobina)a,b, Neutropenia (disminución del recuento absoluto de neutrófilos)a,b, Linfopenia (disminución de linfocitos)a,b, Aumento de creatinina en sangrea,d, Elevación del volumen corpuscular medioa,c Frecuentes Trombocitopenia (disminución de plaquetas)a,b |
Muy frecuentes Anemia (disminución de hemoglobina)a,b, Linfopenia (disminución de linfocitos)a,b Frecuentes Neutropenia (disminución del recuento absoluto de neutrófilos)a,b, Trombocitopenia (disminución de plaquetas)a,b Poco frecuentes Aumento de creatinina en sangrea,d |
a
b
c
d
Elevación en el volumen corpuscular medio desde el valor inicial hasta por encima del LSN (límite superior normal). Los niveles parecían volver a la normalidad una vez interrumpido el tratamiento y no parecían tener consecuencias clínicas.
Los datos extraídos de un estudio doble ciego controlado con placebo mostraron un aumento de la mediana (en porcentaje de cambio con respecto a la línea de base) de hasta el 23%, siendo coherente en el tiempo y volviendo al valor inicial una vez interrumpido el tratamiento, sin secuelas clínicas aparentes. En el inicio el 90% de las pacientes fueron CTCAE grado 0, y el 10% CTCAE grado 1.
Descripción de las reacciones adversas seleccionadas
En la terapia con olaparib se han notificado con frecuencia toxicidades gastrointestinales y son generalmente de grado bajo (CTCAE grado 1 ó 2) e intermitentes, pudiendo ser tratadas interrumpiendo la dosis o reduciéndola y/o con medicamentos concomitantes (por ej., terapia antiemética). No se requiere profilaxis antiemética.
Tanto la anemia, como otras toxicidades hematológicas son generalmente de grado bajo (CTCAE grado 1 ó 2). No obstante, se han notificado acontecimientos CTCAE grado 3 y superior. Se recomienda realizar pruebas al inicio, seguidas de una monitorización mensual, del recuento sanguíneo completo durante los primeros 12 meses de tratamiento y de forma periódica a partir de ese momento, para monitorizar los cambios clínicamente significativos en cualquier parámetro durante el tratamiento que pueda requerir la interrupción o reducción de la dosis y/o tratamiento adicional.
Población pediátrica
No se han realizado estudios en pacientes pediátricos.
Otras poblaciones especiales
Se dispone de datos limitados sobre seguridad en pacientes de edad avanzada (edad > 75 años) y en pacientes no caucásicas.
Notificación de sospechas de reacciones adversas
. Se invita a los sistema nacional de
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento profesionales sanitarios a notificar las sospechas de reacciones adversas a través del notificación incluido en el Apéndice V.
4.9 Sobredosis
No hay tratamiento específico en el caso de una sobredosis con Lynparza y no se han establecido los síntomas de sobredosis. En el caso de una sobredosis, los médicos deben seguir las medidas generales de soporte y deben tratar sintomáticamente.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: otros fármacos antineoplásicos, código ATC: L01XX46 Mecanismo de acción y efectos farmacodinámicos
Lynparza es un inhibidor potente de los enzimas poli (ADP-ribosa) polimerasa (PARP-1, PARP-2, y PARP-3) humanos y se ha demostrado que inhibe el crecimiento de líneas celulares tumorales seleccionadas in vitro y el crecimiento de tumores in vivo, ya sea como tratamiento en monoterapia o en combinación con quimioterapias establecidas.
Los PARP son necesarios para la reparación eficiente de las roturas monocatenarias del ADN y un aspecto importante de la reparación inducida del PARP requiere que, tras la modificación de la cromatina, el PARP se automodifique y se disocie del ADN para facilitar el acceso de los enzimas de reparación por escisión de bases (BER). Cuando Lynparza se une al sitio activo del PARP asociado al ADN, evita la disociación del PARP y lo atrapa en el ADN, bloqueando la reparación. Durante la replicación de células, esto produce roturas bicatenarias (DSB) del ADN cuando las horquillas de replicación alcanzan el complejo PARP-ADN. En células normales, la reparación por recombinación homóloga (HRR), que requiere los genes funcionales BRCA1 y 2, es eficaz a la hora de reparar estas roturas bicatenarias del ADN. En ausencia de BRCA1 ó 2 funcionales, las DSB del ADN no se pueden reparar mediante la HRR. En su lugar, se activan vías alternativas y propensas a los errores, como la unión de extremos no homólogos (NHEJ), que ocasiona una mayor inestabilidad genómica. Tras varias rondas de replicación, la inestabilidad genómica puede alcanzar niveles no tolerables y dar como resultado la muerte de las células cancerosas, ya que las células cancerosas presentan una elevada carga de daños del ADN con respecto a las células normales.
En modelos in vivo BRCA deficientes, la administración de olaparib tras el tratamiento con platino dio como resultado un retraso en la progresión del tumor y un aumento en la supervivencia global frente al tratamiento solo con platino.
Detección de la mutación BRCA
Las pacientes son aptas al tratamiento con Lynparza, si se ha confirmado o se sospecha que presentan una mutación BRCA deletérea (es decir, una mutación que interrumpa el funcionamiento normal del gen) bien germinal o somática (detectada mediante un test adecuadamente validado).
Eficacia clínica
En un ensayo fase II aleatorizado, doble ciego y controlado con placebo (estudio 19), se estudió la seguridad y la eficacia de olaparib como una terapia de mantenimiento en el tratamiento de pacientes con cáncer de ovario seroso de alto grado, incluyendo trompa de Falopio o peritoneal primario, sensible a platino en recaída (PSR), tras un tratamiento con dos o más regímenes basados en platino. El estudio comparó la eficacia del tratamiento de mantenimiento con olaparib administrado hasta la progresión respecto al brazo sin tratamiento de mantenimiento en 265 (136 olaparib y 129 placebo) pacientes con cáncer de ovario seroso PSR que estaban en respuesta (CR [respuesta completa] o PR [respuesta parcial]), confirmada mediante los RECIST y/o por el criterio CA-125 definido por el “Gynecologic Cancer InterGroup” (GCIG) (al menos un 50% de reducción en los niveles de CA-125 desde la última muestra previa al tratamiento, confirmado 28 días después) tras finalizar dos o más tratamientos previos de quimioterapia basada en platino. La variable principal era la PFS (supervivencia libre de progresión) basada en la evaluación del investigador mediante los RECIST 1.0. Las variables secundarias de eficacia incluyeron OS (supervivencia global), DCR (tasa de control de la enfermedad) definidos como confirmados CR/PR + SD (enfermedad estable), HRQoL (calidad de vida relacionada con la salud) y síntomas relacionados con la enfermedad. También se realizaron análisis exploratorios del tiempo hasta la primera terapia siguiente o fallecimiento (TFST) y el tiempo hasta la segunda terapia siguiente o fallecimiento (TSST- una aproximación de PFS2).
Sólo participaron pacientes PSR con enfermedad parcialmente sensible a platino (intervalo libre de platino de 6 a 12 meses) y pacientes con enfermedad sensible a platino (intervalo libre de platino de > 12 meses) que estaban en respuesta tras la finalización de la última quimioterapia basada en platino. Las pacientes no podían haber recibido antes olaparib ni otro tratamiento inhibidor del PARP, pero sí haber recibido antes bevacizumab, excepto en el tratamiento inmediatamente anterior a la aleatorización. El retratamiento con olaparib no se permitió tras progresión en tratamiento con este medicamento.
Las pacientes fueron aleatorizadas en el estudio una media de 40 días después de completar su quimioterapia final de platino. Las pacientes recibieron un promedio de 3 regímenes previos de quimioterapia (intervalo 2-11) y 2,6 quimioterapias previas que contenían platino (intervalo 2-8).
Las pacientes del grupo de olaparib siguieron recibiendo tratamiento más tiempo que las del grupo de placebo. Un total de 54 (39,7%) pacientes recibieron tratamiento durante > 12 meses en el grupo de olaparib en comparación con 14 (10,9%) pacientes en el grupo de placebo.
El estudio alcanzó su objetivo principal de mejora estadísticamente significativa de la PFS para la monoterapia de mantenimiento con olaparib en comparación con placebo en la población global (HR 0,35; IC al 95% 0,25-0,49; p < 0,00001), además, el análisis de subgrupo preplanificado por estado de mutación BRCA, identificó a las pacientes con cáncer de ovario BRCA mutado (n=136, 51,3%) como el sugbgrupo que obtuvo el mayor beneficio clínico de la monoterapia de mantenimiento con olaparib.
En pacientes BRCA mutadas (n=136) hubo una mejora estadísticamente significativa en la PFS, TFST y TSST. La mediana de la mejora de la PFS fue de 6,9 meses respecto al placebo para las pacientes tratadas con olaparib (HR0,18; IC al 95% 0,10-0,31; p < 0,00001; mediana de 11,2 meses frente a 4,3 meses). La evaluación de la PFS por el investigador fue coherente con la revisión ciega radiológica central independiente de este objetivo principal. El tiempo desde la aleatorización hasta el principio de la primera terapia siguiente o fallecimiento (TFST) fue de 9,4 meses mayor para las pacientes tratadas con olaparib (HR 0,33; IC al 95% 0,22-0,50; p < 0,00001; mediana 15,6 meses frente a 6,2 meses). El tiempo desde la aleatorización hasta el principio de la segunda terapia siguiente o fallecimiento (TSST) fUe 8,6 meses mayor para las pacientes tratadas con olaparib (HR 0,44; IC al 95% 0,29-0,67; p=0,00013; mediana de 23,8 meses frente a 15,2 meses). No hubo ninguna diferencia estadísticamente significativa en la OS (HR 0,73; IC al 95% 0,45-1,17; p=0,19; mediana de 34,9 meses frente a 31,9 meses). En la población BRCA mutada, el índice de control de la enfermedad a las 24 semanas era del 57% y el 24% para las pacientes de los grupos de olaparib y placebo, respectivamente.
No se observaron diferencias estadísticamente significativas entre olaparib y placebo en los síntomas notificados por las pacientes o HRQoL, medibles por las tasas de mejora y empeoramiento en el Índice de Síntomas Ováricos FACT/NCCN (FOSI), Índice del Resultado del Ensayo (tOi) y Evaluación Funcional del Tratamiento del Cáncer-Cuestionario sobre el cáncer de ovario (FACT-O total).
Los hallazgos clave de eficacia del estudio 19 para pacientes BRCA mutadas se presentan en la Tabla 2, y las Figuras 1 y 2.
Tabla 2 Resumen de los hallazgos clave de eficacia para pacientes con cáncer de ovario BCRA mutado PSR, en el Estudio 19_
|
PFS |
N (acontecimientos /pacientes) (%) |
Mediana de la PFS (meses) |
HRa |
IC al 95% |
Valor p |
|
Olaparib 400 mg dos veces al día |
26/74 (35%) |
11,2 |
0,18 |
0,10-0,31 |
<0,00001 |
|
Placebo |
46/62 (74%) |
4,3 | |||
|
TSST-una aproximación de la PFS2 |
N |
Mediana del TSST (meses) |
HRa |
IC al 95% |
Valor p |
|
Olaparib 400 mg dos veces al dia |
42/74 (57%) |
23,8 |
0,44 |
0,29-0,67 |
0,00013 |
|
Placebo |
49/62 (79%) |
15,2 | |||
|
OS interina (madurez del 52%) |
N |
Mediana de la OS (meses) |
HRa |
IC al 95% |
Valor p |
|
Olaparib 400 mg dos veces al día |
37/74 (50%) |
34,9 |
0,73 |
0,45-1,17 |
0,19 |
|
Placebob |
34/62 (55%) |
31,9 |
a HR= “Hazard Ratio”. Un valor < 1 favorece olaparib. El análisis se realizó utilizando un modelo de riesgos proporcionales de Cox con factores para el tratamiento, tiempo hasta la progresión de la enfermedad durante la penúltima terapia con platino anterior, respuesta objetiva a la última terapia con platino anterior y origen judío.
b Aproximadamente una cuarta parte de las pacientes tratadas con placebo en el subgrupo BRCA mutadas (14/62; 22,6%) recibieron un inhibidor del PARP posterior.
N Número de acontecimientos/número de pacientes aleatorizadas; OS Supervivencia global; PFS Supervivencia libre de progresión; IC Intervalo de confianza; TSST Tiempo desde la aleatorización hasta el principio de la segunda terapia siguiente o fallecimiento.
Figura 1 Estudio 19: Curva de Kaplan-Meier de la PFS en pacientes BRCA mutadas (madurez
del 53%-evaluación del investigador)
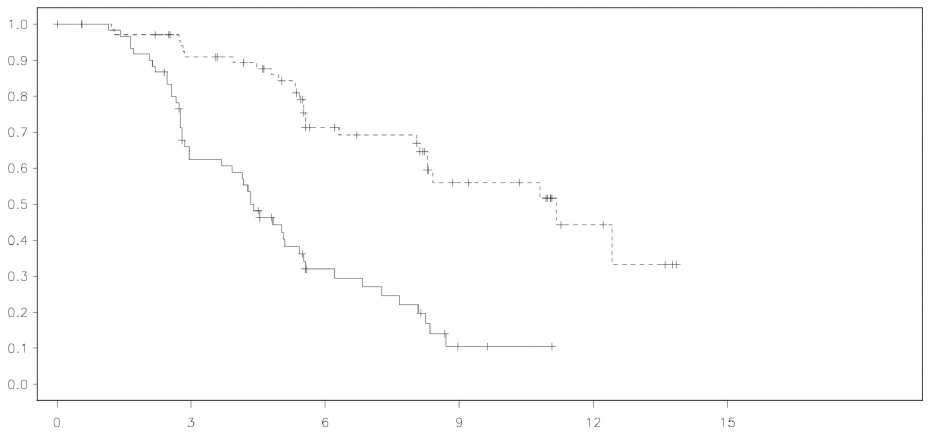
|
meses |
0 |
3 |
6 |
9 |
12 |
15 |
|
n-olaparib |
74 |
59 |
34 |
15 |
5 |
0 |
|
n-placebo |
62 |
35 |
13 |
2 |
0 |
0 |
-----olaparib 400 mg, dos veces al día,_placebo, eje x=tiempo desde la aleatorización en meses, eje
y=PFS (supervivencia libre de progresión), n-olaparib= número de pacientes en riesgo-olaparib, n-placebo=número de pacientes en riesgo-placebo
Figura 2 Estudio 19: Curva de Kaplan-Meier de la OS en pacientes BRCA mutadas (madurez del
52%)
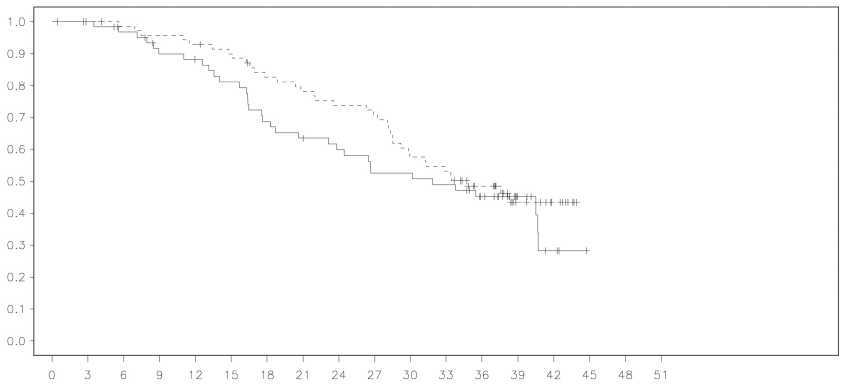
|
meses |
0 |
3 |
6 |
9 |
12 |
15 |
18 |
21 |
24 |
27 |
30 |
33 |
36 |
39 |
42 |
45 |
48 |
51 |
|
n-olaparib |
74 |
71 |
69 |
67 |
65 |
62 |
56 |
53 |
50 |
48 |
39 |
36 |
26 |
12 |
7 |
0 |
0 |
0 |
|
n-placebo |
62 |
62 |
58 |
52 |
50 |
46 |
39 |
36 |
33 |
29 |
29 |
27 |
21 |
10 |
4 |
0 |
0 |
0 |
-----olaparib 400 mg, dos veces al día_placebo, eje x=tiempo desde la aleatorización en meses, eje
y=OS (supervivencia global), n-olaparib= número de pacientes en riesgo-olaparib, n-placebo=número de pacientes en riesgo-placebo
En el Estudio 19, se identificaron 18 pacientes con una mutación somática tumoral BRCA (una mutación en el tumor, pero no en la línea germinal). Los datos limitados para estas pacientes con mutación somática tumoral BRCA (sBRCA) muestran que menos pacientes tratadas con olaparib notificaron eventos de progresión o fallecimiento en comparación con placebo (Tabla 3).
Tabla 3 Resumen de la supervivencia libre de progresión y supervivencia global: población sBRCA mutada en el Estudio 19
|
N eventos/pacientes (%) | |
|
PFS | |
|
Olaparib 400 mg dos veces al día |
3/8 (38%) |
|
Placebo |
6/10 (60%) |
|
OS | |
|
Olaparib 400 mg dos veces al día |
4/8 (50%) |
|
Placebo |
6/10 (60%) |
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Lynparza, en los diferentes grupos de la población pediátrica en carcinoma de ovario (excluyendo rabdomiosarcoma y los tumores de células germinales) (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
La farmacocinética de olaparib, a la dosis de 400 mg dos veces al día en cápsulas, se caracteriza por un aclaramiento plasmático aparente de ~8, 6 L/h, un volumen de distribución aparente de ~167 L y una semivida terminal de 11,9 horas.
Absorción
Tras la administración oral de olaparib mediante la formulación en cápsula, la absorción es rápida alcanzándose las concentraciones plasmáticas máximas normalmente entre 1 a 3 horas después de la dosis. Si se administran varias dosis no hay acumulación marcada, alcanzándose exposiciones de estado estacionario dentro de los ~3 a 4 días.
La administración concomitante con alimentos ralentizó la tasa (tm4x retrasada 2 horas) y aumentó marginalmente la extensión de la absorción de olaparib (AUC aumentó aproximadamente un 20%). Por tanto, se recomienda que las pacientes tomen Lynparza al menos una hora después de las comidas, y se abstengan de comer preferiblemente hasta dos horas más tarde (ver sección 4.2).
Distribución
La unión a proteínas in vitro de olaparib a concentraciones plasmáticas alcanzadas tras la dosificación de 400 mg dos veces al día, es de ~82%.
Olaparib se une moderadamente a ASH (Albúmina Sérica Humana) de forma no saturable (55% aproximadamente) y débilmente (35% aproximadamente) a AAG (Ácido Alfa-1 Glicoproteina).
Biotransformación
In vitro, CYP3A4 ha mostrado ser el principal enzima responsable del metabolismo de olaparib (ver sección 4.5).
Tras la administración oral de olaparib-14C a las pacientes, olaparib sin alterar fue responsable de la mayor parte de la radioactividad circulante en plasma (70%) y fue el componente principal encontrado en orina y
heces (15% y 6% de la dosis respectivamente). El metabolismo de olaparib es extenso. La mayoría del metabolismo fue atribuible a reacciones de oxidación con una serie de componentes producidos bajo posterior conjugación de glucurónico o sulfato. Se detectaron hasta 20, 37 y 20 metabolitos en plasma, orina y heces respectivamente, la mayoría de los cuales representa < 1% del compuesto dosificado. Una fracción de hidroxiciclopropilo por apertura de anillo y dos metabolitos mono-oxigenados (cada uno ~10%) fueron los principales componentes circulantes, siendo uno de los metabolitos mono-oxigenados también el principal metabolito en las excreciones (6% y 5% de la radioactividad urinaria y fecal respectivamente).
In vitro, olaparib produjo poca/nula inhibición de los CYPs 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6 ó 2E1 y no se espera que sea un inhibidor clínicamente significativo dependiente del tiempo de ninguno de estos enzimas P450. Los datos in vitro han demostrado, también, que olaparib no es un sustrato para OATP1B1, OATP1B3, OCT1, BCRP o MRP2, ni un inhibidor de OATP1B3, OAT1 o MRP2.
Eliminación
Tras una dosis única de olaparib-14C, se recuperó el ~86% de la radioactividad administrada dentro de un periodo de recogida de 7 días, ~44% a través de la orina y ~42% a través de las heces. La mayoría del compuesto se excretó como metabolitos.
Poblaciones especiales Insuficiencia renal
En pacientes con insuficiencia renal leve (aclaramiento de creatinina de 51 a 80 ml/min), el AUC incrementó un 24% y la Cmax un 15% comparado con las pacientes con función renal normal. No se requiere ajuste de dosis en pacientes con insuficiencia renal leve.
En pacientes con insuficiencia renal moderada (aclaramiento de creatinina de 31 a 50 ml/min), el AUC incrementó un 44% y la Cmax un 26% comparado con las pacientes con función renal normal. Se recomienda ajustar la dosis de Lynparza en pacientes con insuficiencia renal moderada (ver sección 4.2). No existen datos en pacientes con insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min).
Insuficiencia hepática
No se ha estudiado el efecto de la insuficiencia hepática sobre la exposición a olaparib. No se recomienda el uso de olaparib en pacientes con insuficiencia hepática (bilirrubina sérica >1,5 veces el límite superior normal).
Pacientes de edad avanzada
Se dispone de datos clínicos limitados en pacientes de 75 años y mayores. Un análisis de población de los datos disponibles no ha encontrado ninguna relación entre las concentraciones plasmáticas de olaparib y la edad de la paciente.
Peso
No existen datos en pacientes obesas (IMC > 30 kg/m2) o en pacientes con peso inferior al normal (IMC <18 kg/m2). Un análisis de población de los datos disponibles no ha encontrado evidencias de que el peso de la paciente afecte a las concentraciones plasmáticas de olaparib.
Raza
No existen datos suficientes para evaluar el posible efecto de la raza en la farmacocinética de olaparib, ya que la experiencia clínica es principalmente en pacientes caucásicas (el 94% de las pacientes incluidas en el análisis de población eran caucásicas). En los datos limitados disponibles, no hubo evidencia de una diferencia étnica marcada en la farmacocinética de olaparib entre pacientes japonesas y caucásicas.
Población _ pediátrica
No se han realizado estudios para investigar la farmacocinética de olaparib en pacientes pediátricos.
5.3 Datos preclínicos sobre seguridad
Genotoxicidad
Olaparib no mostró potencial mutagénico, pero sí clastogénico en células de mamíferos in vitro. Cuando se administró oralmente a ratas, olaparib indujo micronúcleos en la médula ósea. Esta clastogenicidad es coherente con la farmacología conocida de olaparib, e indica la posibilidad de genotoxicidad en humanos.
Toxicidad a dosis repetidas
En los estudios de toxicidad a dosis repetidas de hasta 6 meses de duración en ratas y perros, las dosis orales diarias de olaparib fueron bien toleradas. El órgano diana principal para la toxicidad en ambas especies fue la médula ósea, con cambios asociados en los parámetros hematológicos periféricos. Estos hallazgos tuvieron lugar a exposiciones por debajo de las observadas clínicamente y fueron mayormente reversibles dentro de las 4 semanas de la interrupción de la administración. Estudios con células de médula ósea humana también mostraron que la exposición directa a olaparib puede producir toxicidad en células de la médula ósea en ensayos ex vivo.
Toxicología reproductiva
En un estudio de fertilidad en hembras en el que las ratas fueron tratadas hasta la implantación, aunque se observó celo durante un mayor periodo en algunos animales, el apareamiento y la tasa de embarazo no se vieron afectados. No obstante, hubo una ligera reducción en la supervivencia embriofetal.
En estudios de desarrollo embriofetal en rata, y a niveles de dosis que no indujeron toxicidad maternal significativa, olaparib causó una reducción de esta supervivencia embriofetal, menor peso fetal y anormalidades en el desarrollo fetal, incluyendo importantes malformaciones oculares (por ej.,anoftalmia, microftalmia), malformación de vertebras/costillas, y anormalidades viscerales y esqueléticas.
Carcinogenicidad
No se han realizado estudios de carcinogenicidad con olaparib.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Contenido de la cápsula Lauril macrogol-32 glicéridos
Cubierta de la cápsula
Hipromelosa
Dióxido de titanio (E171)
Goma gellan (E418)
Acetato de potasio
Tinta de impresión Shellac
Óxido de hierro negro (E172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30°C.
6.5 Naturaleza y contenido del envase
Frasco de plástico HDPE con cierre a prueba de niños, conteniendo 112 cápsulas duras.
Envase de 448 cápsulas (4 frascos de 112 cápsulas).
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
AstraZeneca AB S-151 85 Sódertalje Suecia
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/959/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 16 de diciembre de 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
ANEXO II
A. FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del (de los) fabricante(s) responsable^) de la liberación de los lotes AstraZeneca UK Limited
SILK ROAD BUSINESS PARK, MACCLESFIELD, CHESHIRE, SK10 2NA, Reino Unido
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2)
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y cualquier actualización posterior publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Obligación de llevar a cabo medidas post-autorización
El TAC deberá completar, dentro del plazo establecido, las siguientes medidas:
|
Descripción |
Fecha de vencimiento |
|
PAES: Con el fin de definir de forma adicional la eficacia a largo plazo de olaparib en pacientes con cáncer de ovario seroso de alto grado, con mutación BRCA, sensible a platino, en recaída, el TAC debe presentar el análisis final de supervivencia global (SG) del estudio D0810C00019, fase II, aleatorizado, doble ciego, multicéntrico. El informe del ensayo clínico debe ser presentado en: |
Junio 2017 |
|
PAES: Con el fin de confirmar la eficacia de olaparib en pacientes con cáncer de ovario |
|
seroso de alto grado, con mutación BRCA, sensible a platino, en recaída, el TAC debe presentar los resultados del estudio D0816C00002, fase II, aleatorizado, doble ciego, controlado con placebo, multicéntrico. | |
|
El informe del ensayo clínico debe ser presentado en: |
Septiembre 2019 |
|
PAES: Con el fin de definir de forma adicional la eficacia de olaparib en pacientes con cáncer de ovario seroso de alto grado, con mutación BRCA somática, sensible a platino, en recaída, el TAC debe realizar y presentar los resultados del estudio de fase IV, abierto, brazo único, no randomizado, multicéntrico en pacientes con cáncer de ovario sensible a platino, en recaída, que están en respuesta completa o parcial después de quimioterapia basada en platino y que conlleva la pérdida de la función de la(s) mutación(es) BRCA germinal(es) o somática(s). |
Septiembre |
|
El informe del ensayo clínico debe ser presentado en: |
2018 |
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CARTÓN
1. NOMBRE DEL MEDICAMENTO
Lynparza 50 mg cápsulas duras olaparib
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura contiene 50 mg de olaparib.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsula dura.
448 cápsulas (4 frascos de 112 cápsulas).
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 30°C.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
AstraZeneca AB SE-151 85 Sódertalje Suecia
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/959/001
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
MEDICAMENTO SUJETO A PRESCRIPCIÓN MÉDICA.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
lynparza 50 mg
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC:
SN:
NN:
INFORMACIÓN MÍNIMA A INCLUIR EN EL ACONDICIONAMIENTO PRIMARIO FRASCO/ETIQUETA
1. NOMBRE DEL MEDICAMENTO
Lynparza 50 mg cápsulas duras olaparib
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura contiene 50 mg de olaparib.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsula dura. 112 cápsulas.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
Cad.:
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 30°C.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
AstraZeneca AB SE-151 85 Sódertalje Suecia
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/959/001
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
No procede._
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
No procede.
B. PROSPECTO
Prospecto: información para el paciente
Lynparza 50 mg cápsulas duras
Olaparib
'VEste medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Lynparza y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Lynparza
3. Cómo tomar Lynparza
4. Posibles efectos adversos
5. Conservación de Lynparza
6. Contenido del envase e información adicional
1. Qué es Lynparza y para qué se utiliza Qué es Lynparza y cómo actúa
Las cápsulas duras de Lynparza contienen el principio activo olaparib. Olaparib es un tipo de medicamento para el cáncer, denominado inhibidor del PARP (poli [adenosina difosfato-ribosa] polimerasa).
En pacientes con mutaciones (cambios) en ciertos genes llamados BRCA (gen del cáncer de mama), las cuales presentan riesgo de desarrollar algunos tipos de cáncer, los inhibidores del PARP son capaces de desencadenar la muerte de células cancerosas bloqueando un enzima que ayuda a reparar el ADN.
Para qué se utiliza Lynparza
Lynparza se utiliza para el tratamiento de un tipo de cáncer de ovario denominado “cáncer de ovario BRCA mutado”. Se emplea después de que el cáncer haya respondido al tratamiento previo con quimioterapia estándar basada en platino. Se realiza una prueba para determinar si usted padece un cáncer BRCA mutado.
2. Qué necesita saber antes de empezar a tomar Lynparza No tome Lynparza
• si es alérgica a olaparib o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
No tome Lynparza si algo de lo anterior es aplicable a usted. Si no está segura, consulte a su médico, farmacéutico o enfermero antes de tomar Lynparza.
Advertencias y precauciones
Consulte a su médico, farmacéutico o enfermero antes de empezar a tomar o durante el tratamiento con Lynparza:
• Si usted tiene un recuento sanguíneo bajo en los análisis. Éste puede ser un recuento bajo de glóbulos rojos (anemia), de glóbulos blancos (neutropenia) o de plaquetas (trombocitopenia). Ver sección 4 para más información sobre estos efectos adversos. Ésta incluye los signos y síntomas a los que usted necesita prestar atención (fiebre o infección, hematomas o sangrado). Raramente, estos pueden ser un signo de un problema más grave de la médula ósea tal como “Síndrome Mielodisplásico” (SMD) o “Leucemia Mieloide Aguda” (LMA). Su médico puede querer analizar su médula ósea para comprobar estos problemas.
• Si usted nota cualquier nuevo síntoma o empeoramiento en la dificultad para respirar, tos o sibilancia (sonido silbante que se produce al respirar). Un pequeño número de pacientes tratadas con Lynparza comunicaron inflamación de los pulmones (neumonitis). La neumonitis es una enfermedad grave que a menudo puede requerir tratamiento en el hospital.
Si alguno de los casos anteriores es aplicable a usted (o no está segura), consulte a su médico, farmacéutico o enfermero.
Pruebas y controles
Su médico le hará análisis de sangre antes y durante el tratamiento con Lynparza.
Usted tendrá un análisis de sangre:
• antes de empezar el tratamiento
• cada mes, durante el primer año de tratamiento
• a intervalos regulares, decididos por su médico, tras el primer año de tratamiento.
Si su recuento sanguíneo desciende a un nivel bajo, puede ser necesario realizar una transfusión sanguínea (en la que le administrarán sangre nueva o hemoderivados de un donante).
Toma de Lynparza con otros medicamentos
Informe a su médico, farmacéutico o enfermero si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento, incluso los adquiridos sin receta y medicamentos a base de plantas. Esto es porque Lynparza puede afectar a la forma de actuar de algunos medicamentos y algunos medicamentos pueden tener efecto sobre Lynparza.
No tome Lynparza si está tomando cualquier otro medicamento contra el cáncer. Informe a su médico, farmacéutico o enfermero si tiene previsto recibir una vacuna o un medicamento que inhiba el sistema inmunitario, ya que puede necesitar ser monitorizada estrechamente.
Informe a su médico o farmacéutico si está tomando cualquiera de los siguientes medicamentos:
• itraconazol, fluconazol - utilizado para las infecciones por hongos
• telitromicina, claritromicina, eritromicina - utilizados para las infecciones bacterianas
• inhibidores de la proteasa potenciados con ritonavir o cobicistat, boceprevir, telaprevir, neviparina, efavirenz - utilizados para las infecciones víricas, incluyendo VIH
• rifampicina, rifapentina, rifabutina - utilizados para las infecciones bacterianas, incluyendo tuberculosis (TB)
• fenitoína, carbamazepina, fenobarbital - utilizados como sedantes o para tratar crisis (convulsiones) y epilepsia
• hierba de San Juan (Hypericum perforatum) - un medicamento a base de plantas utilizado principalmente para la depresión
• digoxina, diltiazem, furosemida, verapamilo, valsartan - utilizados para tratar enfermedades del corazón o hipertensión arterial.
• bosentan - utilizado para tratar la hipertensión arterial pulmonar.
• estatinas, por ejemplo simvastatina, pravastatina - utilizadas para disminuir los niveles de colesterol en la sangre
• dabigatran - utilizado para diluir la sangre
• glibenclamida, metformina, repaglinida - utilizados para tratar la diabetes
• alcaloides ergotamínicos - utilizados para tratar migrañas y dolores de cabeza.
• fentanilo - utilizado para tratar el dolor producido por el cáncer
• pimozida - utilizada para tratar la esquizofrenia
• quetiapina - utilizada para tratar la esquizofrenia y trastorno bipolar
• cisaprida - utilizada para tratar problemas de estómago
• colchicina - utlilizada para tratar la gota
• ciclosporina, sirolimus, tacrolimus - utilizados para suprimir el sistema inmune
• metotrexato - utilizado para tratar el cáncer, la artítris reumatoide y la psoriasis
Toma de Lynparza con bebidas
No beba zumo de pomelo durante el periodo de tiempo que está tomando Lynparza. Esto puede afectar a
la forma en que funciona el medicamento.
Embarazo y lactancia
• No debe tomar Lynparza si está embarazada o cree que pudiera estarlo, pues podría dañar al feto.
• Debe evitar quedarse embarazada mientras toma este medicamento. Usted debe emplear métodos anticonceptivos eficaces mientras toma este medicamento y durante 1 mes después de recibir la última dosis de Lynparza. Se desconoce si Lynparza puede afectar a la eficacia de algunos anticonceptivos orales. Informe a su médico si está tomando un anticonceptivo oral, ya que su médico podría recomendarle también la adición de un método anticonceptivo no hormonal.
• Se debe realizar una prueba de embarazo antes de empezar a tomar Lynparza y a intervalos regulares durante el tratamiento y 1 mes después de recibir la última dosis de Lynparza. Si se queda embarazada durante este periodo, consulte inmediatamente a su médico.
• Se desconoce si Lynparza pasa a la leche materna. No debe dar el pecho si está tomando Lynparza, ni durante un mes después de recibir la última dosis de este medicamento. Si tiene previsto dar el pecho, informe a su médico.
Conducción y uso de máquinas
Lynparza puede afectar a su capacidad para conducir y utilizar máquinas. Si siente mareo, debilidad o cansancio mientras toma Lynparza, no conduzca ni utilice herramientas o máquinas.
3. Cómo tomar Lynparza
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico, farmacéutico o enfermero. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero.
Qué cantidad debe tomar
• La dosis recomendada es de 8 cápsulas (400 mg) tomadas por boca, dos veces al día (un total de 16 cápsulas cada día). Es importante que tome la dosis diaria total recomendada y siga haciéndolo según las instrucciones de su médico, farmacéutico o enfermero. Su médico le puede prescribir una dosis diferente si usted tiene problemas renales.
Cómo tomar
• Tome una dosis (8 cápsulas) de Lynparza por boca con agua, una vez por la mañana y una vez por la noche.
• Tome Lynparza al menos una hora después de tomar los alimentos. No coma preferiblemente hasta dos horas después de tomar Lynparza.
Si experimenta efectos adversos, su médico puede decirle que tome Lynparza a una dosis menor.
Si toma más Lynparza del que debe
Si toma más Lynparza de su dosis habitual, consulte con un médico o vaya a un hospital inmediatamente. Si olvidó tomar Lynparza
Si olvida tomar Lynparza, debe tomar la siguiente dosis a la hora habitual. No tome una dosis doble para compensar las dosis olvidadas.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Es importante que sea consciente de cuáles pueden ser estos efectos adversos.
Su médico también puede recetarle otros medicamentos que ayuden a controlar los efectos adversos.
Informe inmediatamente a su médico si nota alguno de los siguientes efectos adversos - puede necesitar tratamiento médico urgente:
Muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
• fiebre o infección - estos pueden ser signos de un recuento bajo de glóbulos blancos (neutropenia o linfopenia).
• dificultad para respirar, sensación de mucho cansancio, piel pálida o latido cardiaco acelerado -estos pueden ser signos de un recuento bajo de glóbulos rojos (anemia).
Frecuentes (pueden afectar hasta 1 de cada 10 personas):
• hematomas o sangrado durante un periodo superior al normal si se lesiona - estos pueden ser signos de un recuento bajo de plaquetas (trombocitopenia).
Informe inmediatamente a su médico si nota cualquiera de los efectos adversos anteriores.
Otros efectos adversos incluyen:
Muy frecuentes
• dolor de cabeza
• mareo
• pérdida de apetito
• cansancio o debilidad
• náuseas
• vómitos
• cambios en el gusto de los alimentos
• indigestión o ardor de estómago (dispepsia)
• diarrea. Si se agrava, informe a su médico inmediatamente
• aumento de los niveles de creatinina sanguínea identificado mediante un análisis de laboratorio, mostrando el adecuado funcionamiento de sus riñones
• análisis de sangre que muestran un aumento del tamaño de los glóbulos rojos.
Frecuentes
• dolor en la boca (estomatitis)
• dolor en la zona del estómago bajo las costillas.
Si experimenta cualquier efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. Su médico puede recetarle un medicamento para tratar sus síntomas tales como náuseas, vómitos, diarrea y dispepsia.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Lynparza
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase y el frasco después de CAD. La fecha de caducidad es el último día del mes que se indica.
No conservar a temperatura superior a 30°C.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Lynparza
El principio activo es olaparib. Cada cápsula dura contiene 50 mg de olaparib
Los demás componentes (excipientes) son:
• Contenido de la cápsula: lauril macrogol-32 glicéridos.
• Cubierta de la cápsula: hipromelosa, dióxido de titanio (E171), goma gellan (E418), acetato de potasio.
• Tinta de impresión: shellac, óxido de hierro negro (E172).
Aspecto del producto y contenido del envase
Lynparza es una cápsula dura, blanca, opaca, marcada con “OLAPARIB 50 mg” y el logotipo de AstraZeneca en tinta negra.
Lynparza se presenta en frascos de plástico HDPE que contienen 112 cápsulas duras. Un envase contiene 448 cápsulas (4 frascos de 112 cápsulas).
Titular de la autorización de comercialización
AstraZeneca AB SE-151 85 Sodertalje Suecia
Responsable de la fabricación
AstraZeneca UK Limited Silk Road Business Park Macclesfield, Cheshire, SK10 2NA Reino Unido
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien AstraZeneca S.A./N.V. Tel: +32 2 370 48 11 |
Lietuva UAB AstraZeneca Lietuva Tel: +370 5 2660550 |
|
Etarapna Acrpa3eHeKa Etarapua EOOfl Tea.: +359 24455000 |
Luxembourg/Luxemburg AstraZeneca S.A./N.V. Tél/Tel: +32 2 370 48 11 |
|
Ceská republika AstraZeneca Czech Republic s.r.o. Tel: +420 222 807 111 |
Magyarország AstraZeneca Kft. Tel.: +36 1 883 6500 |
|
Danmark AstraZeneca A/S Tlf: +45 43 66 64 62 |
Malta Associated Drug Co. Ltd Tel: +356 2277 8000 |
|
Deutschland AstraZeneca GmbH Tel: +49 41 03 7080 |
Nederland AstraZeneca BV Tel:+31 79 363 2222 |
|
Eesti AstraZeneca Tel: +372 6549 600 |
Norge AstraZeneca AS Tlf: +47 21 00 64 00 |
|
EXXáSa AstraZeneca A.E. T^: +30 210 6871500 |
Osterreich AstraZeneca Osterreich GmbH Tel: +43 1 711 31 0 |
|
España AstraZeneca Farmacéutica Spain, S.A. Tel: +34 91 301 91 00 |
Polska AstraZeneca Pharma Poland Sp. z o.o. Tel.: +48 22 245 73 00 |
|
France AstraZeneca Tél: +33 1 41 29 40 00 |
Portugal AstraZeneca Produtos Farmacéuticos, Lda. Tel: +351 21 434 61 00 |
|
Hrvatska AstraZeneca d.o.o. Tel: +385 1 4628 000 |
Romania AstraZeneca Pharma SRL Tel: +40 21 317 60 41 |
|
Ireland AstraZeneca Pharmaceuticals (Ireland) Ltd Tel: +353 1609 7100 |
Slovenija AstraZeneca UK Limited Tel: +386 1 51 35 600 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika AstraZeneca AB, o.z. Tel: +421 2 5737 7777 |
|
Italia AstraZeneca S.p.A. Tel: +39 02 9801 1 |
Suomi/Finland AstraZeneca Oy Puh/Tel: +358 10 23 010 |
|
Kúnpoq A^skt©p Oap^aKeuxiKq AtS T^: +357 22490305 |
Sverige AstraZeneca AB Tel: +46 8 553 26 000 |
|
Latvija SIA AstraZeneca Latvija Tel: +371 67377100 |
United Kingdom AstraZeneca UK Ltd Tel: +44 1582 836 836 |
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
33