Kogenate Bayer 2000 Ui Polvo Y Disolvente Para Solucion Inyectable
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
KOGENATE Bayer 250 UI polvo y disolvente para solución inyectable KOGENATE Bayer 500 UI polvo y disolvente para solución inyectable KOGENATE Bayer 1000 UI polvo y disolvente para solución inyectable KOGENATE Bayer 2000 UI polvo y disolvente para solución inyectable KOGENATE Bayer 3000 UI polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 250/500/1000/2000/3000 UI de factor VIII de coagulación humano (DCI: octocog alfa).
El factor VIII de coagulación humano se produce mediante técnicas de ADN recombinante (ADN r) en células de riñón de crías de hámster que contienen el gen del factor VIII humano.
• Después de la reconstitución, 1 ml de KOGENATE Bayer 250 UI contiene aproximadamente
100 UI (250 UI/2,5 ml) de factor VIII de coagulación humano (DCI: octocog alfa).
• Después de la reconstitución, 1 ml de KOGENATE Bayer 500 UI contiene aproximadamente
200 UI (500 UI/2,5 ml) de factor VIII de coagulación humano (DCI: octocog alfa).
• Después de la reconstitución, 1 ml de KOGENATE Bayer 1000 UI contiene aproximadamente
400 UI (1000 UI/2,5 ml) de factor VIII de coagulación humano (DCI: octocog alfa).
• Después de la reconstitución, 1 ml de KOGENATE Bayer 2000 UI contiene aproximadamente
400 UI (2000 UI/5 ml) de factor VIII de coagulación humano (DCI: octocog alfa).
• Después de la reconstitución, 1 ml de KOGENATE Bayer 3000 UI contiene aproximadamente
600 UI (3000 UI/5 ml) de factor VIII de coagulación humano (DCI: octocog alfa).
La potencia (UI) se determina utilizando el ensayo de coagulación en una etapa, en comparación con el estándar Mega de la FDA, que ha sido calibrado frente al estándar de la OMS en Unidades Internacionales (UI).
La actividad específica de KOGENATE Bayer es, aproximadamente, de 4000 UI/mg de proteína. Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable (sistema Bio-Set).
Polvo: polvo suelto o sólido friable de color blanco o ligeramente amarillento. Disolvente: agua para preparaciones inyectables, solución transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (déficit congénito de factor VIII). Este medicamento no contiene factor de von Willebrand y por lo tanto no está indicado en la enfermedad de von Willebrand.
Este medicamento está indicado en adultos, adolescentes y niños de cualquier edad.
4.2 Posología y forma de administración
El tratamiento se debe realizar bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia.
Posología
El número de unidades de factor VIII administradas se expresa en Unidades Internacionales (UI), que se corresponden con el estándar actual de la OMS para medicamentos de factor VIII. La actividad del factor VIII en plasma se puede expresar en porcentaje (referido al plasma humano normal) o bien, en Unidades Internacionales (referido al estándar internacional para el factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII equivale a la cantidad de factor VIII presente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis necesaria de factor VIII se basa en el hallazgo empírico de que 1 Unidad Internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad plasmática de factor VIII en 1,5% - 2,5% de la actividad normal. La dosis requerida se determina utilizando las fórmulas siguientes:
I. UI requeridas = peso corporal (kg) * aumento deseado de factor VIII (% del normal) x 0,5
II. Aumento previsto del factor VIII (% del normal) = 2 x UI administradas
peso corporal (kg)
La dosis, frecuencia y duración del tratamiento sustitutivo se debe individualizar según las necesidades del paciente (peso, gravedad del trastorno de la función hemostática, localización e intensidad de la hemorragia, presencia de inhibidores y del nivel deseado de Factor VIII).
La tabla siguiente proporciona una guía de los niveles sanguíneos mínimos de factor VIII en diferentes situaciones clínicas. En el caso de las hemorragias que aparecen en la lista, el nivel de actividad del factor VIII no podrá ser inferior al nivel indicado (en % del normal), en el período correspondiente:
Grado de la hemorragia/Tipo de procedimiento quirúrgico
Nivel de factor VIII requerido (%) (UI/dl)_
Frecuencia de dosificación (horas)/Duración de la terapia (días)_
Hemorragia
Hemartrosis precoz, sangrado muscular o sangrado de la cavidad oral
20 - 40
Hemartrosis más extensa, sangrado muscular o hematoma
Hemorragias con riesgo vital (como sangrado intracraneal, de garganta, sangrado gastrointestinal grave)
Cirugía
Menor
Incluyendo extracciones
dentales
Mayor
30 - 60
60 - 100
Repetir cada 12 - 24 horas. Al menos 1 día hasta que el episodio hemorrágico se haya resuelto, en función del dolor, o hasta la cicatrización de la herida Repetir la perfusión cada 12 -24 horas durante 3 - 4 días o más, hasta que el dolor y discapacidad se hayan resuelto.
Repetir la perfusión cada 8 -24horas hasta que el riesgo desaparezca.
30 - 60
80 - 100 (pre-y
postoperatorio)
Cada 24 horas, al menos 1 día, hasta la cicatrización.
a) Como dosis en bolo Repetir la perfusión cada 8 -24 horas hasta la cicatrización adecuada de la herida; después continuar el tratamiento durante un mínimo de 7 días más para mantener una actividad de factor VIII del 30% al 60% (UI/dl).
b) Como perfusión continua Aumentar la actividad del factor VIII antes de la cirugía con una dosis de carga como perfusión e inmediatamente continuar con una perfusión continua (en UI/kg/h) ajustándola al aclaramiento diario del paciente y al nivel de factor VIII requerido durante un mínimo de
7 días.
La cantidad y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual. En determinadas circunstancias se pueden requerir cantidades superiores a las calculadas, especialmente en el caso de la dosis inicial.
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis a administrar y la frecuencia con la que se debe repetir la perfusión. En el caso concreto de las intervenciones de cirugía mayor, es indispensable controlar con precisión el tratamiento de sustitución mediante pruebas de la coagulación (actividad del factor VIII plasmático). La respuesta individual de cada paciente frente al factor VIII puede variar y presentar semividas y recuperaciones diferentes.
Para el cálculo de la velocidad de perfusión inicial, el aclaramiento se puede obtener realizando una curva de eliminación previa a la cirugía o bien empezar con un valor promedio (3,0-3,5 ml/kg/h) y después ajustar en función del paciente.
Velocidad de perfusión (en UI/kg/h) = Aclaramiento (en ml/h/kg) x nivel deseado de factor VIII (en UI/ml)
Durante la perfusión continua se ha demostrado la estabilidad clínica e in vitro empleando bombas ambulatorias con un reservorio de PVC. KOGENATE Bayer contiene un nivel bajo de polisorbato 80 como excipiente, del cual se sabe que incrementa la tasa de extracción del di- (2-etilhexil) ftalato (DEHF) de los materiales de PVC. Este aspecto se debe tener en cuenta en la administración por perfusión continua.
Profilaxis
En la profilaxis a largo plazo para prevenir hemorragias en pacientes con hemofilia A grave, las dosis habituales son de 20 a 40 UI de KOGENATE Bayer por kg de peso corporal, a intervalos de 2 a 3 días. En algunos casos, especialmente en pacientes más jóvenes, puede ser necesario un intervalo de dosis más corto o dosis mayores.
Poblaciones especiales
Población pediátrica
Se ha establecido la seguridad y eficacia de KOGENATE Bayer en niños de todas las edades. Se han obtenido datos de ensayos clínicos en 61 niños menores de 6 años y datos de estudios no intervencionales en niños de todas las edades.
Pacientes con inhibidores
Se debe controlar en los pacientes el desarrollo de inhibidores del factor VIII. Si no se alcanzan los niveles de actividad plasmática de factor VIII esperados, o si no se controla la hemorragia con una dosis adecuada, se debe realizar una prueba para determinar la presencia de inhibidores del factor VIII. Si el inhibidor está presente en niveles inferiores a 10 Unidades Bethesda (U.B.) por ml, la administración adicional de factor VIII de coagulación de origen recombinante puede neutralizarlo y permitir el tratamiento continuado clínicamente eficaz con KOGENATE Bayer. No obstante, en presencia de un inhibidor, las dosis requeridas son variables y se deben ajustar a la respuesta clínica, y a los valores de la actividad plasmática de factor VIII que se observen. En pacientes con títulos de inhibidores superiores a 10 U.B. o con una respuesta anamnésica alta deberá considerarse la utilización del concentrado de complejo protrombínico (activado) (CCPa) o de preparados de factor VII activado recombinante (rFVIIa). Estos tratamientos deberán ser dirigidos por médicos con experiencia en el tratamiento de pacientes hemofílicos.
Forma de administración
Vía intravenosa.
KOGENATE Bayer se debe administrar por vía intravenosa durante 2 a 5 minutos. La velocidad de administración se determinará en función del grado de bienestar del paciente (velocidad máxima de inyección: 2 ml/min).
Perfusión continua
KOGENATE Bayer se puede administrar mediante perfusión continua. La velocidad de perfusión debe calcularse en función del aclaramiento y del nivel deseado de FVIII.
Ejemplo: para un paciente de 75 Kg con un aclaramiento de 3 ml/Kg/h, la velocidad de perfusión inicial debería ser de 3 UI/Kg/h para obtener un nivel de FVIII del 100%. Para el cálculo de los ml/hora, multiplicar la velocidad de perfusión en UI/Kg/h por los Kg de peso corporal/concentración de la solución (UI/ml).
|
Tabla 2: Ejemplo para el cálculo de la velocidad de perfusión en la perfusión continua después del bolo inicial: |
Nivel plasmático requerido de FVIII |
Velocidad de perfusión UI/h/kg |
Velocidad de perfusión para un paciente de 75 Kg ml/h |
|
Aclaramiento: 3 ml/h/kg |
Concentraciones de la solución de FVIIIr 100 UI/ml 200 UI/ml 400 UI/ml | ||
|
100 % (1 UI/ml) |
3,0 |
2,25 1,125 0,56 | |
|
60% (0,6 UI/ml) |
1,8 |
1,35 0,68 0,34 | |
|
40% (0,4 UI/ml) |
1,2 |
0,9 0,45 0,225 |
Se puede requerir una mayor velocidad de perfusión en situaciones de aclaramiento acelerado durante hemorragias importantes o en caso de daño tisular extenso durante intervenciones quirúrgicas. Transcurridas las 24 horas iniciales de perfusión continua, se debe recalcular el aclaramiento a diario por medio de la ecuación del estado estacionario con el nivel medido de FVIII y la velocidad de perfusión, utilizando la siguiente ecuación:
Aclaramiento = velocidad de perfusión/nivel de FVIII real.
Durante la perfusión continua, las bolsas de perfusión se deben cambiar cada 24 horas.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver la sección 6.6 y el prospecto.
4.3 Contraindicaciones
• Hipersensibilidad al principio activo, o a alguno de los excipientes incluidos en la sección 6.1.
• Reacciones alérgicas conocidas a las proteínas de ratón o hámster.
4.4 Advertencias y precauciones especiales de empleo
Hipersensibilidad
Pueden producirse reacciones de hipersensibilidad de tipo alérgico con la administración del KOGENATE Bayer. Este medicamento contiene trazas de proteínas de ratón y hámster, así como proteínas de origen humano distintas del factor VIII (ver sección 5.1).
Si se producen síntomas de hipersensibilidad, se debe recomendar a los pacientes que interrumpan el uso del medicamento inmediatamente y contacten con su médico.
Se debe informar a los pacientes de los signos precoces de reacciones de hipersensibilidad, tales como ronchas, náuseas, urticaria generalizada, opresión torácica, sibilancia, hipotensión y anafilaxia.
En caso de shock, se debe implementar el tratamiento médico convencional para el shock.
Inhibidores
La formación de anticuerpos neutralizantes al factor VIII (inhibidores) es una complicación conocida en el tratamiento de pacientes con hemofilia A. Estos inhibidores son normalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, y se cuantifican en Unidades Bethesda (U.B.) por ml de plasma mediante el análisis modificado. El riesgo de desarrollo de inhibidores está correlacionado con la exposición al factor VIII y con factores genéticos entre otros, siendo el riesgo mayor en los primeros 20 días de tratamiento. Los inhibidores raramente se forman después de los primeros 100 días de tratamiento.
Se han observado casos recurrentes de inhibidores (con título bajo), después de cambiar de un Factor VIII a otro en los pacientes que tienen un tratamiento previo de más de 100 días de exposición y que tienen antecedentes de desarrollo de inhibidores. Por consiguiente, se recomienda controlar cuidadosamente a todos los pacientes para detectar la aparición de inhibidores tras cambiar de un medicamento a otro.
En general, todos los pacientes tratados con medicamentos con factor VIII de la coagulación deben ser controlados cuidadosamente mediante observación clínica y pruebas de laboratorio adecuadas para determinar la presencia de inhibidores.
Si no se alcanzan los niveles de actividad plasmática de factor VIII esperados, o si no se controla la hemorragia con una dosis adecuada, se deben realizar pruebas de la presencia de inhibidores del factor VIII. En pacientes con niveles elevados de inhibidor, el tratamiento con factor VIII puede no ser eficaz y se deben contemplar otras opciones terapéuticas. El tratamiento de dichos pacientes debe correr a cargo de médicos con experiencia en el tratamiento de la hemofilia y en los inhibidores del factor VIII.
Perfusión continua
En un ensayo clínico sobre el uso de la perfusión continua en cirugía, se utilizó como en cualquier perfusión intravenosa prolongada, heparina para prevenir una tromboflebitis en la zona de perfusión.
Contenido en sodio
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por vial, por lo que se considera esencialmente “exento de sodio”.
Episodios cardiovasculares
Los pacientes hemofílicos con factores de riesgo o enfermedades cardiovasculares pueden tener el mismo riesgo de padecer un episodio cardiovascular que los pacientes no hemofílicos, una vez que la coagulación se ha normalizado mediante el tratamiento con el FVIII.
Si hay factores de riesgo cardiovascular, el aumento de los niveles de factor VIII que se produce tras la administración puede situar al paciente, por lo menos, con el mismo riesgo de oclusión de un vaso o de padecer un infarto de miocardio que el del resto de la población no hemofílica. Por consiguiente, se debe evaluar y controlar la presencia de factores de riesgo cardiaco en los pacientes.
Complicaciones relacionadas con el catéter
Si se requiere un dispositivo de acceso venoso central (DAVC), se debe tener en cuenta el riesgo de complicaciones relacionadas con el DAVC, como por ejemplo, infecciones locales, bacteriemia y trombosis en el lugar de inserción del catéter.
Registro
Se recomienda encarecidamente que cada vez que se administre KOGENATE Bayer a un paciente, se registre el nombre del medicamento y su número de lote con el fin de mantener la trazabilidad entre el paciente y el lote del medicamento.
Las advertencias y precauciones enumeradas se aplican tanto a adultos como a niños.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han notificado interacciones de KOGENATE Bayer con otros medicamentos.
4.6 Fertilidad, embarazo y lactancia
No se han llevado a cabo estudios de reproducción animal con KOGENATE Bayer.
Embarazo y lactancia
Puesto que la hemofilia A es excepcional en mujeres, no se dispone de experiencia sobre el uso de KOGENATE Bayer durante el embarazo y el periodo de lactancia. Por consiguiente, KOGENATE Bayer sólo se utilizará durante el embarazo y la lactancia si está claramente indicado.
Fertilidad
No se dispone de información relativa a la fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de KOGENATE Bayer sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas Resumen del perfil de seguridad
Se han observado reacciones de hipersensibilidad o alérgicas (que puede incluir angioedema, quemazón y escozor en el lugar de perfusión, escalofríos, sofocos, urticaria generalizada, cefalea, ronchas, hipotensión, letargo, náuseas, inquietud, taquicardia, opresión torácica, hormigueo, vómitos, sibilancia) con productos con factor VIII recombinante y en algunos casos pueden evolucionar hasta anafilaxia grave (incluso shock). Concretamente, las reacciones de tipo dérmico pueden ser frecuentes, pero su progreso a anafilaxia grave (incluso shock) se considera raro.
Los pacientes con hemofilia A pueden formar anticuerpos neutralizantes (inhibidores) contra el factor VIII. La afección se puede manifestar como una respuesta clínica insuficiente. En tales casos, se recomienda contactar con un médico especialista en hemofilia.
Tabla de reacciones adversas
La tabla que se presenta a continuación sigue la clasificación de órganos del sistema MedDRA (nivel de SOC y término preferente).
Las frecuencias se han evaluado según la siguiente convención: Muy frecuentes: (>1/10), Frecuentes: (>1/100 a <1/10), Poco frecuentes: (>1/1.000 a <1/100), Raras: (>1/10.000 a <1/1.000), Muy raras:
(< 1/10.000), Frecuencia no conocida: (no puede estimarse a partir de los datos disponibles).
Tabla 3: Frecuencia de reacciones adversas al medicamento
|
Clasificación de órganos del sistema MedDRA |
Frecuencia | ||||
|
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
Muy raras/ no conocida | |
|
Trastornos de la sangre y del sistema linfático |
Formación de inhibidores del FVIII (notificada en los PUPs y MTPs)* |
Formación de inhibidores del FVIII (notificada en PTP en ensayos clínicos y estudios post-comercializaci ón)* | |||
|
Trastornos generales y alteraciones en el lugar de administració n |
Reacción local en el lugar de la perfusión |
Reacción febril relacionada con la perfusión (fiebre) | |||
|
Trastornos del sistema inmunológico |
Reacciones dermatológicas por hipersensibilida d (prurito, urticaria, eritema) |
Reacciones sistémicas de hipersensibilidad (incluyendo reacción anafiláctica, náuseas, alteración de la presión arterial, mareo) | |||
|
Trastornos del sistema nervioso |
Disgeusia | ||||
PUPs = pacientes sin tratamiento anterior.
PTPs = pacientes con tratamiento anterior.
MTPs = pacientes mínimamente tratados.
*Ver sección a continuación.
Descripción de las reacciones adversas seleccionadas
Desarrollo de inhibidores
Se ha notificado el desarrollo de inhibidores en pacientes no tratados y tratados previamente (PUPs / PTPs) (ver sección 4.4).
KOGENATE Bayer se ha utilizado en ensayos clínicos para el tratamiento de episodios hemorrágicos en 37 pacientes no tratados previamente (PUPs) y 23 pacientes pediátricos mínimamente tratados (MTPs, definidos como aquellos pacientes con < 4 días de exposición al tratamiento) con FVIII:C residual < de 2 UI/dl. Cinco de los 37 PUPs (14%) y 4 de los 23 MTPs (17%) tratados con KOGENATE Bayer desarrollaron inhibidores durante los primeros 20 días de exposición. En general, 9 de 60 (15%) desarrollaron inhibidores. Un paciente se perdió en el seguimiento y otro paciente desarrolló un título bajo de inhibidores durante el seguimiento posterior al estudio.
En un estudio observacional en pacientes con hemofilia A grave no tratados previamente, la incidencia de desarrollo de inhibidores fue de 64/183 (37,7%) con KOGENATE Bayer (tras un seguimiento de hasta 75 días de exposición).
En ensayos clínicos con 73 pacientes tratados previamente (PTPs, definidos como aquellos pacientes con > 100 días de exposición al tratamiento), no se observaron inhibidores de novo tras un seguimiento durante 4 años.
En amplios estudios observacionales post-comercialización con más de 1.000 pacientes tratados con KOGENATE Bayer, se observó que menos del 0,2% de los pacientes tratados previamente (PTPs) habían desarrollado inhibidores de-novo.
Población pediátrica
Se espera que la frecuencia, tipo y gravedad de las reacciones adversas en niños sean iguales que en todos los grupos de población salvo en cuanto a la formación de inhibidores.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
No se ha notificado ningún caso de sobredosis con factor VIII de la coagulación de origen recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos: Factor VIII de la coagulación, código ATC: B02BD02. Mecanismo de acción
El complejo de factor VIII/factor de von Willebrand (vWF) está formado por dos moléculas (factor VIII y vWF) con funciones fisiológicas diferentes. Cuando se perfunde a un paciente hemofílico, el factor VIII se une al vWF presente en la circulación del paciente. El factor VIII activado actúa como cofactor del factor IX activado, acelerando la conversión del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Seguidamente, la trombina convierte el fibrinógeno en fibrina, formándose el coágulo. La hemofilia A es una alteración de la coagulación sanguínea hereditaria ligada al sexo y se debe a la presencia de niveles reducidos de factor VIII:C que da lugar a un sangrado profuso en las articulaciones, músculos u órganos internos, ya sea de forma espontánea o a causa de un traumatismo accidental o quirúrgico. El tratamiento de sustitución aumenta los niveles plasmáticos de factor VIII, obteniéndose una corrección temporal del déficit de este factor y de la diátesis hemorrágica.
Efectos farmacodinámicos
La determinación del tiempo de tromboplastina parcial activada (TTPa) es un método de ensayo in vitro convencional para determinar la actividad biológica del factor VIII. El TTPa está prolongado en todos los pacientes hemofílicos. El grado y duración de la normalización del TTPa observados tras la administración de KOGENATE Bayer son similares a los obtenidos con factor VIII derivado del plasma.
En un ensayo clínico con pacientes adultos afectados de hemofilia A sometidos a cirugía mayor se ha demostrado que KOGENATE Bayer se puede administrar mediante perfusión continua en intervenciones quirúrgicas (antes, durante y después de la intervención quirúrgica). En este estudio, se utilizó heparina para prevenir tromboflebitis en la zona de la perfusión, como en cualquier proceso que incluya una perfusión intravenosa durante un tiempo prolongado.
Hipersensibilidad
Durante los ensayos, ningún paciente desarrolló títulos de anticuerpos con repercusión clínica frente a las cantidades mínimas de proteína de ratón y hámster presentes en el medicamento. Sin embargo, en pacientes con cierta predisposición, existe la posibilidad de reacción alérgica a los componentes del medicamento, como por ejemplo, a las pequeñas cantidades de proteína de ratón y hámster (ver secciones 4.3 y 4.4).
Inducción de inmunotolerancia (ITI)
Se han recogido datos sobre inducción de inmunotolerancia en pacientes con hemofilia A que habían desarrollado inhibidores del factor VIII. En la revisión retrospectiva sobre 40 pacientes se seleccionaron 39 en un estudio clínico independiente. Los datos mostraron que KOGENATE Bayer se ha utilizado para inducir inmunotolerancia. Una vez conseguida la inmunotolerancia, los episodios hemorrágicos en esos pacientes se podían prevenir o controlar de nuevo con KOGENATE Bayer. En estas condiciones, el paciente podía continuar un tratamiento profiláctico como terapia de mantenimiento.
5.2 Propiedades farmacocinéticas
Absorción
El análisis de los valores de recuperación in vivo registrados en pacientes tratados previamente ha demostrado un incremento promedio del 2% por UI/Kg de peso corporal para KOGENATE Bayer. Este valor es similar a los valores registrados con el factor VIII derivado del plasma humano.
Distribución y eliminación
Tras la administración de KOGENATE Bayer, la actividad máxima del factor VIII disminuye siguiendo una curva exponencial de dos fases con un promedio de semivida terminal de 15 horas, aproximadamente. Este valor es similar al observado con el factor VIII derivado del plasma, cuya semivida terminal media es de 13 horas, aproximadamente. Otros parámetros farmacocinéticos de KOGENATE Bayer para la dosis de carga como perfusión son: tiempo medio de residencia [TMR (0 - 48)] 22 horas, aproximadamente; aclaramiento: 160 ml/h, aproximadamente. El aclaramiento basal medio para 14 pacientes adultos sometidos a cirugía mayor con perfusión continua es de 188 ml/h, que corresponde a 3,0 ml/Kg/h (intervalo 1,6-4,6 ml/Kg/h).
5.3 Datos preclínicos sobre seguridad
Incluso a dosis varias veces superiores a la dosis clínica recomendada (en relación al peso corporal), no se evidenció ningún efecto tóxico agudo o subagudo de KOGENATE Bayer en animales de laboratorio (ratón, rata, conejo y perro).
No se han llevado a cabo estudios específicos de administración repetida de octocog alfa tales como la toxicidad de reproducción, toxicidad crónica y carcinogénesis, debido a la respuesta inmune a las proteínas heterólogas entre las especies de mamíferos distintas al hombre.
No se han realizado estudios sobre el potencial mutagénico de KOGENATE Bayer, ya que no se ha detectado ningún potencial mutagénico in vivo o in vitro del compuesto precursor de KOGENATE Bayer.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo
Glicina
Cloruro de sodio Cloruro de calcio Histidina Polisorbato 80 Sacarosa
Disolvente
Agua para preparaciones inyectables
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros, excepto con los mencionados en sección 6.6.
Sólo se debe utilizar para la reconstitución y administración el equipo que se suministra (vial conteniendo el polvo con el sistema Bio-Set, jeringa precargada con el disolvente y equipo para punción venosa), ya que se pueden producir fallos en el tratamiento debido a la adsorción del factor VIII recombinante de la coagulación a la superficie interna de algunos equipos de perfusión.
6.3 Periodo de validez
30 meses.
Tras la reconstitución, desde el punto de vista microbiológico, el medicamento se debe utilizar inmediatamente. Si no se utiliza inmediatamente, los tiempos de conservación en uso y las condiciones previas al mismo son responsabilidad del usuario.
No obstante, en estudios in vitro se ha demostrado que la estabilidad química y física en uso es de 24 horas a 30°C en bolsas de PVC para perfusión continua. Una vez reconstituido, se ha comprobado in vitro que la estabilidad química y física en uso es de 3 horas.
No refrigerar una vez reconstituido.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C). No congelar. Conservar el vial y la jeringa precargada en el embalaje exterior para protegerlos de la luz.
Durante el periodo de validez global de 30 meses, el medicamento envasado se puede conservar a temperatura ambiente (inferior a 25°C) durante un periodo máximo de 12 meses. En este caso, el medicamento caduca al finalizar el periodo de 12 meses o en la fecha de caducidad indicada en el vial del medicamento, según cuál sea antes. Se debe anotar la nueva fecha de caducidad en el embalaje exterior.
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase y de los equipos especiales para su utilización, administración o implantación
Cada envase de KOGENATE Bayer contiene:
• un vial y sistema Bio-Set que contiene el polvo (vial de vidrio tipo 1, transparente, de 10 ml con tapón de mezcla de caucho halogenobutílico gris exento de látex y sistema de transferencia con tapón protector [Bio-Set])
• una jeringa precargada con 2,5 ml (para 250 UI, 500 UI, y 1000 UI) o 5 ml (para 2000 UI y 3000 UI) de disolvente (cilindro de vidrio tipo 1, transparente, con tapones de mezcla de caucho bromobutílico gris exento de látex)
• un émbolo
• un equipo para punción venosa
• dos gasas impregnadas en alcohol de un sólo uso
• dos gasas estériles secas
• dos tiras adhesivas
6.6 Precauciones especiales de eliminación y otras manipulaciones
El prospecto incluido en el envase de KOGENATE Bayer contiene las instrucciones para su preparación y administración.
El medicamento reconstituido es una solución transparente e incolora.
El polvo de KOGENATE Bayer se debe reconstituir únicamente con el disolvente suministrado (2,5 ml (para 250 UI, 500 UI, y 1000 UI) o 5 ml (para 2000 UI y 3000 UI) de agua para preparaciones inyectables) en la jeringa precargada y sistema de transferencia integrado (Bio-Set). Para la perfusión, el medicamento se debe preparar en condiciones asépticas. Si alguno de los componentes del envase está abierto o dañado, no use este componente.
Girar suavemente el vial, sin agitar, hasta que se haya disuelto todo el medicamento. Tras la reconstitución, la solución es transparente. Los medicamentos que se administran por vía parenteral se deben inspeccionar visualmente para detectar la presencia de partículas o un cambio de color antes de su administración. No utilizar KOGENATE Bayer si se observan partículas o turbidez en su interior. Una vez reconstituida, la solución se transfiere de nuevo a la jeringa. KOGENATE Bayer se debe reconstituir y administrar con los componentes suministrados en cada envase.
El medicamento reconstituido se debe filtrar antes de la administración para eliminar posibles partículas presentes en la solución. La filtración se puede realizar siguiendo los pasos para la reconstitución y/o administración descritas en el prospecto suministrado con KOGENATE Bayer. Es importante utilizar el equipo de punción venosa suministrado con el medicamento para la administración, ya que contiene un filtro en línea.
En situaciones en las que no se pueda utilizar el equipo de punción venosa suministrado (p. ej., cuando se administre a través de una vía periférica o central), se debe emplear un filtro que sea compatible con KOGENATE Bayer. Estos filtros compatibles son de tipo adaptador luer con carcasa poliacrílica y llevan integrado un filtro tamiz de poliamida con un tamaño de malla de 5 - 20 micras.
El equipo de punción venosa suministrado con el medicamento no se debe usar para la extracción de sangre, porque contiene un filtro en línea. Cuando se deba extraer sangre antes de una perfusión, se usará un equipo de administración sin filtro y, a continuación, se administrará KOGENATE Bayer mediante un filtro de inyección.
Si desea realizar alguna consulta sobre KOGENATE Bayer y uso de otros filtros compatibles, póngase en contacto con Bayer Pharma AG.
Para un solo uso.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG 13342 Berlin Alemania
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
|
EU/1/00/143/004 - |
KOGENATE Bayer 250 UI |
|
EU/1/00/143/005 - |
KOGENATE Bayer 500 UI |
|
EU/1/00/143/006 -EU/1/00/143/010 -EU/1/00/143/012 - |
KOGENATE Bayer 1000 UI KOGENATE Bayer 2000 UI KOGENATE Bayer 3000 UI |
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 04 de agosto de 2000 Fecha de la última renovación: 06 de agosto de 2010
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
1. NOMBRE DEL MEDICAMENTO
KOGENATE Bayer 250 UI polvo y disolvente para solución inyectable KOGENATE Bayer 500 UI polvo y disolvente para solución inyectable KOGENATE Bayer 1000 UI polvo y disolvente para solución inyectable KOGENATE Bayer 2000 UI polvo y disolvente para solución inyectable KOGENATE Bayer 3000 UI polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 250/500/1000/2000/3000 UI de factor VIII de coagulación humano (DCI: octocog alfa).
El factor VIII de coagulación humano se produce mediante técnicas de ADN recombinante (ADN r) en células de riñón de crías de hámster que contienen el gen del factor VIII humano.
• Después de la reconstitución, 1 ml de KOGENATE Bayer contiene aproximadamente 100 UI
(250 UI/2,5 ml) de factor VIII de coagulación humano (DCI: octocog alfa).
• Después de la reconstitución, 1 ml de KOGENATE Bayer 500 UI contiene aproximadamente
200 UI (500 UI/2,5 ml) de factor VIII de coagulación humano (DCI: octocog alfa).
• Después de la reconstitución, 1 ml de KOGENATE Bayer 1000 UI contiene aproximadamente
400 UI (1000 UI/2,5 ml) de factor VIII de coagulación humano (DCI: octocog alfa).
• Después de la reconstitución, 1 ml de KOGENATE Bayer 2000 UI contiene aproximadamente
400 UI (2000 UI/5 ml) de factor VIII de coagulación humano (DCI: octocog alfa).
• Después de la reconstitución, 1 ml de KOGENATE Bayer 3000 UI contiene aproximadamente
600 UI (3000 UI/5 ml) de factor VIII de coagulación humano (DCI: octocog alfa).
La potencia (UI) se determina utilizando el ensayo de coagulación en una etapa, en comparación con el estándar Mega de la FDA, que ha sido calibrado frente al estándar de la OMS en Unidades Internacionales (UI).
La actividad específica de KOGENATE Bayer es, aproximadamente, de 4000 UI/mg de proteína. Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable (adaptador de vial).
Polvo: polvo suelto o sólido friable de color blanco o ligeramente amarillento. Disolvente: agua para preparaciones inyectables, solución transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (déficit congénito de factor VIII). Este medicamento no contiene factor de von Willebrand y por lo tanto no está indicado en la enfermedad de von Willebrand.
Este medicamento está indicado en adultos, adolescentes y niños de cualquier edad.
4.2 Posología y forma de administración
El tratamiento se debe realizar bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia.
Posología
El número de unidades de factor VIII administradas se expresa en Unidades Internacionales (UI), que se corresponden con el estándar actual de la OMS para medicamentos de factor VIII. La actividad del factor VIII en plasma se puede expresar en porcentaje (referido al plasma humano normal) o bien, en Unidades Internacionales (referido al estándar internacional para el factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII equivale a la cantidad de factor VIII presente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis necesaria de factor VIII se basa en el hallazgo empírico de que 1 Unidad Internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad plasmática de factor VIII en 1,5% - 2,5% de la actividad normal. La dosis requerida se determina utilizando las fórmulas siguientes:
I. UI requeridas = peso corporal (kg) * aumento deseado de factor VIII (% del normal) x 0,5
II. Aumento previsto del factor VIII (% del normal) = 2 x UI administradas
peso corporal (kg)
La dosis, frecuencia y duración del tratamiento sustitutivo se debe individualizar según las necesidades del paciente (peso, gravedad del trastorno de la función hemostática, localización e intensidad de la hemorragia, presencia de inhibidores y del nivel deseado de Factor VIII).
La tabla siguiente proporciona una guía de los niveles sanguíneos mínimos de factor VIII en diferentes situaciones clínicas. En el caso de las hemorragias que aparecen en la lista, el nivel de actividad del factor VIII no podrá ser inferior al nivel indicado (en % del normal), en el período correspondiente:
Grado de la hemorragia/Tipo de procedimiento quirúrgico
Nivel de factor VIII requerido (%) (UI/dl)_
Frecuencia de dosificación (horas)/Duración de la terapia (días)_
Hemorragia
Hemartrosis precoz, sangrado muscular o sangrado de la cavidad oral
20 - 40
Hemartrosis más extensa, sangrado muscular o hematoma
Hemorragias con riesgo vital (como sangrado intracraneal, de garganta, sangrado gastrointestinal grave)
Cirugía
Menor
Incluyendo extracciones
dentales
Mayor
30 - 60
60 - 100
Repetir cada 12 - 24 horas. Al menos 1 día hasta que el episodio hemorrágico se haya resuelto, en función del dolor, o hasta la cicatrización de la herida Repetir la perfusión cada 12 -24 horas durante 3 - 4 días o más, hasta que el dolor y discapacidad se hayan resuelto.
Repetir la perfusión cada 8 -24 horas hasta que el riesgo desaparezca.
30 - 60
80 - 100 (pre-y
postoperatorio)
Cada 24 horas, al menos 1 día, hasta la cicatrización.
a) Como dosis en bolo Repetir la perfusión cada 8 -24 horas hasta la cicatrización adecuada de la herida; después continuar el tratamiento durante un mínimo de 7 días más para mantener una actividad de factor VIII del 30% al 60% (UI/dl).
b) Como perfusión continua Aumentar la actividad del factor VIII antes de la cirugía con una dosis de carga como perfusión e inmediatamente continuar con una perfusión continua (en UI/kg/h) ajustándola al aclaramiento diario del paciente y al nivel de factor VIII requerido durante un mínimo de
7 días.
La cantidad y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual. En determinadas circunstancias se pueden requerir cantidades superiores a las calculadas, especialmente en el caso de la dosis inicial.
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis a administrar y la frecuencia con la que se debe repetir la perfusión. En el caso concreto de las intervenciones de cirugía mayor, es indispensable controlar con precisión el tratamiento de sustitución mediante pruebas de la coagulación (actividad del factor VIII plasmático). La respuesta individual de cada paciente frente al factor VIII puede variar y presentar semividas y recuperaciones diferentes.
Para el cálculo de la velocidad de perfusión inicial, el aclaramiento se puede obtener realizando una curva de eliminación previa a la cirugía o bien empezar con un valor promedio (3,0-3,5 ml/kg/h) y después ajustar en función del paciente.
Velocidad de perfusión (en UI/kg/h) = Aclaramiento (en ml/h/kg) x nivel deseado de factor VIII (en UI/ml)
Durante la perfusión continua se ha demostrado la estabilidad clínica e in vitro empleando bombas ambulatorias con un reservorio de PVC. KOGENATE Bayer contiene un nivel bajo de polisorbato 80 como excipiente, del cual se sabe que incrementa la tasa de extracción del di- (2-etilhexil) ftalato (DEHF) de los materiales de PVC. Este aspecto se debe tener en cuenta en la administración por perfusión continua.
Profilaxis
En la profilaxis a largo plazo para prevenir hemorragias en pacientes con hemofilia A grave, las dosis habituales son de 20 a 40 UI de KOGENATE Bayer por kg de peso corporal, a intervalos de 2 a 3 días. En algunos casos, especialmente en pacientes más jóvenes, puede ser necesario un intervalo de dosis más corto o dosis mayores.
Poblaciones especiales
Población pediátrica
Se ha establecido la seguridad y eficacia de KOGENATE Bayer en niños de todas las edades. Se han obtenido datos de ensayos clínicos en 61 niños menores de 6 años y datos de estudios no intervencionales en niños de todas las edades.
Pacientes con inhibidores
Se debe controlar en los pacientes el desarrollo de inhibidores del factor VIII. Si no se alcanzan los niveles de actividad plasmática de factor VIII esperados, o si no se controla la hemorragia con una dosis adecuada, se debe realizar una prueba para determinar la presencia de inhibidores del factor VIII. Si el inhibidor está presente en niveles inferiores a 10 Unidades Bethesda (U.B.) por ml, la administración adicional de factor VIII de coagulación de origen recombinante puede neutralizarlo y permitir el tratamiento continuado clínicamente eficaz con KOGENATE Bayer. No obstante, en presencia de un inhibidor, las dosis requeridas son variables y se deben ajustar a la respuesta clínica, y a los valores de la actividad plasmática de factor VIII que se observen. En pacientes con títulos de inhibidores superiores a 10 U.B. o con una respuesta anamnésica alta deberá considerarse la utilización del concentrado de complejo protrombínico (activado) (CCPa) o de preparados de factor VII activado recombinante (rFVIIa). Estos tratamientos deberán ser dirigidos por médicos con experiencia en el tratamiento de pacientes hemofílicos.
Forma de administración
Vía intravenosa.
KOGENATE Bayer se debe administrar por vía intravenosa durante 2 a 5 minutos. La velocidad de administración se determinará en función del grado de bienestar del paciente (velocidad máxima de inyección: 2 ml/min).
Perfusión continua
KOGENATE Bayer se puede administrar mediante perfusión continua. La velocidad de perfusión debe calcularse en función del aclaramiento y del nivel deseado de FVIII
Ejemplo: para un paciente de 75 Kg con un aclaramiento de 3 ml/Kg/h, la velocidad de perfusión inicial debería ser de 3 UI/Kg/h para obtener un nivel de FVIII del 100%. Para el cálculo de los ml/hora, multiplicar la velocidad de perfusión en UI/Kg/h por los Kg de peso corporal/concentración de la solución (UI/ml).
|
Tabla 2: Ejemplo para el cálculo de la velocidad de perfusión en la perfusión continua después del bolo inicial: |
Nivel plasmático requerido de FVIII |
Velocidad de perfusión UI/h/kg |
Velocidad de perfusión para un paciente de 75 Kg ml/h |
|
Aclaramiento: 3 ml/h/kg |
Concentraciones de la solución de FVIIIr 100 UI/ml 200 UI/ml 400 UI/ml | ||
|
100 % (1 UI/ml) |
3,0 |
2,25 1,125 0,56 | |
|
60% (0,6 UI/ml) |
1,8 |
1,35 0,68 0,34 | |
|
40% (0,4 UI/ml) |
1,2 |
0,9 0,45 0,225 |
Se puede requerir una mayor velocidad de perfusión en situaciones de aclaramiento acelerado durante hemorragias importantes o en caso de daño tisular extenso durante intervenciones quirúrgicas. Transcurridas las 24 horas iniciales de perfusión continua, se debe recalcular el aclaramiento a diario por medio de la ecuación del estado estacionario con el nivel medido de FVIII y la velocidad de perfusión, utilizando la siguiente ecuación:
Aclaramiento = velocidad de perfusión/nivel de FVIII real.
Durante la perfusión continua, las bolsas de perfusión se deben cambiar cada 24 horas.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver la sección 6.6 y el prospecto.
4.3 Contraindicaciones
• Hipersensibilidad al principio activo, o a alguno de los excipientes incluidos en la sección 6.1.
• Reacciones alérgicas conocidas a las proteínas de ratón o hámster.
4.4 Advertencias y precauciones especiales de empleo
Hipersensibilidad
Pueden producirse reacciones de hipersensibilidad de tipo alérgico con la administración del KOGENATE Bayer. Este medicamento contiene trazas de proteínas de ratón y hámster, así como proteínas de origen humano distintas del factor VIII (ver sección 5.1).
Si se producen síntomas de hipersensibilidad, se debe recomendar a los pacientes que interrumpan el uso del medicamento inmediatamente y contacten con su médico.
Se debe informar a los pacientes de los signos precoces de reacciones de hipersensibilidad, tales como ronchas, náuseas, urticaria generalizada, opresión torácica, sibilancia, hipotensión y anafilaxia.
En caso de shock, se debe implementar el tratamiento médico convencional para el shock.
Inhibidores
La formación de anticuerpos neutralizantes al factor VIII (inhibidores) es una complicación conocida en el tratamiento de pacientes con hemofilia A. Estos inhibidores son normalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, y se cuantifican en Unidades Bethesda (U.B.) por ml de plasma mediante el análisis modificado. El riesgo de desarrollo de inhibidores está correlacionado con la exposición al factor VIII y con factores genéticos entre otros, siendo el riesgo mayor en los primeros 20 días de tratamiento. Los inhibidores raramente se forman después de los primeros 100 días de tratamiento.
Se han observado casos recurrentes de inhibidores (con título bajo), después de cambiar de un Factor VIII recombinante a otro en los pacientes que tienen un tratamiento previo de más de 100 días de exposición y que tienen antecedentes de desarrollo de inhibidores. . Por consiguiente, se recomienda controlar cuidadosamente a todos los pacientes para detectar la aparición de inhibidores tras cambiar de un medicamento a otro.
En general, todos los pacientes tratados con medicamentos con factor VIII de la coagulación deben ser controlados cuidadosamente mediante observación clínica y pruebas de laboratorio adecuadas para determinar la presencia de inhibidores. Si no se alcanzan los niveles de actividad plasmática de factor VIII esperados, o si no se controla la hemorragia con una dosis adecuada, se deben realizar pruebas de la presencia de inhibidores del factor VIII. En pacientes con niveles elevados de inhibidor, el tratamiento con factor VIII puede no ser eficaz y se deben contemplar otras opciones terapéuticas.
El tratamiento de dichos pacientes debe correr a cargo de médicos con experiencia en el tratamiento de la hemofilia y en los inhibidores del factor VIII.
Perfusión continua
En un ensayo clínico sobre el uso de la perfusión continua en cirugía, se utilizó como en cualquier perfusión intravenosa prolongada, heparina para prevenir una tromboflebitis en la zona de perfusión.
Contenido en sodio
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por vial, por lo que se considera esencialmente “exento de sodio”.
Episodios cardiovasculares
Los pacientes hemofílicos con factores de riesgo o enfermedades cardiovasculares pueden tener el mismo riesgo de padecer un episodio cardiovascular que los pacientes no hemofílicos, una vez que la coagulación se ha normalizado mediante el tratamiento con el FVIII.
Si hay factores de riesgo cardiovascular, el aumento de los niveles de factor VIII que se produce tras la administración puede situar al paciente, por lo menos, con el mismo riesgo de oclusión de un vaso o de padecer un infarto de miocardio que el del resto de la población no hemofílica. Por consiguiente, se debe evaluar y controlar la presencia de factores de riesgo cardiaco en los pacientes.
Complicaciones relacionadas con el catéter
Si se requiere un dispositivo de acceso venoso central (DAVC), se debe tener en cuenta el riesgo de complicaciones relacionadas con el DAVC, como por ejemplo, infecciones locales, bacteriemia y trombosis en el lugar de inserción del catéter.
Registro
Se recomienda encarecidamente que cada vez que se administre KOGENATE Bayer a un paciente, se registre el nombre del medicamento y su número de lote con el fin de mantener la trazabilidad entre el paciente y el lote del medicamento.
Las advertencias y precauciones enumeradas se aplican tanto a adultos como a niños.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han notificado interacciones de KOGENATE Bayer con otros medicamentos.
4.6 Fertilidad, embarazo y lactancia
No se han llevado a cabo estudios de reproducción animal con KOGENATE Bayer.
Embarazo y lactancia
Puesto que la hemofilia A es excepcional en mujeres, no se dispone de experiencia sobre el uso de KOGENATE Bayer durante el embarazo y el periodo de lactancia. Por consiguiente, KOGENATE Bayer sólo se utilizará durante el embarazo y la lactancia si está claramente indicado.
Fertilidad
No se dispone de información relativa a la fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de KOGENATE Bayer sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas Resumen del perfil de seguridad
Se han observado reacciones de hipersensibilidad o alérgicas (que puede incluir angioedema, quemazón y escozor en el lugar de perfusión, escalofríos, sofocos, urticaria generalizada, cefalea, ronchas, hipotensión, letargo, náuseas, inquietud, taquicardia, opresión torácica, hormigueo, vómitos, sibilancia) con productos con factor VIII recombinante y en algunos casos pueden evolucionar hasta anafilaxia grave (incluso shock). Concretamente, las reacciones de tipo dérmico pueden ser frecuentes, pero su progreso a anafilaxia grave (incluso shock) se considera raro.
Los pacientes con hemofilia A pueden formar anticuerpos neutralizantes (inhibidores) contra el factor VIII. La afección se puede manifestar como una respuesta clínica insuficiente. En tales casos, se recomienda contactar con un médico especialista en hemofilia.
Tabla de reacciones adversas
La tabla que se presenta a continuación sigue la clasificación de órganos del sistema del sistema MedDRA (nivel de SOC y término preferente).
Las frecuencias se han evaluado según la siguiente convención: Muy frecuentes: (>1/10), Frecuentes: (>1/100 a <1/10), Poco frecuentes: (>1/1.000 a <1/100), Raras: (>1/10.000 a <1/1.000), Muy raras:
(< 1/10.000), Frecuencia no conocida: (no puede estimarse a partir de los datos disponibles).
|
Tabla 3: Frecuencia de reacciones adversas al medicament oClasificación de órganos del sistema MedDRA |
Frecuencia | ||||
|
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
Muy raras/ no conocida | |
|
Trastornos de la sangre y del sistema linfático |
Formación de inhibidores del FVIII (notificada en los PUPs y MTPs)* |
Formación de inhibidores del FVIII (notificada en PTP en ensayos clínicos y estudios post-comercializaci ón)* | |||
|
Trastornos generales y alteraciones en el lugar de administració n |
Reacción local en el lugar de la perfusión |
Reacción febril relacionada con la perfusión (fiebre) | |||
|
Trastornos del sistema inmunológico |
Reacciones dermatológicas por hipersensibilida d (prurito, urticaria, eritema) |
Reacciones sistémicas de hipersensibilidad (incluyendo reacción anafiláctica, náuseas, alteración de la presión arterial, mareo) | |||
|
Trastornos del sistema nervioso |
Disgeusia | ||||
PUPs = pacientes sin tratamiento anterior.
PTPs = pacientes con tratamiento anterior.
MTPs = pacientes mínimamente tratados.
*Ver sección a continuación.
Descripción de las reacciones adversas seleccionadas
Desarrollo de inhibidores
Se ha notificado el desarrollo de inhibidores en pacientes no tratados y tratados previamente (PUPs / PTPs) (ver sección 4.4).
KOGENATE Bayer se ha utilizado en ensayos clínicos para el tratamiento de episodios hemorrágicos en 37 pacientes no tratados previamente (PUPs) y 23 pacientes pediátricos mínimamente tratados (MTPs, definidos como aquellos pacientes con < 4 días de exposición al tratamiento) con FVIII:C residual < de 2 Ul/dl. Cinco de los 37 PUPs (14%) y 4 de los 23 MTPs (17%) tratados con KOGENATE Bayer desarrollaron inhibidores durante los primeros 20 días de exposición. En general,
9 de 60 (15%) desarrollaron inhibidores. Un paciente se perdió en el seguimiento y otro paciente desarrolló un título bajo de inhibidores durante el seguimiento posterior al estudio.
En un estudio observacional en pacientes con hemofilia A grave no tratados previamente, la incidencia de desarrollo de inhibidores fue de 64/183 (37,7%) con KOGENATE Bayer (tras un seguimiento de hasta 75 días de exposición).
En ensayos clínicos con 73 pacientes tratados previamente (PTPs, definidos como aquellos pacientes con >100 días de exposición al tratamiento), no se observaron inhibidores de novo tras un seguimiento durante 4 años.
En amplios estudios observacionales post-comercialización con más de 1.000 pacientes tratados con KOGENATE Bayer, se observó que menos del 0,2% de los pacientes tratados previamente (PTPs) habían desarrollado inhibidores de-novo.
Población pediátrica
Se espera que la frecuencia, tipo y gravedad de las reacciones adversas en niños sean iguales que en todos los grupos de población salvo en cuanto a la formación de inhibidores.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
No se ha notificado ningún caso de sobredosis con factor VIII de la coagulación de origen recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos: Factor VIII de la coagulación, código ATC: B02BD02. Mecanismo de acción
El complejo de factor VIII/factor de von Willebrand (vWF) está formado por dos moléculas (factor VIII y vWF) con funciones fisiológicas diferentes. Cuando se perfunde a un paciente hemofílico, el factor VIII se une al vWF presente en la circulación del paciente. El factor VIII activado actúa como cofactor del factor IX activado, acelerando la conversión del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Seguidamente, la trombina convierte el fibrinógeno en fibrina, formándose el coágulo. La hemofilia A es una alteración de la coagulación sanguínea hereditaria ligada al sexo y se debe a la presencia de niveles reducidos de factor VIII:C que da lugar a un sangrado profuso en las articulaciones, músculos u órganos internos, ya sea de forma espontánea o a causa de un traumatismo accidental o quirúrgico. El tratamiento de sustitución aumenta los niveles plasmáticos de factor VIII, obteniéndose una corrección temporal del déficit de este factor y de la diátesis hemorrágica.
Efectos farmacodinámicos
La determinación del tiempo de tromboplastina parcial activada (TTPa) es un método de ensayo in vitro convencional para determinar la actividad biológica del factor VIII. El TTPa está prolongado en todos los pacientes hemofílicos. El grado y duración de la normalización del TTPa observados tras la administración de KOGENATE Bayer son similares a los obtenidos con factor VIII derivado del plasma.
En un ensayo clínico con pacientes adultos afectados de hemofilia A sometidos a cirugía mayor se ha demostrado que KOGENATE Bayer se puede administrar mediante perfusión continua en intervenciones quirúrgicas (antes, durante y después de la intervención quirúrgica). En este estudio, se utilizó heparina para prevenir tromboflebitis en la zona de la perfusión, como en cualquier proceso que incluya una perfusión intravenosa durante un tiempo prolongado.
Hipersensibilidad
Durante los ensayos, ningún paciente desarrolló títulos de anticuerpos con repercusión clínica frente a las cantidades mínimas de proteína de ratón y hámster presentes en el medicamento. Sin embargo, en pacientes con cierta predisposición, existe la posibilidad de reacción alérgica a los componentes del medicamento, como por ejemplo, a las pequeñas cantidades de proteína de ratón y hámster (ver secciones 4.3 y 4.4).
Inducción de inmunotolerancia (ITI)
Se han recogido datos sobre inducción de inmunotolerancia en pacientes con hemofilia A que habían desarrollado inhibidores del factor VIII. En la revisión retrospectiva sobre 40 pacientes se seleccionaron 39 en un estudio clínico independiente. Los datos mostraron que KOGENATE Bayer se ha utilizado para inducir inmunotolerancia. Una vez conseguida la inmunotolerancia, los episodios hemorrágicos en esos pacientes se podían prevenir o controlar de nuevo con KOGENATE Bayer. En estas condiciones, el paciente podía continuar un tratamiento profiláctico como terapia de mantenimiento.
5.2 Propiedades farmacocinéticas
Absorción
El análisis de los valores de recuperación in vivo registrados en pacientes tratados previamente ha demostrado un incremento promedio del 2% por UI/Kg de peso corporal para KOGENATE Bayer. Este valor es similar a los valores registrados con el factor VIII derivado del plasma humano.
Distribución y eliminación
Tras la administración de KOGENATE Bayer, la actividad máxima del factor VIII disminuye siguiendo una curva exponencial de dos fases con un promedio de semivida terminal de 15 horas, aproximadamente. Este valor es similar al observado con el factor VIII derivado del plasma, cuya semivida terminal media es de 13 horas, aproximadamente. Otros parámetros farmacocinéticos de KOGENATE Bayer para la dosis de carga como perfusión son: tiempo medio de residencia [TMR (0 - 48)] 22 horas, aproximadamente; aclaramiento: 160 ml/h, aproximadamente. El aclaramiento basal medio para 14 pacientes adultos sometidos a cirugía mayor con perfusión continua es de 188 ml/h, que corresponde a 3,0 ml/Kg/h (intervalo 1,6-4,6 ml/Kg/h).
5.3 Datos preclínicos sobre seguridad
Incluso a dosis varias veces superiores a la dosis clínica recomendada (en relación al peso corporal), no se evidenció ningún efecto tóxico agudo o subagudo de KOGENATE Bayer en animales de laboratorio (ratón, rata, conejo y perro).
No se han llevado a cabo estudios específicos de administración repetida de octocog alfa tales como la toxicidad de reproducción, toxicidad crónica y carcinogénesis, debido a la respuesta inmune a las proteínas heterólogas entre las especies de mamíferos distintas al hombre.
No se han realizado estudios sobre el potencial mutagénico de KOGENATE Bayer, ya que no se ha detectado ningún potencial mutagénico in vivo o in vitro del compuesto precursor de KOGENATE Bayer.
6. DATOS FARMACÉUTICOS 6.1 Lista de excipientes
Polvo
Glicina
Cloruro de sodio Cloruro de calcio Histidina Polisorbato 80 Sacarosa
Disolvente
Agua para preparaciones inyectables
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros, excepto con los mencionados en sección 6.6.
Sólo se debe utilizar para la reconstitución y administración el equipo que se suministra (vial conteniendo el polvo, jeringa precargada con el disolvente, adaptador de vial y equipo para punción venosa), ya que se pueden producir fallos en el tratamiento debido a la adsorción del factor VIII recombinante de la coagulación a la superficie interna de algunos equipos de perfusión.
6.3 Periodo de validez
30 meses.
Tras la reconstitución, desde el punto de vista microbiológico, el medicamento se debe utilizar inmediatamente. Si no se utiliza inmediatamente, los tiempos de conservación en uso y las condiciones previas al mismo son responsabilidad del usuario.
No obstante, en estudios in vitro se ha demostrado que la estabilidad química y física en uso es de 24 horas a 30°C en bolsas de PVC para perfusión continua. Una vez reconstituido, se ha comprobado in vitro que la estabilidad química y física en uso es de 3 horas.
No refrigerar una vez reconstituido.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C). No congelar. Conservar el vial y la jeringa precargada en el embalaje exterior para protegerlos de la luz.
Durante el periodo de validez global de 30 meses, el medicamento envasado se puede conservar a temperatura ambiente (inferior a 25°C) durante un periodo máximo de 12 meses. En este caso, el medicamento caduca al finalizar el periodo de 12 meses o en la fecha de caducidad indicada en el vial del medicamento, según cuál sea antes. Se debe anotar la nueva fecha de caducidad en el embalaje exterior.
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase y de los equipos especiales para su utilización, administración o implantación
Cada envase de KOGENATE Bayer contiene:
• un vial con el polvo (vial de vidrio tipo 1, transparente, de 10 ml con tapón de mezcla de caucho halogenobutílico de color gris exento de látex y precinto de aluminio).
• una jeringa precargada con 2,5 ml (para 250 UI, 500 UI, y 1000 UI) o 5 ml (para 2000 UI y 3000 UI) de disolvente (cilindro de vidrio tipo 1, transparente, con tapones de mezcla de caucho bromobutílico gris exento de látex)
• un émbolo
• un adaptador de vial
• un equipo para punción venosa
• dos gasas impregnadas en alcohol de un sólo uso
• dos gasas estériles secas
• dos tiras adhesivas
6.6 Precauciones especiales de eliminación y otras manipulaciones
El prospecto incluido en el envase de KOGENATE Bayer contiene las instrucciones para su preparación y administración.
El medicamento reconstituido es una solución transparente e incolora.
El polvo de KOGENATE Bayer se debe reconstituir únicamente con el disolvente suministrado (2,5 ml (para 250 UI, 500 UI, y 1000 UI) o 5 ml (para 2000 UI y 3000 UI) de agua para preparaciones inyectables) en la jeringa precargada y el adaptador de vial. Para la perfusión, el medicamento se debe preparar en condiciones asépticas. Si alguno de los componentes del envase está abierto o dañado, no use este componente.
Girar suavemente el vial, sin agitar, hasta que se haya disuelto todo el medicamento. Tras la reconstitución, la solución es transparente. Los medicamentos que se administran por vía parenteral se deben inspeccionar visualmente para detectar la presencia de partículas o un cambio de color antes de su administración. No utilizar KOGENATE Bayer si se observan partículas o turbidez en su interior.
Una vez reconstituida, la solución se transfiere de nuevo a la jeringa. KOGENATE Bayer se debe reconstituir y administrar con los componentes suministrados en cada envase.
El medicamento reconstituido se debe filtrar antes de la administración para eliminar posibles partículas presentes en la solución. La filtración se consigue utilizando el adaptador de vial.
Para un solo uso.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG 13342 Berlin Alemania
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
|
EU/1/00/143/007 |
- KOGENATE Bayer 250 UI |
|
EU/1/00/143/008 |
- KOGENATE Bayer 500 UI |
|
EU/1/00/143/009 EU/1/00/143/011 EU/1/00/143/013 |
- KOGENATE Bayer 1000 UI - KOGENATE Bayer 2000 UI - KOGENATE Bayer 3000 UI |
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 04 de agosto de 2000 Fecha de la última renovación: 06 de agosto de 2010
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del (de los) fabricantc(s) del principio activo biológico
Bayer Corporation (license holder)
Bayer HealthCare LLC 800 Dwight Way Berkeley CA 94710 EEUU.
Nombre y dirección del (de los) fabricante(s) responsable(s) de la liberación de los lotes
Bayer HealthCare Manufacturing S.r.l.
Via delle Groane 126
20024 Garbagnate Milanese (MI)
Italia
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
1. NOMBRE DEL MEDICAMENTO
KOGENATE Bayer 250 UI polvo y disolvente para solución inyectable KOGENATE Bayer 500 UI polvo y disolvente para solución inyectable KOGENATE Bayer 1000 UI polvo y disolvente para solución inyectable KOGENATE Bayer 2000 UI polvo y disolvente para solución inyectable KOGENATE Bayer 3000 UI polvo y disolvente para solución inyectable
Factor VIII de la coagulación de origen recombinante (octocog alfa)
2. PRINCIPIO(S) ACTIVO(S)
3. LISTA DE EXCIPIENTES
Una vez reconstituido, 1 ml de KOGENATE Bayer 250 UI contiene (250UI / 2,5 ml) = 100 UI de octocog alfa.
|
Una vez reconstituido, 1 ml de KOGENATE Bayer 500 UI contiene (500UI / 2,5 ml) = 200 UI de | ||
|
octocog alfa. | ||
|
Una vez reconstituido, 1 ml de KOGENATE Bayer 1000 UI contiene (1000UI / 2,5 ml) = 400 UI de | ||
|
octocog alfa. | ||
|
Una vez reconstituido, 1 ml de KOGENATE Bayer 2000 UI contiene (2000UI / 5 ml) = 400 UI de | ||
|
octocog alfa. | ||
|
Una vez reconstituido, 1 ml de KOGENATE Bayer 3000 UI contiene (3000UI / 5 ml) = 600 UI de | ||
|
octocog alfa. | ||
Glicina, cloruro de sodio, cloruro de calcio, histidina, polisorbato 80, sacarosa.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Sistema Bio-Set:
1 vial con sistema Bio-Set con polvo para solución inyectable.
1 jeringa precargada con 2,5 ml o 5 ml de agua para inyectables y émbolo independiente.
1 equipo para punción venosa
2 gasas impregnadas en alcohol de un solo uso 2 gasas estériles secas
2 tiras adhesivas 5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía intravenosa, para uso único.
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
CAD (Fin del periodo de 12 meses, si se conserva a temperatura ambiente):.......
No utilizar después de esta fecha.
Se puede conservar a temperaturas de hasta 25°C durante un máximo de 12 meses dentro de la fecha de caducidad indicada en la etiqueta. Anotar la nueva fecha de caducidad en la caja. Una vez reconstituido, el producto se debe utilizar en un plazo máximo de 3 horas. No refrigerar una vez se ha reconstituido.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera (entre 2°C y 8°C). No congelar.
Conservar el vial y la jeringa precargada en el embalaje exterior para protegerlos de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Desechar la solución no utilizada.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG 13342 Berlin Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/00/143/004 - KOGENATE Bayer 250 UI EU/1/00/143/005 - KOGENATE Bayer 500 UI EU/1/00/143/006 - KOGENATE Bayer 1000 UI EU/1/00/143/010 - KOGENATE Bayer 2000 UI EU/1/00/143/012 - KOGENATE Bayer 3000 UI
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
Leer el prospecto antes de usar este medicamento.
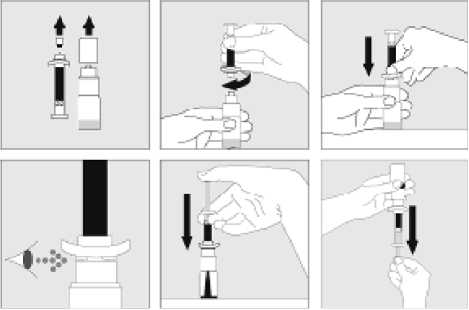
16. INFORMACIÓN EN BRAILLE
KOGENATE Bayer 250 KOGENATE Bayer 500 KOGENATE Bayer 1000 KOGENATE Bayer 2000 KOGENATE Bayer 3000
1. NOMBRE DEL MEDICAMENTO
KOGENATE Bayer 250 UI polvo y disolvente para solución inyectable KOGENATE Bayer 500 UI polvo y disolvente para solución inyectable KOGENATE Bayer 1000 UI polvo y disolvente para solución inyectable KOGENATE Bayer 2000 UI polvo y disolvente para solución inyectable KOGENATE Bayer 3000 UI polvo y disolvente para solución inyectable
Factor VIII de la coagulación de origen recombinante (octocog alfa)
2. PRINCIPIO(S) ACTIVO(S)
Una vez reconstituido, 1 ml de KOGENATE Bayer 250 UI contiene (250UI / 2,5 ml) = 100 UI de octocog alfa.
Una vez reconstituido, 1 ml de KOGENATE Bayer 500 UI contiene (500UI / 2,5 ml) = 200 UI de
|
octocog alfa. | ||
|
Una vez reconstituido, 1 ml de KOGENATE Bayer 1000 UI contiene (1000UI / 2,5 ml) = 400 UI de | ||
|
octocog alfa. | ||
|
Una vez reconstituido, 1 ml de KOGENATE Bayer 2000 UI contiene (2000UI / 5 ml) = 400 UI de | ||
|
octocog alfa. | ||
|
Una vez reconstituido, 1 ml de KOGENATE Bayer 3000 UI contiene (3000UI / 5 ml) = 600 UI de | ||
octocog alfa.
Glicina, cloruro de sodio, cloruro de calcio, histidina, polisorbato 80, sacarosa.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Adaptador de vial:
1 vial con polvo para solución inyectable.
1 jeringa precargada con 2,5 ml o 5 ml de agua para inyectables y émbolo independiente. 1 adaptador de vial
1 equipo para punción venosa
2 gasas impregnadas en alcohol, de un solo uso 2 gasas estériles secas
2 tiras adhesivas 5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía intravenosa, para uso único.
3. LISTA DE EXCIPIENTES
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
CAD (Fin del periodo de 12 meses, si se conserva a temperatura ambiente):.......
No utilizar después de esta fecha.
Se puede conservar a temperaturas de hasta 25°C durante un máximo de 12 meses dentro de la fecha de caducidad indicada en la etiqueta. Anotar la nueva fecha de caducidad en la caja. Una vez reconstituido, el producto se debe utilizar en un plazo máximo de 3 horas. No refrigerar una vez se ha reconstituido.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera (entre 2°C y 8°C). No congelar.
Conservar el vial y la jeringa precargada en el embalaje exterior para protegerlos de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Desechar la solución no utilizada.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG 13342 Berlin Alemania
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/00/143/007 - KOGENATE Bayer 250 UI EU/1/00/143/008 - KOGENATE Bayer 500 UI EU/1/00/143/009 - KOGENATE Bayer 1000 UI EU/1/00/143/011 - KOGENATE Bayer 2000 UI EU/1/00/143/013 - KOGENATE Bayer 3000 UI
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
Leer el prospecto antes de usar este medicamento.
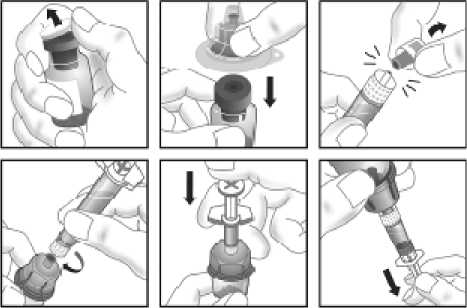
16. INFORMACIÓN EN BRAILLE
KOGENATE Bayer 250 KOGENATE Bayer 500 KOGENATE Bayer 1000 KOGENATE Bayer 2000 KOGENATE Bayer 3000
ETIQUETA DE VIAL_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
KOGENATE Bayer 250 UI polvo y disolvente para solución inyectable KOGENATE Bayer 500 UI polvo y disolvente para solución inyectable KOGENATE Bayer 1000 UI polvo y disolvente para solución inyectable KOGENATE Bayer 2000 UI polvo y disolvente para solución inyectable KOGENATE Bayer 3000 UI polvo y disolvente para solución inyectable
Factor VIII de la coagulación de origen recombinante (octocog alfa)
Vía intravenosa.
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento.
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
250 UI de octocog alfa (una vez reconstituido: 100 UI/ml). 500 UI de octocog alfa (una vez reconstituido: 200 UI/ml). 1000 UI de octocog alfa (una vez reconstituido: 400 UI/ml). 2000 UI de octocog alfa (una vez reconstituido: 400 UI/ml). 3000 UI de octocog alfa (una vez reconstituido: 600 UI/ml).
6. OTROS
Bayer-Logo
JERINGA PRECARGADA CON 2,5 ML Ó 5 ML DE AGUA PARA PREPARACIONES INYECTABLES
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua para preparaciones inyectables
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
2,5 ml [para la reconstitución de las dosis 250/500/100 UI] 5 ml [para la reconstitución de las dosis 2000/3000 UI]
6. OTROS
B. PROSPECTO
Prospecto: información para el usuario
KOGENATE Bayer 250 UI Polvo y disolvente para solución inyectable KOGENATE Bayer 500 UI Polvo y disolvente para solución inyectable KOGENATE Bayer 1000 UI Polvo y disolvente para solución inyectable KOGENATE Bayer 2000 UI Polvo y disolvente para solución inyectable KOGENATE Bayer 3000 UI Polvo y disolvente para solución inyectable Factor VIII de la coagulación de origen recombinante (octocog alfa)
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es KOGENATE Bayer y para qué se utiliza
2. Qué necesita saber antes de empezar a usar KOGENATE Bayer
3. Cómo usar KOGENATE Bayer
4. Posibles efectos adversos
5. Conservación de KOGENATE Bayer
6. Contenido del envase e información adicional
1. Qué es KOGENATE Bayer y para qué se utiliza
KOGENATE Bayer contiene el principio activo Factor VIII de la coagulación humano recombinante (octocog alfa).
KOGENATE Bayer se utiliza para el tratamiento y prevención de la hemorragia en adultos, adolescentes y niños de cualquier edad con hemofilia A (déficit congénito de factor VIII).
Este medicamento no contiene factor de von Willebrand, por lo que no se debe utilizar en la enfermedad de von Willebrand.
2. Qué necesita saber antes de empezar a usar KOGENATE Bayer
No use KOGENATE Bayer
• si es alérgico a octocog alfa o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6 y al final de la sección 2).
• si es alérgico a las proteínas de ratón o hámster.
Si tiene alguna duda sobre lo anterior, consulte a su médico.
Advertencias y precauciones
Tenga especial cuidado con KOGENATE Bayer y consulte a su médico o farmacéutico si:
• siente opresión en el pecho, sensación de mareo, vértigo o náusea, o bien se marea estando de pie, es posible que esté sufriendo una reacción alérgica repentina grave (reacción anafiláctica) rara a este medicamento. Si esto ocurre, interrumpa inmediatamente la administración del medicamento y pida asistencia médica.
• la hemorragia no se llega a controlar con su dosis habitual de este medicamento, consulte a su médico inmediatamente. Es posible que haya desarrollado inhibidores del factor VIII, por lo que su médico realizará pruebas para confirmarlo. Los inhibidores del factor VIII son anticuerpos presentes en la sangre que bloquean el factor VIII que está utilizando y ello hace que el factor VIII sea menos eficaz para prevenir y controlar el sangrado.
• ha desarrollado anteriormente inhibidores del factor VIII y cambia a otro medicamento de factor VIII, es posible que vuelva a desarrollar inhibidores.
• le han indicado que padece una enfermedad cardíaca o que tiene riesgo de padecer una enfermedad cardíaca.
• necesita un dispositivo de acceso venoso central (DAVC) para la administración de KOGENATE Bayer. Debe tener en cuenta el riesgo de complicaciones relacionadas con el DAVC, como por ejemplo, infecciones locales, presencia de bacterias en la sangre (bacteriemia) y formación de un coágulo de sangre en el vaso sanguíneo (trombosis) donde se inserta el catéter.
Su médico debe llevar a cabo las pruebas necesarias para garantizar que su dosis actual del fármaco le proporciona los niveles adecuados de factor VIII.
Uso de KOGENATE Bayer con otros medicamentos
No se conocen interacciones con otros medicamentos. No obstante, por favor, informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Niños y adolescentes
La lista de advertencias y precauciones es aplicable a pacientes de todas las edades, adultos y niños.
Embarazo, lactancia y fertilidad
No se dispone de experiencia sobre la fertilidad ni sobre el uso de KOGENATE Bayer durante el embarazo y el periodo de lactancia. Por consiguiente, si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento.
No es probable que KOGENATE Bayer afecte a la fertilidad de los pacientes de sexo masculino ni femenino, ya que el principio activo aparece de forma natural en el organismo.
Conducción y uso de máquinas
No se han observado efectos sobre la capacidad para conducir o utilizar máquinas.
KOGENATE Bayer contiene sodio
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por vial, por lo que se considera esencialmente “exento de sodio”.
Documentación
Se recomienda que cada vez que utilice KOGENATE Bayer, anote el nombre y el número de lote del medicamento.
3. Cómo usar KOGENATE Bayer
Siga exactamente las instrucciones de administración de este medicamento indicadas en este prospecto o las indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero.
Tratamiento del sangrado
Su médico calculará la dosis de este medicamento y la frecuencia con la que se debe administrar para alcanzar el nivel necesario de actividad del factor VIII en su sangre. Él/Ella debe ajustar siempre la dosis y la frecuencia de administración, en función de sus necesidades. La cantidad de KOGENATE Bayer que debe usar y con qué frecuencia depende de muchos factores como:
• su peso
• la gravedad de su hemofilia
• la localización y gravedad del sangrado
• si tiene inhibidores del factor VIII y la cantidad de ellos
• el nivel requerido de factor VIII.
Prevención del sangrado
Si está utilizando KOGENATE Bayer para prevenir el sangrado (profilaxis), su médico calculará la dosis que le conviene. Esta dosis será normalmente de 20 a 40 UI de octocog alfa por kg de peso corporal, administrada cada 2 ó 3 días. No obstante, en algunos casos, especialmente en pacientes más jóvenes, pueden requerirse intervalos de tratamientos más cortos o dosis superiores.
Pruebas de laboratorio
Es altamente recomendable realizar las pruebas analíticas adecuadas a intervalos apropiados para comprobar que se hayan alcanzado y se mantengan en el plasma los niveles adecuados de factor VIII. En el caso concreto de las intervenciones quirúrgicas importantes, debe realizarse un control estricto del tratamiento sustitutivo mediante pruebas de la coagulación.
Uso en niños y adolescentes
KOGENATE Bayer se puede usar en niños de todas las edades.
Si el sangrado no se controla
Si su nivel de factor VIII no alcanza los niveles esperados en el plasma, o si no se puede controlar el sangrado con una dosificación aparentemente adecuada, es posible que haya desarrollado inhibidores del factor VIII. Un médico especialista comprobará si presenta inhibidores del factor VIII.
Si estima que el efecto de este medicamento es excesivo o insuficiente, comuníqueselo a su médico.
Pacientes con inhibidores
Si su médico le dice que ha desarrollado inhibidores del factor VIII, podrá necesitar una cantidad superior de este medicamento para controlar el sangrado. Si esta dosis superior no lo controla, su médico podrá considerar la utilización de otro medicamento, el concentrado de factor VIIa o bien el concentrado de complejo protrombínico (activado).
Estos tratamientos deben ser recetados por médicos con experiencia en el cuidado de pacientes con hemofilia A. Si desea más información, hable con su médico.
No aumente la dosis de este medicamento que le han prescrito para controlar su sangrado sin consultar con su médico.
Duración del tratamiento
Su médico indicará con qué frecuencia y a qué intervalos se debe administrar este medicamento. Generalmente, el tratamiento sustitutivo con KOGENATE Bayer será necesario de por vida.
Cómo se administra KOGENATE Bayer
Este medicamento se debe inyectar en una vena durante 2 a 5 minutos dependiendo del volumen total y del grado de bienestar y se debe usar en el plazo de 3 horas después de su reconstitución.
Cómo se prepara KOGENATE Bayer para la administración
Utilice únicamente los elementos (vial con polvo y sistema Bio-Set, jeringa precargada con el disolvente y equipo para punción venosa) incluidos en el envase para este medicamento. Si no es posible utilizar estos componentes, consulte a su médico. Si alguno de los componentes del envase está abierto o dañado, no use este componente.
Debe filtrar el medicamento reconstituido antes de la administración para eliminar posibles partículas presentes en la solución. La filtración se puede realizar siguiendo los pasos para la reconstitución y/o administración descritas a continuación. Use el equipo de punción venosa suministrado, ya que contiene un filtro en línea. Si no puede usar el equipo de punción venosa suministrado, utilice un filtro indicado por su médico o enfermero.
No use el equipo de punción venosa suministrado para la extracción de sangre, porque contiene un filtro en línea. Cuando se deba extraer sangre antes de una perfusión, se usará un equipo de administración sin filtro y, a continuación, se administrará este medicamento mediante un filtro de inyección. Si tiene alguna pregunta sobre este medicamento y el uso de otros filtros compatibles, consulte a su médico.
Este medicamento no se debe mezclar con otras soluciones para perfusión. No utilice soluciones si observa partículas o si la solución está turbia. Siga estrictamente las instrucciones de administración que le dé su médico y utilice las instrucciones detalladas de reconstitución y administración recogidas al final de este prospecto.
Si usa más KOGENATE Bayer del que debe
No se han comunicado casos de sobredosis con factor VIII de la coagulación de origen recombinante. Si ha usado más KOGENATE Bayer del que debe, por favor, informe a su médico.
Si olvidó usar KOGENATE Bayer
• Aplique inmediatamente la siguiente dosis y continúe a intervalos regulares siguiendo las indicaciones de su médico.
• No use una dosis doble para compensar las dosis olvidadas.
Si quiere interrumpir el tratamiento con KOGENATE Bayer
No deje de usar KOGENATE Bayer sin consultarlo con su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Los efectos adversos más graves son reacciones de hipersensibilidad o shock anafiláctico (efecto adverso raro).
Si se produce una reacción alérgica o anafiláctica, se debe interrumpir la inyección o perfusión y consultar a su médico inmediatamente.
Lista general de posibles efectos adversos:
Muy frecuentes
(pueden afectar a más de 1 de cada 10 personas):
• formación de anticuerpos neutralizantes al factor VIII (inhibidores) en pacientes sin tratamiento previo
Frecuentes
(pueden afectar hasta 1 de cada 10 personas):
• sarpullido con o sin picor
• reacciones locales en el lugar donde se ha inyectado el medicamento (p.ej., sensación de quemazón, enrojecimiento pasajero)
Poco frecuentes
(pueden afectar hasta 1 de cada 100 personas):
• formación de anticuerpos neutralizantes al factor VIII (inhibidores) en pacientes con tratamiento previo
Raros
(pueden afectar hasta 1 de cada 1.000 personas):
• reacciones alérgicas incluyendo reacción alérgica repentina grave (que puede incluir ronchas, náuseas, urticaria, angioedema, escalofríos, sofocos, cefalea, letargo, sibilancias o dificultad para respirar, inquietud, taquicardia, hormigueo o shock anafiláctico, p. ej. opresión en el pecho, sensación general de malestar, mareo, náuseas y ligera bajada de tensión que puede hacer que se maree al levantarse)
• fiebre
Frecuencia no conocida
(la frecuencia no puede estimarse a partir de los datos disponibles):
• disgeusia (alteración del sentido del gusto)
Si nota cualquiera de los síntomas siguientes durante la inyección o la perfusión:
• opresión en el pecho, o sensación general de malestar
• mareo
• hipotensión leve (ligera disminución de la presión arterial con sensación de mareo al levantarse)
• náusea
puede ser un signo precoz de hipersensibilidad y reacción anafiláctica.
Si se produce una reacción alérgica o anafiláctica, la inyección o perfusión se debe interrumpir y consultar a su médico inmediatamente.
Anticuerpos (Inhibidores)
La formación de anticuerpos neutralizantes al factor VIII (inhibidores) es una complicación conocida en el tratamiento de pacientes con hemofilia A. Su médico considerará realizarle algunas pruebas para controlar el desarrollo de inhibidores.
Reacciones de hipersensibilidad
Durante los estudios clínicos realizados, ningún paciente desarrolló títulos de anticuerpos clínicamente importantes frente a las pequeñas cantidades de proteína de hámster y ratón presentes en el preparado. Existe la posibilidad de reacción alérgica a las sustancias contenidas en este medicamento, como por ejemplo, a las pequeñas cantidades de proteína de ratón y hámster, en pacientes con cierta predisposición. Su médico considerará realizarle algunas pruebas para controlar el desarrollo de inhibidores.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de KOGENATE Bayer
Mantener este medicamento fuera de la vista y del alcance de los niños.
Conservar en nevera (entre 2°C y 8°C). No congelar. Conservar el vial y la jeringa precargada en el embalaje exterior para protegerlos de la luz.
Dentro de la fecha de caducidad indicada en la etiqueta, el medicamento envasado en su embalaje exterior puede conservarse a temperatura ambiente (inferior a 25°C) durante un periodo máximo de 12 meses. En este caso, el medicamento caduca al finalizar el periodo de 12 meses o en la fecha de caducidad indicada en el vial del medicamento, según cuál sea antes. Debe anotar la nueva fecha de caducidad en el embalaje exterior.
No refrigerar la solución después de su reconstitución. La solución reconstituida se debe utilizar en un plazo máximo de 3 horas. Utilizar el contenido una sola vez. Desechar la solución no utilizada.
No utilice este medicamento después de la fecha de caducidad que aparece en las etiquetas y las cajas. La fecha de caducidad es el último día del mes que se indica.
No utilice este medicamento si observa partículas o si la solución está turbia.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de KOGENATE Bayer
Polvo
El principio activo es el factor VIII de la coagulación humano (octocog alfa) producido por tecnología de ADN recombinante. Cada vial de KOGENATE Bayer contiene 250, 500, 1000, 2000 ó 3000 UI de octocog alfa.
Los demás componentes son glicina, cloruro de sodio, cloruro de calcio, histidina, polisorbato 80 y sacarosa (ver el final de la sección 2).
Disolvente
Agua para preparaciones inyectables.
Aspecto del producto y contenido del envase
KOGENATE Bayer se presenta en forma de polvo y disolvente para solución inyectable; se presenta en forma de polvo o polvo compacto, de color blanco a ligeramente amarillo. La jeringa precargada contiene agua para preparaciones inyectables para reconstituir el contenido del vial. Después de la reconstitución la solución es transparente. Los dispositivos necesarios para la reconstitución y administración se proporcionan en cada envase de este medicamento.
Cada envase de KOGENATE Bayer contiene un vial con sistema de transferencia Bio-Set y una jeringa precargada con émbolo independiente, así como un equipo para punción venosa (para inyección intravenosa), dos gasas impregnadas en alcohol, dos gasas estériles secas y dos tiras adhesivas.
Titular de la autorización de comercialización
Bayer Pharma AG 13342 Berlin Alemania
Responsable de la fabricación
Bayer HealthCare Manufacturing S.r.l.
Via delle Groane 126
20024 Garbagnate Milanese (MI)
Italia
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Bayer SA-NV Tél/Tel: +32-(0)2-535 63 11 Etarapun Eaüep Etarapna EOOfl Tea.: 359 02 81 401 01 Ceská republika Bayer s.r.o. Tel: +420 266 101 111 Danmark Bayer A/S Tlf: +45 45 23 50 00 Deutschland Bayer Vital GmbH Tel: +49 (0)214-30 513 48 Eesti Bayer OÜ Tel: +372 655 8565 ELLáóa Bayer EXkáq ABEE Tr(k: +30-210-61 87 500 España Bayer Hispania S.L. Tel: +34-93-495 65 00 France Bayer HealthCare Tél (N° vert): +33-(0)800 87 54 54 Hrvatska Bayer d.o.o. Tel: +385-(0)1-6599 900 Ireland Bayer Limited Tel: +353 1 2999313 Ísland Icepharma hf. Sími: +354 540 8000 Italia Bayer S.p.A. Tel: +39 02 397 81 Kúnpoq NOVAGEM Limited Tr(k: +357 22 48 38 58 Latvija SIA Bayer Tel: +371 67 84 55 63 |
Lietuva UAB Bayer Tel. +37 05 23 36 868 Luxembourg/Luxemburg Bayer SA-NV Tél/Tel: +32-(0)2-535 63 11 Magyarország Bayer Hungária KFT Tel:+36 14 87-41 00 Malta Alfred Gera and Sons Ltd. Tel: +35 621 44 62 05 Nederland Bayer B.V. Tel: +31-(0)297-28 06 66 Norge Bayer AS Tlf: +47 23 13 05 00 Osterreich Bayer Austria Ges.m.b.H. Tel: +43-(0)1-711 46-0 Polska Bayer Sp. z o.o. Tel: +48 22 572 35 00 Portugal Bayer Portugal, Lda. Tel: +351 21 416 42 00 Romania SC Bayer SRL Tel: +40 21 529 59 00 Slovenija Bayer d. o. o. Tel: +386 (0)1 58 14 400 Slovenská republika Bayer spol. s r.o. Tel. +421 2 59 21 31 11 Suomi/Finland Bayer Oy Puh/Tel: +358- 20 785 21 Sverige Bayer AB Tel: +46 (0) 8 580 223 00 United Kingdom Bayer plc Tel: +44-(0)1635 563000 |
Fecha de la última revisión de este prospecto: {MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
Instrucciones detalladas de reconstitución y administración de KOGENATE Bayer utilizando el vial con tapa de reconstitución (Sistema Bio-Set):
1. Lávese cuidadosamente las manos con jabón y agua templada. La solución se debe preparar
sobre una superficie limpia y seca.
2. Caliente el vial sin perforar y la jeringa precargada con sus manos hasta que estén a la misma temperatura. El material no debe superar la temperatura corporal (no pasar de 37 °C).
3.
Retire la cápsula protectora del vial que contiene el polvo moviéndola suavemente de un lado al otro, al tiempo que tira de ella hacia arriba. Retire el tapón unido a la cápsula blanca de la jeringa (A).
4.
Enrosque con suavidad la jeringa en el vial del polvo (B).
5.
Coloque el vial sobre una superficie rígida y antideslizante, y sujételo firmemente con una mano. Seguidamente, con sus dedos pulgar e índice, presione fuertemente la placa de sujeción del extremo de la jeringa (C) hasta tocar el borde superior del Bio-Set.
Esto confirma que el sistema está activado (D).
6.
Conecte el émbolo a la jeringa haciéndolo girar dentro del tapón de goma (E).
7.
Inyecte el disolvente en el vial que contiene el polvo empujando lentamente el émbolo de la jeringa (F).
8.
Disuelva el polvo girando suavemente el vial (G). No agite el vial! Antes de utilizar, compruebe que el polvo se ha disuelto completamente. Inspeccione visualmente la presencia de partículas o un cambio de color antes de la administración. No utilice soluciones turbias o que contengan partículas visibles.
9.
Invierta el conjunto formado por el vial y la jeringa y transfiera la solución a la jeringa tirando lenta y suavemente del émbolo (H). Compruebe que todo el contenido del vial ha pasado a la jeringa. Mantenga la jeringa en posición vertical y presione el émbolo hasta que no quede aire en el interior de la jeringa.
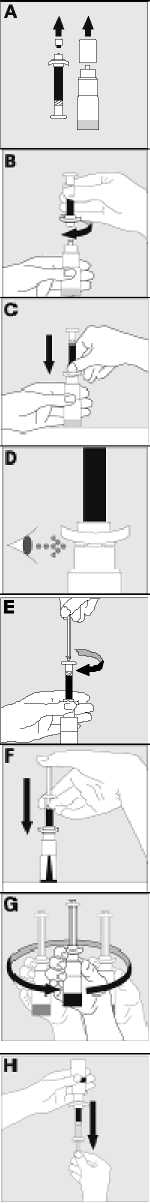
10. Aplique un torniquete. Determine el punto de inyección y desinfecte la piel con una gasa impregnada en alcohol y prepare la zona de inyección asépticamente siguiendo las instrucciones de su médico. Practique la punción venosa y fije el equipo de punción con una tira adhesiva.
11. Desenrosque la jeringa para desconectarla del vial (I).
12. Acople la jeringa al equipo de punción venosa, enroscándola en la dirección
de las agujas del reloj y asegúrese de que no entra sangre en la jeringa (J).
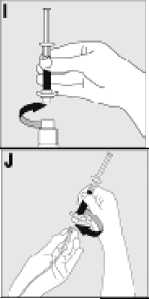
13. Retire el torniquete.
14. Inyecte la solución en la vena durante 2 a 5 minutos, vigilando en todo momento la posición de la aguja. La velocidad de administración se basará en el grado de bienestar del paciente, pero no debe ser mayor de 2 ml/min.
15. Si se necesita administrar una dosis adicional, retire la jeringa vacía haciéndola girar al
contrario de las agujas del reloj. Reconstituya la cantidad deseada de producto, repitiendo los pasos 2 - 9, utilizando una nueva jeringa y conectándola al equipo de punción venosa.
16. Si no se precisan más dosis, retire el equipo de punción venosa y la jeringa. Aplique una gasa durante unos 2 minutos sobre el punto de inyección, manteniendo el brazo extendido. Finalmente, presione sobre el punto de inyección y considere si es necesario colocar una tira adhesiva.
Prospecto: información para el usuario
KOGENATE Bayer 250 UI Polvo y disolvente para solución inyectable KOGENATE Bayer 500 UI Polvo y disolvente para solución inyectable KOGENATE Bayer 1000 UI Polvo y disolvente para solución inyectable KOGENATE Bayer 2000 UI Polvo y disolvente para solución inyectable KOGENATE Bayer 3000 UI Polvo y disolvente para solución inyectable Factor VIII de la coagulación de origen recombinante (octocog alfa)
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es KOGENATE Bayer y para qué se utiliza
2. Qué necesita saber antes de empezar a usar KOGENATE Bayer
3. Cómo usar KOGENATE Bayer
4. Posibles efectos adversos
5. Conservación de KOGENATE Bayer
6. Contenido del envase e información adicional
1. Qué es KOGENATE Bayer y para qué se utiliza
KOGENATE Bayer contiene el principio activo Factor VIII de la coagulación humano recombinante (octocog alfa).
KOGENATE Bayer se utiliza para el tratamiento y prevención de la hemorragia en adultos, adolescentes y niños de cualquier edad con hemofilia A (déficit congénito de factor VIII).
Este medicamento no contiene factor de von Willebrand, por lo que no se debe utilizar en la enfermedad de von Willebrand.
2. Qué necesita saber antes de empezar a usar KOGENATE Bayer
No use KOGENATE Bayer
• si es alérgico a octocog alfa o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6 y al final de la sección 2).
• si es alérgico a las proteínas de ratón o hámster.
Si tiene alguna duda sobre lo anterior, consulte a su médico.
Advertencias y precauciones
Tenga especial cuidado con KOGENATE Bayer y consulte a su médico o farmacéutico si:
• siente opresión en el pecho, sensación de mareo, vértigo o náusea, o bien se marea estando de pie, es posible que esté sufriendo una reacción alérgica repentina grave (reacción anafiláctica) rara a este medicamento. Si esto ocurre, interrumpa inmediatamente la administración del medicamento y pida asistencia médica.
• la hemorragia no se llega a controlar con su dosis habitual de este medicamento, consulte a su médico inmediatamente. Es posible que haya desarrollado inhibidores del factor VIII, por lo que su médico realizará pruebas para confirmarlo. Los inhibidores del factor VIII son anticuerpos presentes en la sangre que bloquean el factor VIII que está utilizando y ello hace que el factor VIII sea menos eficaz para prevenir y controlar el sangrado.
• ha desarrollado anteriormente inhibidores del factor VIII y cambia a otro medicamento de factor VIII, es posible que vuelva a desarrollar inhibidores.
• le han indicado que padece una enfermedad cardíaca o que tiene riesgo de padecer una enfermedad cardíaca.
• necesita un dispositivo de acceso venoso central (DAVC) para la administración de KOGENATE Bayer. Debe tener en cuenta el riesgo de complicaciones relacionadas con el DAVC, como por ejemplo, infecciones locales, presencia de bacterias en la sangre (bacteriemia) y formación de un coágulo de sangre en el vaso sanguíneo (trombosis) donde se inserta el catéter.
Su médico debe llevar a cabo las pruebas necesarias para garantizar que su dosis actual del fármaco le proporciona los niveles adecuados de factor VIII.
Uso de KOGENATE Bayer con otros medicamentos
No se conocen interacciones con otros medicamentos. No obstante, por favor, informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Niños y adolescentes
La lista de advertencias y precauciones es aplicable a pacientes de todas las edades, adultos y niños.
Embarazo, lactancia y fertilidad
No se dispone de experiencia sobre la fertilidad ni sobre el uso de KOGENATE Bayer durante el embarazo y el periodo de lactancia. Por consiguiente, si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento.
No es probable que KOGENATE Bayer afecte a la fertilidad de los pacientes de sexo masculino ni femenino, ya que el principio activo aparece de forma natural en el organismo.
Conducción y uso de máquinas
No se han observado efectos sobre la capacidad para conducir o utilizar máquinas.
KOGENATE Bayer contiene sodio
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por vial, por lo que se considera esencialmente “exento de sodio”.
Documentación
Se recomienda que cada vez que utilice KOGENATE Bayer, anote el nombre y el número de lote del medicamento.
3. Cómo usar KOGENATE Bayer
Siga exactamente las instrucciones de administración de este medicamento indicadas en este prospecto o las indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero.
Tratamiento del sangrado
Su médico calculará la dosis de este medicamento y la frecuencia con la que se debe administrar para alcanzar el nivel necesario de actividad del factor VIII en su sangre. Él/Ella debe ajustar siempre la dosis y la frecuencia de administración, en función de sus necesidades. La cantidad de KOGENATE Bayer que se debe usar y con qué frecuencia depende de muchos factores como:
• su peso
• la gravedad de su hemofilia
• la localización y gravedad del sangrado
• si tiene inhibidores del factor VIII y la cantidad de ellos
• el nivel requerido de factor VIII.
Prevención del sangrado
Si está utilizando KOGENATE Bayer para prevenir el sangrado (profilaxis), su médico calculará la dosis que le conviene. Esta dosis será normalmente de 20 a 40 UI de octocog alfa por kg de peso corporal, administrada cada 2 ó 3 días. No obstante, en algunos casos, especialmente en pacientes más jóvenes, pueden requerirse intervalos de tratamientos más cortos o dosis superiores.
Pruebas de laboratorio
Es altamente recomendable realizar las pruebas analíticas adecuadas a intervalos apropiados para comprobar que se hayan alcanzado y se mantengan en el plasma los niveles adecuados de factor VIII. En el caso concreto de las intervenciones quirúrgicas importantes, debe realizarse un control estricto del tratamiento sustitutivo mediante pruebas de la coagulación.
Uso en niños y adolescentes
KOGENATE Bayer se puede usar en niños de todas las edades.
Si el sangrado no se controla
Si su nivel de factor VIII no alcanza los niveles esperados en el plasma, o si no se puede controlar el sangrado con una dosificación aparentemente adecuada, es posible que haya desarrollado inhibidores del factor VIII. Un médico especialista comprobará si presenta inhibidores del factor VIII.
Si estima que el efecto de este medicamento es excesivo o insuficiente, comuníqueselo a su médico.
Pacientes con inhibidores
Si su médico le dice que ha desarrollado inhibidores del factor VIII, podrá necesitar una cantidad superior de este medicamento para controlar el sangrado. Si esta dosis superior no lo controla, su médico podrá considerar la utilización de otro medicamento, el concentrado de factor VIIa o bien el concentrado de complejo protrombínico (activado).
Estos tratamientos deben ser recetados por médicos con experiencia en el cuidado de pacientes con hemofilia A. Si desea más información, hable con su médico.
No aumente la dosis de este medicamento que le han prescrito para controlar su sangrado sin consultar con su médico.
Duración del tratamiento
Su médico indicará con qué frecuencia y a qué intervalos se debe administrar este medicamento. Generalmente, el tratamiento sustitutivo con KOGENATE Bayer será necesario de por vida.
Cómo se administra KOGENATE Bayer
Este medicamento se debe inyectar en una vena durante 2 a 5 minutos dependiendo del volumen total y del grado de bienestar y se debe usar en el plazo de 3 horas después de su reconstitución.
Cómo se prepara KOGENATE Bayer para la administración
Utilice únicamente los elementos (adaptador de vial, jeringa precargada con el disolvente y equipo para punción venosa) incluidos en el envase para este medicamento. Si no es posible utilizar estos componentes, consulte a su médico. Si alguno de los componentes del envase está abierto o dañado, no use este componente.
Debe filtrar el medicamento reconstituido antes de la administración para eliminar posibles partículas presentes en la solución. La filtración se consigue utilizando el adaptador de vial.
No use el equipo de punción venosa suministrado para la extracción de sangre, porque contiene un filtro en línea.
Este medicamento no se debe mezclar con otras soluciones para perfusión. No utilice soluciones si observa partículas o si la solución está turbia. Siga estrictamente las instrucciones de administración que le dé su médico y utilice las instrucciones detalladas de reconstitución y administración recogidas al final de este prospecto.
Si usa más KOGENATE Bayer del que debe
No se han comunicado casos de sobredosis con factor VIII de la coagulación de origen recombinante. Si ha usado más KOGENATE Bayer del que debe, por favor, informe a su médico.
Si olvidó usar KOGENATE Bayer
• Aplique inmediatamente la siguiente dosis y continúe a intervalos regulares siguiendo las indicaciones de su médico.
• No use una dosis doble para compensar las dosis olvidadas.
Si quiere interrumpir el tratamiento con KOGENATE Bayer
No deje de usar KOGENATE Bayer sin consultarlo con su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Los efectos adversos más graves son reacciones de hipersensibilidad o shock anafiláctico (efecto adverso raro).
Si se produce una reacción alérgica o anafiláctica, se debe interrumpir la inyección o perfusión y consultar a su médico inmediatamente.
Lista general de posibles efectos adversos:
Muy frecuentes
(pueden afectar a más de 1 de cada 10 personas):
• formación de anticuerpos neutralizantes al factor VIII (inhibidores) en pacientes sin tratamiento previo
Frecuentes
(pueden afectar hasta 1 de cada 10 personas):
• sarpullido con o sin picor
• reacciones locales en el lugar donde se ha inyectado el medicamento (p.ej., sensación de quemazón, enrojecimiento pasajero)
Poco frecuentes
(pueden afectar hasta 1 de cada 100 personas):
• formación de anticuerpos neutralizantes al factor VIII (inhibidores) en pacientes con tratamiento previo
Raros
(pueden afectar hasta 1 de cada 1.000 personas):
• reacciones alérgicas incluyendo reacción alérgica repentina grave (que puede incluir ronchas, náuseas, urticaria, angioedema, escalofríos, sofocos, cefalea, letargo, sibilancias o dificultad para respirar, inquietud, taquicardia, hormigueo o shock anafiláctico, p. ej. opresión en el pecho, sensación general de malestar, mareo, náuseas y ligera bajada de tensión que puede hacer que se maree al levantarse)
• fiebre
Frecuencia no conocida
(la frecuencia no puede estimarse a partir de los datos disponibles):
• disgeusia (alteración del sentido del gusto)
Si nota cualquiera de los síntomas siguientes durante la inyección o la perfusión:
• opresión en el pecho, o sensación general de malestar
• mareo
• hipotensión leve (ligera disminución de la presión arterial con sensación de mareo al levantarse)
• náusea
puede ser un signo precoz de hipersensibilidad y reacción anafiláctica.
Si se produce una reacción alérgica o anafiláctica, la inyección o perfusión se debe interrumpir y consultar a su médico inmediatamente.
Anticuerpos (Inhibidores)
La formación de anticuerpos neutralizantes al factor VIII (inhibidores) es una complicación conocida en el tratamiento de pacientes con hemofilia A. Su médico considerará realizarle algunas pruebas para controlar el desarrollo de inhibidores.
Reacciones de hipersensibilidad
Durante los estudios clínicos realizados, ningún paciente desarrolló títulos de anticuerpos clínicamente importantes frente a las pequeñas cantidades de proteína de hámster y ratón presentes en el preparado. Existe la posibilidad de reacción alérgica a las sustancias contenidas en este medicamento, como por ejemplo, a las pequeñas cantidades de proteína de ratón y hámster, en pacientes con cierta predisposición. Su médico considerará realizarle algunas pruebas para controlar el desarrollo de inhibidores.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de KOGENATE Bayer
Mantener este medicamento fuera de la vista y del alcance de los niños.
Dentro de la fecha de caducidad indicada en la etiqueta, el medicamento envasado en su embalaje exterior puede conservarse a temperatura ambiente (inferior a 25°C) durante un periodo máximo de 12 meses. En este caso, el medicamento caduca al finalizar el periodo de 12 meses o en la fecha de caducidad indicada en el vial del medicamento, según cuál sea antes. Debe anotar la nueva fecha de caducidad en el embalaje exterior.
No refrigerar la solución después de su reconstitución. La solución reconstituida se debe utilizar en un plazo máximo de 3 horas. Utilizar el contenido una sola vez. Desechar la solución no utilizada.
No utilice este medicamento después de la fecha de caducidad que aparece en las etiquetas y las cajas. La fecha de caducidad es el último día del mes que se indica.
No utilice este medicamento si observa partículas o si la solución está turbia.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de KOGENATE Bayer
Polvo
El principio activo es el factor VIII de la coagulación humano (octocog alfa) producido por tecnología de ADN recombinante. Cada vial de KOGENATE Bayer contiene 250, 500, 1000, 2000 ó 3000 UI de octocog alfa.
Los demás componentes son glicina, cloruro de sodio, cloruro de calcio, histidina, polisorbato 80 y sacarosa (ver el final de la sección 2).
Disolvente
Agua para preparaciones inyectables.
Aspecto del producto y contenido del envase
KOGENATE Bayer se presenta en forma de polvo y disolvente para solución inyectable; se presenta en forma de polvo o polvo compacto, de color blanco a ligeramente amarillo. La jeringa precargada contiene agua para inyección para reconstituir el contenido del vial. Después de la reconstitución la solución es transparente. Los dispositivos necesarios para la reconstitución y administración se proporcionan en cada envase de, este medicamento.
Cada envase de KOGENATE Bayer contiene un vial con una jeringa precargada con émbolo independiente, así como un adaptador de vial, un equipo para punción venosa (para inyección intravenosa), dos gasas impregnadas en alcohol, dos gasas estériles secas y dos tiras adhesivas.
Titular de la autorización de comercialización
Bayer Pharma AG 13342 Berlin Alemania
Responsable de la fabricación
Bayer HealthCare Manufacturing S.r.l.
Via delle Groane 126
20024 Garbagnate Milanese (MI)
Italia
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Bayer SA-NV Tél/Tel: +32-(0)2-535 63 11 Etarapun Eaüep Etarapna EOOfl Tea.: 359 02 81 401 01 Ceská republika Bayer s.r.o. Tel: +420 266 101 111 Danmark Bayer A/S Tlf: +45 45 23 50 00 Deutschland Bayer Vital GmbH Tel: +49 (0)214-30 513 48 Eesti Bayer OÜ Tel: +372 655 8565 ELLáóa Bayer EXkáq ABEE Tr(k: +30-210-61 87 500 España Bayer Hispania S.L. Tel: +34-93-495 65 00 France Bayer HealthCare Tél (N° vert): +33-(0)800 87 54 54 Hrvatska Bayer d.o.o. Tel: +385-(0)1-6599 900 Ireland Bayer Limited Tel: +353 1 2999313 Ísland Icepharma hf. Sími: +354 540 8000 Italia Bayer S.p.A. Tel: +39 02 397 81 Kúnpoq NOVAGEM Limited TpL +357 22 48 38 58 Latvija SIA Bayer Tel: +371 67 84 55 63 |
Lietuva UAB Bayer Tel. +37 05 23 36 868 Luxembourg/Luxemburg Bayer SA-NV Tél/Tel: +32-(0)2-535 63 11 Magyarország Bayer Hungária KFT Tel:+36 14 87-41 00 Malta Alfred Gera and Sons Ltd. Tel: +35 621 44 62 05 Nederland Bayer B.V. Tel: +31-(0)297-28 06 66 Norge Bayer AS Tlf: +47 23 13 05 00 Osterreich Bayer Austria Ges.m.b.H. Tel: +43-(0)1-711 46-0 Polska Bayer Sp. z o.o. Tel: +48 22 572 35 00 Portugal Bayer Portugal, Lda. Tel: +351 21 416 42 00 Romania SC Bayer SRL Tel: +40 21 529 59 00 Slovenija Bayer d. o. o. Tel: +386 (0)1 58 14 400 Slovenská republika Bayer spol. s r.o. Tel. +421 2 59 21 31 11 Suomi/Finland Bayer Oy Puh/Tel: +358- 20 785 21 Sverige Bayer AB Tel: +46 (0) 8 580 223 00 United Kingdom Bayer plc Tel: +44-(0)1635 563000 |
Fecha de la última revisión de este prospecto: {MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
Instrucciones detalladas de reconstitución y administración de KOGENATE Bayer utilizando el vial con el adaptador de vial:
1. Lávese cuidadosamente las manos con jabón y agua templada.
2. Caliente los dos viales sin perforar y la jeringa con sus manos a una temperatura confortable (no superior a 37 °C).
|
3. Retire la cápsula protectora del vial (A) y limpie el tapón de goma del vial con una gasa con alcohol y deje que se seque al aire antes de su uso. |
A |
|
4. Ponga el vial del producto sobre una superficie sólida y antideslizante. Retire la cubierta de papel del envase de plástico del adaptador de vial. No extraiga el adaptador del envase de plástico. Sujetando el envase del adaptador, colóquelo sobre el vial del producto y presione firmemente hacia abajo (B). El adaptador se ajustará al tapón del vial. No retire el envase del adaptador en este momento. |
i b |
|
5. Sujete verticalmente la jeringa precargada con agua para inyectables, coja el émbolo de la jeringa según el pictograma y acople el émbolo girándolo firmemente en el sentido de las agujas del reloj dentro del tapón de rosca (C). | |
|
6. Sujetando el cuerpo de la jeringa, desprenda el tapón del extremo de ésta (D). No toque el extremo de la jeringa con la mano u otra superficie. Reserve la jeringa para su uso posterior. |
' í |
|
7. Ahora, retire y elimine el envase del adaptador (E). | |
|
8. Acople la jeringa precargada al adaptador del vial enroscándola en el sentido de las agujas del reloj (F). | |
|
9. Inyecte el diluyente presionando lentamente sobre el émbolo de la jeringa (G). |
W G |
|
10. Mueva suavemente el vial hasta que se haya disuelto todo el polvo (H). No agite el vial. Asegúrese de que el polvo se ha disuelto completamente. Inspeccione visualmente la presencia de partículas o un cambio de color antes de la administración. No utilice soluciones turbias o que contengan partículas visibles. |
m- |
11. Sujete el vial por el extremo, por encima del adaptador y de la jeringa (I).
Llene la jeringa tirando lenta y suavemente del émbolo. Compruebe que todo el contenido del vial ha pasado a la jeringa. Mantenga la jeringa en posición vertical y presione el émbolo hasta que no quede aire en el interior de la jeringa.
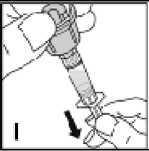
12. Aplique un torniquete.
13. Determine el punto de inyección, límpie la piel con una gasa con alcohol, y prepare el lugar de la inyección de manera antiséptica según lo aconsejado por su médico.
14. Practique la punción venosa y fije el equipo de punción venosa con un esparadrapo.
15. Sujete el adaptador del vial y retire la jeringa del adaptador de vial (éste debe permanecer unido al vial). Acople la jeringa al equipo de punción venosa y asegúrese de que no entra sangre en la jeringa (J).
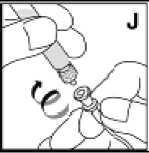
16. Retire el torniquete.
17. Inyecte la solución en la vena durante 2 a 5 minutos, vigilando en todo momento la posición de la aguja. La velocidad de administración se basará en el grado de bienestar del paciente, pero no debe ser mayor de 2 ml/min.
18. Si necesita administrar una segunda dosis, utilice una nueva jeringa con el medicamento reconstituido de la forma indicada anteriormente.
19. Si no se precisa una segunda dosis, retire el equipo de punción venosa y la jeringa. Aplique una gasa durante unos 2 minutos sobre el punto de inyección, manteniendo el brazo extendido. Finalmente, presione sobre el punto de inyección y considere si es necesario colocar una tira adhesiva.
58