Kineret 100 Mg, Solucion Inyectable En Jeringa Precargada
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Kineret 100 mg, solución inyectable en jeringa precargada.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada jeringa precargada contiene 100 mg de anakinra* en 0,67 ml (150 mg/ml).
* Antagonista del receptor humano para la interleucina 1 (r-metHuIL-1ra), producido en células de Escherichia coli por tecnología del ADN recombinante.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución inyectable transparente, entre incolora y blanquecina que puede contener partículas amorfas, relacionadas con el producto, de aspecto entre translúcido y blanco.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Kineret está indicado en adultos para el tratamiento de los signos y síntomas de la artritis reumatoide (AR) en combinación con metotrexato, en aquellos pacientes que no hayan respondido bien a la administración de metotrexato solo.
4.2 Posología y forma de administración
El tratamiento con Kineret deberá ser iniciado y controlado por médicos especialistas con experiencia en el diagnóstico y tratamiento de la artritis reumatoide.
Posología
La dosis recomendada de Kineret es de 100 mg administrados una vez al día en inyección subcutánea. La dosis debe administrarse cada día a la misma hora aproximadamente.
Población de _pacientes de edad avanzada (> 65 años)
No es necesario ajustar la dosis. La posología y administración es la misma que en los adultos de 18 a 64 años.
Población _pediátrica (< 18 años)
No se ha establecido la seguridad y eficacia de Kineret en niños con AR (Artritis Idiopática Juvenil (AIJ)) de 0 a 18 años..
Insuficiencia hepática
No es necesario ajustar la dosis en pacientes con insuficiencia hepática moderada (Child-Pugh Clase B). Kineret debe utilizarse con precaución en pacientes con insuficiencia hepática grave.
Insuficiencia renal
Kineret no debe utilizarse en pacientes con insuficiencia renal grave (CLcr < 30 ml/minuto) (ver sección 4.3). No es necesario ajustar la dosis en pacientes con insuficiencia renal leve (CLcr 50 a 80 ml/minuto). A falta de datos adecuados, Kineret debe usarse con precaución en pacientes con insuficiencia renal moderada (CRcr 30 a 50 ml/minuto).
Forma de administración
Kineret se administra mediante inyección subcutánea.
Kineret se suministra listo para inyectar en una jeringa precargada. La jeringa precargada no debe agitarse. Para las precauciones especiales de eliminación y otras manipulaciones, ver sección 6.6.
Se recomienda alternar el lugar de la inyección para evitar molestias en el mismo. Los signos y síntomas de las reacciones en el lugar de la inyección pueden aliviarse enfriando la zona, calentando el líquido de inyección, aplicando compresas frías (antes y después de la inyección) y utilizando antihistamínicos y corticosteroides de uso tópico después de la inyección.
4.3 Contraindicaciones
Hipersensibilidad al principio activo, o a alguno de sus excipientes incluidos en la sección 6.1 o a proteínas derivadas de E. coli.
No se debe administrar Kineret a pacientes con insuficiencia renal grave (Clcr< 30 ml/minuto) (ver sección 4.2).
No se debe iniciar el tratamiento con Kineret en pacientes con neutropenia (RAN < 1,5 x 109/l) (ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
Reacciones alérgicas
De forma poco frecuente se ha informado de reacciones alérgicas que incluyeron reacciones anafilácticas y angioedema. La mayoría de estas reacciones fueron erupciones maculopapulares o urticariales. En caso de que se produjera una reacción alérgica grave, se deberá interrumpir la administración de Kineret e iniciar el tratamiento apropiado.
Acontecimientos hepáticos
En los estudios clínicos en pacientes con AR y CAPS se han observado con poca frecuencia aumentos transitorios de las enzimas hepáticas. Dichos aumentos no se han asociado a signos o síntomas de daño hepatocelular. En el seguimiento post-comercialización, se han comunicado casos aislados de hepatitis no infecciosa. Estos acontecimientos hepáticos observados tras la comercialización se han comunicado sobre todo en pacientes con factores de predisposición; p. ej., con antecedentes de elevación de las transaminasas previos el tratamiento con Kineret. No se han evaluado la eficacia y la seguridad de Kineret en pacientes con AST/ALT > 1,5 veces el límite superior de la normalidad.
Infecciones graves
La administración de Kineret se ha asociado con una mayor incidencia de infecciones graves (1,8%) comparado con el placebo (0,7%). En un reducido grupo de pacientes con asma, la incidencia de infecciones graves también fue mayor en los pacientes tratados con Kineret (4,5%) comparada con la de los pacientes tratados con placebo (0%); estas infecciones están principalmente relacionadas con las vías respiratorias.
No se ha evaluado la seguridad y eficacia de Kineret en pacientes con infecciones crónicas.
No debe iniciarse el tratamiento con Kineret en pacientes con infecciones activas. El tratamiento con Kineret debe interrumpirse si los pacientes con AR desarrollan una infección grave.
Los médicos deberán actuar con precaución cuando administren Kineret a pacientes con un historial de infecciones recurrentes o con enfermedades subyacentes que puedan predisponerlos a infecciones.
Se desconoce la seguridad de Kineret en pacientes con tuberculosis latente. Se han registrado casos de tuberculosis en pacientes tratados con diversos regímenes de antiinflamatorios biológicos. Los pacientes deben someterse a un estudio para detectar tuberculosis latente antes de iniciar el tratamiento con Kineret. También deben tenerse en cuenta las directrices médicas disponibles a este respecto.
Otros tratamientos con antirreumáticos se han asociado a una reactivación de la hepatitis B. Por tanto, también se debe realizar un estudio de hepatitis vírica en los pacientes, conforme a las directrices publicadas, antes de iniciar el tratamiento con Kineret.
Neutropenia
En estudios controlados con placebo sobre AR, Kineret se ha asociado frecuentemente a neutropenia (RAN < 1,5 x 109/l). Para más información sobre la neutropenia ver sección 4.8.
El tratamiento con Kineret no debe iniciarse en pacientes con neutropenia (RAN < 1,5 x 109/l). Se recomienda valorar el recuento de neutrófilos antes de iniciar el tratamiento con Kineret y mientras dure su administración, mensualmente durante los primeros 6 meses de tratamiento y trimestralmente después. En los pacientes que desarrollen una neutropenia (RAN < 1,5 x 109/l) se vigilará estrechamente el RAN y se interrumpirá la administración de Kineret. La seguridad y la eficacia de Kineret en pacientes con neutropenia no han sido evaluadas.
Inmunosupresión
No se ha estudiado el efecto del tratamiento con Kineret en pacientes con neoplasias preexistentes. Por tanto, no se recomienda el uso de Kineret en estos pacientes.
Vacunas
En un ensayo clínico controlado con placebo (n = 126), no se detectaron diferencias en la respuesta de anticuerpos antitetánicos entre los grupos de tratamiento con Kineret o con placebo cuando se administró la vacuna toxoide tetánico/diftérica conjuntamente con Kineret. No se dispone de datos sobre los efectos de la vacunación con otros antígenos inactivados en pacientes tratados con Kineret.
No se dispone tampoco de datos sobre los efectos de la vacunación con microorganismos vivos, ni sobre la transmisión secundaria de infecciones por vacunas de microorganismos vivos en los pacientes tratados con Kineret. Por tanto, no se deben administrar vacunas con microorganismos vivos conjuntamente con Kineret.
Población de pacientes de edad avanzada (> 65 años)
En los ensayos clínicos se han estudiado un total de 752 pacientes > 65 años, incluyendo 163 pacientes > 75 años. No se apreciaron diferencias globales en la seguridad o eficacia entre estos pacientes y los más jóvenes. Debido a la mayor incidencia de infecciones en las personas de la tercera edad en general, se actuará con cautela al tratar pacientes ancianos.
Tratamiento conjunto con Kineret y antagonistas del Factor de Necrosis Tumoral (TNF)
La administración conjunta de Kineret y etanercept se ha relacionado con un incremento del riesgo de sufrir infección grave y neutropenia comparado con etanercept solo. Este tratamiento combinado no ha demostrado que incremente el beneficio clínico.
La administración conjunta de Kineret y etanercept u otros antagonistas del TNF no está recomendada (ver sección 4.5).
Este medicamento contiene menos 1 mmol de sodio (23 mg) por dosis de 100 mg, por lo que se considera esencialmente "exento de sodio".
4.5 Interacción con otros medicamentos y otras formas de interacción
Las interacciones de Kineret con otros medicamentos no se han investigado en estudios formales. En los ensayos clínicos, no se han observado interacciones entre Kineret y otros medicamentos (incluyendo medicamentos antiinflamatorios no esteroideos, corticoides y fármacos antirreumáticos modificadores de la enfermedad (FARME)).
Tratamiento conjunto con Kineret y antagonistas del TNF
En un ensayo clínico con pacientes que estaban recibiendo metotrexato como tratamiento de base, se observó que el grupo de pacientes tratados con Kineret y etanercept presentaron una mayor incidencia de infecciones graves (7%) y neutropenia que el grupo tratado con etanercept solo y mayor que la observada en ensayos previos en los que Kineret se administró solo. El tratamiento combinado de Kineret y etanercept no ha demostrado que incremente el beneficio clínico.
No se recomienda el uso conjunto de Kineret y etanercept o cualquier otro antagonista del TNF (ver sección 4.4).
Sustratos del citocromo P450
La formación de enzimas del CYP450 se inhibe al aumentar los niveles de citocinas (p. ej., IL-1) durante los procesos inflamatorios crónicos. Por tanto, es de esperar que, en el caso de un antagonista de los receptores de IL-1 como anakinra, la formación de las enzimas del CYP450 se normalice durante el tratamiento. Esto sería clínicamente relevante para los sustratos del CYP450 con un índice terapéutico estrecho (p. ej., warfarina y fenitoína). Al iniciar o terminar el tratamiento con Kineret en pacientes que estén recibiendo este tipo de medicamentos, puede ser pertinente considerar la monitorización del efecto terapéutico o la concentración de estos productos, y puede ser necesario el ajuste individualizado de la dosis del medicamento.
Para información sobre vacunaciones, ver sección 4.4.
4.6 Fertilidad, embarazo y lactancia
Los datos referentes al uso de anakinra en mujeres embarazadas son limitados.No obstante, se han realizado estudios reproductivos con Kineret en ratas y conejos con dosis de hasta 100 veces la dosis humana en la AR y no hay evidencias de que afecte a la fertilidad ni de que se produzcan daños en el feto.
No se recomienda utilizar Kineret durante el embarazo, ni en mujeres en edad fértil que no estén utilizando métodos anticonceptivos.
Se desconoce si anakinra o los metabolitos se excretan en la leche humana. No se puede excluir el riesgo en recién nacidos/niños. Debe interrumpirse la lactancia durante el tratamiento con Kineret.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No procede.
4.8 Reacciones adversas
En los estudios controlados con placebo en pacientes con AR, las reacciones adversas notificadas con mayor frecuencia durante el tratamiento con Kineret fueron reacciones en el lugar de la inyección (RLI), que fueron leves o moderadas en la mayoría de los pacientes. Estas reacciones fueron la causa más frecuente de abandono del estudio entre los pacientes tratados con Kineret. La incidencia de reacciones adversas graves en los sujetos tratados con la dosis recomendada de Kineret (100 mg/día) fue comparable a la del placebo (7,1% frente a un 6,5% en el grupo placebo). La incidencia de infección grave fue mayor en los pacientes tratados con Kineret, comparados con los pacientes tratados con placebo (1,8% frente a 0,7%). El descenso de neutrófilos se produjo con mayor frecuencia entre los pacientes tratados con Kineret comparados con placebo.
Las reacciones adversas se enumeran de acuerdo a la frecuencia y clasificación por órganos y sistemas de MedDRA. Las categorías de frecuencia se definen mediante la siguiente convención: muy frecuentes (>1/10); frecuentes(> 1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles). En cada categoría de frecuencia, las reacciones adversas se recogen por orden de gravedad decreciente.
|
Sistema de Clasificación de Órganos (MedDRA) |
Frecuencia |
Reacción Adversa |
|
Infecciones e infestaciones |
Frecuentes (> 1/100, < 1/10) |
Infecciones graves |
|
Trastornos de la sangre y del sistema linfático |
Frecuentes (> 1/100, < 1/10) |
Neutropenia Trombocitopenia |
|
Trastornos del sistema inmunológico |
Poco frecuentes (> 1/1.000 a < 1/100) |
Reacciones alérgicas que incluyen reacciones anfilácticas, angioedema, urticaria y prurito |
|
Trastornos del sistema nervioso |
Muy frecuentes (> 1/10) |
Dolor de cabeza |
|
Trastornos hepatobiliares |
Poco frecuentes (> 1/1.000 a < 1/100) |
Aumento de las enzimas hepáticas |
|
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles) |
Hepatitis no infecciosa | |
|
Trastornos de la piel y del tejido subcutáneo |
Muy frecuentes (> 1/10) |
Reacción en el lugar de la inyección |
|
Poco frecuentes (> 1/1.000 a < 1/100) |
Erupción | |
|
Exploraciones complementarias |
Muy frecuentes (> 1/10) |
Aumento de los niveles sanguíneos de colesterol |
Infecciones graves
La incidencia de infecciones graves en los estudios en AR realizados con la dosis recomendada (100 mg/día) fue del 1,8% entre los pacientes tratados con Kineret y del 0,7% entre los pacientes tratados con placebo. En estudios hasta 3 años, la tasa de infecciones graves se mantuvo estable en el tiempo. Las infecciones observadas consistieron, principalmente, en afecciones bacterianas, como celulitis, neumonía e infecciones óseas y articulares. La mayoría de los pacientes continuó en el estudio con el medicamento después de que se resolviera la infección.
No se produjeron muertes durante los estudios en AR debido a episodios infecciosos graves.
En estudios clínicos y en la experiencia tras la comercialización, raramente se han observado casos de infecciones oportunistas incluyendo hongos, micobacterias, bacterias y virus. Las infecciones aparecieron en todos los sistemas corporales y en pacientes que recibieron Kineret solo o en combinación con agentes inmunosupresores.
Neutropenia
En los estudios controlados con placebo realizados con Kineret, el tratamiento se asoció con pequeñas reducciones en la media del recuento total de leucocitos y en el recuento absoluto de neutrófilos (RAN). Se comunicó neutropenia (RAN < 1,5 x 109/l) en el 2,4% de los pacientes tratados con Kineret comparados con el 0,4% de los tratados con placebo. Ninguno de estos pacientes sufrió infecciones graves asociadas con neutropenia.
T rombocitopenia
En los estudios clínicos de pacientes con AR, se notificó trombocitopenia en el 1,9% de los pacientes tratados, comparado con el 0,3% de los pacientes en el grupo de placebo. Las trombocitopenias fueron leves, es decir, con recuentos de plaquetas > 75 x109/l. También se ha observado trombocitopenia leve en pacientes con CAPS.
Se ha notificado trombocitopenia en el seguimiento poscomercialización de Kineret, incluyendo informes esporádicos de casos de trombocitopenia grave (es decir, con recuentos de plaquetas < 10 x109/l).
Neoplasias malignas
Los pacientes con AR pueden tener un mayor riesgo (una media de 2 a 3 veces superior) de desarrollar linfoma. En ensayos clínicos, los pacientes tratados con Kineret presentaron una mayor incidencia de linfoma respecto a la tasa esperada en la población general, dicha tasa es coherente con las tasas comunicadas generalmente en pacientes con artritis reumatoide.
En ensayos clínicos, la tasa bruta de incidencia de neoplasias malignas fue la misma entre los pacientes tratados con Kineret y los pacientes tratados con placebo, y no fue diferente de la observada en la población general. Además, la incidencia total de neoplasias no se incrementó en 3 años de exposición de pacientes a Kineret.
Reacciones alérgicas
De forma poco frecuente se ha informado de reacciones alérgicas que incluyen reacciones anafilácticas, angioedema, urticaria, erupción y prurito con Kineret. La mayoría de estas reacciones fueron erupciones maculopapulares o urticariales.
Inmunogenicidad
En ensayos clínicos, alrededor de un 3% de pacientes adultos dieron un resultado positivo, al menos en una ocasión, en el test de detección de anticuerpos con capacidad de neutralizar los efectos biológicos de anakinra. La aparición de anticuerpos fue generalmente transitoria y no se asoció con reacciones adversas o con una disminución de la eficacia. Adicionalmente en un ensayo clínico, el 6% de los pacientes pediátricos dieron un resultado positivo, al menos en una ocasión, en el test de detección de anticuerpos con capacidad de neutralizar los efectos biológicos de anakinra.
Acontecimientos hepáticos
En los estudios clínicos de pacientes con AR se han observado con poca frecuencia aumentos transitorios de las enzimas hepáticas. Dichos aumentos no se han asociado a signos o síntomas de daño hepatocelular. En el seguimiento post-comercialización se han notificado casos aislados de hepatitis no infecciosa. Estos acontecimientos hepáticos observados tras la comercialización se han notificado sobre todo en pacientes con factores de predisposición, p. ej., pacientes con antecedentes de transaminasas elevadas antes de iniciar el tratamiento con Kineret.
Reacciones en el lugar de la inyección
Las reacciones adversas más frecuentemente comunicadas asociadas con la administración de Kineret fueron las RLI. La mayoría de ellas (95%) se describieron como leves o moderadas. Se caracterizaron típicamente por 1 o más de los siguientes signos o síntomas: eritema, equimosis, inflamación y dolor. Con la dosis de 100 mg/día, el 71% de los pacientes con AR desarrolló una RLI, comparado con el 28% entre los pacientes tratados con placebo. La aparición de RLI se produjo comúnmente dentro de las 2 primeras semanas de tratamiento y desapareció dentro de las 4-6 semanas. El desarrollo de RLI en pacientes que no la habían experimentado previamente fue infrecuente después del primer mes de tratamiento.
Aumento de los niveles sanguíneos de colesterol
En los estudios clínicos sobre la AR, en 775 pacientes tratados con dosis diarias de Kineret de 30 mg, 75 mg, 150 mg, 1 mg/kg o 2 mg/kg, se detectó un aumento del 2,4% al 5,3% en los niveles de colesterol total 2 semanas después del inicio del tratamiento con Kineret, sin que se observara una relación dosis-respuesta. El patrón era similar a las 24 semanas de tratamiento con Kineret. El tratamiento con placebo (n = 213) produjo una disminución en los niveles de colesterol total de alrededor del 2,2% en la semana 2 y del 2,3% en la semana 24. No se dispone de datos sobre el LDL o el HDL.
Notificación de sospechas de reacciones adversas:
Es importante notificar las sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
En los ensayos clínicos con pacientes con artritis reumatoide no se observó toxicidad limitante de la dosis.
En los ensayos de sepsis, 1.015 pacientes recibieron Kineret a dosis hasta 2 mg/kg/hora IV (es decir, aprox. 35 veces la dosis recomendada en AR) durante un periodo de tratamiento de 72 horas. El perfil de acontecimientos adversos en estos ensayos no muestra ninguna diferencia significativa con respecto a lo observado en los ensayos en artritis reumatoide.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inmunosupresores, inhibidores de la interleucina. Código ATC: L04AC03
Anakinra neutraliza la actividad biológica de la interleucina-1a (IL-1a) e interleucina-1p (IL-1P) inhibiendo competitivamente su unión al receptor de tipo I de la interleucina-1 (IL-1RI). La interleucina-1 (IL-1) es una citocina pro-inflamatoria clave, que interviene en muchas respuestas celulares entre ellas aquellas importantes en la inflamación sinovial.
La IL-1 se encuentra en el plasma y el líquido sinovial de los pacientes con artritis reumatoide, y se ha indicado una correlación entre las concentraciones de IL-1 en plasma y la actividad de la enfermedad. Anakinra inhibe las respuestas producidas por la IL-1 in vitro, incluyendo la inducción de óxido nítrico y prostaglandina E2 y/o la producción de colagenasa por las células sinoviales, fibroblastos y los condriocitos.
Eficacia clínica y seguridad
Se ha demostrado la seguridad y eficacia de anakinra en combinación con metotrexato en 1790 pacientes de edad > 18 años con AR y distintos grados de severidad de la enfermedad.
La respuesta clínica a anakinra generalmente se observó a las 2 semanas de iniciar el tratamiento, manteniéndose con la administración continuada de anakinra. La máxima respuesta clínica generalmente se observó a las 12 semanas de iniciar el tratamiento.
El tratamiento combinado con anakinra y metotrexato muestra una reducción estadística y clínicamente significativa en la gravedad de los signos y síntomas de la artritis reumatoide en pacientes con respuesta inadecuada a la administración de metotrexato solo (38% frente al 22% de respuestas, usando como criterio de medida el ACR20). Se han observado mejorías significativas en el dolor, en el recuento de articulaciones dolorosas, en la funcionalidad física (puntuación HAQ), en los reactantes de fase aguda y en las valoraciones globales del paciente y del médico.
Se han realizado exploraciones mediante rayos X en un estudio clínico con anakinra. No se ha demostrado un efecto perjudicial sobre el cartílago articular.
Seguridad en pacientes pediátricos con AR (AIJ)
Se evaluó Kineret en un único ensayo randomizado, ciego, multicéntrico, en el que participaron 86 pacientes con artritis reumatoide juvenil poliarticular (ARJ; edades 2-17 años) que recibieron una dosis diaria subcutánea de 1 mg/kg, hasta una dosis máxima de 100 mg. Los 50 pacientes que alcanzaron una respuesta clínica tras un periodo abierto de 12 semanas, fueron randomizadamente designados a recibir Kineret (25 pacientes) o placebo (25 pacientes), administrado diariamente durante otras 16 semanas adicionales. Un subgrupo de estos pacientes continuó el tratamiento abierto con Kineret durante un máximo de 1 año en un estudio de extensión complementario. En estos estudios se observó un perfil de acontecimientos adversos similar al observado en los pacientes adultos con AR. Los datos de este estudio son insuficientes para demostrar la eficacia, por lo que, no se recomienda el uso de Kineret en pacientes pediátricos con ARJ.
Inmunogenicidad Ver sección 4.8.
5.2 Propiedades farmacocinéticas
La biodisponibilidad absoluta de anakinra en sujetos sanos (n = 11) después de inyectar 70 mg en bolo subcutáneo es del 95%. El proceso de absorción es el factor limitante de la velocidad de desaparición de anakinra del plasma después de la inyección subcutánea. En sujetos con AR, las concentraciones plasmáticas máximas de anakinra se produjeron entre las 3 y 7 horas después de la administración subcutánea del medicamento a dosis clínicamente relevantes (1 a 2 mg/kg; n = 18). La concentración plasmática disminuyó sin una fase de distribución discernible y la semivida terminal osciló entre 4 y 6 horas. En los pacientes con AR no se apreció una acumulación inesperada de anakinra después de la administración de dosis diarias subcutáneas durante un periodo de hasta 24 semanas. Las medias (DE) estimadas de aclaramiento (CL/F) y volumen de distribución (Vd/F) para los análisis de datos de población de dos estudios farmacocinéticos (FC) en 35 pacientes con AR fueron de 105 (27) ml/min y 18,5 (11) l, respectivamente. Los datos procedentes de animales y de humanos demostraron que el riñón es el principal órgano responsable de la eliminación de anakinra. El aclaramiento de anakinra en pacientes con AR aumentó con el incremento del aclaramiento de creatinina.
Se estudió la influencia de las covariables demográficas sobre la farmacocinética de anakinra utilizando un análisis farmacocinético poblacional que incluyó 341 pacientes tratados con una inyección diaria subcutánea de anakinra en dosis de 30, 75 y 150 mg en un periodo de hasta 24 semanas. El aclaramiento estimado de anakinra aumentó al aumentar el aclaramiento de creatinina y el peso corporal. El análisis farmacocinético de la población demostró que el aclaramiento plasmático medio después de la administración en bolo subcutáneo fue aproximadamente un 14% mayor en los varones que en las mujeres y aproximadamente un 10% mayor en sujetos < 65 años que en > 65 años. Sin embargo, después de ajustar según el aclaramiento de creatinina y el peso corporal, el sexo y la edad no fueron factores influyentes significativos en el aclaramiento plasmático medio. No se requieren ajustes de la dosis en función de la edad o el sexo del paciente.
Insuficiencia hepática
Se ha realizado un estudio en el que participaron 12 pacientes con insuficiencia hepática (Child-Pugh B) a los que se administró una dosis única intravenosa de 1mg/kg. Los parámetros farmacocinéticos no fueron muy distintos de los hallados en voluntarios sanos, a excepción de una disminución del 30% en el aclaramiento, en comparación con los datos de un estudio en voluntarios sanos. Se observó una disminución correspondiente del aclaramiento de creatinina en la población con insuficiencia hepática. Por consiguiente, la disminución del aclaramiento es más probable que pueda explicarse por una reducción de la función renal en esta población. Estos datos avalan que no se requiera ajuste de la dosis en pacientes con insuficiencia hepática Child-Pugh Clase B. Ver sección 4.2.
Insuficiencia renal
El aclaramiento plasmático medio de Kineret en pacientes con insuficiencia renal_leve (aclaramiento de creatinina 50-80 ml/min) y moderada (aclaramiento de creatinina 30-49 ml/min) se redujo en un 16% y un 50%, respectivamente. Con insuficiencia renal grave y enfermedad renal avanzada (aclaramiento de creatinina < 30 ml/min), el aclaramiento plasmático medio se redujo en un 70% y un 75%, respectivamente. Menos del 2,5% de la dosis de Kineret administrada se eliminó por hemodiálisis o diálisis peritoneal continua ambulatoria. Estos datos avalan que no se requiera ajuste de la dosis en pacientes con insuficiencia renal leve (CLcr 50 a 80 ml/minuto). Ver sección 4.2.
5.3 Datos preclínicos sobre seguridad
Anakinra no produjo ningún efecto observable sobre la fertilidad, desarrollo inicial, desarrollo embrio-fetal o desarrollo peri o post-natal en rata a dosis de hasta 100 veces la dosis en humanos. Tampoco se han observado efectos en el desarrollo embrio-fetal en conejo a dosis 100 veces la humana.
En una serie estándar de pruebas diseñada para identificar riesgos sobre el ADN, anakinra no indujo la aparición de mutaciones genéticas en bacterias o células de mamífero. Anakinra tampoco aumentó la incidencia de anomalías cromosómicas o micronúcleos en las células de la médula ósea del ratón. No se han realizado estudios a largo plazo para evaluar el potencial carcinogénico de anakinra. Los datos obtenidos en ratones que sobreexpresan el IL-1ra y en ratones mutantes carentes de IL-1ra no indicaron un mayor riesgo de desarrollo tumoral.
Un ensayo formal sobre las interacciones toxicológicas y toxicocinéticas en ratas no reveló ninguna prueba de que Kineret altere el perfil toxicológico o farmacocinético del metotrexato.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Ácido cítrico anhidro Cloruro sódico Edetato disódico dihidratado Polisorbato 80 Hidróxido sódico
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 3 años.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
Para el uso ambulatorio, Kineret se puede sacar de la nevera durante 12 horas a temperatura no superior a 25 °C, sin sobrepasar la fecha de caducidad. Tras este periodo, el producto no debe ponerse nuevamente en la nevera y debe desecharse.
6.5 Naturaleza y contenido del envase
0,67 ml de solución inyectable en jeringa precargada (vidrio tipo I) con émbolo (caucho bromobutilo) y una aguja de calibre 29. La jeringa precargada tiene una funda externa para la aguja, de plástico rígido, unida a una cubierta interna. Ningún componente de la jeringa ni de la aguja está hecho con látex de caucho natural.
Envases de 1, 7 o 28 (envase múltiple con 4 paquetes de 7 jeringas precargadas) jeringas precargadas. Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Kineret es una solución estéril que no contiene conservantes. Para un solo uso.
No agitar. Dejar que la jeringa alcance la temperatura ambiente antes de inyectar.
Antes de la administración se debe comprobar si la solución contiene partículas o ha cambiado de color. Solamente se deben inyectar las soluciones que sean transparentes, entre incoloras y blanquecinas y que pueden contener partículas amorfas, relacionadas con el producto, de aspecto entre translúcido y blanco.
La presencia de estas partículas no afecta a la calidad del producto.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con élse realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm Suecia
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/203/001 envase de 1 jeringa precargada EU/1/02/203/002 envase de 7 jeringas precargadas EU/1/02/203/003 envase de 28 jeringas precargadas
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA
AUTORIZACIÓN
Fecha de la primera autorización: 8 Marzo 2002 Fecha de la última renovación: 20 Marzo 2007
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
1. NOMBRE DEL MEDICAMENTO
Kineret 100 mg/0,67 ml, solución inyectable en jeringa precargada.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada jeringa graduada precargada contiene 100 mg de anakinra* en 0,67 ml (150 mg/ml).
* Antagonista del receptor humano para la interleucina 1 (r-metHuIL-1ra), producido en células de Escherichia coli por tecnología del ADN recombinante.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
Solución inyectable transparente, entre incolora y blanquecina que puede contener partículas amorfas, relacionadas con el producto, de aspecto entre translúcido y blanco.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Kineret está indicado en adultos para el tratamiento de los signos y síntomas de la artritis reumatoide (AR) en combinación con metotrexato, en aquellos pacientes que no hayan respondido bien a la administración de metotrexato solo.
Kineret está indicado en adultos, adolescentes, niños y lactantes a partir de los 8 meses de edad, con un peso corporal de 10 kg o superior, para el tratamiento de los síndromes periódicos asociados a criopirina (CAPS), a saber:
- Enfermedad inflamatoria multisistémica de inicio neonatal (NOMID)/síndrome articular, cutáneo y neurológico infantil crónico (CINCA)
- Síndrome de Muckle-Wells (MWS)
- Síndrome autoinflamatorio familiar por frío (FCAS)
4.2 Posología y forma de administración
El tratamiento con Kineret deberá ser iniciado y controlado por médicos especialistas con experiencia en el diagnóstico y tratamiento de la artritis reumatoide y los CAPS, respectivamente.
Posología
AR: adultos
La dosis recomendada de Kineret es de 100 mg administrados una vez al día en inyección subcutánea. La dosis debe administrarse cada día a la misma hora aproximadamente.
CAPS: adultos, adolescentes, niños y lactantes a partir de los 8 meses de edad, con un peso corporal de 10 kg o superior
Dosis inicial:
La dosis inicial recomendada en todos los subtipos de CAPS es de 1-2 mg/kg/día en inyección subcutánea. La respuesta terapéutica se refleja principalmente en la reducción de los síntomas clínicos, como fiebre, erupción, dolor articular y cefalea, pero también de los marcadores séricos (niveles de CRP/SAA) o en la frecuencia de los brotes.
Dosis de mantenimiento en los CAPS leves (FCAS, MWS leve):
Por lo general, la enfermedad puede controlarse adecuadamente manteniendo la dosis inicial recomendada (1-2 mg/kg/día).
Dosis de mantenimiento en los CAPS graves (MWSy NOMID/CINCA):
Puede ser necesario un aumento de la dosis en los primeros 1-2 meses, en función de la respuesta terapéutica. La dosis de mantenimiento habitual en los CAPS graves es de 3-4 mg/kg/día, que puede ajustarse hasta un máximo de 8 mg/kg/día.
Además de evaluar los síntomas clínicos y los marcadores inflamatorios en los CAPS graves, se recomienda valorar la inflamación del SNC, incluyendo el oído interno (RMN o TAC, punción lumbar o audiometría) y los ojos (valoraciones oftalmológicas) después de los 3 primeros meses de tratamiento, y posteriormente cada 6 meses, hasta identificar la dosis de tratamiento eficaz. Cuando los pacientes estén bien controlados clínicamente, el seguimiento del SNC y oftalmológico puede realizarse anualmente.
Población de _pacientes de edad avanzada (> 65 años)
No es necesario ajustar la dosis en los pacientes con AR. La posología y administración es la misma que en los adultos de 18 a 64 años.
Los datos en pacientes de edad avanzada con CAPS son limitados. No cabe esperar que se requieran ajustes de dosis.
Población _pediátrica (< 18 años)
AR: No se ha establecido la eficacia de Kineret en niños con AR (Artritis Idiopática Juvenil (AIJ)) de 0 a 18 años.
CAPS: La posología y forma de administración en niños y lactantes a partir de los 8 meses de edad, con un peso corporal de 10 kg o superior es la misma que para los pacientes adultos con CAPS, basa en el peso corporal. No se dispone de datos en niños menores de 8 meses de edad.
Insuficiencia hepática
No es necesario ajustar la dosis en pacientes con insuficiencia hepática moderada (Child-Pugh Clase B). Kineret debe utilizarse con precaución en pacientes con insuficiencia hepática grave.
Insuficiencia renal
Kineret no debe utilizarse en pacientes con insuficiencia renal grave (CLcr < 30 ml/minuto) (ver sección 4.3). No es necesario ajustar la dosis en pacientes con insuficiencia renal leve (CLcr 50 a 80 ml/minuto). A falta de datos adecuados, Kineret debe usarse con precaución en pacientes con insuficiencia renal moderada (CRcr 30 a 50 ml/minuto).
Forma de administración
Kineret se administra mediante inyección subcutánea.
Kineret se suministra listo para inyectar en una jeringa graduada precargada que permite administrar dosis de entre 20 y 100 mg. Dado que la dosis mínima es de 20 mg, la jeringa no es apta para pacientes pediátricos con un peso corporal inferior a 10 kg. La jeringa precargada no debe agitarse. Para las precauciones especiales de eliminación y otras manipulaciones, ver sección 6.6.
Se recomienda alternar el lugar de la inyección para evitar molestias en el mismo. Los signos y síntomas de las reacciones en el lugar de la inyección pueden aliviarse enfriando la zona, calentando el líquido de inyección, aplicando compresas frías (antes y después de la inyección) y utilizando antihistamínicos y corticosteroides de uso tópico después de la inyección.
4.3 Contraindicaciones
Hipersensibilidad al principio activo, o a alguno de sus excipientes incluidos en la sección 6.1 o a proteínas derivadas de E. coli.
No se debe administrar Kineret a pacientes con insuficiencia renal grave (Clcr< 30 ml/minuto) (ver sección 4.2).
No se debe iniciar el tratamiento con Kineret en pacientes con neutropenia (RAN < 1,5 x 109/l) (ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
Reacciones alérgicas
De forma poco frecuente se ha informado de reacciones alérgicas que incluyeron reacciones anafilácticas y angioedema. La mayoría de estas reacciones fueron erupciones maculopapulares o urticariales. En caso de que se produjera una reacción alérgica grave, se deberá interrumpir la administración de Kineret e iniciar el tratamiento apropiado.
Acontecimientos hepáticos
En los estudios clínicos en pacientes con AR y CAPS se han observado con poca frecuencia aumentos transitorios de las enzimas hepáticas. Dichos aumentos no se han asociado a signos o síntomas de daño hepatocelular. En el seguimiento post-comercialización, se han comunicado casos aislados de hepatitis no infecciosa. Estos acontecimientos hepáticos observados tras la comercialización se han comunicado sobre todo en pacientes con factores de predisposición p. ej., con antecedentes de elevación de las transaminasas previos al tratamiento con Kineret. No se han evaluado la eficacia y la seguridad de Kineret en pacientes con AST/ALT > 1,5 veces el límite superior de la normalidad.
Infecciones graves
La administración de Kineret se ha asociado con una mayor incidencia de infecciones graves (1,8%) comparado con el placebo (0,7%) en pacientes con AR. En un reducido grupo de pacientes con asma, la incidencia de infecciones graves también fue mayor en los pacientes tratados con Kineret (4,5%) comparada con la de los pacientes tratados con placebo (0%). Estas infecciones están principalmente relacionadas con las vías respiratorias.
No se ha evaluado la seguridad y eficacia del tratamiento con Kineret en pacientes con infecciones crónicas y graves.
No debe iniciarse el tratamiento con Kineret en pacientes con infecciones activas. El tratamiento con Kineret debe interrumpirse si los pacientes con AR desarrollan una infección grave. En los pacientes con CAPS tratados con Kineret, existe un riesgo de que se produzcan brotes de la enfermedad al interrumpir el tratamiento. Esto debe tenerse en cuenta si se decide interrumpir el tratamiento con Kineret durante una infección grave.
Los médicos deberán actuar con precaución cuando administren Kineret a pacientes con un historial de infecciones recurrentes o con enfermedades subyacentes que puedan predisponerlos a infecciones.
Se desconoce la seguridad de Kineret en pacientes con tuberculosis latente. Se han registrado casos de tuberculosis en pacientes tratados con diversos regímenes de antiinflamatorios biológicos. Los pacientes deben someterse a un estudio para detectar tuberculosis latente antes de iniciar el tratamiento con Kineret. También deben tenerse en cuenta las directrices médicas disponibles a este respecto.
Otros tratamientos con antirreumáticos se han asociado a una reactivación de la hepatitis B. Por tanto, también se debe realizar un estudio de hepatitis vírica en los pacientes, conforme a las directrices publicadas, antes de iniciar el tratamiento con Kineret.
Neutropenia
En estudios controlados con placebo sobre AR, Kineret se ha asociado frecuentemente a neutropenia (RAN < 1,5 x 109/l). Para más información sobre la neutropenia ver sección 4.8.
El tratamiento con Kineret no debe iniciarse en pacientes con neutropenia (RAN < 1,5 x 109/l). Se recomienda valorar el recuento de neutrófilos antes de iniciar el tratamiento con Kineret y mientras dure su administración, mensualmente durante los primeros 6 meses de tratamiento y trimestralmente después. En los pacientes que desarrollen una neutropenia (RAN < 1,5 x 109/l) se vigilará estrechamente el RAN y se interrumpirá la administración de Kineret. La seguridad y la eficacia de Kineret en pacientes con neutropenia no han sido evaluadas.
Inmunosupresión
No se ha estudiado el efecto del tratamiento con Kineret en pacientes con neoplasias malignas preexistentes. Por tanto, no se recomienda el uso de Kineret en estos pacientes.
Vacunas
En un ensayo clínico controlado con placebo (n = 126), no se detectaron diferencias en la respuesta de anticuerpos antitetánicos entre los grupos de tratamiento con Kineret o con placebo cuando se administró la vacuna toxoide tetánico/diftérica conjuntamente con Kineret. No se dispone de datos sobre los efectos de la vacunación con otros antígenos inactivados en pacientes tratados con Kineret.
No se dispone tampoco de datos sobre los efectos de la vacunación con microorganismos vivos, ni sobre la transmisión secundaria de infecciones por vacunas de microorganismos vivos en los pacientes tratados con Kineret. Por tanto, no se deben administrar vacunas con microorganismos vivos conjuntamente con Kineret.
Población de pacientes de edad avanzada (> 65 años)
En los ensayos clínicos se han estudiado un total de 752 pacientes > 65 años con AR, incluyendo 163 pacientes > 75 años. No se apreciaron diferencias globales en la seguridad o eficacia entre estos pacientes y los más jóvenes. La experiencia en el tratamiento de pacientes de edad avanzada con CAPS es escasa. Debido a la mayor incidencia de infecciones en la población de edad avanzada en general, se actuará con cautela al tratar pacientes ancianos.
Tratamiento conjunto con Kineret y antagonistas del Factor de Necrosis Tumoral (TNF)
La administración conjunta de Kineret y etanercept se ha relacionado con un incremento del riesgo de sufrir infección grave y neutropenia comparado con etanercept solo en pacientes con AR. Este tratamiento combinado no ha demostrado que incremente el beneficio clínico.
La administración conjunta de Kineret y etanercept u otros antagonistas del TNF no está recomendada (ver sección 4.5).
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis de 100 mg, por lo que se considera esencialmente "exento de sodio".
4.5 Interacción con otros medicamentos y otras formas de interacción
Las interacciones de Kineret con otros medicamentos no se han investigado en estudios formales. En los ensayos clínicos, no se han observado interacciones entre Kineret y otros medicamentos (incluyendo medicamentos antiinflamatorios no esteroideos, corticoides y fármacos antirreumáticos modificadores de la enfermedad (FARME)).
Tratamiento conjunto con Kineret y antagonistas del TNF
En un ensayo clínico con pacientes con AR que estaban recibiendo metotrexato como tratamiento de base, se observó que el grupo de pacientes tratados con Kineret y etanercept presentaron una mayor incidencia de infecciones graves (7%) y neutropenia que el grupo tratado con etanercept solo y mayor que la observada en ensayos previos en los que Kineret se administró solo. El tratamiento combinado de Kineret y etanercept no ha demostrado que incremente el beneficio clínico.
No se recomienda el uso conjunto de Kineret y etanercept o cualquier otro antagonista del TNF (ver sección 4.4).
Sustratos del citocromo P450
La formación de enzimas del CYP450 se inhibe al aumentar los niveles de citocinas (p. ej., IL-1) durante los procesos inflamatorios crónicos. Por tanto, es de esperar que, en el caso de un antagonista de los receptores de IL-1 como anakinra, la formación de las enzimas del CYP450 se normalice durante el tratamiento. Esto sería clínicamente relevante para los sustratos del CYP450 con un índice terapéutico estrecho (p. ej., warfarina y fenitoína). Al iniciar o terminar el tratamiento con Kineret en pacientes que estén recibiendo este tipo de medicamentos, puede ser pertinente considerar la monitorización del efecto terapéutico o la concentración de estos productos, y puede ser necesario el ajuste individualizado de la dosis del medicamento.
Para información sobre vacunaciones, ver sección 4.4.
4.6 Fertilidad, embarazo y lactancia
Los datos referentes al uso de anakinra en mujeres embarazadas son limitados.
No obstante, se han realizado estudios de reproducción con Kineret en ratas y conejos con dosis de hasta 100 veces la dosis humana en la AR y no hay evidencias de que afecte a la fertilidad ni de que se produzcan daños en el feto.
No se recomienda utilizar Kineret durante el embarazo, ni en mujeres en edad fértil que no estén utilizando métodos anticonceptivos.
Se desconoce si anakinra/metabolitos se excretan en la leche materna. No se puede excluir el riesgo en recién nacidos/niños. Debe interrumpirse la lactancia durante el tratamiento con Kineret.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No procede.
4.8 Reacciones adversas
En los estudios controlados con placebo en pacientes con AR, las reacciones adversas notificadas con mayor frecuencia durante el tratamiento con Kineret fueron reacciones en el lugar de la inyección (RLI), que fueron leves o moderadas en la mayoría de los pacientes. Estas reacciones fueron la causa más frecuente de abandono del estudio entre los pacientes con AR tratados con Kineret. La incidencia de reacciones adversas graves en los sujetos tratados con la dosis recomendada de Kineret en los estudios sobre la AR (100 mg/día) fue comparable a la del placebo (7,1% frente a un 6,5% en el grupo placebo). La incidencia de infección grave fue mayor en los pacientes tratados con Kineret, comparados con los pacientes tratados con placebo (1,8% frente a 0,7%). El descenso de neutrófilos se produjo con mayor frecuencia entre los pacientes tratados con Kineret comparados con placebo.
Los datos sobre reacciones adversas en pacientes con CAPS se extrajeron de un estudio abierto en el que participaron 43 pacientes con NOMID/CINCA tratados con Kineret durante un máximo de 5 años, con una exposición total a Kineret de 159,8 pacientes-año. Durante el estudio de 5 años de duración,
14 pacientes (32,6%) notificaron 24 acontecimientos graves, de los cuales 11 acontecimientos graves en 4 pacientes (9,3%) se consideraron relacionados con Kineret. Ningún paciente abandonó el tratamiento con Kineret debido a reacciones adversas. Nada indica en las reacciones adversas observadas en este estudio y en el seguimiento posterior a la comercialización que el perfil de seguridad para los pacientes con CAPS sea distinto que para los pacientes con AR. La tabla de reacciones adversas que figura a continuación es aplicable, por tanto, al tratamiento con Kineret de pacientes con AR y de pacientes con CAPS.
Las reacciones adversas se enumeran de acuerdo a la frecuencia y clasificación por órganos y sistemas de MedDRA. Las categorías de frecuencia se definen mediante la siguiente convención: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles). En cada categoría de frecuencia, las reacciones adversas se recogen por orden de gravedad decreciente.
|
Sistema de Clasificación de Órganos (MedDRA) |
Frecuencia |
Reacción Adversa |
|
Infecciones e infestaciones |
Frecuentes (> 1/100, < 1/10) |
Infecciones graves |
|
Trastornos de la sangre y del sistema linfático |
Frecuentes (> 1/100, < 1/10) |
Neutropenia Trombocitopenia |
|
Trastornos del sistema inmunológico |
Poco frecuentes (> 1/1.000 a < 1/100) |
Reacciones alérgicas que incluyen reacciones anafilácticas, angioedema, urticaria y prurito |
|
Trastornos del sistema nervioso |
Muy frecuentes (> 1/10) |
Cefalea |
|
Trastornos hepatobiliares |
Poco frecuentes (> 1/1.000 a < 1/100) |
Aumento de las enzimas hepáticas |
|
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles) |
Hepatitis no infecciosa | |
|
Trastornos de la piel y del tejido subcutáneo |
Muy frecuentes (> 1/10) |
Reacción en el lugar de la inyección |
|
Poco frecuentes (> 1/1.000 a < 1/100) |
Erupción | |
|
Exploraciones complementarias |
Muy frecuentes (> 1/10) |
Aumento de los niveles sanguíneos de colesterol |
Infecciones graves
La incidencia de infecciones graves en los estudios en AR realizados con la dosis recomendada (100 mg/día) fue del 1,8% entre los pacientes tratados con Kineret y del 0,7% entre los pacientes tratados con placebo. En estudios hasta 3 años, la tasa de infecciones graves se mantuvo estable en el tiempo. Las infecciones observadas consistieron, principalmente, en afecciones bacterianas, como celulitis, neumonía e infecciones óseas y articulares. La mayoría de los pacientes continuó en el estudio con el medicamento después de que se resolviera la infección.
En el seguimiento durante 5 años de 43 pacientes con CAPS, la frecuencia de infecciones graves fue de 0,1/año; las más frecuentes fueron neumonía y gastroenteritis. La administración de Kineret se interrumpió temporalmente en un paciente, mientras que los demás continuaron el tratamiento con este medicamento durante las infecciones.
En los estudios en AR o CAPS no se produjeron muertes debidas a infecciones graves.
En estudios clínicos en AR y en la experiencia tras la comercialización, raramente se han observado casos de infecciones oportunistas incluyendo hongos, micobacterias, bacterias y virus. Las infecciones aparecieron en todos los sistemas corporales y en pacientes que recibieron Kineret solo o en combinación con agentes inmunosupresores.
Neutropenia
En los estudios en AR controlados con placebo realizados con Kineret, el tratamiento se asoció con pequeñas reducciones en la media del recuento total de leucocitos y en el recuento absoluto de neutrófilos (RAN). Se notificó neutropenia (RAN < 1,5 x 109/l) en el 2,4% de los pacientes tratados con Kineret comparados con el 0,4% de los tratados con placebo. Ninguno de estos pacientes sufrió infecciones graves asociadas con neutropenia.
En el seguimiento durante 5 años de 43 pacientes con CAPS, se notificó neutropenia en 2 pacientes. Ambos episodios se resolvieron con el tiempo mientras se continuaba el tratamiento con Kineret.
T rombocitopenia
En los estudios clínicos de pacientes con AR, se notificó trombocitopenia en el 1,9% de los pacientes tratados, comparado con el 0,3% de los pacientes en el grupo de placebo. Las trombocitopenias fueron leves, es decir, con recuentos de plaquetas > 75 x109/l. También se ha observado trombocitopenia leve en pacientes con CAPS.
Se ha notificado trombocitopenia en el seguimiento poscomercialización de Kineret, incluyendo informes esporádicos de casos de trombocitopenia grave (es decir, con recuentos de plaquetas < 10 x109/l).
Neoplasias malignas
Los pacientes con AR pueden tener un mayor riesgo (una media de 2 a 3 veces superior) de desarrollar linfoma. En ensayos clínicos, los pacientes tratados con Kineret presentaron una mayor incidencia de linfoma respecto a la tasa esperada en la población general, dicha tasa es coherente con las tasas comunicadas generalmente en pacientes con AR.
En ensayos clínicos, la tasa bruta de incidencia de neoplasias malignas fue la misma entre los pacientes tratados con Kineret y los pacientes tratados con placebo, y no fue diferente de la observada en la población general. Además, la incidencia total de neoplasias malignas no se incrementó en 3 años de exposición de pacientes a Kineret.
Reacciones alérgicas
De forma poco frecuente se han notificado reacciones alérgicas con Kineret, que incluyen reacciones anafilácticas, angioedema, urticaria, erupción y prurito. La mayoría de estas reacciones fueron erupciones maculopapulares o urticariales.
En el seguimiento durante 5 años de 43 pacientes con CAPS, no se produjeron acontecimientos alérgicos graves y ninguno requirió la interrupción del tratamiento con Kineret.
Inmunogenicidad
En ensayos clínicos en AR, alrededor de un 3% de pacientes adultos dieron un resultado positivo, al menos en una ocasión, en el test de detección de anticuerpos con capacidad de neutralizar los efectos biológicos de anakinra. La aparición de anticuerpos fue generalmente transitoria y no se asoció con reacciones adversas clínicas o con una disminución de la eficacia. Adicionalmente en un ensayo clínico, el 6% de los pacientes pediátricos dieron un resultado positivo, al menos en una ocasión, en el test de detección de anticuerpos con capacidad de neutralizar los efectos biológicos de anakinra.
La mayoría de los pacientes con CAPS del estudio 03-AR-0298 desarrollaron anticuerpos anti-anakinra. Esto no se asoció con ningún efecto clínicamente significativo sobre la farmacocinética, la eficacia o la seguridad.
Acontecimientos hepáticos
En los estudios clínicos de pacientes con AR y CAPS se han observado con poca frecuencia aumentos transitorios de las enzimas hepáticas. Dichos aumentos no se han asociado a signos o síntomas de daño hepatocelular. En el seguimiento post-comercialización, se han notificado casos aislados de hepatitis no infecciosa. Estos acontecimientos hepáticos observados tras la comercialización se han notificado sobre todo en pacientes con factores de predisposición, p. ej., pacientes con antecedentes de transaminasas elevadas antes de iniciar el tratamiento con Kineret.
Reacciones en el lugar de la inyección
Las reacciones adversas más frecuentemente comunicadas asociadas con la administración de Kineret fueron las RLI. La mayoría de ellas (95%) se notificaron como leves o moderadas. Se caracterizaron típicamente por 1 o más de los siguientes signos o síntomas: eritema, equimosis, inflamación y dolor. Con la dosis de 100 mg/día, el 71% de los pacientes con AR desarrolló una RLI, comparado con el 28% entre los pacientes tratados con placebo. En el seguimiento durante 5 años de 43 pacientes con CAPS, ningún paciente tuvo que interrumpir el tratamiento con Kineret, temporal o permanentemente, debido a RLI. Las RLI suelen aparecer en las 2 primeras semanas del tratamiento y desaparecen a las 4-6 semanas. El desarrollo de RLI en pacientes que no la habían experimentado previamente fue infrecuente después del primer mes de tratamiento.
Aumento de los niveles sanguíneos de colesterol
En los estudios clínicos sobre la AR, en 775 pacientes tratados con dosis diarias de Kineret de 30 mg, 75 mg, 150 mg, 1 mg/kg o 2 mg/kg, se detectó un aumento del 2,4% al 5,3% en los niveles de colesterol total 2 semanas después del inicio del tratamiento con Kineret, sin que se observara una relación dosis-respuesta. El patrón era similar a las 24 semanas de tratamiento con Kineret. El tratamiento con placebo (n = 213) produjo una disminución en los niveles de colesterol total de alrededor del 2,2% en la semana 2 y del 2,3% en la semana 24. No se dispone de datos sobre el LDL o el HDL.
Población pediátrica
Kineret se ha estudiado durante 5 años en 36 pacientes con CAPS de edades comprendidas entre 8 meses y < 18 años. Con la excepción de las infecciones y sus síntomas relacionados, que se notificaron con más frecuencia en niños menores de 2 años, el perfil de seguridad fue similar en todos los grupos de edad pediátrica. El perfil de seguridad en los pacientes pediátricos fue similar al observado en la población adulta, sin que se observaran nuevas reacciones adversas clínicamente relevantes.
Notificación de sospechas de reacciones adversas:
Es importante notificar las sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
En los ensayos clínicos con pacientes con AR o CAPS no se observó toxicidad limitante de la dosis.
En los ensayos de sepsis, 1015 pacientes recibieron Kineret a dosis de hasta 2 mg/kg/hora IV (es decir, aprox. 35 veces la dosis recomendada en AR) durante un periodo de tratamiento de 72 horas. El perfil de acontecimientos adversos en estos ensayos no muestra ninguna diferencia significativa con respecto a lo observado en los ensayos en artritis reumatoide.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inmunosupresores, inhibidores de la interleucina. Código ATC: L04AC03
Anakinra neutraliza la actividad biológica de la interleucina- 1a (IL-1a) e interleucina-1p (IL-1P) al inhibiendo competitivamente su unión al receptor de tipo I de la interleucina- 1 (IL-1RI). La interleucina-1 (IL-1) es una citocina proinflamatoria clave, que interviene en muchas respuestas celulares entre ellas aquellas importantes en la inflamación sinovial.
La IL-1 se encuentra en el plasma y el líquido sinovial de los pacientes con artritis reumatoide, y se ha indicado una correlación entre las concentraciones de IL-1 en plasma y la actividad de la enfermedad. Anakinra inhibe las respuestas producidas por la IL-1 in vitro, incluyendo la inducción de óxido nítrico y prostaglandina E2 y/o la producción de colagenasa por las células sinoviales, fibroblastos y los condrocitos.
Se han identificado mutaciones espontáneas del gen CIAS1/NLRP3 en la mayoría de los pacientes con CAPS. El gen CIAS1/NLRP3 codifica criopirina, un componente del inflamasoma. Cuando el inflamasoma se activa, se produce la maduración proteolítica y secreción de IL-ip, que tiene un amplio abanico de efectos, entre ellos la inflamación sistémica. Los pacientes con CAPS que no reciben tratamiento se caracterizan por un aumento de CRP, SAA e IL-6 con respecto a los niveles séricos normales. La administración de Kineret disminuye los reactantes de fase aguda, y se ha observado un descenso del nivel de expresión de IL-6. La disminución de los niveles de proteínas de fase aguda se observó ya en las primeras semanas de tratamiento.
Eficacia clínica y seguridad en AR
Se ha demostrado la seguridad y eficacia de anakinra en combinación con metotrexato en 1790 pacientes de edad > 18 años con AR y distintos grados de severidad de la enfermedad.
La respuesta clínica a anakinra generalmente se observó a las 2 semanas de iniciar el tratamiento, manteniéndose con la administración continuada de anakinra. La máxima respuesta clínica generalmente se observó a las 12 semanas de iniciar el tratamiento.
El tratamiento combinado con anakinra y metotrexato muestra una reducción estadística y clínicamente significativa en la gravedad de los signos y síntomas de la artritis reumatoide en pacientes con respuesta inadecuada a la administración de metotrexato solo (38% frente a 22% de respuestas, usando como criterio de medida el ACR20). Se han observado mejorías significativas en el dolor, en el recuento de articulaciones dolorosas, en la funcionalidad física (puntuación HAQ), en los reactantes de fase aguda y en las valoraciones globales del paciente y del médico.
Se han realizado exploraciones mediante rayos X en un estudio clínico con anakinra. No se ha demostrado un efecto perjudicial sobre el cartílago articular.
Eficacia clínica y seguridad en CAPS
Se ha demostrado la seguridad y eficacia de Kineret en pacientes con CAPS y distintos grados de severidad de la enfermedad. En un estudio clínico en el que participaron 43 pacientes adultos y pediátricos (36 pacientes tenían entre 8 meses y < 18 años de edad) con CAPS graves (NOMID/CINCA y MWS), se observó una respuesta clínica a anakinra en los primeros 10 días desde el inicio del tratamiento en todos los pacientes, y se mantuvo durante un periodo de hasta 5 años de administración continuada de Kineret.
El tratamiento con Kineret disminuye significativamente las manifestaciones de los CAPS, incluyendo una reducción de síntomas frecuentes como fiebre, erupción, dolor articular, cefalea, cansancio y enrojecimiento ocular. Se observa una disminución rápida y sostenida de los niveles de biomarcadores inflamatorios: amiloide A sérico (SAA), proteína C reactiva (CRP) y velocidad de sedimentación globular (VSG), y una normalización de los cambios hematológicos inflamatorios. En las formas graves de los CAPS, el tratamiento a largo plazo mejora las manifestaciones inflamatorias sistémicas en órganos como los ojos, el oído interno y el SNC. La audición y la agudeza visual dejan de deteriorarse durante el tratamiento con anakinra.
El análisis de los AA surgidos durante el tratamiento, clasificados por la presencia de la mutación CIAS1, demostró que no existían diferencias importantes entre los grupos con CIAS1 y sin CIAS1 en cuanto a las tasas globales de AA comunicados: 7,4 y 9,2, respectivamente. Se obtuvieron tasas similares para ambos grupos a nivel de órgano o sistema, excepto para los trastornos oculares, con 55 AA (tasa 0,5), de los cuales 35 fueron hiperemia ocular (que también puede ser un síntoma de los CAPS) en el grupo con CIAS1, y 4 AA en el grupo sin CIAS1 (tasa 0,1).
Población pediátrica
En general, los perfiles de eficacia y seguridad de Kineret son comparables entre la población adulta y pediátrica de pacientes con CAPS.
La Agencia Europea de Medicamentos ha eximido de la obligación de presentar los resultados de los ensayos realizados con Kineret en uno o más grupos de la población pediátrica con CAPS y AR (AIJ). Ver sección 4.2 para consultar la información sobre el uso en la población pediátrica.
Seguridad en pacientes pediátricos con AR (AIJ)
Se evaluó Kineret en un único ensayo randomizado, ciego, multicéntrico, en el que participaron 86 pacientes con artritis reumatoide juvenil poliarticular (ARJ; edades 2-17 años) que recibieron una dosis diaria subcutánea de 1 mg/kg, hasta una dosis máxima de 100 mg. Los 50 pacientes que alcanzaron una respuesta clínica tras un periodo abierto de 12 semanas, fueron randomizadamente designados a recibir Kineret (25 pacientes) o placebo (25 pacientes), administrado diariamente durante otras 16 semanas adicionales. Un subgrupo de estos pacientes continuó el tratamiento abierto con Kineret durante un máximo de 1 año en un estudio de extensión complementario. En estos estudios se observó un perfil de acontecimientos adversos similar al observado en los pacientes adultos con AR. Los datos de este estudio son insuficientes para demostrar la eficacia, por lo que, no se recomienda el uso de Kineret en pacientes pediátricos con ARJ.
Inmunogenicidad Ver sección 4.8.
5.2 Propiedades farmacocinéticas
La biodisponibilidad absoluta de anakinra en sujetos sanos (n = 11) después de inyectar 70 mg en bolo subcutáneo es del 95%. El proceso de absorción es el factor limitante de la velocidad de desaparición de anakinra del plasma después de la inyección subcutánea. En sujetos con AR, las concentraciones plasmáticas máximas de anakinra se produjeron entre las 3 y 7 horas después de la administración subcutánea del medicamento a dosis clínicamente relevantes (1 a 2 mg/kg; n = 18). La concentración plasmática disminuyó sin una fase de distribución discernible y la semivida terminal osciló entre 4 y 6 horas. En los pacientes con AR no se apreció una acumulación inesperada de anakinra después de la administración de dosis diarias subcutáneas durante un periodo de hasta 24 semanas. La medias (DE) estimadas de aclaramiento (CL/F) y volumen de distribución (Vd/F) para los análisis de datos de población de dos estudios farmacocinéticos (FC) en 35 pacientes con AR fueron de 105 (27) ml/min y 18,5 (11) l, respectivamente. Los datos procedentes de animales y de humanos demostraron que el riñón es el principal órgano responsable de la eliminación de anakinra. El aclaramiento de anakinra en pacientes con AR aumentó con el incremento del aclaramiento de creatinina.
Se estudió la influencia de las covariables demográficas sobre la farmacocinética de anakinra utilizando un análisis farmacocinético poblacional que incluyó 341 pacientes tratados con una inyección diaria subcutánea de anakinra en dosis de 30, 75 y 150 mg en un periodo de hasta 24 semanas. El aclaramiento estimado de anakinra aumentó al aumentar el aclaramiento de creatinina y el peso corporal. El análisis farmacocinético de la población demostró que el aclaramiento plasmático medio después de la administración en bolo subcutáneo fue aproximadamente un 14% mayor en los varones que en las mujeres y aproximadamente un 10% mayor en sujetos < 65 años que en > 65 años. Sin embargo, después de ajustar según el aclaramiento de creatinina y el peso corporal, el sexo y la edad no fueron factores influyentes significativos en el aclaramiento plasmático medio. No se requieren ajustes de la dosis en función de la edad o el sexo del paciente.
En general, la farmacocinética en los pacientes con CAPS es similar a la de los pacientes con AR. En los pacientes con CAPS, se ha detectado una linealidad aproximada de la dosis con una ligera tendencia a un aumento mayor que el proporcional. Faltan datos farmacocinéticos en niños < 4 años, pero se dispone de experiencia clínica a partir de los 8 meses de edad y, cuando se comienza a la dosis diaria recomendada de 1-2 mg/kg, no se han identificado problemas de seguridad. Faltan datos farmacocinéticos en pacientes de más edad con CAPS. Se ha demostrados su distribución en el líquido cefalorraquídeo.
Insuficiencia hepática
Se ha realizado un estudio en el que participaron 12 pacientes con insuficiencia hepática (Child-Pugh B) a los que se administró una dosis única intravenosa de 1mg/kg. Los parámetros farmacocinéticos no fueron muy distintos de los hallados en voluntarios sanos, a excepción de una disminución del 30% en el aclaramiento, en comparación con los datos de un estudio en voluntarios sanos. Se observó una disminución correspondiente del aclaramiento de creatinina en la población con insuficiencia hepática. Por consiguiente, la disminución del aclaramiento es más probable que pueda explicarse por una reducción de la función renal en esta población. Estos datos avalan que no se requiera ajuste de la dosis en pacientes con insuficiencia hepática Child-Pugh Clase B. Ver sección 4.2.
Insuficiencia renal
El aclaramiento plasmático medio de Kineret en pacientes con insuficiencia renaljeve (aclaramiento de creatinina 50-80 ml/min) y moderada (aclaramiento de creatinina 30-49 ml/min) se redujo en un 16% y un 50%, respectivamente. Con insuficiencia renal grave y enfermedad renal avanzada (aclaramiento de creatinina < 30 ml/min), el aclaramiento plasmático medio se redujo en un 70% y un 75%, respectivamente. Menos del 2,5% de la dosis de Kineret administrada se eliminó por hemodiálisis o diálisis peritoneal continua ambulatoria. Estos datos avalan que no se requiera ajuste de la dosis en pacientes con insuficiencia renal leve (CLcr 50 a 80 ml/minuto). Ver sección 4.2.
5.3 Datos preclínicos sobre seguridad
Anakinra no produjo ningún efecto observable sobre la fertilidad, desarrollo inicial, desarrollo embrio-fetal o desarrollo peri o posnatal en la rata a dosis de hasta 100 veces la dosis en humanos. Tampoco se han observado efectos en el desarrollo embrio-fetal en el conejo a dosis 100 veces la humana.
En una serie estándar de pruebas diseñada para identificar riesgos sobre el ADN, anakinra no indujo la aparición de mutaciones genéticas en bacterias o células de mamífero. Anakinra tampoco aumentó la incidencia de anomalías cromosómicas o micronúcleos en las células de la médula ósea del ratón. No se han realizado estudios a largo plazo para evaluar el potencial carcinogénico de anakinra. Los datos obtenidos en ratones que sobreexpresan el IL-1ra y en ratones mutantes carentes de IL-1ra no indicaron un mayor riesgo de desarrollo tumoral.
Un ensayo formal sobre las interacciones toxicológicas y toxicocinéticas en ratas no reveló ninguna prueba de que Kineret altere el perfil toxicológico o farmacocinético del metotrexato.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Ácido cítrico anhidro Cloruro sódico Edetato disódico dihidratado Polisorbato 80 Hidróxido sódico
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
3 años.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
Para el uso ambulatorio, Kineret se puede sacar de la nevera durante 12 horas a temperatura no superior a 25 °C, sin sobrepasar la fecha de caducidad. Tras este periodo, el producto no debe ponerse nuevamente en la nevera y debe desecharse.
6.5 Naturaleza y contenido del envase
0,67 ml de solución inyectable en jeringa graduada precargada (vidrio tipo I) con émbolo (caucho bromobutilo) y una aguja de calibre 29. La jeringa precargada tiene una funda externa para la aguja, de plástico rígido, unida a una cubierta interna. Ningún componente de la jeringa ni de la aguja está hecho con látex de caucho natural.
Envases de 1, 7 o 28 (envase múltiple con 4 paquetes de 7 jeringas precargadas) jeringas precargadas. Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Kineret es una solución estéril que no contiene conservantes. Para un solo uso.
No agitar. Dejar que la jeringa alcance la temperatura ambiente antes de inyectar.
Antes de la administración se debe comprobar visualmente si la solución contiene partículas o ha cambiado de color. Solamente se deben inyectar las soluciones que sean transparentes, entre incoloras y blanquecinas y que pueden contener partículas amorfas, relacionadas con el producto, de aspecto entre translúcido y blanco.
La presencia de estas partículas no afecta a la calidad del producto.
La jeringa precargada es de un solo uso. Deseche cualquier resto del medicamento no utilizado. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Estocolmo Suecia
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/203/005 envase de 1 jeringa precargada EU/1/02/203/006 envase de 7 jeringas precargadas EU/1/02/203/007 envase de 28 jeringas precargadas
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA
AUTORIZACIÓN
Fecha de la primera autorización: 8 Marzo 2002 Fecha de la última renovación: 20 Marzo 2007
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
A. FABRICANTE(S) DEL (DE LOS) PRINCIPIO(S) ACTIVO(S) BIOLÓGICO(S) Y FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE(S) DEL (DE LOS) PRINCIPIO(S) ACTIVO(S) BIOLÓGICO(S) Y FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante del principio activo biológico
Boehringer Ingelheim RCV GmbH & Co KG Dr. Boehringer-Gasse 5-11 A-1121 Viena Austria
Nombre y dirección del fabricante responsable de la liberación de los lotes
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Estocolmo Suecia
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107 ter, párrafo 7, de la Directiva 2001/83/CE y publicados en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2. de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
Medidas adicionales de minimización de riesgos
El TAC acordará el contenido y el formato de los materiales educativos con las autoridades nacionales competentes de cada uno de los Estados miembros en los que se comercializa Kineret, antes de su comercialización en cualquier otro Estado miembro.
El TAC se asegurará de que todos los médicos que vayan a prescribir KINERET dispongan de los siguientes elementos:
• Material educativo para el personal médico
• Material educativo para pacientes y cuidadores
El material educativo para el personal médico deberá incluir los siguientes elementos clave:
• La importancia de explicar el uso de la nueva jeringa graduada y la correcta técnica de inyección para pacientes y/o cuidadores
• La importancia de facilitar a los pacientes y/o cuidadores el material educativo
El material educativo para pacientes y cuidadores deberá incluir los siguientes elementos clave:
• Instrucciones de uso de la jeringa graduada
• Instrucciones sobre los procedimientos correctos de inyección y la eliminación de las jeringas usadas
• Cómo tratar las reacciones en el lugar de la inyección
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CAJA DE LA JERINGA PRECARGADA
1. NOMBRE DEL MEDICAMENTO
Kineret 100 mg Solución inyectable en jeringa precargada Anakinra
2. PRINCIPIO(S) ACTIVO(S)
Cada jeringa precargada de 0,67 ml contiene 100 mg de anakinra.
3. LISTA DE EXCIPIENTES
Excipientes: ácido cítrico anhidro, cloruro sódico, edetato disódico dihidratado, polisorbato 80, hidróxido sódico, agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable en jeringa precargada de un solo uso. 1 jeringa precargada 7 jeringas precargadas 28 jeringas precargadas
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía subcutánea
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD:
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm
Suecia
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/203/001 - envase unitario EU/1/02/203/002 - envase de 7 unidades EU/1/02/203/003 - envase de 28 unidades
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Kineret 100 mg
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
CAJA CON 7 JERINGAS PRECARGADAS COMO ENVASE INTERMEDIO (SIN BLUE BOX)_
1. NOMBRE DEL MEDICAMENTO
Kineret 100 mg Solución inyectable en jeringa precargada Anakinra
2. PRINCIPIO(S) ACTIVO(S)
Cada jeringa precargada de 0,67 ml contiene 100 mg de anakinra.
3. LISTA DE EXCIPIENTES
Excipientes: ácido cítrico anhidro, cloruro sódico, edetato disódico dihidratado, polisorbato 80, hidróxido sódico, agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable en jeringa precargada de un solo uso.
7 jeringas precargadas
La caja que contiene 7 jeringas precargadas forma parte de un envase múltiple con 28 jeringas precargadas.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía subcutánea
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD:
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm
Suecia
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/203/003
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Kineret 100 mg
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
JERINGAS PRECARGADAS_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Kineret 100 mg inyectable Anakinra
SC
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP.:
4. NÚMERO DE LOTE
Lot:
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
0,67 ml
6 OTROS
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CAJA DE LA JERINGA PRECARGADA
1. NOMBRE DEL MEDICAMENTO
Kineret 100 mg/0,67 ml Solución inyectable en jeringa precargada Anakinra
2. PRINCIPIO(S) ACTIVO(S)
Cada jeringa graduada precargada de 0,67 ml contiene 100 mg de anakinra.
3. LISTA DE EXCIPIENTES
Excipientes: ácido cítrico anhidro, cloruro sódico, edetato disódico dihidratado, polisorbato 80, hidróxido sódico, agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable en jeringa precargada.
1 jeringa GRADUADA precargada 7 jeringas GRADUADAS precargadas
Envase múltiple: 28 (4 x 7) jeringas GRADUADAS precargadas
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Un solo uso Vía subcutánea
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD:
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Estocolmo
Suecia
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/203/005 - envase unitario EU/1/02/203/006 - envase de 7 unidades EU/1/02/203/007 - envase de 28 unidades
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Kineret 100 mg 0,67 ml
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR
CAJA CON 7 JERINGAS PRECARGADAS COMO ENVASE INTERMEDIO (SIN BLUE BOX)_
1. NOMBRE DEL MEDICAMENTO
Kineret 100 mg/0,67 ml Solución inyectable en jeringa precargada Anakinra
2. PRINCIPIO(S) ACTIVO(S)
Cada jeringa graduada precargada de 0,67 ml contiene 100 mg de anakinra.
3. LISTA DE EXCIPIENTES
Excipientes: ácido cítrico anhidro, cloruro sódico, edetato disódico dihidratado, polisorbato 80, hidróxido sódico, agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Solución inyectable en jeringa precargada.
7 jeringas GRADUADAS precargadas
La caja que contiene 7 jeringas precargadas forma parte de un envase múltiple con 28 jeringas precargadas.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Un solo uso Vía subcutánea
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD:
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera. No congelar.
Conservar en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Estocolmo
Suecia
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/02/203/007
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Kineret 100 mg 0,67 ml
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
JERINGAS PRECARGADAS_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Kineret 100 mg/0,67 ml inyectable Anakinra
SC
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
0,67 ml
6 OTROS
B. PROSPECTO
Kineret 100 mg solución inyectable en jeringa precargada
Anakinra
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas, aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Kineret y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Kineret
3. Cómo usar Kineret
4. Posibles efectos adversos
5. Conservación de Kineret
6. Contenido del envase e información adicional
1. Qué es Kineret y para qué se utiliza
Kineret contiene el principio activo anakinra. Es un tipo de citocina (un agente inmunosupresor) que se usa para tratar la artritis reumatoide. Las citocinas son proteínas producidas por nuestro cuerpo que coordinan la comunicación entre las células y ayudan a controlar la actividad celular. En la artritis reumatoide, el cuerpo produce una cantidad excesiva de una citocina llamada interleucina-1. Esto da lugar a efectos nocivos como inflamación y lesiones en los tejidos.
Normalmente, el organismo produce una proteína que bloquea los efectos nocivos de la interleucina-1. En la artritis reumatoide, el cuerpo no produce cantidades suficientes de esta proteína bloqueante. El principio activo de Kineret es anakinra producida por tecnología de ADN recombinante utilizando el microorganismo E. coli. Esta sustancia actúa del mismo modo que su proteína bloqueante natural.
Kineret se usa para tratar los signos y síntomas de la artritis reumatoide (AR) en adultos (a partir de los 18 años de edad) en combinación con otro medicamento llamado metotrexato. Kineret está destinado a pacientes cuya respuesta a la administración de metotrexato por sí sola no es suficiente para controlar la artritis reumatoide.
2. Qué necesita saber antes de empezar a usar Kineret
No use Kineret
- si es alérgico al anakinra o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6)
- si es alérgico a otros productos producidos por tecnología ADN recombinante que empleen el microorganismo E. coli
- si tiene una insuficiencia renal grave (daño en el riñón).
- si padece neutropenia (nivel bajo en el recuento de los glóbulos blancos) determinada tras un análisis de sangre.
Contacte con su médico inmediatamente
- si le aparece un sarpullido por todo el cuerpo, le falta la respiración, tiene pitidos al respirar, pulso rápido o sudoración después de inyectarse Kineret. Pueden ser los síntomas de que es alérgico a Kineret.
Tenga especial cuidado con Kineret
Consulte a su médico antes de empezar a usar Kineret:
- si tiene antecedentes de infecciones recurrentes o si padece asma, ya que Kineret puede empeorar estas afecciones.
- si tiene cáncer. Su médico tendrá que decidir si usted puede recibir Kineret.
- si tiene antecedentes de aumento de los niveles de las enzimas hepáticas
- si necesita vacunarse. Usted no debe vacunarse con vacunas vivas mientras esté siendo tratado con Kineret.
Niños y adolescentes
El uso de Kineret en niños y adolescentes con artritis reumatoide no se ha investigado de forma exhaustiva y, por tanto, no puede recomendarse.
Uso de Kineret con otros medicamentos
Comunique a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento.
Kineret no ha sido probado en mujeres embarazadas.
No se recomienda el uso de Kineret durante el embarazo. Las mujeres en edad fértil que usen Kineret deben utilizar un método anticonceptivo adecuado durante el tratamiento.
Se desconoce si anakinra pasa a la leche materna. Abandone la lactancia materna si está utilizando Kineret.
Kineret contiene sodio
Este medicamento contiene menos de 23 mg (1 mmol) por dosis de 100 mg, por lo que se considera esencialmente "exento de sodio".
3. Cómo usar Kineret
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico. Kineret se inyecta debajo de la piel (inyección subcutánea) una vez al día. Debe procurar ponerse la inyección cada día a la misma hora.
Autoadministración de Kineret
Su médico puede decidir que es más conveniente que se inyecte Kineret usted mismo. Su médico o enfermera le enseñarán cómo hacerlo. No intente inyectarse usted mismo si no le han explicado cómo hacerlo.
Para información sobre cómo inyectarse Kineret usted mismo, por favor lea la sección incluida al final de este prospecto.
Si usa más Kineret del que debiera
No debe tener ningún problema serio si, accidentalmente, usa más Kineret del que necesita. Sin embargo, debe contactar con su médico o farmacéutico si esto ocurriera. Si no se encuentra bien, debe contactar con su médico inmediatamente.
Si olvidó usar Kineret
Si olvidó ponerse una dosis de Kineret, debe contactar con su médico para comentar cuándo debe ponerse la dosis siguiente.
4. Posibles efectos adversos
Al igual que todos los medicamentos, Kineret puede producir efectos adversos, aunque no todas las personas los sufran.
Si se presentan algunas de las siguientes condiciones, informe a su médico inmediatamente:
- Infecciones graves tales como neumonía (infección torácica) o infecciones en la piel pueden darse durante el tratamiento con Kineret. Los síntomas pueden incluir fiebre, tos o enrojecimiento y dolor en la piel.
- Las reacciones alérgicas graves son poco frecuentes. Sin embargo, alguno de los siguientes síntomas puede indicar una reacción alérgica Kineret por la que debería buscar atención médica inmediata. No inyecte más Kineret:
- Hinchazón de la cara, lengua o garganta.
- Problemas para tragar o respirar
- Sensación súbita de taquicardia o sudoración
- Picor en piel o erupción
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
- Enrojecimiento, hinchazón, aparición de cardenales o picazón en el lugar de la inyección. Generalmente estos síntomas son de leves a moderados y más frecuentes al inicio del tratamiento.
- Dolor de cabeza.
- Aumento de los niveles totales de colesterol en sangre.
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas):
- Neutropenia (bajo número de glóbulos blancos) determinada tras un análisis de sangre. La neutropenia puede aumentar el riesgo de que usted contraiga una infección. Los síntomas de una infección pueden ser fiebre o dolor de garganta.
- Infecciones graves como neumonía (infección torácica) o infecciones de la piel.
- Trombocitopenia (bajo número de plaquetas en la sangre).
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas):
- Reacciones alérgicas graves que incluyen hinchazón de la cara, lengua o garganta, problemas para tragar o respirar, sensación súbita de taquicardia o sudoración, picor en piel o erupción.
- Niveles elevados de las enzimas hepáticas detectadas tras un análisis de sangre.
Efectos adversos de frecuencia no conocida (no puede estimarse a partir de los datos disponibles):
- Signos de trastornos hepáticos, como color amarillo de la piel y de los ojos, náuseas, pérdida de apetito, orina de color oscuro y heces de color muy claro.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Kineret
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase después de “CAD”. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2 °C y 8 °C). No congelar.
Conservar en el embalaje original para protegerlo de la luz.
No utilice Kineret si cree que ha estado congelado. Una vez que una jeringa se ha sacado de la nevera y alcanzado temperatura ambiente (hasta 25 °C), debe utilizarse en las 12 horas siguientes o desecharse.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Kineret
- El principio activo es anakinra. Cada jeringa precargada contiene 100 mg de anakinra.
- Los demás componentes (excipientes) son: ácido cítrico anhidro, cloruro sódico, edetato disódico dihidratado, polisorbato 80, hidróxido sódico y agua para preparaciones inyectables.
Aspecto del producto y contenido del envase
Kineret es una solución transparente, entre incolora y blanquecina que se presenta en una jeringa precargada lista para su uso. La solución puede contener partículas de proteína de aspecto entre translúcido y blanco cuya presencia no afecta a la calidad del producto.
Los envases contienen 1, 7 ó 28 (envase múltiple con 4 paquetes de 7 jeringas precargadas) jeringas precargadas.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm Suecia
Fecha de la última revisión de este prospecto:
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
Esta sección contiene información sobre cómo inyectarse Kineret usted mismo. Es importante que no intente administrarse usted mismo la inyección si no ha sido instruido sobre cómo hacerlo por su médico, enfermera o farmacéutico. Si usted tiene alguna duda sobre cómo ponerse la inyección, consúltelo con su médico, enfermera o farmacéutico.
¿Cómo debe usted o la persona que le va a inyectar, utilizar la jeringa precargada de Kineret?
Necesitará ponerse una inyección cada día a la misma hora. Kineret se inyecta debajo de la piel. Esto se conoce como inyección subcutánea.
Material:
Para la autoadministración de una inyección subcutánea, necesitará:
• una jeringa precargada nueva de Kineret y
• algodón con alcohol o similar.
¿Qué debe hacer antes de ponerse la inyección subcutánea de Kineret?
1. Saque su j eringa precargada de Kineret de la nevera.
2. No agite la jeringa precargada.
3. Compruebe la fecha de caducidad indicada en la etiqueta de la jeringa precargada (CAD). No la use si la fecha actual ha sobrepasado el último día del mes indicado.
4. Compruebe el aspecto de Kineret. Tiene que ser una solución transparente, entre incolora y blanquecina. La solución puede contener partículas de proteína de aspecto entre translúcido y blanco cuya presencia no afecta a la calidad del producto. No utilice este medicamento si presenta un color diferente, está turbio o las partículas presentes no son entre translúcidas y blancas.
5. Para una administración más cómoda, dej e la j eringa precargada a temperatura ambiente durante 30 minutos aproximadamente o manténgala con cuidado en su mano cerrada durante unos pocos minutos. No caliente Kineret de ninguna otra forma (por ejemplo no lo ponga en el microondas, ni en agua caliente).
6. No retire el protector de la jeringa hasta que esté preparado para la inyección.
7. Lávese las manos cuidadosamente.
8. Busque una superficie cómoda, limpia y bien iluminada y coloque todo el material que necesite a su alcance.
¿Cómo preparar la inyección de Kineret?
Antes de inyectarse Kineret debe hacer lo siguiente:
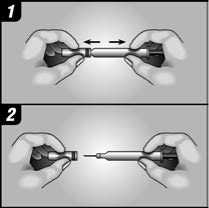
1. Sostenga la jeringa y quite suavemente la cubierta de la aguja sin girarla como se muestra en las figuras 1 y 2. No toque la aguja ni empuje el émbolo.
2. Puede haber una pequeña burbuja de aire en la jeringa precargada. No es necesario eliminarla antes de la inyección. La inyección de la solución con una burbuja de aire no es perjudicial.
3. Ahora ya puede usar la j eringa precargada.
¿Dónde debe ponerse la inyección?
Los lugares más adecuados para ponerse usted mismo la inyección son:
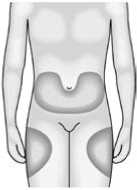
• la parte superior de los muslos y
• el abdomen, excepto la zona alrededor del ombligo.
Cambie el lugar de la inyección de una vez a otra para que no tenga molestias en ninguna zona. Si la inyección se la pone otra persona, también se la puede poner en la parte posterior de los brazos.
¿Cómo ponerse la inyección?
1.
Desinfecte la piel usando un algodón con alcohol y pellizque la piel entre el pulgar y el índice, sin apretar.
2. Inserte completamente la aguja en la piel como le ha enseñado la enfermera o el médico.
3. Inyecte el líquido lenta y regularmente, manteniendo siempre la piel pellizcada.
4. Tras inyectar la solución, retire la aguja y suelte la piel.
5. Cada jeringa solamente debe utilizarse para una inyección.
Recuerde
Si tiene dificultad, pida ayuda y consejo a su médico o enfermera.
Cómo deshacerse de las jeringas usadas
• No vuelva a poner la cubierta en las agujas usadas.
• Mantenga las jeringas usadas fuera del alcance y de la vista de los niños.
• Nunca tire las jeringas precargadas usadas en su cubo de basura habitual.
• Las jeringas precargadas deben eliminarse según los requerimientos locales. Pregunte a su farmacéutico cómo deshacerse de los medicamentos que no necesita. De esta forma ayudará a proteger el medio ambiente.
Prospecto: información para el usuario
Kineret 100 mg/0,67 ml solución inyectable en jeringa precargada
Anakinra
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Kineret y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Kineret
3. Cómo usar Kineret
4. Posibles efectos adversos
5. Conservación de Kineret
6. Contenido del envase e información adicional
1. Qué es Kineret y para qué se utiliza
Kineret contiene el principio activo anakinra. Es un tipo de citocina (un agente inmunosupresor) que se usa para tratar:
• La artritis reumatoide (AR)
• Los síndromes periódicos asociados a criopirina (CAPS), los cuales incluyen las siguientes enfermedades auto-inflamatorias:
- Enfermedad inflamatoria multisistémica de inicio neonatal (NOMID)/síndrome articular, cutáneo y neurológico infantil crónico (CINCA)
- Síndrome de Muckle-Wells (MWS)
- Síndrome autoinflamatorio familiar por frío (FCAS)
Las citocinas son proteínas producidas por nuestro cuerpo que coordinan la comunicación entre las células y ayudan a controlar la actividad celular. En la artritis reumatoide y los CAPS, el cuerpo produce una cantidad excesiva de una citocina llamada interleucina-1. Esto da lugar a efectos nocivos que provocan inflamación y causan los síntomas de la enfermedad. Normalmente, el organismo produce una proteína que bloquea los efectos nocivos de la interleucina-1. El principio activo de Kineret es anakinra, que actúa de forma similar a la proteína natural que bloquea la interleucina -1. Anakinra se obtiene por tecnología del ADN recombinante utilizando el microorganismo E. coli.
En artritis reumatoide (AR), Kineret se usa para tratar los signos y síntomas de la enfermedad en adultos (a partir de los 18 años de edad) en combinación con otro medicamento llamado metotrexato. Kineret está destinado a pacientes cuya respuesta a la administración de metotrexato por sí sola no es suficiente para controlar la artritis reumatoide.
En los síndromes periódicos asociados a criopirina (CAPS), Kineret se usa para tratar los signos y síntomas de la inflamación asociada a esta enfermedad, como erupción, dolor en las articulaciones, fiebre, dolor de cabeza y cansancio, tanto en adultos como en niños (a partir de los 8 meses de edad).
2. Qué necesita saber antes de empezar a usar Kineret
No use Kineret
- si es alérgico a anakinra o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6)
- si es alérgico a otros productos producidos por tecnología ADN recombinante que empleen el microorganismo E. coli
- si tiene una insuficiencia renal grave (daño en el riñón)
- si padece neutropenia (nivel bajo en el recuento de los glóbulos blancos) determinada tras un análisis de sangre.
Contacte con su médico inmediatamente
- si le aparece un sarpullido por todo el cuerpo, le falta la respiración, tiene pitidos al respirar, pulso rápido o sudoración después de inyectarse Kineret. Pueden ser los síntomas de que es alérgico a Kineret.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar Kineret:
- si tiene antecedentes de infecciones recurrentes o si padece asma, ya que Kineret puede empeorar estas afecciones
- si tiene cáncer. Su médico tendrá que decidir si usted puede recibir Kineret
- si tiene antecedentes de aumento de los niveles de las enzimas hepáticas
si necesita vacunarse. Usted no debe vacunarse con vacunas vivas mientras esté siendo tratado con Kineret.
Niños y adolescentes
• AR: El uso de Kineret en niños y adolescentes con artritis reumatoide (AR) no se ha investigado de forma exhaustiva y, por tanto, no puede recomendarse.
• CAPS: El uso de Kineret no se recomienda en niños menores de 8 meses, ya que no se dispone de datos para este grupo de edad.
Uso de Kineret con otros medicamentos
Comunique a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Los medicamentos denominados inhibidores del factor de necrosis tumoral (TNF), como etanercept, no deben utilizarse junto con Kineret ya que esto podría incrementar el riesgo de infecciones.
Cuando comience a usar Kineret, la inflamación crónica de su organismo disminuirá. Esto podría hacer que se deban ajustar las dosis de algunos otros medicamentos, como warfarina o fenitoína.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento.
Kineret no ha sido probado en mujeres embarazadas. No se recomienda el uso de Kineret durante el embarazo. Las mujeres en edad fértil que usen Kineret deben utilizar un método anticonceptivo adecuado durante el tratamiento.
Se desconoce si anakinra pasa a la leche materna. Abandone la lactancia materna si está utilizando Kineret.
Kineret contiene sodio
Este medicamento contiene menos de 23 mg (1 mmol) de sodio por dosis de 100 mg, por lo que se considera esencialmente "exento de sodio"”.
3. Cómo usar Kineret
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico o farmacéutico. Kineret se inyecta debajo de la piel (inyección subcutánea) diariamente. Debe procurar ponerse la inyección cada día a la misma hora.
La dosis recomendada es de 20 a 90 mg o 100 mg. El médico le indicará la dosis que necesita, o si necesita una dosis mayor de 100 mg.
Autoadministración de Kineret
Su médico puede decidir que es más conveniente que se inyecte Kineret usted mismo. Su médico o enfermera le enseñarán cómo hacerlo. No intente inyectarse usted mismo si no le han explicado cómo hacerlo.
Para información sobre cómo inyectarse Kineret usted mismo o inyectárselo a su hijo, por favor lea la sección “Instrucciones para preparar y administrar una inyección de Kineret” incluida al final de este prospecto.
Si usa más Kineret del que debe
No debe tener ningún problema serio si, accidentalmente, usa más Kineret del que necesita. Sin embargo, debe contactar con su médico o farmacéutico si esto ocurriera. Si no se encuentra bien, debe contactar con su médico inmediatamente.
Si olvidó usar Kineret
Si olvidó ponerse una dosis de Kineret, debe contactar con su médico para comentar cuándo debe ponerse la dosis siguiente.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Los posibles efectos adversos son similares tanto si se usa Kineret para la AR como para los CAPS.
Si se presentan algunas de los siguientes efectos adversos, informe a su médico inmediatamente:
- Infecciones graves tales como neumonía (infección torácica) o infecciones en la piel pueden darse durante el tratamiento con Kineret. Los síntomas pueden incluir fiebre, tos o enrojecimiento y dolor en la piel.
- Las reacciones alérgicas graves son poco frecuentes. Sin embargo, alguno de los siguientes síntomas puede indicar una reacción alérgica Kineret por la que debería buscar atención médica inmediata. No inyecte más Kineret:
- Hinchazón de la cara, lengua o garganta.
- Problemas para tragar o respirar
- Sensación súbita de taquicardia o sudoración
- Picor en la piel o erupción
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
- Enrojecimiento, hinchazón, aparición de cardenales o picazón en el lugar de la inyección. Generalmente estos síntomas son leves a moderados y más frecuentes al inicio del tratamiento.
- Dolor de cabeza.
- Aumento de los niveles totales de colesterol en sangre.
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas):
- Neutropenia (bajo número de glóbulos blancos) determinada tras un análisis de sangre. La neutropenia puede aumentar el riesgo de que usted contraiga una infección. Los síntomas de una infección pueden ser fiebre o dolor de garganta.
- Infecciones graves como neumonía (infección torácica) o infecciones de la piel.
- Trombocitopenia (bajo número de plaquetas en la sangre).
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas):
- Reacciones alérgicas graves que incluyen hinchazón de la cara, lengua o garganta, problemas para tragar o respirar, sensación súbita de taquicardia o sudoración, picor en piel o erupción.
- Niveles elevados de las enzimas hepáticas detectadas tras un análisis de sangre.
Efectos adversos de frecuencia no conocida (no puede estimarse a partir de los datos disponibles):
- Signos de trastornos hepáticos, como color amarillo de la piel y de los ojos, náuseas, pérdida de apetito, orina de color oscuro y heces de color muy claro.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Kineret
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta y el envase, después de “CAD”. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2 °C - 8 °C). No congelar.
Conservar en el embalaje original para protegerlo de la luz.
No utilice Kineret si cree que ha estado congelado. Una vez que una jeringa se ha sacado de la nevera y alcanzado temperatura ambiente (hasta 25 °C), debe utilizarse en las 12 horas siguientes o desecharse.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Kineret
- El principio activo es anakinra. Cada jeringa graduada precargada contiene 100 mg de anakinra.
- Los demás componentes (excipientes) son: ácido cítrico anhidro, cloruro sódico, edetato disódico dihidratado, polisorbato 80, hidróxido sódico y agua para preparaciones inyectables.
Aspecto del producto y contenido del envase
Kineret es una solución transparente, entre incolora y blanquecina que se presenta en una jeringa precargada lista para su uso. La solución puede contener partículas de proteína de aspecto entre translúcido y blanco cuya presencia no afecta a la calidad del producto.
Los envases contienen 1, 7 o 28 (envase múltiple con 4 paquetes de 7 jeringas precargadas) jeringas precargadas.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Estocolmo Suecia
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
INSTRUCCIONES PARA PREPARAR Y ADMINISTRAR UNA INYECCIÓN DE KINERET
Esta sección contiene información sobre cómo inyectarse Kineret usted mismo o inyectárselo a su hijo. Es importante que no intente administrarse la inyección usted mismo o administrársela a su hijo si no ha sido instruido sobre cómo hacerlo por un médico, enfermero o farmacéutico. Si usted tiene alguna duda sobre cómo ponerse la inyección, consúltelo con su médico, enfermera o farmacéutico.
¿Cómo debe usted o la persona que le va a inyectar, utilizar la jeringa precargada de Kineret?
Necesitará ponerse una inyección usted mismo o ponérsela a su hijo cada día a la misma hora. Kineret se inyecta debajo de la piel. Esto se conoce como inyección subcutánea.
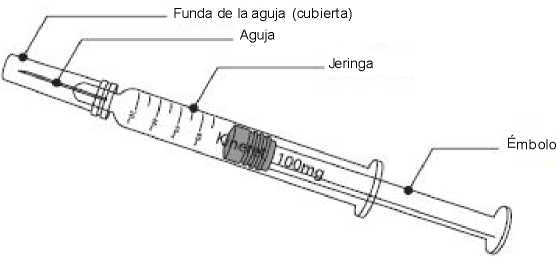
Material:
Para administrarse usted mismo o administrarle a su hijo una inyección subcutánea, necesitará:
• una jeringa precargada de Kineret;
• toallita con alcohol o similar, y
• una gasa o un apósito estériles
¿Qué debe hacer antes de ponerse usted mismo o ponerle a su hijo la inyección subcutánea de
Kineret?
1. Saque la jeringa precargada de Kineret de la nevera.
2. No agite la jeringa precargada.
3. Compruebe la fecha de caducidad indicada en la etiqueta de la jeringa precargada (EXP). No la use si la fecha actual ha sobrepasado el último día del mes indicado.
4. Compruebe el aspecto de Kineret. Tiene que ser una solución transparente, entre incolora y blanquecina. La solución puede contener partículas de proteína de aspecto entre translúcido y blanco cuya presencia no afecta a la calidad del producto. No utilice este medicamento si presenta un color diferente, está turbio o las partículas presentes no son entre translúcidas y blancas.
5. Para una administración más cómoda, dej e la j eringa precargada a temperatura ambiente durante 30 minutos aproximadamente o manténgala con cuidado en su mano cerrada durante unos minutos. No caliente Kineret de ninguna otra forma (por ejemplo, no lo ponga en el microondas ni en agua caliente).
6. No retire el protector de la jeringa hasta que esté preparado para la inyección.
7. Lávese las manos cuidadosamente.
8. Busque una superficie cómoda, limpia y bien iluminada y coloque todo el material que necesite a su alcance.
9 Asegúrese de que conoce la dosis de Kineret que le ha recetado su médico; 20 a 90 mg, 100 mg o mayor de 100 mg.
• Si su médico le ha recetado una dosis de 100 mg, vaya a la sección “¿Cómo preparar una
dosis de 100 mg?”.
• Si su médico le ha recetado una dosis menor, vaya a la sección “¿Cómo preparar una dosis de
20 a 90 mg?”.
¿Cómo preparar una dosis de 100 mg?
Antes de inyectarse Kineret debe hacer lo siguiente:
Funda de la aguja (cubeita)
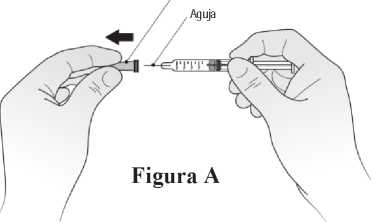
1. Sostenga la jeringa y retire suavemente la cubierta de la aguja sin girarla, como se muestra en la Figura A.
No toque la aguja ni empuje el émbolo. Deseche inmediatamente la cubierta de la aguja.
2. Puede haber una pequeña burbuja de aire en la jeringa precargada. No es necesario eliminarla antes de la inyección. La inyección de la solución con una burbuja de aire no es perjudicial.
3. Ahora ya puede usar la jeringa precargada tal como se describe en la sección “¿Dónde debe ponerse la inyección? “ y en la sección “¿Cómo ponerse la inyección?”.
¿Cómo preparar una dosis de 20 a 90 mg?
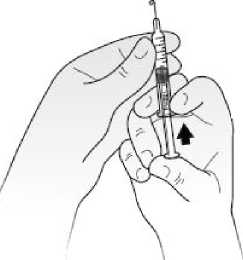
Figura B
Antes de inyectar Kineret debe hacer lo siguiente:
1. Sujete la jeringa y retire suavemente la cubierta de la aguja sin girarla, como se muestra en la Figura A. No toque la aguja ni empuje el émbolo. Deseche inmediatamente la cubierta de la aguja.
2.
Figura C
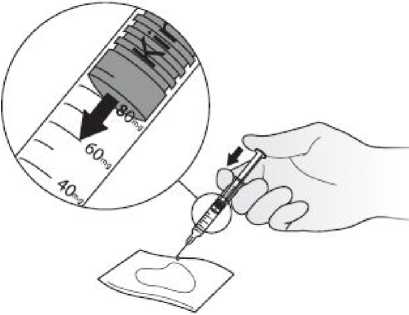
Debe colocar la jeringa en una mano, con la aguja apuntando hacia arriba, como se muestra en la Figura B. Coloque el pulgar en el émbolo y empuje suavemente hasta que vea aparecer una pequeña gota de líquido en la punta de la aguja.
3. Gire la jeringa de forma que la aguja apunte hacia abajo. Coloque una gasa o un apósito estériles en una superficie plana y sujete la jeringa encima, con la aguja apuntando hacia la gasa o el apósito, como se muestra en la Figura C. No deje que la aguja toque la gasa o el apósito.
4. Coloque el pulgar en el émbolo y apriete lentamente hasta que el frontal del émbolo llegue a la marca en la escala que se corresponda con su dosis de Kineret (su médico le habrá indicado cuál es la dosis que usted necesita). El líquido que salga será absorbido por la gasa o el apósito, tal como se muestra en la Figura C.
5. Si no ha podido obtener la dosis correcta, deseche la jeringa y utilice una nueva.
6. Ahora podrá utilizar la jeringa precargada de la forma en que se describe en la sección “¿Dónde debe ponerse la inyección?” y en la sección “¿Cómo ponerse la inyección?”.
¿Dónde debe ponerse la inyección?
Los lugares más adecuados para ponerse la inyección usted mismo o ponérsela a su hijo son (ver la Figura D):
• el abdomen (excepto la zona de alrededor del ombligo)
• la parte superior de los muslos
• la zona superior externa de los glúteos.
• la zona externa superior de los brazos.
Niño
Adulto
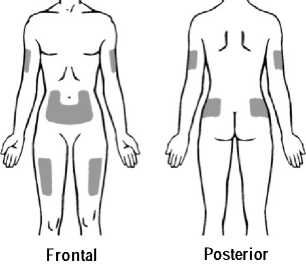
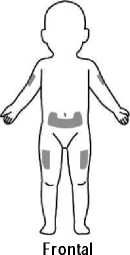
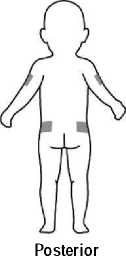
Cambie el lugar de la inyección de una vez a otra para que no tenga molestias en ninguna zona. Si la inyección se la pone otra persona, también se la puede poner en la parte posterior de los brazos.
¿Cómo ponerse la inyección?
1. Desinfecte la piel usando una toallita con alcohol y pellizque la piel entre el pulgar y el índice, sin apretar.
2. Inserte completamente la aguja en la piel como le ha enseñado la enfermera o el médico.
3. Inyecte el líquido lenta y regularmente, manteniendo siempre la piel pellizcada, como se muestra en la Figura E.
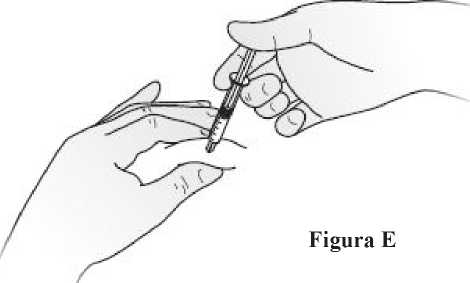
4. Tras inyectar la solución, retire la aguja y suelte la piel.
5. Cualquier resto de medicamento no utilizado debe desecharse. Cada jeringa solamente debe utilizarse para una inyección. No reutilice las jeringas, ya que esto puede provocar una infección.
Recuerde
Si tiene dificultad, pida ayuda y consejo a su médico o enfermero.
Cómo deshacerse de las jeringas usadas y materiales adicionales
• No vuelva a poner la cubierta en las agujas usadas.
• Mantenga las jeringas usadas fuera del alcance y de la vista de los niños.
• Nunca tire las jeringas precargadas usadas en su cubo de basura doméstica.
• Si su dosis es inferior a 100 mg, deberá verter el líquido sobrante de la jeringa en una gasa o un apósito. Tras la inyección, deseche la gasa o el apósito húmedos junto con la jeringa y limpie la superficie con un paño limpio.
• Las jeringas precargadas, así como la gasa o el apósito impregnados de solución Kineret, deben eliminarse de acuerdo con la normativa local. Pregunte a su farmacéutico cómo deshacerse de los medicamentos que no necesita. De esta forma ayudará a proteger el medio ambiente.
54