Jevtana 60 Mg Concentrado Y Disolvente Para Solucion Para Perfusion
ANEXO I
RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
NOMBRE DEL MEDICAMENTO
1.
JEVTANA 60 mg concentrado y disolvente para solución para perfusión.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un ml de concentrado contiene 40 mg de cabazitaxel.
Cada vial de 1,5 ml (volumen nominal) de concentrado contiene 60 mg de cabazitaxel.
Después de la dilución inicial con todo el disolvente, cada ml de solución contiene 10 mg de cabazitaxel.
Nota: tanto el vial del concentrado de JEVTANA 60 mg/1,5 ml (volumen de llenado: 73,2 mg de cabazitaxel/1,83 ml) como el del vial de disolvente (volumen de llenado: 5,67 ml) contienen un sobrellenado para compensar la pérdida de líquido durante la preparación. Este sobrellenado asegura que después de la dilución con el contenido COMPLETO del disolvente proporcionado, haya una solución conteniendo 10 mg/ml de cabazitaxel.
Excipiente con efecto conocido
Cada vial de disolvente contiene 573,3 mg de etanol 96%.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Concentrado y disolvente para solución para perfusión (concentrado estéril).
El concentrado es una solución oleosa, transparente, de color amarillo a amarillo-marronáceo. El disolvente es una solución transparente e incolora.
4. DATOS CLÍNICOS
4.1. Indicaciones terapéuticas
JEVTANA en combinación con prednisona o prednisolona está indicado para el tratamiento de pacientes adultos con cáncer de próstata metastático hormono-resistente, tratados anteriormente con una pauta terapéutica conteniendo docetaxel (ver sección 5.1).
4.2. Posología y forma de administración
El uso de JEVTANA debe estar limitado a unidades especializadas en la administración de citotóxicos y sólo debe ser administrado bajo la supervisión de un médico con experiencia en el manejo de quimioterapia anticancerígena. Debe disponerse de instalaciones y equipo para el tratamiento de reacciones graves de hipersensibilidad como hipotensión y broncoespasmo (ver sección 4.4).
Premedicación
El régimen de premedicación recomendado debe aplicarse al menos 30 minutos antes de cada administración de JEVTANA, con los siguientes medicamentos intravenosos para mitigar el riesgo y la gravedad de la hipersensibilidad:
• antihistamínicos (dexclorfeniramina 5 mg o difenhidramina 25 mg o equivalente),
• corticosteroides (dexametasona 8 mg o equivalente), y
• antagonistas H2 (ranitidina o equivalente) (ver sección 4.4).
Se recomienda profilaxis antiemética y puede administrarse oralmente o intravenosamente, según se necesite.
A lo largo del tratamiento, debe asegurarse la adecuada hidratación del paciente, para prevenir complicaciones como la insuficiencia renal.
Posología
La dosis recomendada de JEVTANA es 25 mg/m2 administrada durante 1 hora en perfusión intravenosa cada 3 semanas, en combinación con prednisona oral o 10 mg diarios de prednisolona a lo largo del tratamiento.
Ajustes de la dosis
Debe modificarse la dosis si los pacientes experimentan las siguientes reacciones adversas (los Grados se refieren al Common Terminology Criteria of Adverse Events [CTCAE 4.0]):
Tabla 1 - Modificaciones de dosis recomendadas para reacciones adversas en pacientes tratados con
cabazitaxel
|
Reacciones adversas |
Modificación de la dosis |
|
Neutropenia prolongada grado > 3 (más de 1 semana) a pesar del tratamiento adecuado incluyendo G-CSF |
Retrasar el tratamiento hasta que el recuento de neutrófilos sea > 1.500 células/mm3, a continuación reducir la dosis de cabazitaxel de 25 mg/m2 a 20 mg/m2. |
|
Neutropenia febril o infección neutropénica |
Retrasar el tratamiento hasta mejoría o resolución, y hasta que el recuento de neutrófilos sea > 1.500 células/mm3, a continuación reducir la dosis de cabazitaxel de 25 mg/m2 a 20 mg/m2. |
|
Diarrea grado > 3 o diarrea persistente a pesar del tratamiento apropiado, incluyendo reconstitución de fluidos y electrolitos |
Retrasar el tratamiento hasta mejoría o resolución, a continuación reducir la dosis de cabazitaxel de 25 mg/m2 a 20 mg/m2. |
|
Neuropatía periférica grado > 2 |
Retrasar el tratamiento hasta mejoría, a continuación reducir la dosis de cabazitaxel de 25 mg/m2 a 20 mg/m2. |
El tratamiento debe discontinuarse si un paciente continúa experimentando cualquiera de las anteriores reacciones con 20 mg/m2.
Poblaciones especiales
Pacientes con insuficiencia hepática
Cabazitaxel se metaboliza en un alto grado en el hígado. En pacientes con insuficiencia hepática leve (bilirrubina total >1 a < 1,5 x límite normal superior (LNS) o AST >1,5 x LNS) debe reducirse la dosis de cabazitaxel a 20 mg/m2. La administración de cabazitaxel a pacientes con insuficiencia hepática leve debe realizarse con precaución y con una vigilancia estrecha de seguridad.
En pacientes con insuficiencia hepática moderada (bilirrubina total >1,5 a < 3,0 x-LNS), la dosis máxima tolerada (DMT) fue de 15 mg/m2. Si el tratamiento está previsto en pacientes con insuficiencia hepática moderada, la dosis de cabazitaxel no debe exceder 15 mg/m2. Sin embargo, los datos de eficacia disponibles a esta dosis son limitados.
No debe administrarse cabazitaxel a pacientes con insuficiencia hepática grave (bilirrubina total >3 x LNS) (ver secciones 4.3, 4.4 y 5.2).
Pacientes con insuficiencia renal
Cabazitaxel se excreta mínimamente a través del riñón. No es necesario ajustar la dosis en pacientes con insuficiencia renal, no requiriendo hemodiálisis. Los pacientes que presentan enfermedad renal en estado terminal (aclaramiento de creatinina (CLcr)< 15 mL/min/1,73 m2) por su afección y la cantidad limitada de datos disponibles deben ser tratados con precaución y monitorizados cuidadosamente durante el tratamiento (ver secciones 4.4 y 5.2).
Pacientes de edad avanzada
No se recomienda ningún ajuste específico de la dosis para el uso de cabazitaxel en pacientes mayores de 65 años (ver también secciones 4.4, 4.8 y 5.2).
Uso concomitante de medicamentos
Deben evitarse los medicamentos concomitantes que son inductores potentes o inhibidores potentes de la actividad CYP3A. Sin embargo, si los pacientes requieren la administración concomitante de un inhibidor potente del CYP3A, se debe considerar una reducción de la dosis de cabazitaxel de un 25 % (ver secciones 4.4 y 4.5).
Población pediátrica
No se ha establecido la seguridad y eficacia de JEVTANA en niños y adolescentes menores de 18 años. No se dispone de datos.
Forma de administración
Para las instrucciones sobre la preparación y administración del producto, ver sección 6.6.
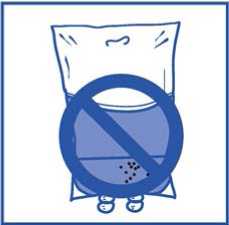
No deben utilizarse envases de perfusión de PVC y sets de perfusión de poliuretano.
JEVTANA no se debe mezclar con ningún otro medicamento de los mencionados en la sección 6.6.
4.3. Contraindicaciones
• Hipersensibilidad a cabazitaxel, a otros taxanos, a polisorbato 80 o a alguno de los excipientes incluidos en la sección 6.1.
• Recuento de neutrófilos menor de 1.500/mm3.
• Insuficiencia hepática grave (bilirrubina total >3 x LNS).
• Vacunación concomitante con la vacuna de la fiebre amarilla (ver sección 4.5).
4.4. Advertencias y precauciones especiales de empleo
Reacciones de hipersensibilidad
Todos los pacientes deben premedicarse antes del inicio de la perfusión de cabazitaxel (ver sección 4.2).
Los pacientes deben ser vigilados estrechamente para detectar reacciones de hipersensibilidad, especialmente durante la primera y segunda perfusión. Las reacciones de hipersensibilidad podrían ocurrir unos minutos después del inicio de la perfusión de cabazitaxel, por lo que debe disponerse de instalaciones y equipo para el tratamiento de la hipotensión y broncoespasmo. Pueden producirse reacciones graves, que podrían incluir erupción/eritema generalizado, hipotensión y broncoespasmo. Las reacciones graves de hipersensibilidad requieren la interrupción inmediata de cabazitaxel y terapia apropiada. Los pacientes con una reacción de hipersensibilidad deben interrumpir el tratamiento con JEVTANA (ver sección 4.3).
Supresión de la médula ósea
Puede ocurrir supresión de le médula ósea que se manifiesta como neutropenia, anemia, trombocitopenia o pancitopenia (ver “Riesgo de neutropenia” y “Anemia” a continuación en sección 4.4).
Riesgo de neutropenia
Los pacientes tratados con cabazitaxel podrían recibir G-CSF en profilaxis, según las directrices de la Sociedad Americana de Oncología Clínica (SAOC) y/o las directrices institucionales vigentes, para reducir el riesgo o tratar las complicaciones de la neutropenia (neutropenia febril, neutropenia prolongada o infección neutropénica). En pacientes con características clínicas de alto riesgo (edad > 65 años, mal estado general, episodios previos de neutropenia febril, campos muy extensos de radiación previa, estado nutricional deficiente, u otras comorbilidades graves) que los predisponen a un aumento de las complicaciones derivadas de la neutropenia prolongada, debe considerarse la profilaxis primaria con G-CSF. El uso de G-CSF ha demostrado limitar la incidencia y gravedad de la neutropenia.
La neutropenia es la reacción adversa más común de cabazitaxel (ver sección 4.8). Es esencial la monitorización semanal de los recuentos sanguíneos completos durante el primer ciclo y antes de cada ciclo posterior de tratamiento de forma que pueda ajustarse la dosis, si fuera necesario.
En caso de neutropenia febril, o neutropenia prolongada debe reducirse la dosis a pesar de que el tratamiento sea el apropiado (ver sección 4.2).
Debe volver a tratarse a los pacientes sólo cuando los neutrófilos se recuperen a un nivel de > 1.500/mm3 (ver sección 4.3).
Trastornos gastrointestinales
Podrían ser manifestaciones tempranas de toxicidad gastrointestinal grave, síntomas tales como dolor abdominal y sensibilidad, fiebre, estreñimiento persistente, diarrea, con o sin neutropenia, y deben ser evaluados y tratados rápidamente. Podría ser necesario el retraso o la interrupción del tratamiento con cabazitaxel.
Riesgo de náuseas, vómitos, diarrea y deshidratación
Si los pacientes experimentan diarrea tras la administración de cabazitaxel, pueden ser tratados con medicamentos antidiarreicos de uso común. Deben tomarse las medidas apropiadas para rehidratar a los pacientes. La diarrea puede aparecer más frecuentemente en pacientes que hayan recibido radiación abdomino-pélvica previa. La deshidratación es más frecuente en pacientes de 65 años o más. Se deben tomar las medidas apropiadas para rehidratar a los pacientes y monitorizar y corregir los niveles séricos de electrolitos, especialmente de potasio. Podría ser necesario retrasar el tratamiento o reducir la dosis para la diarrea grado > 3 (ver sección 4.2). Si los pacientes experimentan náuseas o vómitos, podrían ser tratados con los antieméticos frecuentemente utilizados.
Riesgo de reacciones gastrointestinales graves
Se han notificado en pacientes tratados con cabazitaxel, hemorragia gastrointestinal (GI) y perforación, íleo (ileus), colitis, incluyendo desenlace mortal (ver sección 4.8). Se recomienda precaución con el tratamiento de pacientes con más riesgo de desarrollar complicaciones gastrointestinales: aquellos con neutropenia, pacientes de edad avanzada, uso concomitante de AINEs, terapia antiplaquetaria o anticoagulantes, y pacientes con antecedentes de radioterapia pélvica o enfermedad gastrointestinal, tales como ulceración o sangrado GI.
Neuropatía periférica
Se han observado casos de neuropatía periférica, neuropatía sensorial periférica (por ej. parestesias, disestesias) y neuropatía motora periférica, en pacientes en tratamiento con cabazitaxel. Debe advertirse a los pacientes en tratamiento con cabazitaxel que informen a su médico antes de continuar con el tratamiento si desarrollan síntomas de neuropatía tales como dolor, ardor, hormigueo, falta de sensibilidad, o debilidad. Los médicos deben evaluar la presencia o empeoramiento de la neuropatía antes de cada tratamiento. El tratamiento debe retrasarse hasta la mejora de los síntomas. La dosis de cabazitaxel debe reducirse de 25 mg/m2 a 20 mg/m2 para neuropatía periférica persistente grado > 2 (ver sección 4.2).
Anemia
Se ha observado anemia en pacientes en tratamiento con cabazitaxel (ver sección 4.8). Antes del tratamiento con cabazitaxel y si el paciente tiene síntomas o signos de anemia o pérdida de sangre, se debe comprobar la hemoglobina y el hematocrito. Se recomienda precaución en pacientes con hemoglobina < 10 g/dl y se deben tomar medidas adecuadas según indicación clínica.
Riesgo de insuficiencia renal
Se han notificado alteraciones renales en asociación con sepsis, deshidratación grave por diarrea, vómitos y uropatía obstructiva. Se han observado casos de insuficiencia renal incluyendo casos con desenlace fatal. Si esto ocurre deben tomarse las medidas apropiadas para identificar la causa y tratar a los pacientes intensivamente.
Durante el tratamiento con cabazitaxel debe garantizarse una hidratación adecuada. Debe advertirse al paciente para que notifique inmediatamente cualquier cambio significativo en el volumen diario de orina. Debe medirse la creatinina sérica basal en cada recuento sanguíneo y siempre que el paciente notifique un cambio en la orina producida. Debe interrumpirse el tratamiento con cabazitaxel en caso de cualquier degradación de la función renal a insuficiencia renal > CTCAE 4.0 Grado 3.
Trastornos respiratorios
Se ha notificado neumonía/pneumonitis intersticial y enfermedad pulmonar intersticial, y podría estar asociado a desenlace mortal (ver sección 4.8).
Si se desarrollaran nuevos síntomas pulmonares o hubiera un empeoramiento de los mismos, los pacientes deben ser monitorizados cuidadosamente, examinados inmediatamente, y tratados de manera apropiada. Se recomienda la interrupción del tratamiento con cabazitaxel hasta que el diagnóstico esté disponible. El uso temprano del tratamiento específico para cada situación podría ayudar a mejorar el estado del paciente. Se debe evaluar cuidadosamente el beneficio de la reanudación del tratamiento con cabazitaxel.
Riesgo de arritmias cardiacas
Se han notificado arritmias cardiacas, más frecuentemente taquicardia y fibrilación auricular (ver sección 4.8).
Pacientes de edad avanzada
Los pacientes de edad avanzada (> 65 años) tienen más probabilidad de experimentar ciertas reacciones adversas incluyendo neutropenia y neutropenia febril (ver sección 4.8).
Pacientes con insuficiencia hepática
Está contraindicado el tratamiento con JEVTANA en pacientes con insuficiencia hepática grave (bilirrubina total > 3 x LNS) (ver secciones 4.3 y 5.2).
La dosis se debe reducir en pacientes con insuficiencia hepática leve (bilirrubina total >1 a <
1,5 x LNS o AST >1,5 x LNS) (ver secciones 4.2 y 5.2).
Interacciones
Se debe evitar la administración concomitante con inhibidores potentes del CYP3A, ya que pueden aumentar las concentraciones plasmáticas de cabazitaxel (ver secciones 4.2 y 4.5). Si no se puede evitar la administración concomitante de un inhibidor potente del CYP3A, se debe considerar realizar una vigilancia estrecha de la toxicidad y una reducción de la dosis de cabazitaxel (ver secciones 4.2 y 4.5).
Se debe evitar la administración concomitante con inductores potentes del CYP3A, ya que pueden reducir las concentraciones plasmáticas de cabazitaxel (ver secciones 4.2 y 4.5).
Excipientes
El disolvente contiene 573,3 mg de etanol 96% (15% v/v), equivalente a 14 ml de cerveza o 6 ml de vino.
Nocivo para personas alcohólicas.
A tener en cuenta en grupos de alto riesgo, como pacientes con enfermedad hepática, o epilepsia.
4.5. Interacción con otros medicamentos y otras formas de interacción
Los estudios in vitro han demostrado que cabazitaxel se metaboliza principalmente a través del CYP3A (80%-90%) (ver sección 5.2).
Inhibidores del CYP3A
La administración repetida de ketoconazol (400 mg una vez al día), un inhibidor potente del CYP3A, resultó en una disminución del 20% del aclaramiento de cabazitaxel correspondiente a un aumento del 25% en el AUC. Por tanto, se debe evitar la administración concomitante de inhibidores potentes del CYP3A (por ej., ketoconazol, itraconazol, claritromicina, indinavir, nefazodona, nelfinavir, ritonavir, saquinavir, telitromicina, voriconazol)debido a que podría ocurrir un incremento de las concentraciones plasmáticas de cabazitaxel (ver secciones 4.2 y 4.4).
La administración concomitante de aprepitant, un inhibidor moderado del CYP3A, no tiene ningún efecto en el aclaramiento de cabazitaxel.
Inductores del CYP3A
La administración repetida de rifampicina (600 mg una vez al día), un inductor potente del CYP3A, resultó en un aumento del 21% del aclaramiento de cabazitaxel correspondiente a una disminución del 17% en el AUC. Por tanto, se debe evitar la administración concomitante de inductores potentes del CYP3A (por ej. fenitoína, carbamacepina, rifampicina, rifabutina, rifapentina, fenobarbital) debido a que p odría ocurrir una disminución de las concentraciones plasmáticas de cabazitaxel (ver secciones 4.2 y 4.4). Adicionalmente, los pacientes deben abstenerse de tomar hierba de San Juan o hipérico.
OATP1B1
In vitro, cabazitaxel también ha mostrado inhibir el transporte de proteínas de los Polipéptidos Transportadores de Aniones Orgánicos OATP1B1. Es posible el riesgo de interacción con los sustratos del OATP1B1 (por ej. estatinas, valsartan, repaglinida), particularmente durante la duración de la perfusión (1 hora) y hasta 20 minutos después de la finalización de la perfusión. Se recomienda un intervalo de tiempo de 12 horas antes de la perfusión y al menos de 3 horas después de la finalización de la perfusión, antes de administrar sustratos del OATP1B1.
Vacunas
La administración de vacunas vivas o vivas-atenuadas en pacientes inmunodeprimidos por agentes quimioterápicos puede dar lugar a infecciones graves o fatales. En pacientes en tratamiento con cabazitaxel se debe evitar la vacunación con vacunas vivas-atenuadas. Se pueden administrar vacunas muertas o inactivadas; no obstante, la respuesta a dichas vacunas puede disminuir.
4.6. Fertilidad, embarazo y lactancia
Embarazo
No hay datos sobre el uso de cabazitaxel en mujeres embarazadas. Los estudios en animales han demostrado toxicidad reproductiva en dosis maternotóxicas (ver sección 5.3) y cabazitaxel atraviesa la barrera placentaria (ver sección 5.3). Como otros medicamentos citotóxicos, cabazitaxel puede causar daño fetal cuando se administra en mujeres embarazadas.
No se recomienda el uso de cabazitaxel durante el embarazo y en mujeres en edad fértil que no utilicen anticonceptivos.
Lactancia
Los datos farmacocinéticos disponibles en animales han demostrado excreción de cabazitaxel y sus metabolitos en la leche (ver sección 5.3). No puede excluirse el riesgo para el bebé lactante. Cabazitaxel no se debe utilizar durante la lactancia.
Fertilidad
Los estudios en animales demostraron que cabazitaxel afectaba al sistema reproductor en ratas y perros macho sin ningún efecto funcional sobre la fertilidad (ver sección 5.3). Sin embargo, considerando la actividad farmacológica de los taxanos, su potencial genotóxico y el efecto de muchos compuestos de esta clase sobre la fertilidad en estudios animales, no podría excluirse el efecto sobre la fertilidad de los machos en humanos.
Debido a los potenciales efectos sobre los gametos masculinos y a la potencial exposición vía líquido seminal, los hombres tratados con cabazitaxel deben utilizar métodos anticonceptivos eficaces durante todo el tratamiento y se recomienda continuar utilizándolos hasta 6 meses después de la última dosis de cabazitaxel. Debido a la potencial exposición vía líquido seminal, durante todo el tratamiento los hombres tratados con cabazitaxel deben evitar el contacto con otra persona a través del eyaculado. A los hombres que van a ser tratados con cabazitaxel se les recomienda que consulten sobre la conservación de esperma antes del tratamiento.
4.7. Efectos sobre la capacidad para conducir y utilizar máquinas
Cabazitaxel podría influenciar la capacidad para conducir y utilizar máquinas ya que puede causar fatiga y mareo. Se debe recomendar a los pacientes que no conduzcan o utilicen máquinas si experimentan estas reacciones adversas durante el tratamiento.
4.8. Reacciones adversas
Resumen del perfil de seguridad
La seguridad de JEVTANA en combinación con prednisona o prednisolona se evaluó en 371 pacientes con cáncer de próstata metastático hormono-resistente, que fueron tratados con 25 mg/m2 de cabazitaxel, una vez cada tres semanas, en un ensayo clínico en fase III controlado, abierto, aleatorizado. Los pacientes recibieron una media de 6 ciclos de cabazitaxel.
Las reacciones adversas que se produjeron más frecuentemente (> 10) en todos los grados fue anemia (97,3%), leucopenia (95,7%), neutropenia (93,5%), trombocitopenia (47,4%), y diarrea (46,6%). Las reacciones adversas de grado > 3 que se produjeron más frecuentemente en el grupo de cabazitaxel fueron neutropenia (81,7%), leucopenia (68,2%), anemia (10,5%), neutropenia febril (7,5%), y diarrea (6,2%).
En 68 pacientes (18,3%) tratados con cabazitaxel se interrumpió el tratamiento por la aparición de reacciones adversas. La reacción adversa más frecuente que dio lugar a la interrupción de cabazitaxel fue neutropenia.
Tabla de reacciones adversas
Las reacciones adversas están descritas en la tabla 2, según el sistema de clasificación de órganos y categorías de frecuencia de MedDRA. Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad. La intensidad de las reacciones adversas se clasifica según el CTCAE 4.0 (grado >3 = G>3). Las frecuencias se basan en todos los grados y se definen como: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 2: Reacciones adversas y anomalías hematológicas notificadas con cabazitaxel en combinación con prednisona o prednisolona en el ensayo clínico TROPIC (n=371)
|
Sistema de clasificación de órganos |
Reacción adversa |
Todos los grados n (%) |
Grado > 3 n (%) | |
|
Muy frecuentes |
Frecuentes | |||
|
Infecciones e infestaciones |
Shock séptico |
4 (1,1) |
4 (1,1) | |
|
Sepsis |
4 (1,1) |
4 (1,1) | ||
|
Celulitis |
6 (1,6) |
2 (0,5) | ||
|
Infección del tracto urinario |
27 (7,3) |
4 (1,1) | ||
|
Gripe |
11(3) |
0 | ||
|
Cistitis |
10 (2,7) |
1 (0,3) | ||
|
Infección de vías respiratorias altas |
10 (2,7) |
0 | ||
|
Herpes zoster |
5 (1,3) |
0 | ||
|
Candidiasis |
4 (1,1) |
0 | ||
|
Trastornos de la sangre y del sistema linfático |
Neutropenia3 |
347 (93,5) |
303 (81,7) | |
|
Anemia3 |
361 (97,3) |
39 (10,5) | ||
|
Leucopeniaa |
355 (95,7) |
253 (68,2) | ||
|
T rombocitopeniaa |
176 (47,4) |
15 (4) | ||
|
Neutropenia febril |
28 (7,5) |
28 (7,5) | ||
|
Sistema de clasificación de órganos |
Reacción adversa |
Todos los grados n (%) |
Grado > 3 n (%) | |
|
Muy frecuentes |
Frecuentes | |||
|
Trastornos del sistema inmunológico |
Hipersensibilidad |
5 (1,3) |
0 | |
|
Trastornos del metabolismo y de la nutrición |
Anorexia |
59 (15,9) |
3 (0,8) | |
|
Deshidratación |
18 (4,9) |
8 (2,2) | ||
|
Hiperglucemia |
4 (1,1) |
3 (0,8) | ||
|
Hipopotasemia |
4 (1,1) |
2(0,5) | ||
|
Trastornos psiquiátricos |
Ansiedad |
11 (3) |
0 | |
|
Estado de confusión |
5 (1,3) |
0 | ||
|
Disgeusia |
41 (11,1) |
0 | ||
|
Neuropatía periférica |
30 (8,1) |
2 (0,5) | ||
|
Trastornos del |
Neuropatía sensorial periférica |
20 (5,4) |
1 (0,3) | |
|
sistema nervioso |
Mareo |
30 (8,1) |
0 | |
|
Cefalea |
28 (7,5) |
0 | ||
|
Parestesia |
17 (4,6) |
0 | ||
|
Letargia |
5 (1,3) |
1 (0,3) | ||
|
Hipoestesia |
5 (1,3) |
0 | ||
|
Ciática |
4 (1,1) |
1 (0,3) | ||
|
Conjuntivitis |
5 (1,3) |
0 | ||
|
Trastornos oculares |
Aumento de la lacrimación |
5 (1,3) |
0 | |
|
Trastornos del oído |
Tinnitus |
5 (1,3) |
0 | |
|
y del laberinto |
Vértigo |
5 (1,3) |
0 | |
|
Trastornos cardiacos* |
Fibrilación auricular |
4 (1,1) |
2 (0,5) | |
|
Taquicardia |
6 (1,6) |
0 | ||
|
Hipotensión |
20 (5,4) |
2 (0,5) | ||
|
Trombosis venosa profunda |
8 (2,2) |
7 (1,9) | ||
|
Trastornos |
Hipertensión |
6 (1,6) |
1 (0,3) | |
|
vasculares |
Hipotensión ortostática |
5 (1,3) |
1 (0,3) | |
|
Sofoco |
5 (1,3) |
0 | ||
|
Rubor |
4 (1,1) |
0 | ||
|
Trastornos |
Disnea |
44 (11,9) |
5 (1,3) | |
|
respiratorios, |
Tos |
40 (10,8) |
0 | |
|
torácicos y |
Dolor orofaríngeo |
13 (3,5) |
0 | |
|
mediastínicos |
Neumonía |
9 (2,4) |
6 (1,6) | |
|
Diarrea |
173 (46,6) |
23 (6,2) | ||
|
Náuseas |
127 (34,2) |
7 (1,9) | ||
|
Vómitos |
84 (22,6) |
7 (1,9) | ||
|
Trastornos |
Estreñimiento |
76 (20,5) |
4 (1,1) | |
|
gastrointestinales |
Dolor abdominal |
43 (11,6) |
7 (1,9) | |
|
Dispepsia |
25 (6,7) |
0 | ||
|
Dolor abdominal superior |
20 (5,4) |
0 | ||
|
Sistema de clasificación de órganos |
Reacción adversa |
Todos los grados n (%) |
Grado > 3 n (%) | |
|
Muy frecuentes |
Frecuentes | |||
|
Hemorroides |
14 (3,8) |
0 | ||
|
Reflujo gastroesofágico |
12 (3,2) |
0 | ||
|
Hemorragia rectal |
8 (2,2) |
2 (0,5) | ||
|
Sequedad de boca |
8 (2,2) |
1 (0,3) | ||
|
Distensión abdominal |
5 (1,3) |
1 (0,3) | ||
|
Trastornos de la piel y del tejido subcutáneo |
Alopecia |
37 (10) |
0 | |
|
Sequedad de boca |
9 (2,4) |
0 | ||
|
Eritema |
5 (1,3) |
0 | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor de espalda |
60 (16,2) |
14 (3,8) | |
|
Artralgia |
39 (10,5) |
4 (1,1) | ||
|
Dolor en extremidades |
30 (8,1) |
6 (1,6) | ||
|
Espasmos musculares |
27 (7,3) |
0 | ||
|
Mialgia |
14 (3,8) |
1 (0,3) | ||
|
Dolor músculo esquelético de pecho |
11 (3) |
1 (0,3) | ||
|
Dolor en el costado |
7 (1,9) |
3 (0,8) | ||
|
Trastornos renales y urinarios |
Insuficiencia renal aguda |
8 (2,2) |
6 (1,6) | |
|
Insuficiencia renal |
7 (1,9) |
6 (1,6) | ||
|
Disuria |
25 (6,7) |
0 | ||
|
Cólico renal |
5 (1,3) |
1 (0,3) | ||
|
Hematuria |
62 (16,7) |
7 (1,9) | ||
|
Polaquiuria |
13 (3,5) |
1 (0,3) | ||
|
Hidronefrosis |
9 (2,4) |
3 (0,8) | ||
|
Retención urinaria |
9 (2,4) |
3 (0,8) | ||
|
Incontinencia urinaria |
9 (2,4) |
0 | ||
|
Obstrucción de uréteres |
7 (1,9) |
5 (1,3) | ||
|
Trastornos del aparato reproductor y de la mama |
Dolor pélvico |
7(1,9) |
1 (0,3) | |
|
Trastornos generales y alteraciones en el lugar de administración |
Fatiga |
136 (36,7) |
18 (4,9) | |
|
Astenia |
76 (20,5) |
17 (4,6) | ||
|
Pirexia |
45 (12,1) |
4 (1,1) | ||
|
Edema periférico |
34 (9,2) |
2 (0,5) | ||
|
Inflamación de mucosas |
22 (5,9) |
1 (0,3) | ||
|
Dolor |
20 (5,4) |
4 (1,1) | ||
|
Dolor torácico |
9 (2,4) |
2 (0,5) | ||
|
Edema |
7 (1,9) |
1 (0,3) | ||
|
Escalofríos |
6 (1,6) |
0 | ||
|
Malestar general |
5 (1,3) |
0 | ||
|
Exploraciones complementarias |
Pérdida de peso |
32 (8,6) |
0 | |
|
Aumento de la |
4 (1,1) |
0 | ||
|
Sistema de clasificación de órganos |
Reacción adversa |
Todos los grados n (%) |
Grado > 3 n (%) | |
|
Muy frecuentes |
Frecuentes | |||
|
aspartato aminotransferasa | ||||
|
Aumento de las transaminasas |
4 (1,1) |
0 | ||
a basado en valores de laboratorio * ver detalles en la sección siguiente
Descripción de las reacciones adversas seleccionadas
Neutropenia y acontecimientos clínicos asociados
La incidencia de neutropenia grado > 3, según los datos de laboratorio, fue del 81,7%. La incidencia de las reacciones adversas de grado > 3 neutropenia clínica y neutropenia febril fueron del 21,3% y 7,5% respectivamente. La neutropenia fue la reacción adversa más frecuente que dio lugar a la interrupción del medicamento (2,4%).
Las complicaciones neutropénicas incluían infecciones neutropénicas (0,5%), sepsis neutropénica (0,8%) y shock séptico (1,1%), las cuales en algunos casos tuvieron un desenlace fatal.
El uso de G-CSF ha demostrado limitar la incidencia y gravedad de la neutropenia (ver secciones 4.2 y 4.4).
Trastornos cardiacos y arritmias
Los acontecimientos de todos los grados entre los trastornos cardiacos fueron más frecuentes con cabazitaxel, de los cuales 6 pacientes (1,6%) tuvieron arritmias cardiacas Grado > 3. La incidencia de taquicardia fue de 1,6 %, ninguno fue de Grado > 3. La incidencia de fibrilación auricular fue de 1,1 % en el grupo cabazitaxel. Los acontecimientos de insuficiencia cardiaca fueron más frecuentes con cabazitaxel, notificándose el acontecimiento a término para 2 pacientes (0,5%). Un paciente del grupo cabazitaxel falleció por insuficiencia cardiaca. Se notificó fibrilación ventricular fatal en 1 paciente (0,3%), y parada cardiaca en 2 pacientes (0,5%). Ninguno fue considerado por el investigador como relacionado.
Otras anomalías de laboratorio
La incidencia de anemia grado > 3, aumento de AST, ALT y bilirrubina, basada en las anomalías de laboratorio, fue del 10,5%, 0,7%, 0,9% y 0,6%, respectivamente.
Trastornos gastrointestinales
Se ha observado colitis, enterocolitis, gastritis, enterocolitis neutropénica. También se ha notificado hemorragia gastrointestinal y perforación, íleo (ileus) y obstrucción intestinal (ver sección 4.4).
Trastornos respiratorios
Se han notificado con una frecuencia no conocida (no puede estimarse a partir de los datos disponibles) casos de pneumonía/pneumonitis intersticial y enfermedad pulmonar intersticial, algunas veces mortal (ver sección 4.4).
Población pediátrica Ver sección 4.2.
Otras poblaciones especiales
Pacientes de edad avanzada
Entre los 371 pacientes tratados con cabazitaxel para cáncer de próstata, 240 pacientes tenían 65 años o más incluyendo 70 pacientes de más de 75 años.
Las siguientes reacciones adversas se notificaron en porcentajes > 5% más altos en pacientes de 65 años o más, en comparación con los pacientes jóvenes: fatiga (40,4% frente a 29,8%), neutropenia clínica (24,2% frente a 17,6%), astenia (23,8% frente a 14,5%), pirexia (14,6% frente a 7,6%), mareo
(10,0% frente a 4,6%), infección del tracto urinario (9,6% frente a 3,1%) y deshidratación (6,7% frente a 1,5%), respectivamente.
La incidencia de las siguientes reacciones adversas de grado > 3 fue mayor en pacientes > 65 años de edad, en comparación con los pacientes jóvenes: neutropenia basada en anomalías de laboratorio (86,3% frente a 73,3%), neutropenia clínica (23,8% frente a 16,8%) y neutropenia febril (8,3% frente a 6,1%) (ver secciones 4.2 y 4.4).
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9. Sobredosis
No se conoce ningún antídoto para cabazitaxel. Las complicaciones anticipadas de la sobredosis podrían consistir en exacerbación de las reacciones adversas, así como supresión de la médula ósea y alteraciones gastrointestinales. En caso de sobredosis, el paciente debe mantenerse en una unidad especializada y monitorizarse estrechamente. Los pacientes deben recibir G-CSF terapéutico lo más pronto posible tras descubrir la sobredosis. Deben tomarse otras medidas sintomáticas apropiadas.
5. PROPIEDADES FARMACOLÓGICAS
5.1. Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agentes antineoplásicos, taxanos, código ATC: L01CD04.
Mecanismo de acción
Cabazitaxel es un agente antineoplásico que actúa mediante la interrupción de la red microtubular en las células. Cabazitaxel se une a la tubulina y promueve la unión de la tubulina en los microtúbulos mientras, simultáneamente, inhibe su desmontaje. Esto da lugar a la estabilización de los microtúbulos, lo que resulta en la inhibición de las funciones celulares mitótica e interfase.
Efectos farmacodinámicos
Cabazitaxel demostró un amplio espectro de actividad antitumoral frente a tumores humanos avanzados xenotransplantados en ratones. Cabazitaxel es activo en tumores sensibles a docetaxel. Además, cabazitaxel demostró actividad en modelos tumorales insensibles a la quimioterapia incluyendo docetaxel.
Eficacia y seguridad clínica
La eficacia y seguridad de JEVTANA en combinación con prednisona o prednisolona se evaluó en un ensayo clínico en fase III, aleatorizado, abierto, internacional, multicéntrico, en pacientes con cáncer de próstata metastático hormono-resistente previamente tratados con un régimen que contenía docetaxel.
La supervivencia global (OS) fue la variable principal de la eficacia del ensayo.
Las variables secundarias incluyeron Supervivencia Libre de Progresión [SLP (definida como el tiempo desde la aleatorización hasta la progresión del tumor, progresión del Antígeno Prostético Específico (PSA), progresión del dolor, o muerte debida a cualquier causa, lo que sucediera primero], Tasa de Respuesta Tumoral basada en los Criterios de Evaluación de la Respuesta en Tumores Sólidos (RECIST), progresión del PSA (definida como un aumento > 25% o > 50% en el PSA de los que no responden o de los que responden, respectivamente), respuesta del PSA (disminución del 50%, como mínimo, en los niveles séricos de PSA), progresión del dolor [evaluada utilizando la escala de Intensidad de Dolor Presente (PPI) mediante el cuestionario de McGill-Melzack y una Escala Analgésica (AS)] y respuesta al dolor (definida como una reducción de más de 2 puntos desde el nivel
basal medio de PPI, sin aumento concomitante de AS, o una reducción > 50% del uso analgésico desde la AS media basal sin aumento concomitante del dolor).
Se aleatorizaron un total de 755 pacientes para recibir JEVTANA 25 mg/m2 por vía intravenosa cada 3 semanas, un máximo de 10 ciclos con 10 mg diarios vía oral de prednisona o prednisolona (n=378), o para recibir 12 mg/m2 de mitoxantrona, por vía intravenosa, cada 3 semanas, un máximo de 10 ciclos, con 10 mg diarios vía oral de prednisona o prednisolona (n=377).
Este ensayo incluyó pacientes mayores de 18 años, con cáncer de próstata metastático hormono-resistente, con enfermedad medible por los criterios RECIST o no medible, con niveles incrementados de PSA o aparición de nuevas lesiones, Grupo Oncológico Cooperativo del Este (ECOG)-PS de 0 a 2. Los pacientes tenían que tener neutrófilos >1.500/mm3, plaquetas >100.000/mm3, hemoglobina >10 g/dl, creatinina <1,5 x LNS, bilirrubina total <1 x LNS, AST y ALT <1,5 x LNS.
Los pacientes con historial de insuficiencia cardiaca congestiva o infarto de miocardio en los últimos 6 meses, o los pacientes con arritmias cardíacas incontroladas, angina de pecho y/o hipertensión no se incluyeron en el ensayo.
Los datos demográficos, incluyendo edad, raza y ECOG-PS (de 0 a 2) se distribuyeron de forma equilibrada entre los dos brazos de tratamiento. En el grupo de JEVTANA, la edad media fue de 68 años, rango (46-92), y la distribución racial fue un 83,9 Caucasiana, un 6,9% Asiática/Oriental, un 5,3% Negra y un 4% Otras.
La mediana del número de ciclos fue 6 en el grupo de JEVTANA y 4 en el grupo de mitoxantrona. El porcentaje de pacientes que completó el tratamiento del ensayo (10 ciclos) fue un 29,4% y un 13,5% en el grupo de JEVTANA y en el grupo de comparación, respectivamente.
La supervivencia global fue significativamente más larga con JEVTANA, en comparación con mitoxantrona (15,1 meses versus 12,7 meses, respectivamente), con una reducción del 30% en el riesgo de muerte en comparación con mitoxantrona (ver tabla 3 y figura 1).
Un subgrupo de 59 pacientes recibieron dosis acumulativas de docetaxel < 225 mg/m2 (29 pacientes en el brazo JEVTANA, 30 pacientes en el brazo mitoxantrona). No hubo diferencias significativas en la supervivencia global en este grupo de pacientes (HR (95% IC) 0,96 (0,49-1,86)).
Tabla 3 - Eficacia de JEVTANA en el tratamiento de pacientes con cáncer de próstata metastásico
hormono-resistente
|
JEVTANA + prednisona n=378 |
Mitoxantrona + prednisona n=377 | |
|
Supervivencia global Número de pacientes con muertes (%) |
234 (61,9%) |
279 (74%) |
|
Mediana de supervivencia (meses) |
15,1 (14,1-16,3) |
12,7 (11,6-13,7) |
|
(95% IC) Razón de Riesgos (HR)1 (95% IC) |
0,70 (0,59-0,83) | |
|
Valor de p |
<0,0001 |
1 HR calculado usando el modelo Cox; una razón de riesgos menor de 1 favorece a JEVTANA
Figura 1: Curvas de supervivencia global de Kaplan Meier
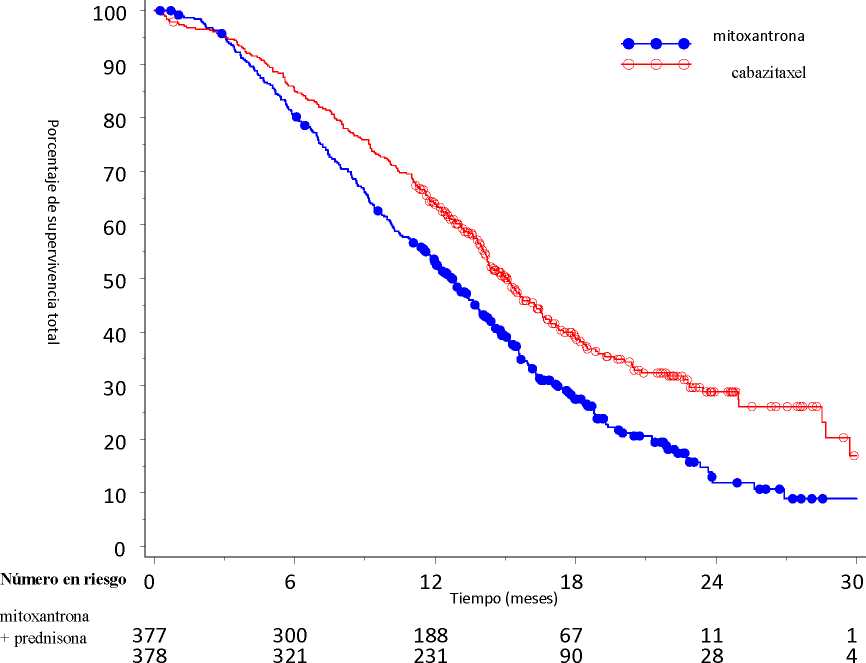
cabazitaxel + prednisona
Hubo una mejora en la SLP en el brazo JEVTANA, en comparación con el brazo de mitoxantrona,
2,8 (2,4-3,0) meses frente a 1,4 (1,4-1,7), respectivamente, HR (95% IC) 0,74 (0,64-0,86), p<0,0001.
Hubo una tasa significativamente más alta de respuesta tumoral del 14,4% (95% IC: 9,6-19,3) en los pacientes del brazo JEVTANA, en comparación con un 4,4% (95% IC: 1,6-7,2) para los pacientes del brazo mitoxantrona, p=0,0005.
Las variables secundarias PSA fueron positivas en el brazo JEVTANA. La mediana de progresión de PSA fue de 6,4 meses (95% IC: 5,1-7,3) para los pacientes del brazo JEVTANA, comparado con 3,1 meses (95% IC: 2,2-4,4) en el brazo mitoxantrona, HR 0,75 meses (95% IC: 0,63-0,90), p=0,0010. La respuesta del PSA fue del 39,2% en los pacientes del brazo JEVTANA (95% IC: 33,9-44,5) frente al 17,8% de los pacientes del brazo mitoxantrona (95% IC: 13,7-22,0), p=0,0002.
No hubo diferencia estadísticamente significativa entre ambos grupos de tratamiento en la progresión del dolor y en la respuesta al dolor.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con JEVTANA en los diferentes grupos de la población pediátrica en la indicación de cáncer de próstata (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2. Propiedades farmacocinéticas
Se realizó un análisis farmacocinético poblacional en 170 pacientes incluyendo pacientes con tumores sólidos avanzados (n=69), cáncer de mama metastásico (n=43) y cáncer de próstata metastásico (n=67). Estos pacientes recibieron cabazitaxel, en dosis de 10 a 30 mg/m2, semanalmente o cada 3 semanas.
Absorción
Después de 1 hora de administración intravenosa de 25 mg/m2 de cabazitaxel en pacientes con cáncer de próstata metastásico (n=67), la Cmáx fue de 226 ng/ml (coeficiente de variación (CV): 107%) y se alcanzó al final de la perfusión de 1 hora (Tmáx). El AUC media fue 991 ng.h/ml (CV: 34%).
No se observó ninguna desviación importante de la proporcionalidad con la dosis de 10 a 30 mg/m2, en pacientes con tumores sólidos avanzados (n=126).
Distribución
El volumen de distribución (Vss) fue 4870 l (2640 l/m2 para un paciente con un área de superficie corporal (BSA) media de 1,84 m2), en estado estacionario.
In vitro, la unión de cabazitaxel a las proteínas séricas humanas fue del 89-92% y no fue saturable hasta 50.000 ng/ml, lo que cubre la concentración máxima observada en los ensayos clínicos. Cabazitaxel se une, principalmente, a la albúmina sérica humana (82,0%) y a lipoproteínas (87,9% para HDL, 69,8% para LDL y 55,8 % para VLDL). In vitro, las tasas de concentración sangre-plasma en sangre humana oscilaron de 0,90 a 0,99, lo que indica que cabazitaxel se distribuyó igualmente entre la sangre y el plasma.
Metabolismo
Cabazitaxel se metaboliza principalmente en el hígado (> 95%), principalmente por la isoenzima CYP3A (80%-90%). Cabazitaxel es el principal compuesto circulante en el plasma humano. Se detectaron siete metabolitos en plasma (incluyendo 3 metabolitos activos derivados de O-demetilaciones), de los cuales el principal supone el 5% de la exposición padre. Se excretan alrededor de 20 metabolitos de cabazitaxel en orina y heces humanas.
Basándose en los estudios in vitro, el riesgo potencial de inhibición por cabazitaxel en concentraciones clínicamente significativas es posible en medicamentos que son el principal substrato del CYP3A.
Sin embargo, un estudio clínico ha mostrado que cabazitaxel (25 mg/m2 administrado como una perfusión única de 1 hora) no modificó los niveles plasmáticos de midazolam, un sustrato sonda del CYP3A. Por tanto, a dosis terapéuticas, no se espera ningún impacto clínico en pacientes en la administración conjunta de sustratos del CYP3A con cabazitaxel.
No hay riesgo potencial de inhibición de medicamentos que son sustrato de otros enzimas CYP (1A2, 2B6, 2C9, 2C8, 2C19, 2E1 y 2D6), ni tampoco riesgo potencial de inducción por cabazitaxel sobre medicamentos que son sustratos de CYP1A, CYP2C9 y CYP3A. Cabazitaxel no inhibió in vitro la vía principal de biotransformación de warfarina a 7-hidroxiwarfarina, que está mediada por CYP2C9. Por tanto, no es de esperar ninguna interacción farmacocinética in vivo de cabazitaxel sobre warfarina.
Cabazitaxel no inhibió in vitro las proteínas multifármaco-resistentes (MRP): MRP1 y MRP2 o los Transportadores de Cationes Orgánicos (OCT1). Cabazitaxel inhibió el transporte de P-glicoproteína (PgP) (digoxina, vinblastina), proteínas resistentes al cáncer de mama (BCRP) (metotrexato), o los Polipéptidos Transportadores de Aniones Orgánicos OATP1B3 (CCK8) en concentraciones al menos 15 veces a las observadas a nivel clínico mientras que inhibió el transporte del OATP1B1 (estradiol-17p-glucuronida) en concentraciones de sólo 5 veces a las observadas a nivel clínico. Por tanto el riesgo de interacción con sustratos de MRP, OCT1, PgP, BCRP y OATP1B3 es improbable in vivo, con la dosis de 25 mg/m2. Es posible el riesgo de interacción con transportadores OATP1B1, particularmente durante la duración de la perfusión (1 hora) y hasta 20 minutos después de la finalización de la perfusión (ver sección 4.5).
Eliminación
Tras 1 hora de perfusión intravenosa de 25 mg/m2 de [14C]-cabazitaxel en pacientes, aproximadamente el 80% de la dosis administrada se eliminó en 2 semanas. Cabazitaxel se excreta principalmente en las heces, como numerosos metabolitos (el 76% de la dosis), mientras que la excreción renal de cabazitaxel y metabolitos supone menos del 4% de la dosis (2,3% como medicamento sin modificar en orina).
Cabazitaxel tuvo un aclaramiento plasmático elevado, 48,5 l/h (26,4 l/h/m2 para pacientes con una BSA media de 1,84 m2) y una semivida terminal larga de 95 horas.
Poblaciones especiales
Pacientes de edad avanzada
En el análisis farmacocinético poblacional en 70 pacientes de 65 años y mayores (57 de 65 a 75 y 13 pacientes de más de 75 años), no se observó ningún efecto de la edad sobre la farmacocinética de cabazitaxel.
Pacientes pediátricos
No se ha establecido la seguridad y efectividad de JEVTANA en niños y adolescentes menores de 18 años.
Insuficiencia hepática
Cabazitaxel se elimina principalmente vía metabolismo hepático.
En un estudio realizado en 43 pacientes con cáncer e insuficiencia hepática, la insuficiencia hepática leve (bilirrubina total >1 a < 1,5 x LNS o AST >1,5 x LNS) o moderada (bilirrubma total > 1,5 a <3,0 x LNS) no mostró ninguna influencia en la farmacocinética de cabazitaxel. La dosis máxima tolerada (DMT) de cabazitaxel fue de 20 y 15 mg/m2, respectivamente.
En 3 pacientes con insuficiencia hepática grave (bilirrubina total > 3 LNS), se observó una disminución del 39% en el aclaramiento cuando se comparó con pacientes con insuficiencia hepática leve, evidenciando algún efecto de la insuficiencia hepática grave en la farmacocinética de cabazitaxel. No se ha establecido la DMT de cabazitaxel en pacientes con insuficiencia hepática grave.
De acuerdo a los datos de seguridad y tolerancia, la dosis de cabazitaxel se debe reducir en pacientes con insuficiencia hepática leve (ver secciones 4.2, 4.4). JEVTANA está contraindicado en pacientes con insuficiencia hepática grave (ver sección 4.3).
Insuficiencia renal
Cabazitaxel se excreta mínimamente a través de los riñones (el 2,3% de la dosis). El análisis farmacocinético poblacional realizado en 170 pacientes que incluía 14 pacientes con insuficiencia renal moderada (aclaramiento de creatinina en el intervalo de 30 a 50 ml/min.) y 59 pacientes con insuficiencia renal leve (aclaramiento de creatinina en el intervalo de 50 a 80 ml/min.) demostró que la insuficiencia renal leve o moderada no tenía efectos significativos sobre la farmacocinética de cabazitaxel. Esto fue confirmado por un estudio comparativo farmacocinético en pacientes con tumores sólidos con función renal normal (8 pacientes), insuficiencia renal moderada (8 pacientes) e insuficiencia renal grave (9 pacientes), que recibieron varios ciclos de cabazitaxel en perfusión única IV hasta 25 mg/m2.
5.3. Datos preclínicos sobre seguridad
Las reacciones adversas no observadas en estudios clínicos, pero sí en perros tras una dosis única, administrada cada 5 días o semanalmente, a niveles de exposición por debajo de los niveles clínicos de exposición y con posible importancia para el uso clínico, fueron necrosis arteriolar/periarterolar en el hígado, hiperplasia del ducto biliar y/o necrosis hepatocelular (ver sección 4.2).
Las reacciones adversas no observadas en los estudios clínicos, pero sí en ratas durante los estudios de toxicidad a dosis repetidas a niveles de exposición por encima de los niveles clínicos de exposición y con posible relevancia para el uso clínico, fueron alteraciones oculares, caracterizadas por inflamación/degeneración subcapsular de las fibras del cristalino. Estos efectos fueron parcialmente reversibles a las 8 semanas.
No se han realizado estudios de carcinogenicidad con cabazitaxel.
Cabazitaxel no indujo mutaciones en el test de mutación reversa en bacterias (Ames). No fue clastogénico en un ensayo in vitro en linfocitos humanos (no indujo aberración cromosómica estructural, sino que aumentó el número de células poliploides) e indujo un aumento de micronúcleos en el ensayo in vivo en ratas. No obstante estos resultados de genotoxicidad son inherentes a la actividad farmacológica del compuesto (inhibición de la despolimerización de tubulina) y se han observado con medicamentos que muestran la misma actividad farmacológica.
Cabazitaxel no afectó a los rendimientos de apareamiento o a la fertilidad de las ratas macho tratadas. Sin embargo, en los estudios de toxicidad de dosis repetidas, se observó degeneración de la vesícula seminal y atrofia del túbulo seminífero en los testículos de las ratas, y se observó degeneración testicular (necrosis mínima de la célula epitelial en el epidídimo) en perros. Las exposiciones en animales fueron similares o menores que las observadas en humanos que recibieron dosis clínicamente relevantes de cabazitaxel.
Cabazitaxel indujo toxicidad embriofetal en ratas hembra tratadas intravenosamente, una vez al día, de los días 6 a 17 de gestación, asociada con toxicidad maternal y consistente en muertes fetales y disminución del peso medio fetal, asociado con retraso en la osificación del esqueleto. Las exposiciones en animales fueron menores que las observadas en humanos que recibieron dosis clínicamente relevantes de cabazitaxel. Cabazitaxel atraviesa la barrera placentaria en ratas.
En ratas, cabazitaxel y sus metabolitos se excretan en leche materna, en una cantidad de hasta el 1,5% de la dosis administrada en 24 horas.
Evaluación del Riesgo Medioambiental (ERA)
Los resultados de los estudios de evaluación del riesgo medioambiental indicaron que el uso de JEVTANA no causa riesgo significativo para el entorno acuático (ver sección 6.6 para la eliminación del producto no utilizado).
6. DATOS FARMACÉUTICOS
6.1. Lista de excipientes
Concentrado Polisorbato 80 Ácido cítrico
Disolvente Etanol 96%
Agua para preparaciones inyectables
6.2. Incompatibilidades
Este medicamento no debe mezclarse con otros medicamentos, excepto los mencionados en la sección 6.6.
No deben utilizarse envases de perfusión de PVC o sets de perfusión de poliuretano para la preparación y administración de la solución para perfusión.
6.3. Periodo de validez
Viales sin abrir 3 años.
Después de la apertura del vial
Los viales de concentrado y disolvente deben utilizarse inmediatamente. Si no se usan inmediatamente, el tiempo y las condiciones de conservación en uso son responsabilidad del usuario.
Después de la dilución inicial del concentrado con el disolvente
Se ha demostrado la estabilidad química y física en uso durante 1 hora, a temperatura ambiente (15°C -30°C). Desde el punto de vista microbiológico, la mezcla concentrado-disolvente debe utilizarse inmediatamente. Si no se utiliza inmediatamente, los tiempos y las condiciones de conservación son responsabilidad del usuario y normalmente no deberían ser más de 24 horas a 2°C - 8°C, al menos que la dilución se haya realizado condiciones asépticas controladas y validadas.
Después de la dilución final en la bolsa/botella de perfusión
Se ha demostrado la estabilidad química y física de la solución de perfusión durante 8 horas a temperatura ambiente (incluyendo 1 hora de tiempo de perfusión) y durante 48 horas en nevera (incluyendo 1 hora de tiempo de perfusión).
Desde un punto de vista microbiológico, la solución de perfusión debe utilizarse inmediatamente. Si no se utiliza inmediatamente, los tiempos y las condiciones de conservación son responsabilidad del usuario y normalmente no deberían ser más de 24 h a 2°C - 8°C, al menos que la dilución se haya realizado condiciones asépticas controladas y validadas.
6.4. Precauciones especiales de conservación
No conservar a temperatura superior a 30°C.
No refrigerar.
Para las condiciones de conservación del medicamento después de la dilución, ver sección 6.3.
6.5. Naturaleza y contenido del envase
Un envase contiene un vial de concentrado y un vial de disolvente:
• Concentrado: 1,5 ml de concentrado en un vial de vidrio transparente (tipo I) de 15 ml cerrado con un tapón de caucho clorobutilo de color gris, sellado con una cápsula de aluminio y cubierto con un tapón expulsor flip-off de plástico de color verde claro. Cada vial contiene 60 mg de cabazitaxel por 1,5 ml de volumen nominal (volumen de llenado: 73,2 mg de cabazitaxel/1,83 ml). Este volumen de llenado se ha establecido durante el desarrollo de JEVTANA para compensar la pérdida de líquido durante la preparación de la premezcla. Este sobrellenado asegura que después de la dilución con el contenido completo del disolvente incluido en JEVTANA, haya un volumen de la premezcla mínimo extraíble de 6 ml conteniendo 10 mg/ml de JEVTANA que se corresponde con la cantidad marcada de 60 mg por vial.
• Disolvente: 4,5 ml de disolvente en un vial de vidrio transparente (tipo I) de 15 ml cerrado con un tapón de caucho clorobutilo de color gris gris, sellado con una cápsula de aluminio dorada y cubierto con un tapón expulsor flip-off de plástico incoloro. Cada vial contiene 4,5 ml de volumen nominal (volumen de llenado: 5,67 ml). Este volumen de llenado se ha establecido durante el desarrollo y el sobrellenado asegura, después de la adición del contenido completo del vial de disolvente al contenido del vial del concentrado de JEVTANA 60 mg, una concentración de la solución de premezcla de JEVTANA de 10 mg/ml.
6.6. Precauciones especiales de eliminación y otras manipulaciones
JEVTANA sólo debe ser preparado y administrado por personal entrenado en el manejo de agentes citotóxicos. Las trabajadoras embarazadas no deben manipular el producto. Como para cualquier otro agente antineoplásico, se debe actuar con precaución cuando se manejan y preparan soluciones de JEVTANA, teniendo en cuenta el uso de dispositivos de seguridad, equipo de protección personal (por ej. guantes) y procedimientos de preparación. Si JEVTANA, en cualquiera de las etapas de su preparación, entrara en contacto con la piel, lavar inmediata y minuciosamente con agua y jabón. Si entrara en contacto con membranas mucosas, lavar inmediata y minuciosamente con agua.
Diluir siempre el concentrado para solución para perfusión con el contenido COMPLETO del disolvente que se proporciona antes de añadirlo a la solución de perfusión.
Lea detenidamente TODA esta sección antes de mezclar y diluir. JEVTANA requiere DOS diluciones antes de la administración. Siga las instrucciones de preparación que se proporcionan a continuación.
Nota: tanto el vial del concentrado de JEVTANA 60 mg/1,5 ml (volumen de llenado: 73,2 mg de cabazitaxel/1,83 ml) como el del vial de disolvente (volumen de llenado: 5,67 ml) contienen un sobrellenado para compensar la pérdida de líquido durante la preparación. Este sobrellenado asegura que después de la dilución con el contenido COMPLETO del disolvente proporcionado, haya una solución conteniendo 10 mg/ml de cabazitaxel.
Para preparar la solución para perfusión, el siguiente proceso de dilución en dos etapas debe realizarse de forma aséptica.
Etapa 1: dilución inicial del concentrado de solución para perfusión con el disolvente proporcionado.
Etapa 1.1
Inspeccionar el vial de concentrado y el disolvente proporcionado. La solución de concentrado y de disolvente deben ser transparentes
|
Vial de concentrado |
Vial de disolvente |
|
(60 mg -1,5 ml) |
Etapa 1.2
Utilizando una jeringa provista de una aguja fija, extraer de forma aséptica el contenido completo del disolvente proporcionado invirtiendo parcialmente el vial
Etapa 1.3
Inyectar el contenido completo en el correspondiente vial de concentrado.
Para limitar todo lo posible la formación de espuma al inyectar el disolvente, dirigir la aguja hacia la pared interior del vial de solución de concentrado e inyectar lentamente.
Una vez reconstituido, la solución resultante contiene 10 mg/ml de cabazitaxel.
|
Mezcla concentrado-disolvente |
Vial de disolvente | |
|
10 mg/ml | ||
Etapa 1.4
Mezcla
concentrado-disolvente 10 mg/ml
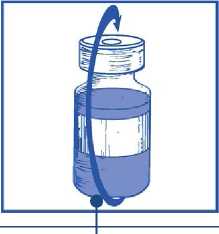
Sacar la jeringa y la aguja y mezclar manualmente y suavemente, mediante inversiones repetidas, hasta que se obtenga una solución transparente y homogénea. Se pueden tardar unos 45 segundos
Etapa 1.5
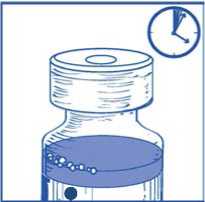
Dejar reposar la solución durante aproximadamente 5 minutos y a continuación comprobar que la solución es homogénea y transparente.
Es normal que persista la espuma pasado este tiempo.
Mezcla
concentrado-disolvente 10 mg/ml
Esta mezcla concentrado-disolvente resultante contiene 10 mg/ml de cabazitaxel (al menos 6 ml de volumen liberado). La segunda dilución se debe realizar inmediatamente (antes de 1 hora) como se detalla en la Etapa 2.
Podría ser necesario más de un vial de mezcla concentrado-disolvente para administrar la dosis prescrita.
Etapa 2: segunda dilución (final) para perfusión Etapa 2.1
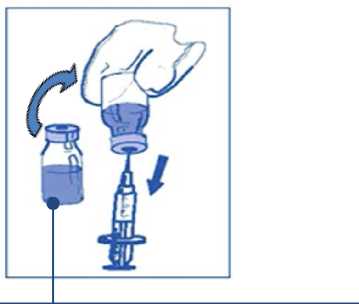
Mezcla
concentrado-disolvente 10 mg/ml
De forma aséptica extraer la cantidad requerida de mezcla concentrado-disolvente (10 mg/ml de cabazitaxel), con una jeringa graduada provista de una aguja fija. Como ejemplo, una dosis de 45 mg de JEVTANA requeriría 4,5 ml de la mezcla de concentrado-disolvente preparada en la Etapa 1.
Como puede seguir habiendo espuma en la pared del vial de esta solución, después de la preparación descrita en la Etapa 1, es preferible
situar la aguja de la jeringa en la mitad del contenido durante la extracción.
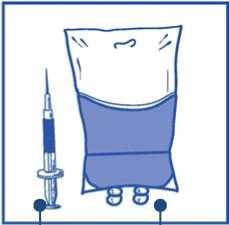
Etapa 2.2
Inyectar en un envase estéril sin PVC de solución de glucosa al 5% o solución de cloruro sódico 9 mg/ml (0,9%) para perfusión. La concentración de la solución para perfusión debe estar entre 0,10 mg/ml y 0,26 mg/ml.
Cantidad requerida de mezc concentrado-disolvente
a
Solución de glucosa al 5% o solución de cloruro sódico 9 mg/ml (0,9%) para perfusión

Etapa 2.3
Sacar la jeringa y mezclar el contenido de la bolsa o botella de perfusión manualmente, mediante movimiento de balanceo.
Etapa 2.4
Al igual que todos los productos parenterales, la solución de perfusión resultante se debe inspeccionar visualmente antes de usarla.. Como la solución de perfusión está sobresaturada, puede cristalizar con el tiempo. En este caso, no se debe utilizar la solución y debe eliminarse.
La solución para perfusión debe utilizarse inmediatamente. No obstante, el tiempo de conservación en uso puede ser más largo bajo las condiciones específicas mencionadas en la sección 6.3 Se recomienda el uso de un filtro en línea de 0,22 micrómetros de tamaño de poro nominal (también denominado 0,2 micrómetros) durante la administración.
No deben utilizarse envases de perfusión de PVC o sets de perfusión de poliuretano para la preparación y administración de JEVTANA.
JEVTANA no se debe mezclar con otros medicamentos que los mencionados.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
sanofi-aventis groupe 54, rue La Boétie F - 75008 París Francia
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/11/676/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de autorización: 17 marzo 2011
Fecha de la última renovación: 19 noviembre 2015
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www. ema.europa.eu.
ANEXO II
A. FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección de los fabricantes responsables de la liberación de los lotes
Sanofi-Aventis Deutschland GmbH
Industriepark Hochst
65926 Frankfurt am Main
Alemania
El prospecto impreso del medicamento debe especificar el nombre y dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (Ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 quater, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias de acuerdo con la versión del PGR incluido en el Módulo 1.8.2. de la Autorización de Comercialización y en cualquier actualización posterior del PGR.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos
resultado de nueva beneficio/riesgo, o o minimización de
• Cuando se modifique el sistema de gestión de riesgos, especialmente como información disponible que pueda conllevar cambios relevantes en el perfil como resultado de la consecución de un hito importante (farmacovigilancia riesgos).
ANEXO III
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR ENVASE EXTERIOR
1. NOMBRE DEL MEDICAMENTO
JEVTANA 60 mg concentrado y disolvente para solución para perfusión cabazitaxel
2. PRINCIPIO(S) ACTIVO(S)
1 ml de concentrado contiene 40 mg de cabazitaxel.
Cada vial de concentrado de 1,5 ml contiene 60 mg de cabazitaxel.
El vial de concentrado (llenado: 73,2 mg de cabazitaxel/1,83 ml) y el vial de disolvente (5,67 ml) contienen un sobrellenado para compensar la pérdida de líquido durante la preparación. Este sobrellenado asegura que después de la dilución inicial con el contenido COMPLETO del vial de disolvente proporcionado, la concentración de cabazitaxel sea 10 mg/ml.
3. LISTA DE EXCIPIENTES
Excipientes
Vial de concentrado: polisorbato 80 y ácido cítrico;
Vial de disolvente: etanol 96% y agua para preparaciones inyectables. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Concentrado y disolvente para solución para perfusión.
1 vial de concentrado de 1,5 ml y 1 vial de disolvente de 4,5 ml.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Viales para un solo uso.
PRECAUCIÓN: se requiere dilución realizada en dos etapas. Lea el prospecto antes de utilizar este medicamento.
Vía intravenosa (perfusión) DESPUÉS de la dilución final.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑO S
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
CITOTÓXICO
FECHA DE CADUCIDAD
CAD
Leer el prospecto para la caducidad de la solución diluida.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
No conservar a temperatura superior a 30°C. No refrigerar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO
UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE
COMERCIALIZACIÓN
sanofi-aventis groupe 54, rue La Boétie F - 75008 París Francia
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/11/676/001
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información Braille.
ETIQUETA DEL VIAL PARA EL CONCENTRADO
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
JEVTANA 60 mg concentrado estéril cabazitaxel
2. FORMA DE ADMINISTRACIÓN
Diluir con TODO el disolvente proporcionado.
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
1,5 ml
10 mg/ml después de la primera dilución
6. OTROS
Solución para perfusión IV después de la dilución final (ver prospecto). Contiene un sobrellenado.
ETIQUETA DEL VIAL PARA EL DISOLVENTE
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
DISOLVENTE para JEVTANA
2. FORMA DE ADMINISTRACIÓN
Utilizar el contenido COMPLETO para la dilución (ver prospecto).
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
4,5 ml (etanol 96% y agua para preparaciones inyectables).
6. OTROS
Este vial contiene un sobrellenado.
B. PROSPECTO
Prospecto: información para el usuario
JEVTANA 60 mg concentrado y disolvente para solución para perfusión
cabazitaxel
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
• Conserve este prospecto, ya que puede tener que volver a leerlo.
• Si tiene alguna duda, consulte a su médico, farmacéutico o enfermera.
• Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermera, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es JEVTANA y para qué se utiliza
2. Qué necesita saber antes de que le administren JEVTANA
3. Cómo usar JEVTANA
4. Posibles efectos adversos
5. Conservación de JEVTANA
6. Contenido del envase e información adicional
1. Qué es JEVTANA y para qué se utiliza
El nombre de su medicamento es JEVTANA. Su denominación común es cabazitaxel. Pertenece a un grupo de medicamentos denominado “taxanos”, utilizados para tratar cánceres.
JEVTANA se utiliza para el tratamiento del cáncer de próstata que ha progresado después de haber recibido otra quimioterapia. Actúa deteniendo el crecimiento de las células y su multiplicación.
Como parte de su tratamiento, tomará también cada día un corticosteroide (prednisona o prednisolona), por vía oral. Pida información a su médico sobre este otro medicamento.
2. Qué necesita saber antes de que le administren JEVTANA No use JEVTANA:
• si es alérgico (hipersensible) a cabazitaxel, a otros taxanos, al polisorbato 80 o a alguno de los demás componentes de este medicamento (incluidos en la sección 6),
• si el número de sus glóbulos blancos es muy bajo (número de neutrófilos menor o igual a 1.500/mm3),
• si tiene problemas graves de hígado,
• si recientemente ha sido o va a ser vacunado contra la fiebre amarilla.
No debe recibir JEVTANA si le sucede alguna de las circunstancias anteriores. Si no está seguro, consulte a su médico antes de recibir JEVTANA.
Advertencias y precauciones
Antes de iniciar el tratamiento con JEVTANA, le harán análisis de sangre para comprobar que tiene suficientes células sanguíneas y que sus riñones e hígado funcionan adecuadamente para recibir JEVTANA.
Informe a su médico inmediatamente si:
• tiene fiebre. Durante el tratamiento con JEVTANA es más probable que se reduzca el número de sus glóbulos blancos. El médico controlará su sangre y su estado general para detectar signos de infecciones. Podría administrarle otros medicamentos para mantener el número de sus células sanguíneas. Las personas con recuentos celulares bajos pueden desarrollar infecciones que pueden poner en peligro la vida. El primer signo de infección podría ser fiebre, por lo que si tiene fiebre, informe a su médico inmediatamente.
• alguna vez ha tenido cualquier alergia. Durante el tratamiento con JEVTANA pueden producirse reacciones alérgicas graves.
• tiene diarrea grave o duradera, se siente mal (náuseas) o está mal (vómitos). Cualquiera de estas situaciones puede producir deshidratación grave. Su médico tendría que ponerle un tratamiento.
• tiene sensación de insensibilidad, hormigueo, ardor o disminución de las sensaciones en manos y pies.
• tiene algún problema de sangrado en el intestino o tiene cambios en el color de sus heces o dolor de estómago. Si el sangrado o el dolor es grave, su médico interrumpirá su tratamiento con JEVTANA. Esto es porque JEVTANA podría incrementar el riesgo de sangrado o desarrollo de perforaciones en la pared intestinal.
• tiene problemas de riñón.
• aparecen problemas de hígado durante el tratamiento.
• nota que el volumen de su orina aumenta o disminuye significativamente.
Si le sucede cualquiera de las circunstancias anteriores, informe a su médico inmediatamente. Su médico podría reducir la dosis de JEVTANA o interrumpir el tratamiento.
Uso de JEVTANA con otros medicamentos
Informe a su médico, farmacéutico o enfermera si está utilizando o ha utilizado recientemente otros medicamentos, incluso los adquiridos sin receta. Esto se debe a que algunos medicamentos pueden afectar la eficacia de JEVTANA o JEVTANA puede afectar la eficacia de otros medicamentos. Estos medicamentos incluyen los siguientes:
- ketoconazol, rifampicina (para infecciones);
- carbamazepina, fenobarbital o fenitoína (para convulsiones);
- Hierba de San Juan o hipérico (Hypericum perforatum) (planta medicinal utilizada para tratar la
depresión y otros problemas);
- estatinas (tales como simvastatina, lovastatina, atorvastatina, rosuvastatina, o pravastatina) (para
reducir el colesterol en su sangre);
- valsartan (para la hipertensión);
- repaglinida
(para la diabetes).
Mientras esté en tratamiento con JEVTANA, consulte con su médico antes de vacunarse.
Embarazo, lactancia y fertilidad
JEVTANA no se debe usar en mujeres embarazadas o en edad fértil que no utilicen anticonceptivos. JEVTANA no se debe utilizar durante la lactancia.
Use preservativos en sus relaciones sexuales si su pareja está o pudiera estar embarazada. JEVTANA podría estar presente en su semen y puede afectar al feto. Se recomienda no engendrar un hijo durante y hasta 6 meses después del tratamiento y solicitar información sobre la conservación del esperma antes del tratamiento, ya que JEVTANA podría alterar la fertilidad masculina.
Conducción y uso de máquinas
Durante el tratamiento con este medicamento podría sentirse cansado o mareado. Si esto sucede, no conduzca ni use herramientas o máquinas hasta que se sienta mejor.
JEVTANA contiene etanol (alcohol)
Este medicamento contiene un 15% v/v de etanol, equivalente a 14 ml de cerveza o 6 ml de vino. Este medicamento podría ser perjudicial para las personas alcohólicas.
Debe tenerse en cuenta en personas pertenecientes a grupos de alto riesgo, como pacientes con problemas de hígado, o epilepsia.
3. Cómo usar JEVTANA
Instrucciones de uso
Antes de recibir JEVTANA le administrarán medicamentos antialérgicos para reducir el riesgo de
reacciones alérgicas.
• JEVTANA será administrado por un médico o una enfermera.
• JEVTANA debe prepararse (diluirse) antes de administrarse. Con este prospecto se proporciona información práctica para la manipulación y administración de JEVTANA para médicos, enfermeras y farmacéuticos.
• JEVTANA se administrará en el hospital mediante un gotero (perfusión) en una de sus venas (vía intravenosa) durante aproximadamente 1 hora.
• Como parte de su tratamiento, tomará también un medicamento corticosteroide (prednisona o prednisolona) por vía oral todos los días.
Cuánto y con qué frecuencia se administra
• La dosis habitual depende de su área de superficie corporal. Su médico calculará su área de superficie corporal en metros cuadrados (m2) y decidirá la dosis que debe recibir.
• Habitualmente recibirá una perfusión cada 3 semanas.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o
enfermera.
4. Posibles efectos adversos
Al igual que todos los medicamentos, JEVTANA puede producir efectos adversos, aunque no todas las personas los sufran. Su médico comentará esto con usted y le explicará los riesgos y los beneficios potenciales de su tratamiento.
Acuda inmediatamente al médico si nota cualquiera de los siguientes efectos adversos:
fiebre (temperatura alta). Esto es muy frecuente (podría afectar a más de 1 de cada 10 personas).
• pérdida grave de fluidos corporales (deshidratación). Esto es frecuente (podría afectar hasta 1 de cada 10 personas). Esto puede ocurrir si tiene diarrea grave o duradera, o fiebre, o si ha estado vomitando.
• dolor de estómago grave o dolor de estómago que no se resuelve. Esto puede suceder si usted tiene una perforación en el estómago, esófago, intestino (perforación gastrointestinal). Esto puede causar la muerte.
Si le sucede alguna de las circunstancias anteriores, comuníquelo inmediatamente a su médico.
Otros efectos adversos incluyen:
Muy frecuentes (pueden afectar a más de 1de cada 10 personas):
• reducción del número de células sanguíneas rojas (anemia), o blancas (que son importantes para combatir las infecciones)
• reducción del número de plaquetas (lo cual resulta en un aumento del riesgo de tener hemorragias)
• pérdida de apetito (anorexia)
• alteración del gusto
• respiración entrecortada
• tos
• molestias de estómago, incluyendo náuseas, vómitos, diarrea o estreñimiento
• dolor abdominal
• pérdida de cabello a corto plazo (en la mayoría de los casos el pelo vuelve a crecer con normalidad)
• dolor de espalda
• dolor de las articulaciones
• sangre en la orina
• cansancio, debilidad o falta de energía.
Frecuentes (pueden afectar hasta 1de cada 10 personas):
• infección del tracto urinario
• escasez de glóbulos blancos asociada con fiebre e infecciones
• sensación de insensibilidad, hormigueo, ardor o disminución de las sensaciones en manos y pies
• mareo
• dolor de cabeza
• aumento o disminución de la tensión arterial
• malestar de estómago, ardor de estómago o eructos
• dolor de estómago
• hemorroides
• espasmos musculares
• orinar con frecuencia o con dolor
• incontinencia urinaria
• problemas o alteración de los riñones
• úlceras en la boca o en los labios
• infecciones o riesgo de infecciones
• nivel de azúcar en sangre elevado
• nivel de potasio en sangre bajo
• confusión mental
• sensación de ansiedad
• sensación rara o pérdida de sensación o dolor en manos y pies
• zumbidos en los oídos
• problemas de equilibrio
• latidos rápidos o irregulares del corazón
• coágulos de sangre en las piernas
• sensación de calor o sofoco en la piel
• dolor de boca o garganta
• hemorragia rectal
• piel enrojecida
• molestias, trastornos o dolores musculares
• inflamación de pies o piernas
• escalofríos.
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
• enfermedad pulmonar intersticial (inflamación de los pulmones causando tos y dificultad para respirar).
Comunicación de efectos adversos:
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de JEVTANA
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase y en la etiqueta de los viales después de CAD. La fecha de caducidad es el último día del mes que se indica.
No conservar a temperatura superior a 30°C. No refrigerar.
En la sección “INFORMACIÓN PRÁCTICA PARA MÉDICOS O PROFESIONALES SANITARIOS SOBRE LA PREPARACIÓN, ADMINISTRACIÓN Y MANIPULACIÓN DE JEVTANA” se incluye información sobre la conservación y el tiempo de uso de JEVTANA, una vez que se ha diluido y está listo para usar.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local. Estas medidas ayudarán a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de JEVTANA
El principio activo es cabazitaxel. Un ml de concentrado contiene 40 mg de cabazitaxel. Cada vial de concentrado contiene 60 mg de cabazitaxel.
Los demás componentes son polisorbato 80 y ácido cítrico en el concentrado, y etanol 96% y agua para preparaciones inyectables en el disolvente (ver sección 2 “JEVTANA contiene etanol (alcohol)”). Nota: tanto el vial del concentrado de JEVTANA 60 mg/1,5 ml (volumen de llenado: 73,2 mg de cabazitaxel/1,83 ml) como el del vial de disolvente (volumen de llenado: 5,67 ml) contienen un sobrellenado para compensar la pérdida de líquido durante la preparación. Este sobrellenado asegura que después de la dilución con el contenido COMPLETO del disolvente proporcionado, haya una solución conteniendo 10 mg/ml de cabazitaxel.
Aspecto del producto y contenido del envase
JEVTANA es un concentrado y disolvente para solución para perfusión (concentrado estéril).
El concentrado es una solución oleosa transparente, de color amarillo a amarillento-marronáceo.
El disolvente es una solución transparente e incolora.
Un envase de JEVTANA contiene:
- Un vial de un solo uso de vidrio transparente, cerrado con un tapón de caucho clorobutilo de color gris, sellado con una cápsula de aluminio, cubierta con un tapón expulsor flip-off de plástico de color verde claro, conteniendo 1,5 ml (volumen nominal) de concentrado.
- Un vial de un solo uso de vidrio transparente, cerrado con un tapón de caucho clorobutilo de color gris, sellado con una cápsula de aluminio dorada, cubierta con un tapón expulsor flip-off de plástico incoloro, conteniendo 4,5 ml (volumen nominal) de disolvente.
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización
sanofi-aventis groupe 54, rue La Boétie F - 75008 París Francia
Responsable de la fabricación
Sanofi-Aventis Deutschland GmbH Industriepark Hochst 65926 Frankfurt am Main Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Sanofi Belgium Tél.: +32 (0)2 710 54 00
Bt^rapna
sanofi-aventis Bulgaria EOOD Tél.: +359 (0)2 970 53 00
Ceská republika
sanofi-aventis, s.r.o.
Tél.: +420 233 086 111
Danmark
sanofi-aventis Denmark A/S Tél.: +45 45 16 70 00
Deutschland
sanofi-aventis Deutschland GmbH Tél.: +49 (0)180 2 222010
Eesti
sanofi-aventis Estonia OÜ Tél.: +372 627 34 88
EXláóa
sanofi-aventis AEBE Tél.: +30 210 900 16 00
España
sanofi-aventis, S.A.
Luxembourg/Luxemburg
Sanofi Belgium
Tél.: +32 (0)2 710 54 00 (Bélgica) Magyarország
sanofi-aventis zrt., Magyarország Tél.: +36 1 505 0050
Malta
Sanofi Malta Ltd.
Tél.: 356 21493022
Nederland
sanofi-aventis Netherlands, B.V.
Tél.: +31 (0)182 557 755
Norge
sanofi-aventis Norge AS Tél.: +47 67 10 71 00
Osterreich
sanofi-aventis GmbH Tél.: +43 1 80 185 - 0
Polska
sanofi-aventis Sp. z.o.o.
Tél.: +48 22 280 00 00
Portugal
Sanofi - Productos Farmacéuticos, Lda.
Romania
Sanofi Romania SRL Tél.: +40 (0)21 317 31 36
France
sanofi-aventis France Tél.: 0 800 222 555
Llamar después al extranjero: +33 1 57 63 23 23 Hrvatska
Sanofi-aventis Croatia d.o.o.
Tel. +385 1 600 34 00
Ireland Slovenija
sanofi-aventis Ireland Ltd. T/A SANOFI sanofi-aventis d.o.o.
Tél.: +353 (0) 1 403 56 00
Ísland Vistor hf.
Tél.: +354 535 7000 Italia
Sanofi S.p.A.
Tél.: +39.800.536389
Kúrcpog
sanofi-aventis Cyprus Ltd. Tél.: +357 22 871600
Latvija
sanofi-aventis Latvia SIA Tél.: +371 67 33 24 51
Tél.: +34 93 485 94 00
Tél.: +351 21 35 89 400
Tél.: +386 1 560 48 00
Slovenská republika
sanofi-aventis Pharma Slovakia s.r.o. Tél.: +421 2 33 100 100
Suomi/Finland
Sanofi Oy
Tél.: +358 (0) 201 200 300
Sverige Sanofi AB
Tél.: +46 (0)8 634 50 00
United Kingdom
Sanofi
Tél.: +44 (0) 845 372 7101
Lietuva
UAB »SANOFI-AVENTIS LIETUVA «
Tél.: +370 5 2755224
Fecha de la última revisión de este prospecto:.
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www. ema.europa.eu.
La siguiente información está destinada únicamente a profesionales sanitarios.
INFORMACIÓN PRÁCTICA PARA MÉDICOS O PROFESIONALES SANITARIOS SOBRE LA PREPARACIÓN, ADMINISTRACIÓN Y MANIPULACIÓN DE JEVTANA 60 mg CONCENTRADO Y DISOLVENTE PARA SOLUCIÓN PARA PERFUSIÓN
Esta información complementa las secciones 3 y 5 para el usuario.
Es importante que lea el contenido completo de este procedimiento antes de preparar la solución para perfusión.
Incompatibilidades
Este medicamento no debe mezclarse con otros medicamentos excepto los utilizados para las diluciones.
Periodo de validez y precauciones especiales de conservación
Para el envase de JEVTANA 60 mg concentrado y disolvente No conservar a temperatura superior a 30°C.
No refrigerar.
Después de la apertura del vial
Los viales de concentrado y disolvente deben utilizarse inmediatamente. Si no se utilizan inmediatamente, el tiempo y las condiciones de conservación son responsabilidad del usuario. Desde un punto de vista microbiológico, el proceso de dilución en dos etapas debe realizarse en condiciones controladas y asépticas (ver a continuación “Precauciones de preparación y administración”).
Después de la dilución inicial de JEVTANA 60 mg concentrado con el contenido completo del vial de disolvente:
se ha demostrado la estabilidad química y física en uso durante 1 hora a temperatura ambiente. Después de la dilución final en la bolsa/botella de perfusión
Se ha demostrado la estabilidad química y física de la solución de perfusión durante 8 horas a temperatura ambiente (15°C - 30°C) incluyendo 1 hora de tiempo de perfusión y durante 48 horas en nevera incluyendo la hora de tiempo de perfusión.
Desde un punto de vista microbiológico, la solución de perfusión debe utilizarse inmediatamente. Si no se utiliza inmediatamente, los tiempos y las condiciones de conservación son responsabilidad del usuario y normalmente no deberían ser más de 24 horas a 2°C - 8°C, a menos que la dilución se haya realizado en condiciones asépticas controladas y validadas.
Precauciones de preparación y administración
Al igual que otros agentes antineoplásicos, debe actuarse con precaución durante la preparación y administración de las soluciones de JEVTANA, teniendo en cuenta el uso de dispositivos de seguridad, equipo de protección personal (por ej. guantes) y procedimientos de preparación.
Si en cualquiera de las etapas de preparación, JEVTANA entrara en contacto con la piel, lavar inmediatamente y minuciosamente con agua y jabón. Si entrara en contacto con membranas mucosas, lavar inmediata y minuciosamente con agua.
JEVTANA sólo debe ser preparado y administrado por personal entrenado en el manejo de agentes citotóxicos. Las trabajadoras embarazadas no deben manipularlo.
Diluir siempre el concentrado para solución para perfusión con el disolvente completo que se proporciona antes de añadirlo a las soluciones de perfusión.
Lea detenidamente TODA esta sección antes de mezclar y diluir. JEVTANA requiere DOS diluciones antes de la administración. Siga las instrucciones de preparación que se proporcionan a continuación.
Nota: tanto el vial del concentrado de JEVTANA 60 mg/1,5 ml (volumen de llenado: 73,2 mg de cabazitaxel/1,83 ml) como el vial de disolvente (volumen de llenado: 5,67 ml) contienen un sobrellenado para compensar la pérdida de líquido durante la preparación. Este sobrellenado asegura que después de la dilución con el contenido COMPLETO del disolvente proporcionado, haya una solución conteniendo 10 mg/ml de cabazitaxel.
Para preparar la solución para perfusión, el siguiente proceso de dilución en dos etapas debe realizarse de forma aséptica.
Etapa 1: dilución inicial del concentrado de solución para perfusión con el disolvente proporcionado.
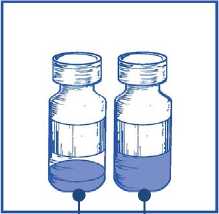
Etapa 1.1
Inspeccionar el vial de concentrado y el disolvente proporcionado. La solución de concentrado y de disolvente deben ser transparentes.
|
Vial de concentrado |
Vial de disolvente |
|
(60mg - 1,5 ml) |

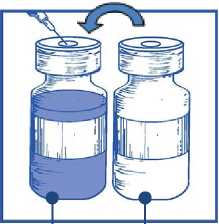
Etapa 1.2
Utilizando una jeringa provista de una aguja fija, extraer de forma aséptica el contenido completo del disolvente proporcionado invirtiendo parcialmente el vial.
Etapa 1.3
Inyectar el contenido completo en el correspondiente vial de concentrado.
Para limitar todo lo posible la formación de espuma al inyectar el disolvente, dirigir la aguja hacia la pared interior del vial de solución de concentrado e inyectar lentamente.
Una vez reconstituido, la solución resultante
|
Mezcla concentrado-disolvente |
Vial de disolvente |
|
10 mg/ml |
Sacar la jeringa y la aguja y mezclar manualmente y suavemente, mediante inversiones repetidas, hasta que se obtenga una solución transparente y homogénea. Se pueden tardar unos 45 segundos.
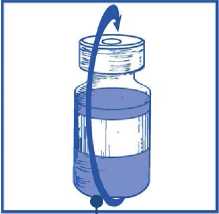
Mezcla
concentrado-disolvente 10 mg/ml
Etapa 1.5
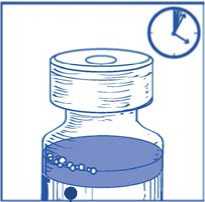
Dejar reposar la solución durante aproximadamente 5 minutos y a continuación comprobar que la solución es homogénea y transparente.
Es normal que persista la espuma pasado este tiempo.
Mezcla concentrado-disolvente 10 mg/ml
Esta mezcla concentrado-disolvente resultante contiene 10 mg/ml de cabazitaxel (al menos 6 ml de volumen liberado). La segunda dilución se debe realizar inmediatamente (antes de 1 hora) como se detalla en la Etapa 2.
Podría ser necesario más de un vial de mezcla concentrado-disolvente para administrar la dosis prescrita.
Etapa 2: segunda dilución (final) para perfusión
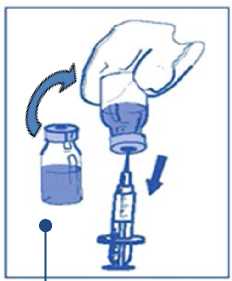
Mezcla concentrado-disolvente 10 mg/ml
Etapa 2.1
De forma aséptica extraer la cantidad requerida de mezcla concentrado-disolvente (10 mg/ml de cabazitaxel), con una jeringa graduada provista de una aguja fija. Como ejemplo, una dosis de 45 mg de JEVTANA requeriría 4,5 ml de la mezcla de concentrado-disolvente preparada en la Etapa 1.
Como puede seguir habiendo espuma en la pared del vial de esta solución, después de la preparación descrita en la Etapa 1, es preferible situar la aguja de la jeringa en la mitad del contenido durante la extracción.
Inyectar en un envase estéril sin PVC de solución de glucosa al 5% o solución de cloruro sódico 9 mg/ml (0,9%) para perfusión. La concentración de la solución para perfusión debe estar entre 0,10 mg/ml y 0,26 mg/ml.
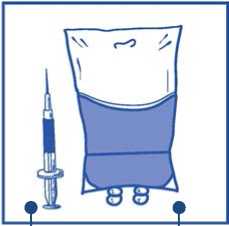
Cantidad requerida de mezcla concentrado-disolvente
Solución de glucosa al 5% o solución de cloruro sódico 9 mg/ml (0,9%) para perfusión

Etapa 2.3
Sacar la jeringa y mezclar el contenido de la bolsa o botella de perfusión manualmente, mediante movimiento de balanceo.
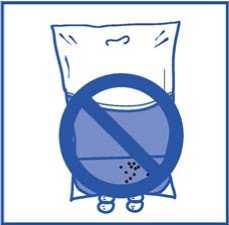
Etapa 2.4
Al igual que todos los productos parenterales, la solución de perfusión resultante debe inspeccionarse visualmente antes de usarla.
Como la solución de perfusión está sobresaturada, puede cristalizar con el tiempo. En este caso, no se debe utilizar la solución y debe eliminarse.
La solución para perfusión debe utilizarse inmediatamente. No obstante, el tiempo de conservación en uso puede ser más largo bajo las condiciones específicas mencionadas en la sección Periodo de validez y precauciones especiales de conservación.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
Método de administración
JEVTANA se administra en perfusión durante 1 hora.
Se recomienda el uso de un filtro en línea de 0,22 micrómetros de tamaño de poro nominal (también denominado 0,2 micrómetros) durante la administración.
No deben utilizarse envases de perfusión de PVC o sets de perfusión de poliuretano para la preparación y administración de la solución para perfusión.
43