Lodotra 2 Mg Comprimidos De Liberacion Modificada
"I
an
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Lodotra 1 mg comprimidos de liberación modificada Lodotra 2 mg comprimidos de liberación modificada Lodotra 5 mg comprimidos de liberación modificada
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Lodotra 1 mg: un comprimido de liberación modificada contiene 1 mg de prednisona
Lodotra 2 mg: un comprimido de liberación modificada contiene 2 mg de prednisona
Lodotra 5 mg: un comprimido de liberación modificada contiene 5 mg de prednisona
42.80 mg de lactosa monohidrato.
41.80 mg de lactosa monohidrato.
38.80 mg de lactosa monohidrato.
-Excipiente con efecto conocido: Lactosa monohidrato Lodotra 1 mg: cada comprimido de liberación modificada contiene Lodotra 2 mg: cada comprimido de liberación modificada contiene Lodotra 5 mg: cada comprimido de liberación modificada contiene Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimidos de liberación modificada.
Lodotra 1 mg: Comprimidos de liberación modificada de color blanco amarillento pálido, cilíndricos, de 5 mm de altura y 9 mm de diámetro, con “NP1” grabado en relieve en una cara.
Lodotra 2 mg: Comprimidos de liberación modificada de color blanco amarillento, cilíndricos, de 5 mm de altura y 9 mm de diámetro, con “NP2” grabado en relieve en una cara.
Lodotra 5 mg: Comprimidos de liberación modificada de color amarillo claro, cilíndricos, de 5 mm de altura y 9 mm de diámetro, con“NP5” grabado en relieve en una cara.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Lodotra está indicado para el tratamiento de la artritis reumatoide de moderada a grave, especialmente cuando va acompañada de síntomas de rigidez matinal, en adultos.
4.2 Posología y forma de administración
Posología
La dosis apropiada depende de la gravedad de la enfermedad y de la respuesta individual del paciente. En general, como inicio de la terapia se recomienda una dosis de 10 mg de prednisona. En ciertos casos, puede requerirse una dosis inicial superior (ejemplo 15 ó 20 mg de prednisona).
Según sean los síntomas clínicos y la respuesta del paciente, la dosis inicial puede reducirse gradualmente a una dosis de mantenimiento más baja.
Al cambiar del régimen normal (administración de glucocorticoides por la mañana) a Lodotra administrada a la hora de acostarse (aproximadamente a las 10 de la noche), debería mantenerse la misma dosis (equivalente en mg de prednisona). Después del cambio, puede ajustarse la dosis en función del estado clínico.
Para dosificaciones no factibles con estas presentaciones, se dispone de otras dosificaciones de este medicamento. En caso de tratamiento de larga duración de la artritis reumatoide, la dosis individual de hasta 10 mg de prednisona al día, debe ajustarse de acuerdo con la gravedad del ciclo de la enfermedad.
.Uí1.
"I ■"
¡m
Dependiendo del resultado del tratamiento, puede reducirse la dosis gradualmente a razón de 1 mg cada 2 -4 semanas hasta alcanzar la dosis de mantenimiento adecuada.
Para suspender la terapia con Lodotra, deberá reducirse la dosis gradualmente a razón de 1 mg cada 2 - 4 semanas, monitorizando si hace falta, los parámetros del eje pituitario-adrenal.
Población pediátrica
No se recomienda el uso en niños y en adolescentes porque no dispone de datos suficientes sobre tolerabilidad y eficacia.
Forma de administración:
Lodotra debe tomarse a la hora de acostarse (aproximadamente a las 10 de la noche), con la cena o inmediatamente después de la misma y tragarse entero con suficiente líquido. Si han transcurrido más de 2 ó 3 horas después de la cena, se recomienda tomar el comprimido con una comida ligera o tentempié (por ejemplo una rebanada de pan con jamón o queso). Lodotra no debe administrarse en ayunas, ya que podría disminuir su biodisponibilidad.
Lodotra está diseñado para liberar la sustancia activa con un retraso de entre 4-6 horas después de su ingesta, de forma que la liberación de la sustancia activa y los efectos farmacológicos se inicien durante la noche.
Lodotra comprimidos de liberación modificada están compuestos de un núcleo de prednisona y un recubrimiento inerte. La liberación retardada de prednisona depende de la integridad del recubrimiento. Por este motivo, los comprimidos de liberación modificada no deben romperse, partirse ni masticarse.
En los pacientes afectados por hipotiroidismo o cirrosis hepática podría ser suficiente una dosis comparativamente más baja o ser preciso reducir la dosis.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Únicamente debería administrarse una farmacoterapia a base de prednisona si fuera absolutamente necesario y tendría que ir acompañada de una terapia anti-infecciosa adecuada, cuando se presenten alguna de las siguientes circunstancias:
- Infecciones virales agudas (herpes zoster, herpes simplex, varicela, queratitis herpética)
- Hepatitis HbsAg positiva crónica activa
- Aproximadamente 8 semanas antes y 2 semanas después de inmunización con vacunas vivas
- Micosis sistémica y parasitosis (por ejemplo nematodos)
- Poliomielitis
- Linfadenitis como consecuencia de una inoculación de BCG
- Infecciones bacterianas agudas y crónicas
- Historia de tuberculosis (precaución con una posible reactivación). Debido a sus propiedades inmunosupresoras los glucocorticoides pueden inducir o agravar las infecciones. Hay que monitorizar con cuidado a estos pacientes, por ejemplo realizando un test de tuberculina. Los pacientes de especial riesgo deben recibir un tratamiento tuberculostático.
Además, una farmacoterapia a base de prednisona sólo debería administrarse en casos de absoluta necesidad e ir acompañada de la terapia adecuada, si es preciso, en las situaciones siguientes:
- Úlceras gastrointestinales
- Osteoporosis grave y osteomalacia
- Hipertensión de difícil control
- Diabetes mellitus grave
ÍTTI
- Trastornos psiquiátricos (también si surgen en la historia previa del paciente)
- Glaucoma de espectro estrecho y amplio
- Úlceras corneales y afecciones de la córnea
Debido al riesgo existente de perforación intestinal, sólo se debería administrar prednisona en el caso de ser absolutamente necesario y con el control adecuado en los casos de:
- Colitis ulcerativa grave con perforación inminente
- Diverticulitis
- Entero-anastomosis (inmediatamente postoperatoria).
Lodotra no puede alcanzar la concentración deseada de prednisona en sangre si se toma en situación de ayunas. Por lo tanto, Lodotra debe tomarse siempre con la cena o inmediatamente después para asegurar la eficacia necesaria. Además, se pueden presentar concentraciones bajas en plasma en un 6%-7% de las dosis administradas de Lodotra cuando estas se administran siguiendo las indicaciones, según se ha observado en los estudios farmacocinéticos y de un 11% en único estudio farmacocinético. Esto debería considerarse si Lodotra no fuera suficientemente eficaz. En estas situaciones, se debería considerar un cambio a una formulación de liberación inmediata.
Lodotra no debe sustituirse por comprimidos de prednisona de liberación inmediata en la misma pauta de administración debido al mecanismo de liberación retardada de Lodotra.
En caso de sustitución, terminación o interrupción del tratamiento prolongado, deberán tenerse en cuenta los siguientes riesgos: recurrencia de la actividad de la afección artrítica reumatoide, fallo adrenal agudo (especialmente en situaciones de stress, por ejemplo durante infecciones, después de accidentes, al aumentar el esfuerzo físico), síndrome de abstinencia de cortisona.
Lodotra no debe administrarse en indicaciones agudas en lugar de prednisona de liberación inmediata debido a sus propiedades farmacológicas.
Durante la administración de Lodotra, se debería considerar una posible necesidad de incremento de insulina o de la medicación con antidiabéticos orales. Por lo tanto, pacientes con diabetes mellitus, deberían tratarse en condiciones de estrecha monitorización.
Durante el tratamiento con Lodotra, será preciso realizar controles regulares de la tensión arterial en pacientes con hipertensión difícil de controlar.
Los pacientes con insuficiencia cardíaca grave tienen que tener una estrecha monitorización debido al riesgo que existe de que empeore su estado.
Es necesario tener precaución cuando se prescriban corticoesteroides, incluyendo prednisona, a pacientes que hayan sufrido un infarto de miocardio reciente debido al riesgo de rotura cardiaca.
Es necesario tener precaución cuando se prescriban corticoesteroides, incluyendo prednisona, a pacientes con insuficiencia renal.
Está documentado que se presentan alteraciones del sueño más frecuentemente con Lodotra que con formulaciones convencionales de liberación inmediata tomadas en la mañana. Si se produce insomnio que no mejora, puede ser aconsejable un cambio a una formulación convencional de liberación inmediata.
El tratamiento con Lodotra también puede enmascarar los signos y síntomas de una infección existente o en desarrollo, haciendo más difícil el diagnóstico.
Incluso con dosis bajas, la administración durante un período de tiempo prolongado de Lodotra incrementa el riesgo de infección. Estas posibles infecciones pueden ser provocadas por microorganismos que en circunstancias normales raramente causan infecciones (las llamadas infecciones “oportunistas”).
ÍTTI
Ciertas enfermedades virales (varicela, sarampión) pueden agravarse en pacientes tratados con glucocorticoides. Los individuos inmunodeprimidos sin infección previa por varicela o sarampión tienen especial riesgo. Si estos individuos, mientras están siendo tratados con Lodotra, han estado en contacto con personas infectadas con varicela o sarampión, habrá que empezar a administrarles un tratamiento preventivo si hace falta.
En aquellos pacientes que padezcan o en los que se sospeche infestación por Strongyloides (lombrices) los glucocorticoides pueden dar lugar a una hiperinfección y enfermedad diseminada por migración generalizada de las larvas.
Son generalmente posibles las vacunaciones con vacunas inactivadas. Sin embargo debe tenerse en cuenta que la respuesta inmune y el consiguiente éxito de la vacunación pueden verse dificultados por las altas dosis de corticoides.
En el caso de una terapia de larga duración con Lodotra, debe realizarse un seguimiento médico regular (incluyendo exámenes oftalmológicos a intervalos de tres meses); si se administran dosis comparativamente altas, deberá asegurarse la administración suficiente de suplementos de potasio y restricción de sodio, controlando al mismo tiempo los niveles de potasio sérico.
Si en el transcurso del tratamiento con Lodotra se producen hechos concretos que hacen aumentar notablemente el estrés físico (accidentes, operación quirúrgica, etc.), podrá ser preciso aumentar temporalmente la dosis.
Según sea la duración del tratamiento y de la dosis administrada, habrá que esperar un impacto negativo sobre el metabolismo de calcio. En consecuencia es aconsejable prever una profilaxis para la osteoporosis y resulta especialmente importante en presencia de otros factores de riesgo (incluyendo entre éstos una predisposición familiar, edad avanzada, situación postmenopáusica, ingestión insuficiente de proteínas y calcio, consumo excesivo de alcohol y tabaco, así como la actividad física reducida). La profilaxis se basa en una administración suficiente de calcio y de vitamina D, así como la actividad física. En el caso de una osteoporosis previamente existente, debería considerarse la administración de una terapia adicional.
Este medicamento contiene lactosa. Los pacientes con intolerancia hereditaria a galactosa, insuficiencia de lactasa de Lapp (insuficiencia observada en ciertas poblaciones de Laponia) o malabsorción de glucosa o galactosa no deben tomar este medicamento.
Al usar altas dosis de prednisolona durante un largo periodo de tiempo (30 mg/día durante un mínimo de 4 semanas), se observaron alteraciones reversibles de la espermatogénesis que persistían durante varios meses después de interrumpir la administración del medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
- Glicósidos cardíacos: es posible que el efecto de los glicósidos aumente como consecuencia de la deficiencia de potasio.
- Saluréticos / laxantes: se aumenta la eliminación del potasio.
- Agentes antidiabéticos: se reduce la bajada de los niveles de azúcar en sangre.
- Derivados de la cumarina: puede aumentar o disminuir la eficacia de los anticoagulantes a base de cumarina.
- Agentes antiinflamatorios y antirreumáticos no esteroideos, salicilatos e indometacina: se aumenta el riesgo de hemorragia gastrointestinal.
- Relajantes musculares no despolarizantes: se puede prolongar la relajación muscular.
- Atropina y otros anticolinérgicos: el uso concomitante con Lodotra puede causar un aumento adicional de la presión intraocular.
- Prazicuantel: los glucocorticoides pueden producir una disminución de las concentraciones del praziquantel en la sangre.
- Cloroquina, hidroxicloroquina, mefloquina: aumenta el riesgo de aparición de miopatías y cardiomiopatías.
ílMt
ÍTTI
- Somatropina: puede reducirse la eficacia de la somatropina.
- Estrógenos (por ejemplo anticonceptivos orales): puede aumentar la eficacia de los glucocorticoides.
- Regaliz: es posible una inhibición del metabolismo de los glucocorticoides.
- Rifampicina, fenitoína, barbitúricos, bupropión y primidona: se reduce la eficacia de los glucocorticoides.
- Ciclosporina: los niveles de ciclosporina en sangre aumentan. Existe un aumento del riesgo de convulsiones.
- Anfotericina B: se puede incrementar el riesgo de hipopotasemia.
- Ciclofosfamida: se pueden incrementar los efectos de la ciclofosfamida.
- Inhibidores ACE: aumenta el riesgo de cambios en el recuento sanguíneo.
- Antiácidos de aluminio y magnesio: se reduce la absorción de glucocorticoides. Sin embargo, debido al mecanismo de liberación retardada de Lodotra, es improbable una interacción de la prednisona con los antiácidos de aluminio/magnesio.
- Impacto en lo referente a métodos de diagnóstico: pueden suprimirse las reacciones cutáneas causadas por los tests de alergia. Puede reducirse el incremento de TSH después de la administración de protirelina.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Durante el embarazo, Lodotra sólo debe utilizarse cuando el beneficio supere los potenciales riesgos. Debe utilizarse la mínima dosis eficaz de Lodotra necesaria para mantener un adecuado control de la enfermedad.
Estudios en animales indican que la administración de dosis farmacológicas de glucocorticoides durante el embarazo puede aumentar el riesgo de retraso en el crecimiento intrauterino, desarrollo de enfermedades cardiovasculares y/o metabólicas en el adulto, y puede tener un efecto sobre la densidad de los receptores de glucocorticoides, y el recambio del neurotransmisor o el desarrollo neuroconductual.
La prednisona ha provocado la formación de paladar hendido en experimentos animales (ver sección 5.3). Actualmente se discute la posibilidad de un riesgo incrementado de formación de paladar hendido en el feto humano como consecuencia de la administración de glucocorticoides durante el primer trimestre del embarazo.
Si los glucocorticoides se administran hacia el final del embarazo existe riesgo de atrofia de la corteza adrenal del feto, que hará precisa una terapia de sustitución en el recién nacido, que deberá reducirse lentamente.
Lactancia
Los glucocorticoides pasan a la lecha materna en pequeñas cantidades (hasta el 0,23 % de una dosis individual). En las dosis de hasta 10 mg diarios, la cantidad que se absorbe a través de la leche materna estará por debajo del nivel de detección. Hasta el momento no se ha informado de daños en lactantes. Sin embargo solo deberán prescribirse glucocorticoides cuando los beneficios previsibles para la madre y el niño superen los riesgos existentes.
Como la relación de la concentración leche / plasma aumenta con dosis por encima de los 10 mg por día (por ejemplo el 25 % de concentración plasmática se encuentran en la leche materna cuando se administran 80 mg de prednisona diaria) se aconseja suspender la lactancia en estos casos.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios para determinar los efectos sobre la capacidad para conducir y utilizar maquinaria.
4.8 Reacciones adversas
La frecuencia y gravedad de las reacciones adversas que se relacionan a continuación depende de la dosis y duración del tratamiento. En el rango de dosis recomendada de Lodotra (terapia con baja dosis de
ÍTTI
corticoides entre 1 y 10 mg diarios), las reacciones adversas que se relacionan se manifiestan con menor frecuencia y menor gravedad que si se superan dosis de 10 mg.
Según sea la duración del tratamiento y la dosis podrán aparecer las siguientes reacciones adversas:
Muy frecuentes (>1/10)
Frecuentes (>1/100 a <1/10)
Poco frecuentes (>1/1.000 a <1/100)
Raras (>1/10.000 a < 1/1.000)
Muy raras (< 1/10.000)
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Trastornos de la sangre y del sistema linfático:
Frecuentes: leucocitosis moderada, linfopenia, eosinopenia, policitemia.
Trastornos cardiacos:
Frecuencia no conocida: taquicardia.
Trastornos del sistema inmunológico:
Frecuentes: defensa inmune reducida, enmascaramiento de infecciones, exacerbación de infecciones latentes.
Raras: reacciones alérgicas.
Infecciones e infestaciones:
Frecuentes: susceptibilidad y gravedad aumentada de las infecciones.
Trastornos endocrinos:
Frecuentes: supresión adrenal e inducción del síndrome de Cushing (síntomas típicos: cara en forma de luna, obesidad de la parte superior del cuerpo y plétora).
Raras: alteraciones en la secreción de hormonas sexuales (amenorrea, impotencia), alteraciones de la función tiroidea.
Trastornos del metabolismo y de la nutrición:
Frecuentes: retención de sodio con edema, incremento de la excreción de potasio (atención a las posibles arritmias), aumento del apetito y ganancia de peso, disminución de la tolerancia a la glucosa, diabetes mellitus, hipercolesterolemia e hipertrigliceridemia.
Frecuencia no conocida: lipomatosis reversible epidural, epicardial o mediastínica, alcalosis hipopotasémica.
Trastornos psiquiátricos:
Frecuentes: insomnio.
Raras: depresión, irritabilidad, euforia, impulsividad, psicosis.
Trastornos del sistema nervioso:
Frecuentes: cefalea.
Raras: pseudotumores cerebrales, manifestación de epilepsia latente e incremento de la predisposición a desarrollar ataques en casos de epilepsia manifiesta.
Trastornos oculares:
Frecuentes: cataratas, especialmente con opacidad subcapsular posterior, glaucoma
Raras: agravamiento de los síntomas asociados con la úlcera corneal, aparición de inflamaciones oculares
de origen viral, fungico y bacteriano.
Frecuencia no conocida: Corioretinopatía serosa central.
Trastornos vasculares:
Poco frecuentes: hipertensión, aumento en el riesgo de la arteroesclerosis y trombosis, vasculitis (también como síndrome de abstinencia después de una terapia de larga duración).
Trastornos gastrointestinales:
Poco frecuentes (no concomitante con AINEs): ulceraciones gastrointestinales, hemorragias gastrointestinales.
Raras: pancreatitis.
Frecuencia no conocida: Náuseas, diarrea, vómitos Trastornos de la piel y del tejido subcutáneo:
Frecuentes: estrías enrojecidas, atrofia, telangiectasia, incremento de la fragilidad capilar, petequias, equimosis.
Poco frecuentes: hipertricosis, acné esteroide, retraso en la curación de heridas, dermatitis similar a la rosácea (perioral), alteraciones de la pigmentación de la piel.
Raras: reacciones de hipersensibilidad, por ejemplo exantema medicamentoso.
Frecuencia no conocida: hirsutismo.
Trastornos musculoesqueléticos y del tejido conjuntivo:
Frecuentes: atrofia muscular y debilidad, osteoporosis (dependiendo de la dosis, puede aparecer incluso con un tratamiento breve).
Raras: osteonecrosis aséptica (de la cabeza humeral y femoral).
Frecuencia no conocida: miopatía esteroidea, rotura tendinosa, fracturas vertebrales y de los huesos largos. Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano.Website: www.notificaRAM.es.
4.9 Sobredosis
No se conocen intoxicaciones agudas por Lodotra. En caso de sobredosis, podría presentarse un aumento de las reacciones adversas, especialmente las de tipo endocrino, metabólico y relacionadas con los electrolitos (ver sección 4.8).
No se conoce antídoto para la prednisona.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Corticoides para uso sistémico. Glucocorticoides, código ATC: H02AB07 La prednisona es un glucocorticoide no fluorado para terapia sistémica.
La prednisona ejerce un efecto dosis-dependiente sobre el metabolismo de casi todos los tejidos. Bajo condiciones fisiológicas estos efectos resultan vitales para mantener la homeostasis del organismo en reposo y en situación de estrés, así como para garantizar el control de las actividades del sistema inmunitario.
Administrado en las dosis típicas prescritas para Lodotra, la prednisona tiene un efecto antiinflamatorio inmediato (antiexudativo y antiproliferativo) y un efecto inmunosupresivo retardado. Al mismo tiempo inhibe la quimiotaxia y la actividad de las células inmunes así como la liberación y el efecto de los mediadores de las reacciones inflamatorias e inmunitarias, por ejemplo los enzimas lisosomales, prostaglandinas y leucotrienos.
La terapia prolongada con dosis elevadas altera la respuesta del sistema inmune y de la corteza adrenal. El efecto mineralotrópico que es pronunciado en la hidrocortisona sigue siendo detectable en la prednisona y puede requerir la monitorización de los niveles de electrolitos séricos.
ÍTTI
En los pacientes afectados de artritis reumatoide, las citoquinas proinflamatorias tales como las interleuquinas IL - 1 y IL - 6 y el factor alfa de necrosis tumoral (TNFa) alcanzan los niveles máximos en plasma a primera hora de la mañana (por ej. IL-6 entre las 7 y las 8 de la mañana). Se observó que la concentración de citoquinas disminuía después de administrar Lodotra y de la subsiguiente liberación durante la noche de la prednisona (se iniciaba la absorción entre las 2-4 de la madrugada y la Cmax se alcanzaba entre las 4 -6 de la mañana).
Se evaluó la eficacia y seguridad de Lodotra en dos estudios aleatorizados, doble-ciego controlados en pacientes con artritis reumatoide activa.
En el primer estudio, un estudio multicéntrico aleatorizado doble ciego, de fase III, de 12 semanas de duración con un total de 288 pacientes previamente tratados con prednisona o prednisolona, el grupo que se cambió a Lodotra a la misma dosis presentó una reducción media del 23% en la duración de la rigidez matinal mientras que la duración en el grupo de referencia no cambió. Los resultados se incluyen en la tabla siguiente.
La variación relativa en la duración de la rigidez matinal después de 12 semanas de tratamiento fue
|
Variación relativa [%] |
Lodotra (n = 125) |
Prednisona IR (n = 129) |
|
Media |
-23 |
0 |
|
(SD) |
(89) |
(89) |
|
Mediana |
-34 |
-13 |
|
(min, máx) |
(-100, 500) |
(-100, 610) |
En una fase de extensión posterior y abierta (9 meses de tratamiento) la variación media relativa en la duración de la rigidez matinal comparada con la basal fue del -50%.
La variación de la duración de la rigidez matinal después de 12 meses de tratamiento con Lodotra:
|
Duración de la rigidez matinal [min] |
Lodotra | |
|
Media (SD) |
N | |
|
0 meses |
156 |
107 |
|
Inicio del estudio |
(97) | |
|
12 meses |
74 |
96 |
|
Final de la fase abierta |
(92) | |
En el mismo estudio, después de 12 semanas de tratamiento, se observó una disminución media de la citoquina proinflamatoria IL-6 del 29% en el grupo tratado con Lodotra, mientras que no se observaron cambios en el grupo comparador que recibió prednisona estándar. Después de 12 meses de tratamiento con Lodotra el nivel de IL-6 permaneció estable.
Las variaciones en los niveles de IL-6 después de 12 meses fueron:
|
IL-6 |
Lodotra | |
|
[IU/L] |
mediana (min, max) |
N |
|
0 meses |
860 |
142 |
|
Inicio del estudio |
(200,23000) | |
|
12 meses |
470 |
103 |
|
Final de la fase abierta |
(200,18300) | |
Valores < 200 UI/L se fijaron en 200 UI/L para el análisis
ÍTTI
La eficacia de Lodotra administrada adicionalmente a los fármacos antirreumáticos modificadores de la enfermedad (FARME/DMARD) fue confirmada en un segundo ensayo aleatorizado, controlado con placebo en pacientes que respondían de forma insuficiente al tratamiento solo con FARME. A las 12 semanas, los pacientes del grupo de Lodotra alcanzaron unas tasas de respuesta a ACR20 y ACR50 significativamente más altas (46,8% y 22,1%, respectivamente) comparado con los pacientes que recibieron placebo (29,4% y 10,1%, respectivamente). También hubo un cambio medio mayor en el índice medio de DAS 28 desde la basal (5,2 en el grupo de Lodotra y 5,1 en el grupo placebo) hasta la semana 12 en el grupo de Lodotra (-1,2 puntos) comparado con el observado en el grupo de placebo (cambio de -0,7 puntos).
Además, después de 12 semanas de tratamiento la duración media de la rigidez matinal era de 86,0 minutos (reducción de 66 minutos) en el grupo de Lodotra y 114,1 minutos (reducción de 42,6 minutos) en el grupo placebo. Lodotra se puede utilizar con seguridad en combinación con otros FARME.
5.2 Propiedades farmacocinéticas
Absorción:
Lodotra son comprimidos de liberación modificada que contienen prednisona. La prednisona se libera entre 4-6 horas después de la administración de Lodotra. Posteriormente, la prednisona se absorbe rápida y casi completamente.
Distribución:
Los niveles plasmáticos máximos se alcanzan aproximadamente a las 6-9 horas después de la administración.
Biotransformación:
Más del 80 % de la prednisona se convierte en prednisolona mediante un metabolismo hepático de primer paso. El ratio de prednisona : prednisolona es de aproximadamente 1:6 a 1:10. La prednisona por sí misma ejerce un efecto farmacológico insignificante. La prednisolona es el metabolito activo. Los compuestos se unen de forma reversible a las proteínas plasmáticas con alta afinidad para la transcortina (corticosteroide que se une a la globulina, CBG) y una afinidad baja para la albúmina plasmática.
A niveles bajos de dosis (hasta 5 mg) aproximadamente el 6 % de la prednisolona se presenta en forma libre. Dentro de este rango, la eliminación metabólica es lineal. En dosis superiores a 10 mg la capacidad de fijación de la transcortina va agotándose progresivamente y aparece más prednisolona libre. Esto puede tener como resultado una eliminación metabólica más rápida.
Eliminación:
La prednisolona se elimina fundamentalmente por metabolismo hepático, aproximadamente un 70 % por glucuronidación y aproximadamente un 30 % mediante sulfatación. También hay una conversión a 11B, 17B-dihidroxiandrosta-1,4-dien-3-ona y a 1,4-pregnadien-20-ol. Los metabolitos no muestran actividad hormonal alguna y se eliminan principalmente por vía renal. En la orina se han detectado restos insignificantes de prednisona y prednisolona inalteradas. La vida media de eliminación en plasma de prednisona/prednisolona es de aproximadamente 3 horas. Se prolonga en pacientes con disfunción hepática grave por lo que se debería considerar una reducción de la dosis en estos casos. La duración de los efectos biológicos de la prednisona / prednisolona supera la duración de su presencia serológica.
Biodisponibilidad:
Un estudio de biodisponibilidad llevado a cabo en 27 individuos sanos realizado en el año 2003 demostró los siguientes resultados en comparación con los comprimidos de prednisona de liberación inmediata:
|
Parámetro |
Lodotra 5 mg: |
Lodotra 5 mg: |
Fármaco de |
|
2,5 horas después de |
inmediatamente |
referencia de 5 mg | |
|
una comida ligera |
después de una comida |
en ayunas |
|
Máxima concentración |
20,2 |
21,8 |
20,7 |
|
plasmática (Cmax): ng/ml |
(18,5; 21,9) |
(20,0; 23,7) |
(19,0; 22,5) |
|
Tiempo de máxima concentración plasmática(tmax): h |
6,0 (4,5; 10,0) |
6,5 (4,5; 9,0) |
2,0 (1,0; 4,0) |
|
4,0 |
3,5 |
0,0 | |
|
Duración de la liberación modificada del fármaco (tlag): h |
(3,5; 5,0) |
(2,0; 5,5) |
(0,0; 0,5) |
|
Área bajo la curva de concentración plasmática respecto al tiempo (AUC 0-®): ng x h/ml |
110 (101; 119) |
123 (114;133) |
109 (101; 118) |
Los valores son medias geométricas de mínimos cuadrados y rango
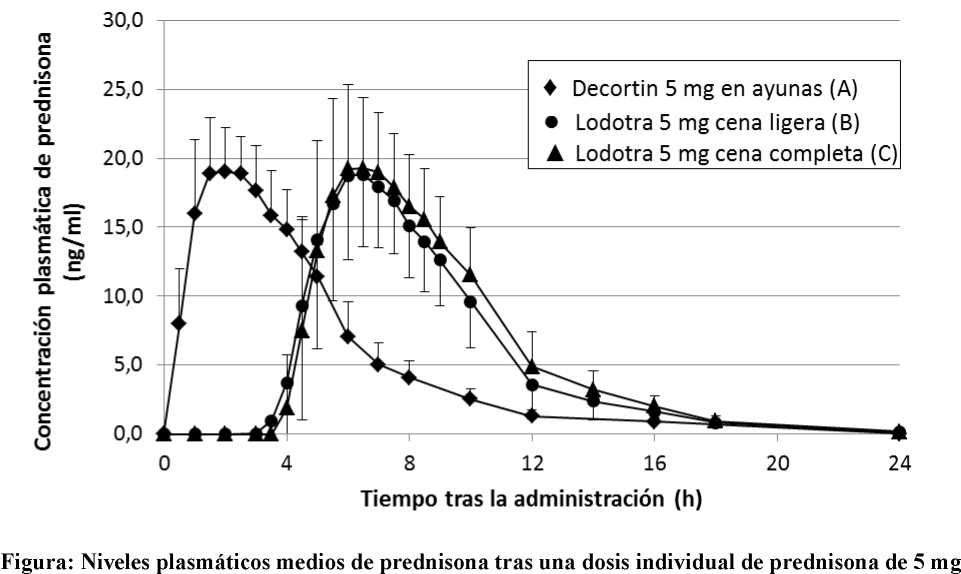
administrada como Lodotra 5 mg o un comprimido de liberación inmediata.
Comprimido de 5 mg de liberación inmediata (A: en ayunas, administrado a las 2 am), Lodotra 5 mg (B: 2,5 horas después de una cena ligera) y Lodotra 5 mg (C: inmediatamente después de una cena completa).
Los perfiles de concentración plasmática de Lodotra son muy similares a los de un comprimido de liberación inmediata, con la importante diferencia de que el perfil de Lodotra se retrasa 4 - 6 horas después de la administración del medicamento. Se han observado bajas concentraciones en plasma en el 6-7% de las dosis.
Se demostró la proporcionalidad de las dosis de Lodotra 1 mg, 2 mg y 5 mg basándose en la AUC y Cmax.
5.3 Datos preclínicos sobre seguridad Toxicidad subcrónica/crónica
Después de una administración diaria intraperitoneal de 33 mg/kg de peso corporal durante 7 a 14 días en ratas se observaron cambios al microscopio óptico y electrónico en las células de los islotes de Langerhans
ÍTTI
de las ratas. En conejos se indujeron daños en el hígado, administrando de 2 a 3 mg/kg de peso corporal por día durante un periodo de 2 a 4 semanas. Se han descrito efectos histotóxicos (mionecrosis) después de varias semanas de administración de 0,5 a 5 mg/kg de peso corporal en cobayas y 4 mg/kg de peso corporal en perros.
Potencial mutagénico y carcinogénico
La toxicidad observada en estudios animales con prednisona se asoció con una actividad farmacológica exagerada. No se han observado efectos genotóxicos de prednisona en ensayos convencionales de genotoxicidad.
Toxicidad en la reproducción
En estudios de reproducción en animales, los glucocorticoides como la prednisona han demostrado inducir malformaciones (paladar hendido, malformaciones esqueléticas). Con la administración parenteral en ratas se han encontrado anomalías menores del cráneo, la mandíbula y la lengua. Se observa un retraso en el crecimiento intrauterino (ver también sección 4.6).
Se considera poco probable que se produzcan efectos similares en pacientes tratados con las dosis terapéuticas.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido:
Sílice coloidal anhidra Croscarmelosa de sodio Lactosa monohidrato Estearato de magnesio Povidona K 29/32 Óxido de hierro rojo E 172
Cubierta del comprimido:
Sílice coloidal anhidra
Hidrógeno fosfato de calcio dihidrato
Glicerol dibehenato
Estearato de magnesio
Povidona K 29/32
Óxido de hierro amarillo E 172
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
2 años.
Validez después de abrir el bote: 14 semanas.
6.4 Precauciones especiales de conservación No conservar a temperatura superior a 25°C.
6.5 Naturaleza y contenido del envase
Envases con 30 y 100 comprimidos de liberación modificada: bote blanco fabricado con polietileno de alta densidad (HDPE). Tapón de rosca (que incluye una cápsula desecante) y tres puntos elevados dispuestos alrededor del borde para facilitar la apertura fabricado con HDPE.
.Uí1.
"I ■"
¡m
Envase con 500 comprimidos de liberación modificada: bote blanco de polietileno de alta densidad (con una pequeña cantidad de LDPE) con un tapón de rosca estándar (sin los tres puntos elevados) compuesto de polipropileno.
Tamaños de envase:
- bote con 30 y 100 comprimidos de liberación modificada
- bote con 30, 100 y 500 comprimidos de liberación modificada (envase hospitalario)
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Mundipharma Pharmaceuticals S.L.
Bahía de Pollensa, 11 28042 Madrid Teléfono: 913821870 Fax: 913821871
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
Concentración
1 mg
2 mg 5 mg
Número de la autorización de comercialización 71.061 71.065 71.067
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Lodotra 1 mg: 1/septiembre/2011 Lodotra 2 mg y 5 mg: 25/agosto/2011
10. FECHA DE LA REVISIÓN DEL TEXTO
Julio 2014
12 de 12