Lutrate Depot Trimestral 22,5 Mg Polvo Y Disolvente Para Suspension De Liberacion Prolongada Inyectable
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Lutrate Depot Trimestral 22,5 mg polvo y disolvente para suspensión de liberación prolongada inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene 22,5 mg de acetato de leuprorelina (equivalente a 21,42 mg de base libre de leuprorelina.
1ml de suspensión reconstituida contiene 11,25 mg de acetato de leuprorelina.
Excipiente(s) con efecto conocido
Cada vial contiene de 1,6 a 2,7 mg (<1mmol) de sodio (como carmelosa de sodio).
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para suspensión de liberación prolongada inyectable.
Polvo: polvo blanco o casi blanco.
Disolvente: solución transparente e incolora (pH 5.0 - 7.0).
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Lutrate Depot Trimestral está indicado para el tratamiento paliativo del cáncer de próstata avanzado hormonodependiente
4.2 Posología y forma de administración
Posología
La dosis habitual recomendada de Lutrate Depot Trimestral es de 22,5 mg en forma de una inyección depot trimestral y administrada como una única inyección intramuscular cada tres meses.
Lutrate Depot Trimestral debe administrarse bajo la supervisión de un profesional sanitario que cuente con la experiencia adecuada para monitorizar la respuesta al tratamiento.
La dosis de Lutrate Depot Trimestral que permite la liberación continua de acetato de leuprorelina durante tres meses está incorporada a una formulación de liberación prolongada. El polvo liofilizado debe reconstituirse y administrarse en forma de inyección intramuscular cada tres meses. No debe administrarse por vía intravenosa o intraarterial. El vial de polvo de microsferas de Lutrate Depot Trimestral debe reconstituirse inmediatamente antes de su administración mediante inyección intramuscular. Como otros medicamentos que se administran de forma regular mediante inyección, el lugar de la inyección debe variar periódicamente.
El tratamiento con Lutrate Depot Trimestral no debe interrumpirse cuando se experimente mejora o remisión.
La respuesta al tratamiento con Lutrate Depot Trimestral debe supervisarse analizando periódicamente los niveles séricos de testosterona y del antígeno prostático específico (PSA).Los estudios clínicos han demostrado que los niveles de testosterona aumentaron durante los primeros 4 días de tratamiento en la mayoría de pacientes no orquiectomizados. A continuación descendieron y alcanzaron niveles de castración en 3-4 semanas. Una vez alcanzados, los niveles de castración (definido como un nivel de testosterona inferior a 0,5 ng/ml) se mantuvieron durante todo el tratamiento con el fármaco.
Si la respuesta de un paciente no es óptima, es aconsejable confirmar que los niveles séricos de testosterona han alcanzado o se mantienen a niveles de castración. En ocasiones pueden producirse elevaciones pasajeras del nivel de fosfatasa ácida al inicio del período de tratamiento, si bien generalmente este nivel retorna a los valores normales o casi normales en la cuarta semana de tratamiento.
Duración del tratamiento
Lutrate Depot Trimestral se administra en forma de inyecciones intramusculares trimestrales.
Como norma, el tratamiento del cáncer de próstata avanzado con Lutrate Depot Trimestral implica un tratamiento prolongado por lo que no debe interrumpirse cuando se produce remisión o mejoría.
Poblaciones especiales
Población pediátrica
No se ha establecido la seguridad y eficacia de Lutrate Depot Trimestral en pacientes pediátricos. Por tanto, el uso de Lutrate Depot Trimestral no está recomendado en niños o adolescentes hasta que se disponga de datos de eficacia y seguridad.
Insuficiencia renal/hepática
No se ha determinado la farmacocinética de Lutrate Depot Trimestral en pacientes con insuficiencia hepática o renal.
Ancianos
En el ensayo clinico de Lutrate Depot Trimestral 22,5 mg, la media de edad de los pacientes estudiados fue de 71,0±9,02 años. Por tanto, la ficha técnica refleja la farmacocinética, eficacia y seguridad de Lutrate Depot Trimestral en esta población.
Forma de administración
Lutrate Depot Trimestral debe administrarse únicamente por vía intramuscular. No administrar el producto por ninguna otra vía. Si por error se administrara el producto por vía subcutánea, el paciente debería ser estrechamente monitorizado puesto que no hay datos disponibles sobre la administración de Lutrate Depot Trimestral por otras vías aparte de la vía intramuscular.
Las instrucciones para la reconstitución del medicamento antes de su administración se describen en la sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo, los análogos de la hormona liberadora de gonadotropina (LHRH) o a alguno de los excipientes incluidos en la sección 6.1. Se han descrito reacciones anafilácticas a la LHRH sintética o a análogos agonistas de la LHRH en la literatura médica.
Orquiectomía previa.
Lutrate Depot Trimestral no debe utilizarse como tratamiento único en pacientes con cáncer de próstata e indicios de compresión espinal o metástasis medular.
Lutrate Depot Trimestral no está indicado para uso en mujeres.
Lutrate Depot Trimestral no está indicado para uso en pacientes pediátricos
4.4 Advertencias y precauciones especiales de empleo
Al inicio del tratamiento con Lutrate Depot Trimestral, como sucede durante el tratamiento con otros agonistas de la LHRH, puede producirse un aumento pasajero de los niveles de testosterona. En algunos casos, este aumento puede estar asociado a un empeoramiento o exacerbación del crecimiento del tumor, que provoca un agravamiento temporal de los síntomas del cáncer de próstata. Estos síntomas generalmente disminuyen al continuar con el tratamiento (ver sección 4.8). En algunos casos la exacerbación puede manifestarse en forma de síntomas sistémicos o neurológicos (por ejemplo, dolor óseo). También se han descrito casos de atrofia testicular y ginecomastia asociados al tratamiento con otros agonistas de la LHRH.
El tratamiento deberá ser suspendido inmediatamente si el paciente desarrolla cualquier signo o síntoma de anafilaxis/reacción anafiláctica (disnea, asma, rinitis, edema angioneurótico o de glotis, hipotensión, urticaria, erupción, prurito o neumonitis intersticial). Los pacientes deben ser informados antes de empezar el tratamiento, advirtiéndoles de interrumpir el tratamiento y consultar con su médico si alguno de los síntomas mencionados aparece. Los pacientes que hayan experimentado alguna reacción de
¿¡*■5
hipersensibilidad a leuprolide deberán ser estrechamente monitorizados y no se les debe volver a administrar Lutrate Depot Trimestral.
En pacientes tratados con acetato de leuprorelina, se han observado casos aislados de obstrucción uretral (con o sin hematuria) y compresión medular o lesiones vertebrales metastásicas, que puedan contribuir a la aparición de parálisis con o sin complicaciones mortales. Los pacientes con riesgo de obstrucción uretral, compresión de la médula espinal o lesiones vertebrales metastásicas deberán ser tratados cuidadosamente y se supervisarán de forma estrecha durante las primeras semanas de tratamiento. Para estos pacientes se deberá considerar la posibilidad de administrar tratamiento profiláctico con antiandrógenos.
Si se producen complicaciones urológicas/neurológicas, éstas deberán tratarse con medidas específicas apropiadas.
Existe riesgo alto de incidencia de depresión (que puede ser severa) en pacientes que reciban tratamiento con agonistas de la GnRH como el acetato de leuprorelina. Los pacientes deben ser informados al respecto y ser tratado adecuadamente si los síntomas aparecen.
En la literatura médica se ha descrito reducción de la densidad ósea en varones que se habían sometido a orquiectomía o que habían recibido tratamiento con un agonista de la LHRH. La adición de tratamiento andiandrogénico a la pauta de tratamiento reduce la pérdida ósea, pero aumenta el riesgo de reacciones adversas tales como problemas de coagulación y edema. Si se emplea un antiandrógeno durante un período prolongado, deberá prestarse la atención debida a las contraindicaciones y precauciones asociadas a su uso prolongado. Los pacientes con riesgo de padecer osteoporosis o con antecedentes clínicos de este trastorno deberán ser tratados cuidadosamente, y serán estrechamente supervisados durante el tratamiento con acetato de leuprorelina (ver sección 4.8).
Se ha descrito disfunción hepática e ictericia con elevación de las enzimas hepáticas con el uso de acetato de leuprorelina. Por lo tanto, se realizará una observación minuciosa y se adoptarán las medidas adecuadas que sean necesarias.
La respuesta al tratamiento con Lutrate Depot Trimestral deberá controlarse mediante parámetros clínicos y analizando periódicamente los niveles séricos de testosterona y PSA.
Los pacientes pueden experimentar cambios metabólicos (por ejemplo, intolerancia a la glucosa o empeoramiento de la diabetes existente), hipertensión, alteraciones de peso y trastornos cardiovasculares. Tal como cabe esperar en este tipo de medicamento, puede aparecer diabetes o empeoramiento de la diabetes existente. Por tanto, los pacientes diabéticos pueden necesitar de una monitorización más
frecuente de los niveles sanguíneos de glucosa durante el tratamiento con Lutrate Depot Trimestral. Los pacientes con elevado riesgo de enfermedad metabólica o cardiovascular deberán ser evaluados cuidadosamente antes de iniciar el tratamiento, y se les someterá a un control adecuado durante el tratamiento de privación de andrógenos. El tratamiento con acetato de leuprorelina causa la supresión del sistema hipófiso-gonadal. Los resultados de las pruebas diagnósticas de las funciones gonadal e hipofisiaria gonadotrópica realizadas durante y después del tratamiento con acetato de leuprorelina pueden verse afectados.
Se ha descrito un aumento del tiempo de protrombina en pacientes en tratamiento con acetato de leuprorelina. El acetato de leuprorelina debe usarse con precaución en pacientes con trastornos conocidos de coagulación, trombocitopenia o en tratamiento con anticoagulantes.
Se han descrito convulsiones con la administración de acetato de leuprorelina. Estos casos se han observado en pacientes con antecedentes de convulsiones, epilepsia, trastornos cardiovasculares, anomalías o tumores del sistema nervioso central, y en pacientes en tratamiento con medicamentos concomitantes que se han asociado a convulsiones, tales como bupropion o inhibidores selectivos de la recaptación de serotonina (ISRS). También se han descrito convulsiones en ausencia de los trastornos mencionados arriba.
El acetato de leuprorelina debe utilizarse con precaución en presencia de enfermedad cardiovascular (incluida insuficiencia cardiaca congestiva), tromboembolia, edema, depresión y apoplejía hipofisaria.
Este medicamento contiene menos de 1mmol de sodio (23mg) por vial. Básicamente está libre de sodio.
El tratamiento de deprivación androgénica puede prolongar el intervalo QT.
En pacientes con antecedentes o con factores de riesgo de la prolongación del intervalo QT y en pacientes que reciben medicamentos concomitantes que podrían prolongar el intervalo QT (ver sección 4.5), los profesionales sanitarios deben evaluar el balance beneficio/riesgo incluyendo el riesgo potencial de Torsade de Pointes antes de iniciar el tratamiento con Lutrate Depot Trimestral.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacción farmacológica basados en la farmacocinética con acetato de leuprorelina. No obstante, dado que se trata de un péptido que es degradado principalmente por peptidasas y no por enzimas del citocromo P-450 como han demostrado estudios específicos, y dado que el fármaco solo se une a proteínas plasmáticas en un 46%, no cabe esperar interacciones farmacocinéticas.
Se debe valorar cuidadosamente el uso concomitante de Lutrate Depot Trimestral con medicamentos que prolongan el intervalo QT o medicamentos capaces de inducir Torsade des Pointes, tales como
antiarrítmicos clase IA (por ejemplo: quinidina, disopiramida) o clase III (por ejemplo: amiodarona, sotalol, dofetilida, ibutilida), metadona, moxifloxacino, antipsicóticos, etc (ver sección 4.4) ya que el tratamiento de deprivación androgénica también puede prolongar el intervalo QT.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Lutrate Depot Trimestral no está indicado para uso en mujeres embarazadas.
La inyección de acetato de leuprorelina puede causar daños fetales cuando se administra a mujeres embarazadas.
Por tanto, existe la posibilidad de aborto espontáneo si el fármaco se administra durante el embarazo. Lactancia
Lutrate Depot Trimestral no debe usarse en mujeres lactantes Fertilidad
Estudios en animales han demostrado toxicidad reproductiva (ver sección 5.3)
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios específicos de los efectos de Lutrate Depot Trimestral en relación a la capacidad para conducir y utilizar máquinas. No obstante, La capacidad para conducir y utilizar máquinas puede verse alterada a causa de alteraciones visuales y mareos.
4.8 Reacciones adversas
El perfil de seguridad de Lutrate Depot Trimestral se basa en los resultados de un ensayo clínico de fase III en el que pacientes con cáncer de próstata recibieron tratamiento con dos dosis secuenciales intramusculares de Lutrate Depot Trimestral con un intervalo de 3 meses y fueron sometidos a seguimiento durante un total de 6 meses. La mayoría de los acontecimientos adversos relacionados con el tratamiento descrito fueron los acontecimientos habituales asociados a la acción farmacológica del acetato de leuprorelina y asociados al tratamiento de supresión de testosterona.
Las reacciones adversas notificadas con mayor frecuencia con Lutrate Depot Trimestral son sofocos (acaloramiento), fatiga, astenia, hiperhidrosis, náuseas y dolor óseo.
A continuación se enumeran las reacciones adversas observadas en investigaciones clínicas según la clasificación por órganos y sistemas, y en orden de incidencia decreciente (muy frecuentes: >1/10;
frecuentes: >1/100 a <1/10; poco frecuentes: 1/1.000 a < 1/100; raras: >1/10.000 a <1/1.000; muy raras: < 1/10.000).
Tabla 1. Número y frecuencia de RAF durante el tratamiento con Lutrate Depot Trimestral 22,5mg. Categoría
Clasificación por órganos y sistemas
Frecuencia: Término preferente
Trastornos del metabolismo y de la nutrición
Frecuentes: Apetito disminuido
Poco frecuentes: Hipercolesterolemia Trastornos psiquiátricos
Frecuentes: Insomnio, Libido disminuida. Uso a largo plazo: Cambios del estado de ánimo,
depresión.
Poco frecuentes: Trastorno del sueño, trastorno emocional, ansiedad, reacción de ira. Uso a corto plazo: Cambios del estado de ánimo, depresión.
Trastornos del sistema nervioso
Frecuentes: Mareo
Poco frecuentes: Disgeusia, hormigueo, cefalea, letargia Trastornos oculares
Poco frecuentes: Visión borrosa Trastornos respiratorios, torácicos y mediastínicos
Frecuentes: Pleuresía
No conocida: Neumonitis, enfermedad pulmonar intersticial.
Trastornos del oído y del laberinto Poco frecuentes: Acúfenos
Trastornos vasculares
Muy frecuentes: Acaloramiento
Frecuentes: Rubefacción
Trastornos gastrointestinales
Frecuentes: Náuseas, diarrea
Poco frecuentes: Dolor en la zona superior del abdomen, estreñimiento.
Trastornos de la piel y el tejido subcutáneo
Frecuentes: Hiperhidrosis, prurito, sudor frío
Poco frecuente: Pápula, erupción, prurito generalizado, sudores nocturnos.
Trastornos musculoesqueléticos y del tejido conjuntivo
Frecuentes: Dolor óseo, artralgia
Poco frecuente: Dolor de espalda, dolor musculoesquelético, Cervicalgia .
Trastornos renales y urinarios
Frecuente: Polaquiuria, nicturia, Dolor en tracto urinario, flujo de orina disminuido.
Trastornos del aparato reproductor y de la mama
Frecuentes: Disfunción eréctil
Poco frecuente: Dolor de pezón, dolor pélvico, atrofia testicular, trastorno testicular.
Trastornos generales y alteraciones en el lugar de administración
Frecuentes: Fatiga, astenia, dolor, reacciones adversas locales (ver tabla 2)
Poco frecuente: Sensación de calor, hiperhidrosis
Exploraciones complementarias
Frecuentes:
Alanina aminotransferasa elevada, Aspartato aminotransferasa elevada, Triglicéridos elevados en sangre, Creatinfosfoquinasa en sangre elevada, Glucosa elevada en sangre.
Poco frecuentes: Calcio elevado en sangre, Creatina elevada en sangre, Lactatodeshidrogenasa elevada en sangre, Potasio disminuido en sangre, Potasio elevado en sangre, Urea elevada en sangre, Intervalo QT del electrocardiograma prolongado (ver sección 4.4 y 4.5), Intervalo QT del electrocardiograma acortado, Inversión de la onda T del electrocardiograma, Glutamiltransferasa gamma elevada, Tasa de filtración glomerular disminuida, Hematocrito disminuido, Prueba hematológica anormal, Hemoglobina disminuida, Volumen globular medio aumentado, Recuento disminuido de hematíes, Volumen residual de orina aumentado
En términos de intensidad, el 84,7% de todos los acontecimientos adversos relacionados con el tratamiento fueron leves o moderados. El AA más común fueron sofocos (77,3%), el 57,7% de los cuales fueron descritos como leves y el 17,2% como moderados. Cinco casos de sofocos (3.1%) fueron descritos como severos.
Se describieron un total de 38 reacciones adversas locales (RAL) en la zona de inyección por 24 pacientes (14.7%) durante el estudio.
Las reacciones adversas locales tras la administración de Lutrate Depot Trimestral 22.5 mg son las descritas generalmente con otros productos similares administrados mediante inyección intramuscular. Las reacciones notificadas con mayor frecuencia son dolor en la zona de inyección, eritema en la zona de inyección e induración de la zona de inyección. Las reacciones poco frecuentes fueron molestia en la zona de inyección, urticaria en la zona de inyección, calor en el lugar de inyección, dolor en el lugar de punción vascular, dolor musculoesquelético y hemorragia en la zona de inyección (Tabla 2)
i**4
'ni®:
Tabla 2. Frecuencia de pacientes con reacciones adversas locales durante el tratamiento con Lutrate Depot Trimestral.
|
SOC Principal* |
Pacientes con RAL relacionadas |
|
Termino Preferente: Trastornos generales y alteraciones en el lugar de administración |
% |
|
Muy frecuentes | |
|
Dolor en la zona de inyección |
10.4 |
|
Frecuentes | |
|
Eritema en la zona de inyección |
3.1 |
|
Induración de la zona de inyección |
2.5 |
|
Poco frecuente | |
|
Molestia en la zona de inyección |
0.6 |
|
Urticaria en la zona de inyección |
0.6 |
|
Calor en el lugar de inyección |
0.6 |
|
Hemorragia en la zona de inyección |
0.6 |
|
Artralgia |
0.6 |
|
Dolor musculoesquelético |
0.6 |
|
Dolor en el lugar de punción |
0.6 |
|
vascular | |
*Los pacientes pueden ser incluidos en más de una categoría, RAL: reacción adversa local; COS: clasificación por órganos y sistemas.
Estos acontecimientos fueron todos descritos como no graves y leves o moderados en severidad. Ningún paciente abandonó el tratamiento a causa de acontecimientos adversos locales.
Otros acontecimientos adversos en general con el tratamiento de acetato de leuprorelina Edema periférico, embolia pulmonar, palpitaciones, mialgia, Pérdida de fuerza muscular, escalofrío, vértigo, erupción , amnesia,alteración visual y alteraciones en la sensibilidad de la piel. Raramente se han comunicado casos de infarto por adenomas hipofisarios preexistente tras la administración de agonistas LHRH de larga y corta acción. Se han comunicado casos raros de trombocitopenia y leucopenia. Se han comunicado cambios en la tolerancia a la glucosa.
Cambios en la densidad ósea
En la literatura médica se ha descrito densidad ósea disminuida en varones que habían sido orquiectomizados o que habían sido tratados con análogos LHRH. Puede esperarse que el tratamiento prolongado con leuprorelina revele signos crecientes de osteoporosis. En relación con el mayor riesgo de facturas debidas a osteoporosis (ver la sección 4.4).
Exacerbación de los signos y síntomas de la enfermedad
El tratamiento con acetato de leuprorelina puede dar lugar a una exacerbación de los signos y síntomas de la enfermedad durante las primeras semanas. En el caso de que afecciones como las metástasis vertebrales y/o la obstrucción del tracto urinario o la hematuria empeorasen, podrían surgir problemas neurológicos como astenia y/o parestesia de las extremidades inferiores o empeoramiento de los síntomas urinarios (ver la sección 4.4).
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaRAM.es.
4.9 Sobredosis
No se dispone de experiencia clínica sobre los efectos de una sobredosis aguda de Lutrate Depot Trimestral o acetato de leuprorelina. En ensayos clínicos que utilizaban acetato de leuprorelina adminsitrado a diario por vía subcutánea a pacientes con cáncer de próstata, dosis de hasta 20 mg/día durante un periodo de hasta dos años no causaron AA distintos de los observados con la dosis de 1mg/día.
En estudios con animales, dosis de hasta 500 veces la dosis recomendada en humanos causaron disnea, reducción de la actividad e irritación local en la zona de inyección. En caso de sobredosis, se supervisará estrechamente el paciente y el tratamiento será sintomático y de soporte.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacéutico: Terapia endocrina. Hormonas y sustancias relacionadas. Análogos de la hormona liberadora de gonadotropina; código ATC: L02AE02.
El nombre químico del acetato de leuprorelina es 5-oxo-L-prolil-L-histidil-L-triptofil-L-seril-L-tyrosil-D-leucil-L-leucil-L-arginil-L-prolil-etilamida.
El acetato de leuprorelina es inactivo cuando se administra por vía oral a causa de una baja permeabilidad de la membrana y una casi incompleta inactivación por enzimas proteolíticas intestinales.
El acetato de leuprorelina tiene un potente efecto agonista de la LHRH cuando se administra durante el tratamiento a corto plazo e intermitente; no obstante, cuando se administra de forma continua y no pulsátil, los análogos de la LHRH inducen inhibición de la secreción de gonadotropinas y supresión de la esteroidogénesis testicular.
Con la unión a los receptores de LHRH hipofisarios, el acetato de leuprorelina provoca un aumento inicial de los niveles circulantes de hormona luteinizante (LH) y hormona foliculoestimulante (FSH), causando un aumento agudo de los niveles de testosterona y dihidrotestosterona. Sin embargo, en un plazo de cinco a ocho días tras su administración, los análogos de la LHRH producen la desensibilización del complejo del receptor de LHRH y/o la regulación negativa de la hipófisis anterior. A causa del menor número de receptores en la superficie celular, la estimulación celular se reduce y se sintetiza y segrega una menor cantidad de gonadotropinas. Finalmente, tras varias semanas de tratamiento con el agonista de la LHRH, la secreción de LH y FSH se suprime por completo. Como consecuencia de ello, las células de Leydig de los testículos dejan de producir testosterona, y la concentración sérica de testosterona disminuye hasta niveles de castración (menos de 0,5 ng/ml) en un plazo de dos a cuatro semanas desde el inicio del tratamiento.
En un estudio clínico abierto, multicéntrico y de dosis múltiple con Lutrate Depot Trimestral 22.5 mg, se reclutó a 163 pacientes con cáncer de próstata. El objetivo fue determinar la eficacia y seguridad de Lutrate Depot Trimestral cuando se administra a pacientes con cáncer de próstata que podrían beneficiarse de la terapia de privación de andrógenos. Lutrate Depot Trimestral se administró por vía intramuscular en 2 dosis con un intervalo de 3 meses.
Los niveles de testosterona se analizaron en diferentes días durante 168 días. El muestreo de testosterona se realizó en los días: 0 (1 y 4 horas), 2, 14, 28, 56, 84 pre-dosis, 84 (1 y 4 horas), 86, 112 y 168. El criterio de valoración principal se definió como valores de testosterona < 0.5 ng/mL y sin datos incompletos en los días 28, 84 y 168. Para cada paciente, si los niveles de testosterona eran superiores a 0,5ng/mL o si había datos incompletos en los días clave (28, 84 y 168), el paciente se clasificó como fallido, salvo que la falta de datos se debiera a un acontecimiento como la muerte sin relación al estudio del medicamento. Específicamente, si en cualquier de los días clave (28, 84 y 168) había una falta de datos debido a un AA relacionado con el estudio del fármaco, el paciente era clasificado como fallido.
Después de la primera administración los niveles medios de testosterona aumentaron de niveles basales (4,09±1,79 ng/mL) a un nivel pico (Cmax) de 6,33±3,40 ng/mL al segundo día. Después de alcanzar su valor máximo, los niveles de testosterona cayeron y un 98.8% (159/161) de los pacientes evaluables alcanzaron la castración médica en el día 28 (definida como niveles de testosterona inferiores a 0,5 ng/mL). Además, en este momento, un 77,0% de los pacientes alcanzaron un criterio más exigente en los niveles de
testosterona < 0,2 ng/mL (Figura 1). En el día 168, un 99,4% de los pacientes evaluables (150/151) presentaron unos niveles de testosterona por debajo de 0,5 ng/mL y un 90,7% por debajo de < 0,2 ng/mL.
Teniendo en cuenta el criterio primario de valoración descrito anteriormente el porcentaje de pacientes que mantuvieron la castración durante el estudio fue del 98,1% (158/161).
Figura 1. Media (±DE) de la concentración plasmática de testosterona durante el tratamiento con 2 inyecciones i.m. secuenciales de Lutrate Depot 22,5 mg Trimestral con un intervalo de 3 meses.
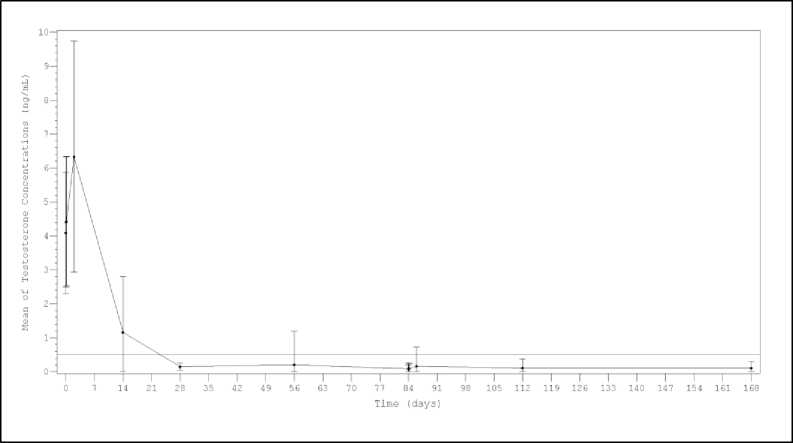
Los resultados del análisis de sensibilidad llevado a cabo considerando escapes de testosterona y datos incompletos como pacientes fallidos, mostraron tasas de castración de alrededor de un 92% a cada punto clave (día 28: 97,5% (157/161); día 56, 93,2% (150/161); día 84predose, 96,9% (156/161); día 84llTOra post-dosis, 91,9% (148/161); día 84^ post-dosis, 91,9% (148/161); día 86, 93,8% (151/161); día 112, 92,5% (149/161) y día 168, 93,2% (150/161)).
La frecuencia de escapes justo después de una segunda administración fue del 6,8% (11/161) y la frecuencia de escapes fue del 6,2% (10/161). Ninguno de los escapes transitorios estuvo asociado al aumento de LH, síntomas clínicos o incremento de PSA.
No se comunicaron efectos adversos relacionados con el tratamiento que fuesen clínicamente indicativos de un pico en los niveles de testosterona (retención urinaria, compresión de la médula espinal o exacerbación del dolor óseo) en ninguno de los pacientes en los que se registró un escape en los niveles de testosterona.
Los criterios secundarios de valoración de la eficacia incluyeron la determinación de las concentraciones séricas de LH, FSH y PSA. En el día 14 después de la primera inyección con Lutrate Depot Trimestral las concentraciones séricas medias de LH y FSH habían descendido por debajo de las concentraciones basales. Las concentraciones permanecieron por debajo de los niveles basales desde el día 28 hasta el fin del estudio. Durante el tratamiento, las concentraciones séricas medias de PSA se redujeron gradualmente (primer mes) y luego permanecieron por debajo del nivel basal de forma constante hasta el fin del estudio. No obstante, como era de esperar, se observó una amplia variación interindividual en las concentraciones de PSA durante todo el estudio.
5.2 Propiedades farmacocinéticas
Absorción
Tras la administración de 2 inyecciones de Lutrate Depot Trimestral con un intervalo de 3 meses en una muestra de pacientes de cáncer de próstata (N=30), la máxima concentración plasmática de acetato de leuprolina fue similar en los 2 ciclos. Después de la primera administración (días 0-84), la Cmax fue de 46,79±18,008 ng/mL. La mediana de tiempo hasta alcanzar la Cmax (Tmax) fue de 0,07 días, correspondiente a 1,68h (intervalo 1,008 - 4,008h).
Distribución
No se han realizado estudios de distribución con Lutrate Depot Trimestral. No obstante, en voluntarios varones sanos, el volumen de distribución medio en estado estacionario de acetato de leuprorelina tras la administración de una dosis en bolo intravenoso (i.v.) de 1,0 mg fue de 27 L. La unión a proteínas plasmáticas in vitro osciló entre el 43% y el 49%.
Eliminación
No se han realizado estudios de eliminación del fármaco con Lutrate Depot Trimestral.
Se espera que la leuprorelina se metabolice en pequeños péptidos inactivos que pueden ser eliminados o catabolizados.
En voluntarios varones sanos, un bolo de 1,0 mg de acetato de leuprorelina administrado por vía intravenosa puso de manifiesto que el aclaramiento sistémico medio era de 7,6 l/h, con una semivida de eliminación terminal de aproximadamente 3 horas según un modelo bicompartimental.
Tras la administración de acetato de leuprorelina a 3 pacientes, menos del 5% de la dosis se recuperó en forma de compuesto original y metabolito M-I en la orina.
Poblaciones especiales
Insuficiencia hepática/renal
No se ha determinado la farmacocinética del medicamento en pacientes con insuficiencia hepática o renal.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas y genotoxicidad realizados con acetato leuprorelina.
Como cabe esperar por sus propiedades farmacológicas conocidas, los estudios preclínicos demostraron efectos sobre el sistema reproductor, que fueron reversibles. En los estudios de toxicidad para la reproducción, el acetato de leuprorelina no mostró teratogenicidad. No obstante, se observó embriotoxicidad/letalidad en conejos.
Los estudios de carcinogenicidad realizados en ratas con administración subcutánea de acetato de leuprorelina (0,6-4 mg/kg/día) demostraron un aumento relacionado con la dosis de los adenomas hipofisarios. Además se observó un aumento significativo pero no relacionada con la dosis de células de los islotes pancreáticos-adenomas en hembras y de adenomas celulares intersticiales testiculares en machos. La mayor incidencia se observó en el grupo de dosis baja. La administración de acetato de leuprorelina provocó la inhibición del crecimiento de ciertos tumores dependientes de hormonas (tumores prostáticos en ratas macho Noble y Dunning y tumores mamarios inducidos por DMBA en ratas hembra). No se observaron tales efectos en los estudios de carcinogenicidad realizados en ratones. No se han realizado estudios de carcinogenicidad con Lutrate Depot Trimestral.
Los estudios con acetato de leuprorelina demostraron que el producto no era mutagénico en una serie de ensayos in vitro e in vivo. No se han realizado estudios de mutagenicidad con Lutrate Depot Trimestral.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Excipientes del liofilizado (vial):
Polisorbato 80 Manitol (E-421)
Carmelosa sódica (E-466)
Trietil citrato Poli(ácido láctico) (PLA)
Excipientes del disolvente (jeringa precargada): Manitol (E-421)
Hidróxido de sodio (para ajuste de pH)
Acido clorhídrico (para ajuste de pH)
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
Para la reconstitución del polvo de Lutrate Depot Trimestral no puede utilizarse ningún otro disolvente estéril que el disolvente que se proporciona para Lutrate Depot Trimestral.
6.3 Periodo de validez
3 años sin abrir.
Una vez reconstituido con el disolvente, la suspensión debe administrarse inmediatamente.
6.4 Precauciones especiales de conservación
Conservar por debajo de 25°C. No congelar.
6.5 Naturaleza y contenido del envase
El kit comercial incluye:
1. Un (1) vial de vidrio de tipo I que contiene 22,5 mg de acetato de leuprorelina en forma de polvo liofilizado, sellado con un tapón de elastómero y un cierre desprendible de aluminio con plástico.
2. Una (1) jeringa de vidrio de tipo I precargada que contiene 2 ml de disolvente, sellada con un tapón de elastómero.
3. Un (1) sistema adaptador de policarbonato/HDPE incluyendo una (1) aguja estéril del calibre 20.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Método de administración
El vial de Lutrate Depot Trimestral deberá ser reconstituido inmediatamente antes de la administración por vía intramuscular. Asegúrese de que se sigue un método aséptico.
La solución reconstituida es una suspensión lechosa de color blanco.
No puede utilizarse otro disolvente para la reconstitución de Lutrate Depot Trimestral.
Reconstituir Lutrate Depot Trimestral conforme a las siguientes instrucciones:
.HP.

2
. I
I
Ajuste el sistema adaptador (de color morado) al vial hasta que oiga un “clic”.

3
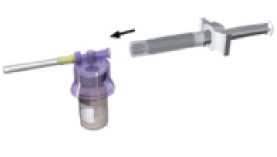
Adapte la pieza blanca a la jeringa que contiene el disolvente Retire el tapón de goma de la jeringa y acople la jeringa al sistema adaptador.
4
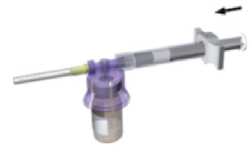
Manteniendo la jeringa y el vial bien acoplados en posición vertical, empuje lentamente el émbolo para transferir todo el diluyente al vial.
7
1'
5
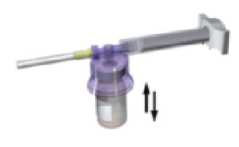
Con la jeringa todavía acoplada al vial, agite suavemente el vial durante un minuto aproximadamente hasta obtener una suspensión lechosa uniforme.
8
6

Coloque el sistema del revés, y tire cuidadosamente del émbolo para extraer el fármaco resuspendido desde el vial a la jeringa.

Retire la jeringa y la aguja del sistema adaptador girando la pieza superior del adaptador en el sentido contrario a las agujas del reloj. El medicamento está listo para ser administrado.

Limpie la zona de inyección con un algodón impregnado en alcohol y deje que se seque la piel. Inyecte la suspensión por vía intramuscular en el cuadrante superior externo del glúteo.
Parte del producto puede acumularse o depositarse en la pared del vial. Esto se considera normal. Durante el desarrollo del producto, el vial se llena con exceso de producto para garantizar que se administra una dosis final de 22,5 mg de acetato de leuprorelina.
El producto está diseñado para administrar una única inyección. El producto restante debe desecharse.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
GP-PHARM, S.A.
Pol. Ind. Els Vinyets - Els Fogars Sector 2 Carretera comarcal 244, km22 08777 Sant Quintí de Mediona España
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Julio 2015
10. FECHA DE LA REVISIÓN DEL TEXTO
Mayo 2015
18 de 18