Lemtrada 12Mg Concentrado Para Solucion Para Perfusion
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
LEMTRADA 12 mg concentrado para solución para perfusión
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene 12 mg de alemtuzumab en 1,2 ml (10 mg/ml).
Alemtuzumab es un anticuerpo monoclonal que se produce mediante tecnología de ADN recombinante en un cultivo en suspensión de células de mamíferos (ovario de hámster chino) en un medio nutriente.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Concentrado para solución para perfusión (concentrado estéril).
Concentrado transparente, de incoloro a ligeramente amarillento con pH 7,0 - 7,4.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
LEMTRADA está indicado en pacientes adultos con esclerosis múltiple remitente recurrente (EMRR) con enfermedad activa definida por manifestaciones clínicas o detectadas por resonancia magnética (ver sección 4.4 y 5.1).
4.2 Posología y forma de administración
El tratamiento con LEMTRADA debe ser iniciado y supervisado por un neurólogo con experiencia en el tratamiento de pacientes con EM. Se debe disponer de los especialistas y los equipos necesarios para el diagnóstico y la gestión puntual de las reacciones adversas más frecuentes, especialmente las infecciones y las enfermedades autoinmunes.
Debe haber recursos disponibles para tratar los casos de hipersensibilidad y/o reacciones anafilácticas.
A los pacientes tratados con LEMTRADA se les debe suministrar la Tarjeta de Paciente y la Guía para el Paciente, así como informarles acerca de los riesgos de LEMTRADA (ver también Prospecto).
Posología
La dosis recomendada de LEMTRADA es de 12 mg/día, administrados por perfusión intravenosa en 2 cursos de tratamiento.
• Curso inicial: 12 mg/día durante 5 días consecutivos (dosis total de 60 mg)
• Segundo curso: 12 mg/día durante 3 días consecutivos (dosis total de 36 mg) administrados 12 meses después del curso inicial de tratamiento.
Las dosis que no se administren no deben administrarse el mismo día que una dosis programada.
Seguimiento de los pacientes
Se recomienda una terapia de 2 cursos de tratamiento (ver posología) con un seguimiento de seguridad de los pacientes desde_el inicio del tratamiento hasta 48 meses después de la última perfusión (ver sección 4.4).
Pretratamiento
Los pacientes deben pre-tratarse con corticoesteroides inmediatamente antes de la administración de LEMTRADA en cada uno de los 3 primeros días de cualquier curso de tratamiento. En los ensayos clínicos, los pacientes fueron pre-tratados con 1.000 mg de metilprednisolona durante los 3 primeros días de cada curso de tratamiento con LEMTRADA.
De forma adicional, puede considerarse el pretratamiento con antihistamínicos y/o antipiréticos antes de la administración de LEMTRADA.
Debe administrarse profilaxis oral para la infección por herpes a todos los pacientes desde el primer día de cada curso de tratamiento hasta, como mínimo, 1 mes después del tratamiento con LEMTRADA (ver también 'Infecciones' en la sección 4.4). En los ensayos clínicos, se administró a los pacientes aciclovir 200 mg dos veces al día o equivalente.
Pacientes de edad avanzada
Los ensayos clínicos no incluyeron pacientes con edades superiores a 55 años. No se ha determinado si responden de forma diferente a los pacientes de menor edad.
Pacientes con insuficiencia hepática o renal
No se ha realizado ningún estudio en pacientes que padezcan insuficiencia hepática o renal.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de LEMTRADA en niños con EM de 0 a 18 años de edad. No existe una recomendación de uso específica para alemtuzumab en niños de 0 a menos de 10 años para el tratamiento de la esclerosis múltiple. No se dispone de datos.
Forma de administración
LEMTRADA debe diluirse antes de la perfusión. La solución diluida debe administrarse por perfusión intravenosa durante un periodo de unas 4 horas.
Para consultar las instrucciones de dilución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Infección por virus de la inmunodeficiencia humana (VIH).
4.4 Advertencias y precauciones especiales de empleo
No se recomienda el uso de LEMTRADA en pacientes con formas no activas de la enfermedad o en aquellos que estén estables con su tratamiento actual.
A los pacientes tratados con LEMTRADA se les debe suministrar el prospecto, la Tarjeta de Paciente y la Guía para el Paciente. Antes del tratamiento deberá informarse al paciente de los riesgos y los beneficios, así como de la necesidad de comprometerse a un seguimiento de 48 meses tras la última administración de LEMTRADA.
Autoinmunidad
El tratamiento puede dar lugar a la formación de autoanticuerpos y aumentar el riesgo de enfermedades de origen autoinmune incluyendo: púrpura trombocitopénica inmune (PTI), trastornos de tiroides o, raramente, nefropatías (por ejemplo, enfermedad por anticuerpos anti-membrana basal glomerular). Se debe tener cuidado en pacientes con enfermedades autoinmunes previas distintas de la EM, aunque los datos disponibles sugieren que no se produce un empeoramiento de las enfermedades autoinmunes preexistentes tras el tratamiento con alemtuzumab.
Púrpura trombocitopénica inmune (PTI)
Se han observado casos graves de PTI en aproximadamente un 1% de los pacientes tratados en ensayos clínicos controlados en EM. En un ensayo clínico controlado en pacientes con EM, un paciente desarrolló PTI que no se detectó antes de la implementación de los requisitos de controles sanguíneos mensuales y falleció de hemorragia intracerebral. La aparición de PTI normalmente se ha dado entre 14 y 36 meses después de la primera administración. Los síntomas de PTI pueden incluir (pero sin limitarse a) tendencia a la formación de hematomas, petequias, sangrado mucocutáneao espontáneo (por ejemplo, epistaxis, hemoptisis), sangrado menstrual irregular o más abundante de lo normal. La hemoptisis puede ser también indicativa de enfermedad anti-MBG (ver a continuación) y debe realizarse un diagnóstico diferencial adecuado. Recuerde al paciente que debe estar alerta ante cualquier síntoma que pueda experimentar y que debe buscar consejo médico en caso de duda.
Antes de iniciar el tratamiento deben realizarse recuentos sanguíneos completos con fórmula leucocitaria completa y a partir de entonces a intervalos mensuales hasta 48 meses después de la última perfusión. Después de este periodo, las pruebas deben realizarse en base a los hallazgos clínicos sugestivos de PTI. Si se sospecha de PTI, debe realizarse un recuento sanguíneo completo de forma inmediata.
Si se confirma la aparición de PTI, deberá iniciarse rápidamente una intervención médica adecuada, incluyendo la consulta inmediata a un especialista. Los datos de los ensayos clínicos de EM han mostrado que el cumplimiento de los requisitos de monitorización hematológica y la educación sobre los signos y síntomas de la PTI han llevado a la detección y el tratamiento tempranos de la PTI, consiguiendo que la mayoría de los casos respondan al tratamiento médico de primera línea.
Se desconoce el riesgo potencial asociado a la reinstauración del tratamiento con LEMTRADA después de la aparición de PTI.
Nefropatías
Se observaron nefropatías, incluyendo la enfermedad por anticuerpos anti-membrana basal glomerular (anti-MBG), en un 0,3% de los pacientes en los ensayos clínicos en EM y, normalmente, ocurrieron en los 39 meses siguientes a la última administración de LEMTRADA. En los ensayos clínicos, hubo dos casos de enfermedad anti-MBG. Ambos casos fueron graves, se identificaron pronto en los controles clínicos y de laboratorio y respondieron bien al tratamiento.
Las manifestaciones clínicas de nefropatía pueden incluir una elevación de la creatinina en suero, hematuria y/o proteinuria. Aunque no se observó en los ensayos clínicos, puede producirse hemorragia alveolar con la enfermedad anti-MBG. La hemoptisis puede ser también indicativa de PTI (ver anteriormente) y debe realizarse un diagnóstico diferencial adecuado. Se debe recordar al paciente que debe estar alerta ante cualquier síntoma que pueda experimentar y que debe buscar consejo médico en caso de duda. La enfermedad anti-MBG puede provocar fallo renal con necesidad de diálisis y/o transplante si no se trata rápidamente y puede ser una amenaza para la vida si no se trata.
Antes de iniciar el tratamiento deben obtenerse niveles de creatina en suero y, a partir de entonces, a intervalos mensuales hasta 48 meses después de la última perfusión. Deben realizarse analíticas de orina con microscopía antes del inicio y a intervalos mensuales a partir de entonces hasta 48 meses después de la última perfusión. La observación de cambios clínicamente significativos respecto al nivel basal en la creatinina en suero, hematuria sin explicación y/o proteinuria requieren evaluación adicional para detectar nefropatías, incluyendo la consulta inmediata a un especialista. La detección y el tratamiento tempranos de las nefropatías pueden reducir el riesgo de resultados desfavorables. Después de este periodo, las pruebas deben realizarse en base a los hallazgos clínicos sugestivos de nefropatías.
Se desconoce el riesgo potencial asociado a la reinstauración del tratamiento con LEMTRADA después de la aparición de nefropatías.
Trastornos de tiroides
Se observaron trastornos autoinmunes de tiroides en un 36% estimado de los pacientes tratados con LEMTRADA 12 mg en los ensayos clínicos en EM en los 48 meses siguientes a la primera administración de LEMTRADA. La incidencia de acontecimientos tiroideos fue superior en los pacientes con antecedentes de trastornos de tiroides tanto en los grupos de tratamiento con LEMTRADA como con interferón beta 1a (IFNB-1a). En pacientes con trastorno de tiroides en curso, LEMTRADA debe administrarse si los posibles beneficios justifican los posibles riesgos. Los trastornos autoinmunes de tiroides observados incluyeron hipertiroidismo o hipotiroidismo. La mayoría de los acontecimientos fueron de intensidad leve a moderada. Antes de la autorización, ocurrieron acontecimientos graves en <1% de los pacientes. Sólo la enfermedad de Basedow (también conocida como enfermedad de Graves), el hipertiroidismo y el hipotiroidismo ocurrieron en más de 1 paciente. La mayoría de los acontecimientos tiroideos fueron tratados con tratamiento médico convencional, aunque algunos pacientes requirieron intervención quirúrgica. En los ensayos clínicos, se permitió que los pacientes que habían desarrollado acontecimientos tiroideos volvieran a recibir tratamiento con LEMTRADA. Aunque la experiencia es limitada, en general los pacientes que volvieron a recibir tratamiento no experimentaron un empeoramiento de la intensidad de los trastornos de tiroides. Debe valorarse de forma individual la repetición del tratamiento con LEMTRADA teniendo en cuenta la condición clínica de cada paciente.
Deben realizarse pruebas de función tiroidea, como los niveles de hormona estimulante del tiroides, antes de iniciar el tratamiento y cada 3 meses a partir de entonces hasta 48 meses después de la última perfusión. Después de este periodo, las pruebas deben realizarse basadas en los hallazgos clínicos que sugieran una disfunción del tiroides.
La enfermedad tiroidea supone un riesgo especial en el caso de mujeres embarazadas (ver sección 4.6).
En los ensayos clínicos, el estado de los anticuerpos contra la peroxidasa tiroidea (anti-TPO) del paciente antes del tratamiento no fue indicativo del desarrollo de un acontecimiento adverso relacionado con el tiroides. La mitad de los pacientes que dieron positivo al inicio y un cuarto de los pacientes que dieron negativo al inicio en anticuerpos anti-TPO desarrollaron un acontecimiento relacionado con el tiroides. La amplia mayoría (aproximadamente el 80%) de los pacientes que presentaron un acontecimiento relacionado con el tiroides tras el tratamiento habían dado negativo en anticuerpos anti-TPO al inicio. Así, independientemente del estado de los anticuerpos anti-TPO en el pretratamiento, los pacientes pueden desarrollar un acontecimiento adverso relacionado con el tiroides y deberán realizarse todas las pruebas -analíticas de forma periódica tal y como se ha descrito anteriormente.
Citopenias
De forma poco frecuente, se han notificado citopenias autoinmunes como neutropenia, anemia hemolítica y pancitopenia en ensayos clínicos en EM. Los resultados de los recuentos sanguíneos completos (ver anteriormente en PTI) deben utilizarse para detectar las citopenias. Si se confirma la aparición de una citopenia, debe iniciarse rápidamente una intervención médica adecuada, incluyendo la consulta a un especialista.
Reacciones asociadas a la perfusión (RAP)
En los ensayos clínicos controlados, las reacciones asociadas a la perfusión (RAP) se definieron como cualquier acontecimiento adverso ocurrido durante o en las 24 horas siguientes a la perfusión de LEMTRADA. La mayoría podían deberse a la liberación de citoquinas durante la perfusión. La mayoría de los pacientes tratados con LEMTRADA en ensayos clínicos controlados en EM experimentaron RAP leves a moderadas durante y/o hasta 24 horas después de la administración de LEMTRADA 12 mg que, a menudo, incluyeron cefalea, erupción, pirexia, náuseas, urticaria, prurito, insomnio, escalofríos, rubefacción, fatiga, disnea, disgeusia, malestar torácico, erupción generalizada, taquicardia, bradicardia, dispepsia, mareo y dolor. En el 3% de los pacientes se produjeron reacciones graves que incluyeron casos de pirexia, urticaria, fibrilación auricular, náuseas, malestar torácico e hipotensión. Pueden aparecer manifestaciones clínicas de anafilaxis similares a las manifestaciones clínicas de las reacciones asociadas a la perfusión, pero tendrán tendencia a ser más severas o potencialmente una amenaza para la vida. Las reacciones atribuidas a anafilaxis se han notificado raramente, en contraste con las reacciones asociadas con la perfusión.
Se recomienda pretratar a los pacientes para mejorar los efectos de las reacciones a la perfusión (ver sección 4.2). La mayoría de los pacientes de los ensayos clínicos recibieron antihistamínicos y/o antipiréticos antes de, al menos, una perfusión de LEMTRADA. Los pacientes pueden sufrir RAP a pesar del pretratamiento. Se recomienda observar si hay reacciones durante la perfusión de LEMTRADA y hasta 2 horas después de ésta. Si se produjera una RAP, proporcione el tratamiento sintomático adecuado según sea necesario. Si la perfusión no fuera bien tolerada, su duración podría extenderse. En caso de reacciones graves a la perfusión, debe considerarse la interrupción inmediata de la perfusión intravenosa. En los ensayos clínicos, fueron muy raras las reacciones graves o de anafilaxia que necesitaron la interrupción del tratamiento.
Los médicos deben conocer el historial cardiaco del paciente ya que las reacciones asociadas a la perfusión pueden incluir síntomas cardiacos como taquicardia.
Debe haber recursos disponibles para tratar las reacciones graves o de anafilaxia.
Infecciones
Se produjeron infecciones en el 71% de los pacientes tratados con LEMTRADA 12 mg en comparación con el 53% de los pacientes tratados con interferón beta-1a [IFNB 1a] (44 mcg 3 veces a la semana) en ensayos clínicos controlados en EM de hasta 2 años de duración, y fueron predominantemente de intensidad leve a moderada. Las infecciones que se dieron con más frecuencia en los pacientes tratados con LEMTRADA que en los pacientes con IFNB 1a incluyeron nasofaringitis, infección del tracto urinario, infección del tracto respiratorio superior, sinusitis, herpes oral, gripe y bronquitis. Se produjeron infecciones graves en el 2,7% de los pacientes tratados con LEMTRADA en comparación con el 1% de los pacientes tratados con IFNB-1a en ensayos clínicos controlados en EM. Las infecciones graves del grupo de LEMTRADA incluyeron: apendicitis, gastroenteritis, neumonía, herpes zóster e infección dental. Las infecciones tuvieron, en general, una duración típica y se resolvieron con tratamiento médico convencional.
Se produjeron infecciones graves de varicela zóster, incluyendo varicela primaria y reactivación de varicela zóster, con más frecuencia en pacientes tratados con LEMTRADA 12 mg (0,3%) en los ensayos clínicos en comparación con los pacientes tratados con IFNB-1a (0%). También se ha notificado infección cervical por virus del papiloma humano (VPH), incluyendo displasia cervical, en pacientes tratadas con LEMTRADA 12 mg (2%). Se recomienda realizar pruebas anuales de detección del VPH a las pacientes.
Se han notificado casos de tuberculosis en pacientes tratados con LEMTRADA e IFNB-1a en los ensayos clínicos controlados. Se han notificado casos de tuberculosis activa y latente en el 0,3% de los pacientes tratados con LEMTRADA, con más frecuencia en las regiones endémicas. Antes de iniciar el tratamiento, es necesario evaluar a todos los pacientes para detectar una posible infección activa o inactiva (“latente”) por tuberculosis, según la normativa local.
Se han notificado casos de listeriosis/’Listeria meningitis en pacientes tratados con LEMTRADA, generalmente en el mes de perfusión de LEMTRADA. Para reducir este riesgo, los pacientes que estén recibiendo LEMTRADA deben evitar la ingestión de carnes crudas o poco hechas, quesos frescos y productos lácteos no pasteurizados durante al menos un mes después del tratamiento con LEMTRADA.
Se produjeron infecciones superficiales por hongos, especialmente candidiasis oral y vaginal, de forma más frecuente en los pacientes tratados con LEMTRADA (12%) que en los pacientes tratados con IFNB-1a (3%) en los ensayos clínicos controlados en EM.
Los médicos deben considerar retrasar el inicio de la administración de LEMTRADA en pacientes con infección activa hasta que ésta esté completamente controlada.
Debe iniciarse la profilaxis con un agente oral contra el herpes desde el primer día de tratamiento con LEMTRADA y hasta, como mínimo, 1 mes después de cada curso de tratamiento. En los ensayos clínicos se administró a los pacientes aciclovir 200 mg dos veces al día o equivalente.
LEMTRADA no se ha administrado para el tratamiento de la EM de forma simultánea o después de tratamientos inmunosupresores o antineoplásicos. Al igual que con otros tratamientos inmunomoduladores, deben tenerse en cuenta los posibles efectos combinados sobre el sistema inmunológico del paciente al considerar la administración de LEMTRADA. El uso simultáneo de LEMTRADA con alguno de estos tratamientos podría aumentar el riesgo de inmunosupresión.
No hay datos disponibles sobre la relación de LEMTRADA con la reactivación del virus de la hepatitis B (VHB) o el virus de la hepatitis C (VHC) ya que los pacientes con infecciones crónicas o activas fueron excluidos de los ensayos clínicos. Debe considerarse el cribado de pacientes con riesgo alto de infección por VHB y/o VHC antes de iniciar el tratamiento con LEMTRADA y se debe tener precaución a la hora de prescribir LEMTRADA a pacientes identificados como portadores de VHB y/o VHC ya que estos pacientes podrían estar en riesgo de daño hepático irreversible por la posible reactivación del virus como consecuencia de su estado preexistente.
Malignidad
Al igual que con otros tratamientos inmunomoduladores, se debe tener precaución al iniciar el tratamiento con LEMTRADA en pacientes con enfermedad maligna preexistente y/o en desarrollo. Actualmente se desconoce si alemtuzumab confiere un mayor riesgo de desarrollar tumores malignos de tiroides, ya que la autoinmunidad del tiroides en sí misma puede ser un factor de riesgo para los tumores malignos de tiroides.
Anticoncepción
Se observaron transferencia placentaria y posible actividad farmacológica de LEMTRADA en ratones durante la gestación y tras el parto. Las mujeres en edad fértil deben utilizar métodos anticonceptivos eficaces durante el tratamiento y hasta 4 meses después de un curso de tratamiento con LEMTRADA (ver sección 4.6).
Vacunas
Se recomienda que los pacientes hayan completado los requisitos locales de inmunización al menos 6 semanas antes del tratamiento con LEMTRADA. No se ha estudiado la capacidad de las vacunas de generar respuesta inmune tras el tratamiento con LEMTRADA.
No se ha estudiado formalmente la seguridad de la inmunización con vacunas víricas basadas en virus vivos tras un curso de tratamiento con LEMTRADA en los ensayos clínicos controlados en EM y no se deben administrar a pacientes con EM que hayan recibido recientemente un curso de tratamiento con LEMTRADA.
Vacunación y pruebas de detección de anticuerpos del virus de la varicela zóster Al igual que con cualquier medicamento modulador del sistema inmune, antes de iniciar un curso de tratamiento con LEMTRADA, los pacientes sin antecedentes de varicela o que no estén vacunados contra el virus de la varicela zóster (VVZ) deben realizarse pruebas de detección de anticuerpos del VVZ. Debe considerarse la vacunación contra el VVZ en los pacientes con anticuerpos negativos antes de iniciar el tratamiento con LEMTRADA. Para permitir el efecto total de la vacunación contra VVZ, posponga el tratamiento con LEMTRADA hasta 6 semanas después de la vacunación.
Pruebas de laboratorio recomendadas para monitorizar a los pacientes
Deben realizarse pruebas de laboratorio a intervalos periódicos durante los 48 meses siguientes al último curso de tratamiento con LEMTRADA para controlar que no haya síntomas tempranos de enfermedad autoinmune:
• Recuento sanguíneo completo con fórmula leucocitaria completa (antes de iniciar el tratamiento y a intervalos mensuales a partir de entonces)
• Niveles de creatinina en suero (antes de iniciar el tratamiento y a intervalos mensuales a partir de entonces)
• Analítica de orina con microscopía (antes de iniciar el tratamiento y a intervalos mensuales a partir de entonces)
• Una prueba de función tiroidea, como nivel de hormona estimulante del tiroides (antes de iniciar el tratamiento y cada 3 meses a partir de entonces)
Después de este periodo, cualquier hallazgo clínico que sugiera una nefropatía o una disfunción del tiroides requerirá pruebas adicionales.
Información del uso de alemtuzumab antes de la autorización de comercialización de LEMTRADA fuera de los estudios patrocinados por la compañía
Las siguientes reacciones adversas se identificaron antes del registro de LEMTRADA, durante el uso de alemtuzumab para el tratamiento de la leucemia linfocítica crónica de células B (LLC-B), así como para el tratamiento de otros trastornos, normalmente a dosis mayores y más frecuentes (por ejemplo, 30 mg) que las recomendadas en el tratamiento de la EM. Dado que estas reacciones fueron notificadas voluntariamente por una población de tamaño desconocido, no siempre es posible calcular de forma fiable su frecuencia o establecer una relación causal con la exposición a alemtuzumab.
Enfermedad autoinmune
Los acontecimientos autoinmunes que se han notificado en pacientes tratados con alemtuzumab incluyeron neutropenia, anemia hemolítica (incluyendo un caso mortal), hemofilia adquirida, enfermedad anti-MBG y enfermedad tiroidea. Se han notificado fenómenos autoinmunes graves y, en ocasiones mortales, incluyendo anemia hemolítica autoinmune, trombocitopenia autoinmune, anemia aplásica, síndrome de Guillain-Barré y polirradiculoneuropatía crónica inflamatoria desmielinizante, en pacientes sin EM tratados con alemtuzumab para otras patologías. Se ha notificado una prueba de Coombs positiva en un paciente oncológico tratado con alemtuzumab. Se ha notificado un acontecimiento mortal de enfermedad de injerto contra huésped asociada a transfusión en un paciente oncológico tratado con alemtuzumab.
Reacciones asociadas a la perfusión
Se han observado RAP graves, y en ocasiones mortales, incluyendo broncoespasmo, hipoxia, síncope, infiltrados pulmonares, síndrome de sufrimiento respiratorio agudo, parada respiratoria, infarto de miocardio, arritmias, insuficiencia cardiaca aguda y parada cardiaca en pacientes sin EM tratados con alemtuzumab con dosis mayores y más frecuentes que las utilizadas para EM. También se ha notificado anafilaxia severa y otras reacciones de hipersensibilidad, incluyendo shock anafiláctico y angioedema.
Infecciones e infestaciones
Se han notificado infecciones graves, y en ocasiones mortales, por virus, bacterias, protozoos y hongos, incluyendo las debidas a infecciones latentes o reactivadas, en pacientes sin EM tratados con alemtuzumab para otras patologías con dosis mayores y más frecuentes que las utilizadas para EM. Se han notificado casos de leucoencefalopatía multifocal progresiva (LMP) en pacientes con LLC-B con o sin tratamiento con alemtuzumab. La frecuencia de LMP en pacientes con LLC-B tratados con alemtuzumab no es mayor que la frecuencia habitual.
Trastornos de la sangre y del sistema linfático
Se han notificado reacciones de sangrado intenso en pacientes sin EM.
Trastornos cardiacos
Se han notificado casos de insuficiencia cardiaca congestiva, cardiomiopatía y fracción de eyección reducida en pacientes sin EM tratados con alemtuzumab para otras patologías y tratados previamente con agentes potencialmente cardiotóxicos.
Trastornos linfoproliferativos asociados a virus de Epstein-Barr
Se han observado trastornos linfoproliferativos asociados a virus de Epstein-Barr fuera de los estudios patrocinados por la compañía.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios formales de interacción medicamentosa con LEMTRADA utilizando la dosis recomendada en pacientes con EM. En un ensayo clínico controlado con pacientes con EM tratados recientemente con interferón beta y acetato de glatirámero fue necesario interrumpir el tratamiento 28 días antes de iniciar el tratamiento con LEMTRADA.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil
Las concentraciones en suero fueron bajas o indetectables dentro de los 30 días, aproximadamente, después de cada curso de tratamiento. Por tanto, las mujeres en edad fértil deben utilizar métodos anticonceptivos eficaces cuando reciban un curso de tratamiento con LEMTRADA y durante los 4 meses posteriores a ese curso de tratamiento.
Embarazo
Hay datos limitados relativos al uso de LEMTRADA en mujeres embarazadas. Sólo debe administrarse LEMTRADA durante el embarazo si los posibles beneficios justifican los riesgos potenciales para el feto.
Se sabe que la IgG humana traspasa la barrera placentaria; alemtuzumab podría traspasar también la barrera placentaria y, por tanto, suponer un riesgo para el feto. Los estudios realizados en animales han mostrado toxicidad en la reproducción (ver sección 5.3). Se desconoce si alemtuzumab puede causar daños fetales si se administra a mujeres embarazadas o si puede afectar a la capacidad reproductora.
Las enfermedades de tiroides (ver sección 4.4 Trastornos de tiroides) suponen un riesgo especial para las mujeres embarazadas. Si no se trata el hipotiroidismo durante el embarazo, aumenta el riesgo de aborto espontáneo y de que el feto resulte afectado con problemas como retraso mental y enanismo. En madres con la enfermedad de Graves, los anticuerpos receptores de la hormona estimulante del tiroides se pueden transmitir al feto en desarrollo y causar una enfermedad de Graves neonatal transitoria.
Lactancia
Se detectó alemtuzumab en la leche y en las crías de ratones en periodo de lactancia.
Se desconoce si alemtuzumab se excreta a la leche humana. No se puede excluir el riesgo para niños lactantes. Por tanto, la lactancia materna debe interrumpirse durante cada curso de tratamiento con LEMTRADA y durante 4 meses después de la última perfusión de cada curso de tratamiento. No obstante, las ventajas de la inmunidad que confiere la leche materna pueden superar los riesgos de una posible exposición del lactante a alemtuzumab.
Fertilidad
No existen datos clínicos adecuados sobre seguridad sobre el efecto de LEMTRADA en la fertilidad. En un subestudio con 13 pacientes varones tratados con alemtuzumab (en tratamiento con 12 mg o 24 mg), no hubo evidencia de aspermia, azoospermia, recuento de espermatozoides sistemáticamente reducido, trastornos de la movilidad o un aumento de anormalidades morfológicas del esperma.
Se sabe que el CD52 está presente en los tejidos reproductivos humanos y de roedores. Los datos sobre animales han mostrado efectos en la fertilidad de ratones humanizados (ver sección 5.3), no obstante se desconoce si existe un posible impacto en la fertilidad humana durante el periodo de exposición, según los datos disponibles.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios sobre los efectos de LEMTRADA sobre la capacidad para conducir y utilizar máquinas.
La mayoría de pacientes experimentan RAP que se producen durante o en las 24 horas siguientes al tratamiento con LEMTRADA. Algunas RAP (por ejemplo, mareo) podrían afectar de forma temporal a la capacidad del paciente para conducir y utilizar máquinas y se debe tener precaución hasta que se resuelvan.
4.8 Reacciones adversas
Resumen del perfil de seguridad
En un análisis conjunto de estudios clínicos controlados, la población estuvo constituida por un total de 1.188 pacientes con EM remitente recurrente (EMRR) tratados con LEMTRADA (12 mg o 24 mg) con un resultado de 2.363 pacientes-años de seguimiento de seguridad y un seguimiento medio de 24 meses.
Las reacciones adversas más importantes son autoinmunidad (PTI, trastornos de tiroides, nefropatías, citopenias), RAP e infecciones. Se describen en la sección 4.4.
Las reacciones adversas más frecuentes con LEMTRADA (en >20% de los pacientes) son erupción, cefalea, pirexia e infecciones del tracto respiratorio.
Tabla de las reacciones adversas
La tabla siguiente se basa en los datos de seguridad acumulados hasta 24 meses de pacientes con EMRR tratados con LEMTRADA 12 mg/día durante 5 días consecutivos al inicio del estudio y durante 3 días consecutivos en el mes 12 del estudio. Las reacciones adversas que se dieron en un >0,5% de los pacientes se incluyen por grupo sistémico y según el término preferido del diccionario MedDRA. Las frecuencias se definen según la siguiente convención: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100). Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 1: Reacciones adversas en el estudio 1, 2 y 3 observadas en el >0,5% de los pacientes tratados con LEMTRADA 12 mg
|
Sistema de Clasificación de Órganos |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Infecciones e infestaciones |
Infección del tracto respiratorio superior, infección del tracto urinario |
Infecciones del tracto respiratorio inferior, herpes zóster, gastroenteritis, herpes oral, candidiasis oral, candidiasis vulvovaginal, gripe, infección de oído |
Infección dental, herpes genital, onicomicosis |
|
Trastornos de la sangre y del sistema linfático |
Linfopenia, leucopenia |
Linfadenopatía |
Púrpura trombocitopénica inmune, trombocitopenia con disminución de hemoglobina, hematocrito disminuido |
|
Trastornos del sistema inmunológico |
Síndrome de liberación de citoquinas | ||
|
Trastornos endocrinos |
Enfermedad de Basedow, hipertiroidismo, tiroiditis autoinmune, hipotiroidismo, bocio, anticuerpos antitiroideos positivos | ||
|
Trastornos psiquiátricos |
Insomnio*, ansiedad |
Depresión | |
|
Trastornos del sistema nervioso |
Cefalea* |
Recaída de EM, mareo*, hipoestesia, parestesia, temblor, disgeusia* |
Alteración sensitiva, hiperestesia |
|
Trastornos oculares |
Visión borrosa |
Conjuntivitis | |
|
Trastornos del oído y del laberinto |
Vértigo | ||
|
Trastornos cardiacos |
Taquicardia*, bradicardia*, palpitaciones | ||
|
Trastornos vasculares |
Rubefacción* |
Hipotensión*, hipertensión | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea*, tos, epistaxis, dolor orofaríngeo |
Opresión en la garganta, hipo, irritación de garganta | |
|
Trastornos gastrointestinales |
Náusea* |
Dolor abdominal, vómitos, diarrea, dispepsia*, estomatitis |
Estreñimiento, enfermedad por reflujo gastroesofágico, hemorragia gingival, disfagia |
|
Trastornos hepatobiliares |
Aspartato aminotransferasa elevada | ||
|
Trastornos de la piel y del tejido subcutáneo |
Urticaria*, erupción*, prurito* |
Erupción generalizada*, eritema, equimosis, alopecia, hiperhidrosis, acné |
Ampollas, sudores nocturnos |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Mialgia, debilidad muscular, artralgia, dolor de espalda, dolor en una extremidad, espasmos musculares, cervicalgia | ||
|
Trastornos renales y urinarios |
Proteinuria, hematuria | ||
|
Trastornos del aparato reproductor y de la mama |
Menorragia, menstruación irregular |
Displasia cervical, amenorrea | |
|
Trastornos generales y alteraciones en el lugar de administración |
Pirexia*, fatiga* |
Malestar torácico*, escalofríos*, dolor*, edema periférico, astenia, enfermedad de tipo gripal, malestar general, dolor en el lugar de infusión | |
|
Exploraciones complementarias |
Disminución de peso | ||
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Contusión |
Descripción de reacciones adversas seleccionadas
Los términos marcados con un asterisco (*) en la Tabla 1 incluyen reacciones adversas notificadas como Reacciones Asociadas a la Perfusión. Las RAP también incluyen fibrilación auricular y anafilaxia que ocurren por debajo del corte del 0,5% para los acontecimientos relacionados (ver sección 4.4).
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
En los ensayos clínicos controlados, dos pacientes con EM recibieron de forma accidental hasta 60 mg de LEMTRADA (es decir, la dosis total para el curso inicial de tratamiento) en una sola perfusión y experimentaron reacciones graves (cefalea, erupción, además de hipotensión o taquicardia sinusal). Las dosis de LEMTRADA superiores a las probadas en los estudios clínicos pueden aumentar la intensidad y/o duración de las reacciones adversas asociadas a la perfusión o sus efectos inmunes.
No se conoce un antídoto para la sobredosis de alemtuzumab. El tratamiento consiste en interrumpir la administración del medicamento y proporcionar tratamiento de apoyo.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inmunosupresor selectivo, código ATC: L04AA34.
Mecanismo de acción
Alemtuzumab es un anticuerpo monoclonal humanizado derivado de ADN recombinante dirigido contra la glicoproteína de superficie celular CD 52 de 21-28 kD. Alemtuzumab es un anticuerpo IgG1 kappa con regiones constantes y marco de variable humana y regiones complementarias-determinantes de un anticuerpo monoclonal murino (rata). El anticuerpo tiene un peso molecular aproximado de 150 kD.
Alemtuzumab se une al CD52, un antígeno de superficie celular presente en grandes cantidades en los linfocitos T (CD3+) y B (CD19+) y, en menores cantidades, en los linfocitos citolíticos naturales (natural killer), monocitos y macrófagos. El antígeno CD52 se expresa poco o nada en los neutrófilos, células plasmáticas o células madre de la médula ósea. Alemtuzumab actúa a través de la citolisis celular dependiente de anticuerpos y la lisis mediada por el complemento tras la unión de la superficie celular con los linfocitos T y B.
No se ha elucidado por completo el mecanismo por el cual LEMTRADA ejerce sus efectos terapéuticos en la EM. No obstante, las investigaciones sugieren efectos inmunomoduladores a través de la depleción y repoblación de linfocitos, incluyendo:
- Alteraciones en el número, las proporciones y las propiedades de algunos grupos de linfocitos tras el tratamiento
- Aumento de la representación de grupos de linfocitos T reguladores
- Aumento de la representación de linfocitos T y B de memoria
- Efectos transitorios en la inmunidad innata de los componentes (es decir, neutrófilos, macrófagos y linfocitos citolíticos naturales (natural killer)
La reducción en el nivel de células B y T circulantes por LEMTRADA y posterior repoblación podría reducir la posibilidad de recaídas que, finalmente, retrasan la evolución de la enfermedad.
Efectos farmacodinámicos
LEMTRADA reduce los linfocitos T y B circulantes tras cada curso de tratamiento y los valores más bajos se observan 1 mes después del curso de tratamiento (el momento más temprano tras el tratamiento en estudios de fase 3). Los linfocitos se reponen con el tiempo, la recuperación total de las células B se completa en 6 meses. Los recuentos de linfocitos CD3+ y CD4+ aumentan hacia la normalidad más lentamente, pero en general no vuelven a los niveles iniciales 12 meses después del tratamiento. En aproximadamente el 40% de los pacientes, los recuentos de linfocitos totales alcanzan el límite normal más bajo (LNB) 6 meses después de cada curso de tratamiento y aproximadamente el 80% de los pacientes alcanzan el LNB de linfocitos 12 meses después de cada curso de tratamiento.
Los neutrófilos, monocitos, eosinófilos, basófilos y linfocitos citolíticos naturales (natural killer) solo se ven afectados por LEMTRADA de forma transitoria.
Eficacia clínica y seguridad
La seguridad y eficacia de LEMTRADA se evaluaron en 3 ensayos clínicos comparativos directos, aleatorizados, ciegos para el evaluador, en pacientes con EMRR.
Para los estudios 1 y 2, el diseño/demografía y los resultados del estudio se muestran en la Tabla 2 y la Tabla 3 respectivamente.
|
Tabla 2: Diseño del estudio y datos iniciales para los estudios 1 y 2 | ||
|
Estudio 1 |
Estudio 2 | |
|
Nombre del estudio |
CAMMS323 (CARE-MS I) |
CAMMS32400507 (CARE-MS II) |
|
Diseño del estudio | ||
|
Historial de la enfermedad |
Pacientes con EM activa, definida como al menos 2 recaídas en los 2 años anteriores. | |
|
Seguimiento |
2 años | |
|
Población del estudio |
Pacientes no tratados previamente |
Pacientes con respuesta inadecuada al tratamiento anterior* |
|
Datos iniciales | ||
|
Media de edad (años) |
33 |
35 |
|
Duración media/mediana de la enfermedad |
2/1,6 años |
4,5/3,8 años |
|
Duración media del tratamiento de EM anterior (>1 fármaco utilizado) |
Ninguno |
36 meses |
|
% que han recibido >2 tratamientos de EM anteriores |
No procede. |
28% |
|
Valor medio de EDSS al inicio |
2,0 |
2,7 |
* Definido como pacientes que hayan experimentado al menos 1 recaída durante el tratamiento con interferón beta o acetato de glatirámero tras haber estado en tratamiento con el medicamento durante, al menos, 6 meses.
|
Tabla 3: Criterios clínicos y de RMN de los estudios 1 y 2 | ||||
|
Estudio 1 |
Estudio 2 | |||
|
Nombre del estudio |
CAMMS323 (CARE-MS I) |
CAMMS32400507 (CARE-MS II) | ||
|
Criterios clínicos |
LEMTRADA 12 mg (N=376) |
SC IFNB-1a (N=187) |
LEMTRADA 12 mg (N=426) |
SC IFNB-1a (N=202) |
|
Índice de recaídas1 Índice anualizado de recaídas (ARR) (95% CI) |
0,18 (0,13, 0,23) |
0,39 (0,29, 0,53) |
0,26 (0,21, 0,33) |
0,52 (0,41, 0,66) |
|
Cociente de tasas (95% CI) Reducción del riesgo |
0,45 (0,32, 0,63) 54,9 (p<0,0001) |
0,51 (0,39, 0,65) 49,4 (p<0,0001) | ||
|
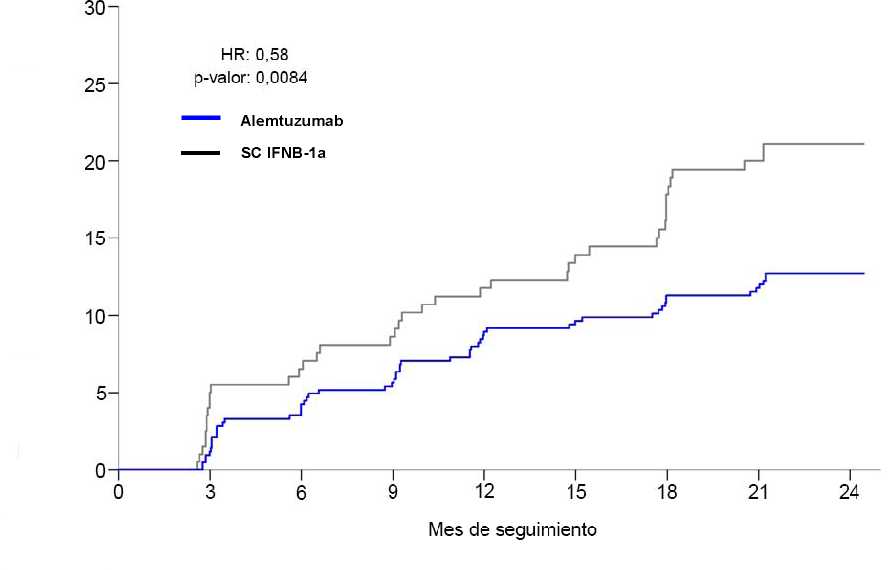
Discapacidad2 (acumulación de discapacidad sostenida [SAD] >6 meses1) Pacientes con SAD de 6 meses (95% CI) |
8,0% (5,7, 11,2) |
11,1% (7,3, 16,7) |
12,7% (9,9, 16,3) |
21,1% (15,9, 27,7) |
|
Cociente de riesgo (95% CI) |
0,70 (0,40, 1,23) (p=0,22) |
0,58 (0,38, 0,87) (p=0,0084) | ||
|
Pacientes sin recaídas en el Año 2 (95% CI) |
77,6% (72,9, 81,60) (p<0,0001) |
58,7% (51,1, 65,50) |
65,4% (60,6, 69,7) (p<0,0001) |
46,7% (39,5, 53,5) |
|
Cambio respecto al valor inicial en EDSS en el Año 2 Estimación (95% CI) |
-0,14 (-0,25, -0,02) (p=0,42) |
-0,14 (-0,29, 0,01) |
-0,17 (-0,29, -0,05) (p<0,0001) |
0,24 (0,07, 0,41) |
|
Criterios de RMN (0-2 años) | ||||
|
Cambio medio % en el volumen de la lesión RMN-T2 |
-9,3 (-19,6, -0,2) (p=0,31) |
-6,5 (-20,7, 2,5) |
-1,3 (p=0,14) |
-1,2 |
|
Pacientes con lesiones T2 nuevas o más grandes a lo largo del Año 2 |
48,5% (p=0,035) |
57,6% |
46,2% (p<0,0001) |
67,9% |
|
Pacientes con lesiones realzadas con gadolinio a lo largo del Año 2 |
15,4% (p=0,001) |
27,0% |
18,5% (p<0,0001) |
34,2% |
|
Pacientes con lesiones T1 hipointensas a lo largo del Año 2 |
24,0% (p=0,055) |
31,4% |
19,9% (p<0,0001) |
38,0% |
|
Cambio medio % en la fracción parenquimal del cerebro |
-0,867 (p<0,0001) |
-1,488 |
-0,615 (p=0,012) |
-0,810 |
|
1 Variables principales: ARR y SAD. Se declaraba que el estudio había tenido éxito si se cumplía al menos uno de las dos variables principales. 2 El tiempo transcurrido hasta el inicio del SAD (acumulación sostenida de la discapacidad) se definió como un incremento de, al menos, 1 punto en la Escala Expandida del Estado de Discapacidad (EDSS, por sus siglas en inglés) a partir de un valor inicial de EDSS > 1,0 (1,5 puntos de aumento para pacientes con valor inicial de EDSS de 0) que se mantuvo durante 6 meses. | ||||
Figura 1: Tiempo hasta la acumulación de discapacidad sostenida durante 6 meses en el Estudio 2
Porcentaje de pacientes con SAD
Gravedad de la recaída
En línea con el efecto en el índice de recaídas, los análisis complementarios del Estudio 1 (CAMMS323) mostraron que LEMTRADA 12 mg/día produjo un número significativamente menor de pacientes tratados con LEMTRADA que sufrieran recaídas graves (61% reducción, p=0,0056) y un número significativamente menor de recaídas que requirieran tratamiento con esteroides (58% reducción, p<0,0001) en comparación con IFNB-1a.
Los análisis complementarios del Estudio 2 (CAMMS32400507) mostraron que LEMTRADA 12 mg/día produjo un número significativamente menor de pacientes tratados con LEMTRADA que sufrieran recaídas graves (48% reducción, p=0,01216) y un número significativamente menor de recaídas que requirieran tratamiento con esteroides (56% reducción, p<0,0001) u hospitalización (55 % reducción, p=0,0045) en comparación con IFNB-1a.
Reducción mantenida de la discapacidad (RMD)
El tiempo transcurrido hasta el inicio de la RMD se definió como un descenso de, al menos, un punto en la escala expandida de estado de discapacidad (EDSS) a partir de un valor inicial de EDSS > 2 que se mantuvo durante 6 meses. RMD es la medida de la mejora mantenida de la discapacidad. El 29% de los pacientes tratados con LEMTRADA alcanzaron la RMD en el estudio 2, mientras que solo el 13% de los pacientes tratados con IFNB-1a subcutáneo la alcanzaron. La diferencia era estadísticamente significativa
(p=0,0002).
El Estudio 3 (fase 2 estudio CAMMS223) evaluó la seguridad y la eficacia de LEMTRADA en pacientes con EMRR a lo largo de 5 años. Los pacientes tenían una EDSS entre 0-3,0, al menos 2 episodios clínicos de EM en los 2 años anteriores y >1 lesión realzada con gadolinio al inicio del estudio. Los pacientes no habían recibido tratamiento anterior para la EM. Los pacientes fueron tratados con LEMTRADA 12 mg/día (N=108) o 24 mg/día (N=108) administrado una vez al día durante 5 días en el Mes 0 y durante 3 días en el Mes 12 o con IFNB-1a subcutáneo 44 pg (N=107) administrado 3 veces a la semana durante 3 años.
Cuarenta y seis pacientes recibieron un tercer curso de tratamiento con LEMTRADA 12 mg/día o 24/mg día durante 3 días en el Mes 24.
A los 3 años, LEMTRADA redujo el riesgo de SAD de 6 meses en un 76% (cociente de riesgo 0,24 [95% CI: 0,110, 0,545], p<0,0006) y redujo el ARR un 67% (cociente de tasas 0,33 [95% CI: 0,196, 0,552], p<0,0001) en comparación con IFNB-1a subcutáneo. Alemtuzumab 12 mg/día produjo unos valores de EDSS significativamente menores (mejora comparada con los datos iniciales) a lo largo de 2 años de seguimiento, en comparación con IFNB-1a (p<0,0001).
A los 5 años, LEMTRADA redujo el riesgo de SAD 69% (cociente de riesgo 0,31 [95% CI: 0,161, 0,598], p=0,0005) y redujo el ARR un 66% (cociente de tasas 0,34 [95% CI: 0,202, 0,569], p<0,0001) en comparación con IFNB-1a subcutáneo.
En un seguimiento abierto de los ensayos clínicos con LEMTRADA, algunos pacientes recibieron tratamiento adicional "según necesidades" con LEMTRADA tras la evidencia documentada de una reanudación de la actividad de la EM. Los cursos adicionales de tratamiento con LEMTRADA se administraron con 12 mg/día durante 3 días consecutivos (dosis total de 36 mg) al menos 12 meses después del curso de tratamiento anterior. Los beneficios y riesgos de >2 cursos de tratamiento no se han establecido por completo, pero los resultados sugieren que el perfil de seguridad no parece cambiar con los cursos de tratamiento adicionales. Si van a administrarse fases de tratamiento adicionales, deberán administrarse al menos 12 meses después de la fase anterior.
Inmunogenicidad
Al igual que con todas las proteínas terapéuticas, hay posibilidad de inmunogenicidad. Los datos reflejan el porcentaje de pacientes cuyos resultados se consideraron positivos para anticuerpos de alemtuzumab mediante una prueba de inmunoadsorción enzimática (ELISA) y confirmados por un ensayo de unión competitiva. Las muestras positivas se volvieron a evaluar para buscar evidencia de inhibición in vitro mediante un ensayo de citometría de flujo. Se tomaron muestras de suero a los pacientes de los ensayos clínicos controlados de EM 1, 3 y 12 meses después de cada curso de tratamiento para determinar si había anticuerpos anti-alemtuzumab. Aproximadamente el 85% de los pacientes que recibieron LEMTRADA dieron positivo en las pruebas de anticuerpos anti-alemtuzumab durante el estudio y el 92% de estos pacientes dieron positivo también en las pruebas de anticuerpos que inhiben la fijación de LEMTRADA in vitro. Los pacientes que desarrollaron anticuerpos anti-alemtuzumab lo hicieron a los 15 meses de la exposición inicial. No hay relación entre la presencia de anticuerpos anti-alemtuzumab o anticuerpos anti-alemtuzumab inhibidores y una reducción de la eficacia, un cambio en la farmacodinámica, o la aparición de reacciones adversas, incluyendo las reacciones asociadas a la perfusión.
La incidencia de anticuerpos depende en gran parte de la sensibilidad y la especificidad del ensayo.
Además, la incidencia observada de positivos para anticuerpos (incluyendo anticuerpos inhibidores) en un ensayo puede verse influida por varios factores como la metodología del ensayo, la manipulación de las muestras, el horario de manipulación de las muestras, fármacos utilizados de forma simultánea y enfermedades subyacentes. Por estos motivos, la comparación de la incidencia de anticuerpos de LEMTRADA con la incidencia de anticuerpos de otros productos puede ser engañosa.
Población pediátrica.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con alemtuzumab en niños de 0 a menos de 10 años en tratamiento por esclerosis múltiple (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con LEMTRADA en uno o más grupos de la población pediátrica con EMRR (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
La farmacocinética de LEMTRADA se evaluó en un total de 216 pacientes con EMRR que recibieron perfusiones intravenosas de 12 mg/días o 24 mg/día durante 5 días consecutivos, seguidos de 3 días consecutivos 12 meses después del curso de tratamiento inicial. Las concentraciones en suero aumentaron con cada dosis consecutiva en los cursos de tratamiento. Las mayores concentraciones se observaron tras la última perfusión de un curso. La administración de 12 mg/día provocó una Cmáx media de 3014 ng/ml el Día 5 del curso de tratamiento inicial y de 2276 ng/ml el Día 3 del segundo curso de tratamiento. La vida media de las partículas alfa se aproximó a 4-5 días y fue comparable entre cursos de tratamiento produciendo concentraciones bajas o indetectables en suero a los 30 días, aproximadamente, de cada curso de tratamiento.
Alemtuzumab es una proteína cuya ruta metabólica se espera que sea la degradación en pequeños péptidos y aminoácidos individuales por parte de enzimas proteolíticas ampliamente distribuidas. No se han realizado estudios clásicos de biotransformación.
No se pueden sacar conclusiones a partir de los datos disponibles sobre el efecto de la raza y el sexo en la farmacocinética de LEMTRADA. La farmacocinética de LEMTRADA no se ha estudiado en pacientes de más de 55 años.
5.3 Datos preclínicos sobre seguridad
Carcinogénesis y mutagénesis
No se han realizado estudios para valorar el potencial carcinogénico o mutagénico de alemtuzumab. Fertilidad y reproducción
El tratamiento con alemtuzumab intravenoso en dosis de hasta 10 mg/kg/día, administrado durante 5 días consecutivos (AUC de 7,1 veces la exposición humana en la dosis diaria recomendada) no tuvo efecto en la fertilidad ni la función reproductora en ratones transgénicos macho huCD52. El número de espermatozoides normales se vio significativamente reducido (<10%) en relación con los controles y que el porcentaje de espermatozoides anormales (cabezas separadas o sin cabeza) aumentó significativamente (hasta el 3%). No obstante, estos cambios no afectaron a la fertilidad y fueron, por tanto, considerados como no adversos.
En ratones hembra con dosis de alemtuzumab intravenoso de hasta 10 mg/kg/día IV (AUC de 4,7 veces la exposición humana en la dosis diaria recomendada) durante 5 días consecutivos antes de su apareamiento con ratones salvajes macho, el promedio de cuerpos lúteos y lugares de implantación por ratón se redujo significativamente en comparación con los animales tratados con vehículo. Se observó una reducción de la ganancia de peso gestacional en relación con los controles con vehículos en ratones hembra embarazadas con dosis de 10 mg/kg/día.
En un estudio de toxicidad para la reproducción en ratones hembra embarazadas expuestas a dosis intravenosas de alemtuzumab de hasta 10 mg/kg/día (AUC 2,4 veces la exposición humana a la dosis recomendada de 12 mg/día) durante 5 días consecutivos durante la gestación, los resultados mostraron un aumento significativo en el número de madres con todos los embriones muertos o reabsorbidos, junto con una reducción simultánea del número de madres con fetos viables. No se observaron variaciones o malformaciones externas, de tejidos blandos o esqueléticos con dosis de hasta 10 mg/kg/día.
Se observaron transferencia placentaria y posible actividad farmacológica de alemtuzumab en ratones durante la gestación y tras el parto. En los estudios con ratones, se observaron alteraciones en los recuentos de linfocitos en crías expuestas a alemtuzumab durante la gestación con dosis de hasta 3 mg/kg/día durante 5 días consecutivos (AUC de 0,6 veces la exposición humana en la dosis recomendada de 12 mg/día). El desarrollo cognitivo, físico y sexual de las crías expuestas a alemtuzumab durante la lactancia no se vio afectado con dosis de hasta 10 mg/kg/día.
DATOS FARMACÉUTICOS
6.
6.1 Lista de excipientes
Fosfato disódico dihidratado (E339)
Edetato disódico dihidratado Cloruro de potasio (E508)
Dihidrógeno fosfato de potasio (E340)
Polisorbato 80 (E433)
Cloruro de sodio
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros, exceptuando con los mencionados en la sección 6.6.
6.3 Periodo de validez
Concentrado 3 años
Solución diluida
Se ha demostrado la estabilidad química y física en uso durante 8 horas a 2°C - 8°C.
Desde el punto de vista microbiológico, se recomienda utilizar el producto inmediatamente. Si no se utiliza inmediatamente, las condiciones y los tiempos de conservación empleados antes de su uso son responsabilidad del usuario y no deben superar las 8 horas a 2°C - 8°C, protegido de la luz.
6.4 Precauciones especiales de conservación
Concentrado.
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Para las condiciones de conservación tras la dilución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
LEMTRADA se suministra en un vial de vidrio transparente de 2 ml con un tapón de goma de butilo y un sello de aluminio con cápsula de cierre de plástico fácil de abrir.
Tamaño de envase: caja de cartón con 1 vial.
6.6 Precauciones especiales de eliminación y otras manipulaciones
El contenido del vial debe examinarse visualmente antes de cada administración para descartar la existencia de partículas o decoloración. Si el concentrado contiene partículas o presenta decoloración, no debe utilizarse.
No agitar los viales antes de utilizarlos.
Para administración intravenosa, extraer 1,2 ml de LEMTRADA del vial en una jeringa mediante una técnica aséptica. Inyectar en 100 ml de una solución para perfusión de 9 mg/ml (0,9%) de cloruro de sodio o una solución para perfusión de glucosa (5%). Este medicamento no debe diluirse con otros disolventes.
La bolsa debe invertirse suavemente para mezclar la solución.
LEMTRADA no contiene conservantes antimicrobianos así que se debe tener cuidado para garantizar la esterilidad de la solución preparada. Se recomienda administrar el producto diluido de forma inmediata. Cada vial está destinado a un solo uso.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Genzyme Therapeutics Ltd 4620 Kingsgate Cascade Way
Oxford Business Park South
Oxford
OX4 2SU
Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/869/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización 12 septiembre 2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante del principio activo biológico.
Boehringer Ingelheim Pharma GmbH & Co. KG Birkendorfer Strabe 65 88397 Biberach an der Riss ALEMANIA
Nombre y dirección de los fabricantes responsables de la liberación de los lotes
Genzyme Limited 37 Hollands Road Haverhill Suffolk CB9 8PU Reino Unido
Genzyme Ireland Limited IDA Industrial Park Old Kilmeaden Road Waterford Irlanda
El prospecto impreso del medicamento debe especificar el nombre y dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará el primer informe periódico de seguridad para este medicamento en un plazo de 6 meses después de la autorización. Posteriormente, el titular de la autorización de comercialización presentará informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 ter, (párrafo 7), de la Directiva 2001/83/CE y publicados en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva
información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
• Medidas adicionales de minimización de riesgos
Antes de su comercialización en cada Estado Miembro, el Titular de la Autorización de Comercialización (TAC) deberá acordar un programa educacional para profesionales sanitarios y pacientes con la Autoridad Nacional Competente.
El TAC deberá asegurarse de que, según el acuerdo con las Autoridades Nacionales Competentes de cada Estado Miembro donde se comercializa LEMTRADA, en su comercialización y después de esta, todos los médicos que vayan a prescribir LEMTRADA reciban materiales educacionales actualizados para el médico con los siguientes elementos:
• Ficha técnica o resumen de las características del producto
• Guía para profesionales sanitarios
• Lista de comprobación para el médico que emite la receta
• Guía para el Paciente
• Tarjeta de alerta de Paciente
La guía para profesionales sanitarios debe contener los siguientes mensajes clave:
1. Una descripción de los riesgos asociados al uso de LEMTRADA, concretamente:
• Púrpura trombocitopénica inmune (PTI)
• Nefropatías, incluyendo enfermedad de anticuerpos anti-membrana basal glomerular
• Trastornos de tiroides
2. Recomendaciones sobre cómo reducir estos riesgos a través del asesoramiento, la supervisión y la gestión adecuados del paciente.
3. Una sección de “Preguntas frecuentes”
La lista de comprobación para el médico que emite la receta debe contener los siguientes mensajes clave:
1. Lista de las pruebas que hay que realizar antes del cribado inicial del paciente
2. Curso de vacunación que hay que completar 6 semanas antes del tratamiento
3. Premedicación y pruebas de salud general, embarazo y anticoncepción inmediatamente antes del tratamiento
4. Actividades de supervisión durante el tratamiento y hasta 4 años después del último
5. Una mención específica al hecho de que el paciente ha sido informado y comprende los riesgos de
los trastornos autoinmunes, las infecciones graves y tumores malignos, y las medidas para reducirlos
La Guía para el Paciente debe contener los siguientes mensajes clave:
1. Una descripción de los riesgos asociados al uso de LEMTRADA, concretamente:
• Púrpura trombocitopénica inmune (PTI)
• Nefropatías, incluyendo enfermedad de anticuerpos anti-membrana basal glomerular
• Trastornos de tiroides
• Infecciones graves
2. Una descripción de los signos y síntomas de los riesgos autoinmunes
3. Una descripción de la mejor forma de proceder en caso de aparición de los signos y síntomas de dichos riesgos (por ejemplo, cómo ponerse en contacto con los médicos)
4. Recomendaciones para planificar la programación de supervisión
La Tarjeta de alerta de Paciente debe contener los siguientes mensajes clave:
1. Un mensaje de advertencia para los profesionales sanitarios que traten al paciente en cualquier momento, incluyendo en condiciones de emergencia, que indique que el paciente ha sido tratado con LEMTRADA
2. El tratamiento con LEMTRADA puede aumentar el riesgo de:
• Púrpura trombocitopénica inmune (PTI)
• Nefropatías, incluyendo enfermedad de anticuerpos anti-membrana basal glomerular
• Trastornos de tiroides
• Infecciones graves
3. Detalles de contacto del médico que emite la receta de LEMTRADA
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
LEMTRADA 12 mg concentrado para solución para perfusión alemtuzumab
Cada vial contiene 12 mg de alemtuzumab en 1,2 ml (10 mg/ml).
E339, edetato disódico dihidratado, E508, E340, E433, cloruro de sodio, agua para preparaciones inyectables
Concentrado para solución para perfusión 1 vial
12 mg/1,2 ml
Leer el prospecto antes de utilizar este medicamento. Vía intravenosa
Administrar antes de 8 horas después la dilución.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar el vial en el embalaje exterior para protegerlo de la luz. Conservar en nevera.
No congelar-ni agitar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Genzyme Therapeutics Ltd 4620 Kingsgate Cascade Way
Oxford Business Park South
Oxford
OX4 2SU
Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/869/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
<Se acepta la justificación para no incluir la información en Braille>
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETA/VIAL
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
LEMTRADA 12 mg concentrado estéril
alemtuzumab
IV
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
1,2 ml
6. OTROS
B. PROSPECTO
Prospecto: información para el paciente
LEMTRADA 12 mg concentrado para solución para perfusión
alemtuzumab
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de que se le administre este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Si experimenta efectos adversos, consulte a su médico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es LEMTRADA y para qué se utiliza
2. Qué necesita saber antes de que se le administre LEMTRADA
3. Cómo se administrará LEMTRADA
4. Posibles efectos adversos
5. Conservación de LEMTRADA
6. Contenido del envase e información adicional 1. Qué es LEMTRADA y para qué se utiliza
LEMTRADA contiene el principio activo alemtuzumab que se utiliza para tratar una forma de esclerosis múltiple (EM) en adultos, denominada esclerosis múltiple remitente-recurrente (EMRR). LEMTRADA no cura la EM pero puede reducir el número de brotes. También puede ralentizar o revertir algunos de los signos y síntomas de la EM. En los ensayos clínicos, los pacientes tratados con LEMTRADA sufrieron menos recaídas y fue menos probable que experimentaran un empeoramiento de la discapacidad en comparación con los pacientes a los que se trató con un interferón beta inyectado varias veces a la semana.
¿Qué es la esclerosis múltiple?
La EM es una enfermedad autoinmune que afecta al sistema nervioso central (cerebro y médula espinal). En la EM, el sistema inmunológico ataca por equivocación la capa protectora (mielina) que cubre las fibras nerviosas, causando inflamación. Cuando la inflamación provoca síntomas, suele denominarse “ataque” o “brote”. En las EMRR los pacientes experimentan brotes seguidos de periodos de recuperación.
Los síntomas que se experimentan dependen de la parte del sistema nervioso central que esté afectada. El daño causado a los nervios durante esta inflamación puede ser reversible pero, conforme la enfermedad progrese, los daños se pueden acumular y convertirse en permanentes.
Cómo funciona LEMTRADA
LEMTRADA regula el sistema inmunológico para limitar sus ataques al sistema nervioso.
2. Qué necesita saber antes de que se le administre LEMTRADA NO use LEMTRADA si:
- es alérgico a alemtuzumab o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6)
- es portador del virus de la inmunodeficiencia humana (VIH).
Advertencias y precauciones
Consulte a su médico antes de que se le administre LEMTRADA. Tras finalizar un curso de tratamiento con LEMTRADA podría tener un riesgo más elevado de desarrollar otras enfermedades autoinmunes o de experimentar infecciones graves. Es importante que entienda estos riesgos y sepa cómo detectarlos. Se le proporcionará una Tarjeta de Paciente y una Guía para el Paciente con más información. Es importante que tenga la Tarjeta de Paciente con usted durante el tratamiento y hasta 4 años después de la última perfusión de LEMTRADA, ya que pueden producirse efectos adversos años después del tratamiento. Cuando esté en tratamiento médico, aunque no sea para EM, muestre la Tarjeta de Paciente al médico.
Su médico le realizará análisis de sangre antes de comenzar el tratamiento con LEMTRADA. Estos análisis se realizan para saber si puede tomar LEMTRADA. Su médico también se asegurará de que no padece determinadas enfermedades o trastornos médicos antes de comenzar el tratamiento con LEMTRADA.
• Enfermedades autoinmunes
El tratamiento con LEMTRADA puede aumentar el riesgo de enfermedades autoinmunes. Estas enfermedades se caracterizan porque el sistema inmunológico ataca, por equivocación, al propio cuerpo. A continuación, se proporciona información sobre algunas enfermedades específicas que se han observado en pacientes con EM que han sido tratados con LEMTRADA.
Las enfermedades autoinmunes pueden darse muchos años después del tratamiento con LEMTRADA. Por ello, es necesario realizar análisis de sangre y orina hasta 4 años después de la última perfusión. Los análisis son necesarios aunque se encuentre bien y los síntomas de la EM estén bajo control. Además, hay determinados signos y síntomas que debería vigilar usted mismo. En la sección 4 - enfermedades autoinmunes se dan detalles sobre los signos y síntomas, los análisis y las acciones que deberá llevar a cabo.
En la guía para el paciente de LEMTRADA se puede encontrar más información de utilidad sobre estas enfermedades autoinmunes (y las pruebas asociadas).
o Púrpura trombocitopénica inmune (PTI)
Con poca frecuencia, los pacientes han desarrollado un trastorno hemorrágico provocado por un nivel bajo de plaquetas, denominado púrpura trombocitopénica inmune (PTI). Este trastorno debe detectarse y tratarse con rapidez ya que, de lo contrario, sus efectos pueden ser graves o incluso mortales. Los signos y síntomas de la PTI se describen en la sección 4.
o Enfermedad renal (como enfermedad anti-MBG.)
En raras ocasiones, los pacientes han experimentado problemas relacionados con trastornos autoinmunes en los riñones, como por ejemplo la enfermedad antimembrana basal glomerular (enfermedad anti-MBG). Los signos y síntomas de la enfermedad renal se describen en la sección 4. Si no se trata, puede producir fallo renal con necesidad de diálisis o transplante y podría provocar la muerte.
o Trastornos del tiroides
Con mucha frecuencia, los pacientes han experimentado un trastorno autoinmune de la glándula tiroidea que afecta a su capacidad para generar o controlar hormonas importantes para el metabolismo.
LEMTRADA puede provocar distintos tipos de trastornos del tiroides, incluyendo:
• Glándula tiroides hiperactiva (hipertiroidismo) cuando el tiroides produce demasiada hormona
• Glándula tiroides hipoactiva (hipotiroidismo) cuando el tiroides no produce suficiente hormona
Los signos y síntomas de los trastornos del tiroides se describen en la sección 4.
Si desarrolla un trastorno del tiroides, en la mayoría de los casos necesitará tratamiento durante el resto de su vida con fármacos que controlen el trastorno y, en algunos casos, puede que sea necesario extirpar la glándula tiroides.
Es muy importante seguir el tratamiento adecuado para el trastorno del tiroides, especialmente si queda embarazada tras el uso de LEMTRADA. Un trastorno de tiroides sin tratar podría dañar al feto o al bebé tras el nacimiento.
o Otras enfermedades autoinmunes
En raras ocasiones, algunos pacientes han experimentado enfermedades autoinmunes relacionadas con los glóbulos rojos o los glóbulos blancos. Estas enfermedades se pueden diagnosticar a partir de los análisis de sangre que se le realizarán con regularidad tras el tratamiento con LEMTRADA. Si desarrolla una de estas enfermedades su médico se lo indicará y tomará las medidas adecuadas para tratarla.
• Reacciones a la perfusión
La mayoría de pacientes tratados con LEMTRADA experimentarán efectos adversos en el momento de la perfusión o en las 24 horas siguientes. Para intentar reducir las reacciones a la perfusión, el médico le tratará con otros fármacos (ver sección 4 - reacciones a la perfusión).
• Infecciones
Los pacientes tratados con LEMTRADA tienen un mayor riesgo de sufrir una infección grave (ver sección 4 -infecciones). En general, las infecciones pueden tratarse con medicamentos estándar.
Para reducir la posibilidad de sufrir una infección, su médico comprobará si los demás medicamentos que toma pueden estar afectando a su sistema inmunológico. Por esta razón, es importante informar al médico de todos los medicamentos que esté tomando.
También, si tiene una infección antes de comenzar el tratamiento con LEMTRADA, su médico considerará el retrasar el tratamiento hasta que la infección esté bajo control o resuelta.
Los pacientes tratados con Lemtrada tienen mayor riesgo de desarrollar una infección por herpes (por ejemplo, úlcera bucal). En general, una vez que un paciente ha tenido una infección por herpes, tiene un mayor riesgo de desarrollar otra. También es posible desarrollar una infección por herpes por primera vez. Se recomienda que el médico prescriba un tratamiento apropiado para reducir las posibilidades de desarrollar una infección por herpes, que debe tomarse durante los días en que se administre el tratamiento con LEMTRADA y durante el mes siguiente al tratamiento.
Además, también pueden producirse infecciones que pueden provocar anormalidades del cuello del útero (cérvix). Por ello, se recomienda que todas las pacientes se realicen una revisión anual, tipo frotis cervical. Su médico le indicará qué pruebas necesita.
Los pacientes tratados con LEMTRADA tienen también un riesgo alto de desarrollar listeriosis/’Listeria meningitis. Para reducir este riesgo, se debe evitar la ingestión de carnes crudas o poco hechas, quesos frescos y productos lácteos no pasteurizados durante al menos 1 mes después del tratamiento con LEMTRADA.
Si vive en una zona donde es común la tuberculosis, puede que tenga un mayor riesgo de padecerla. Su médico programará pruebas para la detección de la tuberculosis.
Si es portador del virus de la hepatitis B o hepatitis C (que afectan al hígado), es necesario un mayor cuidado antes de recibir LEMTRADA ya que se desconoce si el tratamiento puede conducir a la activación de la infección, que podría posteriormente, dañar el hígado.
• Diagnóstico previo de cáncer
Si se le diagnosticó cáncer en el pasado, informe a su médico.
• Vacunas
Se desconoce si LEMTRADA afecta a la respuesta a las vacunas. Si no ha completado las vacunaciones estándar necesarias, el médico valorará si debe recibirlas antes del tratamiento con LEMTRADA. En particular, su médico considerará vacunarle de varicela si aún no la ha padecido. Cualquier vacunación deberá administrarse, al menos, 6 semanas antes de comenzar un curso de tratamiento con LEMTRADA.
NO debe recibir determinados tipos de vacunas (vacunas de virus vivos) si ha recibido LEMTRADA recientemente.
Niños y adolescentes
LEMTRADA no debe utilizarse en niños y adolescentes menores de 18 años ya que no se ha estudiado en pacientes de EM menores de esa edad.
Uso de LEMTRADA con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento (incluyendo vacunaciones o medicamentos a base de plantas).
Aparte de LEMTRADA, existen otros tratamientos (incluyendo los de EM o para tratar otras enfermedades) que pueden afectar al sistema inmunológico y, por tanto, afectar a su capacidad de combatir las infecciones. Si está utilizando alguno de estos medicamentos, puede que su médico le pida que deje de tomarlo antes de iniciar el tratamiento con LEMTRADA.
Embarazo
Si está embarazada, cree que podría estarlo o tiene intención de quedarse embarazada, consulte a su médico antes de que se le administre este medicamento.
Las mujeres en edad fértil deberán utilizar métodos anticonceptivos eficaces durante cada curso del tratamiento con LEMTRADA y en los 4 meses siguientes tras cada curso.
Deberá tenerse un cuidado especial si se queda embarazada tras el tratamiento con LEMTRADA y experimenta un trastorno de tiroides durante el embarazo. Los trastornos de tiroides pueden causar daños en el bebé (ver sección 2 Advertencias y precauciones - enfermedades autoinmunes).
Lactancia
Se desconoce si LEMTRADA pasa al bebé a través de la leche materna, pero existe una posibilidad de que esto ocurra. Se recomienda interrumpir la lactancia materna durante cada curso del tratamiento con LEMTRADA y durante 4 meses tras cada curso del tratamiento. No obstante, la leche materna puede ser beneficiosa (puede proteger al bebé de infecciones), así que consulte a su médico si piensa dar el pecho a su bebé y le aconsejará lo mejor para usted y su bebé.
Fertilidad
LEMTRADA podría permanecer en su cuerpo durante el curso de tratamiento y hasta 4 meses después. Se desconoce si LEMTRADA tiene efectos en la fertilidad durante este periodo. Consulte con su médico si está pensando en tener hijos.
Conducción y uso de máquinas
Muchos pacientes experimentan efectos adversos en el momento de la perfusión con LEMTRADA o en las 24 horas siguientes y algunos de estos efectos, por ejemplo, mareo, podrían hacer que conducir o utilizar máquinas no resultara seguro. Si le ocurriera, interrumpa estas actividades hasta que se encuentre mejor.
LEMTRADA contiene potasio y sodio
Este medicamento contiene menos de 1 mmol de potasio (39 mg) por perfusión, considerándose por tanto libre de potasio.
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por perfusión, considerándose por tanto libre de sodio.
Cómo se administrará LEMTRADA
3.
Su médico le explicará cómo se administra LEMTRADA. En caso de duda pregunte a su médico.
Durante el primer curso de tratamiento recibirá una perfusión al día durante 5 días (curso 1).
Un año después recibirá una perfusión al día durante 3 días (curso 2).
Entre los dos cursos no recibirá tratamiento con LEMTRADA.
La dosis diaria máxima es de una perfusión.
LEMTRADA se le administrará por perfusión en la vena. Cada perfusión dura unas 4 horas. En la mayoría de los pacientes, 2 cursos de tratamiento reducen la actividad de la EM durante 2 años. La supervisión para detectar efectos adversos y las pruebas periódicas debe continuar durante 4 años después de la última perfusión.
Para ayudarle a comprender mejor la duración de los efectos del tratamiento y la duración del seguimiento necesario, consulte el siguiente diagrama.
Comenzar a realizar pruebas de sangre y orina antes del tratamiento
-► y seguir durante 4 años después de la última perfusión
Curso de tratamiento 1 Curso de tratamiento 2
mu ni
Tratamiento de 5 días Tratamiento de 3 días
Año 1 Año 2 Año 3 Año 4 Año 5
NOTA Para la mayoría de los pacientes, 2 cursos funcionarán durante 2 años o más.
|
TRATAMIENTO | |||
|
SUPERVISIÓN | |||
Seguimiento tras el tratamiento con LEMTRADA
Una vez que haya recibido LEMTRADA, deberá someterse a pruebas periódicas para que cualquier posible efecto adverso se pueda diagnosticar y tratar con rapidez. Estas pruebas deben continuar hasta 4 años después de la última perfusión y se describen en la sección 4, efectos adversos más importantes.
Si recibe más LEMTRADA del que debe
Los pacientes a los que, accidentalmente, se les ha administrado demasiado LEMTRADA en una perfusión han experimentado reacciones graves, como dolor de cabeza, erupción, presión arterial baja o aumento de la frecuencia cardiaca. Dosis mayores de la recomendada pueden provocar reacciones a la perfusión más graves o de mayor duración (ver sección 4) o un efecto mayor en el sistema inmunológico. El tratamiento consiste en interrumpir la administración de LEMTRADA y tratar los síntomas.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, LEMTRADA puede producir efectos adversos, aunque no todas las personas los sufran.
Los efectos adversos más importantes son las enfermedades autoinmunes descritas en la sección 2, entre las que se incluyen:
• PTI (trastorno hemorrágico), (poco frecuente - puede afectar hasta 1 de cada 100 personas): puede presentarse como pequeñas manchas de color rojo, rosa o morado dispersas por la piel; propensión a los cardenales; hemorragias en caso de corte más difíciles de detener; periodos menstruales más abundantes, más largos o más frecuentes
de lo normal; sangrado entre periodos menstruales; sangrado de nueva aparición o más largo de lo habitual de las encías o de la nariz; o tos con sangre • trastornos del riñón, (raros - pueden afectar hasta 1 de cada 1.000 personas): pueden presentarse como sangre en la orina (la orina puede ser roja o de color té) o como hinchazón de piernas o pies. También puede provocar daño en los pulmones y provocar tos con sangre.
Si experimenta cualquiera de estos signos o síntomas relacionados con trastornos de los riñones o hemorrágicos, informe inmediatamente a su médico. Si no puede ponerse en contacto con su médico, busque atención médica inmediata._
• trastornos del tiroides, (muy frecuentes - pueden afectar a más de 1 de cada 10 personas): pueden presentarse como sudoración excesiva; aumento o pérdida de peso sin explicación; hinchazón ocular; nerviosismo; ritmo cardiaco acelerado; sensación de frío; empeoramiento del cansancio; o estreñimiento de nueva aparición
• trastornos de los glóbulos rojos y de los glóbulos blancos, (raros - pueden afectar hasta 1 de cada 1.000 personas) diagnosticados a partir de los análisis de sangre.
Resumen de las pruebas que se le realizarán para controlar la aparición de condiciones autoinmunes:
|
Prueba |
¿Cuándo? |
Duración |
|
Análisis de sangre (para diagnosticar todos los efectos adversos graves importantes mencionados con anterioridad) |
Antes de comenzar el tratamiento y cada mes después del tratamiento |
Hasta 4 años después de la última perfusión de LEMTRADA |
|
Análisis de orina (prueba adicional para diagnosticar trastornos del riñón) |
Antes del inicio del tratamiento y cada mes cuando éste finalice |
Hasta 4 años después de la última perfusión de LEMTRADA |
Todos estos efectos adversos graves pueden aparecer años después de la administración de LEMTRADA. Si experimenta cualquiera de estos signos o síntomas, informe inmediatamente a su médico. También se le realizarán análisis de sangre y orina, de forma periódica, para asegurar que si desarrolla alguna de estas enfermedades, sea tratada con rapidez.
Después de este periodo, si experimenta síntomas de PTI, trastornos de tiroides o riñón, su médico le realizará más pruebas. Debe seguir atento a los signos y síntomas de efectos adversos más allá de los cuatro años tal como se indica en la Guía para el Paciente, y también debe seguir llevando la Tarjeta de Paciente con usted.
Otro efecto adverso importante es un aumento del riesgo de padecer infecciones (consulte a continuación información sobre la frecuencia con que los pacientes experimentan infecciones). En la mayoría de los casos, estas infecciones son leves pero pueden producirse infecciones graves.
Si experimenta cualquiera de estos signos de infección, informe inmediatamente a su médico • fiebre y/o escalofríos
_• inflamación de las glándulas_
Para ayudarle a reducir el riesgo de algunas infecciones, puede que su médico considere administrarle vacunación frente a la varicela y/u otras vacunaciones que piense que pueda necesitar (ver sección 2: Qué necesita saber antes de empezar a usar LEMTRADA - Vacunas). Puede que su médico también le prescriba un medicamento para las úlceras bucales (ver sección 2: Qué necesita saber antes de empezar a usar LEMTRADA - Infecciones).
Los efectos adversos más frecuentes son las reacciones a la perfusión (vea, a continuación, información sobre la frecuencia con la que las experimentan los pacientes), que pueden producirse en el momento de la perfusión o en las 24 horas siguientes. En la mayoría de los casos, estas reacciones son leves pero pueden producirse reacciones graves. En ocasiones, pueden producirse reacciones alérgicas.
Para intentar reducir las reacciones a la perfusión, el médico le administrará medicación (corticosteroides) antes de cada una de las 3 primeras perfusiones de un curso de LEMTRADA. Para limitar estas reacciones también se pueden administrar otros tratamientos antes de la perfusión o cuando se experimenten los síntomas. Además, se le vigilará durante la perfusión y hasta 2 horas después de que esta haya finalizado. En caso de reacciones graves, la perfusión se puede ralentizar o incluso detenerse.
Consulte la guía para el paciente de LEMTRADA para obtener más información sobre estos eventos.
Estos son los efectos adversos que puede experimentar
Muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
• Reacciones a la perfusión que pueden ocurrir en el momento de la perfusión o en las 24 horas siguientes: dolor de cabeza, erupción, fiebre, malestar, urticaria, picor, enrojecimiento de la cara y cuello, sensación de cansancio
• Infecciones: infecciones de las vías respiratorias como resfriados y sinusitis, cistitis
• Descenso del número de glóbulos blancos (linfocitos)
Frecuentes (pueden afectar hasta 1 de cada 10 personas):
• Reacciones a la perfusión que pueden ocurrir en el momento de la perfusión o en las 24 horas siguientes: cambios en la frecuencia cardiaca, indigestión, escalofríos, malestar torácico, dolor, mareo, alteración del gusto, dificultad para dormir, dificultad para respirar o falta de aire, erupción por el cuerpo, presión arterial baja.
• Infecciones: tos, infección de oído, enfermedad tipo gripal, bronquitis, neumonía, muguet, candidiasis vaginal, herpes, varicela, úlceras bucales, glándulas hinchadas o aumentadas
• dolor en el lugar de la perfusión, dolor de espalda, cuello, brazos o piernas, dolor muscular, espasmos musculares, dolor articular, dolor de boca o garganta
• Inflamación de boca/encías/lengua
• malestar general, debilidad, vómitos, diarrea, dolor abdominal, gripe intestinal
• ardor de estómago
• anormalidades que se pueden encontrar durante la exploración: sangre o proteínas en la orina, frecuencia cardiaca disminuida, latido cardiaco irregular o anormal, presión arterial alta
• Brote de EM
• temblor, pérdida de la sensibilidad, sensación de ardor o cosquilleo
• glándula tiroides hiperactiva o hipoactiva, o bocio (inflamación de la glándula tiroides del cuello)
• entumecimiento de brazos y/o piernas
• problemas de visión
• sensación de ansiedad
• menstruación irregular, prolongada o anormalmente abundante
• acné, enrojecimiento de la piel, sudoración excesiva
• sangrados de nariz, cardenales
• pérdida del pelo
Poco frecuentes (pueden afectar hasta 1 de cada 100 personas)
• Infecciones: herpes genital, infección ocular, infección de dientes
• problemas de coagulación, anemia
• pie de atleta
• frotis vaginal anormal
• depresión
• aumento de la sensibilidad
• dificultad para tragar
• hipo
• disminución de peso
• estreñimiento
• sangrado de encías
• prueba anormal del hígado
• ampollas
Muestre la Tarjeta de Paciente y este prospecto a cualquier médico relacionado con su tratamiento y no solo al neurólogo.
También encontrará esta información en la Tarjeta de Paciente y la guía para el paciente que le ha proporcionado su médico.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de LEMTRADA
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la caja y la etiqueta del vial después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
Se recomienda utilizar el producto inmediatamente después de su dilución debido al posible riesgo de contaminación microbiana. Si no se utiliza de forma inmediata, las condiciones y los tiempos de conservación empleados antes de su uso son responsabilidad del usuario y no deberán superar las 8 horas a una temperatura de 2oC a 8oC, protegido de la luz.
No utilice este medicamento si observa partículas en el líquido y/o si el líquido del vial presenta alteración del color.
Los medicamentos no se deben tirar por los desagües. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de LEMTRADA El principio activo es alemtuzumab.
Cada vial contiene 12 mg de alemtuzumab en 1,2 ml.
Los demás componentes son:
• fosfato disódico dihidratado (E339)
• edetato disódico dihidratado
• cloruro de potasio (E508)
• dihidrógeno fosfato de potasio (E340)
polisorbato 80 (E433) cloruro sódico
agua para preparaciones inyectables
Aspecto de LEMTRADA y contenido del envase
LEMTRADA es un concentrado para solución para perfusión (concentrado estéril) transparente e incoloro o ligeramente amarillo que se incluye en un vial de vidrio con tapón.
Hay 1 vial en cada caja.
Titular de la autorización de comercialización
Genzyme Therapeutics Ltd, 4620 Kingsgate, Cascade Way, Oxford Business Park South, Oxford, OX4 2SU, Reino Unido
Responsable de la fabricación
Genzyme Ltd., 37 Hollands Road, Haverhill, Suffolk CB9 8PU, Reino Unido.
Genzyme Ireland Limited, IDA Industrial Park, Old Kilmeaden Road, Waterford, Irlanda.
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien/ Luxemburg/Luxembourg Sanofi Belgium Tél/Tel: + 32 2 710 54 00 |
Lietuva UAB „SANOFI-AVENTIS LIETUVA1 Tel. +370 5 275 5224 |
|
EKarapHH Sanofi-Aventis Bulgaria EOOD Tea: +359 2 9705300 |
Magyarország sanofi-aventis Zrt Tel: +36 1 505 0050 |
|
Ceská republika sanofi-aventis, s.r.o. Tel: +420 233086 111 |
Malta Sanofi-Aventis Malta Ltd Tel: +356 21493022 |
|
Danmark sanofi-aventis Denmark A/S Tlf: +45 45 16 70 00 |
Nederland Genzyme Europe B.V. Tel: +31 35 699 1200 |
|
Deutschland Genzyme Therapeutics Ltd. Tel: +49 (0) 6102 3674 451 |
Norge sanofi-aventis Norge AS Tlf: + 47 67 10 71 00 |
|
Eesti sanofi-aventis Estonia OÜ Tel. +372 6 273 488 |
Ósterreich sanofi-aventis GmbH Tel: + 43 1 80 185 - 0 |
|
Ekkáda/Kúnpo^ sanofi-aventis AEBE (EkkáSa) Tpk: +30 210 900 1600 |
Polska sanofi-aventis Sp. z o.o. Tel.: +48 22 280 00 00 |
|
España Genzyme, S.L.U. Tel: +34 93 485 94 00 sanofi-aventis, S.A. Tel: +34 93 485 94 00 |
Portugal Sanofi - Produtos Farmacéuticos, Lda. Tel: +351 21 422 0100 |
|
France Genzyme S.A.S. Tél : +33 (0) 825 825 863 |
Romanía sanofi-aventis Romania S.R.L. Tel: +40 (0) 21 317 31 36 |
|
Hrvatska sanofi-aventis Croatia d.o.o. Tel: +385 1 6003 400 |
Slovenija sanofi-aventis d.o.o. Tel: +386 1 560 4800 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika sanofi-aventis Pharma Slovakia s.r.o. Tel.: +421 2 33 100 100 |
|
Ireland Genzyme Therapeutics Ltd. (United Kingdom) Tel: +44 (0) 1865 405200 |
Suomi/Finland sanofi-aventis Oy Puh/Tel: + 358 201 200 300 |
Sverige
Italia
Genzyme Srl Tel: +39 059 349 811
Latvija
sanofi-aventis Latvia SIA Tel: +371 67 33 24 51
sanofi-aventis AB Tel: +46 (0)8 634 50 00
United Kingdom
Genzyme Therapeutics Ltd. (United Kingdom)
Tel: +44 (0) 1865 405200
Fecha de la última revisión de este prospecto: .
Otras fuentes de información
Para ayudar a formar a los pacientes sobre los posibles efectos adversos y las instrucciones de qué hacer en caso de que ocurran determinados efectos adversos, están disponibles los siguientes materiales de minimización de riesgos:
1 Tarjeta de Paciente: Para que el paciente la presente a otros profesionales sanitarios para advertirles del
uso de LEMTRADA en el paciente
2 Guía para el Paciente: Para más información sobre reacciones autoinmunes, infecciones y otro tipo de
información.
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Esta información está destinada únicamente a profesionales del sector sanitario:
Información sobre minimización de riesgos - condiciones autoinmunes
• Es extremadamente importante que el paciente comprenda el compromiso de realizarse las pruebas periódicas (durante los 4 años siguientes a la última perfusión) aunque no tenga síntomas y la enfermedad esté bien controlada.
• Junto con el paciente, necesita planificar y gestionar los controles periódicos.
• Los pacientes no cumplidores pueden necesitar asesoramiento adicional para resaltar los riesgos de saltarse las pruebas periódicas programadas.
• Debe controlar los resultados de las pruebas y permanecer alerta ante cualquier síntoma de reacción adversa.
• Revise el prospecto y la guía para el paciente de LEMTRADA con su paciente. Recuerde al paciente que debe estar alerta ante cualquier síntoma relacionado con condiciones autoinmunes y que debe buscar consejo médico en caso de duda.
También hay materiales educacionales disponibles para los profesionales sanitarios:
• Guía de LEMTRADA para profesionales sanitarios
• Módulo de formación de LEMTRADA
• Check-list para el médico prescriptor de LEMTRADA
Lea la Ficha Técnica (disponible en el sitio web de la EMA mencionado anteriormente) para obtener más información.
Información para preparar la administración de LEMTRADA y supervisar al paciente
• Los pacientes deben ser premedicados con corticosteroides inmediatamente antes de la perfusión con LEMTRADA durante los 3 primeros días de cualquier curso de tratamiento. También puede considerarse el pretratamiento con antihistamínicos y/o antipiréticos antes de la administración de LEMTRADA.
• Durante el tratamiento y hasta 1 mes después, debe administrarse un agente antiherpético oral a todos los pacientes. En los ensayos clínicos, se administraron 200 mg de aciclovir dos veces al día o equivalente a los pacientes.
• Las pruebas basales completas y cribados están descritos en el apartado 4 de la Ficha Técnica.
• El contenido del vial debe examinarse visualmente antes de cada administración para descartar la existencia de partículas o decoloración. Si el concentrado contiene partículas o presenta decoloración, no debe utilizarse.
NO AGITAR LOS VIALES ANTES DE UTILIZARLOS.
• Siga técnicas asépticas para extraer 1,2 ml de LEMTRADA del vial e inyecte en 100 ml de una solución para perfusión de 9 mg/ml (0,9%) de cloruro de sodio o una solución para perfusión de glucosa (5%). La bolsa debe invertirse suavemente para mezclar la solución. LEMTRADA no contiene conservantes así que se debe tener cuidado para garantizar la esterilidad de la solución preparada.
• Administre la solución para perfusión de LEMTRADA por vía intravenosa durante un periodo de unas 4 horas.
• No se deben añadir otros medicamentos a la solución para perfusión de LEMTRADA ni tampoco perfundirlos simultáneamente a través de la misma vía intravenosa.
• Se recomienda utilizar el producto inmediatamente después de la dilución, debido al posible riesgo de contaminación microbiana. Si no se utiliza de forma inmediata, las condiciones y los tiempos de conservación empleados antes de su uso son responsabilidad del usuario y no deberán superar las 8 horas a una temperatura de 2oC a 8oC, protegido de la luz.
• Deben seguirse los procedimientos adecuados de manipulación y eliminación. La eliminación de cualquier resto o material de desecho se realizará de acuerdo con la normativa local.
• Después de cada perfusión, se debe observar al paciente durante 2 horas para detectar posibles reacciones asociadas a la perfusión. Se puede iniciar tratamiento sintomático si fuera necesario -ver Ficha Técnica. Siga realizando pruebas analíticas al paciente cada mes para detectar enfermedades autoinmunes, hasta 4 años después de la última perfusión. Consulte la guía de LEMTRADA para profesionales sanitarios para obtener más información, o lea la Ficha Técnica disponible en el sitio web de la EMA mencionado anteriormente.
ANEXO IV
Conclusiones científicas y motivos para la modificación de las condiciones de las autorizaciones de comercialización
Conclusiones científicas
Teniendo en cuenta lo dispuesto en el Informe de Evaluación del Comité para la Evaluación de Riesgos en Farmacovigilancia (PRAC) sobre los informes periódicos de seguridad (IPS) para alemtuzumab, las conclusiones científicas del Comité de Medicamentos de Uso Humano (CHMP) son las siguientes:
Listeriosis/Listeria meningitis
Los medicamentos que tienen un efecto modulante sobre el sistema inmunitario como Lemtrada podrían estar asociados con un aumento del riesgo de infecciones oportunistas. Se identificaron un total de 5 casos notificados, todos ellos procedentes de la Unión Europea. Un paciente con esclerosis múltiple tratado con alemtuzumab en un ensayo clínico, incluido en el estudio CAMMS223, desarrolló meningitis por listeria, y cuatro casos poscomercialización espontáneos desarrollaron listeriosis sistémica o meningitis por Listeria monocytogenes.
Bradicardia como reacción adversa relacionada con la perfusión
Se notificaron setenta y uno casos (en 55 pacientes) de bradicardia (dos de los cuales fueron evaluados como graves, el resto como no graves) en ensayos clínicos. Un total de 1.505 pacientes tratados con alemtuzumab estuvieron expuestos en estos ensayos. Además, se notificaron treinta y nueve casos de bradicardia (ocho de los cuales fueron evaluados como graves, el resto como no graves) en notificaciones poscomercialización a partir del 01 de mayo 2015. Cada uno de los diez casos graves que implicaron bradicardia sucedieron en el contexto de reacciones asociadas con la perfusión.
Por tanto, a la vista de los datos presentados en el IPS revisado, el PRAC considera que los cambios en la información del producto de los medicamentos que contienen alemtuzumab están justificados.
El CHMP está de acuerdo con las conclusiones científicas realizadas por el PRAC.
Motivos para la modificación de las condiciones de la Autorización de Comercialización
De acuerdo con las conclusiones científicas para alemtuzumab el CHMP considera que el balance beneficio-riesgo de los medicamentos que contienen alemtuzumab no se modifica sujeto a los cambios propuestos en la información del producto.
El CHMP recomienda que se modifiquen las condiciones de la Autorización de Comercialización.
44