Imnovid 4 Mg Capsulas Duras
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la identificación de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Imnovid 1 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula dura contiene 1 mg de pomalidomida.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura.
Imnovid 1 mg cápsula dura: tapa opaca de color azul oscuro y cuerpo opaco de color amarillo marcadas con “POML” en tinta blanca y “1 mg” en tinta negra, de tamaño 4, cápsulas de gelatina dura.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Imnovid en combinación con dexametasona está indicado en el tratamiento de los pacientes adultos con mieloma múltiple resistente al tratamiento o recidivante que hayan recibido al menos dos tratamientos previos, incluyendo lenalidomida y bortezomib, y que hayan experimentado una progresión de la enfermedad en el último tratamiento.
4.2 Posología y forma de administración
El tratamiento debe iniciarse y monitorizarse bajo la supervisión de médicos con experiencia en el tratamiento del mieloma múltiple.
Posología
La dosis inicial recomendada es de 4 mg de Imnovid una vez al día por vía oral, en los días del 1 al 21 de ciclos repetidos de 28 días. La dosis recomendada de dexametasona es de 40 mg por vía oral una vez al día, en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
La posología se mantiene o modifica en función de los resultados clínicos y de laboratorio.
El tratamiento debe suspenderse si existe evidencia de progresión de la enfermedad.
Modificación o interrupción de la dosis de _pomalidomida
Las instrucciones para la interrupción y reducción de la dosis de pomalidomida relacionadas con reacciones adversas hematológicas se indican en la siguiente tabla:
|
Toxicidad |
Modificación de la dosis |
|
NeutroDenia • RAN* <0,5 x 109/l o neutropenia febril (fiebre >38,5°C y RAN <1 x 109/l) |
Interrumpir el tratamiento con pomalidomida, control semanal del hemograma completo. |
|
• RAN vuelve a >1 x 109/1 |
Reanudar el tratamiento con 3 mg de pomalidomida al día. |
|
• Con cada disminución posterior a <0,5 x 1071 |
Interrumpir el tratamiento con pomalidomida |
|
• RAN vuelve a >1 x 109/l |
Reanudar el tratamiento con 1 mg menos de pomalidomida que la dosis previa |
|
T rombocitopenia • Recuento de plaquetas <25 x 109/l |
Interrumpir el tratamiento con pomalidomida, control semanal del hemograma completo |
|
• Recuento de plaquetas vuelve a >50 x 109/l |
Reanudar el tratamiento con 3 mg de pomalidomida al día |
|
• Con cada disminución posterior a <25 x 1071 |
Interrumpir el tratamiento con pomalidomida |
|
• Recuento de plaquetas vuelve a >50 x 109/l |
Reanudar el tratamiento con 1 mg menos de pomalidomida que la dosis previa |
*RAN - Recuento absoluto de neutrófilos
Para iniciar un nuevo ciclo de pomalidomida, el recuento de neutrófilos debe ser >1 x 109/l y el recuento de plaquetas debe ser >50 x 109/l.
En caso de neutropenia, el médico debe considerar el uso de factores de crecimiento.
En el caso de otras reacciones adversas de grado 3 o 4 relacionadas con pomalidomida, el médico debe considerar interrumpir el tratamiento y reanudarlo con un 1 mg menos que la dosis previa una vez que se haya disminuido la reacción adversa a un grado inferior o igual a 2.
Si la reacción adversa ocurre tras disminuciones de la dosis a 1 mg, entonces debe suspenderse el tratamiento con este medicamento.
Se debe considerar la interrupción o suspensión de pomalidomida en caso de exantema de grado 2-3. Se debe suspender el tratamiento con pomalidomida en caso de angioedema, exantema de grado 4 y exantema ampolloso o exfoliativo, y no se debe reanudar una vez suspendido por estas reacciones.
Si se administran inhibidores potentes del CYP1A2 (p. ej., ciprofloxacino, enoxacino y fluvoxamina) de forma concomitante con pomalidomida, se debe reducir la dosis de pomalidomida en un 50 %.
|
Toxicidad |
Modificación de la dosis |
|
Dispepsia = grado 1-2 Dispepsia > grado 3 |
Mantener la dosis y tratar con antihistamínicos H2 o equivalentes. Reducir la dosis en un nivel de dosis si los síntomas persisten. Interrumpir la administración hasta que se controlen los síntomas. Añadir antihistamínicos H2 o equivalentes y reducir la dosis en un nivel cuando se reanude su administración. |
|
Edema > grado 3 |
Usar diuréticos según sea necesario y reducir la dosis en un nivel de dosis. |
|
Confusión o cambios en el estado de ánimo > grado 2 |
Interrumpir la administración hasta que desaparezcan los síntomas. Cuando se reanude su administración reducir la dosis en un nivel de dosis. |
|
Debilidad muscular > grado 2 |
Interrumpir la administración hasta que la debilidad muscular sea < grado 1. Reiniciar la dosis con una reducción de un nivel. |
|
Hiperglucemia > grado 3 |
Reducir la dosis en un nivel de dosis. Tratar con insulina o hipoglucemiantes orales según sea necesario. |
|
Pancreatitis aguda |
Suspensión de dexametasona del régimen de tratamiento del paciente. |
|
Otros efectos adversos relacionados con dexametasona > grado 3 |
Interrumpir la administración de dexametasona hasta que los efectos adversos sean de grado < 2. Reanudar su administración con una reducción de un nivel de la dosis. |
Niveles de reducción de la dosis de dexametasona:
Niveles de reducción de la dosis (<75 años): dosis inicial 40 mg; nivel de dosis -1 20 mg; nivel de dosis -2 10 mg en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
Niveles de reducción de la dosis (>75 años): dosis inicial 20 mg; nivel de dosis -1 12 mg; nivel de dosis -2 8 mg en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
Si la recuperación de las toxicidades tarda más de 14 días, la dosis de dexametasona se reducirá en un nivel de dosis.
Poblaciones especiales Población _ pediátrica
No hay un uso relevante de Imnovid en niños de 0 a 17 años para la indicación del mieloma múltiple.
Pacientes de edad avanzada
No se requiere ningún ajuste de dosis de pomalidomida. En pacientes de >75 años , la dosis inicial de dexametasona es de 20 mg una vez al día en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
Insuficiencia renal
No se requiere ningún ajuste de dosis de pomalidomida en los pacientes con insuficiencia renal. Los días de hemodiálisis, los pacientes deben tomar la dosis de pomalidomida después de la hemodiálisis.
Insuficiencia hepática
Los pacientes con una concentración sérica de bilirrubina total >2,0 mg/dl se excluyeron de los estudios clínicos. La insuficiencia hepática tiene un efecto modesto sobre la farmacocinética de pomalidomida (ver sección 5.2). No se requiere ajustar la dosis inicial de pomalidomida en pacientes con insuficiencia hepática según definen los criterios de Child-Pugh. Sin embargo, los pacientes con insuficiencia hepática deben ser monitorizados cuidadosamente por si presentan reacciones adversas y se debe reducir la dosis o suspender la administración de pomalidomida según sea necesario.
Forma de administración Vía oral.
Imnovid debe tomarse a la misma hora cada día. Las cápsulas no deben abrirse, romperse ni masticarse (ver sección 6.6). Este medicamento debe tomarse entero, preferiblemente con agua, con o sin alimentos. Si el paciente olvida tomar una dosis de Imnovid un día, el paciente debe entonces tomar la próxima dosis al día siguiente a la hora habitual. Los pacientes no deben ajustar la dosis para compensar una dosis olvidada en días anteriores.
Se recomienda presionar solo en un extremo de la cápsula para sacarla del blíster y reducir así el riesgo de deformación o rotura de la cápsula.
4.3 Contraindicaciones
- Embarazo.
- Mujeres con capacidad de gestación, a menos que se cumplan todas las condiciones del Programa de Prevención de Embarazo (ver secciones 4.4 y 4.6).
- Pacientes varones incapaces de seguir o cumplir las medidas anticonceptivas requeridas (ver sección 4.4).
- Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo Teratogenicidad
Pomalidomida no debe tomarse durante el embarazo ya que es esperable un efecto teratogénico. Pomalidomida está relacionada estructuralmente con talidomida. Talidomida es un teratógeno conocido en humanos, que causa defectos congénitos de nacimiento graves que pueden poner en peligro la vida del niño. Pomalidomida tiene un efecto teratogénico en ratas y conejos cuando se administra durante el periodo de mayor organogénesis (ver sección 5.3).
Todas las pacientes deben cumplir las condiciones del Programa de Prevención de Embarazo a menos que exista evidencia fiable de que la paciente no tiene capacidad de gestación.
Criterios para definir a las mujeres que no tienen capacidad de gestación
Se considera que una paciente o la pareja de un paciente varón no tiene capacidad de gestación si cumple por lo menos uno de los siguientes criterios:
• Edad >50 años y con amenorrea natural durante >1 año*.
• Insuficiencia ovárica prematura confirmada por un ginecólogo especialista.
• Salpingooforectomía bilateral o histerectomía previas.
• Genotipo XY, síndrome de Turner, agenesia uterina.
*La amenorrea que pueda aparecer después de un tratamiento oncológico o durante la lactancia no descarta la capacidad de gestación.
Asesoramiento
En mujeres con capacidad de gestación, pomalidomida está contraindicada a menos que la paciente cumpla todas las condiciones que se indican a continuación:
• Comprende el riesgo teratogénico esperado para el feto.
• Comprende la necesidad de utilizar métodos anticonceptivos eficaces, sin interrupción, desde 4 semanas antes de iniciar el tratamiento, durante la duración completa del mismo y 4 semanas después de finalizarlo.
• Incluso si una mujer con capacidad de gestación tiene amenorrea, debe seguir todos los consejos sobre anticoncepción eficaz.
• Debe ser capaz de cumplir las medidas anticonceptivas eficaces.
• Está informada y comprende las potenciales consecuencias del embarazo, y la necesidad de consultar rápidamente a un especialista si hay riesgo de embarazo.
• Comprende la necesidad de comenzar el tratamiento tan pronto como se le dispense pomalidomida y tras haber obtenido un resultado negativo en la prueba de embarazo.
• Comprende la necesidad de realizar pruebas de embarazo y acepta hacérselas cada 4 semanas, excepto en el caso de que se haya sometido previamente a una ligadura de trompas de eficacia confirmada.
• Confirma que comprende los peligros y las precauciones necesarias asociadas al uso de pomalidomida.
El médico prescriptor debe comprobar que, en el caso de las mujeres con capacidad de gestación:
• La paciente cumple las condiciones del Programa de Prevención de Embarazo, incluida la confirmación de que tiene un nivel de comprensión adecuado.
• La paciente ha aceptado las condiciones mencionadas anteriormente.
En el caso de pacientes varones que toman pomalidomida, los datos farmacocinéticos han demostrado que pomalidomida está presente en el semen humano. Como medida de precaución, todos los pacientes varones que tomen pomalidomida deben cumplir los siguientes requisitos:
• Comprende el riesgo teratogénico esperado si tiene relaciones sexuales con una mujer embarazada o con una mujer con capacidad de gestación.
• Comprende la necesidad del uso de preservativos si tiene relaciones sexuales con una mujer embarazada o con una mujer con capacidad de gestación que no utiliza métodos anticonceptivos eficaces, durante el tratamiento y durante los 7 días posteriores a la interrupción de la dosis y/o el cese del tratamiento. Los varones vasectomizados deben utilizar preservativos si tienen relaciones sexuales con una mujer embarazada o con una mujer con capacidad de gestación ya que pomalidomida puede estar presente en el semen aún en ausencia de espermatozoides.
• Comprende que si su pareja se queda embarazada mientras él está tomando pomalidomida o durante los 7 días posteriores a la suspensión del tratamiento con pomalidomida, debe informar inmediatamente a su médico y es recomendable derivar a su pareja a un médico especialista o con experiencia en teratología para su evaluación y asesoramiento.
Anticoncepción
Las mujeres con capacidad de gestación deben usar un método anticonceptivo eficaz desde 4 semanas antes del tratamiento, durante el tratamiento y hasta 4 semanas después del tratamiento con pomalidomida, e incluso en el caso de interrupción de la administración, a menos que la paciente se comprometa a mantener una abstinencia sexual absoluta y continua, que será confirmada mensualmente. Si la paciente no utiliza un método anticonceptivo eficaz, debe ser derivada a un profesional sanitario debidamente capacitado con objeto de que reciba asesoramiento para empezar a utilizar métodos anticonceptivos.
Los siguientes métodos pueden considerarse ejemplos de métodos anticonceptivos adecuados:
• Implante
• Sistema de liberación intrauterino de levonorgestrel
• Sistemas “depot” de liberación de acetato de medroxiprogesterona
• Ligadura de trompas
• Relaciones sexuales sólo con varones vasectomizados; la eficacia de la vasectomía debe confirmarse mediante dos análisis de semen negativos
• Inhibidores de la ovulación que contienen progestágeno solo (p. ej., desogestrel)
Debido al riesgo aumentado de tromboembolismo venoso (TEV) en pacientes con mieloma múltiple que toman pomalidomida y dexametasona, no se recomienda el uso concomitante de anticonceptivos orales combinados (ver también sección 4.5). Si una paciente está tomando anticonceptivos orales combinados, debe cambiar a uno de los métodos anticonceptivos eficaces enumerados anteriormente. El riesgo aumentado de tromboembolismo venoso se mantiene durante un periodo de 4 a 6 semanas después de suspender el tratamiento con anticonceptivos orales combinados. La eficacia de los anticonceptivos esteroideos puede verse reducida durante el tratamiento concomitante con dexametasona (ver sección 4.5).
Los implantes y los sistemas de liberación intrauterinos de levonorgestrel se asocian con un mayor riesgo de infección en el momento de la colocación y con hemorragia vaginal irregular. En especial en las pacientes con neutropenia debe considerarse el uso profiláctico de antibióticos.
La colocación de dispositivos intrauterinos de liberación de cobre no está recomendada, debido al potencial riesgo de infección en el momento de su colocación y a la pérdida de sangre menstrual, que pueden suponer un peligro para las pacientes con neutropenia grave o trombocitopenia grave.
Pruebas de embarazo
Las mujeres con capacidad de gestación deben efectuarse pruebas de embarazo con una sensibilidad mínima de 25 mUI/ml bajo supervisión médica y conforme a la práctica habitual, tal como se explica a continuación. Este requisito incluye a las mujeres con capacidad de gestación que practican una abstinencia sexual absoluta y continua. Idealmente, la prueba de embarazo, la prescripción y la dispensación deben realizarse el mismo día. Pomalidomida se debe dispensar a las mujeres con capacidad de gestación en un plazo de siete días tras la prescripción.
Antes de iniciar el tratamiento
Debe efectuarse una prueba de embarazo bajo supervisión médica durante la consulta, en el momento de prescribir pomalidomida o en los tres días anteriores a la visita al médico prescriptor, siempre que la paciente haya estado usando un método anticonceptivo eficaz durante al menos 4 semanas. La prueba debe garantizar que la paciente no esté embarazada cuando inicie el tratamiento con pomalidomida.
Seguimiento y _finalización del tratamiento
Se debe repetir cada 4 semanas una prueba de embarazo bajo supervisión médica, y realizar otra 4 semanas después de la finalización del tratamiento, excepto en el caso de que la paciente se haya sometido a una ligadura de trompas de eficacia confirmada. Estas pruebas de embarazo deben efectuarse el mismo día de la consulta en que se prescriba el medicamento o en los tres días anteriores a la visita al médico prescriptor.
Varones
Pomalidomida está presente en el semen humano durante el tratamiento. Como medida de precaución, y teniendo en cuenta las poblaciones especiales con un tiempo de eliminación potencialmente prolongado, como la insuficiencia renal, todos los pacientes varones que tomen pomalidomida, incluyendo a aquellos que se hayan sometido a una vasectomía, deben usar preservativos durante todo el tratamiento, durante la interrupción de la administración y hasta 7 días después del final del tratamiento, si su pareja está embarazada o tiene capacidad de gestación y no está usando ningún método anticonceptivo.
Los pacientes varones no deben donar semen o esperma durante el tratamiento (periodos de interrupción de la dosis incluidos) ni en el plazo de 7 días después de la suspensión del tratamiento con pomalidomida.
Precauciones adicionales
Se debe indicar a los pacientes que no den nunca este medicamento a otra persona y que devuelvan las cápsulas sin usar al farmacéutico al final del tratamiento.
Los pacientes no deben donar sangre, semen o esperma durante el tratamiento (periodos de interrupción de la dosis incluidos) ni en el plazo de 7 días después de la suspensión del tratamiento con pomalidomida.
Material educativo, restricciones de prescripción y dispensación
Con objeto de ayudar a los pacientes a evitar la exposición fetal a pomalidomida, el Titular de la Autorización de Comercialización distribuirá material educativo a los profesionales sanitarios, destinado a reforzar las advertencias acerca de la teratogenicidad esperada de pomalidomida y a proporcionar asesoramiento sobre anticoncepción antes de iniciar el tratamiento y sobre la necesidad de realizar pruebas de embarazo. El médico debe informar a la paciente acerca del riesgo teratogénico esperado y de las estrictas medidas de prevención de embarazo, especificadas en el Programa de Prevención de Embarazo así como proporcionar a la paciente un folleto informativo adecuado, una tarjeta de paciente y/o una herramienta equivalente conforme al sistema nacional implementado de tarjeta del paciente. En colaboración con cada autoridad nacional competente, se ha implementado un sistema de distribución nacional controlada. El sistema de distribución controlada incluye el uso de una tarjeta del paciente y/o un herramienta equivalente para el control de la prescripción y/o dispensación, así como la recogida de datos detallados en relación con la indicación terapéutica, para monitorizar el uso en una indicación no autorizada dentro del territorio nacional. Idealmente, la prueba de embarazo, la prescripción y la dispensación deben realizarse el mismo día. La dispensación de pomalidomida en mujeres con capacidad de gestación debe hacerse dentro de los 7 días de prescripción y tras haber obtenido un resultado negativo en la prueba de embarazo supervisada por un médico. Las prescripciones a mujeres con capacidad de gestación pueden tener una caducidad de 4 semanas y las prescripciones para el resto de pacientes pueden tener una caducidad de 12 semanas.
Eventos hematológicos
La reacción adversa hematológica de Grado 3 o 4 notificada con mayor frecuencia en pacientes con mieloma múltiple en recaída/refractario fue la neutropenia, seguido de anemia y trombocitopenia. Se debe monitorizar a los pacientes en busca de posibles reacciones adversas hematológicas, especialmente neutropenia. Se debe advertir a los pacientes que informen rápidamente acerca de los episodios febriles que presenten. Los médicos deben estar atentos a los signos de hemorragia en los pacientes, incluyendo epistaxis, especialmente en el caso de medicación concomitante conocida por aumentar el riesgo de sangrado (ver sección 4.8). Debe efectuarse a los pacientes un hemograma completo semanal en el momento basal, durante las primeras 8 semanas y después mensualmente. Puede ser necesaria una modificación de la dosis (ver sección 4.2). Los pacientes pueden requerir el uso de hemoderivados y/o factores de crecimiento.
Eventos tromboembólicos
Se han observado eventos tromboembólicos venosos (predominantemente trombosis venosa profunda y embolia pulmonar) y eventos trombóticos arteriales (infarto de miocardio y accidente cerebrovascular) en pacientes tratados con pomalidomida en combinación con dexametasona. Los pacientes con factores de riesgo conocidos de tromboembolismo, incluida una trombosis previa, deben estar estrechamente monitorizados. Se deben tomar medidas para intentar minimizar todos los factores de riesgo modificables (p. ej. tabaquismo, hipertensión e hiperlipidemia). Se aconseja a médicos y pacientes que estén atentos a los signos y síntomas de tromboembolismo. Se debe advertir a los pacientes que soliciten atención médica si presentan síntomas como respiración entrecortada, dolor torácico o edema de las extremidades. Es recomendable el uso de terapia anticoagulante (si no está contraindicada), como el ácido acetilsalicílico, warfarina, heparina o clopidogrel, especialmente en pacientes con factores de riesgo trombótico adicionales. Después de una cuidadosa evaluación de los factores de riesgo subyacentes del paciente individual, se debe tomar una decisión respecto al uso de medidas profilácticas. En los estudios clínicos los pacientes recibieron ácido aceltilsalicílico profiláctico o terapia antitrombótica alternativa. El uso de agentes eritropoyéticos conlleva un riesgo de eventos trombóticos incluyendo tromboembolismo. Por lo tanto, deben emplearse con precaución los agentes eritropoyéticos así como otros agentes que puedan aumentar el riesgo de eventos tromboembólicos.
Neuropatía periférica
Se excluyó de los estudios clínicos con pomalidomida a los pacientes con neuropatía periférica en curso de Grado >2. Se deben adoptar las precauciones adecuadas al considerar el tratamiento de estos pacientes con pomalidomida.
Disfunción cardíaca significativa
Se excluyó de los estudios clínicos con pomalidomida a los pacientes con una disfunción cardíaca significativa (insuficiencia cardíaca congestiva [Clase III o IV de la NY Heart Association]; infarto de miocardio dentro de los 12 meses desde el inicio del estudio; angina de pecho inestable o mal controlada). Se han notificado acontecimientos de insuficiencia cardiaca, que incluyen insuficiencia cardiaca congestiva y edema pulmonar (ver sección 4.8), especialmente en pacientes con enfermedad cardiaca preexistente o factores de riesgo cardiacos. Se deben adoptar las precauciones adecuadas al considerar el tratamiento de estos pacientes con pomalidomida, incluido el control periódico para detectar la presencia de signos y síntomas de insuficiencia cardiaca.
Síndrome de lisis tumoral
Puede producirse un síndrome de lisis tumoral. Los pacientes con mayor riesgo de sufrir un síndrome de lisis tumoral son aquellos que presentan una carga tumoral elevada antes del tratamiento. Se debe monitorizar estrechamente a estos pacientes y se deben adoptar las precauciones adecuadas.
Segundas neoplasias malignas primarias
Se han notificado segundas neoplasias malignas primarias, como cáncer de piel no melanoma, en pacientes en tratamiento con pomalidomida (ver sección 4.8). Los médicos deben evaluar cuidadosamente a los pacientes antes y durante el tratamiento, utilizando pruebas estándar de detección de cáncer por si aparecieran segundas neoplasias malignas primarias e instaurar el tratamiento indicado.
Reacciones alérgicas
Se han notificado angioedema y reacciones dermatológicas graves (ver sección 4.8). Se excluyó de los estudios clínicos a los pacientes con antecedentes de reacciones alérgicas graves asociadas a talidomida o lenalidomida. Estos pacientes pueden presentar un mayor riesgo de reacciones de hipersensibilidad y no deben tomar pomalidomida. Se debe considerar la interrupción o suspensión de pomalidomida si se presenta exantema de Grado 2 o 3. Se debe suspender definitivamente el tratamiento con pomalidomida si se presenta angioedema, exantema de Grado 4 y exantema ampolloso o exfoliativo.
Mareo y confusión
Se han notificado mareo y estados de confusión con pomalidomida. Los pacientes deben evitar las situaciones en que el mareo o la confusión puedan representar un problema y no tomar otros medicamentos que puedan causar mareo o confusión sin solicitar antes consejo médico.
Enfermedad pulmonar intersticial (EPI)
Se han observado EPI y acontecimientos asociados que incluyen casos de neumonitis con pomalidomida. Se debe realizar una evaluación cuidadosa de los pacientes que presenten un inicio repentino o empeoramiento inexplicable de los síntomas pulmonares para descartar la EPI. Se debe interrumpir la administración de pomalidomida durante la investigación de estos síntomas Y, si se confirma la EPI, debe iniciarse un tratamiento adecuado. Únicamente se debe reanudar pomalidomida después de una evaluación exhaustiva de los beneficios y los riesgos.
Trastornos hepáticos
Se han observado concentraciones notablemente elevadas de alanina aminotransferasa y bilirrubina en los pacientes tratados con pomalidomida (ver sección 4.8). Se han notificado también casos de hepatitis que provocaron la suspensión de pomalidomida. Se recomienda controlar periódicamente la función hepática durante los primeros 6 meses de tratamiento con pomalidomida y, posteriormente, cuando esté clínicamente indicado.
Infecciones
Se han notificado rara vez casos de reactivación de la hepatitis B en pacientes en tratamiento con pomalidomida combinado con dexametasona que habían sido previamente infectados por el virus de la hepatitis B (VHB). Algunos de estos casos han evolucionado a fallo hepático agudo, dando lugar a la suspensión de pomalidomida. Se debe determinar el estado del virus de la hepatitis B antes de iniciar el tratamiento con pomalidomida. Se recomienda que los pacientes que den un resultado positivo en la prueba de infección por VHB se pongan en contacto con un médico con experiencia en el tratamiento de la hepatitis B. Se debe tener precaución cuando se administre pomalidomida en combinación con dexametasona en pacientes previamente infectados por el VHB, incluidos los pacientes anti-Bc positivos pero con HBsAg negativos. Se debe monitorizar estrechamente a estos pacientes para detectar signos y síntomas de infección activa por el VHB durante todo el tratamiento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Efecto de Imnovid sobre otros medicamentos
No se espera que pomalidomida pueda causar interacciones medicamentosas farmacocinéticas clínicamente relevantes debido a la inhibición o inducción de la isoenzima P450, o inhibición de transportadores cuando se administra de forma concomitante con sustratos de estas enzimas o transportadores. No se ha evaluado clínicamente el potencial de estas interacciones medicamentosas, incluyendo el posible impacto de pomalidomida en la farmacocinética de los anticonceptivos orales combinados (ver sección 4.4 Teratogenicidad).
Efecto de otros medicamentos sobre Imnovid
Pomalidomida se metaboliza parcialmente por CYP1A2 y CYP3A4/5.También es un sustrato de la glicoproteína P. La administración concomitante de pomalidomida con ketoconazol, inhibidor potente del CYP3A4/5 y de la Gp-P, o con el inductor potente del CYP3A4/5, carbamazepina, no demostró ningún efecto clínicamente relevante a la exposición a pomalidomida. La administración concomitante de pomalidomida con el inhibidor potente del CYP1A2 fluvoxamina en presencia de ketoconazol, incrementó la exposición media a pomalidomida en un 107 %, con un intervalo de confianza del 90 % [del 91 % al 124 %], frente a pomalidomida más ketoconazol. En un segundo estudio realizado para evaluar la contribución a los cambios del metabolismo de un inhibidor del CYP1A2 solo, la administración conjunta de fluvoxamina sola con pomalidomida aumentó la exposición media a pomalidomida en un 125 % con un intervalo de confianza del 90 % [del 98 % al 157 %] frente a pomalidomida administrada en monoterapia. Si se administran inhibidores potentes del CYP1A2 (p. ej., ciprofloxacino, enoxacino y fluvoxamina) de forma concomitante con pomalidomida, se debe reducir la dosis de pomalidomida en un 50 %.
Dexametasona
La administración concomitante de múltiples dosis de hasta 4 mg de pomalidomida con dexametasona de 20 mg a 40 mg (un inductor leve a moderado de varias enzimas CYP, incluido el CYP3A) en pacientes con mieloma múltiple no tuvo ningún efecto sobre la farmacocinética de pomalidomida frente a pomalidomida administrada sola.
Se desconoce el efecto de dexametasona sobre la warfarina. Se aconseja realizar una monitorización rigurosa de la concentración de warfarina durante el tratamiento.
4.6 Fertilidad, embarazo y lactancia
Mujeres con capacidad de gestación / Anticonceptivos en varones y mujeres
Las mujeres con capacidad de gestación deben utilizar métodos anticonceptivos efectivos. Si una mujer tratada con pomalidomida se queda embarazada, se debe suspender el tratamiento y derivar a la paciente a un médico especialista o con experiencia en teratología, para su evaluación y asesoramiento. Si un paciente varón toma pomalidomida y su pareja se queda embarazada, se recomienda derivar a la mujer a un médico especialista o con experiencia en teratología, para su evaluación y asesoramiento. Pomalidomida está presente en el semen humano. Como medida de precaución, todos los pacientes varones que tomen pomalidomida deben usar preservativos durante todo el tratamiento, durante la interrupción de la administración y hasta 7 días después del final del tratamiento, si su pareja está embarazada o tiene capacidad de gestación y no está usando ningún método anticonceptivo (ver secciones 4.3 y 4.4).
Embarazo
Se espera un efecto teratogénico de pomalidomida en humanos. Pomalidomida está contraindicada durante el embarazo y en mujeres con capacidad de gestación, a menos que se cumplan todas las condiciones para la prevención del embarazo, ver sección 4.3 y sección 4.4.
Lactancia
Se desconoce si pomalidomida se excreta en la leche materna. Se detectó la presencia de pomalidomida en la leche de ratas que estaban siendo amamantadas tras administrar el medicamento a la madre.
Debido a las posibles reacciones adversas en lactantes asociadas a pomalidomida, se debe decidir si es
necesario suspender la lactancia o suspender el tratamiento tras considerar la importancia del tratamiento para la madre.
Fertilidad
Se sabe que pomalidomida tiene un efecto negativo sobre la fertilidad y que es teratogénica en los animales. Tras su administración a conejas embarazadas, pomalidomida atravesó la placenta y se detectó en la sangre fetal. Ver sección 5.3.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Imnovid sobre la capacidad para conducir y utilizar máquinas es pequeña o moderada. Se han notificado casos de fatiga, disminución del nivel de conciencia, confusión y mareo relacionados con el uso de pomalidomida. Si notan estos efectos, se debe advertir a los pacientes de que no deben conducir automóviles, utilizar máquinas o realizar cualquier actividad peligrosa durante su tratamiento con pomalidomida.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Las reacciones adversas notificadas con mayor frecuencia en los estudios clínicos han sido los trastornos de la sangre y del sistema linfático, incluyendo anemia (45,7 %), neutropenia (45,3 %) y trombocitopenia (27 %); trastornos generales y alteraciones en el lugar de administración, incluyendo fatiga (28,3 %), pirexia (21 %) y edema periférico (13 %); e infecciones e infestaciones incluyendo neumonía (10,7 %). Las reacciones adversas relacionadas con neuropatía periférica fueron notificadas en el 12,3 % de los pacientes y las reacciones adversas de embolismo o tromboembolismo venoso fueron notificadas en el 3,3 % de los pacientes. Las reacciones adversas de grado 3 o 4 más frecuentes estaban relacionadas con trastornos de la sangre y del sistema linfático, incluyendo neutropenia (41,7 %), anemia (27 %) y trombocitopenia (20,7 %); infecciones e infestaciones, incluyendo neumonía (9 %); y trastornos generales y alteraciones en el lugar de administración, incluyendo fatiga (4,7 %), pirexia (3 %) y edema periférico (1,3 %). La reacción adversa grave notificada con mayor frecuencia fue la neumonía (9,3 %). Otras reacciones adversas graves notificadas incluyen neutropenia febril (4,0 %), neutropenia (2,0 %), trombocitopenia (1,7 %) y reacciones adversas de TEV (1,7 %).
Se observó que las reacciones adversas tendían a ocurrir con mayor frecuencia dentro de los primeros 2 ciclos de tratamiento con pomalidomida.
Tabla de reacciones adversas
En el estudio aleatorizado CC-4047-MM-003, un total de 302 pacientes con mieloma múltiple en recaída y refractario fueron tratados con 4 mg de pomalidomida administrada una vez al día durante 21 días en cada ciclo de 28 días, en combinación con una dosis baja semanal de dexametasona.
Las reacciones adversas observadas en los pacientes tratados con pomalidomida y dexametasona se incluyen a continuación, según el sistema de clasificación por órganos y la frecuencia (SOC por sus siglas en inglés) para todas las reacciones adversas y para las reacciones adversas de Grado 3 o 4.
Las frecuencias de las reacciones adversas son las notificadas en el grupo de pomalidomida más dexametasona del estudio CC-4047-MM-003 (n=302) y de los datos poscomercialización. Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de SOC (por sus siglas en inglés) y de frecuencia. Las frecuencias se definen, según las guías actuales, como: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); y poco frecuentes (>1/1.000 a <1/100).
|
Sistema de clasificación de órganos / Término preferente |
Todas las reacciones adversas/Frecuencia |
Reacciones adversas de Grado 3-4/Frecuencia |
|
Infecciones e |
Muy frecuentes |
Frecuentes |
|
infestaciones |
Neumonía (infecciones bacterianas, |
Sepsis neutropénica |
|
víricas y fungicas, incluidas las |
Neumonía (infecciones | |
|
infecciones oportunistas) |
bacterianas, víricas y fungicas, incluidas las infecciones | |
|
Frecuentes |
oportunistas) | |
|
Sepsis neutropénica |
Bronconeumonía | |
|
Bronconeumonía |
Infección del tracto respiratorio | |
|
Bronquitis |
Infección del tracto respiratorio | |
|
Infección del tracto respiratorio Infección del tracto respiratorio |
superior | |
|
superior |
Poco frecuentes | |
|
Nasofaringitis |
Bronquitis | |
|
Herpes zóster |
Herpes zóster | |
|
Frecuencia no conocida |
Frecuencia no conocida | |
|
Reactivación de la hepatitis B |
Reactivación de la hepatitis B | |
|
Neoplasias benignas, |
Poco frecuentes |
Poco frecuentes |
|
malignas y no |
Carcinoma de piel de células |
Carcinoma de piel de células |
|
especificadas (incl |
basales |
basales |
|
quistes y pólipos) |
Carcinoma de piel de células |
Carcinoma de piel de células |
|
escamosas |
escamosas | |
|
Trastornos de la |
Muy frecuentes |
Muy frecuentes |
|
sangre y del sistema |
Neutropenia |
Neutropenia |
|
linfático |
Trombocitopenia |
Trombocitopenia |
|
Leucopenia Anemia |
Anemia | |
|
Frecuentes |
Frecuentes | |
|
Neutropenia febril |
Neutropenia febril | |
|
Pancitopenia* |
Leucopenia Pancitopenia* | |
|
Trastornos del |
Muy frecuentes |
Frecuentes |
|
metabolismo y de la |
Disminución del apetito |
Hiperpotasemia |
|
nutrición |
Hiponatremia | |
|
Frecuentes Hiperpotasemia |
Hiperuricemia* | |
|
Hiponatremia |
Poco frecuentes | |
|
Hiperuricemia* Poco frecuentes Síndrome de lisis tumoral* |
Disminución del apetito Síndrome de lisis tumoral* | |
|
Trastornos |
Frecuentes |
Frecuentes |
|
psiquiátricos |
Estado de confusión |
Estado de confusión |
|
Sistema de clasificación de órganos / Término preferente |
Todas las reacciones adversas/Frecuencia |
Reacciones adversas de Grado 3-4/Frecuencia |
|
Trastornos del |
Frecuentes |
Frecuentes |
|
sistema nervioso |
Disminución del nivel de |
Disminución del nivel de |
|
conciencia Neuropatía sensitiva periférica |
conciencia | |
|
Mareo |
Poco frecuentes | |
|
Temblor |
Neuropatía sensitiva periférica | |
|
Hemorragia intracraneal* |
Mareo Temblor | |
|
Poco frecuentes |
Accidente cerebrovascular* | |
|
Accidente cerebrovascular* |
Hemorragia intracraneal* | |
|
Trastornos del oído y |
Frecuentes |
Frecuentes |
|
del laberinto |
Vértigo |
Vértigo |
|
Trastonos vasculares |
Frecuentes |
Poco frecuentes |
|
Trombosis venosa profunda |
Trombosis venosa profunda | |
|
Trastornos cardiacos |
Frecuentes |
Frecuentes |
|
Insuficiencia cardíaca* |
Insuficiencia cardíaca* | |
|
Fibrilación auricular* Infarto de miocardio* |
Fibrilación auricular* Poco frecuentes Infarto de miocardio* | |
|
Trastornos del |
Frecuentes |
Poco frecuentes |
|
sistema inmunológico |
Angioedema* |
Angioedema* |
|
Urticaria* |
Urticaria* | |
|
Trastornos |
Muy frecuentes |
Frecuentes |
|
respiratorios, |
Disnea |
Disnea |
|
torácicos y |
Tos | |
|
mediastínicos |
Poco frecuentes | |
|
Frecuentes |
Embolia pulmonar | |
|
Embolia pulmonar |
Tos | |
|
Epistaxis* |
Epistaxis* | |
|
Enfermedad pulmonar intersticial* |
Enfermedad pulmonar intersticial* | |
|
Trastornos |
Muy frecuentes |
Frecuentes |
|
gastrointestinales |
Diarrea |
Diarrea |
|
Náuseas |
Vómitos | |
|
Estreñimiento |
Estreñimiento | |
|
Frecuentes |
Poco frecuentes | |
|
Vómitos |
Náuseas | |
|
Hemorragia gastrointestinal |
Hemorragia gastrointestinal | |
|
Trastornos |
Poco frecuentes |
Poco frecuentes |
|
hepatobiliares |
Hiperbilirrubinemia Hepatitis* |
Hiperbilirrubinemia |
|
Trastornos de la piel y |
Frecuentes |
Frecuentes |
|
del tejido subcutáneo |
Erupción Prurito |
Erupción |
|
Sistema de clasificación de órganos / Término preferente |
Todas las reacciones adversas/Frecuencia |
Reacciones adversas de Grado 3-4/Frecuencia |
|
Trastornos |
Muy frecuentes |
Frecuentes |
|
musculoesqueléticos y |
Dolor óseo |
Dolor óseo |
|
del tejido conjuntivo |
Espasmos musculares |
Poco frecuentes Espasmos musculares |
|
Trastornos renales y |
Frecuentes |
Frecuentes |
|
urinarios |
Insuficiencia renal Retención urinaria |
Insuficiencia renal Poco frecuentes Retención urinaria |
|
Trastornos del |
Frecuentes |
Frecuentes |
|
aparato reproductor y de la mama |
Dolor pélvico |
Dolor pélvico |
|
Trastornos generales |
Muy frecuentes |
Frecuentes |
|
y alteraciones en el |
Fatiga |
Fatiga |
|
lugar de |
Pirexia |
Pirexia |
|
administración |
Edema periférico |
Edema periférico |
|
Exploraciones |
Frecuentes |
Frecuentes |
|
complementarias |
Disminución del recuento de |
Disminución del recuento de |
|
neutrófilos |
neutrófilos | |
|
Disminución del recuento de |
Disminución del recuento de | |
|
leucocitos |
leucocitos | |
|
Disminución del recuento de |
Disminución del recuento de | |
|
plaquetas |
plaquetas | |
|
Aumento de la alanina |
Aumento de la alanina | |
|
aminotransferasa Aumento del ácido úrico en sangre* |
aminotransferasa Poco frecuentes Aumento del ácido úrico en sangre* |
*Identificados a partir de los datos poscomercialización; las frecuencias se basan en los datos de los ensayos clínicos.
Descripción de reacciones adversas seleccionadas
Teratogenicidad
Pomalidomida está relacionada estructuralmente con talidomida. Talidomida es un principio activo con acción teratogénica conocida en humanos, que causa defectos de nacimiento graves que pueden poner en peligro la vida del niño. Pomalidomida tiene un efecto teratogénico en ratas y conejos cuando se administra durante el periodo de mayor organogénesis (ver secciones 4.6 y 5.3). Si se toma pomalidomida durante el embarazo, se espera un efecto teratogénico de pomalidomida en los seres humanos (ver sección 4.4).
Neutropenia y trombocitopenia
El 45,3 % de los pacientes que recibieron pomalidomida más dosis bajas de dexametasona (Pom + LD-Dex) experimentó neutropenia, frente al 19,5 % de los pacientes que recibieron dosis altas de dexametasona (HD-Dex). La neutropenia fue de grado 3 o 4 en un 41,7 % de los pacientes que recibieron Pom + LD-Dex frente al 14,8 % de los que recibieron HD-Dex. En los pacientes tratados con Pom + LD-Dex, la neutropenia fue grave en una minoría (2,0 % de los pacientes), no tuvo como consecuencia la suspensión del tratamiento, y se asoció con la interrupción del tratamiento en un 21,0 % de los pacientes y con una reducción de la dosis en un 7,7 % de los pacientes.
El 6,7 % de los pacientes que recibieron Pom + LD-Dex experimentó neutropenia febril (NF), frente a ninguno de aquellos que recibieron HD-Dex. Todos los casos fueron notificados como de grado 3 o 4. La NF fue notificada como grave en un 4,0 % de los pacientes. Asimismo, la NF se asoció con una interrupción de la dosis en un 3,7 % de los pacientes, con una reducción de la dosis en un 1,3 % de los pacientes y con ningún caso de suspensión del tratamiento.
El 27,0 % de los pacientes que recibieron Pom + LD-Dex experimentó trombocitopenia frente a un
26,8 % de los pacientes que recibieronHD-Dex. La trombocitopenia fue de grado 3 o 4 en un 20,7 % de los pacientes que recibieron Pom + LD-Dex frente a un 24,2 % de los que recibieron HD-Dex. En los pacientes tratados con Pom + LD-Dex, la trombocitopenia fue grave en un 1,7 % de los pacientes, conllevó una reducción de la dosis en un 6,3 % de los pacientes, una interrupción de la dosis en un 8 % de los pacientes y la suspensión del tratamiento en un 0,7 % de los pacientes (ver secciones 4.2 y 4.4).
Infección
La infección fue la toxicidad no hematológica más frecuente, con una incidencia del 55,0 % en los pacientes que recibieron Pom + LD-Dex frente al 48,3 % en los pacientes que recibieron HD-Dex. Aproximadamente la mitad de dichas infecciones fueron de grado 3 o 4; un 24,0 % en los pacientes tratados con Pom + LD-Dex y un 22,8 % en los pacientes tratados con HD-Dex.
Entre los pacientes tratados con Pom + LD-Dex las infecciones notificadas con mayor frecuencia fueron la neumonía y la infección del tracto respiratorio superior (en un 10,7 % y un 9,3 % de los pacientes, respectivamente); con un 24,3 % de las infecciones notificadas clasificadas como graves o mortales (grado 5) en el 2,7 % de los pacientes tratados. En los pacientes tratados con Pom + LD-Dex las infecciones conllevaron la suspensión de la dosis en un 2,0 % de los pacientes, la interrupción del tratamiento en un 14,3 % de los pacientes y la reducción de la dosis en un 1,3 % de los pacientes.
Eventos tromboembólicos
El 3,3 % de los pacientes que recibieron Pom + LD-Dex y un 2,0 % de los pacientes que recibieron HD-Dex experimentaron un embolismo o tromboembolismo venoso. El 1,3 % de los pacientes que recibieron Pom + LD-Dex experimentó reacciones de grado 3 o 4 frente a ningún caso entre los pacientes que recibieron HD-Dex. En los pacientes tratados con Pom + LD-Dex, el TEV se notificó como grave en el 1,7 %, no se notificó ninguna reacción adversa mortal en los estudios clínicos y el TEV no se asoció con suspensión de la dosis.
La profilaxis con ácido acetilsalicílico (y otros anticoagulantes en pacientes de alto riesgo) fue obligatoria para todos los pacientes participantes en los estudios clínicos. Se recomienda terapia anticoagulante (a no ser que esté contraindicada) (ver sección 4.4).
Neuropatía _ periférica
Se excluyó de los estudios clínicos a los pacientes con neuropatía periférica en curso de grado >2. El 12,3 % de los pacientes que recibieron Pom + LD-Dex experimentaron una neuropatía periférica, la mayoría de grado 1 o 2, frente a un 10,7 % de los pacientes que recibieron HD-Dex. El 1,0 % de los pacientes que recibieron Pom + LD-Dex experimentó reacciones de grado 3 o 4 frente al 1,3 % de los pacientes que recibieron HD-Dex. En los estudios clínicos, ninguna de las neuropatías periféricas se notificó como grave en los pacientes tratados con Pom + LD-Dex y la neuropatía periférica condujo a la suspensión de la dosis en un 0,3 % de los pacientes (ver sección 4.4).
La mediana del tiempo hasta la aparición de la neuropatía fue de 2,1 semanas, con una variación de 0,1 a 48,3 semanas. La mediana del tiempo hasta la aparición fue inferior en los pacientes que recibieron HD-Dex comparado con los pacientes tratados con Pom+LD-Dex (1,3 semanas frente a 2,1 semanas).
La mediana del tiempo hasta la desaparición de los síntomas fue de 22,4 semanas en los pacientes que recibieron Pom+LD-Dex y de 13,6 semanas en los pacientes que recibieron HD-Dex. El límite inferior
del intervalo de confianza del 95 % fue de 5,3 semanas en los pacientes tratados con Pom+LD-Dex y de
2,0 semanas en los pacientes que recibieron HD-Dex.
Hemorragia
Se han notificado trastornos hemorrágicos con pomalidomida, especialmente en pacientes con factores de riesgo tales como medicamentos concomitantes que aumentan el riesgo de hemorragia. Los eventos hemorrágicos incluyen epistaxis, hemorragia intracraneal y hemorragia gastrointestinal.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los
profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Se han evaluado dosis de pomalidomida de hasta 50 mg en dosis única en voluntarios sanos y 10 mg en múltiples dosis una vez al día en pacientes con mieloma múltiple sin que se haya notificado ningún caso de efecto adverso grave relacionado con sobredosis. Pomalidomida se eliminó mediante hemodiálisis. En caso de sobredosis, se recomienda terapia de soporte.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agente inmunomodulador, código ATC: L04AX06 Mecanismo de acción
Pomalidomida es un medicamento con actividad tumoricida directa contra el mieloma, actividad inmunomoduladora y capaz de inhibir el apoyo de las células del estroma para el crecimiento de las células cancerosas del mieloma múltiple. En concreto, pomalidomida inhibe la proliferación e induce la apoptosis de las células hematopoyéticas tumorales. Además, pomalidomida inhibe la proliferación de las líneas celulares de mieloma múltiple resistentes a lenalidomida y presenta un efecto sinérgico con dexametasona tanto en las líneas celulares de mieloma múltiple resistentes a lenalidomida como en las sensibles a lenalidomida para inducir la apoptosis de las células tumorales. Pomalidomida potencia la inmunidad celular mediada por los linfocitos T y por los linfocitos natural killer (NK) e inhibe la producción de citocinas proinflamatorias (p. ej. TNF-a e IL-6) por los monocitos. Pomalidomida también inhibe la angiogénesis mediante el bloqueo de la migración y adhesión de células endoteliales.
Eficacia clínica y seguridad
Se evaluó la eficacia y seguridad de pomalidomida en combinación con dexametasona en un ensayo de fase III, multicéntrico, aleatorizado y abierto (CC-4047-MM-003), donde se comparó el tratamiento de pomalidomida más una dosis baja de dexametasona (Pom + LD-Dex) frente a una dosis alta de dexametasona sola (HD-Dex) en pacientes adultos con mieloma múltiple en recaída o refractario que ya habían recibido al menos dos tratamientos previos, incluyendo lenalidomida y bortezomib, y que habían mostrado progresión de la enfermedad durante el último tratamiento. En el estudio participaron un total de 455 pacientes: 302 en el grupo Pom + LD-Dex y 153 en el grupo HD-Dex. La mayoría de los pacientes eran varones (59 %) y caucásicos (79 %); la mediana de edad de toda la población era de 64 años (mín, máx: 35, 87 años).
Los pacientes del grupo Pom + LD-Dex tomaron 4 mg de pomalidomida por vía oral en los días del 1 al 21 en cada ciclo de 28 días. Se administró la dosis de LD-Dex (40 mg) una vez al día en los días 1, 8, 15 y 22 de un ciclo de 28 días. En el grupo HD-Dex, los pacientes tomaron dexametasona (40 mg) una vez al día en los días del 1 al 4, del 9 al 12 y del 17 al 20 en un ciclo de 28 días. Los pacientes mayores de 75 años iniciaron el tratamiento con dexametasona 20 mg. Los pacientes continuaron con el tratamiento hasta la progresión de la enfermedad.
La variable principal de eficacia fue la supervivencia libre de progresión (SLP) de acuerdo con los criterios del InternationalMyeloma Working Group (IMWG). Para la población por intención de tratar (IDT), la mediana del tiempo de SLP según revisión del Independent Review Adjudication Committee (IRAC) basada en los criterios del IMWG fue de 15,7 semanas (IC de 95 %: 13,0; 20,1) en el grupo Pom + LD-Dex; la tasa estimada de supervivencia libre de eventos a las 26 semanas fue del 35,99 %
(± 3,46 %). En el grupo HD-Dex, la mediana del tiempo de SLP fue de 8,0 semanas (IC de 95 %: 7,0; 9,0); la tasa estimada de supervivencia libre de eventos a las 26 semanas fue del 12,15 % (± 3,63 %).
Se evaluó la SLP en varios subgrupos relevantes: sexo, raza, estatus de rendimiento ECOG, factores de estratificación (edad, población con enfermedad, terapias antimieloma previas [2, >2]), parámetros de relevancia pronóstica seleccionados (niveles basales de beta-2 microglobulina, niveles basales de albúmina, insuficiencia renal basal y riesgo citogenético) y exposición y refractariedad a terapias antimieloma previas. Independientemente del subgrupo evaluado, la SLP fue generalmente consistente con la observada en la población por IDT en ambos grupos de tratamiento.
En la tabla 1 se resumen los resultados de la supervivencia libre de progresión para la población por IDT. La figura 1 presenta la curva de Kaplan-Meier de la SLP para la población por IDT.
Tabla 1: Tiempo de Supervivencia Libre de Progresión según revisión del IRAC basada en
los criterios IMWG (prueba de log-rank estratificada) (población por IDT)
|
Pom+LD-Dex (N=302) |
HD-Dex (N=153) | |
|
Supervivencia libre de progresión (SLP), n |
302 (100,0) |
153 (100,0) |
|
Censurado, n (%) |
138 (45,7) |
50 (32,7) |
|
Progresión/muerte, n (%) |
164 (54,3) |
103 (67,3) |
|
Tiempo de supervivencia libre de progresión (semanas) | ||
|
Mediana3 |
15,7 |
8,0 |
|
[IC de 95 % bilateral]b |
[13,0; 20,1] |
[7,0 ; 9,0] |
|
Razón de riesgo (Hazard Ratio) (Pom+LD-Dex: HD-Dex) [IC de 95 % bilateral]0 |
0,45 [0,35; 0,59] | |
|
Valor P (bilateral) de la prueba de log-rankd |
<0,001 | |
Nota: IC = intervalo de confianza; IRAC = Comité Independiente Revisor de Evaluación; NE = no estimable a La mediana está basada en la estimación de Kaplan-Meier.
b Intervalo de confianza de 95 % de la mediana del tiempo de supervivencia libre de progresión.
c Basado en el modelo de riesgos proporcionales de Cox que compara las funciones de riesgo asociadas con los grupos de tratamiento, estratificados por edad (<75 frente a >75), población con la enfermedad (refractario a lenalidomida y bortezomib frente a no refractario a estos dos medicamentos), y número de terapias antimieloma previas (=2 frente a >2).
d El valor P está basado en la prueba de log-rank estratificada con los mismos factores de estratificación arriba mencionados para el modelo de Cox.
Fecha de corte: 7 de septiembre de 2012
Supervivencia Libre de Progresión según la revisión de respuesta del IRAC basada en los criterios IMWG (prueba de log-rank estratificada) (población por IDT)
Figura 1:
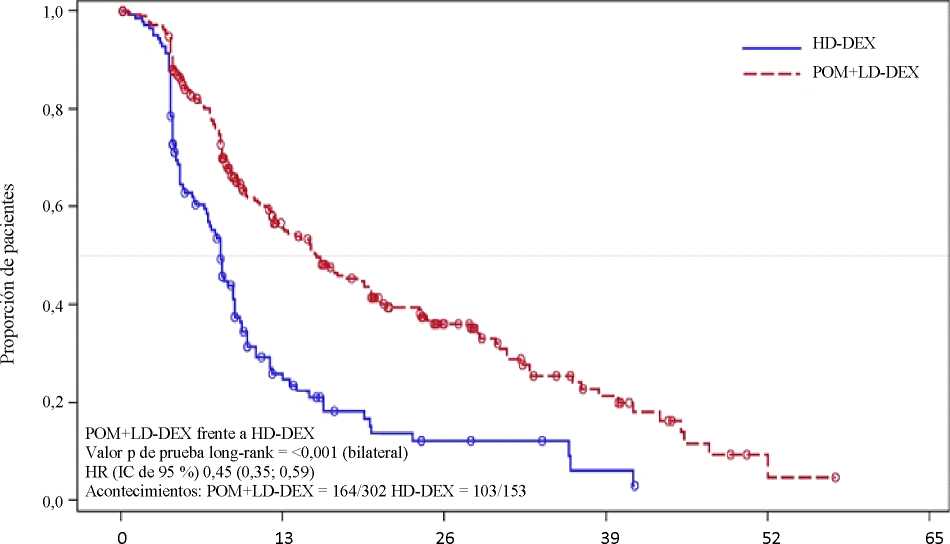
Fecha de corte: 7 de septiembre de 2012
Supervivencia Libre de Progresión (semanas)
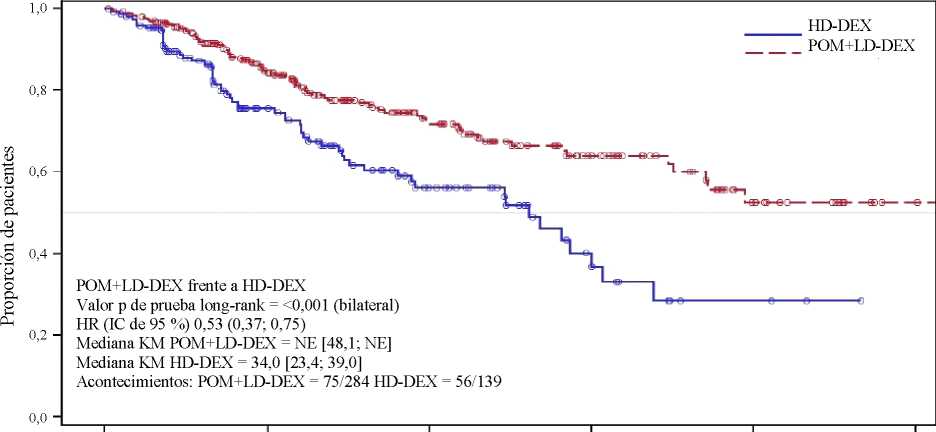
La variable secundaria clave fue la supervivencia global (SG). Un total de 226 pacientes (74,8 %) del grupo Pom + LD-Dex y 95 pacientes (62,1 %) del grupo HD-Dex vivían en el momento de la fecha de corte (7 de septiembre de 2012). La mediana del tiempo de supervivencia global según las estimaciones Kaplan-Meier no fue alcanzado por el grupo Pom + LD Dex, pero podría esperarse que fuera al menos de 48 semanas, que corresponde al umbral más bajo del IC de 95 %. La mediana del tiempo de SG del grupo HD-Dex fue de 34 semanas (IC de 95 %: 23,4; 39,9). La tasa libre de eventos al año fue del 52,6 % (± 5,72 %) para el grupo Pom + LD-Dex y del 28,4 % (± 7,51 %) para el grupo HD-Dex. La diferencia en términos de SG entre los dos grupos de tratamiento fue estadísticamente significativa (p <0,001).
La Tabla 2 resume los resultados de supervivencia global para la población por IDT. La curva Kaplan-Meier de SG para la población por IDT se presenta en la Figura 2.
Basándose en los resultados de las variables SLP y SG, el Comité de Monitorización de Datos para este estudio recomendó que se completara el estudio y que los pacientes en el grupo HD-Dex fueran cruzados al grupo Pom + LD-Dex.
|
Estadísticas |
Pom+LD-Dex (N=302) |
HD-Dex (N=153) | |
|
N |
302 (100,0) |
153 (100,0) | |
|
Censurado |
n (%) |
226 (74,8) |
95 (62,1) |
|
Muerto |
n (%) |
76 (25,2) |
58 (37,9) |
|
Tiempo de supervivencia (semanas) |
Mediana3 |
NE |
34,0 |
|
IC de 95 % bilateralb |
[ 48,1; NE] |
[ 23,4; 39,9] | |
|
Razón de riesgo (Hazard Ratio) (Pom+LD-Dex:HD-Dex) [IC de 95 % bilateral0] |
0,53 [ 0,37; 0,74] | ||
|
Valor P (bilateral) de la prueba de log-rankd |
<0,001 | ||
Nota: IC = intervalo de confianza; NE = no estimable. a La mediana está basada en la estimación de Kaplan-Meier.
b Intervalo de confianza de 95 % sobre la mediana del tiempo de supervivencia global.
c Basado en el modelo de riesgos proporcionales de Cox que compara las funciones de riesgo asociadas con los grupos de tratamiento. d El valor P está basado en la prueba de log-rank no estratificada.
Fecha de corte: 7 de septiembre de 2012
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Imnovid en todos los grupos de la población pediátrica en mieloma múltiple (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Pomalidomida se absorbe alcanzando una concentración plasmática máxima (Cmáx) a las 2 o 3 horas y, por lo menos, un 73 % se absorbe después de administrar una dosis única por vía oral. El área bajo la curva (AUC) de pomalidomida aumenta, aproximadamente, lineal y proporcionalmente con los incrementos de la dosis. Tras la administración de múltiples dosis, pomalidomida tiene una ratio de acumulación del 27 al 31 % en el AUC.
Figura 2: Curva de Kaplan-Meier de la Supervivencia Global (Población por IDT)
0 13
Fecha de corte: 7 de septiembre de 2012
26 39 52
Superviviencia global (semanas)
65
La administración conjunta con una comida rica en grasas y rica en calorías reduce la tasa de absorción, disminuyendo la Cmáx plasmática media en, aproximadamente un 27 %, pero con un efecto mínimo sobre la extensión de la absorción global con una disminución del 8 % en el AUC media. Por tanto, pomalidomida puede administrarse con o sin alimentos.
Distribución
Pomalidomida tiene un volumen de distribución aparente (Vd) medio de entre 62 y 138 litros en estado estable. En el semen de sujetos sanos pomalidomida se distribuye a una concentración de aproximadamente el 67 % del nivel de plasma a las 4 horas posteriores a la administración (aproximadamente Tmáx) tras 4 días de administración de 2 mg una vez al día. La unión in vitro de los enantiómeros de pomalidomida a las proteínas plasmáticas en humanos oscila entre el 12 % y el 44 % y no es dependiente de la concentración.
Biotransformación
En sujetos sanos que han recibido una dosis única por vía oral de [14C]-pomalidomida (2 mg), pomalidomida es la mayor sustancia circulante (aproximadamente el 70 % de la radioactividad del plasma) in vivo. No se hallaron metabolitos a >10 % relativos a la radioactividad total o relacionada en plasma.
Las rutas metabólicas principales de la radioactividad excretada son los procesos de hidroxilación con la posterior glucuronidación, o hidrólisis. Los estudios in vitro identificaron al CYP1A2 y al CYP3A4 como las enzimas primarias implicadas en la hidroxilación de pomalidomida mediada por CYP, con contribuciones menores adicionales del CYP2C19 y CYP2D6. Pomalidomida es también un sustrato de la glicoproteína-P in vitro. La administración concomitante de pomalidomida con el potente inhibidor del CYP3A4/5 y de la Gp-P, ketoconazol, o con el potente inductor del CYP3A4/5, carbamazepina, no demostró ningún efecto clínicamente relevante en la exposición a pomalidomida. La administración concomitante de pomalidomida con el inhibidor potente del CYP1A2, fluvoxamina, en presencia de ketoconazol, incrementó la exposición media a pomalidomida en un 107 %, con un intervalo de confianza del 90 % [del 91 % al 124 %], frente a pomalidomida más ketoconazol. En un segundo estudio realizado para evaluar la contribución a los cambios del metabolismo de un inhibidor del CYP1A2 solo, la administración conjunta de fluvoxamina sola con pomalidomida aumentó la exposición media a pomalidomida en un 125 % con un intervalo de confianza del 90 % [del 98 % al 157 %] frente a pomalidomida administrada en monoterapia. Si se administran inhibidores potentes del CYP1A2 (p. ej., ciprofloxacino, enoxacino y fluvoxamina) de forma concomitante con pomalidomida, se debe reducir la dosis de pomalidomida en un 50 %. La administración de pomalidomida a fumadores, sabiendo que el tabaquismo induce la isoforma CYP1A2, no tuvo ningún efecto clínicamente relevante en la exposición a pomalidomida frente a la exposición a pomalidomida observada en los no fumadores.
Según los datos in vitro, pomalidomida no es un inhibidor o inductor de las isoenzimas del citocromo P-450 y no inhibió ninguno de los fármacos transportadores que fueron estudiados. No se espera que pomalidomida pueda causar interacciones medicamentosas clínicamente relevantes cuando se administra de forma concomitante con sustratos de estas rutas.
Eliminación
En sujetos sanos pomalidomida se elimina con una mediana de semivida plasmática de aproximadamente 9,5 horas y de unas 7,5 horas en pacientes con mieloma múltiple. Pomalidomida tiene una media de aclaramiento corporal total (Cl/F) de aproximadamente 7-10 l/h.
Tras una dosis única por vía oral de [14C]-pomalidomida (2 mg) en sujetos sanos, aproximadamente el 73 % y el 15 % de la dosis radioactiva se eliminó por la orina y las heces, respectivamente, con aproximadamente el 2 % y el 8 % del radiocarbono administrado eliminado como pomalidomida en orina y heces.
Pomalidomida se metaboliza ampliamente antes de la excreción, con los metabolitos resultantes eliminados principalmente por la orina. Los tres metabolitos predominantes en la orina (formados mediante hidrólisis o hidroxilación con posterior glucuronidación) representan, aproximadamente, el 23 %, 17 % y 12 %, respectivamente, de la dosis en la orina.
Los metabolitos dependientes del CYP representan aproximadamente el 43 % de la radiactividad total excretada, mientras que los metabolitos hidrolíticos no dependientes del CYP representan el 25 %, y la excreción de pomalidomida inalterada representa el 10 % (2 % en orina y 8 % en heces).
Farmacocinética poblacional
Según análisis de farmacocinética poblacional basado en un modelo bicompartimental, los sujetos sanos y los pacientes con mieloma múltiple mostraron aclaramiento aparente (Cl/F) y volumen de distribución aparente en el compartimento central (WF) comparables. En tejidos periféricos, pomalidomida fue absorbida preferentemente por los tumores con un aclaramiento de distribución aparente en el compartimento periférico (Q/F) y un volumen de distribución aparente en el compartimento periférico (V3/F) 3,7 veces y 8 veces mayor, respectivamente, comparado con los sujetos sanos.
Población pediátrica
No existen datos disponibles sobre la administración de pomalidomida en niños o adolescentes (<18 años).
Población de edad avanzada
Según análisis de farmacocinética poblacional en sujetos sanos y en pacientes con mieloma múltiple, no se observó una influencia significativa de la edad (19-83 años) en el aclaramiento oral de pomalidomida. En los estudios clínicos los pacientes de edad avanzada (>65 años) expuestos a pomalidomida no requirieron ningún ajuste de dosis. Por favor, ver sección 4.2.
Insuficiencia renal
Los análisis de farmacocinética poblacional mostraron que los parámetros farmacocinéticos de pomalidomida no se vieron afectados de forma destacable en los pacientes con insuficiencia renal (definida mediante el aclaramiento de la creatinina o el filtrado glomerular estimado [FGe]) en comparación con los pacientes con la función renal normal (CrCl >60 ml/minuto). La exposición media a pomalidomida normalizada según el AUC fue del 98,2 % con un intervalo de confianza del 90 %
[77,4 % al 120,6 %] en los pacientes con insuficiencia renal moderada (FGe >30 a <45 ml/minuto/1,73 m2) en comparación con los pacientes con la función renal normal. La exposición media a pomalidomida normalizada según el AUC fue del 100,2 % con un intervalo de confianza del 90 % [79,7 % al 127,0 %] en los pacientes con insuficiencia renal grave que no precisaban diálisis (CrCl <30 o FGe <30 ml/minuto/1,73 m2) en comparación con los pacientes con la función renal normal. La exposición media a pomalidomida normalizada según el AUC aumentó en un 35,8 % con un IC del 90 % [7,5 % al 70,0 %] en los pacientes con insuficiencia renal grave que precisaban diálisis (CrCl <30 ml/minuto con necesidad de diálisis) en comparación con los pacientes con la función renal normal. Los cambios medios en la exposición a pomalidomida en cada uno de estos grupos de insuficiencia renal no son de una magnitud que requiera un ajuste de la dosis.
Insuficiencia hepática
Los parámetros farmacocinéticos cambiaron modestamente en los pacientes con insuficiencia hepática (definida mediante los criterios de Child-Pugh) en comparación con los sujetos sanos. La exposición media a pomalidomida aumentó en un 51 % con un intervalo de confianza del 90 % [del 9 % al 110 %] en los pacientes con insuficiencia hepática leve en comparación con los sujetos sanos. La exposición media a pomalidomida aumentó en un 58% con un intervalo de confianza del 90 % [del 13 % al 119 %] en los pacientes con insuficiencia hepática moderada en comparación con los sujetos sanos. La exposición media a pomalidomida aumentó en un 72 % con un intervalo de confianza del 90 % [del 24 % al 138 %] en los pacientes con insuficiencia hepática grave en comparación con los sujetos sanos. Los aumentos medios en la exposición a pomalidomida en cada uno de estos grupos de insuciencia hepática no son de una magnitud que requiera un ajuste del esquema de tratamiento o de la dosis (ver sección 4.2).
Estudios de toxicidad de dosis repetidas
La administración crónica de pomalidomida en ratas en dosis de 50, 250 y 1000 mg/kg/día durante 6 meses fue bien tolerada. No se detectó ningún efecto adverso hasta los 1000 mg/kg/día (una tasa de exposición 175 veces más elevada que la dosis clínica de 4 mg).
Se evaluó pomalidomida en monos en estudios de dosis repetidas de hasta 9 meses de duración. En estos estudios, los monos mostraron una mayor sensibilidad a los efectos de pomalidomida que las ratas. Las toxicidades primarias observadas en monos estuvieron relacionadas con los sistemas hematopoyético/linforreticular. En el estudio de 9 meses en monos con dosis de 0,05, 0,1 y 1 mg/kg/día se observó morbilidad y eutanasia temprana de 6 animales a dosis de 1 mg/kg/día que fueron atribuidas a los efectos inmunosupresores (infección por estafilococos, reducción de los linfocitos en sangre periférica, inflamación crónica del intestino grueso, reducción histológica de los linfocitos e hipocelularidad de la médula ósea) a exposiciones elevadas de pomalidomida (15 veces la tasa de exposición comparada con una dosis clínica de 4 mg). Dichos efectos inmunosupresores provocaron la eutanasia temprana de 4 monos debido a su mal estado de salud (heces líquidas, inapetencia, ingesta de alimentos reducida y pérdida de peso); la evaluación histopatológica de estos animales demostró inflamación crónica del intestino grueso y atrofia vellosa del intestino delgado. Se observó infección por estafilococos en 4 monos; 3 de éstos respondieron al tratamiento con antibióticos y uno murió sin tratamiento. Además, resultados consistentes con la leucemia mielógena aguda llevaron a la eutanasia de un mono; las observaciones clínicas y la patología clínica y/o alteraciones de la médula ósea observadas en este animal eran consistentes con inmunosupresión. La proliferación mínima o leve en los conductos biliares con incrementos asociados de la ALP y de la GGT también se observaron a dosis de 1 mg/kg/día. La evaluación de los animales recuperados indicó que todos los resultados relacionados con el tratamiento eran reversibles a las 8 semanas del cese de la administración, excepto la proliferación de los conductos biliares intrahepáticos observada en 1 animal en el grupo de 1 mg/kg/día. El nivel sin efecto adverso observado (NOAEL) fue de 0,1 mg/kg/día (una tasa de exposición relativa de 0,5 veces comparada con la dosis clínica de 4 mg).
Genotoxicidad/carcinogenicidad
Pomalidomida no resultó mutagénica en los ensayos de mutaciones bacterianas y de los mamíferos, y no indujo aberraciones cromosómicas en los linfocitos de sangre periférica en humanos así como tampoco a la formación de micronúcleos en eritrocitos policromáticos en la médula ósea de ratas a las que les fueron administradas dosis de hasta 2000 mg/kg/día. No se han realizado estudios de carcinogenicidad.
Fertilidad y desarrollo embrionario temprano
En un estudio de fertilidad y desarrollo embrionario temprano en ratas, se administró pomalidomida a los machos y las hembras a dosis de 25, 250 y 1000 mg/kg/día. El examen uterino en el día de gestación 13 mostró una reducción de la cantidad media de embriones viables y un aumento en la pérdida postimplantación con todos los niveles de dosis. Por consiguiente, el NOAEL en estos eventos observados fue <25 mg/kg/día (con un AUC24h de 39960 ng-h/ml (nanogramo^hora/mililitros) para la dosis más baja evaluada y una tasa de exposición 99 veces relativa a la dosis clínica de 4 mg). Cuando los machos tratados en este estudio fueron apareados con hembras no tratadas, todos los parámetros uterinos fueron comparables a los controles. Según estos resultados, los efectos observados fueron atribuidos al tratamiento de las hembras.
Desarrollo embriofetal
Pomalidomida resultó ser teratógena en ratas y conejos cuando se administró durante el periodo de mayor organogénesis. En el estudio de toxicidad sobre el desarrollo embriofetal de la rata, se observaron malformaciones relacionadas con la ausencia de vejiga urinaria, ausencia de la glándula tiroidea, así como la fusión y la desalineación de los elementos vertebrales torácicos y lumbares (arcos centrales y/o neurales) a todos los niveles de dosis (25, 50 y 1000 mg/kg/día).
En este estudio no se observó toxicidad materna. Por ello, el NOAEL materno fue de 1000 mg/kg/día, y el NOAEL para la toxicidad de desarrollo fue <25 mg/kg/día (con un AUC24h de 34340 ng-h/ml en el día de gestación 17 para la dosis más baja evaluada y la tasa de exposición fue de 85 veces comparada
con la dosis clínica de 4 mg). En conejos, pomalidomida a dosis entre los 10 y 250 mg/kg/día produjo malformaciones en el desarrollo embriofetal. Se observaron aumentos de las anomalías cardíacas a todas las dosis con aumentos significativos a 250 mg/kg/día. A dosis de entre 100 y 250 mg/kg/día se registró un ligero aumento de la pérdida post-implantación y un ligero descenso en el peso del feto. A dosis de 250 mg/kg/día, las malformaciones fetales incluyeron anomalías en las extremidades (extremidades anteriores y posteriores dobladas y/o giradas, ausencia de dígito o dígito libre) y malformaciones esqueléticas asociadas (metacarpiano no osificado, metacarpiano y falange no alineados, ausencia de dígito, falange no osificada y tibia corta no osificada o doblada); dilatación moderada de los ventrículos laterales del cerebro; ubicación anormal de la arteria subclavia derecha; ausencia de los lóbulos intermedios pulmonares; par de riñones desplazados hacia abajo; morfología hepática alterada; ausencia de osificación de la pelvis u osificación incompleta; aumento medio de las costillas torácicas supernumerarias y reducción media de los tarsales osificados. Además, se observó una ligera reducción en el incremento del peso materno, una reducción significativa de los triglicéridos, y una reducción significativa del peso absoluto y relativo del bazo a dosis de entre 100 y 250 mg/kg/día. El NOAEL materno fue de 10 mg/kg/día y el NOAEL del desarrollo fue de <10 mg/kg/día (con un AUC24h de 418 ng-h/ml en el día de gestación 19 para la dosis más baja evaluada, similar a la obtenida con una dosis clínica de 4 mg).
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Contenido de la cápsula:
Manitol
Almidón pregelatinizado Estearil fumarato de sodio
Cubierta de la cápsula:
La cubierta de la cápsula de 1 mg contiene gelatina, dióxido de titanio (E171), indigotina (E132), óxido de hierro amarillo (E172) y tinta blanca y negra.
Tinta de impresión:
La cubierta de la cápsula de 1 mg contiene: tinta blanca - goma laca shellac, dióxido de titanio (E171), simeticona, propilenglicol (E1520) e hidróxido de amonio (E527); tinta negra - goma laca shellac, óxido de hierro negro (E172), propilenglicol (E1520) e hidróxido de amonio (E527).
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
4 años.
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Las cápsulas están envasadas en blísteres de polivinilcloruro (PVC) / policlorotrifluoroetileno (PCTFE) con lámina de aluminio.
Tamaño del envase de 21 cápsulas.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Las cápsulas no se deben abrir o triturar. En el caso de que el polvo de pomalidomida entre en contacto con la piel, debe lavar abundantemente la piel con agua y jabón inmediatamente. En el caso de que el polvo de pomalidomida entre en contacto con las membranas mucosas, debe lavarlas abundantemente con agua a presión.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local. El medicamento no utilizado debe devolverse al farmacéutico al final del tratamiento.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd.
1 Longwalk Road Stockley Park Uxbridge UB11 1DB Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/850/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 05/agosto/2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la identificación de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Imnovid 2 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula dura contiene 2 mg de pomalidomida.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura.
Imnovid 2 mg cápsula dura: tapa opaca de color azul oscuro y cuerpo opaco de color naranja marcadas con “POML 2 mg” en tinta blanca, de tamaño 2, cápsulas de gelatina dura.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Imnovid en combinación con dexametasona está indicado en el tratamiento de los pacientes adultos con mieloma múltiple resistente al tratamiento o recidivante que hayan recibido al menos dos tratamientos previos, incluyendo lenalidomida y bortezomib, y que hayan experimentado una progresión de la enfermedad en el último tratamiento.
4.2 Posología y forma de administración
El tratamiento debe iniciarse y monitorizarse bajo la supervisión de médicos con experiencia en el tratamiento del mieloma múltiple.
Posología
La dosis inicial recomendada es de 4 mg de Imnovid una vez al día por vía oral, en los días del 1 al 21 de ciclos repetidos de 28 días. La dosis recomendada de dexametasona es de 40 mg por vía oral una vez al día, en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
La posología se mantiene o modifica en función de los resultados clínicos y de laboratorio.
El tratamiento debe suspenderse si existe evidencia de progresión de la enfermedad.
Modificación o interrupción de la dosis de _pomalidomida
Las instrucciones para la interrupción y reducción de la dosis de pomalidomida relacionadas con reacciones adversas hematológicas se indican en la siguiente tabla:
|
Toxicidad |
Modificación de la dosis |
|
NeutroDenia • RAN* <0,5 x 109/l o neutropenia febril (fiebre >38,5°C y RAN <1 x 109/l) |
Interrumpir el tratamiento con pomalidomida, control semanal del hemograma completo. |
|
• RAN vuelve a >1 x 109/1 |
Reanudar el tratamiento con 3 mg de pomalidomida al día. |
|
• Con cada disminución posterior a <0,5 x 1071 |
Interrumpir el tratamiento con pomalidomida |
|
• RAN vuelve a >1 x 109/l |
Reanudar el tratamiento con 1 mg menos de pomalidomida que la dosis previa |
|
T rombocitopenia • Recuento de plaquetas <25 x 109/l |
Interrumpir el tratamiento con pomalidomida, control semanal del hemograma completo |
|
• Recuento de plaquetas vuelve a >50 x 109/l |
Reanudar el tratamiento con 3 mg de pomalidomida al día |
|
• Con cada disminución posterior a <25 x 1071 |
Interrumpir el tratamiento con pomalidomida |
|
• Recuento de plaquetas vuelve a >50 x 109/l |
Reanudar el tratamiento con 1 mg menos de pomalidomida que la dosis previa |
*RAN - Recuento absoluto de neutrófilos
Para iniciar un nuevo ciclo de pomalidomida, el recuento de neutrófilos debe ser >1 x 109/l y el recuento de plaquetas debe ser >50 x 109/l.
En caso de neutropenia, el médico debe considerar el uso de factores de crecimiento.
En el caso de otras reacciones adversas de grado 3 o 4 relacionadas con pomalidomida, el médico debe considerar interrumpir el tratamiento y reanudarlo con un 1 mg menos que la dosis previa una vez que se haya disminuido la reacción adversa a un grado inferior o igual a 2.
Si la reacción adversa ocurre tras disminuciones de la dosis a 1 mg, entonces debe suspenderse el tratamiento con este medicamento.
Se debe considerar la interrupción o suspensión de pomalidomida en caso de exantema de grado 2-3. Se debe suspender el tratamiento con pomalidomida en caso de angioedema, exantema de grado 4 y exantema ampolloso o exfoliativo, y no se debe reanudar una vez suspendido por estas reacciones.
Si se administran inhibidores potentes del CYP1A2 (p. ej., ciprofloxacino, enoxacino y fluvoxamina) de forma concomitante con pomalidomida, se debe reducir la dosis de pomalidomida en un 50 %.
|
Toxicidad |
Modificación de la dosis |
|
Dispepsia = grado 1-2 Dispepsia > grado 3 |
Mantener la dosis y tratar con antihistamínicos H2 o equivalentes. Reducir la dosis en un nivel de dosis si los síntomas persisten. Interrumpir la administración hasta que se controlen los síntomas. Añadir antihistamínicos H2 o equivalentes y reducir la dosis en un nivel cuando se reanude su administración. |
|
Edema > grado 3 |
Usar diuréticos según sea necesario y reducir la dosis en un nivel de dosis. |
|
Confusión o cambios en el estado de ánimo > grado 2 |
Interrumpir la administración hasta que desaparezcan los síntomas. Cuando se reanude su administración reducir la dosis en un nivel de dosis. |
|
Debilidad muscular > grado 2 |
Interrumpir la administración hasta que la debilidad muscular sea < grado 1. Reiniciar la dosis con una reducción de un nivel. |
|
Hiperglucemia > grado 3 |
Reducir la dosis en un nivel de dosis. Tratar con insulina o hipoglucemiantes orales según sea necesario. |
|
Pancreatitis aguda |
Suspensión de dexametasona del régimen de tratamiento del paciente. |
|
Otros efectos adversos relacionados con dexametasona > grado 3 |
Interrumpir la administración de dexametasona hasta que los efectos adversos sean de grado < 2. Reanudar su administración con una reducción de un nivel de la dosis. |
Niveles de reducción de la dosis de dexametasona:
Niveles de reducción de la dosis (<75 años): dosis inicial 40 mg; nivel de dosis -1 20 mg; nivel de dosis -2 10 mg en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
Niveles de reducción de la dosis (>75 años): dosis inicial 20 mg; nivel de dosis -1 12 mg; nivel de dosis -2 8 mg en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
Si la recuperación de las toxicidades tarda más de 14 días, la dosis de dexametasona se reducirá en un nivel de dosis.
Poblaciones especiales Población _ pediátrica
No hay un uso relevante de Imnovid en niños de 0 a 17 años para la indicación del mieloma múltiple.
Pacientes de edad avanzada
No se requiere ningún ajuste de dosis de pomalidomida. En pacientes de >75 años, la dosis inicial de dexametasona es de 20 mg una vez al día en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
Insuficiencia renal
No se requiere ningún ajuste de dosis de pomalidomida en los pacientes con insuficiencia renal. Los días de hemodiálisis, los pacientes deben tomar la dosis de pomalidomida después de la hemodiálisis.
Insuficiencia hepática
Los pacientes con una concentración sérica de bilirrubina total >2,0 mg/dl se excluyeron de los estudios clínicos. La insuficiencia hepática tiene un efecto modesto sobre la farmacocinética de pomalidomida (ver sección 5.2). No se requiere ajustar la dosis inicial de pomalidomida en pacientes con insuficiencia hepática según definen los criterios de Child-Pugh. Sin embargo, los pacientes con insuficiencia hepática deben ser monitorizados cuidadosamente por si presentan reacciones adversas y se debe reducir la dosis o suspender la administración de pomalidomida según sea necesario.
Forma de administración Vía oral.
Imnovid debe tomarse a la misma hora cada día. Las cápsulas no deben abrirse, romperse ni masticarse (ver sección 6.6). Este medicamento debe tomarse entero, preferiblemente con agua, con o sin alimentos. Si el paciente olvida tomar una dosis de Imnovid un día, el paciente debe entonces tomar la próxima dosis al día siguiente a la hora habitual. Los pacientes no deben ajustar la dosis para compensar una dosis olvidada en días anteriores.
Se recomienda presionar solo en un extremo de la cápsula para sacarla del blíster y reducir así el riesgo de deformación o rotura de la cápsula.
4.3 Contraindicaciones
- Embarazo.
- Mujeres con capacidad de gestación, a menos que se cumplan todas las condiciones del Programa de Prevención de Embarazo (ver secciones 4.4 y 4.6).
- Pacientes varones incapaces de seguir o cumplir las medidas anticonceptivas requeridas (ver sección 4.4).
- Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo Teratogenicidad
Pomalidomida no debe tomarse durante el embarazo ya que es esperable un efecto teratogénico. Pomalidomida está relacionada estructuralmente con talidomida. Talidomida es un teratógeno conocido en humanos, que causa defectos congénitos de nacimiento graves que pueden poner en peligro la vida del niño. Pomalidomida tiene un efecto teratogénico en ratas y conejos cuando se administra durante el periodo de mayor organogénesis (ver sección 5.3).
Todas las pacientes deben cumplir las condiciones del Programa de Prevención de Embarazo a menos que exista evidencia fiable de que la paciente no tiene capacidad de gestación.
Criterios para definir a las mujeres que no tienen capacidad de gestación
Se considera que una paciente o la pareja de un paciente varón no tiene capacidad de gestación si cumple por lo menos uno de los siguientes criterios:
• Edad >50 años y con amenorrea natural durante >1 año*.
• Insuficiencia ovárica prematura confirmada por un ginecólogo especialista.
• Salpingooforectomía bilateral o histerectomía previas.
• Genotipo XY, síndrome de Turner, agenesia uterina.
*La amenorrea que pueda aparecer después de un tratamiento oncológico o durante la lactancia no descarta la capacidad de gestación.
Asesoramiento
En mujeres con capacidad de gestación, pomalidomida está contraindicada a menos que la paciente cumpla todas las condiciones que se indican a continuación:
• Comprende el riesgo teratogénico esperado para el feto.
• Comprende la necesidad de utilizar métodos anticonceptivos eficaces, sin interrupción, desde 4 semanas antes de iniciar el tratamiento, durante la duración completa del mismo y 4 semanas después de finalizarlo.
• Incluso si una mujer con capacidad de gestación tiene amenorrea, debe seguir todos los consejos sobre anticoncepción eficaz.
• Debe ser capaz de cumplir las medidas anticonceptivas eficaces.
• Está informada y comprende las potenciales consecuencias del embarazo, y la necesidad de consultar rápidamente a un especialista si hay riesgo de embarazo.
• Comprende la necesidad de comenzar el tratamiento tan pronto como se le dispense pomalidomida y tras haber obtenido un resultado negativo en la prueba de embarazo.
• Comprende la necesidad de realizar pruebas de embarazo y acepta hacérselas cada 4 semanas, excepto en el caso de que se haya sometido previamente a una ligadura de trompas de eficacia confirmada.
• Confirma que comprende los peligros y las precauciones necesarias asociadas al uso de pomalidomida.
El médico prescriptor debe comprobar que, en el caso de las mujeres con capacidad de gestación:
• La paciente cumple las condiciones del Programa de Prevención de Embarazo, incluida la confirmación de que tiene un nivel de comprensión adecuado.
• La paciente ha aceptado las condiciones mencionadas anteriormente.
En el caso de pacientes varones que toman pomalidomida, los datos farmacocinéticos han demostrado que pomalidomida está presente en el semen humano. Como medida de precaución, todos los pacientes varones que tomen pomalidomida deben cumplir los siguientes requisitos:
• Comprende el riesgo teratogénico esperado si tiene relaciones sexuales con una mujer embarazada o con una mujer con capacidad de gestación.
• Comprende la necesidad del uso de preservativos si tiene relaciones sexuales con una mujer embarazada o con una mujer con capacidad de gestación que no utiliza métodos anticonceptivos eficaces, durante el tratamiento y durante los 7 días posteriores a la interrupción de la dosis y/o el cese del tratamiento. Los varones vasectomizados deben utilizar preservativos si tienen relaciones sexuales con una mujer embarazada o con una mujer con capacidad de gestación ya que pomalidomida puede estar presente en el semen aún en ausencia de espermatozoides.
• Comprende que si su pareja se queda embarazada mientras él está tomando pomalidomida o durante los 7 días posteriores a la suspensión del tratamiento con pomalidomida, debe informar inmediatamente a su médico y es recomendable derivar a su pareja a un médico especialista o con experiencia en teratología para su evaluación y asesoramiento.
Anticoncepción
Las mujeres con capacidad de gestación deben usar un método anticonceptivo eficaz desde 4 semanas antes del tratamiento, durante el tratamiento y hasta 4 semanas después del tratamiento con pomalidomida, e incluso en el caso de interrupción de la administración, a menos que la paciente se comprometa a mantener una abstinencia sexual absoluta y continua, que será confirmada mensualmente. Si la paciente no utiliza un método anticonceptivo eficaz, debe ser derivada a un profesional sanitario debidamente capacitado con objeto de que reciba asesoramiento para empezar a utilizar métodos anticonceptivos.
Los siguientes métodos pueden considerarse ejemplos de métodos anticonceptivos adecuados:
• Implante
• Sistema de liberación intrauterino de levonorgestrel
• Sistemas “depot” de liberación de acetato de medroxiprogesterona
• Ligadura de trompas
• Relaciones sexuales sólo con varones vasectomizados; la eficacia de la vasectomía debe confirmarse mediante dos análisis de semen negativos
• Inhibidores de la ovulación que contienen progestágeno solo (p. ej., desogestrel)
Debido al riesgo aumentado de tromboembolismo venoso (TEV) en pacientes con mieloma múltiple que toman pomalidomida y dexametasona, no se recomienda el uso concomitante de anticonceptivos orales combinados (ver también sección 4.5). Si una paciente está tomando anticonceptivos orales combinados, debe cambiar a uno de los métodos anticonceptivos eficaces enumerados anteriormente. El riesgo aumentado de tromboembolismo venoso se mantiene durante un periodo de 4 a 6 semanas después de suspender el tratamiento con anticonceptivos orales combinados. La eficacia de los anticonceptivos esteroideos puede verse reducida durante el tratamiento concomitante con dexametasona (ver sección 4.5).
Los implantes y los sistemas de liberación intrauterinos de levonorgestrel se asocian con un mayor riesgo de infección en el momento de la colocación y con hemorragia vaginal irregular. En especial en las pacientes con neutropenia debe considerarse el uso profiláctico de antibióticos.
La colocación de dispositivos intrauterinos de liberación de cobre no está recomendada, debido al potencial riesgo de infección en el momento de su colocación y a la pérdida de sangre menstrual, que pueden suponer un peligro para las pacientes con neutropenia grave o trombocitopenia grave.
Pruebas de embarazo
Las mujeres con capacidad de gestación deben efectuarse pruebas de embarazo con una sensibilidad mínima de 25 mUI/ml bajo supervisión médica y conforme a la práctica habitual, tal como se explica a continuación. Este requisito incluye a las mujeres con capacidad de gestación que practican una abstinencia sexual absoluta y continua. Idealmente, la prueba de embarazo, la prescripción y la dispensación deben realizarse el mismo día. Pomalidomida se debe dispensar a las mujeres con capacidad de gestación en un plazo de siete días tras la prescripción.
Antes de iniciar el tratamiento
Debe efectuarse una prueba de embarazo bajo supervisión médica durante la consulta, en el momento de prescribir pomalidomida o en los tres días anteriores a la visita al médico prescriptor, siempre que la paciente haya estado usando un método anticonceptivo eficaz durante al menos 4 semanas. La prueba debe garantizar que la paciente no esté embarazada cuando inicie el tratamiento con pomalidomida.
Seguimiento y _finalización del tratamiento
Se debe repetir cada 4 semanas una prueba de embarazo bajo supervisión médica, y realizar otra 4 semanas después de la finalización del tratamiento, excepto en el caso de que la paciente se haya sometido a una ligadura de trompas de eficacia confirmada. Estas pruebas de embarazo deben efectuarse el mismo día de la consulta en que se prescriba el medicamento o en los tres días anteriores a la visita al médico prescriptor.
Varones
Pomalidomida está presente en el semen humano durante el tratamiento. Como medida de precaución, y teniendo en cuenta las poblaciones especiales con un tiempo de eliminación potencialmente prolongado, como la insuficiencia renal, todos los pacientes varones que tomen pomalidomida, incluyendo a aquellos que se hayan sometido a una vasectomía, deben usar preservativos durante todo el tratamiento, durante la interrupción de la administración y hasta 7 días después del final del tratamiento, si su pareja está embarazada o tiene capacidad de gestación y no está usando ningún método anticonceptivo.
Los pacientes varones no deben donar semen o esperma durante el tratamiento (periodos de interrupción de la dosis incluidos) ni en el plazo de 7 días después de la suspensión del tratamiento con pomalidomida.
Precauciones adicionales
Se debe indicar a los pacientes que no den nunca este medicamento a otra persona y que devuelvan las cápsulas sin usar al farmacéutico al final del tratamiento.
Los pacientes no deben donar sangre, semen o esperma durante el tratamiento (periodos de interrupción de la dosis incluidos) ni en el plazo de 7 días después de la suspensión del tratamiento con pomalidomida.
Material educativo, restricciones de prescripción y dispensación
Con objeto de ayudar a los pacientes a evitar la exposición fetal a pomalidomida, el Titular de la Autorización de Comercialización distribuirá material educativo a los profesionales sanitarios, destinado a reforzar las advertencias acerca de la teratogenicidad esperada de pomalidomida y a proporcionar asesoramiento sobre anticoncepción antes de iniciar el tratamiento y sobre la necesidad de realizar pruebas de embarazo. El médico debe informar a la paciente acerca del riesgo teratogénico esperado y de las estrictas medidas de prevención de embarazo, especificadas en el Programa de Prevención de Embarazo así como proporcionar a la paciente un folleto informativo adecuado, una tarjeta de paciente y/o una herramienta equivalente conforme al sistema nacional implementado de tarjeta del paciente. En colaboración con cada autoridad nacional competente, se ha implementado un sistema de distribución nacional controlada. El sistema de distribución controlada incluye el uso de una tarjeta del paciente y/o un herramienta equivalente para el control de la prescripción y/o dispensación, así como la recogida de datos detallados en relación con la indicación terapéutica, para monitorizar el uso en una indicación no autorizada dentro del territorio nacional. Idealmente, la prueba de embarazo, la prescripción y la dispensación deben realizarse el mismo día. La dispensación de pomalidomida en mujeres con capacidad de gestación debe hacerse dentro de los 7 días de prescripción y tras haber obtenido un resultado negativo en la prueba de embarazo supervisada por un médico. Las prescripciones a mujeres con capacidad de gestación pueden tener una caducidad de 4 semanas y las prescripciones para el resto de pacientes pueden tener una caducidad de 12 semanas.
Eventos hematológicos
La reacción adversa hematológica de Grado 3 o 4 notificada con mayor frecuencia en pacientes con mieloma múltiple en recaída/refractario fue la neutropenia, seguido de anemia y trombocitopenia. Se debe monitorizar a los pacientes en busca de posibles reacciones adversas hematológicas, especialmente neutropenia. Se debe advertir a los pacientes que informen rápidamente acerca de los episodios febriles que presenten. Los médicos deben estar atentos a los signos de hemorragia en los pacientes, incluyendo epistaxis, especialmente en el caso de medicación concomitante conocida por aumentar el riesgo de sangrado (ver sección 4.8). Debe efectuarse a los pacientes un hemograma completo semanal en el momento basal, durante las primeras 8 semanas y después mensualmente. Puede ser necesaria una modificación de la dosis (ver sección 4.2). Los pacientes pueden requerir el uso de hemoderivados y/o factores de crecimiento.
Eventos tromboembólicos
Se han observado eventos tromboembólicos venosos (predominantemente trombosis venosa profunda y embolia pulmonar) y eventos trombóticos arteriales (infarto de miocardio y accidente cerebrovascular) en pacientes tratados con pomalidomida en combinación con dexametasona. Los pacientes con factores de riesgo conocidos de tromboembolismo, incluida una trombosis previa, deben estar estrechamente monitorizados. Se deben tomar medidas para intentar minimizar todos los factores de riesgo modificables (p. ej. tabaquismo, hipertensión e hiperlipidemia). Se aconseja a médicos y pacientes que estén atentos a los signos y síntomas de tromboembolismo. Se debe advertir a los pacientes que soliciten atención médica si presentan síntomas como respiración entrecortada, dolor torácico o edema de las extremidades. Es recomendable el uso de terapia anticoagulante (si no está contraindicada), como el ácido acetilsalicílico, warfarina, heparina o clopidogrel, especialmente en pacientes con factores de riesgo trombótico adicionales. Después de una cuidadosa evaluación de los factores de riesgo subyacentes del paciente individual, se debe tomar una decisión respecto al uso de medidas profilácticas. En los estudios clínicos los pacientes recibieron ácido aceltilsalicílico profiláctico o terapia antitrombótica alternativa. El uso de agentes eritropoyéticos conlleva un riesgo de eventos trombóticos incluyendo tromboembolismo. Por lo tanto, deben emplearse con precaución los agentes eritropoyéticos así como otros agentes que puedan aumentar el riesgo de eventos tromboembólicos.
Neuropatía periférica
Se excluyó de los estudios clínicos con pomalidomida a los pacientes con neuropatía periférica en curso de Grado >2. Se deben adoptar las precauciones adecuadas al considerar el tratamiento de estos pacientes con pomalidomida.
Disfunción cardíaca significativa
Se excluyó de los estudios clínicos con pomalidomida a los pacientes con una disfunción cardíaca significativa (insuficiencia cardíaca congestiva [Clase III o IV de la NY Heart Association]; infarto de miocardio dentro de los 12 meses desde el inicio del estudio; angina de pecho inestable o mal controlada). Se han notificado acontecimientos de insuficiencia cardiaca, que incluyen insuficiencia cardiaca congestiva y edema pulmonar (ver sección 4.8), especialmente en pacientes con enfermedad cardiaca preexistente o factores de riesgo cardiacos. Se deben adoptar las precauciones adecuadas al considerar el tratamiento de estos pacientes con pomalidomida, incluido el control periódico para detectar la presencia de signos y síntomas de insuficiencia cardiaca.
Síndrome de lisis tumoral
Puede producirse un síndrome de lisis tumoral. Los pacientes con mayor riesgo de sufrir un síndrome de lisis tumoral son aquellos que presentan una carga tumoral elevada antes del tratamiento. Se debe monitorizar estrechamente a estos pacientes y se deben adoptar las precauciones adecuadas.
Segundas neoplasias malignas primarias
Se han notificado segundas neoplasias malignas primarias, como cáncer de piel no melanoma, en pacientes en tratamiento con pomalidomida (ver sección 4.8). Los médicos deben evaluar cuidadosamente a los pacientes antes y durante el tratamiento, utilizando pruebas estándar de detección de cáncer por si aparecieran segundas neoplasias malignas primarias e instaurar el tratamiento indicado.
Reacciones alérgicas
Se han notificado angioedema y reacciones dermatológicas graves (ver sección 4.8). Se excluyó de los estudios clínicos a los pacientes con antecedentes de reacciones alérgicas graves asociadas a talidomida o lenalidomida. Estos pacientes pueden presentar un mayor riesgo de reacciones de hipersensibilidad y no deben tomar pomalidomida. Se debe considerar la interrupción o suspensión de pomalidomida si se presenta exantema de Grado 2 o 3. Se debe suspender definitivamente el tratamiento con pomalidomida si se presenta angioedema, exantema de Grado 4 y exantema ampolloso o exfoliativo.
Mareo y confusión
Se han notificado mareo y estados de confusión con pomalidomida. Los pacientes deben evitar las situaciones en que el mareo o la confusión puedan representar un problema y no tomar otros medicamentos que puedan causar mareo o confusión sin solicitar antes consejo médico.
Enfermedad pulmonar intersticial (EPI)
Se han observado EPI y acontecimientos asociados que incluyen casos de neumonitis con pomalidomida. Se debe realizar una evaluación cuidadosa de los pacientes que presenten un inicio repentino o empeoramiento inexplicable de los síntomas pulmonares para descartar la EPI. Se debe interrumpir la administración de pomalidomida durante la investigación de estos síntomas Y, si se confirma la EPI, debe iniciarse un tratamiento adecuado. Únicamente se debe reanudar pomalidomida después de una evaluación exhaustiva de los beneficios y los riesgos.
Trastornos hepáticos
Se han observado concentraciones notablemente elevadas de alanina aminotransferasa y bilirrubina en los pacientes tratados con pomalidomida (ver sección 4.8). Se han notificado también casos de hepatitis que provocaron la suspensión de pomalidomida. Se recomienda controlar periódicamente la función hepática durante los primeros 6 meses de tratamiento con pomalidomida y, posteriormente, cuando esté clínicamente indicado.
Infecciones
Se han notificado rara vez casos de reactivación de la hepatitis B en pacientes en tratamiento con pomalidomida combinado con dexametasona que habían sido previamente infectados por el virus de la hepatitis B (VHB). Algunos de estos casos han evolucionado a fallo hepático agudo, dando lugar a la suspensión de pomalidomida. Se debe determinar el estado del virus de la hepatitis B antes de iniciar el tratamiento con pomalidomida. Se recomienda que los pacientes que den un resultado positivo en la prueba de infección por VHB se pongan en contacto con un médico con experiencia en el tratamiento de la hepatitis B. Se debe tener precaución cuando se administre pomalidomida en combinación con dexametasona en pacientes previamente infectados por el VHB, incluidos los pacientes anti-Bc positivos pero con HBsAg negativos. Se debe monitorizar estrechamente a estos pacientes para detectar signos y síntomas de infección activa por el VHB durante todo el tratamiento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Efecto de Imnovid sobre otros medicamentos
No se espera que pomalidomida pueda causar interacciones medicamentosas farmacocinéticas clínicamente relevantes debido a la inhibición o inducción de la isoenzima P450, o inhibición de transportadores cuando se administra de forma concomitante con sustratos de estas enzimas o transportadores. No se ha evaluado clínicamente el potencial de estas interacciones medicamentosas, incluyendo el posible impacto de pomalidomida en la farmacocinética de los anticonceptivos orales combinados (ver sección 4.4 Teratogenicidad).
Efecto de otros medicamentos sobre Imnovid
Pomalidomida se metaboliza parcialmente por CYP1A2 y CYP3A4/5.También es un sustrato de la glicoproteína P. La administración concomitante de pomalidomida con ketoconazol, inhibidor potente del CYP3A4/5 y de la Gp-P, o con el inductor potente del CYP3A4/5, carbamazepina, no demostró ningún efecto clínicamente relevante a la exposición a pomalidomida. La administración concomitante de pomalidomida con el inhibidor potente del CYP1A2 fluvoxamina en presencia de ketoconazol, incrementó la exposición media a pomalidomida en un 107 %, con un intervalo de confianza del 90 % [del 91 % al 124 %], frente a pomalidomida más ketoconazol. En un segundo estudio realizado para evaluar la contribución a los cambios del metabolismo de un inhibidor del CYP1A2 solo, la administración conjunta de fluvoxamina sola con pomalidomida aumentó la exposición media a pomalidomida en un 125 % con un intervalo de confianza del 90 % [del 98 % al 157 %] frente a pomalidomida administrada en monoterapia. Si se administran inhibidores potentes del CYP1A2 (p. ej., ciprofloxacino, enoxacino y fluvoxamina) de forma concomitante con pomalidomida, se debe reducir la dosis de pomalidomida en un 50 %.
Dexametasona
La administración concomitante de múltiples dosis de hasta 4 mg de pomalidomida con dexametasona de 20 mg a 40 mg (un inductor leve a moderado de varias enzimas CYP, incluido el CYP3A) en pacientes con mieloma múltiple no tuvo ningún efecto sobre la farmacocinética de pomalidomida frente a pomalidomida administrada sola.
Se desconoce el efecto de dexametasona sobre la warfarina. Se aconseja realizar una monitorización rigurosa de la concentración de warfarina durante el tratamiento.
4.6 Fertilidad, embarazo y lactancia
Mujeres con capacidad de gestación / Anticonceptivos en varones y mujeres
Las mujeres con capacidad de gestación deben utilizar métodos anticonceptivos efectivos. Si una mujer tratada con pomalidomida se queda embarazada, se debe suspender el tratamiento y derivar a la paciente a un médico especialista o con experiencia en teratología, para su evaluación y asesoramiento. Si un paciente varón toma pomalidomida y su pareja se queda embarazada, se recomienda derivar a la mujer a un médico especialista o con experiencia en teratología, para su evaluación y asesoramiento. Pomalidomida está presente en el semen humano. Como medida de precaución, todos los pacientes varones que tomen pomalidomida deben usar preservativos durante todo el tratamiento, durante la interrupción de la administración y hasta 7 días después del final del tratamiento, si su pareja está embarazada o tiene capacidad de gestación y no está usando ningún método anticonceptivo (ver secciones 4.3 y 4.4).
Embarazo
Se espera un efecto teratogénico de pomalidomida en humanos. Pomalidomida está contraindicada durante el embarazo y en mujeres con capacidad de gestación, a menos que se cumplan todas las condiciones para la prevención del embarazo, ver sección 4.3 y sección 4.4.
Lactancia
Se desconoce si pomalidomida se excreta en la leche materna. Se detectó la presencia de pomalidomida en la leche de ratas que estaban siendo amamantadas tras administrar el medicamento a la madre.
Debido a las posibles reacciones adversas en lactantes asociadas a pomalidomida, se debe decidir si es
necesario suspender la lactancia o suspender el tratamiento tras considerar la importancia del tratamiento para la madre.
Fertilidad
Se sabe que pomalidomida tiene un efecto negativo sobre la fertilidad y que es teratogénica en los animales. Tras su administración a conejas embarazadas, pomalidomida atravesó la placenta y se detectó en la sangre fetal. Ver sección 5.3.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Imnovid sobre la capacidad para conducir y utilizar máquinas es pequeña o moderada. Se han notificado casos de fatiga, disminución del nivel de conciencia, confusión y mareo relacionados con el uso de pomalidomida. Si notan estos efectos, se debe advertir a los pacientes de que no deben conducir automóviles, utilizar máquinas o realizar cualquier actividad peligrosa durante su tratamiento con pomalidomida.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Las reacciones adversas notificadas con mayor frecuencia en los estudios clínicos han sido los trastornos de la sangre y del sistema linfático, incluyendo anemia (45,7 %), neutropenia (45,3 %) y trombocitopenia (27 %); trastornos generales y alteraciones en el lugar de administración, incluyendo fatiga (28,3 %), pirexia (21 %) y edema periférico (13 %); e infecciones e infestaciones incluyendo neumonía (10,7 %). Las reacciones adversas relacionadas con neuropatía periférica fueron notificadas en el 12,3 % de los pacientes y las reacciones adversas de embolismo o tromboembolismo venoso fueron notificadas en el 3,3 % de los pacientes. Las reacciones adversas de grado 3 o 4 más frecuentes estaban relacionadas con trastornos de la sangre y del sistema linfático, incluyendo neutropenia (41,7 %), anemia (27 %) y trombocitopenia (20,7 %); infecciones e infestaciones, incluyendo neumonía (9 %); y trastornos generales y alteraciones en el lugar de administración, incluyendo fatiga (4,7 %), pirexia (3 %) y edema periférico (1,3 %). La reacción adversa grave notificada con mayor frecuencia fue la neumonía (9,3 %). Otras reacciones adversas graves notificadas incluyen neutropenia febril (4,0 %), neutropenia (2,0 %), trombocitopenia (1,7 %) y reacciones adversas de TEV (1,7 %).
Se observó que las reacciones adversas tendían a ocurrir con mayor frecuencia dentro de los primeros 2 ciclos de tratamiento con pomalidomida.
Tabla de reacciones adversas
En el estudio aleatorizado CC-4047-MM-003, un total de 302 pacientes con mieloma múltiple en recaída y refractario fueron tratados con 4 mg de pomalidomida administrada una vez al día durante 21 días en cada ciclo de 28 días, en combinación con una dosis baja semanal de dexametasona.
Las reacciones adversas observadas en los pacientes tratados con pomalidomida y dexametasona se incluyen a continuación, según el sistema de clasificación por órganos y la frecuencia (SOC por sus siglas en inglés) para todas las reacciones adversas y para las reacciones adversas de Grado 3 o 4.
Las frecuencias de las reacciones adversas son las notificadas en el grupo de pomalidomida más dexametasona del estudio CC-4047-MM-003 (n=302) y de los datos poscomercialización. Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de SOC (por sus siglas en inglés) y de frecuencia. Las frecuencias se definen, según las guías actuales, como: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); y poco frecuentes (>1/1.000 a <1/100).
|
Sistema de clasificación de órganos / Término preferente |
Todas las reacciones adversas/Frecuencia |
Reacciones adversas de Grado 3-4/Frecuencia |
|
Infecciones e |
Muy frecuentes |
Frecuentes |
|
infestaciones |
Neumonía (infecciones bacterianas, |
Sepsis neutropénica |
|
víricas y fungicas, incluidas las |
Neumonía (infecciones | |
|
infecciones oportunistas) |
bacterianas, víricas y fungicas, incluidas las infecciones | |
|
Frecuentes |
oportunistas) | |
|
Sepsis neutropénica |
Bronconeumonía | |
|
Bronconeumonía |
Infección del tracto respiratorio | |
|
Bronquitis |
Infección del tracto respiratorio | |
|
Infección del tracto respiratorio Infección del tracto respiratorio |
superior | |
|
superior |
Poco frecuentes | |
|
Nasofaringitis |
Bronquitis | |
|
Herpes zóster |
Herpes zóster | |
|
Frecuencia no conocida |
Frecuencia no conocida | |
|
Reactivación de la hepatitis B |
Reactivación de la hepatitis B | |
|
Neoplasias benignas, |
Poco frecuentes |
Poco frecuentes |
|
malignas y no |
Carcinoma de piel de células |
Carcinoma de piel de células |
|
especificadas (incl |
basales |
basales |
|
quistes y pólipos) |
Carcinoma de piel de células |
Carcinoma de piel de células |
|
escamosas |
escamosas | |
|
Trastornos de la |
Muy frecuentes |
Muy frecuentes |
|
sangre y del sistema |
Neutropenia |
Neutropenia |
|
linfático |
Trombocitopenia |
Trombocitopenia |
|
Leucopenia Anemia |
Anemia Frecuentes | |
|
Frecuentes |
Neutropenia febril | |
|
Neutropenia febril |
Leucopenia | |
|
Pancitopenia* |
Pancitopenia* | |
|
Trastornos del |
Muy frecuentes |
Frecuentes |
|
metabolismo y de la |
Disminución del apetito |
Hiperpotasemia |
|
nutrición |
Hiponatremia | |
|
Frecuentes Hiperpotasemia |
Hiperuricemia* | |
|
Hiponatremia |
Poco frecuentes | |
|
Hiperuricemia* Poco frecuentes Síndrome de lisis tumoral* |
Disminución del apetito Síndrome de lisis tumoral* | |
|
Trastornos |
Frecuentes |
Frecuentes |
|
psiquiátricos |
Estado de confusión |
Estado de confusión |
|
Sistema de clasificación de órganos / Término preferente |
Todas las reacciones adversas/Frecuencia |
Reacciones adversas de Grado 3-4/Frecuencia |
|
Trastornos del |
Frecuentes |
Frecuentes |
|
sistema nervioso |
Disminución del nivel de |
Disminución del nivel de |
|
conciencia Neuropatía sensitiva periférica |
conciencia | |
|
Mareo |
Poco frecuentes | |
|
Temblor |
Neuropatía sensitiva periférica | |
|
Hemorragia intracraneal* |
Mareo Temblor | |
|
Poco frecuentes |
Accidente cerebrovascular* | |
|
Accidente cerebrovascular* |
Hemorragia intracraneal* | |
|
Trastornos del oído y |
Frecuentes |
Frecuentes |
|
del laberinto |
Vértigo |
Vértigo |
|
Trastonos vasculares |
Frecuentes |
Poco frecuentes |
|
Trombosis venosa profunda |
Trombosis venosa profunda | |
|
Trastornos cardiacos |
Frecuentes |
Frecuentes |
|
Insuficiencia cardíaca* |
Insuficiencia cardíaca* | |
|
Fibrilación auricular* Infarto de miocardio* |
Fibrilación auricular* Poco frecuentes Infarto de miocardio* | |
|
Trastornos del |
Frecuentes |
Poco frecuentes |
|
sistema inmunológico |
Angioedema* |
Angioedema* |
|
Urticaria* |
Urticaria* | |
|
Trastornos |
Muy frecuentes |
Frecuentes |
|
respiratorios, |
Disnea |
Disnea |
|
torácicos y |
Tos | |
|
mediastínicos |
Poco frecuentes | |
|
Frecuentes |
Embolia pulmonar | |
|
Embolia pulmonar |
Tos | |
|
Epistaxis* |
Epistaxis* | |
|
Enfermedad pulmonar intersticial* |
Enfermedad pulmonar intersticial* | |
|
Trastornos |
Muy frecuentes |
Frecuentes |
|
gastrointestinales |
Diarrea |
Diarrea |
|
Náuseas |
Vómitos | |
|
Estreñimiento |
Estreñimiento | |
|
Frecuentes |
Poco frecuentes | |
|
Vómitos |
Náuseas | |
|
Hemorragia gastrointestinal |
Hemorragia gastrointestinal | |
|
Trastornos |
Poco frecuentes |
Poco frecuentes |
|
hepatobiliares |
Hiperbilirrubinemia Hepatitis* |
Hiperbilirrubinemia |
|
Trastornos de la piel y |
Frecuentes |
Frecuentes |
|
del tejido subcutáneo |
Erupción Prurito |
Erupción |
|
Sistema de clasificación de órganos / Término preferente |
Todas las reacciones adversas/Frecuencia |
Reacciones adversas de Grado 3-4/Frecuencia |
|
Trastornos |
Muy frecuentes |
Frecuentes |
|
musculoesqueléticos y |
Dolor óseo |
Dolor óseo |
|
del tejido conjuntivo |
Espasmos musculares |
Poco frecuentes Espasmos musculares |
|
Trastornos renales y |
Frecuentes |
Frecuentes |
|
urinarios |
Insuficiencia renal Retención urinaria |
Insuficiencia renal Poco frecuentes Retención urinaria |
|
Trastornos del |
Frecuentes |
Frecuentes |
|
aparato reproductor y de la mama |
Dolor pélvico |
Dolor pélvico |
|
Trastornos generales |
Muy frecuentes |
Frecuentes |
|
y alteraciones en el |
Fatiga |
Fatiga |
|
lugar de |
Pirexia |
Pirexia |
|
administración |
Edema periférico |
Edema periférico |
|
Exploraciones |
Frecuentes |
Frecuentes |
|
complementarias |
Disminución del recuento de |
Disminución del recuento de |
|
neutrófilos |
neutrófilos | |
|
Disminución del recuento de |
Disminución del recuento de | |
|
leucocitos |
leucocitos | |
|
Disminución del recuento de |
Disminución del recuento de | |
|
plaquetas |
plaquetas | |
|
Aumento de la alanina |
Aumento de la alanina | |
|
aminotransferasa Aumento del ácido úrico en sangre* |
aminotransferasa Poco frecuentes Aumento del ácido úrico en sangre* |
*Identificados a partir de los datos poscomercialización; las frecuencias se basan en los datos de los ensayos clínicos.
Descripción de reacciones adversas seleccionadas
Teratogenicidad
Pomalidomida está relacionada estructuralmente con talidomida. Talidomida es un principio activo con acción teratogénica conocida en humanos, que causa defectos de nacimiento graves que pueden poner en peligro la vida del niño. Pomalidomida tiene un efecto teratogénico en ratas y conejos cuando se administra durante el periodo de mayor organogénesis (ver secciones 4.6 y 5.3). Si se toma pomalidomida durante el embarazo, se espera un efecto teratogénico de pomalidomida en los seres humanos (ver sección 4.4).
Neutropenia y trombocitopenia
El 45,3 % de los pacientes que recibieron pomalidomida más dosis bajas de dexametasona (Pom + LD-Dex) experimentó neutropenia, frente al 19,5 % de los pacientes que recibieron dosis altas de dexametasona (HD-Dex). La neutropenia fue de grado 3 o 4 en un 41,7 % de los pacientes que recibieron Pom + LD-Dex frente al 14,8 % de los que recibieron HD-Dex. En los pacientes tratados con Pom + LD-Dex, la neutropenia fue grave en una minoría (2,0 % de los pacientes), no tuvo como consecuencia la suspensión del tratamiento, y se asoció con la interrupción del tratamiento en un 21,0 % de los pacientes y con una reducción de la dosis en un 7,7 % de los pacientes.
El 6,7 % de los pacientes que recibieron Pom + LD-Dex experimentó neutropenia febril (NF), frente a ninguno de aquellos que recibieron HD-Dex. Todos los casos fueron notificados como de grado 3 o 4. La NF fue notificada como grave en un 4,0 % de los pacientes. Asimismo, la NF se asoció con una interrupción de la dosis en un 3,7 % de los pacientes, con una reducción de la dosis en un 1,3 % de los pacientes y con ningún caso de suspensión del tratamiento.
El 27,0 % de los pacientes que recibieron Pom + LD-Dex experimentó trombocitopenia frente a un
26,8 % de los pacientes que recibieronHD-Dex. La trombocitopenia fue de grado 3 o 4 en un 20,7 % de los pacientes que recibieron Pom + LD-Dex frente a un 24,2 % de los que recibieron HD-Dex. En los pacientes tratados con Pom + LD-Dex, la trombocitopenia fue grave en un 1,7 % de los pacientes, conllevó una reducción de la dosis en un 6,3 % de los pacientes, una interrupción de la dosis en un 8 % de los pacientes y la suspensión del tratamiento en un 0,7 % de los pacientes (ver secciones 4.2 y 4.4).
Infección
La infección fue la toxicidad no hematológica más frecuente, con una incidencia del 55,0 % en los pacientes que recibieron Pom + LD-Dex frente al 48,3 % en los pacientes que recibieron HD-Dex. Aproximadamente la mitad de dichas infecciones fueron de grado 3 o 4; un 24,0 % en los pacientes tratados con Pom + LD-Dex y un 22,8 % en los pacientes tratados con HD-Dex.
Entre los pacientes tratados con Pom + LD-Dex las infecciones notificadas con mayor frecuencia fueron la neumonía y la infección del tracto respiratorio superior (en un 10,7 % y un 9,3 % de los pacientes, respectivamente); con un 24,3 % de las infecciones notificadas clasificadas como graves o mortales (grado 5) en el 2,7 % de los pacientes tratados. En los pacientes tratados con Pom + LD-Dex las infecciones conllevaron la suspensión de la dosis en un 2,0 % de los pacientes, la interrupción del tratamiento en un 14,3 % de los pacientes y la reducción de la dosis en un 1,3 % de los pacientes.
Eventos tromboembólicos
El 3,3 % de los pacientes que recibieron Pom + LD-Dex y un 2,0 % de los pacientes que recibieron HD-Dex experimentaron un embolismo o tromboembolismo venoso. El 1,3 % de los pacientes que recibieron Pom + LD-Dex experimentó reacciones de grado 3 o 4 frente a ningún caso entre los pacientes que recibieron HD-Dex. En los pacientes tratados con Pom + LD-Dex, el TEV se notificó como grave en el 1,7 %, no se notificó ninguna reacción adversa mortal en los estudios clínicos y el TEV no se asoció con suspensión de la dosis.
La profilaxis con ácido acetilsalicílico (y otros anticoagulantes en pacientes de alto riesgo) fue obligatoria para todos los pacientes participantes en los estudios clínicos. Se recomienda terapia anticoagulante (a no ser que esté contraindicada) (ver sección 4.4).
Neuropatía _ periférica
Se excluyó de los estudios clínicos a los pacientes con neuropatía periférica en curso de grado >2. El 12,3 % de los pacientes que recibieron Pom + LD-Dex experimentaron una neuropatía periférica, la mayoría de grado 1 o 2, frente a un 10,7 % de los pacientes que recibieron HD-Dex. El 1,0 % de los pacientes que recibieron Pom + LD-Dex experimentó reacciones de grado 3 o 4 frente al 1,3 % de los pacientes que recibieron HD-Dex. En los estudios clínicos, ninguna de las neuropatías periféricas se notificó como grave en los pacientes tratados con Pom + LD-Dex y la neuropatía periférica condujo a la suspensión de la dosis en un 0,3 % de los pacientes (ver sección 4.4).
La mediana del tiempo hasta la aparición de la neuropatía fue de 2,1 semanas, con una variación de 0,1 a 48,3 semanas. La mediana del tiempo hasta la aparición fue inferior en los pacientes que recibieron HD-Dex comparado con los pacientes tratados con Pom+LD-Dex (1,3 semanas frente a 2,1 semanas).
La mediana del tiempo hasta la desaparición de los síntomas fue de 22,4 semanas en los pacientes que recibieron Pom+LD-Dex y de 13,6 semanas en los pacientes que recibieron HD-Dex. El límite inferior
del intervalo de confianza del 95 % fue de 5,3 semanas en los pacientes tratados con Pom+LD-Dex y de
2,0 semanas en los pacientes que recibieron HD-Dex.
Hemorragia
Se han notificado trastornos hemorrágicos con pomalidomida, especialmente en pacientes con factores de riesgo tales como medicamentos concomitantes que aumentan el riesgo de hemorragia. Los eventos hemorrágicos incluyen epistaxis, hemorragia intracraneal y hemorragia gastrointestinal.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los
profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Se han evaluado dosis de pomalidomida de hasta 50 mg en dosis única en voluntarios sanos y 10 mg en múltiples dosis una vez al día en pacientes con mieloma múltiple sin que se haya notificado ningún caso de efecto adverso grave relacionado con sobredosis. Pomalidomida se eliminó mediante hemodiálisis. En caso de sobredosis, se recomienda terapia de soporte.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agente inmunomodulador, código ATC: L04AX06 Mecanismo de acción
Pomalidomida es un medicamento con actividad tumoricida directa contra el mieloma, actividad inmunomoduladora y capaz de inhibir el apoyo de las células del estroma para el crecimiento de las células cancerosas del mieloma múltiple. En concreto, pomalidomida inhibe la proliferación e induce la apoptosis de las células hematopoyéticas tumorales. Además, pomalidomida inhibe la proliferación de las líneas celulares de mieloma múltiple resistentes a lenalidomida y presenta un efecto sinérgico con dexametasona tanto en las líneas celulares de mieloma múltiple resistentes a lenalidomida como en las sensibles a lenalidomida para inducir la apoptosis de las células tumorales. Pomalidomida potencia la inmunidad celular mediada por los linfocitos T y por los linfocitos natural killer (NK) e inhibe la producción de citocinas proinflamatorias (p. ej. TNF-a e IL-6) por los monocitos. Pomalidomida también inhibe la angiogénesis mediante el bloqueo de la migración y adhesión de células endoteliales.
Eficacia clínica y seguridad
Se evaluó la eficacia y seguridad de pomalidomida en combinación con dexametasona en un ensayo de fase III, multicéntrico, aleatorizado y abierto (CC-4047-MM-003), donde se comparó el tratamiento de pomalidomida más una dosis baja de dexametasona (Pom + LD-Dex) frente a una dosis alta de dexametasona sola (HD-Dex) en pacientes adultos con mieloma múltiple en recaída o refractario que ya habían recibido al menos dos tratamientos previos, incluyendo lenalidomida y bortezomib, y que habían mostrado progresión de la enfermedad durante el último tratamiento. En el estudio participaron un total de 455 pacientes: 302 en el grupo Pom + LD-Dex y 153 en el grupo HD-Dex. La mayoría de los pacientes eran varones (59 %) y caucásicos (79 %); la mediana de edad de toda la población era de 64 años (mín, máx: 35, 87 años).
Los pacientes del grupo Pom + LD-Dex tomaron 4 mg de pomalidomida por vía oral en los días del 1 al 21 en cada ciclo de 28 días. Se administró la dosis de LD-Dex (40 mg) una vez al día en los días 1, 8, 15 y 22 de un ciclo de 28 días. En el grupo HD-Dex, los pacientes tomaron dexametasona (40 mg) una vez al día en los días del 1 al 4, del 9 al 12 y del 17 al 20 en un ciclo de 28 días. Los pacientes mayores de 75 años iniciaron el tratamiento con dexametasona 20 mg. Los pacientes continuaron con el tratamiento hasta la progresión de la enfermedad.
La variable principal de eficacia fue la supervivencia libre de progresión (SLP) de acuerdo con los criterios del InternationalMyeloma Working Group (IMWG). Para la población por intención de tratar (IDT), la mediana del tiempo de SLP según revisión del Independent Review Adjudication Committee (IRAC) basada en los criterios del IMWG fue de 15,7 semanas (IC de 95 %: 13,0; 20,1) en el grupo Pom + LD-Dex; la tasa estimada de supervivencia libre de eventos a las 26 semanas fue del 35,99 %
(± 3,46 %). En el grupo HD-Dex, la mediana del tiempo de SLP fue de 8,0 semanas (IC de 95 %: 7,0; 9,0); la tasa estimada de supervivencia libre de eventos a las 26 semanas fue del 12,15 % (± 3,63 %).
Se evaluó la SLP en varios subgrupos relevantes: sexo, raza, estatus de rendimiento ECOG, factores de estratificación (edad, población con enfermedad, terapias antimieloma previas [2, >2]), parámetros de relevancia pronóstica seleccionados (niveles basales de beta-2 microglobulina, niveles basales de albúmina, insuficiencia renal basal y riesgo citogenético) y exposición y refractariedad a terapias antimieloma previas. Independientemente del subgrupo evaluado, la SLP fue generalmente consistente con la observada en la población por IDT en ambos grupos de tratamiento.
En la tabla 1 se resumen los resultados de la supervivencia libre de progresión para la población por IDT. La figura 1 presenta la curva de Kaplan-Meier de la SLP para la población por IDT.
Tabla 1: Tiempo de Supervivencia Libre de Progresión según revisión del IRAC basada en
los criterios IMWG (prueba de log-rank estratificada) (población por IDT)
|
Pom+LD-Dex (N=302) |
HD-Dex (N=153) | |
|
Supervivencia libre de progresión (SLP), n |
302 (100,0) |
153 (100,0) |
|
Censurado, n (%) |
138 (45,7) |
50 (32,7) |
|
Progresión/muerte, n (%) |
164 (54,3) |
103 (67,3) |
|
Tiempo de supervivencia libre de progresión (semanas) | ||
|
Mediana3 |
15,7 |
8,0 |
|
[IC de 95 % bilateral]b |
[13,0; 20,1] |
[7,0 ; 9,0] |
|
Razón de riesgo (Hazard Ratio) (Pom+LD-Dex: HD-Dex) [IC de 95 % bilateral]0 |
0,45 [0,35; 0,59] | |
|
Valor P (bilateral) de la prueba de log-rankd |
<0,001 | |
Nota: IC = intervalo de confianza; IRAC = Comité Independiente Revisor de Evaluación; NE = no estimable a La mediana está basada en la estimación de Kaplan-Meier.
b Intervalo de confianza de 95 % de la mediana del tiempo de supervivencia libre de progresión.
c Basado en el modelo de riesgos proporcionales de Cox que compara las funciones de riesgo asociadas con los grupos de tratamiento, estratificados por edad (<75 frente a >75), población con la enfermedad (refractario a lenalidomida y bortezomib frente a no refractario a estos dos medicamentos), y número de terapias antimieloma previas (=2 frente a >2).
d El valor P está basado en la prueba de log-rank estratificada con los mismos factores de estratificación arriba mencionados para el modelo de Cox.
Fecha de corte: 7 de septiembre de 2012
Supervivencia Libre de Progresión según la revisión de respuesta del IRAC basada en los criterios IMWG (prueba de log-rank estratificada) (población por IDT)
Figura 1:
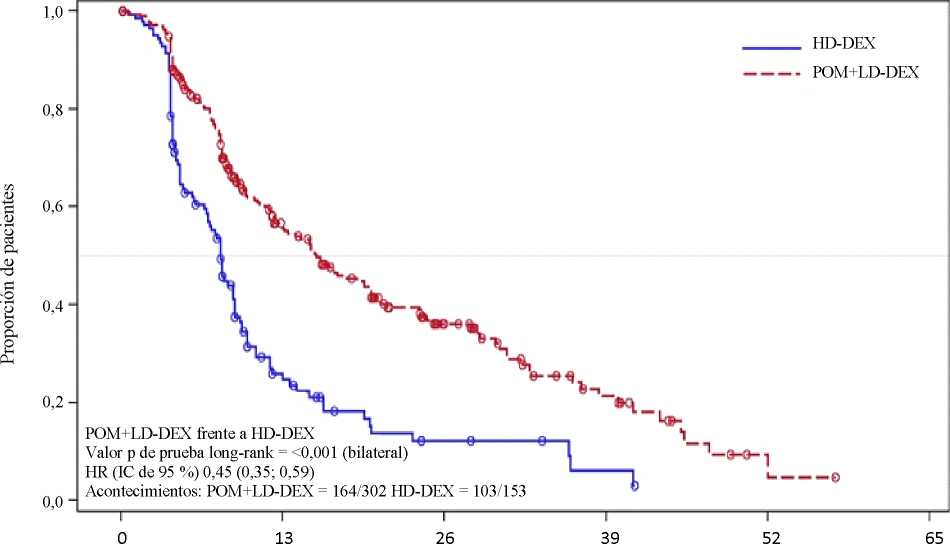
Fecha de corte: 7 de septiembre de 2012
Supervivencia Libre de Progresión (semanas)
La variable secundaria clave fue la supervivencia global (SG). Un total de 226 pacientes (74,8 %) del grupo Pom + LD-Dex y 95 pacientes (62,1 %) del grupo HD-Dex vivían en el momento de la fecha de corte (7 de septiembre de 2012). La mediana del tiempo de supervivencia global según las estimaciones Kaplan-Meier no fue alcanzado por el grupo Pom + LD Dex, pero podría esperarse que fuera al menos de 48 semanas, que corresponde al umbral más bajo del IC de 95 %. La mediana del tiempo de SG del grupo HD-Dex fue de 34 semanas (IC de 95 %: 23,4; 39,9). La tasa libre de eventos al año fue del 52,6 % (± 5,72 %) para el grupo Pom + LD-Dex y del 28,4 % (± 7,51 %) para el grupo HD-Dex. La diferencia en términos de SG entre los dos grupos de tratamiento fue estadísticamente significativa (p <0,001).
La Tabla 2 resume los resultados de supervivencia global para la población por IDT. La curva Kaplan-Meier de SG para la población por IDT se presenta en la Figura 2.
Basándose en los resultados de las variables SLP y SG, el Comité de Monitorización de Datos para este estudio recomendó que se completara el estudio y que los pacientes en el grupo HD-Dex fueran cruzados al grupo Pom + LD-Dex.
|
Estadísticas |
Pom+LD-Dex (N=302) |
HD-Dex (N=153) | |
|
N |
302 (100,0) |
153 (100,0) | |
|
Censurado |
n (%) |
226 (74,8) |
95 (62,1) |
|
Muerto |
n (%) |
76 (25,2) |
58 (37,9) |
|
Tiempo de supervivencia (semanas) |
Mediana3 |
NE |
34,0 |
|
IC de 95 % bilateralb |
[ 48,1; NE] |
[ 23,4; 39,9] | |
|
Razón de riesgo (Hazard Ratio) (Pom+LD-Dex:HD-Dex) [IC de 95 % bilateral0] |
0,53 [ 0,37; 0,74] | ||
|
Valor P (bilateral) de la prueba de log-rankd |
<0,001 | ||
Nota: IC = intervalo de confianza; NE = no estimable. a La mediana está basada en la estimación de Kaplan-Meier.
b Intervalo de confianza de 95 % sobre la mediana del tiempo de supervivencia global.
c Basado en el modelo de riesgos proporcionales de Cox que compara las funciones de riesgo asociadas con los grupos de tratamiento. d El valor P está basado en la prueba de log-rank no estratificada.
Fecha de corte: 7 de septiembre de 2012
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Imnovid en todos los grupos de la población pediátrica en mieloma múltiple (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Pomalidomida se absorbe alcanzando una concentración plasmática máxima (Cmáx) a las 2 o 3 horas y, por lo menos, un 73 % se absorbe después de administrar una dosis única por vía oral. El área bajo la curva (AUC) de pomalidomida aumenta, aproximadamente, lineal y proporcionalmente con los
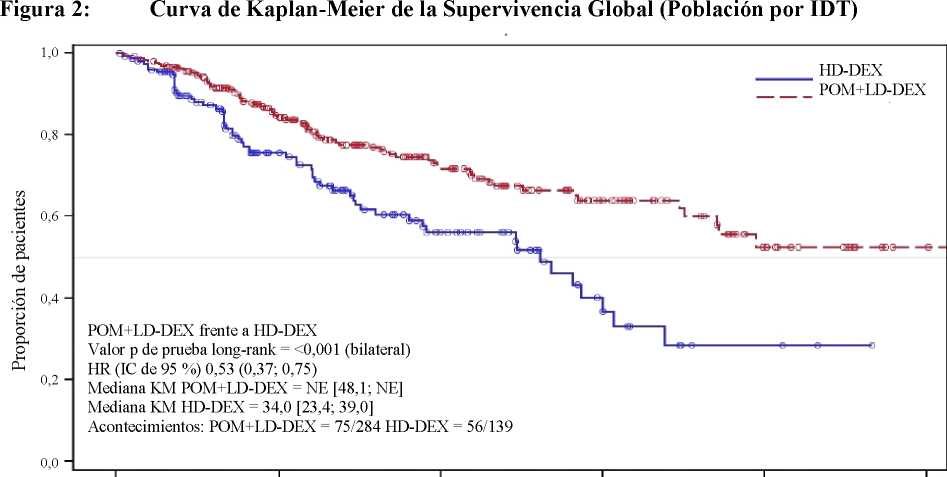
0 13
Fecha de corte: 7 de septiembre de 2012
26 39 52
Superviviencia global (semanas)
65
incrementos de la dosis. Tras la administración de múltiples dosis, pomalidomida tiene una ratio de acumulación del 27 al 31 % en el AUC.
La administración conjunta con una comida rica en grasas y rica en calorías reduce la tasa de absorción, disminuyendo la Cmáx plasmática media en, aproximadamente un 27 %, pero con un efecto mínimo sobre la extensión de la absorción global con una disminución del 8 % en el AUC media. Por tanto, pomalidomida puede administrarse con o sin alimentos.
Distribución
Pomalidomida tiene un volumen de distribución aparente (Vd) medio de entre 62 y 138 litros en estado estable. En el semen de sujetos sanos pomalidomida se distribuye a una concentración de aproximadamente el 67 % del nivel de plasma a las 4 horas posteriores a la administración (aproximadamente Tmáx) tras 4 días de administración de 2 mg una vez al día. La unión in vitro de los enantiómeros de pomalidomida a las proteínas plasmáticas en humanos oscila entre el 12 % y el 44 % y no es dependiente de la concentración.
Biotransformación
En sujetos sanos que han recibido una dosis única por vía oral de [14C]-pomalidomida (2 mg), pomalidomida es la mayor sustancia circulante (aproximadamente el 70 % de la radioactividad del plasma) in vivo. No se hallaron metabolitos a >10 % relativos a la radioactividad total o relacionada en plasma.
Las rutas metabólicas principales de la radioactividad excretada son los procesos de hidroxilación con la posterior glucuronidación, o hidrólisis. Los estudios in vitro identificaron al CYP1A2 y al CYP3A4 como las enzimas primarias implicadas en la hidroxilación de pomalidomida mediada por CYP, con contribuciones menores adicionales del CYP2C19 y CYP2D6. Pomalidomida es también un sustrato de la glicoproteína-P in vitro. La administración concomitante de pomalidomida con el potente inhibidor del CYP3A4/5 y de la Gp-P, ketoconazol, o con el potente inductor del CYP3A4/5, carbamazepina, no demostró ningún efecto clínicamente relevante en la exposición a pomalidomida. La administración concomitante de pomalidomida con el inhibidor potente del CYP1A2, fluvoxamina, en presencia de ketoconazol, incrementó la exposición media a pomalidomida en un 107 %, con un intervalo de confianza del 90 % [del 91 % al 124 %], frente a pomalidomida más ketoconazol. En un segundo estudio realizado para evaluar la contribución a los cambios del metabolismo de un inhibidor del CYP1A2 solo, la administración conjunta de fluvoxamina sola con pomalidomida aumentó la exposición media a pomalidomida en un 125 % con un intervalo de confianza del 90 % [del 98 % al 157 %] frente a pomalidomida administrada en monoterapia. Si se administran inhibidores potentes del CYP1A2 (p. ej., ciprofloxacino, enoxacino y fluvoxamina) de forma concomitante con pomalidomida, se debe reducir la dosis de pomalidomida en un 50 %. La administración de pomalidomida a fumadores, sabiendo que el tabaquismo induce la isoforma CYP1A2, no tuvo ningún efecto clínicamente relevante en la exposición a pomalidomida frente a la exposición a pomalidomida observada en los no fumadores.
Según los datos in vitro, pomalidomida no es un inhibidor o inductor de las isoenzimas del citocromo P-450 y no inhibió ninguno de los fármacos transportadores que fueron estudiados. No se espera que pomalidomida pueda causar interacciones medicamentosas clínicamente relevantes cuando se administra de forma concomitante con sustratos de estas rutas.
Eliminación
En sujetos sanos pomalidomida se elimina con una mediana de semivida plasmática de aproximadamente 9,5 horas y de unas 7,5 horas en pacientes con mieloma múltiple. Pomalidomida tiene una media de aclaramiento corporal total (Cl/F) de aproximadamente 7-10 l/h.
Tras una dosis única por vía oral de [14C]-pomalidomida (2 mg) en sujetos sanos, aproximadamente el 73 % y el 15 % de la dosis radioactiva se eliminó por la orina y las heces, respectivamente, con aproximadamente el 2 % y el 8 % del radiocarbono administrado eliminado como pomalidomida en orina y heces.
Pomalidomida se metaboliza ampliamente antes de la excreción, con los metabolitos resultantes eliminados principalmente por la orina. Los tres metabolitos predominantes en la orina (formados mediante hidrólisis o hidroxilación con posterior glucuronidación) representan, aproximadamente, el
23 %, 17 % y 12 %, respectivamente, de la dosis en la orina.
Los metabolitos dependientes del CYP representan aproximadamente el 43 % de la radiactividad total excretada, mientras que los metabolitos hidrolíticos no dependientes del CYP representan el 25 %, y la excreción de pomalidomida inalterada representa el 10 % (2 % en orina y 8 % en heces).
Farmacocinética poblacional
Según análisis de farmacocinética poblacional basado en un modelo bicompartimental, los sujetos sanos y los pacientes con mieloma múltiple mostraron aclaramiento aparente (Cl/F) y volumen de distribución aparente en el compartimento central (WF) comparables. En tejidos periféricos, pomalidomida fue absorbida preferentemente por los tumores con un aclaramiento de distribución aparente en el compartimento periférico (Q/F) y un volumen de distribución aparente en el compartimento periférico (V3/F) 3,7 veces y 8 veces mayor, respectivamente, comparado con los sujetos sanos.
Población pediátrica
No existen datos disponibles sobre la administración de pomalidomida en niños o adolescentes (<18 años).
Población de edad avanzada
Según análisis de farmacocinética poblacional en sujetos sanos y en pacientes con mieloma múltiple, no se observó una influencia significativa de la edad (19-83 años) en el aclaramiento oral de pomalidomida. En los estudios clínicos los pacientes de edad avanzada (>65 años) expuestos a pomalidomida no requirieron ningún ajuste de dosis. Por favor, ver sección 4.2.
Insuficiencia renal
Los análisis de farmacocinética poblacional mostraron que los parámetros farmacocinéticos de pomalidomida no se vieron afectados de forma destacable en los pacientes con insuficiencia renal (definida mediante el aclaramiento de la creatinina o el filtrado glomerular estimado [FGe]) en comparación con los pacientes con la función renal normal (CrCl >60 ml/minuto). La exposición media a pomalidomida normalizada según el AUC fue del 98,2 % con un intervalo de confianza del 90 %
[77,4 % al 120,6 %] en los pacientes con insuficiencia renal moderada (FGe >30 a <45 ml/minuto/1,73 m2) en comparación con los pacientes con la función renal normal. La exposición media a pomalidomida normalizada según el AUC fue del 100,2 % con un intervalo de confianza del 90 % [79,7 % al 127,0 %] en los pacientes con insuficiencia renal grave que no precisaban diálisis (CrCl <30 o FGe <30 ml/minuto/1,73 m2) en comparación con los pacientes con la función renal normal. La exposición media a pomalidomida normalizada según el AUC aumentó en un 35,8 % con un IC del 90 % [7,5 % al 70,0 %] en los pacientes con insuficiencia renal grave que precisaban diálisis (CrCl <30 ml/minuto con necesidad de diálisis) en comparación con los pacientes con la función renal normal. Los cambios medios en la exposición a pomalidomida en cada uno de estos grupos de insuficiencia renal no son de una magnitud que requiera un ajuste de la dosis.
Insuficiencia hepática
Los parámetros farmacocinéticos cambiaron modestamente en los pacientes con insuficiencia hepática (definida mediante los criterios de Child-Pugh) en comparación con los sujetos sanos. La exposición media a pomalidomida aumentó en un 51 % con un intervalo de confianza del 90 % [del 9 % al 110 %] en los pacientes con insuficiencia hepática leve en comparación con los sujetos sanos. La exposición media a pomalidomida aumentó en un 58% con un intervalo de confianza del 90 % [del 13 % al 119 %] en los pacientes con insuficiencia hepática moderada en comparación con los sujetos sanos. La exposición media a pomalidomida aumentó en un 72 % con un intervalo de confianza del 90 % [del
24 % al 138 %] en los pacientes con insuficiencia hepática grave en comparación con los sujetos sanos. Los aumentos medios en la exposición a pomalidomida en cada uno de estos grupos de insuciencia hepática no son de una magnitud que requiera un ajuste del esquema de tratamiento o de la dosis (ver sección 4.2).
Estudios de toxicidad de dosis repetidas
La administración crónica de pomalidomida en ratas en dosis de 50, 250 y 1000 mg/kg/día durante 6 meses fue bien tolerada. No se detectó ningún efecto adverso hasta los 1000 mg/kg/día (una tasa de exposición 175 veces más elevada que la dosis clínica de 4 mg).
Se evaluó pomalidomida en monos en estudios de dosis repetidas de hasta 9 meses de duración. En estos estudios, los monos mostraron una mayor sensibilidad a los efectos de pomalidomida que las ratas. Las toxicidades primarias observadas en monos estuvieron relacionadas con los sistemas hematopoyético/linforreticular. En el estudio de 9 meses en monos con dosis de 0,05, 0,1 y 1 mg/kg/día se observó morbilidad y eutanasia temprana de 6 animales a dosis de 1 mg/kg/día que fueron atribuidas a los efectos inmunosupresores (infección por estafilococos, reducción de los linfocitos en sangre periférica, inflamación crónica del intestino grueso, reducción histológica de los linfocitos e hipocelularidad de la médula ósea) a exposiciones elevadas de pomalidomida (15 veces la tasa de exposición comparada con una dosis clínica de 4 mg). Dichos efectos inmunosupresores provocaron la eutanasia temprana de 4 monos debido a su mal estado de salud (heces líquidas, inapetencia, ingesta de alimentos reducida y pérdida de peso); la evaluación histopatológica de estos animales demostró inflamación crónica del intestino grueso y atrofia vellosa del intestino delgado. Se observó infección por estafilococos en 4 monos; 3 de éstos respondieron al tratamiento con antibióticos y uno murió sin tratamiento. Además, resultados consistentes con la leucemia mielógena aguda llevaron a la eutanasia de un mono; las observaciones clínicas y la patología clínica y/o alteraciones de la médula ósea observadas en este animal eran consistentes con inmunosupresión. La proliferación mínima o leve en los conductos biliares con incrementos asociados de la ALP y de la GGT también se observaron a dosis de 1 mg/kg/día. La evaluación de los animales recuperados indicó que todos los resultados relacionados con el tratamiento eran reversibles a las 8 semanas del cese de la administración, excepto la proliferación de los conductos biliares intrahepáticos observada en 1 animal en el grupo de 1 mg/kg/día. El nivel sin efecto adverso observado (NOAEL) fue de 0,1 mg/kg/día (una tasa de exposición relativa de 0,5 veces comparada con la dosis clínica de 4 mg).
Genotoxicidad/carcinogenicidad
Pomalidomida no resultó mutagénica en los ensayos de mutaciones bacterianas y de los mamíferos, y no indujo aberraciones cromosómicas en los linfocitos de sangre periférica en humanos así como tampoco a la formación de micronúcleos en eritrocitos policromáticos en la médula ósea de ratas a las que les fueron administradas dosis de hasta 2000 mg/kg/día. No se han realizado estudios de carcinogenicidad.
Fertilidad y desarrollo embrionario temprano
En un estudio de fertilidad y desarrollo embrionario temprano en ratas, se administró pomalidomida a los machos y las hembras a dosis de 25, 250 y 1000 mg/kg/día. El examen uterino en el día de gestación 13 mostró una reducción de la cantidad media de embriones viables y un aumento en la pérdida postimplantación con todos los niveles de dosis. Por consiguiente, el NOAEL en estos eventos observados fue <25 mg/kg/día (con un AUC24h de 39960 ng-h/ml (nanogramo^hora/mililitros) para la dosis más baja evaluada y una tasa de exposición 99 veces relativa a la dosis clínica de 4 mg). Cuando los machos tratados en este estudio fueron apareados con hembras no tratadas, todos los parámetros uterinos fueron comparables a los controles. Según estos resultados, los efectos observados fueron atribuidos al tratamiento de las hembras.
Desarrollo embriofetal
Pomalidomida resultó ser teratógena en ratas y conejos cuando se administró durante el periodo de mayor organogénesis. En el estudio de toxicidad sobre el desarrollo embriofetal de la rata, se observaron malformaciones relacionadas con la ausencia de vejiga urinaria, ausencia de la glándula tiroidea, así como la fusión y la desalineación de los elementos vertebrales torácicos y lumbares (arcos centrales y/o neurales) a todos los niveles de dosis (25, 50 y 1000 mg/kg/día).
En este estudio no se observó toxicidad materna. Por ello, el NOAEL materno fue de 1000 mg/kg/día, y el NOAEL para la toxicidad de desarrollo fue <25 mg/kg/día (con un AUC24h de 34340 ng-h/ml en el día de gestación 17 para la dosis más baja evaluada y la tasa de exposición fue de 85 veces comparada
con la dosis clínica de 4 mg). En conejos, pomalidomida a dosis entre los 10 y 250 mg/kg/día produjo malformaciones en el desarrollo embriofetal. Se observaron aumentos de las anomalías cardíacas a todas las dosis con aumentos significativos a 250 mg/kg/día. A dosis de entre 100 y 250 mg/kg/día se registró un ligero aumento de la pérdida post-implantación y un ligero descenso en el peso del feto. A dosis de 250 mg/kg/día, las malformaciones fetales incluyeron anomalías en las extremidades (extremidades anteriores y posteriores dobladas y/o giradas, ausencia de dígito o dígito libre) y malformaciones esqueléticas asociadas (metacarpiano no osificado, metacarpiano y falange no alineados, ausencia de dígito, falange no osificada y tibia corta no osificada o doblada); dilatación moderada de los ventrículos laterales del cerebro; ubicación anormal de la arteria subclavia derecha; ausencia de los lóbulos intermedios pulmonares; par de riñones desplazados hacia abajo; morfología hepática alterada; ausencia de osificación de la pelvis u osificación incompleta; aumento medio de las costillas torácicas supernumerarias y reducción media de los tarsales osificados. Además, se observó una ligera reducción en el incremento del peso materno, una reducción significativa de los triglicéridos, y una reducción significativa del peso absoluto y relativo del bazo a dosis de entre 100 y 250 mg/kg/día. El NOAEL materno fue de 10 mg/kg/día y el NOAEL del desarrollo fue de <10 mg/kg/día (con un AUC24h de 418 ng-h/ml en el día de gestación 19 para la dosis más baja evaluada, similar a la obtenida con una dosis clínica de 4 mg).
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Contenido de la cápsula:
Manitol
Almidón pregelatinizado Estearil fumarato de sodio
Cubierta de la cápsula:
La cubierta de la cápsula de 2 mg contiene gelatina, dióxido de titanio (E171), indigotina (E132), óxido de hierro amarillo (E172), eritrosina (E172) y tinta blanca.
Tinta de impresión:
La cubierta de la cápsula de 2 mg contiene: tinta blanca - goma laca shellac, dióxido de titanio (E171), simeticona, propilenglicol (E1520) e hidróxido de amonio (E527).
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
4 años.
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Las cápsulas están envasadas en blísteres de polivinilcloruro (PVC) / policlorotrifluoroetileno (PCTFE) con lámina de aluminio.
Tamaño del envase de 21 cápsulas.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Las cápsulas no se deben abrir o triturar. En el caso de que el polvo de pomalidomida entre en contacto con la piel, debe lavar abundantemente la piel con agua y jabón inmediatamente. En el caso de que el polvo de pomalidomida entre en contacto con las membranas mucosas, debe lavarlas abundantemente con agua a presión.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local. El medicamento no utilizado debe devolverse al farmacéutico al final del tratamiento.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd.
1 Longwalk Road Stockley Park Uxbridge UB11 1DB Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/850/002
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 05/agosto/2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la identificación de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Imnovid 3 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula dura contiene 3 mg de pomalidomida.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura.
Imnovid 3 mg cápsula dura: tapa opaca de color azul oscuro y cuerpo opaco de color verde marcadas con “POML 3 mg” en tinta blanca, de tamaño 2, cápsulas de gelatina dura.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Imnovid en combinación con dexametasona está indicado en el tratamiento de los pacientes adultos con mieloma múltiple resistente al tratamiento o recidivante que hayan recibido al menos dos tratamientos previos, incluyendo lenalidomida y bortezomib, y que hayan experimentado una progresión de la enfermedad en el último tratamiento.
4.2 Posología y forma de administración
El tratamiento debe iniciarse y monitorizarse bajo la supervisión de médicos con experiencia en el tratamiento del mieloma múltiple.
Posología
La dosis inicial recomendada es de 4 mg de Imnovid una vez al día por vía oral, en los días del 1 al 21 de ciclos repetidos de 28 días. La dosis recomendada de dexametasona es de 40 mg por vía oral una vez al día, en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
La posología se mantiene o modifica en función de los resultados clínicos y de laboratorio.
El tratamiento debe suspenderse si existe evidencia de progresión de la enfermedad.
Modificación o interrupción de la dosis de _pomalidomida
Las instrucciones para la interrupción y reducción de la dosis de pomalidomida relacionadas con reacciones adversas hematológicas se indican en la siguiente tabla:
|
Toxicidad |
Modificación de la dosis |
|
NeutroDenia • RAN* <0,5 x 109/l o neutropenia febril (fiebre >38,5°C y RAN <1 x 109/l) |
Interrumpir el tratamiento con pomalidomida, control semanal del hemograma completo. |
|
• RAN vuelve a >1 x 109/1 |
Reanudar el tratamiento con 3 mg de pomalidomida al día. |
|
• Con cada disminución posterior a <0,5 x 1071 |
Interrumpir el tratamiento con pomalidomida |
|
• RAN vuelve a >1 x 109/l |
Reanudar el tratamiento con 1 mg menos de pomalidomida que la dosis previa |
|
T rombocitopenia • Recuento de plaquetas <25 x 109/l |
Interrumpir el tratamiento con pomalidomida, control semanal del hemograma completo |
|
• Recuento de plaquetas vuelve a >50 x 109/l |
Reanudar el tratamiento con 3 mg de pomalidomida al día |
|
• Con cada disminución posterior a <25 x 1071 |
Interrumpir el tratamiento con pomalidomida |
|
• Recuento de plaquetas vuelve a >50 x 109/l |
Reanudar el tratamiento con 1 mg menos de pomalidomida que la dosis previa |
*RAN - Recuento absoluto de neutrófilos
Para iniciar un nuevo ciclo de pomalidomida, el recuento de neutrófilos debe ser >1 x 109/l y el recuento de plaquetas debe ser >50 x 109/l.
En caso de neutropenia, el médico debe considerar el uso de factores de crecimiento.
En el caso de otras reacciones adversas de grado 3 o 4 relacionadas con pomalidomida, el médico debe considerar interrumpir el tratamiento y reanudarlo con un 1 mg menos que la dosis previa una vez que se haya disminuido la reacción adversa a un grado inferior o igual a 2.
Si la reacción adversa ocurre tras disminuciones de la dosis a 1 mg, entonces debe suspenderse el tratamiento con este medicamento.
Se debe considerar la interrupción o suspensión de pomalidomida en caso de exantema de grado 2-3. Se debe suspender el tratamiento con pomalidomida en caso de angioedema, exantema de grado 4 y exantema ampolloso o exfoliativo, y no se debe reanudar una vez suspendido por estas reacciones.
Si se administran inhibidores potentes del CYP1A2 (p. ej., ciprofloxacino, enoxacino y fluvoxamina) de forma concomitante con pomalidomida, se debe reducir la dosis de pomalidomida en un 50 %.
|
Toxicidad |
Modificación de la dosis |
|
Dispepsia = grado 1-2 Dispepsia > grado 3 |
Mantener la dosis y tratar con antihistamínicos H2 o equivalentes. Reducir la dosis en un nivel de dosis si los síntomas persisten. Interrumpir la administración hasta que se controlen los síntomas. Añadir antihistamínicos H2 o equivalentes y reducir la dosis en un nivel cuando se reanude su administración. |
|
Edema > grado 3 |
Usar diuréticos según sea necesario y reducir la dosis en un nivel de dosis. |
|
Confusión o cambios en el estado de ánimo > grado 2 |
Interrumpir la administración hasta que desaparezcan los síntomas. Cuando se reanude su administración reducir la dosis en un nivel de dosis. |
|
Debilidad muscular > grado 2 |
Interrumpir la administración hasta que la debilidad muscular sea < grado 1. Reiniciar la dosis con una reducción de un nivel. |
|
Hiperglucemia > grado 3 |
Reducir la dosis en un nivel de dosis. Tratar con insulina o hipoglucemiantes orales según sea necesario. |
|
Pancreatitis aguda |
Suspensión de dexametasona del régimen de tratamiento del paciente. |
|
Otros efectos adversos relacionados con dexametasona > grado 3 |
Interrumpir la administración de dexametasona hasta que los efectos adversos sean de grado < 2. Reanudar su administración con una reducción de un nivel de la dosis. |
Niveles de reducción de la dosis de dexametasona:
Niveles de reducción de la dosis (<75 años): dosis inicial 40 mg; nivel de dosis -1 20 mg; nivel de dosis -2 10 mg en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
Niveles de reducción de la dosis (>75 años): dosis inicial 20 mg; nivel de dosis -1 12 mg; nivel de dosis -2 8 mg en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
Si la recuperación de las toxicidades tarda más de 14 días, la dosis de dexametasona se reducirá en un nivel de dosis.
Poblaciones especiales Población _ pediátrica
No hay un uso relevante de Imnovid en niños de 0 a 17 años para la indicación del mieloma múltiple.
Pacientes de edad avanzada
No se requiere ningún ajuste de dosis de pomalidomida. En pacientes de >75 años, la dosis inicial de dexametasona es de 20 mg una vez al día en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
Insuficiencia renal
No se requiere ningún ajuste de dosis de pomalidomida en los pacientes con insuficiencia renal. Los días de hemodiálisis, los pacientes deben tomar la dosis de pomalidomida después de la hemodiálisis.
Insuficiencia hepática
Los pacientes con una concentración sérica de bilirrubina total >2,0 mg/dl se excluyeron de los estudios clínicos. La insuficiencia hepática tiene un efecto modesto sobre la farmacocinética de pomalidomida (ver sección 5.2). No se requiere ajustar la dosis inicial de pomalidomida en pacientes con insuficiencia hepática según definen los criterios de Child-Pugh. Sin embargo, los pacientes con insuficiencia hepática deben ser monitorizados cuidadosamente por si presentan reacciones adversas y se debe reducir la dosis o suspender la administración de pomalidomida según sea necesario.
Forma de administración Vía oral.
Imnovid debe tomarse a la misma hora cada día. Las cápsulas no deben abrirse, romperse ni masticarse (ver sección 6.6). Este medicamento debe tomarse entero, preferiblemente con agua, con o sin alimentos. Si el paciente olvida tomar una dosis de Imnovid un día, el paciente debe entonces tomar la próxima dosis al día siguiente a la hora habitual. Los pacientes no deben ajustar la dosis para compensar una dosis olvidada en días anteriores.
Se recomienda presionar solo en un extremo de la cápsula para sacarla del blíster y reducir así el riesgo de deformación o rotura de la cápsula.
4.3 Contraindicaciones
- Embarazo.
- Mujeres con capacidad de gestación, a menos que se cumplan todas las condiciones del Programa de Prevención de Embarazo (ver secciones 4.4 y 4.6).
- Pacientes varones incapaces de seguir o cumplir las medidas anticonceptivas requeridas (ver sección 4.4).
- Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo Teratogenicidad
Pomalidomida no debe tomarse durante el embarazo ya que es esperable un efecto teratogénico. Pomalidomida está relacionada estructuralmente con talidomida. Talidomida es un teratógeno conocido en humanos, que causa defectos congénitos de nacimiento graves que pueden poner en peligro la vida del niño. Pomalidomida tiene un efecto teratogénico en ratas y conejos cuando se administra durante el periodo de mayor organogénesis (ver sección 5.3).
Todas las pacientes deben cumplir las condiciones del Programa de Prevención de Embarazo a menos que exista evidencia fiable de que la paciente no tiene capacidad de gestación.
Criterios para definir a las mujeres que no tienen capacidad de gestación
Se considera que una paciente o la pareja de un paciente varón no tiene capacidad de gestación si cumple por lo menos uno de los siguientes criterios:
• Edad >50 años y con amenorrea natural durante >1 año*.
• Insuficiencia ovárica prematura confirmada por un ginecólogo especialista.
• Salpingooforectomía bilateral o histerectomía previas.
• Genotipo XY, síndrome de Turner, agenesia uterina.
*La amenorrea que pueda aparecer después de un tratamiento oncológico o durante la lactancia no descarta la capacidad de gestación.
Asesoramiento
En mujeres con capacidad de gestación, pomalidomida está contraindicada a menos que la paciente cumpla todas las condiciones que se indican a continuación:
• Comprende el riesgo teratogénico esperado para el feto.
• Comprende la necesidad de utilizar métodos anticonceptivos eficaces, sin interrupción, desde 4 semanas antes de iniciar el tratamiento, durante la duración completa del mismo y 4 semanas después de finalizarlo.
• Incluso si una mujer con capacidad de gestación tiene amenorrea, debe seguir todos los consejos sobre anticoncepción eficaz.
• Debe ser capaz de cumplir las medidas anticonceptivas eficaces.
• Está informada y comprende las potenciales consecuencias del embarazo, y la necesidad de consultar rápidamente a un especialista si hay riesgo de embarazo.
• Comprende la necesidad de comenzar el tratamiento tan pronto como se le dispense pomalidomida y tras haber obtenido un resultado negativo en la prueba de embarazo.
• Comprende la necesidad de realizar pruebas de embarazo y acepta hacérselas cada 4 semanas, excepto en el caso de que se haya sometido previamente a una ligadura de trompas de eficacia confirmada.
• Confirma que comprende los peligros y las precauciones necesarias asociadas al uso de pomalidomida.
El médico prescriptor debe comprobar que, en el caso de las mujeres con capacidad de gestación:
• La paciente cumple las condiciones del Programa de Prevención de Embarazo, incluida la confirmación de que tiene un nivel de comprensión adecuado.
• La paciente ha aceptado las condiciones mencionadas anteriormente.
En el caso de pacientes varones que toman pomalidomida, los datos farmacocinéticos han demostrado que pomalidomida está presente en el semen humano. Como medida de precaución, todos los pacientes varones que tomen pomalidomida deben cumplir los siguientes requisitos:
• Comprende el riesgo teratogénico esperado si tiene relaciones sexuales con una mujer embarazada o con una mujer con capacidad de gestación.
• Comprende la necesidad del uso de preservativos si tiene relaciones sexuales con una mujer embarazada o con una mujer con capacidad de gestación que no utiliza métodos anticonceptivos eficaces, durante el tratamiento y durante los 7 días posteriores a la interrupción de la dosis y/o el cese del tratamiento. Los varones vasectomizados deben utilizar preservativos si tienen relaciones sexuales con una mujer embarazada o con una mujer con capacidad de gestación ya que pomalidomida puede estar presente en el semen aún en ausencia de espermatozoides.
• Comprende que si su pareja se queda embarazada mientras él está tomando pomalidomida o durante los 7 días posteriores a la suspensión del tratamiento con pomalidomida, debe informar inmediatamente a su médico y es recomendable derivar a su pareja a un médico especialista o con experiencia en teratología para su evaluación y asesoramiento.
Anticoncepción
Las mujeres con capacidad de gestación deben usar un método anticonceptivo eficaz desde 4 semanas antes del tratamiento, durante el tratamiento y hasta 4 semanas después del tratamiento con pomalidomida, e incluso en el caso de interrupción de la administración, a menos que la paciente se comprometa a mantener una abstinencia sexual absoluta y continua, que será confirmada mensualmente. Si la paciente no utiliza un método anticonceptivo eficaz, debe ser derivada a un profesional sanitario debidamente capacitado con objeto de que reciba asesoramiento para empezar a utilizar métodos anticonceptivos.
Los siguientes métodos pueden considerarse ejemplos de métodos anticonceptivos adecuados:
• Implante
• Sistema de liberación intrauterino de levonorgestrel
• Sistemas “depot” de liberación de acetato de medroxiprogesterona
• Ligadura de trompas
• Relaciones sexuales sólo con varones vasectomizados; la eficacia de la vasectomía debe confirmarse mediante dos análisis de semen negativos
• Inhibidores de la ovulación que contienen progestágeno solo (p. ej., desogestrel)
Debido al riesgo aumentado de tromboembolismo venoso (TEV) en pacientes con mieloma múltiple que toman pomalidomida y dexametasona, no se recomienda el uso concomitante de anticonceptivos orales combinados (ver también sección 4.5). Si una paciente está tomando anticonceptivos orales combinados, debe cambiar a uno de los métodos anticonceptivos eficaces enumerados anteriormente. El riesgo aumentado de tromboembolismo venoso se mantiene durante un periodo de 4 a 6 semanas después de suspender el tratamiento con anticonceptivos orales combinados. La eficacia de los anticonceptivos esteroideos puede verse reducida durante el tratamiento concomitante con dexametasona (ver sección 4.5).
Los implantes y los sistemas de liberación intrauterinos de levonorgestrel se asocian con un mayor riesgo de infección en el momento de la colocación y con hemorragia vaginal irregular. En especial en las pacientes con neutropenia debe considerarse el uso profiláctico de antibióticos.
La colocación de dispositivos intrauterinos de liberación de cobre no está recomendada, debido al potencial riesgo de infección en el momento de su colocación y a la pérdida de sangre menstrual, que pueden suponer un peligro para las pacientes con neutropenia grave o trombocitopenia grave.
Pruebas de embarazo
Las mujeres con capacidad de gestación deben efectuarse pruebas de embarazo con una sensibilidad mínima de 25 mUI/ml bajo supervisión médica y conforme a la práctica habitual, tal como se explica a continuación. Este requisito incluye a las mujeres con capacidad de gestación que practican una abstinencia sexual absoluta y continua. Idealmente, la prueba de embarazo, la prescripción y la dispensación deben realizarse el mismo día. Pomalidomida se debe dispensar a las mujeres con capacidad de gestación en un plazo de siete días tras la prescripción.
Antes de iniciar el tratamiento
Debe efectuarse una prueba de embarazo bajo supervisión médica durante la consulta, en el momento de prescribir pomalidomida o en los tres días anteriores a la visita al médico prescriptor, siempre que la paciente haya estado usando un método anticonceptivo eficaz durante al menos 4 semanas. La prueba debe garantizar que la paciente no esté embarazada cuando inicie el tratamiento con pomalidomida.
Seguimiento y _finalización del tratamiento
Se debe repetir cada 4 semanas una prueba de embarazo bajo supervisión médica, y realizar otra 4 semanas después de la finalización del tratamiento, excepto en el caso de que la paciente se haya sometido a una ligadura de trompas de eficacia confirmada. Estas pruebas de embarazo deben efectuarse el mismo día de la consulta en que se prescriba el medicamento o en los tres días anteriores a la visita al médico prescriptor.
Varones
Pomalidomida está presente en el semen humano durante el tratamiento. Como medida de precaución, y teniendo en cuenta las poblaciones especiales con un tiempo de eliminación potencialmente prolongado, como la insuficiencia renal, todos los pacientes varones que tomen pomalidomida, incluyendo a aquellos que se hayan sometido a una vasectomía, deben usar preservativos durante todo el tratamiento, durante la interrupción de la administración y hasta 7 días después del final del tratamiento, si su pareja está embarazada o tiene capacidad de gestación y no está usando ningún método anticonceptivo.
Los pacientes varones no deben donar semen o esperma durante el tratamiento (periodos de interrupción de la dosis incluidos) ni en el plazo de 7 días después de la suspensión del tratamiento con pomalidomida.
Precauciones adicionales
Se debe indicar a los pacientes que no den nunca este medicamento a otra persona y que devuelvan las cápsulas sin usar al farmacéutico al final del tratamiento.
Los pacientes no deben donar sangre, semen o esperma durante el tratamiento (periodos de interrupción de la dosis incluidos) ni en el plazo de 7 días después de la suspensión del tratamiento con pomalidomida.
Material educativo, restricciones de prescripción y dispensación
Con objeto de ayudar a los pacientes a evitar la exposición fetal a pomalidomida, el Titular de la Autorización de Comercialización distribuirá material educativo a los profesionales sanitarios, destinado a reforzar las advertencias acerca de la teratogenicidad esperada de pomalidomida y a proporcionar asesoramiento sobre anticoncepción antes de iniciar el tratamiento y sobre la necesidad de realizar pruebas de embarazo. El médico debe informar a la paciente acerca del riesgo teratogénico esperado y de las estrictas medidas de prevención de embarazo, especificadas en el Programa de Prevención de Embarazo así como proporcionar a la paciente un folleto informativo adecuado, una tarjeta de paciente y/o una herramienta equivalente conforme al sistema nacional implementado de tarjeta del paciente. En colaboración con cada autoridad nacional competente, se ha implementado un sistema de distribución nacional controlada. El sistema de distribución controlada incluye el uso de una tarjeta del paciente y/o un herramienta equivalente para el control de la prescripción y/o dispensación, así como la recogida de datos detallados en relación con la indicación terapéutica, para monitorizar el uso en una indicación no autorizada dentro del territorio nacional. Idealmente, la prueba de embarazo, la prescripción y la dispensación deben realizarse el mismo día. La dispensación de pomalidomida en mujeres con capacidad de gestación debe hacerse dentro de los 7 días de prescripción y tras haber obtenido un resultado negativo en la prueba de embarazo supervisada por un médico. Las prescripciones a mujeres con capacidad de gestación pueden tener una caducidad de 4 semanas y las prescripciones para el resto de pacientes pueden tener una caducidad de 12 semanas.
Eventos hematológicos
La reacción adversa hematológica de Grado 3 o 4 notificada con mayor frecuencia en pacientes con mieloma múltiple en recaída/refractario fue la neutropenia, seguido de anemia y trombocitopenia. Se debe monitorizar a los pacientes en busca de posibles reacciones adversas hematológicas, especialmente neutropenia. Se debe advertir a los pacientes que informen rápidamente acerca de los episodios febriles que presenten. Los médicos deben estar atentos a los signos de hemorragia en los pacientes, incluyendo epistaxis, especialmente en el caso de medicación concomitante conocida por aumentar el riesgo de sangrado (ver sección 4.8). Debe efectuarse a los pacientes un hemograma completo semanal en el momento basal, durante las primeras 8 semanas y después mensualmente. Puede ser necesaria una modificación de la dosis (ver sección 4.2). Los pacientes pueden requerir el uso de hemoderivados y/o factores de crecimiento.
Eventos tromboembólicos
Se han observado eventos tromboembólicos venosos (predominantemente trombosis venosa profunda y embolia pulmonar) y eventos trombóticos arteriales (infarto de miocardio y accidente cerebrovascular) en pacientes tratados con pomalidomida en combinación con dexametasona. Los pacientes con factores de riesgo conocidos de tromboembolismo, incluida una trombosis previa, deben estar estrechamente monitorizados. Se deben tomar medidas para intentar minimizar todos los factores de riesgo modificables (p. ej. tabaquismo, hipertensión e hiperlipidemia). Se aconseja a médicos y pacientes que estén atentos a los signos y síntomas de tromboembolismo. Se debe advertir a los pacientes que soliciten atención médica si presentan síntomas como respiración entrecortada, dolor torácico o edema de las extremidades. Es recomendable el uso de terapia anticoagulante (si no está contraindicada), como el ácido acetilsalicílico, warfarina, heparina o clopidogrel, especialmente en pacientes con factores de riesgo trombótico adicionales. Después de una cuidadosa evaluación de los factores de riesgo subyacentes del paciente individual, se debe tomar una decisión respecto al uso de medidas profilácticas. En los estudios clínicos los pacientes recibieron ácido aceltilsalicílico profiláctico o terapia antitrombótica alternativa. El uso de agentes eritropoyéticos conlleva un riesgo de eventos trombóticos incluyendo tromboembolismo. Por lo tanto, deben emplearse con precaución los agentes eritropoyéticos así como otros agentes que puedan aumentar el riesgo de eventos tromboembólicos.
Neuropatía periférica
Se excluyó de los estudios clínicos con pomalidomida a los pacientes con neuropatía periférica en curso de Grado >2. Se deben adoptar las precauciones adecuadas al considerar el tratamiento de estos pacientes con pomalidomida.
Disfunción cardíaca significativa
Se excluyó de los estudios clínicos con pomalidomida a los pacientes con una disfunción cardíaca significativa (insuficiencia cardíaca congestiva [Clase III o IV de la NY Heart Association]; infarto de miocardio dentro de los 12 meses desde el inicio del estudio; angina de pecho inestable o mal controlada). Se han notificado acontecimientos de insuficiencia cardiaca, que incluyen insuficiencia cardiaca congestiva y edema pulmonar (ver sección 4.8), especialmente en pacientes con enfermedad cardiaca preexistente o factores de riesgo cardiacos. Se deben adoptar las precauciones adecuadas al considerar el tratamiento de estos pacientes con pomalidomida, incluido el control periódico para detectar la presencia de signos y síntomas de insuficiencia cardiaca.
Síndrome de lisis tumoral
Puede producirse un síndrome de lisis tumoral. Los pacientes con mayor riesgo de sufrir un síndrome de lisis tumoral son aquellos que presentan una carga tumoral elevada antes del tratamiento. Se debe monitorizar estrechamente a estos pacientes y se deben adoptar las precauciones adecuadas.
Segundas neoplasias malignas primarias
Se han notificado segundas neoplasias malignas primarias, como cáncer de piel no melanoma, en pacientes en tratamiento con pomalidomida (ver sección 4.8). Los médicos deben evaluar cuidadosamente a los pacientes antes y durante el tratamiento, utilizando pruebas estándar de detección de cáncer por si aparecieran segundas neoplasias malignas primarias e instaurar el tratamiento indicado.
Reacciones alérgicas
Se han notificado angioedema y reacciones dermatológicas graves (ver sección 4.8). Se excluyó de los estudios clínicos a los pacientes con antecedentes de reacciones alérgicas graves asociadas a talidomida o lenalidomida. Estos pacientes pueden presentar un mayor riesgo de reacciones de hipersensibilidad y no deben tomar pomalidomida. Se debe considerar la interrupción o suspensión de pomalidomida si se presenta exantema de Grado 2 o 3. Se debe suspender definitivamente el tratamiento con pomalidomida si se presenta angioedema, exantema de Grado 4 y exantema ampolloso o exfoliativo.
Mareo y confusión
Se han notificado mareo y estados de confusión con pomalidomida. Los pacientes deben evitar las situaciones en que el mareo o la confusión puedan representar un problema y no tomar otros medicamentos que puedan causar mareo o confusión sin solicitar antes consejo médico.
Enfermedad pulmonar intersticial (EPI)
Se han observado EPI y acontecimientos asociados que incluyen casos de neumonitis con pomalidomida. Se debe realizar una evaluación cuidadosa de los pacientes que presenten un inicio repentino o empeoramiento inexplicable de los síntomas pulmonares para descartar la EPI. Se debe interrumpir la administración de pomalidomida durante la investigación de estos síntomas Y, si se confirma la EPI, debe iniciarse un tratamiento adecuado. Únicamente se debe reanudar pomalidomida después de una evaluación exhaustiva de los beneficios y los riesgos.
Trastornos hepáticos
Se han observado concentraciones notablemente elevadas de alanina aminotransferasa y bilirrubina en los pacientes tratados con pomalidomida (ver sección 4.8). Se han notificado también casos de hepatitis que provocaron la suspensión de pomalidomida. Se recomienda controlar periódicamente la función hepática durante los primeros 6 meses de tratamiento con pomalidomida y, posteriormente, cuando esté clínicamente indicado.
Infecciones
Se han notificado rara vez casos de reactivación de la hepatitis B en pacientes en tratamiento con pomalidomida combinado con dexametasona que habían sido previamente infectados por el virus de la hepatitis B (VHB). Algunos de estos casos han evolucionado a fallo hepático agudo, dando lugar a la suspensión de pomalidomida. Se debe determinar el estado del virus de la hepatitis B antes de iniciar el tratamiento con pomalidomida. Se recomienda que los pacientes que den un resultado positivo en la prueba de infección por VHB se pongan en contacto con un médico con experiencia en el tratamiento de la hepatitis B. Se debe tener precaución cuando se administre pomalidomida en combinación con dexametasona en pacientes previamente infectados por el VHB, incluidos los pacientes anti-Bc positivos pero con HBsAg negativos. Se debe monitorizar estrechamente a estos pacientes para detectar signos y síntomas de infección activa por el VHB durante todo el tratamiento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Efecto de Imnovid sobre otros medicamentos
No se espera que pomalidomida pueda causar interacciones medicamentosas farmacocinéticas clínicamente relevantes debido a la inhibición o inducción de la isoenzima P450, o inhibición de transportadores cuando se administra de forma concomitante con sustratos de estas enzimas o transportadores. No se ha evaluado clínicamente el potencial de estas interacciones medicamentosas, incluyendo el posible impacto de pomalidomida en la farmacocinética de los anticonceptivos orales combinados (ver sección 4.4 Teratogenicidad).
Efecto de otros medicamentos sobre Imnovid
Pomalidomida se metaboliza parcialmente por CYP1A2 y CYP3A4/5.También es un sustrato de la glicoproteína P. La administración concomitante de pomalidomida con ketoconazol, inhibidor potente del CYP3A4/5 y de la Gp-P, o con el inductor potente del CYP3A4/5, carbamazepina, no demostró ningún efecto clínicamente relevante a la exposición a pomalidomida. La administración concomitante de pomalidomida con el inhibidor potente del CYP1A2 fluvoxamina en presencia de ketoconazol, incrementó la exposición media a pomalidomida en un 107 %, con un intervalo de confianza del 90 % [del 91 % al 124 %], frente a pomalidomida más ketoconazol. En un segundo estudio realizado para evaluar la contribución a los cambios del metabolismo de un inhibidor del CYP1A2 solo, la administración conjunta de fluvoxamina sola con pomalidomida aumentó la exposición media a pomalidomida en un 125 % con un intervalo de confianza del 90 % [del 98 % al 157 %] frente a pomalidomida administrada en monoterapia. Si se administran inhibidores potentes del CYP1A2 (p. ej., ciprofloxacino, enoxacino y fluvoxamina) de forma concomitante con pomalidomida, se debe reducir la dosis de pomalidomida en un 50 %.
Dexametasona
La administración concomitante de múltiples dosis de hasta 4 mg de pomalidomida con dexametasona de 20 mg a 40 mg (un inductor leve a moderado de varias enzimas CYP, incluido el CYP3A) en pacientes con mieloma múltiple no tuvo ningún efecto sobre la farmacocinética de pomalidomida frente a pomalidomida administrada sola.
Se desconoce el efecto de dexametasona sobre la warfarina. Se aconseja realizar una monitorización rigurosa de la concentración de warfarina durante el tratamiento.
4.6 Fertilidad, embarazo y lactancia
Mujeres con capacidad de gestación / Anticonceptivos en varones y mujeres
Las mujeres con capacidad de gestación deben utilizar métodos anticonceptivos efectivos. Si una mujer tratada con pomalidomida se queda embarazada, se debe suspender el tratamiento y derivar a la paciente a un médico especialista o con experiencia en teratología, para su evaluación y asesoramiento. Si un paciente varón toma pomalidomida y su pareja se queda embarazada, se recomienda derivar a la mujer a un médico especialista o con experiencia en teratología, para su evaluación y asesoramiento. Pomalidomida está presente en el semen humano. Como medida de precaución, todos los pacientes varones que tomen pomalidomida deben usar preservativos durante todo el tratamiento, durante la interrupción de la administración y hasta 7 días después del final del tratamiento, si su pareja está embarazada o tiene capacidad de gestación y no está usando ningún método anticonceptivo (ver secciones 4.3 y 4.4).
Embarazo
Se espera un efecto teratogénico de pomalidomida en humanos. Pomalidomida está contraindicada durante el embarazo y en mujeres con capacidad de gestación, a menos que se cumplan todas las condiciones para la prevención del embarazo, ver sección 4.3 y sección 4.4.
Lactancia
Se desconoce si pomalidomida se excreta en la leche materna. Se detectó la presencia de pomalidomida en la leche de ratas que estaban siendo amamantadas tras administrar el medicamento a la madre.
Debido a las posibles reacciones adversas en lactantes asociadas a pomalidomida, se debe decidir si es
necesario suspender la lactancia o suspender el tratamiento tras considerar la importancia del tratamiento para la madre.
Fertilidad
Se sabe que pomalidomida tiene un efecto negativo sobre la fertilidad y que es teratogénica en los animales. Tras su administración a conejas embarazadas, pomalidomida atravesó la placenta y se detectó en la sangre fetal. Ver sección 5.3.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Imnovid sobre la capacidad para conducir y utilizar máquinas es pequeña o moderada. Se han notificado casos de fatiga, disminución del nivel de conciencia, confusión y mareo relacionados con el uso de pomalidomida. Si notan estos efectos, se debe advertir a los pacientes de que no deben conducir automóviles, utilizar máquinas o realizar cualquier actividad peligrosa durante su tratamiento con pomalidomida.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Las reacciones adversas notificadas con mayor frecuencia en los estudios clínicos han sido los trastornos de la sangre y del sistema linfático, incluyendo anemia (45,7 %), neutropenia (45,3 %) y trombocitopenia (27 %); trastornos generales y alteraciones en el lugar de administración, incluyendo fatiga (28,3 %), pirexia (21 %) y edema periférico (13 %); e infecciones e infestaciones incluyendo neumonía (10,7 %). Las reacciones adversas relacionadas con neuropatía periférica fueron notificadas en el 12,3 % de los pacientes y las reacciones adversas de embolismo o tromboembolismo venoso fueron notificadas en el 3,3 % de los pacientes. Las reacciones adversas de grado 3 o 4 más frecuentes estaban relacionadas con trastornos de la sangre y del sistema linfático, incluyendo neutropenia (41,7 %), anemia (27 %) y trombocitopenia (20,7 %); infecciones e infestaciones, incluyendo neumonía (9 %); y trastornos generales y alteraciones en el lugar de administración, incluyendo fatiga (4,7 %), pirexia (3 %) y edema periférico (1,3 %). La reacción adversa grave notificada con mayor frecuencia fue la neumonía (9,3 %). Otras reacciones adversas graves notificadas incluyen neutropenia febril (4,0 %), neutropenia (2,0 %), trombocitopenia (1,7 %) y reacciones adversas de TEV (1,7 %).
Se observó que las reacciones adversas tendían a ocurrir con mayor frecuencia dentro de los primeros 2 ciclos de tratamiento con pomalidomida.
Tabla de reacciones adversas
En el estudio aleatorizado CC-4047-MM-003, un total de 302 pacientes con mieloma múltiple en recaída y refractario fueron tratados con 4 mg de pomalidomida administrada una vez al día durante 21 días en cada ciclo de 28 días, en combinación con una dosis baja semanal de dexametasona.
Las reacciones adversas observadas en los pacientes tratados con pomalidomida y dexametasona se incluyen a continuación, según el sistema de clasificación por órganos y la frecuencia (SOC por sus siglas en inglés) para todas las reacciones adversas y para las reacciones adversas de Grado 3 o 4.
Las frecuencias de las reacciones adversas son las notificadas en el grupo de pomalidomida más dexametasona del estudio CC-4047-MM-003 (n=302) y de los datos poscomercialización. Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de SOC (por sus siglas en inglés) y de frecuencia. Las frecuencias se definen, según las guías actuales, como: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); y poco frecuentes (>1/1.000 a <1/100).
|
Sistema de clasificación de órganos / Término preferente |
Todas las reacciones adversas/Frecuencia |
Reacciones adversas de Grado 3-4/Frecuencia |
|
Infecciones e |
Muy frecuentes |
Frecuentes |
|
infestaciones |
Neumonía (infecciones bacterianas, |
Sepsis neutropénica |
|
víricas y fungicas, incluidas las |
Neumonía (infecciones | |
|
infecciones oportunistas) |
bacterianas, víricas y fungicas, incluidas las infecciones | |
|
Frecuentes |
oportunistas) | |
|
Sepsis neutropénica |
Bronconeumonía | |
|
Bronconeumonía |
Infección del tracto respiratorio | |
|
Bronquitis |
Infección del tracto respiratorio | |
|
Infección del tracto respiratorio Infección del tracto respiratorio |
superior | |
|
superior |
Poco frecuentes | |
|
Nasofaringitis |
Bronquitis | |
|
Herpes zóster |
Herpes zóster | |
|
Frecuencia no conocida |
Frecuencia no conocida | |
|
Reactivación de la hepatitis B |
Reactivación de la hepatitis B | |
|
Neoplasias benignas, |
Poco frecuentes |
Poco frecuentes |
|
malignas y no |
Carcinoma de piel de células |
Carcinoma de piel de células |
|
especificadas (incl |
basales |
basales |
|
quistes y pólipos) |
Carcinoma de piel de células |
Carcinoma de piel de células |
|
escamosas |
escamosas | |
|
Trastornos de la |
Muy frecuentes |
Muy frecuentes |
|
sangre y del sistema |
Neutropenia |
Neutropenia |
|
linfático |
Trombocitopenia |
Trombocitopenia |
|
Leucopenia Anemia |
Anemia Frecuentes | |
|
Frecuentes |
Neutropenia febril | |
|
Neutropenia febril |
Leucopenia | |
|
Pancitopenia* |
Pancitopenia* | |
|
Trastornos del |
Muy frecuentes |
Frecuentes |
|
metabolismo y de la |
Disminución del apetito |
Hiperpotasemia |
|
nutrición |
Hiponatremia | |
|
Frecuentes Hiperpotasemia |
Hiperuricemia* | |
|
Hiponatremia |
Poco frecuentes | |
|
Hiperuricemia* Poco frecuentes Síndrome de lisis tumoral* |
Disminución del apetito Síndrome de lisis tumoral* | |
|
Trastornos |
Frecuentes |
Frecuentes |
|
psiquiátricos |
Estado de confusión |
Estado de confusión |
|
Sistema de clasificación de órganos / Término preferente |
Todas las reacciones adversas/Frecuencia |
Reacciones adversas de Grado 3-4/Frecuencia |
|
Trastornos del |
Frecuentes |
Frecuentes |
|
sistema nervioso |
Disminución del nivel de |
Disminución del nivel de |
|
conciencia Neuropatía sensitiva periférica |
conciencia | |
|
Mareo |
Poco frecuentes | |
|
Temblor |
Neuropatía sensitiva periférica | |
|
Hemorragia intracraneal* |
Mareo Temblor | |
|
Poco frecuentes |
Accidente cerebrovascular* | |
|
Accidente cerebrovascular* |
Hemorragia intracraneal* | |
|
Trastornos del oído y |
Frecuentes |
Frecuentes |
|
del laberinto |
Vértigo |
Vértigo |
|
Trastonos vasculares |
Frecuentes |
Poco frecuentes |
|
Trombosis venosa profunda |
Trombosis venosa profunda | |
|
Trastornos cardiacos |
Frecuentes |
Frecuentes |
|
Insuficiencia cardíaca* |
Insuficiencia cardíaca* | |
|
Fibrilación auricular* Infarto de miocardio* |
Fibrilación auricular* Poco frecuentes Infarto de miocardio* | |
|
Trastornos del |
Frecuentes |
Poco frecuentes |
|
sistema inmunológico |
Angioedema* |
Angioedema* |
|
Urticaria* |
Urticaria* | |
|
Trastornos |
Muy frecuentes |
Frecuentes |
|
respiratorios, |
Disnea |
Disnea |
|
torácicos y |
Tos | |
|
mediastínicos |
Poco frecuentes | |
|
Frecuentes |
Embolia pulmonar | |
|
Embolia pulmonar |
Tos | |
|
Epistaxis* |
Epistaxis* | |
|
Enfermedad pulmonar intersticial* |
Enfermedad pulmonar intersticial* | |
|
Trastornos |
Muy frecuentes |
Frecuentes |
|
gastrointestinales |
Diarrea |
Diarrea |
|
Náuseas |
Vómitos | |
|
Estreñimiento |
Estreñimiento | |
|
Frecuentes |
Poco frecuentes | |
|
Vómitos |
Náuseas | |
|
Hemorragia gastrointestinal |
Hemorragia gastrointestinal | |
|
Trastornos |
Poco frecuentes |
Poco frecuentes |
|
hepatobiliares |
Hiperbilirrubinemia Hepatitis* |
Hiperbilirrubinemia |
|
Trastornos de la piel y |
Frecuentes |
Frecuentes |
|
del tejido subcutáneo |
Erupción Prurito |
Erupción |
|
Sistema de clasificación de órganos / Término preferente |
Todas las reacciones adversas/Frecuencia |
Reacciones adversas de Grado 3-4/Frecuencia |
|
Trastornos |
Muy frecuentes |
Frecuentes |
|
musculoesqueléticos y |
Dolor óseo |
Dolor óseo |
|
del tejido conjuntivo |
Espasmos musculares |
Poco frecuentes Espasmos musculares |
|
Trastornos renales y |
Frecuentes |
Frecuentes |
|
urinarios |
Insuficiencia renal Retención urinaria |
Insuficiencia renal Poco frecuentes Retención urinaria |
|
Trastornos del |
Frecuentes |
Frecuentes |
|
aparato reproductor y de la mama |
Dolor pélvico |
Dolor pélvico |
|
Trastornos generales |
Muy frecuentes |
Frecuentes |
|
y alteraciones en el |
Fatiga |
Fatiga |
|
lugar de |
Pirexia |
Pirexia |
|
administración |
Edema periférico |
Edema periférico |
|
Exploraciones |
Frecuentes |
Frecuentes |
|
complementarias |
Disminución del recuento de |
Disminución del recuento de |
|
neutrófilos |
neutrófilos | |
|
Disminución del recuento de |
Disminución del recuento de | |
|
leucocitos |
leucocitos | |
|
Disminución del recuento de |
Disminución del recuento de | |
|
plaquetas |
plaquetas | |
|
Aumento de la alanina |
Aumento de la alanina | |
|
aminotransferasa Aumento del ácido úrico en sangre* |
aminotransferasa Poco frecuentes Aumento del ácido úrico en sangre* |
*Identificados a partir de los datos poscomercialización; las frecuencias se basan en los datos de los ensayos clínicos.
Descripción de reacciones adversas seleccionadas
Teratogenicidad
Pomalidomida está relacionada estructuralmente con talidomida. Talidomida es un principio activo con acción teratogénica conocida en humanos, que causa defectos de nacimiento graves que pueden poner en peligro la vida del niño. Pomalidomida tiene un efecto teratogénico en ratas y conejos cuando se administra durante el periodo de mayor organogénesis (ver secciones 4.6 y 5.3). Si se toma pomalidomida durante el embarazo, se espera un efecto teratogénico de pomalidomida en los seres humanos (ver sección 4.4).
Neutropenia y trombocitopenia
El 45,3 % de los pacientes que recibieron pomalidomida más dosis bajas de dexametasona (Pom + LD-Dex) experimentó neutropenia, frente al 19,5 % de los pacientes que recibieron dosis altas de dexametasona (HD-Dex). La neutropenia fue de grado 3 o 4 en un 41,7 % de los pacientes que recibieron Pom + LD-Dex frente al 14,8 % de los que recibieron HD-Dex. En los pacientes tratados con Pom + LD-Dex, la neutropenia fue grave en una minoría (2,0 % de los pacientes), no tuvo como consecuencia la suspensión del tratamiento, y se asoció con la interrupción del tratamiento en un 21,0 % de los pacientes y con una reducción de la dosis en un 7,7 % de los pacientes.
El 6,7 % de los pacientes que recibieron Pom + LD-Dex experimentó neutropenia febril (NF), frente a ninguno de aquellos que recibieron HD-Dex. Todos los casos fueron notificados como de grado 3 o 4. La NF fue notificada como grave en un 4,0 % de los pacientes. Asimismo, la NF se asoció con una interrupción de la dosis en un 3,7 % de los pacientes, con una reducción de la dosis en un 1,3 % de los pacientes y con ningún caso de suspensión del tratamiento.
El 27,0 % de los pacientes que recibieron Pom + LD-Dex experimentó trombocitopenia frente a un
26,8 % de los pacientes que recibieronHD-Dex. La trombocitopenia fue de grado 3 o 4 en un 20,7 % de los pacientes que recibieron Pom + LD-Dex frente a un 24,2 % de los que recibieron HD-Dex. En los pacientes tratados con Pom + LD-Dex, la trombocitopenia fue grave en un 1,7 % de los pacientes, conllevó una reducción de la dosis en un 6,3 % de los pacientes, una interrupción de la dosis en un 8 % de los pacientes y la suspensión del tratamiento en un 0,7 % de los pacientes (ver secciones 4.2 y 4.4).
Infección
La infección fue la toxicidad no hematológica más frecuente, con una incidencia del 55,0 % en los pacientes que recibieron Pom + LD-Dex frente al 48,3 % en los pacientes que recibieron HD-Dex. Aproximadamente la mitad de dichas infecciones fueron de grado 3 o 4; un 24,0 % en los pacientes tratados con Pom + LD-Dex y un 22,8 % en los pacientes tratados con HD-Dex.
Entre los pacientes tratados con Pom + LD-Dex las infecciones notificadas con mayor frecuencia fueron la neumonía y la infección del tracto respiratorio superior (en un 10,7 % y un 9,3 % de los pacientes, respectivamente); con un 24,3 % de las infecciones notificadas clasificadas como graves o mortales (grado 5) en el 2,7 % de los pacientes tratados. En los pacientes tratados con Pom + LD-Dex las infecciones conllevaron la suspensión de la dosis en un 2,0 % de los pacientes, la interrupción del tratamiento en un 14,3 % de los pacientes y la reducción de la dosis en un 1,3 % de los pacientes.
Eventos tromboembólicos
El 3,3 % de los pacientes que recibieron Pom + LD-Dex y un 2,0 % de los pacientes que recibieron HD-Dex experimentaron un embolismo o tromboembolismo venoso. El 1,3 % de los pacientes que recibieron Pom + LD-Dex experimentó reacciones de grado 3 o 4 frente a ningún caso entre los pacientes que recibieron HD-Dex. En los pacientes tratados con Pom + LD-Dex, el TEV se notificó como grave en el 1,7 %, no se notificó ninguna reacción adversa mortal en los estudios clínicos y el TEV no se asoció con suspensión de la dosis.
La profilaxis con ácido acetilsalicílico (y otros anticoagulantes en pacientes de alto riesgo) fue obligatoria para todos los pacientes participantes en los estudios clínicos. Se recomienda terapia anticoagulante (a no ser que esté contraindicada) (ver sección 4.4).
Neuropatía _ periférica
Se excluyó de los estudios clínicos a los pacientes con neuropatía periférica en curso de grado >2. El 12,3 % de los pacientes que recibieron Pom + LD-Dex experimentaron una neuropatía periférica, la mayoría de grado 1 o 2, frente a un 10,7 % de los pacientes que recibieron HD-Dex. El 1,0 % de los pacientes que recibieron Pom + LD-Dex experimentó reacciones de grado 3 o 4 frente al 1,3 % de los pacientes que recibieron HD-Dex. En los estudios clínicos, ninguna de las neuropatías periféricas se notificó como grave en los pacientes tratados con Pom + LD-Dex y la neuropatía periférica condujo a la suspensión de la dosis en un 0,3 % de los pacientes (ver sección 4.4).
La mediana del tiempo hasta la aparición de la neuropatía fue de 2,1 semanas, con una variación de 0,1 a 48,3 semanas. La mediana del tiempo hasta la aparición fue inferior en los pacientes que recibieron HD-Dex comparado con los pacientes tratados con Pom+LD-Dex (1,3 semanas frente a 2,1 semanas).
La mediana del tiempo hasta la desaparición de los síntomas fue de 22,4 semanas en los pacientes que recibieron Pom+LD-Dex y de 13,6 semanas en los pacientes que recibieron HD-Dex. El límite inferior
del intervalo de confianza del 95 % fue de 5,3 semanas en los pacientes tratados con Pom+LD-Dex y de
2,0 semanas en los pacientes que recibieron HD-Dex.
Hemorragia
Se han notificado trastornos hemorrágicos con pomalidomida, especialmente en pacientes con factores de riesgo tales como medicamentos concomitantes que aumentan el riesgo de hemorragia. Los eventos hemorrágicos incluyen epistaxis, hemorragia intracraneal y hemorragia gastrointestinal.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los
profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Se han evaluado dosis de pomalidomida de hasta 50 mg en dosis única en voluntarios sanos y 10 mg en múltiples dosis una vez al día en pacientes con mieloma múltiple sin que se haya notificado ningún caso de efecto adverso grave relacionado con sobredosis. Pomalidomida se eliminó mediante hemodiálisis. En caso de sobredosis, se recomienda terapia de soporte.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agente inmunomodulador, código ATC: L04AX06 Mecanismo de acción
Pomalidomida es un medicamento con actividad tumoricida directa contra el mieloma, actividad inmunomoduladora y capaz de inhibir el apoyo de las células del estroma para el crecimiento de las células cancerosas del mieloma múltiple. En concreto, pomalidomida inhibe la proliferación e induce la apoptosis de las células hematopoyéticas tumorales. Además, pomalidomida inhibe la proliferación de las líneas celulares de mieloma múltiple resistentes a lenalidomida y presenta un efecto sinérgico con dexametasona tanto en las líneas celulares de mieloma múltiple resistentes a lenalidomida como en las sensibles a lenalidomida para inducir la apoptosis de las células tumorales. Pomalidomida potencia la inmunidad celular mediada por los linfocitos T y por los linfocitos natural killer (NK) e inhibe la producción de citocinas proinflamatorias (p. ej. TNF-a e IL-6) por los monocitos. Pomalidomida también inhibe la angiogénesis mediante el bloqueo de la migración y adhesión de células endoteliales.
Eficacia clínica y seguridad
Se evaluó la eficacia y seguridad de pomalidomida en combinación con dexametasona en un ensayo de fase III, multicéntrico, aleatorizado y abierto (CC-4047-MM-003), donde se comparó el tratamiento de pomalidomida más una dosis baja de dexametasona (Pom + LD-Dex) frente a una dosis alta de dexametasona sola (HD-Dex) en pacientes adultos con mieloma múltiple en recaída o refractario que ya habían recibido al menos dos tratamientos previos, incluyendo lenalidomida y bortezomib, y que habían mostrado progresión de la enfermedad durante el último tratamiento. En el estudio participaron un total de 455 pacientes: 302 en el grupo Pom + LD-Dex y 153 en el grupo HD-Dex. La mayoría de los pacientes eran varones (59 %) y caucásicos (79 %); la mediana de edad de toda la población era de 64 años (mín, máx: 35, 87 años).
Los pacientes del grupo Pom + LD-Dex tomaron 4 mg de pomalidomida por vía oral en los días del 1 al 21 en cada ciclo de 28 días. Se administró la dosis de LD-Dex (40 mg) una vez al día en los días 1, 8, 15 y 22 de un ciclo de 28 días. En el grupo HD-Dex, los pacientes tomaron dexametasona (40 mg) una vez al día en los días del 1 al 4, del 9 al 12 y del 17 al 20 en un ciclo de 28 días. Los pacientes mayores de 75 años iniciaron el tratamiento con dexametasona 20 mg. Los pacientes continuaron con el tratamiento hasta la progresión de la enfermedad.
La variable principal de eficacia fue la supervivencia libre de progresión (SLP) de acuerdo con los criterios del InternationalMyeloma Working Group (IMWG). Para la población por intención de tratar (IDT), la mediana del tiempo de SLP según revisión del Independent Review Adjudication Committee (IRAC) basada en los criterios del IMWG fue de 15,7 semanas (IC de 95 %: 13,0; 20,1) en el grupo Pom + LD-Dex; la tasa estimada de supervivencia libre de eventos a las 26 semanas fue del 35,99 %
(± 3,46 %). En el grupo HD-Dex, la mediana del tiempo de SLP fue de 8,0 semanas (IC de 95 %: 7,0; 9,0); la tasa estimada de supervivencia libre de eventos a las 26 semanas fue del 12,15 % (± 3,63 %).
Se evaluó la SLP en varios subgrupos relevantes: sexo, raza, estatus de rendimiento ECOG, factores de estratificación (edad, población con enfermedad, terapias antimieloma previas [2, >2]), parámetros de relevancia pronóstica seleccionados (niveles basales de beta-2 microglobulina, niveles basales de albúmina, insuficiencia renal basal y riesgo citogenético) y exposición y refractariedad a terapias antimieloma previas. Independientemente del subgrupo evaluado, la SLP fue generalmente consistente con la observada en la población por IDT en ambos grupos de tratamiento.
En la tabla 1 se resumen los resultados de la supervivencia libre de progresión para la población por IDT. La figura 1 presenta la curva de Kaplan-Meier de la SLP para la población por IDT.
Tabla 1: Tiempo de Supervivencia Libre de Progresión según revisión del IRAC basada en
los criterios IMWG (prueba de log-rank estratificada) (población por IDT)
|
Pom+LD-Dex (N=302) |
HD-Dex (N=153) | |
|
Supervivencia libre de progresión (SLP), n |
302 (100,0) |
153 (100,0) |
|
Censurado, n (%) |
138 (45,7) |
50 (32,7) |
|
Progresión/muerte, n (%) |
164 (54,3) |
103 (67,3) |
|
Tiempo de supervivencia libre de progresión (semanas) | ||
|
Mediana3 |
15,7 |
8,0 |
|
[IC de 95 % bilateral]b |
[13,0; 20,1] |
[7,0 ; 9,0] |
|
Razón de riesgo (Hazard Ratio) (Pom+LD-Dex: HD-Dex) [IC de 95 % bilateral]0 |
0,45 [0,35; 0,59] | |
|
Valor P (bilateral) de la prueba de log-rankd |
<0,001 | |
Nota: IC = intervalo de confianza; IRAC = Comité Independiente Revisor de Evaluación; NE = no estimable a La mediana está basada en la estimación de Kaplan-Meier.
b Intervalo de confianza de 95 % de la mediana del tiempo de supervivencia libre de progresión.
c Basado en el modelo de riesgos proporcionales de Cox que compara las funciones de riesgo asociadas con los grupos de tratamiento, estratificados por edad (<75 frente a >75), población con la enfermedad (refractario a lenalidomida y bortezomib frente a no refractario a estos dos medicamentos), y número de terapias antimieloma previas (=2 frente a >2).
d El valor P está basado en la prueba de log-rank estratificada con los mismos factores de estratificación arriba mencionados para el modelo de Cox.
Fecha de corte: 7 de septiembre de 2012
Supervivencia Libre de Progresión según la revisión de respuesta del IRAC basada en los criterios IMWG (prueba de log-rank estratificada) (población por IDT)
Figura 1:

Fecha de corte: 7 de septiembre de 2012
Supervivencia Libre de Progresión (semanas)
La variable secundaria clave fue la supervivencia global (SG). Un total de 226 pacientes (74,8 %) del grupo Pom + LD-Dex y 95 pacientes (62,1 %) del grupo HD-Dex vivían en el momento de la fecha de corte (7 de septiembre de 2012). La mediana del tiempo de supervivencia global según las estimaciones Kaplan-Meier no fue alcanzado por el grupo Pom + LD Dex, pero podría esperarse que fuera al menos de 48 semanas, que corresponde al umbral más bajo del IC de 95 %. La mediana del tiempo de SG del grupo HD-Dex fue de 34 semanas (IC de 95 %: 23,4; 39,9). La tasa libre de eventos al año fue del
52,6 % (± 5,72 %) para el grupo Pom + LD-Dex y del 28,4 % (± 7,51 %) para el grupo HD-Dex. La diferencia en términos de SG entre los dos grupos de tratamiento fue estadísticamente significativa (p <0,001).
La Tabla 2 resume los resultados de supervivencia global para la población por IDT. La curva Kaplan-Meier de SG para la población por IDT se presenta en la Figura 2.
Basándose en los resultados de las variables SLP y SG, el Comité de Monitorización de Datos para este estudio recomendó que se completara el estudio y que los pacientes en el grupo HD-Dex fueran cruzados al grupo Pom + LD-Dex.
|
Estadísticas |
Pom+LD-Dex (N=302) |
HD-Dex (N=153) | |
|
N |
302 (100,0) |
153 (100,0) | |
|
Censurado |
n (%) |
226 (74,8) |
95 (62,1) |
|
Muerto |
n (%) |
76 (25,2) |
58 (37,9) |
|
Tiempo de supervivencia (semanas) |
Mediana3 |
NE |
34,0 |
|
IC de 95 % bilateralb |
[ 48,1; NE] |
[ 23,4; 39,9] | |
|
Razón de riesgo (Hazard Ratio) (Pom+LD-Dex:HD-Dex) [IC de 95 % bilateral0] |
0,53 [ 0,37; 0,74] | ||
|
Valor P (bilateral) de la prueba de log-rankd |
<0,001 | ||
Nota: IC = intervalo de confianza; NE = no estimable. a La mediana está basada en la estimación de Kaplan-Meier.
b Intervalo de confianza de 95 % sobre la mediana del tiempo de supervivencia global.
c Basado en el modelo de riesgos proporcionales de Cox que compara las funciones de riesgo asociadas con los grupos de tratamiento. d El valor P está basado en la prueba de log-rank no estratificada.
Fecha de corte: 7 de septiembre de 2012
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Imnovid en todos los grupos de la población pediátrica en mieloma múltiple (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Pomalidomida se absorbe alcanzando una concentración plasmática máxima (Cmáx) a las 2 o 3 horas y, por lo menos, un 73 % se absorbe después de administrar una dosis única por vía oral. El área bajo la curva (AUC) de pomalidomida aumenta, aproximadamente, lineal y proporcionalmente con los

0 13
Fecha de corte: 7 de septiembre de 2012
26 39 52
Superviviencia global (semanas)
65
incrementos de la dosis. Tras la administración de múltiples dosis, pomalidomida tiene una ratio de acumulación del 27 al 31 % en el AUC.
La administración conjunta con una comida rica en grasas y rica en calorías reduce la tasa de absorción, disminuyendo la Cmáx plasmática media en, aproximadamente un 27 %, pero con un efecto mínimo sobre la extensión de la absorción global con una disminución del 8 % en el AUC media. Por tanto, pomalidomida puede administrarse con o sin alimentos.
Distribución
Pomalidomida tiene un volumen de distribución aparente (Vd) medio de entre 62 y 138 litros en estado estable. En el semen de sujetos sanos pomalidomida se distribuye a una concentración de aproximadamente el 67 % del nivel de plasma a las 4 horas posteriores a la administración (aproximadamente Tmáx) tras 4 días de administración de 2 mg una vez al día. La unión in vitro de los enantiómeros de pomalidomida a las proteínas plasmáticas en humanos oscila entre el 12 % y el 44 % y no es dependiente de la concentración.
Biotransformación
En sujetos sanos que han recibido una dosis única por vía oral de [14C]-pomalidomida (2 mg), pomalidomida es la mayor sustancia circulante (aproximadamente el 70 % de la radioactividad del plasma) in vivo. No se hallaron metabolitos a >10 % relativos a la radioactividad total o relacionada en plasma.
Las rutas metabólicas principales de la radioactividad excretada son los procesos de hidroxilación con la posterior glucuronidación, o hidrólisis. Los estudios in vitro identificaron al CYP1A2 y al CYP3A4 como las enzimas primarias implicadas en la hidroxilación de pomalidomida mediada por CYP, con contribuciones menores adicionales del CYP2C19 y CYP2D6. Pomalidomida es también un sustrato de la glicoproteína-P in vitro. La administración concomitante de pomalidomida con el potente inhibidor del CYP3A4/5 y de la Gp-P, ketoconazol, o con el potente inductor del CYP3A4/5, carbamazepina, no demostró ningún efecto clínicamente relevante en la exposición a pomalidomida. La administración concomitante de pomalidomida con el inhibidor potente del CYP1A2, fluvoxamina, en presencia de ketoconazol, incrementó la exposición media a pomalidomida en un 107 %, con un intervalo de confianza del 90 % [del 91 % al 124 %], frente a pomalidomida más ketoconazol. En un segundo estudio realizado para evaluar la contribución a los cambios del metabolismo de un inhibidor del CYP1A2 solo, la administración conjunta de fluvoxamina sola con pomalidomida aumentó la exposición media a pomalidomida en un 125 % con un intervalo de confianza del 90 % [del 98 % al 157 %] frente a pomalidomida administrada en monoterapia. Si se administran inhibidores potentes del CYP1A2 (p. ej., ciprofloxacino, enoxacino y fluvoxamina) de forma concomitante con pomalidomida, se debe reducir la dosis de pomalidomida en un 50 %. La administración de pomalidomida a fumadores, sabiendo que el tabaquismo induce la isoforma CYP1A2, no tuvo ningún efecto clínicamente relevante en la exposición a pomalidomida frente a la exposición a pomalidomida observada en los no fumadores.
Según los datos in vitro, pomalidomida no es un inhibidor o inductor de las isoenzimas del citocromo P-450 y no inhibió ninguno de los fármacos transportadores que fueron estudiados. No se espera que pomalidomida pueda causar interacciones medicamentosas clínicamente relevantes cuando se administra de forma concomitante con sustratos de estas rutas.
Eliminación
En sujetos sanos pomalidomida se elimina con una mediana de semivida plasmática de aproximadamente 9,5 horas y de unas 7,5 horas en pacientes con mieloma múltiple. Pomalidomida tiene una media de aclaramiento corporal total (Cl/F) de aproximadamente 7-10 l/h.
Tras una dosis única por vía oral de [14C]-pomalidomida (2 mg) en sujetos sanos, aproximadamente el 73 % y el 15 % de la dosis radioactiva se eliminó por la orina y las heces, respectivamente, con aproximadamente el 2 % y el 8 % del radiocarbono administrado eliminado como pomalidomida en orina y heces.
Pomalidomida se metaboliza ampliamente antes de la excreción, con los metabolitos resultantes eliminados principalmente por la orina. Los tres metabolitos predominantes en la orina (formados mediante hidrólisis o hidroxilación con posterior glucuronidación) representan, aproximadamente, el
23 %, 17 % y 12 %, respectivamente, de la dosis en la orina.
Los metabolitos dependientes del CYP representan aproximadamente el 43 % de la radiactividad total excretada, mientras que los metabolitos hidrolíticos no dependientes del CYP representan el 25 %, y la excreción de pomalidomida inalterada representa el 10 % (2 % en orina y 8 % en heces).
Farmacocinética poblacional
Según análisis de farmacocinética poblacional basado en un modelo bicompartimental, los sujetos sanos y los pacientes con mieloma múltiple mostraron aclaramiento aparente (Cl/F) y volumen de distribución aparente en el compartimento central (WF) comparables. En tejidos periféricos, pomalidomida fue absorbida preferentemente por los tumores con un aclaramiento de distribución aparente en el compartimento periférico (Q/F) y un volumen de distribución aparente en el compartimento periférico (V3/F) 3,7 veces y 8 veces mayor, respectivamente, comparado con los sujetos sanos.
Población pediátrica
No existen datos disponibles sobre la administración de pomalidomida en niños o adolescentes (<18 años).
Población de edad avanzada
Según análisis de farmacocinética poblacional en sujetos sanos y en pacientes con mieloma múltiple, no se observó una influencia significativa de la edad (19-83 años) en el aclaramiento oral de pomalidomida. En los estudios clínicos los pacientes de edad avanzada (>65 años) expuestos a pomalidomida no requirieron ningún ajuste de dosis. Por favor, ver sección 4.2.
Insuficiencia renal
Los análisis de farmacocinética poblacional mostraron que los parámetros farmacocinéticos de pomalidomida no se vieron afectados de forma destacable en los pacientes con insuficiencia renal (definida mediante el aclaramiento de la creatinina o el filtrado glomerular estimado [FGe]) en comparación con los pacientes con la función renal normal (CrCl >60 ml/minuto). La exposición media a pomalidomida normalizada según el AUC fue del 98,2 % con un intervalo de confianza del 90 %
[77,4 % al 120,6 %] en los pacientes con insuficiencia renal moderada (FGe >30 a <45 ml/minuto/1,73 m2) en comparación con los pacientes con la función renal normal. La exposición media a pomalidomida normalizada según el AUC fue del 100,2 % con un intervalo de confianza del 90 % [79,7 % al 127,0 %] en los pacientes con insuficiencia renal grave que no precisaban diálisis (CrCl <30 o FGe <30 ml/minuto/1,73 m2) en comparación con los pacientes con la función renal normal. La exposición media a pomalidomida normalizada según el AUC aumentó en un 35,8 % con un IC del 90 % [7,5 % al 70,0 %] en los pacientes con insuficiencia renal grave que precisaban diálisis (CrCl <30 ml/minuto con necesidad de diálisis) en comparación con los pacientes con la función renal normal. Los cambios medios en la exposición a pomalidomida en cada uno de estos grupos de insuficiencia renal no son de una magnitud que requiera un ajuste de la dosis.
Insuficiencia hepática
Los parámetros farmacocinéticos cambiaron modestamente en los pacientes con insuficiencia hepática (definida mediante los criterios de Child-Pugh) en comparación con los sujetos sanos. La exposición media a pomalidomida aumentó en un 51 % con un intervalo de confianza del 90 % [del 9 % al 110 %] en los pacientes con insuficiencia hepática leve en comparación con los sujetos sanos. La exposición media a pomalidomida aumentó en un 58% con un intervalo de confianza del 90 % [del 13 % al 119 %] en los pacientes con insuficiencia hepática moderada en comparación con los sujetos sanos. La exposición media a pomalidomida aumentó en un 72 % con un intervalo de confianza del 90 % [del
24 % al 138 %] en los pacientes con insuficiencia hepática grave en comparación con los sujetos sanos. Los aumentos medios en la exposición a pomalidomida en cada uno de estos grupos de insuciencia hepática no son de una magnitud que requiera un ajuste del esquema de tratamiento o de la dosis (ver sección 4.2).
Estudios de toxicidad de dosis repetidas
La administración crónica de pomalidomida en ratas en dosis de 50, 250 y 1000 mg/kg/día durante 6 meses fue bien tolerada. No se detectó ningún efecto adverso hasta los 1000 mg/kg/día (una tasa de exposición 175 veces más elevada que la dosis clínica de 4 mg).
Se evaluó pomalidomida en monos en estudios de dosis repetidas de hasta 9 meses de duración. En estos estudios, los monos mostraron una mayor sensibilidad a los efectos de pomalidomida que las ratas. Las toxicidades primarias observadas en monos estuvieron relacionadas con los sistemas hematopoyético/linforreticular. En el estudio de 9 meses en monos con dosis de 0,05, 0,1 y 1 mg/kg/día se observó morbilidad y eutanasia temprana de 6 animales a dosis de 1 mg/kg/día que fueron atribuidas a los efectos inmunosupresores (infección por estafilococos, reducción de los linfocitos en sangre periférica, inflamación crónica del intestino grueso, reducción histológica de los linfocitos e hipocelularidad de la médula ósea) a exposiciones elevadas de pomalidomida (15 veces la tasa de exposición comparada con una dosis clínica de 4 mg). Dichos efectos inmunosupresores provocaron la eutanasia temprana de 4 monos debido a su mal estado de salud (heces líquidas, inapetencia, ingesta de alimentos reducida y pérdida de peso); la evaluación histopatológica de estos animales demostró inflamación crónica del intestino grueso y atrofia vellosa del intestino delgado. Se observó infección por estafilococos en 4 monos; 3 de éstos respondieron al tratamiento con antibióticos y uno murió sin tratamiento. Además, resultados consistentes con la leucemia mielógena aguda llevaron a la eutanasia de un mono; las observaciones clínicas y la patología clínica y/o alteraciones de la médula ósea observadas en este animal eran consistentes con inmunosupresión. La proliferación mínima o leve en los conductos biliares con incrementos asociados de la ALP y de la GGT también se observaron a dosis de 1 mg/kg/día. La evaluación de los animales recuperados indicó que todos los resultados relacionados con el tratamiento eran reversibles a las 8 semanas del cese de la administración, excepto la proliferación de los conductos biliares intrahepáticos observada en 1 animal en el grupo de 1 mg/kg/día. El nivel sin efecto adverso observado (NOAEL) fue de 0,1 mg/kg/día (una tasa de exposición relativa de 0,5 veces comparada con la dosis clínica de 4 mg).
Genotoxicidad/carcinogenicidad
Pomalidomida no resultó mutagénica en los ensayos de mutaciones bacterianas y de los mamíferos, y no indujo aberraciones cromosómicas en los linfocitos de sangre periférica en humanos así como tampoco a la formación de micronúcleos en eritrocitos policromáticos en la médula ósea de ratas a las que les fueron administradas dosis de hasta 2000 mg/kg/día. No se han realizado estudios de carcinogenicidad.
Fertilidad y desarrollo embrionario temprano
En un estudio de fertilidad y desarrollo embrionario temprano en ratas, se administró pomalidomida a los machos y las hembras a dosis de 25, 250 y 1000 mg/kg/día. El examen uterino en el día de gestación 13 mostró una reducción de la cantidad media de embriones viables y un aumento en la pérdida postimplantación con todos los niveles de dosis. Por consiguiente, el NOAEL en estos eventos observados fue <25 mg/kg/día (con un AUC24h de 39960 ng-h/ml (nanogramo^hora/mililitros) para la dosis más baja evaluada y una tasa de exposición 99 veces relativa a la dosis clínica de 4 mg). Cuando los machos tratados en este estudio fueron apareados con hembras no tratadas, todos los parámetros uterinos fueron comparables a los controles. Según estos resultados, los efectos observados fueron atribuidos al tratamiento de las hembras.
Desarrollo embriofetal
Pomalidomida resultó ser teratógena en ratas y conejos cuando se administró durante el periodo de mayor organogénesis. En el estudio de toxicidad sobre el desarrollo embriofetal de la rata, se observaron malformaciones relacionadas con la ausencia de vejiga urinaria, ausencia de la glándula tiroidea, así como la fusión y la desalineación de los elementos vertebrales torácicos y lumbares (arcos centrales y/o neurales) a todos los niveles de dosis (25, 50 y 1000 mg/kg/día).
En este estudio no se observó toxicidad materna. Por ello, el NOAEL materno fue de 1000 mg/kg/día, y el NOAEL para la toxicidad de desarrollo fue <25 mg/kg/día (con un AUC24h de 34340 ng-h/ml en el día de gestación 17 para la dosis más baja evaluada y la tasa de exposición fue de 85 veces comparada
con la dosis clínica de 4 mg). En conejos, pomalidomida a dosis entre los 10 y 250 mg/kg/día produjo malformaciones en el desarrollo embriofetal. Se observaron aumentos de las anomalías cardíacas a todas las dosis con aumentos significativos a 250 mg/kg/día. A dosis de entre 100 y 250 mg/kg/día se registró un ligero aumento de la pérdida post-implantación y un ligero descenso en el peso del feto. A dosis de 250 mg/kg/día, las malformaciones fetales incluyeron anomalías en las extremidades (extremidades anteriores y posteriores dobladas y/o giradas, ausencia de dígito o dígito libre) y malformaciones esqueléticas asociadas (metacarpiano no osificado, metacarpiano y falange no alineados, ausencia de dígito, falange no osificada y tibia corta no osificada o doblada); dilatación moderada de los ventrículos laterales del cerebro; ubicación anormal de la arteria subclavia derecha; ausencia de los lóbulos intermedios pulmonares; par de riñones desplazados hacia abajo; morfología hepática alterada; ausencia de osificación de la pelvis u osificación incompleta; aumento medio de las costillas torácicas supernumerarias y reducción media de los tarsales osificados. Además, se observó una ligera reducción en el incremento del peso materno, una reducción significativa de los triglicéridos, y una reducción significativa del peso absoluto y relativo del bazo a dosis de entre 100 y 250 mg/kg/día. El NOAEL materno fue de 10 mg/kg/día y el NOAEL del desarrollo fue de <10 mg/kg/día (con un AUC24h de 418 ng-h/ml en el día de gestación 19 para la dosis más baja evaluada, similar a la obtenida con una dosis clínica de 4 mg).
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Contenido de la cápsula:
Manitol
Almidón pregelatinizado Estearil fumarato de sodio
Cubierta de la cápsula:
La cubierta de la cápsula de 3 mg contiene gelatina, dióxido de titanio (E171), indigotina (E132), óxido de hierro amarillo (E172) y tinta blanca.
Tinta de impresión:
La cubierta de la cápsula de 3 mg contiene: tinta blanca - goma laca shellac, dióxido de titanio (E171), simeticona, propilenglicol (E1520) e hidróxido de amonio (E527).
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
4 años.
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Las cápsulas están envasadas en blísteres de polivinilcloruro (PVC) / policlorotrifluoroetileno (PCTFE) con lámina de aluminio.
Tamaño del envase de 21 cápsulas.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Las cápsulas no se deben abrir o triturar. En el caso de que el polvo de pomalidomida entre en contacto con la piel, debe lavar abundantemente la piel con agua y jabón inmediatamente. En el caso de que el polvo de pomalidomida entre en contacto con las membranas mucosas, debe lavarlas abundantemente con agua a presión.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local. El medicamento no utilizado debe devolverse al farmacéutico al final del tratamiento.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd.
1 Longwalk Road Stockley Park Uxbridge UB11 1DB Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/850/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 05/agosto/2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la identificación de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Imnovid 4 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula dura contiene 4 mg de pomalidomida.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura.
Imnovid 4 mg cápsula dura: tapa opaca de color azul oscuro y cuerpo opaco de color azul marcadas con “POML 4 mg” en tinta blanca, de tamaño 2, cápsulas de gelatina dura.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Imnovid en combinación con dexametasona está indicado en el tratamiento de los pacientes adultos con mieloma múltiple resistente al tratamiento o recidivante que hayan recibido al menos dos tratamientos previos, incluyendo lenalidomida y bortezomib, y que hayan experimentado una progresión de la enfermedad en el último tratamiento.
4.2 Posología y forma de administración
El tratamiento debe iniciarse y monitorizarse bajo la supervisión de médicos con experiencia en el tratamiento del mieloma múltiple.
Posología
La dosis inicial recomendada es de 4 mg de Imnovid una vez al día por vía oral, en los días del 1 al 21 de ciclos repetidos de 28 días. La dosis recomendada de dexametasona es de 40 mg por vía oral una vez al día, en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
La posología se mantiene o modifica en función de los resultados clínicos y de laboratorio.
El tratamiento debe suspenderse si existe evidencia de progresión de la enfermedad.
Modificación o interrupción de la dosis de _pomalidomida
Las instrucciones para la interrupción y reducción de la dosis de pomalidomida relacionadas con reacciones adversas hematológicas se indican en la siguiente tabla:
|
Toxicidad |
Modificación de la dosis |
|
NeutroDenia • RAN* <0,5 x 109/l o neutropenia febril (fiebre >38,5°C y RAN <1 x 109/l) |
Interrumpir el tratamiento con pomalidomida, control semanal del hemograma completo. |
|
• RAN vuelve a >1 x 109/1 |
Reanudar el tratamiento con 3 mg de pomalidomida al día. |
|
• Con cada disminución posterior a <0,5 x 1071 |
Interrumpir el tratamiento con pomalidomida |
|
• RAN vuelve a >1 x 109/l |
Reanudar el tratamiento con 1 mg menos de pomalidomida que la dosis previa |
|
T rombocitopenia • Recuento de plaquetas <25 x 109/l |
Interrumpir el tratamiento con pomalidomida, control semanal del hemograma completo |
|
• Recuento de plaquetas vuelve a >50 x 109/l |
Reanudar el tratamiento con 3 mg de pomalidomida al día |
|
• Con cada disminución posterior a <25 x 1071 |
Interrumpir el tratamiento con pomalidomida |
|
• Recuento de plaquetas vuelve a >50 x 109/l |
Reanudar el tratamiento con 1 mg menos de pomalidomida que la dosis previa |
*RAN - Recuento absoluto de neutrófilos
Para iniciar un nuevo ciclo de pomalidomida, el recuento de neutrófilos debe ser >1 x 109/l y el recuento de plaquetas debe ser >50 x 109/l.
En caso de neutropenia, el médico debe considerar el uso de factores de crecimiento.
En el caso de otras reacciones adversas de grado 3 o 4 relacionadas con pomalidomida, el médico debe considerar interrumpir el tratamiento y reanudarlo con un 1 mg menos que la dosis previa una vez que se haya disminuido la reacción adversa a un grado inferior o igual a 2.
Si la reacción adversa ocurre tras disminuciones de la dosis a 1 mg, entonces debe suspenderse el tratamiento con este medicamento.
Se debe considerar la interrupción o suspensión de pomalidomida en caso de exantema de grado 2-3. Se debe suspender el tratamiento con pomalidomida en caso de angioedema, exantema de grado 4 y exantema ampolloso o exfoliativo, y no se debe reanudar una vez suspendido por estas reacciones.
Si se administran inhibidores potentes del CYP1A2 (p. ej., ciprofloxacino, enoxacino y fluvoxamina) de forma concomitante con pomalidomida, se debe reducir la dosis de pomalidomida en un 50 %.
|
Toxicidad |
Modificación de la dosis |
|
Dispepsia = grado 1-2 Dispepsia > grado 3 |
Mantener la dosis y tratar con antihistamínicos H2 o equivalentes. Reducir la dosis en un nivel de dosis si los síntomas persisten. Interrumpir la administración hasta que se controlen los síntomas. Añadir antihistamínicos H2 o equivalentes y reducir la dosis en un nivel cuando se reanude su administración. |
|
Edema > grado 3 |
Usar diuréticos según sea necesario y reducir la dosis en un nivel de dosis. |
|
Confusión o cambios en el estado de ánimo > grado 2 |
Interrumpir la administración hasta que desaparezcan los síntomas. Cuando se reanude su administración reducir la dosis en un nivel de dosis. |
|
Debilidad muscular > grado 2 |
Interrumpir la administración hasta que la debilidad muscular sea < grado 1. Reiniciar la dosis con una reducción de un nivel. |
|
Hiperglucemia > grado 3 |
Reducir la dosis en un nivel de dosis. Tratar con insulina o hipoglucemiantes orales según sea necesario. |
|
Pancreatitis aguda |
Suspensión de dexametasona del régimen de tratamiento del paciente. |
|
Otros efectos adversos relacionados con dexametasona > grado 3 |
Interrumpir la administración de dexametasona hasta que los efectos adversos sean de grado < 2. Reanudar su administración con una reducción de un nivel de la dosis. |
Niveles de reducción de la dosis de dexametasona:
Niveles de reducción de la dosis (<75 años): dosis inicial 40 mg; nivel de dosis -1 20 mg; nivel de dosis -2 10 mg en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
Niveles de reducción de la dosis (>75 años): dosis inicial 20 mg; nivel de dosis -1 12 mg; nivel de dosis -2 8 mg en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
Si la recuperación de las toxicidades tarda más de 14 días, la dosis de dexametasona se reducirá en un nivel de dosis.
Poblaciones especiales Población _ pediátrica
No hay un uso relevante de Imnovid en niños de 0 a 17 años para la indicación del mieloma múltiple.
Pacientes de edad avanzada
No se requiere ningún ajuste de dosis de pomalidomida. En pacientes de >75 años, la dosis inicial de dexametasona es de 20 mg una vez al día en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días.
Insuficiencia renal
No se requiere ningún ajuste de dosis de pomalidomida en los pacientes con insuficiencia renal. Los días de hemodiálisis, los pacientes deben tomar la dosis de pomalidomida después de la hemodiálisis.
Insuficiencia hepática
Los pacientes con una concentración sérica de bilirrubina total >2,0 mg/dl se excluyeron de los estudios clínicos. La insuficiencia hepática tiene un efecto modesto sobre la farmacocinética de pomalidomida (ver sección 5.2). No se requiere ajustar la dosis inicial de pomalidomida en pacientes con insuficiencia hepática según definen los criterios de Child-Pugh. Sin embargo, los pacientes con insuficiencia hepática deben ser monitorizados cuidadosamente por si presentan reacciones adversas y se debe reducir la dosis o suspender la administración de pomalidomida según sea necesario.
Forma de administración Vía oral.
Imnovid debe tomarse a la misma hora cada día. Las cápsulas no deben abrirse, romperse ni masticarse (ver sección 6.6). Este medicamento debe tomarse entero, preferiblemente con agua, con o sin alimentos. Si el paciente olvida tomar una dosis de Imnovid un día, el paciente debe entonces tomar la próxima dosis al día siguiente a la hora habitual. Los pacientes no deben ajustar la dosis para compensar una dosis olvidada en días anteriores.
Se recomienda presionar solo en un extremo de la cápsula para sacarla del blíster y reducir así el riesgo de deformación o rotura de la cápsula.
4.3 Contraindicaciones
- Embarazo.
- Mujeres con capacidad de gestación, a menos que se cumplan todas las condiciones del Programa de Prevención de Embarazo (ver secciones 4.4 y 4.6).
- Pacientes varones incapaces de seguir o cumplir las medidas anticonceptivas requeridas (ver sección 4.4).
- Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo Teratogenicidad
Pomalidomida no debe tomarse durante el embarazo ya que es esperable un efecto teratogénico. Pomalidomida está relacionada estructuralmente con talidomida. Talidomida es un teratógeno conocido en humanos, que causa defectos congénitos de nacimiento graves que pueden poner en peligro la vida del niño. Pomalidomida tiene un efecto teratogénico en ratas y conejos cuando se administra durante el periodo de mayor organogénesis (ver sección 5.3).
Todas las pacientes deben cumplir las condiciones del Programa de Prevención de Embarazo a menos que exista evidencia fiable de que la paciente no tiene capacidad de gestación.
Criterios para definir a las mujeres que no tienen capacidad de gestación
Se considera que una paciente o la pareja de un paciente varón no tiene capacidad de gestación si cumple por lo menos uno de los siguientes criterios:
• Edad >50 años y con amenorrea natural durante >1 año*.
• Insuficiencia ovárica prematura confirmada por un ginecólogo especialista.
• Salpingooforectomía bilateral o histerectomía previas.
• Genotipo XY, síndrome de Turner, agenesia uterina.
*La amenorrea que pueda aparecer después de un tratamiento oncológico o durante la lactancia no descarta la capacidad de gestación.
Asesoramiento
En mujeres con capacidad de gestación, pomalidomida está contraindicada a menos que la paciente cumpla todas las condiciones que se indican a continuación:
• Comprende el riesgo teratogénico esperado para el feto.
• Comprende la necesidad de utilizar métodos anticonceptivos eficaces, sin interrupción, desde 4 semanas antes de iniciar el tratamiento, durante la duración completa del mismo y 4 semanas después de finalizarlo.
• Incluso si una mujer con capacidad de gestación tiene amenorrea, debe seguir todos los consejos sobre anticoncepción eficaz.
• Debe ser capaz de cumplir las medidas anticonceptivas eficaces.
• Está informada y comprende las potenciales consecuencias del embarazo, y la necesidad de consultar rápidamente a un especialista si hay riesgo de embarazo.
• Comprende la necesidad de comenzar el tratamiento tan pronto como se le dispense pomalidomida y tras haber obtenido un resultado negativo en la prueba de embarazo.
• Comprende la necesidad de realizar pruebas de embarazo y acepta hacérselas cada 4 semanas, excepto en el caso de que se haya sometido previamente a una ligadura de trompas de eficacia confirmada.
• Confirma que comprende los peligros y las precauciones necesarias asociadas al uso de pomalidomida.
El médico prescriptor debe comprobar que, en el caso de las mujeres con capacidad de gestación:
• La paciente cumple las condiciones del Programa de Prevención de Embarazo, incluida la confirmación de que tiene un nivel de comprensión adecuado.
• La paciente ha aceptado las condiciones mencionadas anteriormente.
En el caso de pacientes varones que toman pomalidomida, los datos farmacocinéticos han demostrado que pomalidomida está presente en el semen humano. Como medida de precaución, todos los pacientes varones que tomen pomalidomida deben cumplir los siguientes requisitos:
• Comprende el riesgo teratogénico esperado si tiene relaciones sexuales con una mujer embarazada o con una mujer con capacidad de gestación.
• Comprende la necesidad del uso de preservativos si tiene relaciones sexuales con una mujer embarazada o con una mujer con capacidad de gestación que no utiliza métodos anticonceptivos eficaces, durante el tratamiento y durante los 7 días posteriores a la interrupción de la dosis y/o el cese del tratamiento. Los varones vasectomizados deben utilizar preservativos si tienen relaciones sexuales con una mujer embarazada o con una mujer con capacidad de gestación ya que pomalidomida puede estar presente en el semen aún en ausencia de espermatozoides.
• Comprende que si su pareja se queda embarazada mientras él está tomando pomalidomida o durante los 7 días posteriores a la suspensión del tratamiento con pomalidomida, debe informar inmediatamente a su médico y es recomendable derivar a su pareja a un médico especialista o con experiencia en teratología para su evaluación y asesoramiento.
Anticoncepción
Las mujeres con capacidad de gestación deben usar un método anticonceptivo eficaz desde 4 semanas antes del tratamiento, durante el tratamiento y hasta 4 semanas después del tratamiento con pomalidomida, e incluso en el caso de interrupción de la administración, a menos que la paciente se comprometa a mantener una abstinencia sexual absoluta y continua, que será confirmada mensualmente. Si la paciente no utiliza un método anticonceptivo eficaz, debe ser derivada a un profesional sanitario debidamente capacitado con objeto de que reciba asesoramiento para empezar a utilizar métodos anticonceptivos.
Los siguientes métodos pueden considerarse ejemplos de métodos anticonceptivos adecuados:
• Implante
• Sistema de liberación intrauterino de levonorgestrel
• Sistemas “depot” de liberación de acetato de medroxiprogesterona
• Ligadura de trompas
• Relaciones sexuales sólo con varones vasectomizados; la eficacia de la vasectomía debe confirmarse mediante dos análisis de semen negativos
• Inhibidores de la ovulación que contienen progestágeno solo (p. ej., desogestrel)
Debido al riesgo aumentado de tromboembolismo venoso (TEV) en pacientes con mieloma múltiple que toman pomalidomida y dexametasona, no se recomienda el uso concomitante de anticonceptivos orales combinados (ver también sección 4.5). Si una paciente está tomando anticonceptivos orales combinados, debe cambiar a uno de los métodos anticonceptivos eficaces enumerados anteriormente. El riesgo aumentado de tromboembolismo venoso se mantiene durante un periodo de 4 a 6 semanas después de suspender el tratamiento con anticonceptivos orales combinados. La eficacia de los anticonceptivos esteroideos puede verse reducida durante el tratamiento concomitante con dexametasona (ver sección 4.5).
Los implantes y los sistemas de liberación intrauterinos de levonorgestrel se asocian con un mayor riesgo de infección en el momento de la colocación y con hemorragia vaginal irregular. En especial en las pacientes con neutropenia debe considerarse el uso profiláctico de antibióticos.
La colocación de dispositivos intrauterinos de liberación de cobre no está recomendada, debido al potencial riesgo de infección en el momento de su colocación y a la pérdida de sangre menstrual, que pueden suponer un peligro para las pacientes con neutropenia grave o trombocitopenia grave.
Pruebas de embarazo
Las mujeres con capacidad de gestación deben efectuarse pruebas de embarazo con una sensibilidad mínima de 25 mUI/ml bajo supervisión médica y conforme a la práctica habitual, tal como se explica a continuación. Este requisito incluye a las mujeres con capacidad de gestación que practican una abstinencia sexual absoluta y continua. Idealmente, la prueba de embarazo, la prescripción y la dispensación deben realizarse el mismo día. Pomalidomida se debe dispensar a las mujeres con capacidad de gestación en un plazo de siete días tras la prescripción.
Antes de iniciar el tratamiento
Debe efectuarse una prueba de embarazo bajo supervisión médica durante la consulta, en el momento de prescribir pomalidomida o en los tres días anteriores a la visita al médico prescriptor, siempre que la paciente haya estado usando un método anticonceptivo eficaz durante al menos 4 semanas. La prueba debe garantizar que la paciente no esté embarazada cuando inicie el tratamiento con pomalidomida.
Seguimiento y _finalización del tratamiento
Se debe repetir cada 4 semanas una prueba de embarazo bajo supervisión médica, y realizar otra 4 semanas después de la finalización del tratamiento, excepto en el caso de que la paciente se haya sometido a una ligadura de trompas de eficacia confirmada. Estas pruebas de embarazo deben efectuarse el mismo día de la consulta en que se prescriba el medicamento o en los tres días anteriores a la visita al médico prescriptor.
Varones
Pomalidomida está presente en el semen humano durante el tratamiento. Como medida de precaución, y teniendo en cuenta las poblaciones especiales con un tiempo de eliminación potencialmente prolongado, como la insuficiencia renal, todos los pacientes varones que tomen pomalidomida, incluyendo a aquellos que se hayan sometido a una vasectomía, deben usar preservativos durante todo el tratamiento, durante la interrupción de la administración y hasta 7 días después del final del tratamiento, si su pareja está embarazada o tiene capacidad de gestación y no está usando ningún método anticonceptivo.
Los pacientes varones no deben donar semen o esperma durante el tratamiento (periodos de interrupción de la dosis incluidos) ni en el plazo de 7 días después de la suspensión del tratamiento con pomalidomida.
Precauciones adicionales
Se debe indicar a los pacientes que no den nunca este medicamento a otra persona y que devuelvan las cápsulas sin usar al farmacéutico al final del tratamiento.
Los pacientes no deben donar sangre, semen o esperma durante el tratamiento (periodos de interrupción de la dosis incluidos) ni en el plazo de 7 días después de la suspensión del tratamiento con pomalidomida.
Material educativo, restricciones de prescripción y dispensación
Con objeto de ayudar a los pacientes a evitar la exposición fetal a pomalidomida, el Titular de la Autorización de Comercialización distribuirá material educativo a los profesionales sanitarios, destinado a reforzar las advertencias acerca de la teratogenicidad esperada de pomalidomida y a proporcionar asesoramiento sobre anticoncepción antes de iniciar el tratamiento y sobre la necesidad de realizar pruebas de embarazo. El médico debe informar a la paciente acerca del riesgo teratogénico esperado y de las estrictas medidas de prevención de embarazo, especificadas en el Programa de Prevención de Embarazo así como proporcionar a la paciente un folleto informativo adecuado, una tarjeta de paciente y/o una herramienta equivalente conforme al sistema nacional implementado de tarjeta del paciente. En colaboración con cada autoridad nacional competente, se ha implementado un sistema de distribución nacional controlada. El sistema de distribución controlada incluye el uso de una tarjeta del paciente y/o un herramienta equivalente para el control de la prescripción y/o dispensación, así como la recogida de datos detallados en relación con la indicación terapéutica, para monitorizar el uso en una indicación no autorizada dentro del territorio nacional. Idealmente, la prueba de embarazo, la prescripción y la dispensación deben realizarse el mismo día. La dispensación de pomalidomida en mujeres con capacidad de gestación debe hacerse dentro de los 7 días de prescripción y tras haber obtenido un resultado negativo en la prueba de embarazo supervisada por un médico. Las prescripciones a mujeres con capacidad de gestación pueden tener una caducidad de 4 semanas y las prescripciones para el resto de pacientes pueden tener una caducidad de 12 semanas.
Eventos hematológicos
La reacción adversa hematológica de Grado 3 o 4 notificada con mayor frecuencia en pacientes con mieloma múltiple en recaída/refractario fue la neutropenia, seguido de anemia y trombocitopenia. Se debe monitorizar a los pacientes en busca de posibles reacciones adversas hematológicas, especialmente neutropenia. Se debe advertir a los pacientes que informen rápidamente acerca de los episodios febriles que presenten. Los médicos deben estar atentos a los signos de hemorragia en los pacientes, incluyendo epistaxis, especialmente en el caso de medicación concomitante conocida por aumentar el riesgo de sangrado (ver sección 4.8). Debe efectuarse a los pacientes un hemograma completo semanal en el momento basal, durante las primeras 8 semanas y después mensualmente. Puede ser necesaria una modificación de la dosis (ver sección 4.2). Los pacientes pueden requerir el uso de hemoderivados y/o factores de crecimiento.
Eventos tromboembólicos
Se han observado eventos tromboembólicos venosos (predominantemente trombosis venosa profunda y embolia pulmonar) y eventos trombóticos arteriales (infarto de miocardio y accidente cerebrovascular) en pacientes tratados con pomalidomida en combinación con dexametasona. Los pacientes con factores de riesgo conocidos de tromboembolismo, incluida una trombosis previa, deben estar estrechamente monitorizados. Se deben tomar medidas para intentar minimizar todos los factores de riesgo modificables (p. ej. tabaquismo, hipertensión e hiperlipidemia). Se aconseja a médicos y pacientes que estén atentos a los signos y síntomas de tromboembolismo. Se debe advertir a los pacientes que soliciten atención médica si presentan síntomas como respiración entrecortada, dolor torácico o edema de las extremidades. Es recomendable el uso de terapia anticoagulante (si no está contraindicada), como el ácido acetilsalicílico, warfarina, heparina o clopidogrel, especialmente en pacientes con factores de riesgo trombótico adicionales. Después de una cuidadosa evaluación de los factores de riesgo subyacentes del paciente individual, se debe tomar una decisión respecto al uso de medidas profilácticas. En los estudios clínicos los pacientes recibieron ácido aceltilsalicílico profiláctico o terapia antitrombótica alternativa. El uso de agentes eritropoyéticos conlleva un riesgo de eventos trombóticos incluyendo tromboembolismo. Por lo tanto, deben emplearse con precaución los agentes eritropoyéticos así como otros agentes que puedan aumentar el riesgo de eventos tromboembólicos.
Neuropatía periférica
Se excluyó de los estudios clínicos con pomalidomida a los pacientes con neuropatía periférica en curso de Grado >2. Se deben adoptar las precauciones adecuadas al considerar el tratamiento de estos pacientes con pomalidomida.
Disfunción cardíaca significativa
Se excluyó de los estudios clínicos con pomalidomida a los pacientes con una disfunción cardíaca significativa (insuficiencia cardíaca congestiva [Clase III o IV de la NY Heart Association]; infarto de miocardio dentro de los 12 meses desde el inicio del estudio; angina de pecho inestable o mal controlada). Se han notificado acontecimientos de insuficiencia cardiaca, que incluyen insuficiencia cardiaca congestiva y edema pulmonar (ver sección 4.8), especialmente en pacientes con enfermedad cardiaca preexistente o factores de riesgo cardiacos. Se deben adoptar las precauciones adecuadas al considerar el tratamiento de estos pacientes con pomalidomida, incluido el control periódico para detectar la presencia de signos y síntomas de insuficiencia cardiaca.
Síndrome de lisis tumoral
Puede producirse un síndrome de lisis tumoral. Los pacientes con mayor riesgo de sufrir un síndrome de lisis tumoral son aquellos que presentan una carga tumoral elevada antes del tratamiento. Se debe monitorizar estrechamente a estos pacientes y se deben adoptar las precauciones adecuadas.
Segundas neoplasias malignas primarias
Se han notificado segundas neoplasias malignas primarias, como cáncer de piel no melanoma, en pacientes en tratamiento con pomalidomida (ver sección 4.8). Los médicos deben evaluar cuidadosamente a los pacientes antes y durante el tratamiento, utilizando pruebas estándar de detección de cáncer por si aparecieran segundas neoplasias malignas primarias e instaurar el tratamiento indicado.
Reacciones alérgicas
Se han notificado angioedema y reacciones dermatológicas graves (ver sección 4.8). Se excluyó de los estudios clínicos a los pacientes con antecedentes de reacciones alérgicas graves asociadas a talidomida o lenalidomida. Estos pacientes pueden presentar un mayor riesgo de reacciones de hipersensibilidad y no deben tomar pomalidomida. Se debe considerar la interrupción o suspensión de pomalidomida si se presenta exantema de Grado 2 o 3. Se debe suspender definitivamente el tratamiento con pomalidomida si se presenta angioedema, exantema de Grado 4 y exantema ampolloso o exfoliativo.
Mareo y confusión
Se han notificado mareo y estados de confusión con pomalidomida. Los pacientes deben evitar las situaciones en que el mareo o la confusión puedan representar un problema y no tomar otros medicamentos que puedan causar mareo o confusión sin solicitar antes consejo médico.
Enfermedad pulmonar intersticial (EPI)
Se han observado EPI y acontecimientos asociados que incluyen casos de neumonitis con pomalidomida. Se debe realizar una evaluación cuidadosa de los pacientes que presenten un inicio repentino o empeoramiento inexplicable de los síntomas pulmonares para descartar la EPI. Se debe interrumpir la administración de pomalidomida durante la investigación de estos síntomas Y, si se confirma la EPI, debe iniciarse un tratamiento adecuado. Únicamente se debe reanudar pomalidomida después de una evaluación exhaustiva de los beneficios y los riesgos.
Trastornos hepáticos
Se han observado concentraciones notablemente elevadas de alanina aminotransferasa y bilirrubina en los pacientes tratados con pomalidomida (ver sección 4.8). Se han notificado también casos de hepatitis que provocaron la suspensión de pomalidomida. Se recomienda controlar periódicamente la función hepática durante los primeros 6 meses de tratamiento con pomalidomida y, posteriormente, cuando esté clínicamente indicado.
Infecciones
Se han notificado rara vez casos de reactivación de la hepatitis B en pacientes en tratamiento con pomalidomida combinado con dexametasona que habían sido previamente infectados por el virus de la hepatitis B (VHB). Algunos de estos casos han evolucionado a fallo hepático agudo, dando lugar a la suspensión de pomalidomida. Se debe determinar el estado del virus de la hepatitis B antes de iniciar el tratamiento con pomalidomida. Se recomienda que los pacientes que den un resultado positivo en la prueba de infección por VHB se pongan en contacto con un médico con experiencia en el tratamiento de la hepatitis B. Se debe tener precaución cuando se administre pomalidomida en combinación con dexametasona en pacientes previamente infectados por el VHB, incluidos los pacientes anti-Bc positivos pero con HBsAg negativos. Se debe monitorizar estrechamente a estos pacientes para detectar signos y síntomas de infección activa por el VHB durante todo el tratamiento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Efecto de Imnovid sobre otros medicamentos
No se espera que pomalidomida pueda causar interacciones medicamentosas farmacocinéticas clínicamente relevantes debido a la inhibición o inducción de la isoenzima P450, o inhibición de transportadores cuando se administra de forma concomitante con sustratos de estas enzimas o transportadores. No se ha evaluado clínicamente el potencial de estas interacciones medicamentosas, incluyendo el posible impacto de pomalidomida en la farmacocinética de los anticonceptivos orales combinados (ver sección 4.4 Teratogenicidad).
Efecto de otros medicamentos sobre Imnovid
Pomalidomida se metaboliza parcialmente por CYP1A2 y CYP3A4/5.También es un sustrato de la glicoproteína P. La administración concomitante de pomalidomida con ketoconazol, inhibidor potente del CYP3A4/5 y de la Gp-P, o con el inductor potente del CYP3A4/5, carbamazepina, no demostró ningún efecto clínicamente relevante a la exposición a pomalidomida. La administración concomitante de pomalidomida con el inhibidor potente del CYP1A2 fluvoxamina en presencia de ketoconazol, incrementó la exposición media a pomalidomida en un 107 %, con un intervalo de confianza del 90 % [del 91 % al 124 %], frente a pomalidomida más ketoconazol. En un segundo estudio realizado para evaluar la contribución a los cambios del metabolismo de un inhibidor del CYP1A2 solo, la administración conjunta de fluvoxamina sola con pomalidomida aumentó la exposición media a pomalidomida en un 125 % con un intervalo de confianza del 90 % [del 98 % al 157 %] frente a pomalidomida administrada en monoterapia. Si se administran inhibidores potentes del CYP1A2 (p. ej., ciprofloxacino, enoxacino y fluvoxamina) de forma concomitante con pomalidomida, se debe reducir la dosis de pomalidomida en un 50 %.
Dexametasona
La administración concomitante de múltiples dosis de hasta 4 mg de pomalidomida con dexametasona de 20 mg a 40 mg (un inductor leve a moderado de varias enzimas CYP, incluido el CYP3A) en pacientes con mieloma múltiple no tuvo ningún efecto sobre la farmacocinética de pomalidomida frente a pomalidomida administrada sola.
Se desconoce el efecto de dexametasona sobre la warfarina. Se aconseja realizar una monitorización rigurosa de la concentración de warfarina durante el tratamiento.
4.6 Fertilidad, embarazo y lactancia
Mujeres con capacidad de gestación / Anticonceptivos en varones y mujeres
Las mujeres con capacidad de gestación deben utilizar métodos anticonceptivos efectivos. Si una mujer tratada con pomalidomida se queda embarazada, se debe suspender el tratamiento y derivar a la paciente a un médico especialista o con experiencia en teratología, para su evaluación y asesoramiento. Si un paciente varón toma pomalidomida y su pareja se queda embarazada, se recomienda derivar a la mujer a un médico especialista o con experiencia en teratología, para su evaluación y asesoramiento. Pomalidomida está presente en el semen humano. Como medida de precaución, todos los pacientes varones que tomen pomalidomida deben usar preservativos durante todo el tratamiento, durante la interrupción de la administración y hasta 7 días después del final del tratamiento, si su pareja está embarazada o tiene capacidad de gestación y no está usando ningún método anticonceptivo (ver secciones 4.3 y 4.4).
Embarazo
Se espera un efecto teratogénico de pomalidomida en humanos. Pomalidomida está contraindicada durante el embarazo y en mujeres con capacidad de gestación, a menos que se cumplan todas las condiciones para la prevención del embarazo, ver sección 4.3 y sección 4.4.
Lactancia
Se desconoce si pomalidomida se excreta en la leche materna. Se detectó la presencia de pomalidomida en la leche de ratas que estaban siendo amamantadas tras administrar el medicamento a la madre.
Debido a las posibles reacciones adversas en lactantes asociadas a pomalidomida, se debe decidir si es
necesario suspender la lactancia o suspender el tratamiento tras considerar la importancia del tratamiento para la madre.
Fertilidad
Se sabe que pomalidomida tiene un efecto negativo sobre la fertilidad y que es teratogénica en los animales. Tras su administración a conejas embarazadas, pomalidomida atravesó la placenta y se detectó en la sangre fetal. Ver sección 5.3.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Imnovid sobre la capacidad para conducir y utilizar máquinas es pequeña o moderada. Se han notificado casos de fatiga, disminución del nivel de conciencia, confusión y mareo relacionados con el uso de pomalidomida. Si notan estos efectos, se debe advertir a los pacientes de que no deben conducir automóviles, utilizar máquinas o realizar cualquier actividad peligrosa durante su tratamiento con pomalidomida.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Las reacciones adversas notificadas con mayor frecuencia en los estudios clínicos han sido los trastornos de la sangre y del sistema linfático, incluyendo anemia (45,7 %), neutropenia (45,3 %) y trombocitopenia (27 %); trastornos generales y alteraciones en el lugar de administración, incluyendo fatiga (28,3 %), pirexia (21 %) y edema periférico (13 %); e infecciones e infestaciones incluyendo neumonía (10,7 %). Las reacciones adversas relacionadas con neuropatía periférica fueron notificadas en el 12,3 % de los pacientes y las reacciones adversas de embolismo o tromboembolismo venoso fueron notificadas en el 3,3 % de los pacientes. Las reacciones adversas de grado 3 o 4 más frecuentes estaban relacionadas con trastornos de la sangre y del sistema linfático, incluyendo neutropenia (41,7 %), anemia (27 %) y trombocitopenia (20,7 %); infecciones e infestaciones, incluyendo neumonía (9 %); y trastornos generales y alteraciones en el lugar de administración, incluyendo fatiga (4,7 %), pirexia (3 %) y edema periférico (1,3 %). La reacción adversa grave notificada con mayor frecuencia fue la neumonía (9,3 %). Otras reacciones adversas graves notificadas incluyen neutropenia febril (4,0 %), neutropenia (2,0 %), trombocitopenia (1,7 %) y reacciones adversas de TEV (1,7 %).
Se observó que las reacciones adversas tendían a ocurrir con mayor frecuencia dentro de los primeros 2 ciclos de tratamiento con pomalidomida.
Tabla de reacciones adversas
En el estudio aleatorizado CC-4047-MM-003, un total de 302 pacientes con mieloma múltiple en recaída y refractario fueron tratados con 4 mg de pomalidomida administrada una vez al día durante 21 días en cada ciclo de 28 días, en combinación con una dosis baja semanal de dexametasona.
Las reacciones adversas observadas en los pacientes tratados con pomalidomida y dexametasona se incluyen a continuación, según el sistema de clasificación por órganos y la frecuencia (SOC por sus siglas en inglés) para todas las reacciones adversas y para las reacciones adversas de Grado 3 o 4.
Las frecuencias de las reacciones adversas son las notificadas en el grupo de pomalidomida más dexametasona del estudio CC-4047-MM-003 (n=302) y de los datos poscomercialización. Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de SOC (por sus siglas en inglés) y de frecuencia. Las frecuencias se definen, según las guías actuales, como: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); y poco frecuentes (>1/1.000 a <1/100).
|
Sistema de clasificación de órganos / Término preferente |
Todas las reacciones adversas/Frecuencia |
Reacciones adversas de Grado 3-4/Frecuencia |
|
Infecciones e |
Muy frecuentes |
Frecuentes |
|
infestaciones |
Neumonía (infecciones bacterianas, |
Sepsis neutropénica |
|
víricas y fungicas, incluidas las |
Neumonía (infecciones | |
|
infecciones oportunistas) |
bacterianas, víricas y fungicas, incluidas las infecciones | |
|
Frecuentes |
oportunistas) | |
|
Sepsis neutropénica |
Bronconeumonía | |
|
Bronconeumonía |
Infección del tracto respiratorio | |
|
Bronquitis |
Infección del tracto respiratorio | |
|
Infección del tracto respiratorio Infección del tracto respiratorio |
superior | |
|
superior |
Poco frecuentes | |
|
Nasofaringitis |
Bronquitis | |
|
Herpes zóster |
Herpes zóster | |
|
Frecuencia no conocida |
Frecuencia no conocida | |
|
Reactivación de la hepatitis B |
Reactivación de la hepatitis B | |
|
Neoplasias benignas, |
Poco frecuentes |
Poco frecuentes |
|
malignas y no |
Carcinoma de piel de células |
Carcinoma de piel de células |
|
especificadas (incl |
basales |
basales |
|
quistes y pólipos) |
Carcinoma de piel de células |
Carcinoma de piel de células |
|
escamosas |
escamosas | |
|
Trastornos de la |
Muy frecuentes |
Muy frecuentes |
|
sangre y del sistema |
Neutropenia |
Neutropenia |
|
linfático |
Trombocitopenia |
Trombocitopenia |
|
Leucopenia Anemia |
Anemia Frecuentes | |
|
Frecuentes |
Neutropenia febril | |
|
Neutropenia febril |
Leucopenia | |
|
Pancitopenia* |
Pancitopenia* | |
|
Trastornos del |
Muy frecuentes |
Frecuentes |
|
metabolismo y de la |
Disminución del apetito |
Hiperpotasemia |
|
nutrición |
Hiponatremia | |
|
Frecuentes Hiperpotasemia |
Hiperuricemia* | |
|
Hiponatremia |
Poco frecuentes | |
|
Hiperuricemia* Poco frecuentes Síndrome de lisis tumoral* |
Disminución del apetito Síndrome de lisis tumoral* | |
|
Trastornos |
Frecuentes |
Frecuentes |
|
psiquiátricos |
Estado de confusión |
Estado de confusión |
|
Sistema de clasificación de órganos / Término preferente |
Todas las reacciones adversas/Frecuencia |
Reacciones adversas de Grado 3-4/Frecuencia |
|
Trastornos del |
Frecuentes |
Frecuentes |
|
sistema nervioso |
Disminución del nivel de |
Disminución del nivel de |
|
conciencia Neuropatía sensitiva periférica |
conciencia | |
|
Mareo |
Poco frecuentes | |
|
Temblor |
Neuropatía sensitiva periférica | |
|
Hemorragia intracraneal* |
Mareo Temblor | |
|
Poco frecuentes |
Accidente cerebrovascular* | |
|
Accidente cerebrovascular* |
Hemorragia intracraneal* | |
|
Trastornos del oído y |
Frecuentes |
Frecuentes |
|
del laberinto |
Vértigo |
Vértigo |
|
Trastonos vasculares |
Frecuentes |
Poco frecuentes |
|
Trombosis venosa profunda |
Trombosis venosa profunda | |
|
Trastornos cardiacos |
Frecuentes |
Frecuentes |
|
Insuficiencia cardíaca* |
Insuficiencia cardíaca* | |
|
Fibrilación auricular* Infarto de miocardio* |
Fibrilación auricular* Poco frecuentes Infarto de miocardio* | |
|
Trastornos del |
Frecuentes |
Poco frecuentes |
|
sistema inmunológico |
Angioedema* |
Angioedema* |
|
Urticaria* |
Urticaria* | |
|
Trastornos |
Muy frecuentes |
Frecuentes |
|
respiratorios, |
Disnea |
Disnea |
|
torácicos y |
Tos | |
|
mediastínicos |
Poco frecuentes | |
|
Frecuentes |
Embolia pulmonar | |
|
Embolia pulmonar |
Tos | |
|
Epistaxis* |
Epistaxis* | |
|
Enfermedad pulmonar intersticial* |
Enfermedad pulmonar intersticial* | |
|
Trastornos |
Muy frecuentes | |
|
gastrointestinales |
Diarrea |
Frecuentes |
|
Náuseas |
Diarrea | |
|
Estreñimiento Frecuentes |
Vómitos Estreñimiento | |
|
Vómitos |
Poco frecuentes | |
|
Hemorragia gastrointestinal |
Náuseas Hemorragia gastrointestinal | |
|
Trastornos |
Poco frecuentes |
Poco frecuentes |
|
hepatobiliares |
Hiperbilirrubinemia Hepatitis* |
Hiperbilirrubinemia |
|
Trastornos de la piel y |
Frecuentes |
Frecuentes |
|
del tejido subcutáneo |
Erupción Prurito |
Erupción |
|
Sistema de clasificación de órganos / Término preferente |
Todas las reacciones adversas/Frecuencia |
Reacciones adversas de Grado 3-4/Frecuencia |
|
Trastornos |
Muy frecuentes |
Frecuentes |
|
musculoesqueléticos y |
Dolor óseo |
Dolor óseo |
|
del tejido conjuntivo |
Espasmos musculares |
Poco frecuentes Espasmos musculares |
|
Trastornos renales y |
Frecuentes |
Frecuentes |
|
urinarios |
Insuficiencia renal Retención urinaria |
Insuficiencia renal Poco frecuentes Retención urinaria |
|
Trastornos del |
Frecuentes |
Frecuentes |
|
aparato reproductor y de la mama |
Dolor pélvico |
Dolor pélvico |
|
Trastornos generales |
Muy frecuentes |
Frecuentes |
|
y alteraciones en el |
Fatiga |
Fatiga |
|
lugar de |
Pirexia |
Pirexia |
|
administración |
Edema periférico |
Edema periférico |
|
Exploraciones |
Frecuentes |
Frecuentes |
|
complementarias |
Disminución del recuento de |
Disminución del recuento de |
|
neutrófilos |
neutrófilos | |
|
Disminución del recuento de |
Disminución del recuento de | |
|
leucocitos |
leucocitos | |
|
Disminución del recuento de |
Disminución del recuento de | |
|
plaquetas |
plaquetas | |
|
Aumento de la alanina |
Aumento de la alanina | |
|
aminotransferasa Aumento del ácido úrico en sangre* |
aminotransferasa Poco frecuentes Aumento del ácido úrico en sangre* |
*Identificados a partir de los datos poscomercialización; las frecuencias se basan en los datos de los ensayos clínicos.
Descripción de reacciones adversas seleccionadas
Teratogenicidad
Pomalidomida está relacionada estructuralmente con talidomida. Talidomida es un principio activo con acción teratogénica conocida en humanos, que causa defectos de nacimiento graves que pueden poner en peligro la vida del niño. Pomalidomida tiene un efecto teratogénico en ratas y conejos cuando se administra durante el periodo de mayor organogénesis (ver secciones 4.6 y 5.3). Si se toma pomalidomida durante el embarazo, se espera un efecto teratogénico de pomalidomida en los seres humanos (ver sección 4.4).
Neutropenia y trombocitopenia
El 45,3 % de los pacientes que recibieron pomalidomida más dosis bajas de dexametasona (Pom + LD-Dex) experimentó neutropenia, frente al 19,5 % de los pacientes que recibieron dosis altas de dexametasona (HD-Dex). La neutropenia fue de grado 3 o 4 en un 41,7 % de los pacientes que recibieron Pom + LD-Dex frente al 14,8 % de los que recibieron HD-Dex. En los pacientes tratados con Pom + LD-Dex, la neutropenia fue grave en una minoría (2,0 % de los pacientes), no tuvo como consecuencia la suspensión del tratamiento, y se asoció con la interrupción del tratamiento en un 21,0 % de los pacientes y con una reducción de la dosis en un 7,7 % de los pacientes.
El 6,7 % de los pacientes que recibieron Pom + LD-Dex experimentó neutropenia febril (NF), frente a ninguno de aquellos que recibieron HD-Dex. Todos los casos fueron notificados como de grado 3 o 4. La NF fue notificada como grave en un 4,0 % de los pacientes. Asimismo, la NF se asoció con una interrupción de la dosis en un 3,7 % de los pacientes, con una reducción de la dosis en un 1,3 % de los pacientes y con ningún caso de suspensión del tratamiento.
El 27,0 % de los pacientes que recibieron Pom + LD-Dex experimentó trombocitopenia frente a un
26,8 % de los pacientes que recibieronHD-Dex. La trombocitopenia fue de grado 3 o 4 en un 20,7 % de los pacientes que recibieron Pom + LD-Dex frente a un 24,2 % de los que recibieron HD-Dex. En los pacientes tratados con Pom + LD-Dex, la trombocitopenia fue grave en un 1,7 % de los pacientes, conllevó una reducción de la dosis en un 6,3 % de los pacientes, una interrupción de la dosis en un 8 % de los pacientes y la suspensión del tratamiento en un 0,7 % de los pacientes (ver secciones 4.2 y 4.4).
Infección
La infección fue la toxicidad no hematológica más frecuente, con una incidencia del 55,0 % en los pacientes que recibieron Pom + LD-Dex frente al 48,3 % en los pacientes que recibieron HD-Dex. Aproximadamente la mitad de dichas infecciones fueron de grado 3 o 4; un 24,0 % en los pacientes tratados con Pom + LD-Dex y un 22,8 % en los pacientes tratados con HD-Dex.
Entre los pacientes tratados con Pom + LD-Dex las infecciones notificadas con mayor frecuencia fueron la neumonía y la infección del tracto respiratorio superior (en un 10,7 % y un 9,3 % de los pacientes, respectivamente); con un 24,3 % de las infecciones notificadas clasificadas como graves o mortales (grado 5) en el 2,7 % de los pacientes tratados. En los pacientes tratados con Pom + LD-Dex las infecciones conllevaron la suspensión de la dosis en un 2,0 % de los pacientes, la interrupción del tratamiento en un 14,3 % de los pacientes y la reducción de la dosis en un 1,3 % de los pacientes.
Eventos tromboembólicos
El 3,3 % de los pacientes que recibieron Pom + LD-Dex y un 2,0 % de los pacientes que recibieron HD-Dex experimentaron un embolismo o tromboembolismo venoso. El 1,3 % de los pacientes que recibieron Pom + LD-Dex experimentó reacciones de grado 3 o 4 frente a ningún caso entre los pacientes que recibieron HD-Dex. En los pacientes tratados con Pom + LD-Dex, el TEV se notificó como grave en el 1,7 %, no se notificó ninguna reacción adversa mortal en los estudios clínicos y el TEV no se asoció con suspensión de la dosis.
La profilaxis con ácido acetilsalicílico (y otros anticoagulantes en pacientes de alto riesgo) fue obligatoria para todos los pacientes participantes en los estudios clínicos. Se recomienda terapia anticoagulante (a no ser que esté contraindicada) (ver sección 4.4).
Neuropatía _ periférica
Se excluyó de los estudios clínicos a los pacientes con neuropatía periférica en curso de grado >2. El 12,3 % de los pacientes que recibieron Pom + LD-Dex experimentaron una neuropatía periférica, la mayoría de grado 1 o 2, frente a un 10,7 % de los pacientes que recibieron HD-Dex. El 1,0 % de los pacientes que recibieron Pom + LD-Dex experimentó reacciones de grado 3 o 4 frente al 1,3 % de los pacientes que recibieron HD-Dex. En los estudios clínicos, ninguna de las neuropatías periféricas se notificó como grave en los pacientes tratados con Pom + LD-Dex y la neuropatía periférica condujo a la suspensión de la dosis en un 0,3 % de los pacientes (ver sección 4.4).
La mediana del tiempo hasta la aparición de la neuropatía fue de 2,1 semanas, con una variación de 0,1 a 48,3 semanas. La mediana del tiempo hasta la aparición fue inferior en los pacientes que recibieron HD-Dex comparado con los pacientes tratados con Pom+LD-Dex (1,3 semanas frente a 2,1 semanas).
La mediana del tiempo hasta la desaparición de los síntomas fue de 22,4 semanas en los pacientes que recibieron Pom+LD-Dex y de 13,6 semanas en los pacientes que recibieron HD-Dex. El límite inferior
del intervalo de confianza del 95 % fue de 5,3 semanas en los pacientes tratados con Pom+LD-Dex y de
2,0 semanas en los pacientes que recibieron HD-Dex.
Hemorragia
Se han notificado trastornos hemorrágicos con pomalidomida, especialmente en pacientes con factores de riesgo tales como medicamentos concomitantes que aumentan el riesgo de hemorragia. Los eventos hemorrágicos incluyen epistaxis, hemorragia intracraneal y hemorragia gastrointestinal.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los
profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Se han evaluado dosis de pomalidomida de hasta 50 mg en dosis única en voluntarios sanos y 10 mg en múltiples dosis una vez al día en pacientes con mieloma múltiple sin que se haya notificado ningún caso de efecto adverso grave relacionado con sobredosis. Pomalidomida se eliminó mediante hemodiálisis. En caso de sobredosis, se recomienda terapia de soporte.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agente inmunomodulador, código ATC: L04AX06 Mecanismo de acción
Pomalidomida es un medicamento con actividad tumoricida directa contra el mieloma, actividad inmunomoduladora y capaz de inhibir el apoyo de las células del estroma para el crecimiento de las células cancerosas del mieloma múltiple. En concreto, pomalidomida inhibe la proliferación e induce la apoptosis de las células hematopoyéticas tumorales. Además, pomalidomida inhibe la proliferación de las líneas celulares de mieloma múltiple resistentes a lenalidomida y presenta un efecto sinérgico con dexametasona tanto en las líneas celulares de mieloma múltiple resistentes a lenalidomida como en las sensibles a lenalidomida para inducir la apoptosis de las células tumorales. Pomalidomida potencia la inmunidad celular mediada por los linfocitos T y por los linfocitos natural killer (NK) e inhibe la producción de citocinas proinflamatorias (p. ej. TNF-a e IL-6) por los monocitos. Pomalidomida también inhibe la angiogénesis mediante el bloqueo de la migración y adhesión de células endoteliales.
Eficacia clínica y seguridad
Se evaluó la eficacia y seguridad de pomalidomida en combinación con dexametasona en un ensayo de fase III, multicéntrico, aleatorizado y abierto (CC-4047-MM-003), donde se comparó el tratamiento de pomalidomida más una dosis baja de dexametasona (Pom + LD-Dex) frente a una dosis alta de dexametasona sola (HD-Dex) en pacientes adultos con mieloma múltiple en recaída o refractario que ya habían recibido al menos dos tratamientos previos, incluyendo lenalidomida y bortezomib, y que habían mostrado progresión de la enfermedad durante el último tratamiento. En el estudio participaron un total de 455 pacientes: 302 en el grupo Pom + LD-Dex y 153 en el grupo HD-Dex. La mayoría de los pacientes eran varones (59 %) y caucásicos (79 %); la mediana de edad de toda la población era de 64 años (mín, máx: 35, 87 años).
Los pacientes del grupo Pom + LD-Dex tomaron 4 mg de pomalidomida por vía oral en los días del 1 al 21 en cada ciclo de 28 días. Se administró la dosis de LD-Dex (40 mg) una vez al día en los días 1, 8, 15 y 22 de un ciclo de 28 días. En el grupo HD-Dex, los pacientes tomaron dexametasona (40 mg) una vez al día en los días del 1 al 4, del 9 al 12 y del 17 al 20 en un ciclo de 28 días. Los pacientes mayores de 75 años iniciaron el tratamiento con dexametasona 20 mg. Los pacientes continuaron con el tratamiento hasta la progresión de la enfermedad.
La variable principal de eficacia fue la supervivencia libre de progresión (SLP) de acuerdo con los criterios del InternationalMyeloma Working Group (IMWG). Para la población por intención de tratar (IDT), la mediana del tiempo de SLP según revisión del Independent Review Adjudication Committee (IRAC) basada en los criterios del IMWG fue de 15,7 semanas (IC de 95 %: 13,0; 20,1) en el grupo Pom + LD-Dex; la tasa estimada de supervivencia libre de eventos a las 26 semanas fue del 35,99 %
(± 3,46 %). En el grupo HD-Dex, la mediana del tiempo de SLP fue de 8,0 semanas (IC de 95 %: 7,0; 9,0); la tasa estimada de supervivencia libre de eventos a las 26 semanas fue del 12,15 % (± 3,63 %).
Se evaluó la SLP en varios subgrupos relevantes: sexo, raza, estatus de rendimiento ECOG, factores de estratificación (edad, población con enfermedad, terapias antimieloma previas [2, >2]), parámetros de relevancia pronóstica seleccionados (niveles basales de beta-2 microglobulina, niveles basales de albúmina, insuficiencia renal basal y riesgo citogenético) y exposición y refractariedad a terapias antimieloma previas. Independientemente del subgrupo evaluado, la SLP fue generalmente consistente con la observada en la población por IDT en ambos grupos de tratamiento.
En la tabla 1 se resumen los resultados de la supervivencia libre de progresión para la población por IDT. La figura 1 presenta la curva de Kaplan-Meier de la SLP para la población por IDT.
Tabla 1: Tiempo de Supervivencia Libre de Progresión según revisión del IRAC basada en
los criterios IMWG (prueba de log-rank estratificada) (población por IDT)
|
Pom+LD-Dex (N=302) |
HD-Dex (N=153) | |
|
Supervivencia libre de progresión (SLP), n |
302 (100,0) |
153 (100,0) |
|
Censurado, n (%) |
138 (45,7) |
50 (32,7) |
|
Progresión/muerte, n (%) |
164 (54,3) |
103 (67,3) |
|
Tiempo de supervivencia libre de progresión (semanas) | ||
|
Mediana3 |
15,7 |
8,0 |
|
[IC de 95 % bilateral]b |
[13,0; 20,1] |
[7,0 ; 9,0] |
|
Razón de riesgo (Hazard Ratio) (Pom+LD-Dex: HD-Dex) [IC de 95 % bilateral]0 |
0,45 [0,35; 0,59] | |
|
Valor P (bilateral) de la prueba de log-rankd |
<0,001 | |
Nota: IC = intervalo de confianza; IRAC = Comité Independiente Revisor de Evaluación; NE = no estimable a La mediana está basada en la estimación de Kaplan-Meier.
b Intervalo de confianza de 95 % de la mediana del tiempo de supervivencia libre de progresión.
c Basado en el modelo de riesgos proporcionales de Cox que compara las funciones de riesgo asociadas con los grupos de tratamiento, estratificados por edad (<75 frente a >75), población con la enfermedad (refractario a lenalidomida y bortezomib frente a no refractario a estos dos medicamentos), y número de terapias antimieloma previas (=2 frente a >2).
d El valor P está basado en la prueba de log-rank estratificada con los mismos factores de estratificación arriba mencionados para el modelo de Cox.
Fecha de corte: 7 de septiembre de 2012
Supervivencia Libre de Progresión según la revisión de respuesta del IRAC basada en los criterios IMWG (prueba de log-rank estratificada) (población por IDT)
Figura 1:

Fecha de corte: 7 de septiembre de 2012
Supervivencia Libre de Progresión (semanas)
La variable secundaria clave fue la supervivencia global (SG). Un total de 226 pacientes (74,8 %) del grupo Pom + LD-Dex y 95 pacientes (62,1 %) del grupo HD-Dex vivían en el momento de la fecha de corte (7 de septiembre de 2012). La mediana del tiempo de supervivencia global según las estimaciones Kaplan-Meier no fue alcanzado por el grupo Pom + LD Dex, pero podría esperarse que fuera al menos de 48 semanas, que corresponde al umbral más bajo del IC de 95 %. La mediana del tiempo de SG del grupo HD-Dex fue de 34 semanas (IC de 95 %: 23,4; 39,9). La tasa libre de eventos al año fue del
52,6 % (± 5,72 %) para el grupo Pom + LD-Dex y del 28,4 % (± 7,51 %) para el grupo HD-Dex. La diferencia en términos de SG entre los dos grupos de tratamiento fue estadísticamente significativa (p <0,001).
La Tabla 2 resume los resultados de supervivencia global para la población por IDT. La curva Kaplan-Meier de SG para la población por IDT se presenta en la Figura 2.
Basándose en los resultados de las variables SLP y SG, el Comité de Monitorización de Datos para este estudio recomendó que se completara el estudio y que los pacientes en el grupo HD-Dex fueran cruzados al grupo Pom + LD-Dex.
|
Estadísticas |
Pom+LD-Dex (N=302) |
HD-Dex (N=153) | |
|
N |
302 (100,0) |
153 (100,0) | |
|
Censurado |
n (%) |
226 (74,8) |
95 (62,1) |
|
Muerto |
n (%) |
76 (25,2) |
58 (37,9) |
|
Tiempo de supervivencia (semanas) |
Mediana3 |
NE |
34,0 |
|
IC de 95 % bilateralb |
[ 48,1; NE] |
[ 23,4; 39,9] | |
|
Razón de riesgo (Hazard Ratio) (Pom+LD-Dex:HD-Dex) [IC de 95 % bilateral0] |
0,53 [ 0,37; 0,74] | ||
|
Valor P (bilateral) de la prueba de log-rankd |
<0,001 | ||
Nota: IC = intervalo de confianza; NE = no estimable. a La mediana está basada en la estimación de Kaplan-Meier.
b Intervalo de confianza de 95 % sobre la mediana del tiempo de supervivencia global.
c Basado en el modelo de riesgos proporcionales de Cox que compara las funciones de riesgo asociadas con los grupos de tratamiento. d El valor P está basado en la prueba de log-rank no estratificada.
Fecha de corte: 7 de septiembre de 2012
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Imnovid en todos los grupos de la población pediátrica en mieloma múltiple (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Pomalidomida se absorbe alcanzando una concentración plasmática máxima (Cmáx) a las 2 o 3 horas y, por lo menos, un 73 % se absorbe después de administrar una dosis única por vía oral. El área bajo la curva (AUC) de pomalidomida aumenta, aproximadamente, lineal y proporcionalmente con los

0 13
Fecha de corte: 7 de septiembre de 2012
26 39 52
Superviviencia global (semanas)
65
incrementos de la dosis. Tras la administración de múltiples dosis, pomalidomida tiene una ratio de acumulación del 27 al 31 % en el AUC.
La administración conjunta con una comida rica en grasas y rica en calorías reduce la tasa de absorción, disminuyendo la Cmáx plasmática media en, aproximadamente un 27 %, pero con un efecto mínimo sobre la extensión de la absorción global con una disminución del 8 % en el AUC media. Por tanto, pomalidomida puede administrarse con o sin alimentos.
Distribución
Pomalidomida tiene un volumen de distribución aparente (Vd) medio de entre 62 y 138 litros en estado estable. En el semen de sujetos sanos pomalidomida se distribuye a una concentración de aproximadamente el 67 % del nivel de plasma a las 4 horas posteriores a la administración (aproximadamente Tmáx) tras 4 días de administración de 2 mg una vez al día. La unión in vitro de los enantiómeros de pomalidomida a las proteínas plasmáticas en humanos oscila entre el 12 % y el 44 % y no es dependiente de la concentración.
Biotransformación
En sujetos sanos que han recibido una dosis única por vía oral de [14C]-pomalidomida (2 mg), pomalidomida es la mayor sustancia circulante (aproximadamente el 70 % de la radioactividad del plasma) in vivo. No se hallaron metabolitos a >10 % relativos a la radioactividad total o relacionada en plasma.
Las rutas metabólicas principales de la radioactividad excretada son los procesos de hidroxilación con la posterior glucuronidación, o hidrólisis. Los estudios in vitro identificaron al CYP1A2 y al CYP3A4 como las enzimas primarias implicadas en la hidroxilación de pomalidomida mediada por CYP, con contribuciones menores adicionales del CYP2C19 y CYP2D6. Pomalidomida es también un sustrato de la glicoproteína-P in vitro. La administración concomitante de pomalidomida con el potente inhibidor del CYP3A4/5 y de la Gp-P, ketoconazol, o con el potente inductor del CYP3A4/5, carbamazepina, no demostró ningún efecto clínicamente relevante en la exposición a pomalidomida. La administración concomitante de pomalidomida con el inhibidor potente del CYP1A2, fluvoxamina, en presencia de ketoconazol, incrementó la exposición media a pomalidomida en un 107 %, con un intervalo de confianza del 90 % [del 91 % al 124 %], frente a pomalidomida más ketoconazol. En un segundo estudio realizado para evaluar la contribución a los cambios del metabolismo de un inhibidor del CYP1A2 solo, la administración conjunta de fluvoxamina sola con pomalidomida aumentó la exposición media a pomalidomida en un 125 % con un intervalo de confianza del 90 % [del 98 % al 157 %] frente a pomalidomida administrada en monoterapia. Si se administran inhibidores potentes del CYP1A2 (p. ej., ciprofloxacino, enoxacino y fluvoxamina) de forma concomitante con pomalidomida, se debe reducir la dosis de pomalidomida en un 50 %. La administración de pomalidomida a fumadores, sabiendo que el tabaquismo induce la isoforma CYP1A2, no tuvo ningún efecto clínicamente relevante en la exposición a pomalidomida frente a la exposición a pomalidomida observada en los no fumadores.
Según los datos in vitro, pomalidomida no es un inhibidor o inductor de las isoenzimas del citocromo P-450 y no inhibió ninguno de los fármacos transportadores que fueron estudiados. No se espera que pomalidomida pueda causar interacciones medicamentosas clínicamente relevantes cuando se administra de forma concomitante con sustratos de estas rutas.
Eliminación
En sujetos sanos pomalidomida se elimina con una mediana de semivida plasmática de aproximadamente 9,5 horas y de unas 7,5 horas en pacientes con mieloma múltiple. Pomalidomida tiene una media de aclaramiento corporal total (Cl/F) de aproximadamente 7-10 l/h.
Tras una dosis única por vía oral de [14C]-pomalidomida (2 mg) en sujetos sanos, aproximadamente el 73 % y el 15 % de la dosis radioactiva se eliminó por la orina y las heces, respectivamente, con aproximadamente el 2 % y el 8 % del radiocarbono administrado eliminado como pomalidomida en orina y heces.
Pomalidomida se metaboliza ampliamente antes de la excreción, con los metabolitos resultantes eliminados principalmente por la orina. Los tres metabolitos predominantes en la orina (formados mediante hidrólisis o hidroxilación con posterior glucuronidación) representan, aproximadamente, el
23 %, 17 % y 12 %, respectivamente, de la dosis en la orina.
Los metabolitos dependientes del CYP representan aproximadamente el 43 % de la radiactividad total excretada, mientras que los metabolitos hidrolíticos no dependientes del CYP representan el 25 %, y la excreción de pomalidomida inalterada representa el 10 % (2 % en orina y 8 % en heces).
Farmacocinética poblacional
Según análisis de farmacocinética poblacional basado en un modelo bicompartimental, los sujetos sanos y los pacientes con mieloma múltiple mostraron aclaramiento aparente (Cl/F) y volumen de distribución aparente en el compartimento central (WF) comparables. En tejidos periféricos, pomalidomida fue absorbida preferentemente por los tumores con un aclaramiento de distribución aparente en el compartimento periférico (Q/F) y un volumen de distribución aparente en el compartimento periférico (V3/F) 3,7 veces y 8 veces mayor, respectivamente, comparado con los sujetos sanos.
Población pediátrica
No existen datos disponibles sobre la administración de pomalidomida en niños o adolescentes (<18 años).
Población de edad avanzada
Según análisis de farmacocinética poblacional en sujetos sanos y en pacientes con mieloma múltiple, no se observó una influencia significativa de la edad (19-83 años) en el aclaramiento oral de pomalidomida. En los estudios clínicos los pacientes de edad avanzada (>65 años) expuestos a pomalidomida no requirieron ningún ajuste de dosis. Por favor, ver sección 4.2.
Insuficiencia renal
Los análisis de farmacocinética poblacional mostraron que los parámetros farmacocinéticos de pomalidomida no se vieron afectados de forma destacable en los pacientes con insuficiencia renal (definida mediante el aclaramiento de la creatinina o el filtrado glomerular estimado [FGe]) en comparación con los pacientes con la función renal normal (CrCl >60 ml/minuto). La exposición media a pomalidomida normalizada según el AUC fue del 98,2 % con un intervalo de confianza del 90 %
[77,4 % al 120,6 %] en los pacientes con insuficiencia renal moderada (FGe >30 a <45 ml/minuto/1,73 m2) en comparación con los pacientes con la función renal normal. La exposición media a pomalidomida normalizada según el AUC fue del 100,2 % con un intervalo de confianza del 90 % [79,7 % al 127,0 %] en los pacientes con insuficiencia renal grave que no precisaban diálisis (CrCl <30 o FGe <30 ml/minuto/1,73 m2) en comparación con los pacientes con la función renal normal. La exposición media a pomalidomida normalizada según el AUC aumentó en un 35,8 % con un IC del 90 % [7,5 % al 70,0 %] en los pacientes con insuficiencia renal grave que precisaban diálisis (CrCl <30 ml/minuto con necesidad de diálisis) en comparación con los pacientes con la función renal normal. Los cambios medios en la exposición a pomalidomida en cada uno de estos grupos de insuficiencia renal no son de una magnitud que requiera un ajuste de la dosis.
Insuficiencia hepática
Los parámetros farmacocinéticos cambiaron modestamente en los pacientes con insuficiencia hepática (definida mediante los criterios de Child-Pugh) en comparación con los sujetos sanos. La exposición media a pomalidomida aumentó en un 51 % con un intervalo de confianza del 90 % [del 9 % al 110 %] en los pacientes con insuficiencia hepática leve en comparación con los sujetos sanos. La exposición media a pomalidomida aumentó en un 58% con un intervalo de confianza del 90 % [del 13 % al 119 %] en los pacientes con insuficiencia hepática moderada en comparación con los sujetos sanos. La exposición media a pomalidomida aumentó en un 72 % con un intervalo de confianza del 90 % [del
24 % al 138 %] en los pacientes con insuficiencia hepática grave en comparación con los sujetos sanos. Los aumentos medios en la exposición a pomalidomida en cada uno de estos grupos de insuciencia hepática no son de una magnitud que requiera un ajuste del esquema de tratamiento o de la dosis (ver sección 4.2).
5.3 Datos preclínicos sobre seguridad
Estudios de toxicidad de dosis repetidas
La administración crónica de pomalidomida en ratas en dosis de 50, 250 y 1000 mg/kg/día durante 6 meses fue bien tolerada. No se detectó ningún efecto adverso hasta los 1000 mg/kg/día (una tasa de exposición 175 veces más elevada que la dosis clínica de 4 mg).
Se evaluó pomalidomida en monos en estudios de dosis repetidas de hasta 9 meses de duración. En estos estudios, los monos mostraron una mayor sensibilidad a los efectos de pomalidomida que las ratas. Las toxicidades primarias observadas en monos estuvieron relacionadas con los sistemas hematopoyético/linforreticular. En el estudio de 9 meses en monos con dosis de 0,05, 0,1 y 1 mg/kg/día se observó morbilidad y eutanasia temprana de 6 animales a dosis de 1 mg/kg/día que fueron atribuidas a los efectos inmunosupresores (infección por estafilococos, reducción de los linfocitos en sangre periférica, inflamación crónica del intestino grueso, reducción histológica de los linfocitos e hipocelularidad de la médula ósea) a exposiciones elevadas de pomalidomida (15 veces la tasa de exposición comparada con una dosis clínica de 4 mg). Dichos efectos inmunosupresores provocaron la eutanasia temprana de 4 monos debido a su mal estado de salud (heces líquidas, inapetencia, ingesta de alimentos reducida y pérdida de peso); la evaluación histopatológica de estos animales demostró inflamación crónica del intestino grueso y atrofia vellosa del intestino delgado. Se observó infección por estafilococos en 4 monos; 3 de éstos respondieron al tratamiento con antibióticos y uno murió sin tratamiento. Además, resultados consistentes con la leucemia mielógena aguda llevaron a la eutanasia de un mono; las observaciones clínicas y la patología clínica y/o alteraciones de la médula ósea observadas en este animal eran consistentes con inmunosupresión. La proliferación mínima o leve en los conductos biliares con incrementos asociados de la ALP y de la GGT también se observaron a dosis de 1 mg/kg/día. La evaluación de los animales recuperados indicó que todos los resultados relacionados con el tratamiento eran reversibles a las 8 semanas del cese de la administración, excepto la proliferación de los conductos biliares intrahepáticos observada en 1 animal en el grupo de 1 mg/kg/día. El nivel sin efecto adverso observado (NOAEL) fue de 0,1 mg/kg/día (una tasa de exposición relativa de 0,5 veces comparada con la dosis clínica de 4 mg).
Genotoxicidad/carcinogenicidad
Pomalidomida no resultó mutagénica en los ensayos de mutaciones bacterianas y de los mamíferos, y no indujo aberraciones cromosómicas en los linfocitos de sangre periférica en humanos así como tampoco a la formación de micronúcleos en eritrocitos policromáticos en la médula ósea de ratas a las que les fueron administradas dosis de hasta 2000 mg/kg/día. No se han realizado estudios de carcinogenicidad.
Fertilidad y desarrollo embrionario temprano
En un estudio de fertilidad y desarrollo embrionario temprano en ratas, se administró pomalidomida a los machos y las hembras a dosis de 25, 250 y 1000 mg/kg/día. El examen uterino en el día de gestación 13 mostró una reducción de la cantidad media de embriones viables y un aumento en la pérdida postimplantación con todos los niveles de dosis. Por consiguiente, el NOAEL en estos eventos observados fue <25 mg/kg/día (con un AUC24h de 39960 ng-h/ml (nanogramo^hora/mililitros) para la dosis más baja evaluada y una tasa de exposición 99 veces relativa a la dosis clínica de 4 mg). Cuando los machos tratados en este estudio fueron apareados con hembras no tratadas, todos los parámetros uterinos fueron comparables a los controles. Según estos resultados, los efectos observados fueron atribuidos al tratamiento de las hembras.
Desarrollo embriofetal
Pomalidomida resultó ser teratógena en ratas y conejos cuando se administró durante el periodo de mayor organogénesis. En el estudio de toxicidad sobre el desarrollo embriofetal de la rata, se observaron malformaciones relacionadas con la ausencia de vejiga urinaria, ausencia de la glándula tiroidea, así como la fusión y la desalineación de los elementos vertebrales torácicos y lumbares (arcos centrales y/o neurales) a todos los niveles de dosis (25, 50 y 1000 mg/kg/día).
En este estudio no se observó toxicidad materna. Por ello, el NOAEL materno fue de 1000 mg/kg/día, y el NOAEL para la toxicidad de desarrollo fue <25 mg/kg/día (con un AUC24h de 34340 ng-h/ml en el día de gestación 17 para la dosis más baja evaluada y la tasa de exposición fue de 85 veces comparada con la dosis clínica de 4 mg). En conejos, pomalidomida a dosis entre los 10 y 250 mg/kg/día produjo malformaciones en el desarrollo embriofetal. Se observaron aumentos de las anomalías cardíacas a todas las dosis con aumentos significativos a 250 mg/kg/día. A dosis de entre 100 y 250 mg/kg/día se registró un ligero aumento de la pérdida post-implantación y un ligero descenso en el peso del feto. A dosis de 250 mg/kg/día, las malformaciones fetales incluyeron anomalías en las extremidades (extremidades anteriores y posteriores dobladas y/o giradas, ausencia de dígito o dígito libre) y malformaciones esqueléticas asociadas (metacarpiano no osificado, metacarpiano y falange no alineados, ausencia de dígito, falange no osificada y tibia corta no osificada o doblada); dilatación moderada de los ventrículos laterales del cerebro; ubicación anormal de la arteria subclavia derecha; ausencia de los lóbulos intermedios pulmonares; par de riñones desplazados hacia abajo; morfología hepática alterada; ausencia de osificación de la pelvis u osificación incompleta; aumento medio de las costillas torácicas supernumerarias y reducción media de los tarsales osificados. Además, se observó una ligera reducción en el incremento del peso materno, una reducción significativa de los triglicéridos, y una reducción significativa del peso absoluto y relativo del bazo a dosis de entre 100 y 250 mg/kg/día. El NOAEL materno fue de 10 mg/kg/día y el NOAEL del desarrollo fue de <10 mg/kg/día (con un AUC24h de 418 ng-h/ml en el día de gestación 19 para la dosis más baja evaluada, similar a la obtenida con una dosis clínica de 4 mg).
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Contenido de la cápsula:
Manitol
Almidón pregelatinizado Estearil fumarato de sodio
Cubierta de la cápsula:
La cubierta de la cápsula de 4 mg contiene gelatina, dióxido de titanio (E171), indigotina (E132), FCF azul brillante (E133), indigotina (E132) y tinta blanca.
Tinta de impresión:
La cubierta de la cápsula de 4 mg contiene: tinta blanca - goma laca shellac, dióxido de titanio (E171), simeticona, propilenglicol (E1520) e hidróxido de amonio (E527).
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
4 años.
6.4 Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Las cápsulas están envasadas en blísteres de polivinilcloruro (PVC) / policlorotrifluoroetileno (PCTFE) con lámina de aluminio.
Tamaño del envase de 21 cápsulas.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Las cápsulas no se deben abrir o triturar. En el caso de que el polvo de pomalidomida entre en contacto con la piel, debe lavar abundantemente la piel con agua y jabón inmediatamente. En el caso de que el polvo de pomalidomida entre en contacto con las membranas mucosas, debe lavarlas abundantemente con agua a presión.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local. El medicamento no utilizado debe devolverse al farmacéutico al final del tratamiento.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd.
1 Longwalk Road Stockley Park Uxbridge UB11 1DB Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/850/004
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 05/agosto/2013
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
ANEXO II
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante responsable de la liberación de los lotes
Celgene Europe Limited 1 Longwalk Road Stockley Park Uxbridge UB11 1DB Reino Unido
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (Ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• - Informes periódicos de seguridad (IPS)
Los requerimientos para la presentatición de los informaes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 quater, apartado 7, de la Directiva 2001/83/CE y cualquier actualización posterior publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2. de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
• Medidas adicionales de minimización de riesgos
1. El TAC acordará las características del sistema de distribución controlada con las autoridades nacionales competentes y debe implementar dicho programa a nivel nacional a fin de garantizar que:
• Antes del lanzamiento, todos los médicos que tengan la intención de prescribir pomalidomida y todos los farmacéuticos que puedan dispensar pomalidomida reciban directamente la “Información para Profesionales Sanitarios” ("Direct Healthcare Professional Communication”), como se describe más adelante.
• Antes de la prescripción (y si procede, de acuerdo con las autoridades nacionales competentes, antes de la dispensación), todos los profesionales sanitarios que tengan la intención de prescribir (y dispensar) pomalidomida reciban un pack de información para el médico que contenga los siguientes elementos:
o El material informativo para el profesional sanitario o Folletos educativos para los pacientes o Tarjetas del paciente
o Ficha Técnica o Resumen de las Características del Producto (RCP), Prospecto y Etiquetado
2. El TAC implementará un Programa de Prevención de Embarazo (PPE) en cada Estado Miembro. Los detalles del PPE deben ser acordados con las Autoridades Nacionales Competentes en cada Estado Miembro, para que el programa se ponga en práctica antes de la comercialización del producto.
3. El TAC debe acordar el texto final de la “Información para Profesionales Sanitarios” y el contenido del pack de información para el médico con las autoridades nacionales competentes en cada Estado Miembro y asegurar que contienen los elementos esenciales descritos más adelante.
4. El TAC debe acordar la implementación del sistema de tarjeta del paciente en cada Estado
Miembro.
5. El TAC debe acordar también con cada Estado Miembro antes del lanzamiento del producto:
• La implementación a nivel nacional de medidas para asegurar la efectividad y el cumplimento del PPE.
Elementos esenciales a incluir
Información para Profesionales Sanitarios
La carta de “Información para Profesionales Sanitarios” constará de dos partes:
• Un texto base acordado por el Comité de Medicamentos de Uso Humano (CHMP).
• Los requisitos específicos nacionales acordados con las autoridades nacionales competentes relacionados con:
o La distribución del producto
o La garantía de que se hayan tomado todas las medidas adecuadas antes de la dispensación de pomalidomida
El material informativo para el profesional sanitario
El material informativo para el profesional sanitario constará de los siguientes elementos:
• Breve resumen de antecedentes de pomalidomida y su indicación aprobada
• Posología
• La necesidad de evitar la exposición fetal debido a la teratogenicidad de pomalidomida en animales y al efecto teratogénico previsto de pomalidomida en humanos
• Las obligaciones del profesional sanitario en relación con la prescripción de pomalidomida
o La necesidad de proporcionar asesoramiento integral a los pacientes
o Los pacientes deben ser capaces de cumplir los requisitos para la utilización segura de pomalidomida
o La necesidad de proporcionar a los pacientes un folleto educativo adecuado para el paciente y una tarjeta del paciente
• Consejos sobre la seguridad relevantes para todos los pacientes
o Descripción y manejo de la neutropenia y la trombocitopenia incluyendo las tasas de incidencia de los ensayos clínicos
o Descripción y manejo del riesgo de tromboembolismo incluyendo las tasas de incidencia de los ensayos clínicos y de la experiencia postcomercialización
o Descripción y manejo de infecciones, mareo y confusión, síndrome de lisis tumoral, reacciones alérgicas, trastornos hepáticos, insuficiencia cardiaca y enfermedad pulmonar intersticial
o
o Eliminación de residuos o excedentes del medicamento
o Disposiciones nacionales específicas para la dispensación de una receta de pomalidomida
o Explicación de que el riesgo de neuropatía con el uso durante un tiempo prolongado es desconocido
• Descripción del PPE y clasificación de los pacientes basada en el sexo y en la capacidad de gestación
o Algoritmo para la implementación del PPE
o Definición de las mujeres con capacidad de gestación y medidas que el médico debe tomar en caso de duda
• Consejos sobre la seguridad para las mujeres con capacidad de gestación
o La necesidad de evitar la exposición fetal
o Descripción del PPE
o La necesidad de una anticoncepción efectiva (incluso si la mujer tiene amenorrea) y definición de anticoncepción efectiva
■ Calendario de las pruebas de embarazo
■ Asesoramiento sobre las pruebas adecuadas
■ Antes de comenzar el tratamiento
■ Durante el tratamiento, dependiendo del método anticonceptivo
■ Después de finalizar el tratamiento
o Necesidad de interrumpir inmediatamente el tratamiento con pomalidomida ante la sospecha de embarazo
o Necesidad de informar inmediatamente al médico a cargo del tratamiento ante la sospecha de embarazo
• Consejos sobre la seguridad para los hombres
o La necesidad de evitar la exposición fetal
o La necesidad de utilizar preservativos si la pareja sexual es una mujer embarazada o con capacidad de gestación (incluso aunque el varón se haya sometido a una vasectomía)
■ Durante el tratamiento con pomalidomida
■ Durante una semana después de la dosis final
o No debe donar semen o esperma durante el tratamiento (períodos de interrupción de la dosis incluidos) ni en el plazo de 7 días después de la discontinuación del tratamiento con pomalidomida
o Si su pareja se queda embarazada mientras él está tomando pomalidomida o poco después de que él haya suspendido la toma de pomalidomida, debe informar inmediatamente al médico a cargo del tratamiento
• Medidas en caso de embarazo
o Instrucciones de interrumpir inmediatamente el tratamiento con pomalidomida ante la sospecha de embarazo
o Necesidad de derivar a la paciente a un médico especialista o con experiencia en teratología y su diagnóstico, para evaluación y asesoramiento.
o Información de contacto local para notificar cualquier sospecha de embarazo.
o Formulario de notificación de embarazo
• Formulario de constancia de información al paciente para comprobar que los pacientes reciben el asesoramiento apropiado sobre el tratamiento, los métodos anticonceptivos y la prevención del embarazo, adecuado a su sexo y capacidad de gestación
• Formularios de notificación de reacciones adversas
Folletos educativos para los pacientes
Los 3 tipos de folletos educativos para los pacientes deben ser:
• Folleto para las pacientes con capacidad de gestación y sus parejas
• Folleto para las pacientes que no tienen capacidad de gestación
• Folleto para los pacientes hombres
Todos los folletos para los pacientes deben contener los siguientes puntos:
• Pomalidomida es teratogénica en los animales y se espera que sea teratogénica en humanos
• Pomalidomida puede causar neutropenia y trombocitopenia, y la necesidad de someterse periódicamente a un análisis de sangre
• Descripción de la tarjeta del paciente, y la necesidad de su uso
• Eliminación de los residuos o excedentes del medicamento sin usar
• Indicaciones sobre la manipulación de pomalidomida para los pacientes, cuidadores y familiares
• Disposiciones nacionales, u otras específicas, aplicables a la dispensación de una receta de pomalidomida
• El paciente no debe dar pomalidomida a ninguna otra persona
• El paciente no debe donar sangre durante el tratamiento (períodos de interrupción de la dosis incluidos) ni en el plazo de 7 días después de la interrupción del tratamiento con pomalidomida.
• El paciente debe informar a su médico acerca de cualquier reacción adversa.
Se debe proporcionar también la siguiente información en el folleto adecuado:
Folleto para las pacientes con capacidad de gestación
• La necesidad de evitar la exposición fetal
• Descripción del PPE
• La necesidad de una anticoncepción efectiva y definición de anticoncepción efectiva
• Calendario de las pruebas de embarazo
o Antes de comenzar el tratamiento
o Durante el tratamiento (períodos de interrupción de la dosis incluidos), cada 4 semanas, excepto en el caso de que la paciente se haya sometido previamente a una ligadura de trompas de eficacia confirmada o Después de finalizar el tratamiento
• La necesidad de interrumpir inmediatamente el tratamiento con pomalidomida ante la sospecha de embarazo
• La necesidad de contactar inmediatamente con su médico ante la sospecha de embarazo
Folleto para los pacientes hombres
• La necesidad de evitar la exposición fetal
• La necesidad de utilizar preservativos si la pareja sexual es una mujer embarazada o con capacidad de gestación (incluso aunque el varón se haya sometido a una vasectomía)
o Durante el tratamiento con pomalidomida (períodos de interrupción de la dosis incluidos)
o En el plazo de 7 días después de la dosis final
• Si su pareja se queda embarazada, el paciente debe informar inmediatamente a su médico a cargo del tratamiento
• No debe donar semen o esperma durante el tratamiento (períodos de interrupción de la dosis incluidos) ni en el plazo de 7 días después de la interrupción del tratamiento con pomalidomida
Tarjeta del paciente
La tarjeta del paciente debe contener los siguientes puntos:
• La verificación de que se ha dado el asesoramiento adecuado
• La documentación acerca del estado de la capacidad de gestación
• Las fechas y los resultados de las pruebas de embarazo
• Obligación de llevar a cabo medidas posautorización
El TAC deberá llevar a cabo, dentro del plazo establecido, las siguientes medidas:
|
Descripción |
Fecha límite |
|
Realizar un registro post-autorización no intervencionista de pacientes tratados con pomalidomida para el mieloma múltiple en recaída y refractario con el objetivo de monitorizar la incidencia de reacciones adversas y monitorizar la implantación y cumplimiento del Programa de Prevención de Embarazo de Celgene así como su uso para indicaciones no autorizadas y sistema de distribución controlado en cada país de acuerdo con las autoridades nacionales competentes relevantes. |
Informe final del estudio clínico: 30 de abril del 2020 |
99
ANEXO III
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO_
Imnovid 1 mg cápsulas duras pomalidomida
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula contiene 1 mg de pomalidomida.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
21 cápsulas duras.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
ADVERTENCIA: Riesgo de graves defectos congénitos. No utilizar durante el embarazo o la lactancia. Debe seguir el Programa de Prevención de Embarazo de Imnovid.
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
El medicamento sin usar debe devolverse al farmacéutico.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd. 1 Longwalk Road Stockley Park Uxbridge UB11 1DB Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/850/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
MEDICAMENTO SUJETO A PRESCRIPCIÓN MÉDICA
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Imnovid 1 mg
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
1. NOMBRE DEL MEDICAMENTO_
Imnovid 1 mg cápsulas duras pomalidomida
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd.
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lote
5. OTROS
1. NOMBRE DEL MEDICAMENTO_
Imnovid 2 mg cápsulas duras pomalidomida
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula contiene 2 mg de pomalidomida.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
21 cápsulas duras.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
ADVERTENCIA: Riesgo de graves defectos congénitos. No utilizar durante el embarazo o la lactancia. Debe seguir el Programa de Prevención de Embarazo de Imnovid.
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
El medicamento sin usar debe devolverse al farmacéutico.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd. 1 Longwalk Road Stockley Park Uxbridge UB11 1DB Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/850/002
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
MEDICAMENTO SUJETO A PRESCRIPCIÓN MÉDICA
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Imnovid 2 mg
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
1. NOMBRE DEL MEDICAMENTO_
Imnovid 2 mg cápsulas duras pomalidomida
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd.
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lote
5. OTROS
1. NOMBRE DEL MEDICAMENTO_
Imnovid 3 mg cápsulas duras pomalidomida
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula contiene 3 mg de pomalidomida.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
21 cápsulas duras.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
ADVERTENCIA: Riesgo de graves defectos congénitos. No utilizar durante el embarazo o la lactancia. Debe seguir el Programa de Prevención de Embarazo de Imnovid.
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
El medicamento sin usar debe devolverse al farmacéutico.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd. 1 Longwalk Road Stockley Park Uxbridge UB11 1DB Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/850/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
MEDICAMENTO SUJETO A PRESCRIPCIÓN MÉDICA
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Imnovid 3 mg
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
1. NOMBRE DEL MEDICAMENTO_
Imnovid 3 mg cápsulas duras pomalidomida
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd.
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lote
5. OTROS
1. NOMBRE DEL MEDICAMENTO_
Imnovid 4 mg cápsulas duras pomalidomida
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula contiene 4 mg de pomalidomida.
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
21 cápsulas duras.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
ADVERTENCIA: Riesgo de graves defectos congénitos. No utilizar durante el embarazo o la lactancia. Debe seguir el Programa de Prevención de Embarazo de Imnovid.
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
El medicamento sin usar debe devolverse al farmacéutico.
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd. 1 Longwalk Road Stockley Park Uxbridge UB11 1DB Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/850/004
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
MEDICAMENTO SUJETO A PRESCRIPCIÓN MÉDICA
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Imnovid 4 mg
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
1. NOMBRE DEL MEDICAMENTO_
Imnovid 4 mg cápsulas duras pomalidomida
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Celgene Europe Ltd.
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lote
5. OTROS
B. PROSPECTO
Imnovid 1 mg cápsulas duras Imnovid 2 mg cápsulas duras Imnovid 3 mg cápsulas duras Imnovid 4 mg cápsulas duras Pomalidomida
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Se espera que Imnovid cause graves defectos congénitos y que pueda ocasionar la muerte del feto. No tome este medicamento si está embarazada o pudiera estarlo. Debe seguir las medidas de anticoncepción descritas en este prospecto.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Imnovid y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Imnovid
3. Cómo tomar Imnovid
4. Posibles efectos adversos
5. Conservación de Imnovid
6. Contenido del envase e información adicional
1. Qué es Imnovid y para qué se utiliza Qué es Imnovid
Imnovid contiene el principio activo “pomalidomida”. Este medicamento está relacionado con la talidomida y pertenece a un grupo de medicamentos que afectan al sistema inmunitario (las defensas naturales del organismo).
Para qué se utiliza Imnovid
Imnovid se utiliza con otro medicamento llamado “dexametasona” (un medicamento antiinflamatorio) para tratar a adultos con un tipo de cáncer llamado “mieloma múltiple”. Se utiliza en personas que han sufrido un empeoramiento de su mieloma, a pesar de haber recibido al menos otros dos tipos de tratamientos, incluyendo los medicamentos lenalidomida y bortezomib.
Qué es el mieloma múltiple
El mieloma múltiple es un tipo de cáncer que afecta a un tipo concreto de glóbulos blancos (denominados “células plasmáticas”). Estas células crecen sin control y se acumulan en la médula ósea, dañando los huesos y los riñones.
El mieloma múltiple generalmente no tiene cura. Sin embargo, el tratamiento puede reducir los signos y los síntomas de la enfermedad o hacerlos desaparecer durante un periodo de tiempo. Cuando esto ocurre, se le denomina “respuesta”.
Cómo actúa Imnovid
La combinación de Imnovid y dexametasona actúa de diversas formas:
• detiene el desarrollo de las células del mieloma;
• estimula el sistema inmunitario para que ataque a las células cancerosas;
• detiene la formación de vasos sanguíneos que alimentan las células cancerosas.
La combinación de Imnovid y dexametasona puede detener la progresión del mieloma múltiple:
• Por lo general, la combinación de Imnovid y dexametasona evitó la reaparición del mieloma múltiple durante un periodo de hasta 16 semanas en comparación a las 8 semanas observadas en los pacientes que tomaban dexametasona únicamente.
2. Qué necesita saber antes de empezar a tomar Imnovid No tome Imnovid:
• si está embarazada, cree que podría estar embarazada o tiene intención de quedarse embarazada, ya que se espera que Imnovid sea perjudicial para el feto. (Los hombres y mujeres que estén tomando este medicamento deben leer la sección “Embarazo, anticoncepción y lactancia -información para mujeres y hombres” que aparece más abajo);
• si puede quedarse embarazada, a menos que esté tomando todas las medidas necesarias para evitar un embarazo (ver “Embarazo, anticoncepción y lactancia - información para mujeres y hombres”). Si puede quedarse embarazada, su médico anotará con cada receta que se han tomado todas las medidas necesarias y le proporcionará esta confirmación;
• si es alérgico a pomalidomida o a alguno de los demás componentes de este medicamento (incluidos en la sección 6). Si cree que podría ser alérgico, consulte a su médico.
Si no está seguro de si alguna de estas situaciones descritas es aplicable a usted, consulte a su médico, farmacéutico o enfermero antes de tomar Imnovid.
Advertencias y precauciones
Consulte a su médico, farmacéutico o enfermero antes de empezar a tomar Imnovid si:
• alguna vez ha tenido coágulos de sangre en el pasado. Durante el tratamiento con Imnovid usted tiene un mayor riesgo de desarrollar coágulos de sangre en sus venas o arterias. Su médico le puede recomendar someterse a tratamientos adicionales (p. ej., warfarina) o reducir su dosis de Imnovid para tener menos probabilidades de desarrollar coágulos sanguíneos;
• alguna vez ha sufrido una reacción alérgica, tales como erupción cutánea, picor, hinchazón, mareos o problemas respiratorios mientras tomaba medicamentos relacionados denominados “talidomida” o “lenalidomida”;
• usted ha sufrido un ataque al corazón, tiene insuficiencia cardiaca, tiene dificultad para respirar o, si es fumador, tiene la presión arterial alta o los niveles de colesterol altos;
• usted tiene una cantidad total de tumor alta en el cuerpo, incluida la médula ósea. Esto podría dar lugar a una enfermedad en la que los tumores se descomponen y producen niveles inusuales de sustancias químicas en la sangre que, a su vez, pueden originar insuficiencia renal. También puede experimentar latidos del corazón irregulares. Esta enfermedad se llama síndrome de lisis tumoral;
• usted sufre o ha sufrido neuropatía (daño neurológico que causa hormigueo o dolor en sus pies o sus manos);
• usted tiene o ha tenido infección por el virus de la hepatitis B. El tratamiento con Imnovid puede volver a activar el virus de la hepatitis B en los pacientes portadores del virus, lo que da lugar a que la infección aparezca de nuevo (recurrencia). Su médico debe comprobar si alguna vez ha tenido una infección por el virus de la hepatitis B.
Es importante señalar que los pacientes con mieloma múltiple tratados con pomalidomida pueden desarrollar otros tipos de cáncer, por lo que su médico debe evaluar cuidadosamente los beneficios y los riesgos al recetarle este medicamento.
Al final del tratamiento, debe devolver al farmacéutico todas las cápsulas sin usar.
Embarazo, anticoncepción y lactancia: información para hombres y mujeres
Debe seguir las siguientes indicaciones recogidas en el Programa de Prevención de Embarazo de pomalidomida. Los hombres y mujeres que estén tomando Imnovid no deben engendrar hijos o quedarse embarazadas. El motivo es que se espera que pomalidomida sea perjudicial para el feto. Usted y su pareja deben usar métodos anticonceptivos eficaces mientras estén tomando este medicamento. Mujeres
No tome Imnovid si está embarazada, cree que podría estar embarazada o tiene intención de quedarse embarazada. El motivo es que se espera que este medicamento sea perjudicial para el feto. Antes de comenzar el tratamiento, debe informar a su médico si existe la posibilidad de que pueda quedarse embarazada, aunque crea que esto sea poco probable.
Si puede quedarse embarazada:
• debe usar métodos anticonceptivos eficaces desde 4 semanas antes de iniciar el tratamiento, durante todo el tiempo que esté tomando el tratamiento y hasta 4 semanas después de finalizarlo. Su médico le aconsejará sobre los métodos anticonceptivos más adecuados;
• cada vez que su médico le prescribe una receta, este se asegurará de que ha entendido las medidas necesarias que deben tomarse para prevenir el embarazo;
• su médico programará pruebas de embarazo antes del tratamiento, cada 4 semanas durante el tratamiento y 4 semanas después de finalizar el tratamiento.
Si, a pesar de las medidas de prevención, se queda embarazada:
• debe suspender el tratamiento inmediatamente e informar a su médico de inmediato. Lactancia:
Se desconoce si Imnovid pasa a la leche materna en humanos. Informe a su médico si está dando o si tiene intención de dar el pecho. Su médico le aconsejará si puede continuar o debe abandonar la lactancia.
Hombres
Imnovid pasa al semen humano.
• Si su pareja está embarazada o puede quedarse embarazada, debe usar preservativos durante todo el tiempo que esté tomando el tratamiento y hasta 7 días después de finalizarlo.
• Si su pareja se queda embarazada mientras usted está tomando Imnovid, informe a su médico inmediatamente. Su pareja también debe informar a su médico inmediatamente.
No debe donar semen o esperma durante el tratamiento y hasta 7 días después de finalizarlo.
Donación de sangre y análisis de sangre
No debe donar sangre durante el tratamiento y hasta 7 días después de haber finalizado el mismo.
Antes de iniciar el tratamiento con Imnovid y durante el mismo, le harán análisis de sangre periódicos. Esto se debe a que su medicamento puede provocar una disminución en el número de células sanguíneas que ayudan a luchar contra las infecciones (glóbulos blancos) y en el número de células que ayudan a parar el sangrado (plaquetas).
Su médico le pedirá que se haga un análisis de sangre:
• antes del tratamiento;
• cada semana durante las 8 primeras semanas de tratamiento;
• por lo menos una vez al mes mientras siga tomando Imnovid.
Su médico puede ajustar la dosis de Imnovid o interrumpir su tratamiento, dependiendo de los resultados de estas pruebas. Su médico también puede ajustar la dosis o interrumpir este medicamento debido a su estado de salud general.
Niños y adolescentes
No está recomendado el uso de Imnovid en niños y adolescentes menores de 18 años.
Otros medicamentos e Imnovid
Informe a su médico, farmacéutico o enfermero si está tomando, ha tomado recientemente o pudiera tener que tomar cualquier otro medicamento. Esto se debe a que Imnovid puede afectar a la forma en que funcionan otros medicamentos. Además, algunos medicamentos pueden afectar a la forma en que funciona Imnovid.
En particular, informe a su médico, farmacéutico o enfermero antes de tomar Imnovid si está tomando alguno de los siguientes medicamentos:
• algunos antifúngicos como ketoconazol
• algunos antibióticos (p. ej. , ciprofloxacino, enoxacino)
• ciertos antidepresivos como fluvoxamina.
Conducción y uso de máquinas
Algunas personas experimentan cansancio, desmayos, confusión o disminución del estado de vigilia mientras toman Imnovid. Si esto le ocurre a usted, no conduzca ni utilice herramientas o maquinaria.
3. Cómo tomar Imnovid
Imnovid se lo debe administrar un médico con experiencia en el tratamiento del mieloma múltiple. Imnovid se toma en combinación con otro medicamento denominado dexametasona. Consulte el prospecto que se adjunta con dexametasona para obtener información adicional sobre su uso y sus efectos.
Siga exactamente las instrucciones de administración de sus medicamentos indicadas por su médico. En caso de duda, pregunte a su médico, farmacéutico o enfermero.
Imnovid y dexametasona se toman en ciclos de tratamiento.
• Cada ciclo dura 28 días (4 semanas).
Cuánto tomar
Imnovid
La dosis recomendada de Imnovid es de 4 mg una vez al día. Para cada ciclo de 4 semanas, debe tomar Imnovid una vez al día durante 3 semanas seguido de una semana de descanso. Esto significa:
• Días del 1 al 21: tome Imnovid una vez al día.
• Días del 22 al 28: no tome Imnovid.
Dexametasona
La dosis inicial normal de dexametasona es de 40 mg al día. Para cada ciclo de 4 semanas, solo debe tomar una dosis de dexametasona el primer día de cada semana. Esto significa:
• Días 1, 8, 15 y 22 de cada ciclo: tome una dosis de dexametasona.
• Días del 2 al 7, del 9 al 14, del 16 al 21 y del 23 al 28: no tome dexametasona.
Pacientes de edad avanzada
Para pacientes mayores de 75 años la dosis inicial normal de dexametasona se reduce a 20 mg al día. Después de finalizar cada ciclo, comience uno nuevo.
Su médico puede tener que reducir la dosis de Imnovid o dexametasona o interrumpirle el tratamiento en función de los resultados de su analítica de sangre y de su estado general, de si está tomando otros medicamentos (p. ej., ciprofloxacino, enoxacino y fluvoxamina) y si experimenta efectos adversos (especialmente erupción cutánea o hinchazón) como consecuencia del tratamiento. Si usted sufre problemas hepáticos o renales su médico realizará un cuidadoso seguimiento de su enfermedad mientras reciba este medicamento.
Cómo y cuándo tomar Imnovid
• No rompa, abra ni mastique las cápsulas. Si los polvos de una cápsula rota de Imnovid entran en contacto con la piel, lave la piel inmediatamente y abundantemente con agua y jabón.
• Trague las cápsulas enteras, preferiblemente con agua.
• Puede tomar las cápsulas con o sin alimentos.
• Debe tomar Imnovid aproximadamente a la misma hora cada día.
Para sacar la cápsula del blíster, presione solo un extremo de la cápsula para que salga a través de la lámina. No presione en el centro de la cápsula ya que podría romperla.

Su médico le aconsejará sobre cómo y cuándo tomar Imnovid si tiene problemas renales y está recibiendo tratamiento con diálisis.
Duración del tratamiento con Imnovid
Debe continuar los ciclos de tratamiento hasta que su médico le comunique que suspenda el tratamiento. Si toma más Imnovid del que debe
Si toma más Imnovid del que debe, informe a su médico o acuda al hospital inmediatamente. Traiga el envase del medicamento con usted.
Si olvidó tomar Imnovid
Si olvidó tomar Imnovid el día que debía, tome la próxima cápsula al día siguiente a la hora habitual.
No tome más cápsulas para compensar la dosis de Imnovid que olvidó el día anterior.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Efectos adversos graves que pueden afectar a más de 1 de cada 10 personas
Si experimenta alguno de los siguientes efectos adversos graves, interrumpa el tratamiento con Imnovid y acuda a un médico inmediatamente, porque podría requerir tratamiento médico de urgencia.
• Fiebre, dolor de garganta, tos o cualquier otro signo de infección (debido a la disminución en el número de glóbulos blancos que se ocupan de luchar frente a la infección).
• Hemorragia o moratones sin causa aparente, incluyendo hemorragias nasales y hemorragia intestinal o estomacal (debido a los efectos sobre las células sanguíneas llamadas “plaquetas”).
• Dolor en el pecho o en las piernas e hinchazón, especialmente en la parte inferior de la pierna o las pantorrillas (producido por coágulos de sangre).
• Dificultad respiratoria (debido a una infección grave en el pecho, inflamación del pulmón, insuficiencia cardíaca o coágulos de sangre).
• Hinchazón de la cara, labios, lengua y garganta, que puede causar dificultad respiratoria (debido a un tipo grave de reacción alérgica denominado angioedema).
Otros efectos adversos graves menos frecuentes
• Recurrencia de la infección por el virus de la hepatitis B, que puede causar amarillamiento de la piel y de los ojos, orina de color marrón oscuro, dolor abdominal en el lado derecho, fiebre, náuseas o malestar. Informe a su médico inmediatamente si observa alguno de estos síntomas.
• Ciertos tipos de cáncer de piel (carcinoma de células escamosas y carcinoma de células basales), que pueden producir cambios en el aspecto de la piel o bultos en la piel. Si observa cambios en el aspecto de la piel mientras toma Imnovid, informe a su médico lo antes posible.
Otros efectos adversos
Muy frecuentes: pueden afectar a más de 1 de cada 10 personas
• Infección de los pulmones.
• Una disminución del número de glóbulos rojos lo que puede producir anemia que da lugar a cansancio y debilidad.
• Pérdida de apetito.
• Dificultad respiratoria (disnea).
• Estreñimiento, diarrea o náuseas.
• Espasmos musculares, dolor de huesos.
• Hinchazón generalizada que incluye hinchazón de brazos y piernas.
Frecuentes: pueden afectar hasta 1 de cada 10 personas
• Sangrado en el interior del cráneo
• Infección de nariz, senos paranasales (sinusitis) y garganta.
• Latido cardiaco rápido e irregular (fibrilación auricular).
• Ataque al corazón (dolor de pecho que se extiende a los brazos, el cuello y la mandíbula, sensación de sudoración y dificultad respiratoria, sensación de náuseas o vómitos).
• Habones (urticaria).
• Una reducción del número de glóbulos rojos y blancos y de las plaquetas al mismo tiempo (pancitopenia). Será más propenso a las hemorragias y a los moratones. Puede sentirse cansado y débil, así como tener dificultades para respirar. Tendrá también una mayor predisposición a coger infecciones.
• Infección de la sangre causada por bacterias.
• Niveles altos de potasio en sangre que pueden producir un ritmo cardíaco anormal.
• Niveles bajos de sodio en sangre que pueden producir cansancio y confusión, contracciones musculares, ataques (convulsiones epilépticas) o coma.
• Niveles altos de ácido úrico en sangre, que pueden producir un tipo de artritis llamado gota.
• Confusión.
• Pérdida de la consciencia.
• Entumecimiento, hormigueo o sensación de escozor en la piel, dolores de manos o pies, mareo, temblor.
• Sensación de que la cabeza le da vueltas, lo que le dificulta estar de pie y moverse con normalidad.
• Vómitos.
• Erupciones cutáneas.
• Picor en la piel.
• Insuficiencia renal.
• Incapacidad para orinar.
• Dolor en la pelvis.
• Resultados anómalos en las pruebas hepáticas.
• Herpes zóster.
Poco frecuentes: pueden afectar hasta 1 de cada 100 personas
• Ictus.
• Inflamación del hígado (hepatitis) que puede producir picor en la piel, coloración amarillenta en la piel y en la parte blanca de los ojos (ictericia), heces de color claro, orina de color oscuro y dolor abdominal.
• La degradación de las células tumorales tiene como resultado la liberación de compuestos tóxicos en el torrente sanguíneo (síndrome de lisis tumoral). Puede derivar en problemas renales.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Imnovid
Mantener este medicamento fuera de la vista y del alcance de los niños.
No requiere condiciones especiales de conservación.
No utilice este medicamento después de la fecha de caducidad que aparece en el blíster y la caja después de “CAD”. La fecha de caducidad es el último día del mes que se indica.
No utilice Imnovid si observa indicios visibles de deterioro o signos de manipulación indebida del medicamento.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de Imnovid
• El principio activo es pomalidomida.
• Los demás componentes son manitol, almidón pregelatinizado y estearil fumarato de sodio.
Imnovid 1 mg cápsula dura:
• Cada cápsula contiene 1 mg de pomalidomida.
• La cubierta de la cápsula contiene: gelatina, dióxido de titanio (E171), indigotina (E132), óxido de hierro amarillo (E172) y tinta blanca y negra.
• La tinta de impresión contiene: goma laca shellac, dióxido de titanio (E171), simeticona, propilenglicol (E1520) e hidróxido de amonio (E527) (tinta blanca) y goma laca shellac, óxido de hierro negro (E172), propilenglicol (E1520) e hidróxido de amonio (E527) (tinta negra).
Imnovid 2 mg cápsula dura:
• cada cápsula contiene 2 mg de pomalidomida.
• La cubierta de la cápsula contiene: gelatina, dióxido de titanio (E171), indigotina (E132), óxido de hierro amarillo (E172), eritrosina (E127) y tinta blanca.
• La tinta de impresión contiene: tinta blanca - goma laca shellac, dióxido de titanio (E171), simeticona, propilenglicol (E1520) e hidróxido de amonio (E527).
Imnovid 3 mg cápsula dura:
• Cada cápsula contiene 3 mg de pomalidomida.
• La cubierta de la cápsula contiene: gelatina, dióxido de titanio (E171), indigotina (E132), óxido de hierro amarillo (E172) y tinta blanca.
• La tinta de impresión contiene: tinta blanca - goma laca shellac, dióxido de titanio (E171), simeticona, propilenglicol (E1520) e hidróxido de amonio (E527).
Imnovid 4 mg cápsula dura:
• Cada cápsula contiene 4 mg de pomalidomida.
• La cubierta de la cápsula contiene: gelatina, dióxido de titanio (E171), indigotina (E132), azul brillante FCF (E133)y tinta blanca.
• La tinta de impresión contiene: tinta blanca - goma laca shellac, dióxido de titanio (E171), simeticona, propilenglicol (E1520) e hidróxido de amonio (E527).
Aspecto de Imnovid y contenido del envase
cuerpo opaco de color amarillo con la cuerpo opaco de color naranja con la cuerpo opaco de color verde con la cuerpo opaco de color azul con la
Imnovid 1 mg cápsula dura: tapa opaca de color azul oscuro y inscripción “POML 1 mg”.
Imnovid 2 mg cápsula dura: tapa opaca de color azul oscuro y inscripción “POML 2 mg”.
Imnovid 3 mg cápsula dura: tapa opaca de color azul oscuro y inscripción “POML 3 mg”.
Imnovid 4 mg cápsula dura: tapa opaca de color azul oscuro y inscripción “POML 4 mg”.
Cada envase contiene 21 cápsulas.
Titular de la autorización de comercialización y responsable de la fabricación
Celgene Europe Ltd.
1 Longwalk Road Stockley Park Uxbridge UB11 1DB Reino Unido
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu. También existen enlaces a otras páginas web sobre enfermedades raras y medicamentos huérfanos.
122