Ilaris 150 Mg Polvo Para Solucion Inyectable
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Ilaris 150 mg polvo para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un vial contiene 150 mg de canakinumab*.
Después de la reconstitución, cada ml de la solución contiene 150 mg de canakinumab.
* anticuerpo monoclonal completamente humano obtenido mediante la tecnología del ADN recombinante en células de hibridoma Sp2/0 de ratón
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo para solución inyectable.
El polvo es blanco.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Síndromes Periódicos Asociados a la Criopirina
Ilaris está indicado para el tratamiento de los Síndromes Periódicos Asociados a la Criopirina (CAPS) en adultos, adolescentes y niños a partir de 2 años con un peso corporal de 7,5 kg o superior, incluidos:
- Síndrome de Muckle-Wells (MWS),
- Enfermedad Neonatal Multisistémica Inflamatoria (NOMID) / Síndrome Infantil Neurológico Cutáneo y Articular Crónico (CINCA),
- Manifestaciones graves del Síndrome Autoinflamatorio Familiar inducido por el frío (FCAS) / Urticaria Familiar Fría (FCU) que presente signos y síntomas más allá de la erupción de tipo urticaria inducido por el frío.
Enfermedad de Still
Ilaris está indicado para el tratamiento de la enfermedad de Still activa incluyendo la Enfermedad de Still del Adulto (ESA) y la artritis idiopática juvenil sistémica (AIJS) en pacientes de 2 años de edad o mayores que no hayan respondido adecuadamente al tratamiento previo con antiinflamatorios no esteroideos (AINEs) y corticosteroides sistémicos. Ilaris puede ser administrado en monoterapia o en combinación con metotrexato.
Gota artrítica
Ilaris está indicado para el tratamiento sintomático de pacientes adultos con ataques frecuentes de gota artrítica (al menos 3 ataques en los 12 meses previos) en los cuales está contraindicado el tratamiento con medicamentos antiinflamatorios no esteroideos (AINEs) y colchicina, no está tolerado, o no responden adecuadamente, y en los cuales no son adecuadas las series repetidas de corticoides (ver sección 5.1).
4.2 Posología y forma de administración
CAPS y AIJS
Para CAPS y enfermedad de Still, el tratamiento debe ser iniciado y supervisado por un médico especialista con experiencia en el diagnóstico y tratamiento de la indicación.
Para gota artrítica, el médico debe tener experiencia en el uso de medicamentos biológicos e Ilaris debe ser administrado por un profesional sanitario.
Después de recibir una formación adecuada con respecto a la técnica correcta de inyección, el paciente o sus cuidadores pueden inyectar Ilaris si el médico lo considera apropiado y con seguimiento médico si fuera preciso (ver sección 6.6.).
Posología
CAPS: Adultos, adolescentes y niños a partir de 2 años de edad La dosis inicial recomendada de Ilaris para pacientes con CAPS es:
Adultos, adolescentes y niños > de 4 años de edad:
- 150 mg para pacientes cuyo peso corporal sea > 40 kg
- 2 mg/kg para pacientes cuyo peso corporal sea > 15 kg y < 40 kg
- 4 mg/kg para pacientes cuyo peso corporal sea > 7,5 kg y < 15 kg Niños desde 2 a < 4 años de edad:
- 4 mg/kg para pacientes cuyo peso corporal sea > 7,5 kg
Se administra cada ocho semanas como una dosis única mediante una inyección subcutánea.
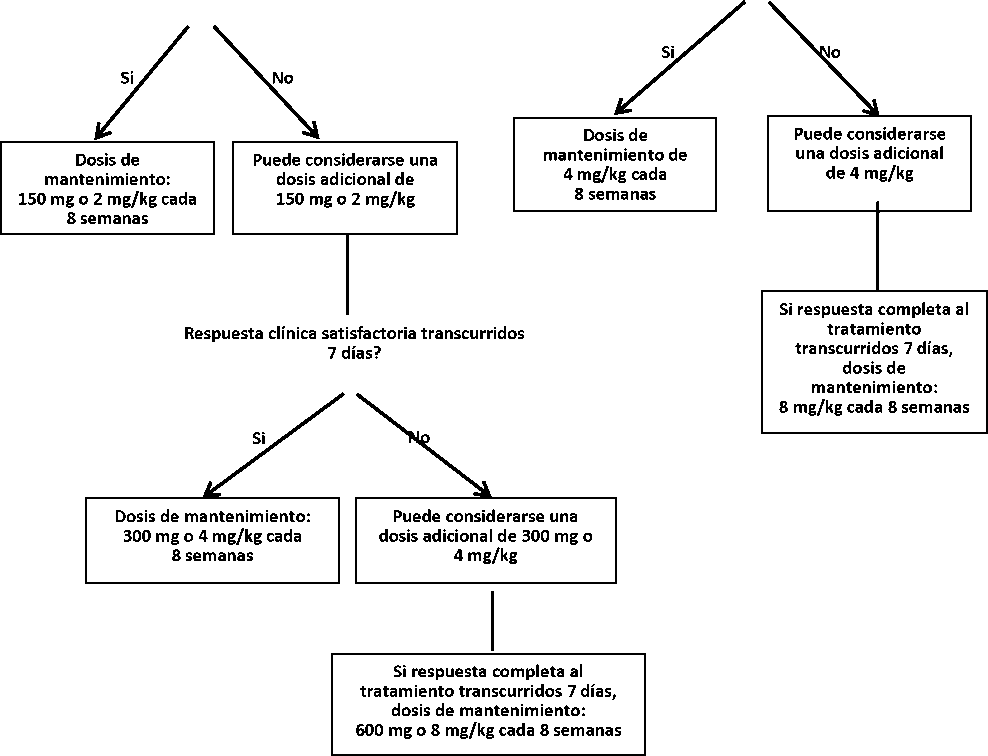
En pacientes con una dosis inicial de 150 mg o 2 mg/kg,si no se consigue una respuesta clínica satisfactoria (resolución de la erupción y otros síntomas inflamatorios generalizados) una vez transcurridos 7 días desde el inicio del tratamiento, puede considerarse una segunda dosis de Ilaris de 150 mg ó 2 mg/kg. Si, en lo sucesivo, se consigue una respuesta completa al tratamiento, se debe mantener el régimen con dosis elevadas de 300 mg o 4 mg/kg cada 8 semanas. Si no se consigue una respuesta clínica satisfactoria una vez transcurridos 7 días de esta dosis aumentada, puede considerarse una tercera dosis de Ilaris a 300 mg o 4 mg/kg. Si, en lo sucesivo, se obtiene una respuesta completa al tratamiento, se debe considerar el régimen con dosis elevadas de 600 mg o 8 mg/kg cada 8 semanas, en base a la valoración clínica individual.
Para pacientes con una dosis inicial de 4 mg/kg, si no se consigue una respuesta clínica satisfactoria una vez transcurridos 7 días del inicio del tratamiento, puede considerarse una segunda dosis de Ilaris 4 mg/kg. Si, en lo sucesivo, se obtiene una respuesta clínica completa, se debe considerar el mantenimiento del régimen con dosis elevadas de 8 mg/kg cada 8 semanas, en base a la valoración clínica individual.
La experiencia clínica con intervalos de dosis inferiores a 4 semanas o con dosis superiores a 600 mg o 8 mg/kg es limitada.
150 mg o 2 mg/kg
4 mg/kg
Adultos y niños >4 años de edad >15 kg
Niños de 2-< 4 años de edad o niños >4 años de edad >7.5 kg y < 15 kg
Respuesta clínica satisfactoria transcurridos 7 días?
Respuesta clínica satisfactoria transcurridos 7 días?
Enfermedad de Still (ESA y AIJS)
La dosis recomendada de Ilaris para pacientes con enfermedad de Still (ESA y AIJS) con peso corporal > 7,5 kg es de 4 mg/kg (hasta un máximo de 300 mg), administrado por inyección subcutanea cada cuatro semanas. En pacientes sin mejoría clínica el médico que lo trate considerará si debe continuar con el tratamiento de Ilaris.
Gota artrítica
Se debe iniciar u optimizar el manejo de la hiperuricemia con tratamientos adecuados para disminuir el urato (ULT, de sus singlas en inglés). Ilaris se debe utilizar como un tratamiento a demanda para tratar los ataques de gota artrítica.
La dosis recomendada de Ilaris para pacientes adultos con gota artrítica es 150 mg administrados subcutáneamente como una única dosis durante un ataque. Para un efecto máximo, se debe administrar Ilaris tan pronto como sea posible después del inicio de un ataque de gota artrítica.
Los pacientes que no responden al tratamiento inicial no deben volver a ser tratados con Ilaris. En pacientes respondedores y que requieren un retratamiento, debe haber un intervalo de al menos 12 semanas antes de que se administre una nueva dosis de Ilaris (ver sección 5.2).
Poblaciones especiales Población pediátrica CAPS
No se ha establecido la seguridad y eficacia de Ilaris en pacientes menores de 2 años de edad con CAPS. Los datos actualmente disponibles están descritos en las secciones 4.8, 5.1 y 5.2, sin embargo no se puede hacer una recomendación posológica.
AIJS
No se han establecido la seguridad y la eficacia de Ilaris en pacientes menores de 2 años de edad con AIJS.
Gota artrítica
No existe una recomendación de uso específica para Ilaris en la población pediátrica para la indicación de gota artrítica.
Pacientes de edad avanzada No se requiere ajuste de dosis.
Insuficiencia hepática
Ilaris no ha sido estudiado en pacientes con insuficiencia hepática.
Insuficiencia renal
No se requiere un ajuste de la dosis en pacientes con insuficiencia renal. No obstante, la experiencia clínica en estos pacientes es limitada.
Forma de administración
Ilaris se debe administrar por inyección subcutánea. Para consultar las instrucciones de uso y manejo de la solución reconstituida ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Infecciones graves activas (ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
Infecciones
Ilaris está asociado con un aumento en la incidencia de infecciones graves. Por consiguiente, debe controlarse estrechamente la aparición de signos y síntomas de infección en los pacientes durante y después del tratamiento con Ilaris. Los médicos deben tener precaución al administrar Ilaris en pacientes con infecciones, con antecedentes de infecciones recurrentes, o con condiciones subyacentes que puedan predisponerles a sufrir infecciones.
Tratamiento de CAPS y Enfermedad de Still (ESA y AIJS)
Ilaris no se debe iniciar o continuar en pacientes con infecciones graves que requieran intervención médica.
Tratamiento de gota artrítica
Ilaris no se debe administrar durante una infección activa.
No se recomienda el uso concomitante de Ilaris con inhibidores del factor de necrosis tumoral (TNF) ya que podría aumentar el riesgo de infecciones graves (ver sección 4.5).
Durante el tratamiento con Ilaris se han notificado casos aislados de infecciones oportunistas o poco habituales (como aspergilosis, infecciones micobacterianas atípicas, herpes zoster). No se puede excluir una relación causal de Ilaris con estos acontecimientos.
En aproximadamente el 12% de los pacientes con CAPS sometidos a una prueba cutánea PPD (derivado proteínico purificado) en los ensayos clínicos, la prueba de seguimiento durante el tratamiento con Ilaris dio un resultados positivo sin que existiera evidencia clínica de una infección tuberculosa latente o activa.
Se desconoce si el uso de inhibidores de interleucina-1 (IL-1) tales como Ilaris incrementa el riesgo de reactivación de tuberculosis. Antes de iniciar el tratamiento, debe evaluarse la existencia de tuberculosis activa y latente en todos los pacientes. Especialmente en pacientes adultos, esta evaluación debe incluir una historia médica detallada. Se deben realizar las pruebas diagnósticas adecuadas (p. ej. prueba cutánea de la tuberculina, la prueba de interferón gamma o radiografía de tórax) a todos los pacientes (deben aplicar las recomendaciones locales). Deben controlarse los síntomas y signos de tuberculosis en los pacientes durante y después del tratamiento con Ilaris. Todos los pacientes deben ser instruidos para pedir consejo médico si aparecen signos o síntomas de tuberculosis (p. ej. tos persistente, pérdida de peso, temperatura subfebril) durante el tratamiento con Ilaris. En el caso de la conversión de un resultado de negativo a positivo en la prueba cutánea PPD, en pacientes con un riesgo alto deben considerarse otras medidas alternativas para la determinación de la infección por tuberculosis.
Neutropenia y leucopenia
Se ha observado neutropenia (recuento absoluto de neutrófilos [RAN] < 1,5 x 109/l) y leucopenia con medicamentos que inhiben la IL-1, incluyendo Ilaris. No debe iniciarse el tratamiento con Ilaris en pacientes con neutropenia o leucopenia. Se recomienda controlar el recuento de glóbulos blancos (RGB) incluyendo el recuento de neutrófilos antes de iniciar el tratamiento y de nuevo, después de 1 o 2 meses. Para el tratamiento crónico o repetido, también se recomienda controlar el recuento RGB periódicamente durante el tratamiento. Si un paciente sufre neutropenia o leucopenia, debe controlarse estrechamente el RGB y se debe considerar la interrupción del tratamiento.
Neoplasias
Se han reportado neoplasias en pacientes tratados con Ilaris. Se desconoce el riesgo de desarrollo de neoplasias con el tratamiento con antiinterleukinas (IL)-1.
Reacciones de hipersensibilidad
Se han notificado reacciones de hipersensibilidad con Ilaris. La mayoría de los casos fueron de carácter leve. Durante el desarrollo clínico de Ilaris en más de 2.300 pacientes, no se describieron reacciones anafilácticas o anafilactoides. Sin embargo, no debe excluirse el riesgo de reacciones de hipersensibilidad graves, que no resulta infrecuente con proteínas inyectables (ver sección 4.3).
Función hepática
En los estudios clínicos se han notificado casos de elevaciones transitorias y asintomáticas de los niveles séricos de transaminasas y bilirrubina (ver sección 4.8).
Vacunas
No se dispone de datos sobre el riesgo de transmisión de la infección secundaria a la administración de vacunas vivas (atenuadas) en pacientes tratados con Ilaris. Por lo tanto, no deben administrarse vacunas vivas de forma concomitante con Ilaris a menos que los beneficios superen claramente los riesgos (ver sección 4.5).
Antes de iniciar el tratamiento con Ilaris se recomienda que los pacientes adultos y pediátricos reciban todas las vacunas, según estén indicadas, incluyendo la vacuna pneumocócica y la vacuna de la gripe inactivada (ver sección 4.5).
Mutación en el gen NLRP3 en pacientes con CAPS
La experiencia clínica en pacientes con CAPS sin mutación confirmada en el gen NLRP3 es limitada.
El síndrome de activación macrofágica en pacientes con Enfermedad de Still El síndrome de activación macrofágica (SAM) es un conocido trastorno potencialmente mortal que puede desarrollarse en pacientes con enfermedades reumáticas, especialmente enfermedad de Still. Si se produce SAM, o si se sospecha, debe evaluarse y tratarse lo antes posible. Los médicos deben estar atentos a los síntomas de la infección o empeoramiento de la enfermedad de Still, ya que éstos se sabe que desencadenan SAM. Basándose en la experiencia de ensayos clínicos, Ilaris no parece aumentar la incidencia de SAM en pacientes con AIJS, pero no se puede llegar a ninguna conclusión definitiva.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han investigado las interacciones entre Ilaris y otros medicamentos con estudios formales.
Se ha descrito una incidencia aumentada de infecciones graves con otro bloqueante de la IL-1 en combinación con inhibidores del TNF. No se recomienda el uso de Ilaris con inhibidores del TNF ya que esto puede aumentar el riesgo de infecciones graves.
La expresión de las enzimas hepáticas CYP450 puede ser suprimida por las citocinas que estimulan la inflamación crónica, tales como interleucina-1 beta (IL-1 beta). De este modo, la expresión de CYP450 puede revertirse cuando se introduce un tratamiento con un inhibidor potente de la citocina, como canakinumab. Esto es clínicamente relevante para sustratos del CYP450 de estrecho margen terapéutico para los que la dosis se ajusta individualmente. Al iniciar el tratamiento con canakinumab en pacientes que reciben este tipo de medicamentos, es preciso monitorizar el efecto o la concentración del principio activo y ajustar la dosis individual si fuese necesario.
No se dispone de datos sobre los efectos de la administración de vacunas vivas o la transmisión de la infección secundaria a la administración de una vacuna viva en pacientes que reciben Ilaris. Por ello, no deben administrarse vacunas vivas concomitantemente con Ilaris a menos que los beneficios superen claramente los riesgos. Si está indicada la administración de vacunas vivas después del inicio del tratamiento con Ilaris, la recomendación es esperar durante al menos 3 meses después de la última inyección de Ilaris y antes de la próxima dosis (ver sección 4.4).
Los resultados de un estudio en voluntarios adultos sanos demostraron que una única dosis de Ilaris de 300 mg no afectó a la inducción ni a la persistencia de las respuestas de anticuerpos tras la vacunación con la vacuna de la gripe o vacuna meningocócica a base de proteínas glicosiladas.
Los resultados de un ensayo abierto de 56 semanas en pacientes con CAPS de hasta 4 años de edad, demostraron que todos los pacientes que recibieron vacunas no vivas según la práctica clínica habitual para los niños, desarrollaron niveles protectores de anticuerpos.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil / Contracepción en hombres y mujeres
Las mujeres deben utilizar un método anticonceptivo efectivo durante el tratamiento con Ilaris y hasta 3 meses después de la última dosis.
Embarazo
Se dispone de datos limitados sobre la utilización de canakinumab en mujeres embarazadas. Los estudios en animales no muestran efectos dañinos directos o indirectos por lo que respecta a la toxicidad reproductiva (ver sección 5.3). Se desconoce el riesgo para el feto/madre. Las mujeres embarazadas o que desean quedar embarazadas solo deberían tratarse después de un exhaustivo análisis de beneficio/riesgo.
Los estudios en animales indican que canakinumab atraviesa la placenta y se detecta en el feto. No se dispone de datos en humanos, sin embargo como canakinumab es una inmunoglobulina de la clase G (IgG1), es de esperar que se produzca transferencia transplacentaria humana. Se desconoce el impacto clínico. Sin embargo, no se recomienda la administración de vacunas vivas a recién nacidos expuestos a canakinumab in utero durante las 16 semanas siguientes a la última dosis de Ilaris de la madre antes del parto. Las mujeres que reciben canakinumab durante el embarazo deben ser instruidas para que informen al pediatra antes de que administren alguna vacuna a su bebé recién nacido.
Lactancia
Se desconoce si canakinumab se excreta en la leche materna. La decisión de dar el pecho durante el tratamiento con Ilaris sólo debe tomarse después de un análisis exhaustivo del beneficio/riesgo.
Los estudios en animales han demostrado que un anticuerpo murino que actúa sobre la IL-1 beta murina no tuvo efectos adversos sobre el desarrollo en crías de ratón y a los que el anticuerpo les fue transferido (ver sección 5.3).
Fertilidad
No se han realizados estudios formales en relación con el efecto potencial de Ilaris sobre la fertilidad en humanos.
Canakinumab no mostró efecto sobre la fertilidad en el macho en monos macho babuinos (C.jacchus). Un anticuerpo murino anti murino IL-1 beta no mostró efectos sobre la fertilidad en ratones macho y hembra (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Ilaris sobre la capacidad para conducir y utilizar máquinas es pequeña. El tratamiento con Ilaris puede producir mareo/vértigo o astenia (ver sección 4.8). Los pacientes que experimenten estos síntomas durante el tratamiento con Ilaris deben esperar a que el síntoma remita antes de conducir o manejar maquinaria.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Más de 2.400 sujetos incluyendo aproximadamente 380 niños (de 2 a 17 años de edad) han recibido tratamiento con Ilaris en estudios intervencionales en pacientes con CAPS, AIJS, gota artrítica o con otras patologías mediadas por IL-1 beta y voluntarios sanos. Las reacciones adversas más frecuentes fueron infecciones predominantemente en el tracto respiratorio. Se han observado infecciones graves. La mayoría de estos acontecimientos fueron de leves a moderados. Con el tratamiento a más largo plazo no se vio impacto en el tipo o la frecuencia de las reacciones adversas.
Se han notificado reacciones de hipersensibilidad en pacientes tratados con Ilaris (ver secciones 4.3 y 4.4).
Se han notificado infecciones oportunistas en pacientes tratados con Ilaris (ver sección 4.4).
CAPS
En los ensayos clínicos un total de 211 pacientes de CAPS adultos y pediátricos (incluidos FCAS/FCU, MWS, y NOMID/CINCA) han recibido tratamiento con Ilaris. La seguridad de Ilaris comparada con placebo se investigó en un ensayo pivotal fase III que consistió en un periodo abierto de 8 semanas (Parte I), un periodo aleatorizado, doble ciego, controlado con placebo y opción de retirada de 24 semanas (Parte II), y un periodo abierto de 16 semanas con Ilaris (Parte III). Todos los pacientes recibieron 150 mg de Ilaris por vía subcutánea ó 2 mg/kg si el peso corporal estaba comprendido entre > 15 kg y < 40 kg.
Enfermedad de Still
Un total de 324 pacientes con AIJS de 2 a < 20 años de edad recibieron Ilaris en ensayos clínicos, incluyendo 293 pacientes de 2 a < 16 años de edad, 21 pacientes de 16 a < 18 años de edad y 10 pacientes de 18 a < 20 años de edad. Se estudió la seguridad de Ilaris en comparación con el placebo en dos estudios pivotales fase III (ver sección 5.1).
Gota artrítica
Más de 700 pacientes con gota artrítica han sido tratados con Ilaris a dosis que van de 10 mg a 300 mg en ensayos clínicos con control activo, doble-ciego, aleatorizado de 24 semanas de duración. Más de 250 pacientes han sido tratados con una dosis recomendada de 150 mg en ensayos clínicos de Fase II y III (ver sección 5.1).
Tabla de reacciones adversas
Las reacciones adversas se presentan según el sistema de clasificación por órganos y sistemas MedDRA. Dentro de cada órgano y sistema, las reacciones adversas se ordenan por frecuencia, las más comunes primero. Las categorías de frecuencia se definen utilizando los siguientes criterios: muy frecuentes (> 1/10); frecuentes (> 1/100 a < 1/10); poco frecuentes (> 1/1.000 a < 1/100); raras (> 1/10.000 a < 1/1.000); muy raras (< 1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
Tabla 1 Tabla de reacciones adversas en CAPS, AIJS y gota artrítica
|
Clasificación de órganos del sistema MedDRA |
CAPS |
AIJS |
Gota artrítica |
|
Infecciones e infestaciones | |||
|
Muy frecuentes |
Nasofaringitis |
Neumonía Gastroenteritis Infección del tracto urinario Infección vírica Sinusitis Rinitis Faringitis Tonsilitis Nasofaringitis Infección del tracto respiratorio superior |
Neumonía Bronquitis Gastroenteritis Infección del tracto urinario Síntomas gripales Celulitis Sinusitis Infección de oído Faringitis Nasofaringitis Infección del tracto respiratorio superior |
|
Frecuentes |
Infección del tracto urinario Infección del tracto respiratorio superior Infección vírica | ||
|
Trastornos del sistema nervioso | |||
|
Frecuentes |
Mareo/vértigo |
Mareo/vértigo | |
|
Trastornos gastrointestinales | |||
|
Muy frecuentes |
Dolor abdominal (superior) | ||
|
Poco frecuentes |
Enfermedad por reflujo gastroesofágico | ||
|
Trastornos de la |
piel y del tejido subcutáneo | ||
|
Muy frecuentes |
Reacción en el lugar de inyección |
Reacción en el lugar de inyección | |
|
Frecuentes |
Reacción en el lugar de inyección | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |||
|
Muy frecuentes |
Artralgia | ||
|
Frecuentes |
Dolor muscular |
Dolor de espalda | |
|
Trastornos generales y alteraciones en el lugar de administración | |||
|
Frecuentes |
Fatiga/astenia | ||
|
Exploraciones complementarias | |||
|
Muy frecuentes |
Disminución del aclaramiento renal* Proteinuria # Leucopenia | ||
|
Frecuentes |
Neutropenia | ||
|
* en base al aclaramiento de creatinina estimado, la mayoría fueron transitorios # la mayoría fueron trazas transitorias hasta 1 + proteína urinaria positiva por varilla | |||
En un subgrupo de pacientes con AIJS adultos jóvenes de 16 a 20 años de edad (n=31), el perfil de seguridad de Ilaris fue consistente con el observado en pacientes con AIJS menores de 16 años de edad. Se espera que el perfil de seguridad en pacientes con ESA sea similar al de pacientes con AIJS de acuerdo con los informes de la literatura.
Descripción de reacciones adversas seleccionadas
Datos a largo plazo y anomalías analíticas en pacientes con CAPS
Durante los ensayos clínicos con Ilaris en pacientes con CAPS aumentaron los valores medios de hemoglobina y los de leucocitos, neutrófilos y plaquetas disminuyeron.
Raramente, se han observado elevaciones de las transaminasas en pacientes de CAPS.
En pacientes con CAPS tratados con Ilaris se han observado elevaciones moderadas y asintomáticas de la bilirrubina sérica sin incremento concomitante de transaminasas.
En los estudios abiertos, a largo plazo, con escalado de dosis, en el grupo de dosis de 600 mg o 8 mg/kg se notificaron más frecuentemente casos de infecciones (gastroenteritis, infección del tracto respiratorio, infección del tracto respiratorio superior), vómitos y mareo, que en otros grupos de dosis.
Anomalías analíticas en pacientes con AIJS Hematología
En el programa global de AIJS, se notificó una disminución transitoria del recuento de glóbulos blancos (RGB) < 0,8 x LLN en 33 pacientes (16,5%).
En el programa global de AIJS, se notificó una disminución transitoria en el recuento absoluto de neutrófilos (RAN) de menos de 1 x 109/l en 12 pacientes (6,0%).
En el programa global de AIJS, se observaron disminuciones transitorias en el recuento de plaquetas (<LIN) en 19 pacientes (9,5%).
ALT / AST
En el programa global de AIJS, se notificaron una ALT alta y/o una AST > 3 x por encima del límite normal (LSN) en 19 pacientes (9,5%).
Anomalías analíticas en _pacientes con gota artrítica Hematología
Se notificó disminución del recuento de glóbulos blancos (RGB)< 0,8 x límite inferior de la normalidad (LIN) en 6,7% de los pacientes tratados con Ilaris comparado con el 1,4% de los pacientes tratados con acetónido de triamcinolona. La disminución del recuento absoluto de neutrófilos (RAN) a menos de 1 x 109/l fue reportado en el 2% de los pacientes en los ensayos comparativos. Se observaron casos aislados de recuento de RAN < 0,5 x 109/l (ver sección 4.4).
En los ensayos clínicos con control activo se observó una disminución leve (< LIN y > 75 x 109/l) y transitoria en el recuento de plaquetas de una incidencia mayor (12,7%) con Ilaris versus el comparador (7,7%) en pacientes con gota artrítica.
Acido úrico
En los ensayos clínicos comparativos en gota artrítica se observó un incremento transitorio de ácido úrico (0,7 mg/dl a las 12 semanas y 0,5 mg/dl a las 24 semanas) después del tratamiento con Ilaris. En otro ensayo, entre pacientes que estaban iniciando tratamiento con ULT, se observó incremento de ácido úrico. El incremento de ácido úrico no se observó en los ensayos clínicos en la población que no tenía gota (ver sección 5.1).
ALT/AST
Se observó un incremento medio y mediano en transaminasa (ALT) de 3,0 U/l y 2,0 U/l, respectivamente, y de aspartato transaminasa (AST) de 2,7 U/l y 2,0 U/l, respectivamente, desde el inicial a la finalización del ensayo en el grupo tratado con Ilaris versus el grupo(s) tratado con acetónido de triamcinolona, sin embargo la incidencia de cambios clínicamente significativos (> 3 x el límite superior normal (LSN)) fue mayor en pacientes tratados con acetónido de triamcinolona (2,5% para ambos AST y ALT) comparado con los pacientes tratados con Ilaris (1,6% para ALT y 0,8% para AST).
Triglicéridos
En los ensayos clínicos de gota artrítica con controlador activo, hubo un incremento medio en triglicéridos de 33,5 mg/dl en los pacientes tratados con Ilaris comparado con una modesta disminución de -3,1 mg/dl con acetónido de triamcinolona. La incidencia de pacientes con una elevación de triglicéridos > 5 x límite superior de la normalidad (LSN) fue 2,4% con Ilaris y 0,7% con acetónido de triamcinolona. Se desconoce el significado clínico de este hallazgo.
Población pediátrica
En los estudios se incluyeron 80 pacientes pediátricos con CAPS (2-17 años de edad). En general, no hubo diferencias clínicas significativas en el perfil de seguridad y tolerancia de Ilaris en los pacientes pediátricos comparados con la población general de CAPS (compuesta por pacientes adultos y pediátricos, N=211), incluyendo la frecuencia total y la gravedad de los episodios de infección. Las infecciones del tracto respiratorio superior fueron las infecciones notificadas con mayor frecuencia.
Adicionalmente, se evaluaron 6 pacientes pediátricos menores de 2 años de edad en un ensayo clínico abierto de pequeño tamaño. El perfil de seguridad de Ilaris fue similar al observado en pacientes a partir de 2 años de edad.
Pacientes de edad avanzada
No se ha observado diferencias significativas en el perfil de seguridad en pacientes > 65 años de edad. Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Los casos notificados por sobredosis son limitados. En los primeros ensayos clínicos, pacientes y voluntarios sanos recibieron dosis tan altas como 10 mg/kg, administradas por vía intravenosa o subcutánea, sin evidencia de toxicidad aguda.
En caso de sobredosis, se recomienda controlar la aparición de signos o síntomas de reacciones adversas en el paciente y e instaurar, en caso necesario, un adecuado tratamiento sintomático inmediatamente.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inmunosupresores, inhibidores de interleucina, código ATC: L04AC08 Mecanismo de acción
Canakinumab es un anticuerpo monoclonal anti-interleucina humana-1 beta (IL-1 beta) del isotipo IgG1/K completamente humano. Canakinumab se une con alta afinidad específicamente a la IL-1 beta humana y neutraliza su actividad biológica mediante el bloqueo de la interacción con los receptores IL-1, lo que permite prevenir la activación del gen inducida por IL-1 beta y la producción de mediadores inflamatorios.
Efectos farmacodinámicos
CAPS
En estudios clínicos, los pacientes de CAPS que presentan una sobreproducción incontrolada de IL-1 beta muestran una respuesta rápida al tratamiento con canakinumab, es decir, los parámetros de laboratorio tales como niveles altos de la proteína C-reactiva (PCR) y de la proteína A amiloidea (AAS), el recuento elevado de neutrófilos y plaquetas y la leucocitosis se normalizaron rápidamente.
Enfermedad de Still (ESA y AIJS)
La Enfermedad de Still del Adulto y la Artritis Idiopática Juvenil Sistémica son enfermedades graves autoinflamatorias producidas por la inmunidad innata a través de las citoquinas pro-inflamatorias, siendo la principal, IL-1-beta.
Las características comunes de ESA y AIJS incluyen fiebre, erupción cutánea, hepatoesplenomegalia, linfadenopatía, poliserositis y artritis. El tratamiento con canakinumab mejoró rápidamente y de forma sostenida las características articulares y sistémicas de AIJS con una reducción significativa del número de articulaciones inflamadas, rápida resolución de la fiebre y la reducción de reactantes de la fase aguda en la mayoría de los pacientes (ver Eficacia clínica y de seguridad).
Gota artrítica
Un ataque de gota artrítica es producido por cristales de urato (urato monosódico monohidratado) en las articulaciones y en los tejidos de alrededor de éstas, que desencadenan la producción de IL-1 beta por parte de los macrófagos residentes, mediante el complejo «NALP3 inflamasoma». La activación de macrófagos y la sobreproducción de IL-1 beta concomitante producen una respuesta inflamatoria dolorosa aguda. Otros activadores del sistema inmune innato, tales como agonistas endógenos de los receptores de tipo «toll-like» pueden contribuir a la activación transcripcional del gen IL-1 beta, iniciando un ataque de gota artrítica. Después del tratamiento con canakinumab, los marcadores inflamatorios CRP o SAA y los signos de inflamación aguda (p. ej. dolor, hinchazón, enrojecimiento) en la articulación afectada remiten rápidamente.
Eficacia clínica y seguridad
CAPS
La eficacia y la seguridad de Ilaris han sido demostradas en pacientes con diversos grados de gravedad de la patología y distintos fenotipos de CAPS (incluidos FCAS/FCU, MWS, y NOMID/CINCA). En el estudio pivotal sólo se incluyeron pacientes con mutación NLRP3 confirmada.
En el estudio de fase I/II, el tratamiento con Ilaris mostró un inicio de acción rápido ya sea con la remisión o con una mejoría clínicamente significativa de los síntomas en un día tras la administración. Los parámetros de laboratorio tales como los niveles altos de PCR y AAS, el recuento elevado de neutrófilos y plaquetas, se normalizaron rápidamente al cabo de unos días de la inyección de Ilaris.
El estudio pivotal consistió en un estudio multicéntrico de 48 semanas y tres partes: un periodo abierto de 8 semanas (Parte I), un periodo aleatorizado, doble ciego, controlado con placebo, de 24 semanas y opción de retirada (Parte II), seguido de un periodo abierto de 16 semanas (Parte III). El objetivo del estudio fue valorar la eficacia, seguridad y tolerabilidad de Ilaris (150 mg ó 2 mg/kg cada 8 semanas) en pacientes con CAPS.
- Parte I: Se observó una respuesta clínica y de biomarcadores completa a Ilaris (definida por el objetivo compuesto de la valoración global de la autoinflamación y la enfermedad cutánea por el médico < mínima y los valores de PCR o AAS < 10 mg/litro) en el 97% de los pacientes durante los 7 primeros días de tratamiento. Se apreciaron mejorías significativas en la valoración clínica por parte del médico de la actividad de la enfermedad autoinflamatoria: en la valoración global de la actividad de la enfermedad autoinflamatoria, la valoración de la enfermedad cutánea (erupción cutánea de tipo urticariforme), artralgias, mialgias, cefalea/migraña, conjuntivitis, fatiga/malestar general, valoración de otros síntomas relacionados y valoración de los síntomas por parte del paciente.
- Parte II: En el periodo con opción de retirada del estudio pivotal, la variable principal se definió como la proporción de pacientes con recaída/agravamiento: ninguno de los pacientes (0%) tratados con Ilaris empeoró frente al 81% de los pacientes asignados al grupo placebo.
- Parte III: Los pacientes tratados con placebo en la Parte II que empeoraron, recuperaron y mantuvieron la respuesta clínica y serológica tras su incorporación en la fase de extensión abierta con Ilaris.
Tabla 2 Resumen tabulado de la eficacia en el estudio pivotal fase III, controlado con placebo y opción de retirada (Part II)
|
Estudio fase III, pivotal controlado con placebo y opción de retirada (Parte II) | |||
|
Ilaris N=15 N(%) |
Placebo N=16 N(%) |
p-valor | |
|
Variable principal (agravamiento) Proporción de pacientes con empeoramiento en la |
0 (0%) |
13 (81%) |
< 0,001 |
|
Parte II Marcadores de la inflamación* Proteína C reactiva, mg/l |
1,10 (0,40) |
19,93 (10,50) |
< 0,001 |
|
Proteína A amiloidea, mg/l |
2,27 (-0,20) |
71,09 (14,35) |
0,002 |
|
* cambio medio (mediana) desde el inicio de la Parte II | |||
Se realizaron dos estudios de fase III a largo plazo, abiertos y no controlados. Uno fue un estudio de seguridad, tolerancia y eficacia con canakinumab en pacientes con CAPS. La duración total del tratamiento oscilaba desde los 6 meses a los 2 años. El otro fue un estudio abierto con canakinumab para evaluar la eficacia y seguridad en pacientes japoneses con CAPS durante 24 semanas, con una fase de extensión hasta 48 semanas. El objetivo primario fue evaluar la proporción de pacientes sin recaídas a las 24 semanas, incluyendo aquellos pacientes a los que se les incrementó la dosis.
En el análisis agrupado de la eficacia para estos dos estudios, el 65,6% de los pacientes que no habían sido tratados previamente con canakinumab alcanzaron una respuesta completa con 150 mg o 2 mg/kg, mientras que el 85,2% de los pacientes alcanzaron una respuesta completa a cualquier dosis. De los pacientes tratados con 600 mg o 8 mg/kg (o incluso superior), el 43,8% alcanzaron una respuesta completa. Menos pacientes de 2 a < 4 años de edad alcanzaron una respuesta completa (57,1%) comparado con pacientes pediátricos de más edad o pacientes adultos. De los pacientes que alcanzaron una respuesta completa, el 89,3% mantuvieron la respuesta sin recaídas.
La experiencia de los pacientes individuales que alcanzaron una respuesta completa después de un escalado de dosis de 600 mg (8 mg/kg) cada 8 semanas sugiere que para pacientes que no alcanzan una respuesta completa o que no mantienen una respuesta completa con las dosis recomendadas (150 mg o 2 mg/kg para pacientes > 15 kg y < 40 kg), una dosis superior puede ser beneficiosa. En pacientes de 2 a < 4 años de edad y en pacientes con síntomas NOMID/CINCA se administró con frecuencia una dosis incrementada comparado con FCAS y MWS.
Población pediátrica
Los estudios de CAPS con Ilaris comprenden un total de 80 pacientes pediátricos de edades comprendidas entre 2 y 17 años (aproximadamente la mitad de ellos han sido tratados en base a mg/kg). En general, no hubo diferencias clínicamente significativas en el perfil de eficacia, seguridad y tolerabilidad de Ilaris en pacientes pediátricos comparado con la población general de CAPS. La mayoría de los pacientes pediátricos alcanzaron la mejora en los síntomas clínicos y los marcadores objetivos de la inflamación (p.ej. SAA y CRP).
Se realizó un ensayo abierto de 56 semanas para evaluar la eficacia, seguridad y tolerancia de Ilaris en pacientes pediátricos con CAPS <4 años de edad. Se evaluaron 17 pacientes (incluyendo 6 pacientes menores de 2 años), utilizando dosis de inicio basadas en el peso de 2-8 mg/kg. En el ensayo también se evaluó el efecto de canakinumab sobre el desarrollo de anticuerpos a las vacunas estándares para niños. No se observaron diferencias en cuanto a seguridad o eficacia en pacientes menores de 2 años comparado con pacientes a partir de 2 años de edad. Todos los pacientes que recibieron vacunas no vivas según la práctica clínica habitual infantil (N=7), desarrollaron niveles protectores de anticuerpos.
Enfermedad de Still AIJS
Se evaluó la eficacia de Ilaris para el tratamiento de SJIA activo en dos estudios pivotales (G2305 y G2301). Los pacientes reclutados fueron de 2 a < 20 años de edad (edad media de 8,5 años y duración media de la enfermedad de 3,5 años al inicio del estudio) y tenían la enfermedad activa definida como > 2 articulaciones con artritis activa, fiebre y elevación de la CRP.
Estudio G2305
El estudio G2305 fue un estudio aleatorizado, doble ciego, controlado con placebo, de 4 semanas para la evaluación de la eficacia a corto plazo de Ilaris en 84 pacientes aleatorizados que recibieron una dosis única de 4 mg/kg (hasta 300 mg) o de placebo Ilaris. El objetivo primario fue el porcentaje de pacientes que en el día 15 lograron una mejoría mínima del 30% en el criterio de respuesta American College of Rheumatology (ACR) adaptado para incluir ausencia de fiebre. El tratamiento con Ilaris mejoró todos los resultados de respuesta ACR pediátricos en comparación con placebo en los días 15 y 29 (Tabla 3).
Tabla 3 Respuesta ACR pediátricos y estado de la enfermedad en el día 15 y 29
|
Día 15 |
Día 29 | |||
|
Ilaris |
Placebo |
Ilaris |
Placebo | |
|
N=43 |
N=41 |
N=43 |
N=41 | |
|
ACR30 |
84% |
10% |
81% |
10% |
|
ACR50 |
67% |
5% |
79% |
5% |
|
ACR70 |
61% |
2% |
67% |
2% |
|
ACR90 |
42% |
0% |
47% |
2% |
|
ACR100 |
33% |
0% |
33% |
2% |
|
Enfermedad inactiva |
33% |
0% |
30% |
0% |
La diferencia de tratamiento en todos los resultados ACR fue significativa (p < 0.0001)
Los resultados de los componentes del ACR adaptado para pediatría que incluye manifestaciones sistémicas y artríticas, fueron consistentes con los resultados generales de respuesta ACR. El cambio medio en el número de articulaciones con artritis activa y el rango de movimiento limitado, en el día 15, eran del 67% y del 73% para Ilaris (N=43) respectivamente, en comparación con un cambio medio del 0% y 0% para placebo (N=41). El cambio medio en la puntuación de dolor del paciente (0-100 mm escala analógica visual) en el día 15 fue de -50,0 mm para Ilaris (N=43) en comparación con +4,5 mm para placebo (N=25). El cambio medio en la puntuación de dolor entre los pacientes tratados con Ilaris fue consistente en el día 29.
Estudio G2301
Estudio G2301 fue un estudio aleatorizado, doble ciego, controlado con placebo, con diseño de retirada en la prevención de brotes con Ilaris. El estudio consistió en dos partes con dos objetivos primarios independientes (retirada de esteroides con éxito y tiempo hasta el brote). En la parte I (abierto) se reclutaron 177 pacientes y recibieron 4 mg/kg (hasta 300 mg) de Ilaris administrado cada 4 semanas durante un máximo de 32 semanas. Los pacientes en la parte II (doble ciego) recibieron
4 mg/kg de Ilaris o placebo cada 4 semanas hasta que se produjeron 37 brotes.
Retirada de corticoides:
Del total de 128 pacientes que entraron en la parte I tomando corticoides, 92 intentaron disminuir los corticoides. Cincuenta y siete (62%) de los 92 pacientes que intentaron disminuir la dosis lo consiguieron y 42 (46%), discontinuaron los corticoides.
Tiempo hasta el brote:
Los pacientes que toman Ilaris en la parte II tenían un 64% menos de riesgo de brotes, en comparación con el grupo placebo (tasa de riesgo de 0,36, IC del 95%: 0,17 a 0,75, p = 0,0032). Sesenta y tres de los 100 pacientes que entraron en la segunda parte, ya fueran asignados a placebo o canakinumab, no experimentaron un brote durante el período de observación (hasta un máximo de 80 semanas).
Relacionada con la salud y la calidad de vida los resultados de los estudios G2305 y G2301 El tratamiento con Ilaris mejoró clínicamente de manera significativa la función física de los pacientes y la calidad de vida. En el estudio G2305, la mejoría media del Cuestionario de Evaluación de Salud Infantil de Mínimos Cuadrados fue de 0,69 para Ilaris vs placebo, lo que representa una diferencia mínima clínicamente importante de 3,6 veces de 0,19 (p=0,0002). La mejoría media al final de la primera parte del estudio G2301 fue de 0,88 (79%). Se notificaron mejorías estadísticamente significativas en el Cuestionario de Salud Infantil - en los resultados PF50 de Ilaris vs placebo en el estudio G2305 (p=0,0012 físicamente, p=0,0017 estado psicosocial).
Análisis de Eficacia
Para evaluar el mantenimiento de la eficacia se combinaron los datos de los estudios G2305, G2301 y el estudio de extensión de las primeras 12 semanas de tratamiento con Ilaris. Estos datos mostraron mejoras similares desde el inicio hasta la semana 12 en las respuestas ACR adaptado pediátrico y de sus manifestaciones a los observados en el estudio controlado con placebo (G2305). En la semana 12, las respuestas ACR adaptado pediátrico 30, 50, 70, 90 y 100 fueron del 70%, 69%, 61%, 49% y 30%, respectivamente, y el 28% de los pacientes tenían enfermedad inactiva (N=178).
La eficacia observada en los estudios G2305 y G2301 se mantuvo en el estudio abierto de extensión a largo plazo en marcha (datos disponibles a través de la mediana de 49 semanas de seguimiento). En este estudio, 25 pacientes que presentaron una respuesta importante de ACR durante un mínimo de
5 meses, se les redujo su dosis a 2 mg/kg de Ilaris cada 4 semanas y se le dio la dosis reducida (media de 32 semanas, 8-124 semanas) los que mantuvieron una respuesta ACR pediátrico 100 a lo largo del tiempo.
Existe evidencia, aunque limitada, de los ensayos clínicos que indica que los pacientes que no responden a tocilizumab o anakinra, pueden responder a canakinumab.
AIJS en adultos jóvenes y ESA
La eficacia de Ilaris en un subgrupo de pacientes con AIJS adultos jóvenes de 16 a 20 años de edad fue consistente con la eficacia observada en pacientes menores de 16 años de edad. Se espera que el perfil de eficacia en pacientes con ESA sea similar al de pacientes con AIJS de acuerdo con los informes de la literatura.
Gota artrítica
La eficacia de Ilaris para el tratamiento de los ataques agudos de gota artrítica se demostró en dos estudios multicéntricos, controlados con activo, doble ciego, aleatorizados, en pacientes con gota artrítica frecuente (> 3 ataques en los 12 meses previos) que no podían utilizar AINEs o colchicina (debido a contraindicación, intolerancia o falta de eficacia). Estos estudios eran de 12 semanas seguidos de una extensión de 12 semanas doble ciego. Un total 225 pacientes fueron tratados con 150 mg de Ilaris subcutáneo y 229 pacientes fueron tratados con 40 mg de acetónido de triamcinolona (TA) al iniciar el tratamiento y después de experimentar un nuevo ataque. El número medio de ataques de gota artrítica en los 12 meses previos era 6,5. Más del 85% de los pacientes tuvieron comorbilidad, incluyendo hipertensión (60%), diabetes (15%), cardiopatía isquémica (12%), y enfermedad renal crónica en fase > 3 (25%). Aproximadamente un tercio de los pacientes incluidos (76 [33,8%] en el grupo Ilaris y 84 [36,7%] en el grupo acetónido de triamcinolona) tenían incapacidad documentada (intolerancia, contraindicación o ausencia de respuesta) para utilizar tanto AINEs como colchicina. Al inicio del estudio se notificó tratamiento concomitante con ULTs en el 42% de los pacientes.
Las dos variables principales de evaluación fueron: (i) intensidad del dolor de la gota artrítica (escala analógica visual, EAV) a las 72 horas post-dosis, y (ii) tiempo hasta un nuevo ataque de gota artrítica.
Para la población general del estudio, la intensidad del dolor fue menor estadísticamente significativa para Ilaris 150 mg comparado con acetónido de triamcinolona a las 72 horas. Ilaris también disminuyó el riesgo de ataques posteriores (ver Tabla 4).
Los resultados de eficacia en pacientes que no pueden utilizar ni AINEs ni colchicina y que no estaban en tratamiento con ULT, no respondieron al tratamiento con ULT o estaban contraindicados de ULT (N=101) fueron consistentes en la población general del estudio con una diferencia estadísticamente significativa comparado a acetónido de triamcinolona en la intensidad del dolor a las 72 horas (-10,2 mm, p=0,0208) y en la disminución del riesgo de ataques posteriores(Cociente del riesgo 0,39, p=0,0047 a las 24 semanas).
En la Tabla 4 se presentan los resultados de eficacia para un subgrupo más restringido limitado a los pacientes que están utilizando ULT (N=62). El tratamiento con Ilaris produjo una reducción del dolor y disminuyó el riesgo de ataques posteriores en pacientes que estaban utilizando ULT y que no podían utilizar ni AINEs ni colchicina, a pesar de que la diferencia observada durante el tratamiento comparado con acetónido de triamcinolona fue menos pronunciada que con la población de estudio general.
Tabla 4 Eficacia de la población general y en un subgrupo de pacientes que utilizan ULT y que no pueden utilizar ni AINEs ni colchicina
Variable de eficacia Población general del Incapacidad de utilizar tanto
estudio; AINEs como colchicina con
N=454 ULT
_N=62_
Tratamiento de los ataques de gota artrítica medidos por la intensidad del dolor (EAV) a las
Diferencia estimada del promedio de -10,7 -3,8
Mínimos Cuadrados a acetónido de
triamcinolona
IC (-15,4, -6,0) (-16,7, 9,1)
Valor p, 1 cola p < 0,0001* p=0,2798
Reducción del riesgo de ataques posteriores de gota artrítica medida por el tiempo hasta un
nuevo ataque (24 semanas)
Cociente de riesgo a acetónido de 0,44 0,71
triamcinolona
IC (0,32, 0,60) (0,29, 1,77)
Valor p, 1 cola p < 0,0001* p=0,2337
* Representa un valor-p significativamente < 0,025_
Los resultados de eficacia mostraron una incidencia mayor de efectos adversos para canakinumab comparado con acetónido de triamcinolona con el 66% vs el 53% de los pacientes notificando algún efecto adverso y el 20% vs el 10% de los pacientes notificando una infección como reacción adversa después de las 24 semanas.
Pacientes de edad avanzada
En global, el perfil de eficacia, seguridad y tolerabilidad de Ilaris en pacientes de edad avanzada de > 65 años de edad fue comparable a los pacientes < 65 años de edad.
Pacientes en tratamiento con reductores de uratos (ULT)
En los ensayos clínicos, Ilaris se ha administrado de forma segura con ULT. En el conjunto de la población de los estudios, los pacientes con ULT tuvieron una diferencia menos pronunciada durante el tratamiento tanto en la reducción del dolor como en la reducción del riesgo de ataques posteriores de gota artrítica comparado con pacientes que no estaban en tratamiento con ULT.
Inmugenecidad
En aproximadamente 1,5%, 3% y 2% de los pacientes tratados con Ilaris para CAPS, AIJS y gota artrítica respectivamente, se observaron anticuerpos contra Ilaris. No se detectaron anticuerpos neutralizantes. No se observó una correlación aparente entre el desarrollo de anticuerpos y la respuesta clínica o efectos adversos.
Este medicamento se ha autorizado para CAPS en «circunstancias excepcionales». Esta modalidad de aprobación significa que debido a la rareza de la enfermedad no ha sido posible obtener información completa de este medicamento. La Agencia Europea de Medicamentos revisará anualmente la información nueva de este medicamento que pueda estar disponible, y esta Ficha Técnica o Resumen de las Características del Producto (RCP) se actualizará cuando sea necesario.
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Ilaris en uno o más grupos de la población pediátrica con Síndromes Periódicos Asociados a la Criopirina (CAPS) y Artritis idiopática juvenil (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Ilaris en los diferentes grupos de la población pediátrica en gota artrítica (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 Propiedades farmacocinéticas
CAPS
Absorción
La concentración sérica máxima (Cmax) de canakinumab se produjo aproximadamente a los 7 días tras una administración única de 150 mg por vía subcutánea a pacientes de CAPS adultos. La semivida terminal media fue de 26 días. Los valores medios de Cmax y AUCinf después de una única dosis subcutánea de 150 mg en un paciente adulto común de CAPS (70 kg) fueron 15,9 pg/ml y 708 pg*d/ml. Se estimó que la biodisponibilidad absoluta de canakinumab administrado por vía subcutánea era del 66%. Los parámetros relativos a la exposición (tales como AUC y Cmáx) se incrementaron de forma proporcional a la dosis en el intervalo de dosis comprendidas entre 0,30 a 10,0 mg/kg por perfusión intravenosa y entre 150 a 600 mg por inyección subcutánea. Los valores previstos de exposición en estado estacionario (Cmin,ss, Cmaxss, AUC,ss,8w) tras la administración de 150 mg subcutánea (ó 2 mg/kg, respectivamente) fueron ligeramente superiores en la categoría de peso 40-70 kg (6,6 pg/ml, 24,3 pg/ml, 767 pg*d/ml) en comparación con las categorías de peso < 40 kg (4,0 pg/ml, 19,9 pg/ml, 566 pg*d/ml) y > 70 kg (4,6 pg/ml, 17,8 pg/ml, 545 pg*d/ml). El coeficiente de acumulación esperado fue de 1,3 veces tras 6 meses de administración subcutánea de 150 mg de canakinumab cada 8 semanas.
Distribución
Canakinumab se une a la IL-1 beta sérica. El volumen de distribución (Vss) de canakinumab varió según el peso corporal. Se estimó que es de 6,2 litros en un paciente de CAPS de 70 kg de peso.
Eliminación
El aclaramiento aparente (CL/F) de canakinumab se incrementa con el peso corporal. Se estimó que era 0,17 l/d en un paciente CAPS de 70 kg de peso y 0,11 l/d en un paciente AIJS de 33 kg de peso. Tras tener en cuenta las diferencias de peso corporal, no se observaron diferencias clínicamente significativas en las propiedades farmacocinéticas de canakinumab entre los pacientes CAPS y los AIJS.
No se observó aclaramiento acelerado o modificación con el tiempo de las propiedades farmacocinéticas de canakinumab tras la administración de dosis repetidas. Tampoco se observaron diferencias por edad y sexo en los parámetros farmacocinéticos una vez efectuada la corrección en función del peso.
Enfermedad de Still (ESA y AIJS)
La biodisponibilidad en pacientes con AIJS no se ha determinado de forma independiente. El aclaramiento aparente por kg de peso corporal (CL/F por kg) fue comparable entre la población de AIJS y la de CAPS (0,004 l/d por kg). El volumen aparente de distribución por kg (V/F por kg) fue de 0,14 l/kg.
El coeficiente de acumulación de canakinumab fue de 1,6 veces tras la administración subcutánea de 4 mg/kg de canakinumab cada 4 semanas en pacientes con AIJS. El estado estacionario se alcanzó a los 110 días. La media prevista total (±SD) para Cminss, Cmaxss y AUC,ss4w fue 14,7±8,8 pg/ml, 36,5±14,9 pg/ml y 696,1±326,5 pg*d/ml, respectivamente.
Las AUCss4w en cada grupo de edad fueron de 692, 615, 707 y 742 pg*d/ml de 2-3, 4-5, 6-11, y 12-19 años respectivamente. Cuando se estratificó por peso, se observó un menor exposición media (30-40%) para Cminss (11,4 vs 19 pg/ml) y AUCss (594 vs 880 pg*d/ml) para la categoría de menor peso (< 40 kg) vs la categoría de más peso (> 40 kg).
De acuerdo con el análisis del modelo farmacocinético poblacional, la farmacocinética de canakinumab en pacientes con AIJS adultos jóvenes de 16 a 20 años de edad fue similar a la de pacientes menores de 16 años de edad. Las exposiciones predecibles de canakinumab en estado estacionario al nivel de dosis de 4 mg/kg (máximo 300 mg) en pacientes mayores de 20 años de edad fueron comparables a las observadas en pacientes con AIJS menores de 20 años de edad.
Pacientes con gota artrítica
La biodisponibilidad en pacientes con gota artrítica no ha sido determinada independientemente. El aclaramiento aparente por kg de peso corporal (CL/F por kg) fue comparable entre la población de gota y de CAPS (0,004 l/d/kg). La exposición media en un paciente común de gota artrítica (93 kg) después de una única dosis subcutánea de 150 mg (Cmax: 10,8 pg/ml y AUCinf: 495 pg*d/ml) fue menor que en un paciente común de CAPS de 70 kg (15,9 pg/ml y 708 pg*d/ml). Esto es consistente con el incremento observado de CL/F con el peso corporal.
El coeficiente de acumulación de canakinumab fue de 1,1 veces tras la administración subcutánea de 150 mg de canakinumab cada 12 semanas.
Población pediátrica
Las concentraciones máximas de canakinumab se alcanzaron entre los 2 y los 7 días (Tmax) después de una administración subcutánea única de 150 mg ó 2 mg/kg de canakinumab en pacientes pediátricos a partir de 4 años de edad. La semivida terminal osciló entre 22,9 y 25,7 días, similar a las propiedades farmacocinéticas observadas en adultos. En base al análisis de los modelos de población farmacocinético, la farmacocinética de canakinumab en niños de 2 a < 4 años fue similar a la de los pacientes de 4 años de edad o mayores. El coeficiente de absorción subcutánea se estima que disminuye con la edad y aparentemente es más rápido en pacientes más jóvenes. En consecuencia, Tmax más corta (3,6 días) en pacientes jóvenes con AIJS (2-3 años) en comparación con pacientes mayores de AISJ (12-19 años; Tmax 6 días). No afecta la biodisponibilidad (AUCss).
Un análisis farmacocinético adicional mostró que la farmacocinética de canakinumab en 6 pacientes pediátricos con CAPS, menores de 2 años de edad, era similar a la farmacocinética en pacientes pediátricos entre 2-4 años de edad. De acuerdo al análisis de los modelos de población farmacocinético, la exposición esperada después de una dosis de 2 mg/kg fue comparable entre los grupos de edad pediátrica con CAPS, pero fue aproximadamente el 40% inferior en pacientes pediátricos de muy bajo peso corporal (p.ej., 10 kg) comparado con pacientes adultos (dosis de 150 mg). Esto es consistente con las observaciones de exposiciones más elevadas en grupos de peso corporal más elevado en pacientes con CAPS.
Las propiedades farmacocinéticas de poblaciones pediátricas CAPS y AIJS son similares.
Población en edad avanzada
No se observaron cambios en los parámetros farmacocinéticos en base al aclaramiento o al volumen de distribución entre pacientes de edad avanzada y pacientes adultos < 65 años de edad.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios de reactividad cruzada, toxicidad a dosis repetidas, inmunotoxicidad, toxicidad para la reproducción y estudios de toxicidad juvenil realizados con canakinumab o con un anticuerpo murino anti IL-1 beta murina.
Como canakinumbab se une a la IL-1 beta del hombre y monos babuinos (C. jacchus) con afinidad similar, la seguridad de canakinumab se ha investigado en estos animales. No se observaron efectos adversos de canakinumab después de la administración dos veces por semana durante 26 semanas a monos babuinos o en el estudio de toxicidad de desarrollo embriofetal en monas embarazadas. Las concentraciones plasmáticas que están bien toleradas en animales exceden como mínimo 42 veces (Cmax) y 78 veces (Cavg) las concentraciones plasmáticas en pacientes pediátricos con CAPS (peso corporal 10 kg) tratados con dosis clínicas de canakinumab hasta 8 mg/kg por vía subcutánea cada 8 semanas. Las concentraciones plasmáticas que se toleran bien en animales son 62 veces superiores (Cmáx) y 104 veces (Cavg) a las concentraciones plasmáticas en los pacientes pediátricos con SJIA tratados con hasta 4 mg / kg por vía subcutánea cada 4 semanas. Además, no se detectaron anticuerpos frente a canakinumab en estos estudios. No se demostró que existiera reactividad cruzada inespecífica cuando se aplicó canakinumab a tejidos humanos normales.
No se han realizado estudios de carcinogénesis formales con canakinumab.
En el estudio de desarrollo embrionario en monos babuinos, canakinumab no mostró toxicidad materna, embrionaria o potencial teratogénico cuando se administró durante la organogénesis.
No se apreciaron efectos no deseados de un anticuerpo murino anti IL-1 beta murina en el conjunto global de estudios de reproducción y en animales jóvenes realizados en ratones. El anticuerpo anti-murino IL-1 beta no provocó acontecimientos adversos en el crecimiento fetal o neonatal al administrarse durante la parte final de la gestación, parto y lactancia (ver sección 4.6). La dosis alta utilizada para estos estudios excedió la dosis máxima efectiva en términos de supresión y de la actvidad de IL-1 beta.
En un estudio inmunotoxicológico en ratones con un anticuerpo murino anti 1L-1 beta murino mostró que la neutralización de IL-1 beta no tiene efectos sobre los parámetros inmunes y no deteriora la función inmune de los ratones.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Sacarosa
Histidina
Histidina hidrocloruro monohidrato Polisorbato 80
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 3 años.
Desde un punto de vista microbiológico, el producto debería utilizarse inmediatamente tras la reconstitución. Si no se usa inmediatamente, el tiempo y las condiciones antes de uso estarán bajo responsabilidad exclusiva del usuario y no deberían superar 24 horas a 2°C - 8°C.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
150 mg de polvo para solución inyectable en un vial (vidrio tipo I) con un tapón (recubierto de goma de clorobutilo) y una cápsula flip-off (aluminio).
Envases con 1 vial o envases múltiples con 4 (4x1) viales.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especialies de eliminación y otras manipulaciones
Ilaris 150 mg polvo para solución inyectable se suministra en un vial de un solo uso para uso individual. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Instrucciones para la reconstitución
Con técnica aséptica, reconstituir el contenido de cada vial de Ilaris a temperatura ambiente (normalmente entre 15°C y 25°C) mediante la inyección lenta de 1,0 ml de agua para inyección con una jeringa de 1 ml y una aguja de 18 G x 2 pulgadas (50 mm). Remover lentamente el vial con un ángulo de 45° aproximadamente durante 1 minuto y dejar reposar durante 5 minutos. A continuación, girar suavemente el vial de abajo a arriba unas 10 veces. Si es posible, debe evitarse tocar el tapón de goma con los dedos. Dejar reposar alrededor de 15 minutos a temperatura ambiente para obtener una solución de transparente a opalescente. No agitar. No utilizar si se observan partículas en la solución.
Dar ligeros golpecitos en las paredes laterales del vial para eliminar cualquier posible residuo de líquido del tapón. La solución debería estar libre de partículas visibles y ser de transparente a opalescente. La solución puede ser incolora o presentar un ligero color pardo amarillo. Si la solución presenta un color pardo evidente, no debe utilizarse. Si no se utiliza inmediatamente después de la reconstitución, la solución debería mantenerse entre 2°C y 8°C y utilizarse en las próximas 24 horas.
Instrucciones para la administración
Extraer con cuidado la cantidad de volumen requerido en función de la dosis que deba administrarse (0,2 ml a 1,0 ml) e inyectar por vía subcutánea con una aguja de 27 G x 0,5 pulgadas (13 mm).
Se recomiendan los siguientes lugares de inyección: muslo superior, abdomen, brazo superior o glúteos. Deben evitarse las áreas de piel lesionada, con hematomas o con erupciones. Debe evitarse inyectar en el tejido cicatricial puesto que podría resultar en una exposición insuficiente a Ilaris.
Eliminación
Los pacientes y el personal sanitario al cuidado deben ser formados en relación a la eliminación de los viales, jeringas y agujas de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/09/564/001 -002
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 23 de octubre de 2009 Fecha de la última renovación: 19 de junio de 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Ilaris 150 mg polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un vial contiene 150 mg de canakinumab*.
Después de la reconstitución, cada ml de la solución contiene 150 mg de canakinumab.
* anticuerpo monoclonal completamente humano obtenido mediante la tecnología del ADN recombinante en células de hibridoma Sp2/0 de ratón
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable. El polvo es blanco.
El disolvente es transparente e incoloro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Síndromes Periódicos Asociados a la Criopirina
Ilaris está indicado para el tratamiento de los Síndromes Periódicos Asociados a la Criopirina (CAPS) en adultos, adolescentes y niños a partir de 2 años con un peso corporal de 7,5 kg o superior, incluidos:
- Síndrome de Muckle-Wells (MWS),
- Enfermedad Neonatal Multisistémica Inflamatoria (NOMID) / Síndrome Infantil Neurológico Cutáneo y Articular Crónico (CINCA),
- Manifestaciones graves del Síndrome Autoinflamatorio Familiar inducido por el frío (FCAS) / Urticaria Familiar Fría (FCU) que presente signos y síntomas más allá de la erupción de tipo urticaria inducido por el frío.
Enfermedad de Still
Ilaris está indicado para el tratamiento de la enfermedad de Still activa incluyendo la Enfermedad de Still del Adulto (ESA) y la artritis idiopática juvenil sistémica (AIJS) en pacientes de 2 años de edad o mayores que no hayan respondido adecuadamente al tratamiento previo con antiinflamatorios no esteroideos (AINEs) y corticosteroides sistémicos. Ilaris puede ser administrado en monoterapia o en combinación con metotrexato.
Gota artrítica
Ilaris está indicado para el tratamiento sintomático de pacientes adultos con ataques frecuentes de gota artrítica (al menos 3 ataques en los 12 meses previos) en los cuales está contraindicado el tratamiento con medicamentos antiinflamatorios no esteroideos (AINEs) y colchicina, no está tolerado, o no responden adecuadamente, y en los cuales no son adecuadas las series repetidas de corticoides (ver sección 5.1).
4.2 Posología y forma de administración
Para CAPS y enfermedad de Still, el tratamiento debe ser iniciado y supervisado por un médico especialista con experiencia en el diagnóstico y tratamiento de la indicación.
Para gota artrítica, el médico debe tener experiencia en el uso de medicamentos biológicos e Ilaris debe ser administrado por un profesional sanitario.
Después de recibir una formación adecuada con respecto a la técnica correcta de inyección, el paciente o sus cuidadores pueden inyectar Ilaris si el médico lo considera apropiado y con seguimiento médico si fuera preciso (ver sección 6.6.).
Posología
CAPS: Adultos, adolescentes y niños a partir de 2 años de edad La dosis inicial recomendada de Ilaris para pacientes con CAPS es:
Adultos, adolescentes y niños > de 4 años de edad:
- 150 mg para pacientes cuyo peso corporal sea > 40 kg
- 2 mg/kg para pacientes cuyo peso corporal sea > 15 kg y < 40 kg
- 4 mg/kg para pacientes cuyo peso corporal sea > 7,5 kg y < 15 kg Niños desde 2 a < 4 años de edad:
- 4 mg/kg para pacientes cuyo peso corporal sea > 7,5 kg
Se administra cada ocho semanas como una dosis única mediante una inyección subcutánea.
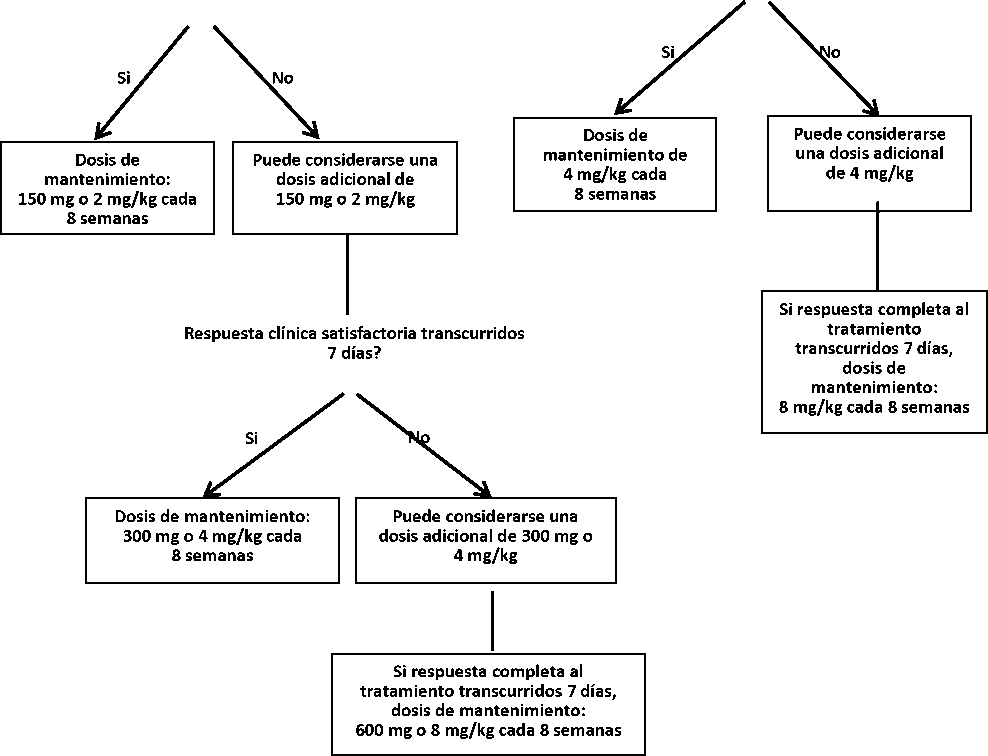
En pacientes con una dosis inicial de 150 mg o 2 mg/kg,si no se consigue una respuesta clínica satisfactoria (resolución de la erupción y otros síntomas inflamatorios generalizados) una vez transcurridos 7 días desde el inicio del tratamiento, puede considerarse una segunda dosis de Ilaris de 150 mg ó 2 mg/kg. Si, en lo sucesivo, se consigue una respuesta completa al tratamiento, se debe mantener el régimen con dosis elevadas de 300 mg o 4 mg/kg cada 8 semanas. Si no se consigue una respuesta clínica satisfactoria una vez transcurridos 7 días de esta dosis aumentada, puede considerarse una tercera dosis de Ilaris a 300 mg o 4 mg/kg. Si, en lo sucesivo, se obtiene una respuesta completa al tratamiento, se debe considerar el régimen con dosis elevadas de 600 mg o 8 mg/kg cada 8 semanas, en base a la valoración clínica individual.
Para pacientes con una dosis inicial de 4 mg/kg, si no se consigue una respuesta clínica satisfactoria una vez transcurridos 7 días del inicio del tratamiento, puede considerarse una segunda dosis de Ilaris 4 mg/kg. Si, en lo sucesivo, se obtiene una respuesta clínica completa, se debe considerar el mantenimiento del régimen con dosis elevadas de 8 mg/kg cada 8 semanas, en base a la valoración clínica individual.
La experiencia clínica con intervalos de dosis inferiores a 4 semanas o con dosis superiores a 600 mg o 8 mg/kg es limitada.
150 mg o 2 mg/kg
4 mg/kg
Adultos y niños >4 años de edad >15 kg
Niños de 2-< 4 años de edad o niños >4 años de edad >7.5 kg y < 15 kg
Respuesta clínica satisfactoria transcurridos 7 días?
Respuesta clínica satisfactoria transcurridos 7 días?
Enfermedad de Still (ESA y AIJS)
La dosis recomendada de Ilaris para pacientes con enfermedad de Still (ESA y AIJS) con peso corporal > 7,5 kg es de 4 mg/kg (hasta un máximo de 300 mg), administrado por inyección subcutanea cada cuatro semanas. En pacientes sin mejoría clínica el médico que lo trate considerará si debe continuar con el tratamiento de Ilaris.
Gota artrítica
Se debe iniciar u optimizar el manejo de la hiperuricemia con tratamientos adecuados para disminuir el urato (ULT, de sus singlas en inglés). Ilaris se debe utilizar como un tratamiento a demanda para tratar los ataques de gota artrítica.
La dosis recomendada de Ilaris para pacientes adultos con gota artrítica es 150 mg administrados subcutáneamente como una única dosis durante un ataque. Para un efecto máximo, se debe administrar Ilaris tan pronto como sea posible después del inicio de un ataque de gota artrítica.
Los pacientes que no responden al tratamiento inicial no deben volver a ser tratados con Ilaris. En pacientes respondedores y que requieren un retratamiento, debe haber un intervalo de al menos 12 semanas antes de que se administre una nueva dosis de Ilaris (ver sección 5.2).
Poblaciones especiales Población pediátrica CAPS
No se ha establecido la seguridad y eficacia de Ilaris en pacientes menores de 2 años de edad con CAPS. Los datos actualmente disponibles están descritos en las secciones 4.8, 5.1 y 5.2, sin embargo no se puede hacer una recomendación posológica.
AIJS
No se han establecido la seguridad y la eficacia de Ilaris en pacientes menores de 2 años de edad con AIJS.
Gota artrítica
No existe una recomendación de uso específica para Ilaris en la población pediátrica para la indicación de gota artrítica.
Pacientes de edad avanzada No se requiere ajuste de dosis.
Insuficiencia hepática
Ilaris no ha sido estudiado en pacientes con insuficiencia hepática.
Insuficiencia renal
No se requiere un ajuste de la dosis en pacientes con insuficiencia renal. No obstante, la experiencia clínica en estos pacientes es limitada.
Forma de administración
Ilaris se debe administrar por inyección subcutánea. Para consultar las instrucciones de uso y manejo de la solución reconstituida ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Infecciones graves activas (ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
Infecciones
Ilaris está asociado con un aumento en la incidencia de infecciones graves. Por consiguiente, debe controlarse estrechamente la aparición de signos y síntomas de infección en los pacientes durante y después del tratamiento con Ilaris. Los médicos deben tener precaución al administrar Ilaris en pacientes con infecciones, con antecedentes de infecciones recurrentes, o con condiciones subyacentes que puedan predisponerles a sufrir infecciones.
Tratamiento de CAPS y Enfermedad de Still (ESA y AIJS)
Ilaris no se debe iniciar o continuar en pacientes con infecciones graves que requieran intervención médica.
Tratamiento de gota artrítica
Ilaris no se debe administrar durante una infección activa.
No se recomienda el uso concomitante de Ilaris con inhibidores del factor de necrosis tumoral (TNF) ya que podría aumentar el riesgo de infecciones graves (ver sección 4.5).
Durante el tratamiento con Ilaris se han notificado casos aislados de infecciones oportunistas o poco habituales (como aspergilosis, infecciones micobacterianas atípicas, herpes zoster). No se puede excluir una relación causal de Ilaris con estos acontecimientos.
En aproximadamente el 12% de los pacientes con CAPS sometidos a una prueba cutánea PPD (derivado proteínico purificado) en los ensayos clínicos, la prueba de seguimiento durante el tratamiento con Ilaris dio un resultados positivo sin que existiera evidencia clínica de una infección tuberculosa latente o activa.
Se desconoce si el uso de inhibidores de interleucina-1 (IL-1) tales como Ilaris incrementa el riesgo de reactivación de tuberculosis. Antes de iniciar el tratamiento, debe evaluarse la existencia de tuberculosis activa y latente en todos los pacientes. Especialmente en pacientes adultos, esta evaluación debe incluir una historia médica detallada. Se deben realizar las pruebas diagnósticas adecuadas (p. ej. prueba cutánea de la tuberculina, la prueba de interferón gamma o radiografía de tórax) a todos los pacientes (deben aplicar las recomendaciones locales). Deben controlarse los síntomas y signos de tuberculosis en los pacientes durante y después del tratamiento con Ilaris. Todos los pacientes deben ser instruidos para pedir consejo médico si aparecen signos o síntomas de tuberculosis (p. ej. tos persistente, pérdida de peso, temperatura subfebril) durante el tratamiento con Ilaris. En el caso de la conversión de un resultado de negativo a positivo en la prueba cutánea PPD, en pacientes con un riesgo alto deben considerarse otras medidas alternativas para la determinación de la infección por tuberculosis.
Neutropenia y leucopenia
Se ha observado neutropenia (recuento absoluto de neutrófilos [RAN] < 1,5 x 109/l) y leucopenia con medicamentos que inhiben la IL-1, incluyendo Ilaris. No debe iniciarse el tratamiento con Ilaris en pacientes con neutropenia o leucopenia. Se recomienda controlar el recuento de glóbulos blancos (RGB) incluyendo el recuento de neutrófilos antes de iniciar el tratamiento y de nuevo, después de 1 o 2 meses. Para el tratamiento crónico o repetido, también se recomienda controlar el recuento RGB periódicamente durante el tratamiento. Si un paciente sufre neutropenia o leucopenia, debe controlarse estrechamente el RGB y se debe considerar la interrupción del tratamiento.
Neoplasias
Se han reportado neoplasias en pacientes tratados con Ilaris. Se desconoce el riesgo de desarrollo de neoplasias con el tratamiento con antiinterleukinas (IL)-1.
Reacciones de hipersensibilidad
Se han notificado reacciones de hipersensibilidad con Ilaris. La mayoría de los casos fueron de carácter leve. Durante el desarrollo clínico de Ilaris en más de 2.300 pacientes, no se describieron reacciones anafilácticas o anafilactoides. Sin embargo, no debe excluirse el riesgo de reacciones de hipersensibilidad graves, que no resulta infrecuente con proteínas inyectables (ver sección 4.3).
Función hepática
En los estudios clínicos se han notificado casos de elevaciones transitorias y asintomáticas de los niveles séricos de transaminasas y bilirrubina (ver sección 4.8).
Vacunas
No se dispone de datos sobre el riesgo de transmisión de la infección secundaria a la administración de vacunas vivas (atenuadas) en pacientes tratados con Ilaris. Por lo tanto, no deben administrarse vacunas vivas de forma concomitante con Ilaris a menos que los beneficios superen claramente los riesgos (ver sección 4.5).
Antes de iniciar el tratamiento con Ilaris se recomienda que los pacientes adultos y pediátricos reciban todas las vacunas, según estén indicadas, incluyendo la vacuna pneumocócica y la vacuna de la gripe inactivada (ver sección 4.5).
Mutación en el gen NLRP3 en pacientes con CAPS
La experiencia clínica en pacientes con CAPS sin mutación confirmada en el gen NLRP3 es limitada.
El síndrome de activación macrofágica en pacientes con Enfermedad de Still El síndrome de activación macrofágica (SAM) es un conocido trastorno potencialmente mortal que puede desarrollarse en pacientes con enfermedades reumáticas, especialmente enfermedad de Still. Si se produce SAM, o si se sospecha, debe evaluarse y tratarse lo antes posible. Los médicos deben estar atentos a los síntomas de la infección o empeoramiento de la enfermedad de Still, ya que éstos se sabe que desencadenan SAM. Basándose en la experiencia de ensayos clínicos, Ilaris no parece aumentar la incidencia de SAM en pacientes con AIJS, pero no se puede llegar a ninguna conclusión definitiva.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han investigado las interacciones entre Ilaris y otros medicamentos con estudios formales.
Se ha descrito una incidencia aumentada de infecciones graves con otro bloqueante de la IL-1 en combinación con inhibidores del TNF. No se recomienda el uso de Ilaris con inhibidores del TNF ya que esto puede aumentar el riesgo de infecciones graves.
La expresión de las enzimas hepáticas CYP450 puede ser suprimida por las citocinas que estimulan la inflamación crónica, tales como interleucina-1 beta (IL-1 beta). De este modo, la expresión de CYP450 puede revertirse cuando se introduce un tratamiento con un inhibidor potente de la citocina, como canakinumab. Esto es clínicamente relevante para sustratos del CYP450 de estrecho margen terapéutico para los que la dosis se ajusta individualmente. Al iniciar el tratamiento con canakinumab en pacientes que reciben este tipo de medicamentos, es preciso monitorizar el efecto o la concentración del principio activo y ajustar la dosis individual si fuese necesario.
No se dispone de datos sobre los efectos de la administración de vacunas vivas o la transmisión de la infección secundaria a la administración de una vacuna viva en pacientes que reciben Ilaris. Por ello, no deben administrarse vacunas vivas concomitantemente con Ilaris a menos que los beneficios superen claramente los riesgos. Si está indicada la administración de vacunas vivas después del inicio del tratamiento con Ilaris, la recomendación es esperar durante al menos 3 meses después de la última inyección de Ilaris y antes de la próxima dosis (ver sección 4.4).
Los resultados de un estudio en voluntarios adultos sanos demostraron que una única dosis de Ilaris de 300 mg no afectó a la inducción ni a la persistencia de las respuestas de anticuerpos tras la vacunación con la vacuna de la gripe o vacuna meningocócica a base de proteínas glicosiladas.
Los resultados de un ensayo abierto de 56 semanas en pacientes con CAPS de hasta 4 años de edad, demostraron que todos los pacientes que recibieron vacunas no vivas según la práctica clínica habitual para los niños, desarrollaron niveles protectores de anticuerpos.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil / Contracepción en hombres y mujeres
Las mujeres deben utilizar un método anticonceptivo efectivo durante el tratamiento con Ilaris y hasta 3 meses después de la última dosis.
Embarazo
Se dispone de datos limitados sobre la utilización de canakinumab en mujeres embarazadas. Los estudios en animales no muestran efectos dañinos directos o indirectos por lo que respecta a la toxicidad reproductiva (ver sección 5.3). Se desconoce el riesgo para el feto/madre. Las mujeres embarazadas o que desean quedar embarazadas solo deberían tratarse después de un exhaustivo análisis de beneficio/riesgo.
Los estudios en animales indican que canakinumab atraviesa la placenta y se detecta en el feto. No se dispone de datos en humanos, sin embargo como canakinumab es una inmunoglobulina de la clase G (IgG1), es de esperar que se produzca transferencia transplacentaria humana. Se desconoce el impacto clínico. Sin embargo, no se recomienda la administración de vacunas vivas a recién nacidos expuestos a canakinumab in utero durante las 16 semanas siguientes a la última dosis de Ilaris de la madre antes del parto. Las mujeres que reciben canakinumab durante el embarazo deben ser instruidas para que informen al pediatra antes de que administren alguna vacuna a su bebé recién nacido.
Lactancia
Se desconoce si canakinumab se excreta en la leche materna. La decisión de dar el pecho durante el tratamiento con Ilaris sólo debe tomarse después de un análisis exhaustivo del beneficio/riesgo.
Los estudios en animales han demostrado que un anticuerpo murino que actúa sobre la IL-1 beta murina no tuvo efectos adversos sobre el desarrollo en crías de ratón y a los que el anticuerpo les fue transferido (ver sección 5.3).
Fertilidad
No se han realizados estudios formales en relación con el efecto potencial de Ilaris sobre la fertilidad en humanos.
Canakinumab no mostró efecto sobre la fertilidad en el macho en monos macho babuinos (C.jacchus). Un anticuerpo murino anti murino IL-1 beta no mostró efectos sobre la fertilidad en ratones macho y hembra (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Ilaris sobre la capacidad para conducir y utilizar máquinas es pequeña. El tratamiento con Ilaris puede producir mareo/vértigo o astenia (ver sección 4.8). Los pacientes que experimenten estos síntomas durante el tratamiento con Ilaris deben esperar a que el síntoma remita antes de conducir o manejar maquinaria.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Más de 2.400 sujetos incluyendo aproximadamente 380 niños (de 2 a 17 años de edad) han recibido tratamiento con Ilaris en estudios intervencionales en pacientes con CAPS, AIJS, gota artrítica o con otras patologías mediadas por IL-1 beta y voluntarios sanos. Las reacciones adversas más frecuentes fueron infecciones predominantemente en el tracto respiratorio. Se han observado infecciones graves. La mayoría de estos acontecimientos fueron de leves a moderados. Con el tratamiento a más largo plazo no se vio impacto en el tipo o la frecuencia de las reacciones adversas.
Se han notificado reacciones de hipersensibilidad en pacientes tratados con Ilaris (ver secciones 4.3 y 4.4).
Se han notificado infecciones oportunistas en pacientes tratados con Ilaris (ver sección 4.4).
CAPS
En los ensayos clínicos un total de 211 pacientes de CAPS adultos y pediátricos (incluidos FCAS/FCU, MWS, y NOMID/CINCA) han recibido tratamiento con Ilaris. La seguridad de Ilaris comparada con placebo se investigó en un ensayo pivotal fase III que consistió en un periodo abierto de 8 semanas (Parte I), un periodo aleatorizado, doble ciego, controlado con placebo y opción de retirada de 24 semanas (Parte II), y un periodo abierto de 16 semanas con Ilaris (Parte III). Todos los pacientes recibieron 150 mg de Ilaris por vía subcutánea ó 2 mg/kg si el peso corporal estaba comprendido entre > 15 kg y < 40 kg.
Enfermedad de Still
Un total de 324 pacientes con AIJS de 2 a < 20 años de edad recibieron Ilaris en ensayos clínicos, incluyendo 293 pacientes de 2 a < 16 años de edad, 21 pacientes de 16 a < 18 años de edad y 10 pacientes de 18 a < 20 años de edad. Se estudió la seguridad de Ilaris en comparación con el placebo en dos estudios pivotales fase III (ver sección 5.1).
Gota artrítica
Más de 700 pacientes con gota artrítica han sido tratados con Ilaris a dosis que van de 10 mg a 300 mg en ensayos clínicos con control activo, doble-ciego, aleatorizado de 24 semanas de duración. Más de 250 pacientes han sido tratados con una dosis recomendada de 150 mg en ensayos clínicos de Fase II y III (ver sección 5.1).
Tabla de reacciones adversas
Las reacciones adversas se presentan según el sistema de clasificación por órganos y sistemas MedDRA. Dentro de cada órgano y sistema, las reacciones adversas se ordenan por frecuencia, las más comunes primero. Las categorías de frecuencia se definen utilizando los siguientes criterios: muy frecuentes (> 1/10); frecuentes (> 1/100 a < 1/10); poco frecuentes (> 1/1.000 a < 1/100); raras (> 1/10.000 a < 1/1.000); muy raras (< 1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
Tabla 1 Tabla de reacciones adversas en CAPS, AIJS y gota artrítica
|
Clasificación de órganos del sistema MedDRA |
CAPS |
AIJS |
Gota artrítica |
|
Infecciones e infestaciones | |||
|
Muy frecuentes |
Nasofaringitis |
Neumonía Gastroenteritis Infección del tracto urinario Infección vírica Sinusitis Rinitis Faringitis Tonsilitis Nasofaringitis Infección del tracto respiratorio superior |
Neumonía Bronquitis Gastroenteritis Infección del tracto urinario Síntomas gripales Celulitis Sinusitis Infección de oído Faringitis Nasofaringitis Infección del tracto respiratorio superior |
|
Frecuentes |
Infección del tracto urinario Infección del tracto respiratorio superior Infección vírica | ||
|
Trastornos del sistema nervioso | |||
|
Frecuentes |
Mareo/vértigo |
Mareo/vértigo | |
|
Trastornos gastrointestinales | |||
|
Muy frecuentes |
Dolor abdominal (superior) | ||
|
Poco frecuentes |
Enfermedad por reflujo gastroesofágico | ||
|
Trastornos de la |
piel y del tejido subcutáneo | ||
|
Muy frecuentes |
Reacción en el lugar de inyección |
Reacción en el lugar de inyección | |
|
Frecuentes |
Reacción en el lugar de inyección | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |||
|
Muy frecuentes |
Artralgia | ||
|
Frecuentes |
Dolor muscular |
Dolor de espalda | |
|
Trastornos generales y alteraciones en el lugar de administración | |||
|
Frecuentes |
Fatiga/astenia | ||
|
Exploraciones complementarias | |||
|
Muy frecuentes |
Disminución del aclaramiento renal* Proteinuria # Leucopenia | ||
|
Frecuentes |
Neutropenia | ||
|
* en base al aclaramiento de creatinina estimado, la mayoría fueron transitorios # la mayoría fueron trazas transitorias hasta 1 + proteína urinaria positiva por varilla | |||
En un subgrupo de pacientes con AIJS adultos jóvenes de 16 a 20 años de edad (n=31), el perfil de seguridad de Ilaris fue consistente con el observado en pacientes con AIJS menores de 16 años de edad. Se espera que el perfil de seguridad en pacientes con ESA sea similar al de pacientes con AIJS de acuerdo con los informes de la literatura.
Descripción de reacciones adversas seleccionadas
Datos a largo plazo y anomalías analíticas en pacientes con CAPS
Durante los ensayos clínicos con Ilaris en pacientes con CAPS aumentaron los valores medios de hemoglobina y los de leucocitos, neutrófilos y plaquetas disminuyeron.
Raramente, se han observado elevaciones de las transaminasas en pacientes de CAPS.
En pacientes con CAPS tratados con Ilaris se han observado elevaciones moderadas y asintomáticas de la bilirrubina sérica sin incremento concomitante de transaminasas.
En los estudios abiertos, a largo plazo, con escalado de dosis, en el grupo de dosis de 600 mg o 8 mg/kg se notificaron más frecuentemente casos de infecciones (gastroenteritis, infección del tracto respiratorio, infección del tracto respiratorio superior), vómitos y mareo, que en otros grupos de dosis.
Anomalías analíticas en pacientes con AIJS Hematología
En el programa global de AIJS, se notificó una disminución transitoria del recuento de glóbulos blancos (RGB) < 0,8 x LLN en 33 pacientes (16,5%).
En el programa global de AIJS, se notificó una disminución transitoria en el recuento absoluto de neutrófilos (RAN) de menos de 1 x 109/l en 12 pacientes (6,0%).
En el programa global de AIJS, se observaron disminuciones transitorias en el recuento de plaquetas (<LIN) en 19 pacientes (9,5%).
ALT / AST
En el programa global de AIJS, se notificaron una ALT alta y/o una AST > 3 x por encima del límite normal (LSN) en 19 pacientes (9,5%).
Anomalías analíticas en _pacientes con gota artrítica Hematología
Se notificó disminución del recuento de glóbulos blancos (RGB)< 0,8 x límite inferior de la normalidad (LIN) en 6,7% de los pacientes tratados con Ilaris comparado con el 1,4% de los pacientes tratados con acetónido de triamcinolona. La disminución del recuento absoluto de neutrófilos (RAN) a menos de 1 x 109/l fue reportado en el 2% de los pacientes en los ensayos comparativos. Se observaron casos aislados de recuento de RAN < 0,5 x 109/l (ver sección 4.4).
En los ensayos clínicos con control activo se observó una disminución leve (< LIN y > 75 x 109/l) y transitoria en el recuento de plaquetas de una incidencia mayor (12,7%) con Ilaris versus el comparador (7,7%) en pacientes con gota artrítica.
Acido úrico
En los ensayos clínicos comparativos en gota artrítica se observó un incremento transitorio de ácido úrico (0,7 mg/dl a las 12 semanas y 0,5 mg/dl a las 24 semanas) después del tratamiento con Ilaris. En otro ensayo, entre pacientes que estaban iniciando tratamiento con ULT, se observó incremento de ácido úrico. El incremento de ácido úrico no se observó en los ensayos clínicos en la población que no tenía gota (ver sección 5.1).
ALT/AST
Se observó un incremento medio y mediano en transaminasa (ALT) de 3,0 U/l y 2,0 U/l, respectivamente, y de aspartato transaminasa (AST) de 2,7 U/l y 2,0 U/l, respectivamente, desde el inicial a la finalización del ensayo en el grupo tratado con Ilaris versus el grupo(s) tratado con acetónido de triamcinolona, sin embargo la incidencia de cambios clínicamente significativos (> 3 x el límite superior normal (LSN)) fue mayor en pacientes tratados con acetónido de triamcinolona (2,5% para ambos AST y ALT) comparado con los pacientes tratados con Ilaris (1,6% para ALT y 0,8% para AST).
Triglicéridos
En los ensayos clínicos de gota artrítica con controlador activo, hubo un incremento medio en triglicéridos de 33,5 mg/dl en los pacientes tratados con Ilaris comparado con una modesta disminución de -3,1 mg/dl con acetónido de triamcinolona. La incidencia de pacientes con una elevación de triglicéridos > 5 x límite superior de la normalidad (LSN) fue 2,4% con Ilaris y 0,7% con acetónido de triamcinolona. Se desconoce el significado clínico de este hallazgo.
Población pediátrica
En los estudios se incluyeron 80 pacientes pediátricos con CAPS (2-17 años de edad). En general, no hubo diferencias clínicas significativas en el perfil de seguridad y tolerancia de Ilaris en los pacientes pediátricos comparados con la población general de CAPS (compuesta por pacientes adultos y pediátricos, N=211), incluyendo la frecuencia total y la gravedad de los episodios de infección. Las infecciones del tracto respiratorio superior fueron las infecciones notificadas con mayor frecuencia.
Adicionalmente, se evaluaron 6 pacientes pediátricos menores de 2 años de edad en un ensayo clínico abierto de pequeño tamaño. El perfil de seguridad de Ilaris fue similar al observado en pacientes a partir de 2 años de edad.
Pacientes de edad avanzada
No se ha observado diferencias significativas en el perfil de seguridad en pacientes > 65 años de edad. Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Los casos notificados por sobredosis son limitados. En los primeros ensayos clínicos, pacientes y voluntarios sanos recibieron dosis tan altas como 10 mg/kg, administradas por vía intravenosa o subcutánea, sin evidencia de toxicidad aguda.
En caso de sobredosis, se recomienda controlar la aparición de signos o síntomas de reacciones adversas en el paciente y e instaurar, en caso necesario, un adecuado tratamiento sintomático inmediatamente.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Inmunosupresores, inhibidores de interleucina, código ATC: L04AC08 Mecanismo de acción
Canakinumab es un anticuerpo monoclonal anti-interleucina humana-1 beta (IL-1 beta) del isotipo IgG1/K completamente humano. Canakinumab se une con alta afinidad específicamente a la IL-1 beta humana y neutraliza su actividad biológica mediante el bloqueo de la interacción con los receptores IL-1, lo que permite prevenir la activación del gen inducida por IL-1 beta y la producción de mediadores inflamatorios.
Efectos farmacodinámicos
CAPS
En estudios clínicos, los pacientes de CAPS que presentan una sobreproducción incontrolada de IL-1 beta muestran una respuesta rápida al tratamiento con canakinumab, es decir, los parámetros de laboratorio tales como niveles altos de la proteína C-reactiva (PCR) y de la proteína A amiloidea (AAS), el recuento elevado de neutrófilos y plaquetas y la leucocitosis se normalizaron rápidamente.
Enfermedad de Still (ESA y AIJS)
La Enfermedad de Still del Adulto y la Artritis Idiopática Juvenil Sistémica son enfermedades graves autoinflamatorias producidas por la inmunidad innata a través de las citoquinas pro-inflamatorias, siendo la principal, IL-1-beta.
Las características comunes de ESA y AIJS incluyen fiebre, erupción cutánea, hepatoesplenomegalia, linfadenopatía, poliserositis y artritis. El tratamiento con canakinumab mejoró rápidamente y de forma sostenida las características articulares y sistémicas de AIJS con una reducción significativa del número de articulaciones inflamadas, rápida resolución de la fiebre y la reducción de reactantes de la fase aguda en la mayoría de los pacientes (ver Eficacia clínica y de seguridad).
Gota artrítica
Un ataque de gota artrítica es producido por cristales de urato (urato monosódico monohidratado) en las articulaciones y en los tejidos de alrededor de éstas, que desencadenan la producción de IL-1 beta por parte de los macrófagos residentes, mediante el complejo «NALP3 inflamasoma». La activación de macrófagos y la sobreproducción de IL-1 beta concomitante producen una respuesta inflamatoria dolorosa aguda. Otros activadores del sistema inmune innato, tales como agonistas endógenos de los receptores de tipo «toll-like» pueden contribuir a la activación transcripcional del gen IL-1 beta, iniciando un ataque de gota artrítica. Después del tratamiento con canakinumab, los marcadores inflamatorios CRP o SAA y los signos de inflamación aguda (p. ej. dolor, hinchazón, enrojecimiento) en la articulación afectada remiten rápidamente.
Eficacia clínica y seguridad
CAPS
La eficacia y la seguridad de Ilaris han sido demostradas en pacientes con diversos grados de gravedad de la patología y distintos fenotipos de CAPS (incluidos FCAS/FCU, MWS, y NOMID/CINCA). En el estudio pivotal sólo se incluyeron pacientes con mutación NLRP3 confirmada.
En el estudio de fase I/II, el tratamiento con Ilaris mostró un inicio de acción rápido ya sea con la remisión o con una mejoría clínicamente significativa de los síntomas en un día tras la administración. Los parámetros de laboratorio tales como los niveles altos de PCR y AAS, el recuento elevado de neutrófilos y plaquetas, se normalizaron rápidamente al cabo de unos días de la inyección de Ilaris.
El estudio pivotal consistió en un estudio multicéntrico de 48 semanas y tres partes: un periodo abierto de 8 semanas (Parte I), un periodo aleatorizado, doble ciego, controlado con placebo, de 24 semanas y opción de retirada (Parte II), seguido de un periodo abierto de 16 semanas (Parte III). El objetivo del estudio fue valorar la eficacia, seguridad y tolerabilidad de Ilaris (150 mg ó 2 mg/kg cada 8 semanas) en pacientes con CAPS.
- Parte I: Se observó una respuesta clínica y de biomarcadores completa a Ilaris (definida por el objetivo compuesto de la valoración global de la autoinflamación y la enfermedad cutánea por el médico < mínima y los valores de PCR o AAS < 10 mg/litro) en el 97% de los pacientes durante los 7 primeros días de tratamiento. Se apreciaron mejorías significativas en la valoración clínica por parte del médico de la actividad de la enfermedad autoinflamatoria: en la valoración global de la actividad de la enfermedad autoinflamatoria, la valoración de la enfermedad cutánea (erupción cutánea de tipo urticariforme), artralgias, mialgias, cefalea/migraña, conjuntivitis, fatiga/malestar general, valoración de otros síntomas relacionados y valoración de los síntomas por parte del paciente.
- Parte II: En el periodo con opción de retirada del estudio pivotal, la variable principal se definió como la proporción de pacientes con recaída/agravamiento: ninguno de los pacientes (0%) tratados con Ilaris empeoró frente al 81% de los pacientes asignados al grupo placebo.
- Parte III: Los pacientes tratados con placebo en la Parte II que empeoraron, recuperaron y mantuvieron la respuesta clínica y serológica tras su incorporación en la fase de extensión abierta con Ilaris.
Tabla 2 Resumen tabulado de la eficacia en el estudio pivotal fase III, controlado con placebo y opción de retirada (Part II)
|
Estudio fase III, pivotal controlado con placebo y opción de retirada (Parte II) | |||
|
Ilaris N=15 N(%) |
Placebo N=16 N(%) |
p-valor | |
|
Variable principal (agravamiento) Proporción de pacientes con empeoramiento en la |
0 (0%) |
13 (81%) |
< 0,001 |
|
Parte II Marcadores de la inflamación* Proteína C reactiva, mg/l |
1,10 (0,40) |
19,93 (10,50) |
< 0,001 |
|
Proteína A amiloidea, mg/l |
2,27 (-0,20) |
71,09 (14,35) |
0,002 |
|
* cambio medio (mediana) desde el inicio de la Parte II | |||
Se realizaron dos estudios de fase III a largo plazo, abiertos y no controlados. Uno fue un estudio de seguridad, tolerancia y eficacia con canakinumab en pacientes con CAPS. La duración total del tratamiento oscilaba desde los 6 meses a los 2 años. El otro fue un estudio abierto con canakinumab para evaluar la eficacia y seguridad en pacientes japoneses con CAPS durante 24 semanas, con una fase de extensión hasta 48 semanas. El objetivo primario fue evaluar la proporción de pacientes sin recaídas a las 24 semanas, incluyendo aquellos pacientes a los que se les incrementó la dosis.
En el análisis agrupado de la eficacia para estos dos estudios, el 65,6% de los pacientes que no habían sido tratados previamente con canakinumab alcanzaron una respuesta completa con 150 mg o 2 mg/kg, mientras que el 85,2% de los pacientes alcanzaron una respuesta completa a cualquier dosis. De los pacientes tratados con 600 mg o 8 mg/kg (o incluso superior), el 43,8% alcanzaron una respuesta completa. Menos pacientes de 2 a < 4 años de edad alcanzaron una respuesta completa (57,1%) comparado con pacientes pediátricos de más edad o pacientes adultos. De los pacientes que alcanzaron una respuesta completa, el 89,3% mantuvieron la respuesta sin recaídas.
La experiencia de los pacientes individuales que alcanzaron una respuesta completa después de un escalado de dosis de 600 mg (8 mg/kg) cada 8 semanas sugiere que para pacientes que no alcanzan una respuesta completa o que no mantienen una respuesta completa con las dosis recomendadas (150 mg o 2 mg/kg para pacientes > 15 kg y < 40 kg), una dosis superior puede ser beneficiosa. En pacientes de 2 a < 4 años de edad y en pacientes con síntomas NOMID/CINCA se administró con frecuencia una dosis incrementada comparado con FCAS y MWS.
Población pediátrica
Los estudios de CAPS con Ilaris comprenden un total de 80 pacientes pediátricos de edades comprendidas entre 2 y 17 años (aproximadamente la mitad de ellos han sido tratados en base a mg/kg). En general, no hubo diferencias clínicamente significativas en el perfil de eficacia, seguridad y tolerabilidad de Ilaris en pacientes pediátricos comparado con la población general de CAPS. La mayoría de los pacientes pediátricos alcanzaron la mejora en los síntomas clínicos y los marcadores objetivos de la inflamación (p.ej. SAA y CRP).
Se realizó un ensayo abierto de 56 semanas para evaluar la eficacia, seguridad y tolerancia de Ilaris en pacientes pediátricos con CAPS <4 años de edad. Se evaluaron 17 pacientes (incluyendo 6 pacientes menores de 2 años), utilizando dosis de inicio basadas en el peso de 2-8 mg/kg. En el ensayo también se evaluó el efecto de canakinumab sobre el desarrollo de anticuerpos a las vacunas estándares para niños. No se observaron diferencias en cuanto a seguridad o eficacia en pacientes menores de 2 años comparado con pacientes a partir de 2 años de edad. Todos los pacientes que recibieron vacunas no vivas según la práctica clínica habitual infantil (N=7), desarrollaron niveles protectores de anticuerpos.
Enfermedad de Still AIJS
Se evaluó la eficacia de Ilaris para el tratamiento de SJIA activo en dos estudios pivotales (G2305 y G2301). Los pacientes reclutados fueron de 2 a < 20 años de edad (edad media de 8,5 años y duración media de la enfermedad de 3,5 años al inicio del estudio) y tenían la enfermedad activa definida como > 2 articulaciones con artritis activa, fiebre y elevación de la CRP.
Estudio G2305
El estudio G2305 fue un estudio aleatorizado, doble ciego, controlado con placebo, de 4 semanas para la evaluación de la eficacia a corto plazo de Ilaris en 84 pacientes aleatorizados que recibieron una dosis única de 4 mg/kg (hasta 300 mg) o de placebo Ilaris. El objetivo primario fue el porcentaje de pacientes que en el día 15 lograron una mejoría mínima del 30% en el criterio de respuesta American College of Rheumatology (ACR) adaptado para incluir ausencia de fiebre. El tratamiento con Ilaris mejoró todos los resultados de respuesta ACR pediátricos en comparación con placebo en los días 15 y 29 (Tabla 3).
Tabla 3 Respuesta ACR pediátricos y estado de la enfermedad en el día 15 y 29
|
Día 15 |
Día 29 | |||
|
Ilaris |
Placebo |
Ilaris |
Placebo | |
|
N=43 |
N=41 |
N=43 |
N=41 | |
|
ACR30 |
84% |
10% |
81% |
10% |
|
ACR50 |
67% |
5% |
79% |
5% |
|
ACR70 |
61% |
2% |
67% |
2% |
|
ACR90 |
42% |
0% |
47% |
2% |
|
ACR100 |
33% |
0% |
33% |
2% |
|
Enfermedad inactiva |
33% |
0% |
30% |
0% |
La diferencia de tratamiento en todos los resultados ACR fue significativa (p < 0.0001)
Los resultados de los componentes del ACR adaptado para pediatría que incluye manifestaciones sistémicas y artríticas, fueron consistentes con los resultados generales de respuesta ACR. El cambio medio en el número de articulaciones con artritis activa y el rango de movimiento limitado, en el día 15, eran del 67% y del 73% para Ilaris (N=43) respectivamente, en comparación con un cambio medio del 0% y 0% para placebo (N=41). El cambio medio en la puntuación de dolor del paciente (0-100 mm escala analógica visual) en el día 15 fue de -50,0 mm para Ilaris (N=43) en comparación con +4,5 mm para placebo (N=25). El cambio medio en la puntuación de dolor entre los pacientes tratados con Ilaris fue consistente en el día 29.
Estudio G2301
Estudio G2301 fue un estudio aleatorizado, doble ciego, controlado con placebo, con diseño de retirada en la prevención de brotes con Ilaris. El estudio consistió en dos partes con dos objetivos primarios independientes (retirada de esteroides con éxito y tiempo hasta el brote). En la parte I (abierto) se reclutaron 177 pacientes y recibieron 4 mg/kg (hasta 300 mg) de Ilaris administrado cada 4 semanas durante un máximo de 32 semanas. Los pacientes en la parte II (doble ciego) recibieron
4 mg/kg de Ilaris o placebo cada 4 semanas hasta que se produjeron 37 brotes.
Retirada de corticoides:
Del total de 128 pacientes que entraron en la parte I tomando corticoides, 92 intentaron disminuir los corticoides. Cincuenta y siete (62%) de los 92 pacientes que intentaron disminuir la dosis lo consiguieron y 42 (46%), discontinuaron los corticoides.
Tiempo hasta el brote:
Los pacientes que toman Ilaris en la parte II tenían un 64% menos de riesgo de brotes, en comparación con el grupo placebo (tasa de riesgo de 0,36, IC del 95%: 0,17 a 0,75, p = 0,0032). Sesenta y tres de los 100 pacientes que entraron en la segunda parte, ya fueran asignados a placebo o canakinumab, no experimentaron un brote durante el período de observación (hasta un máximo de 80 semanas).
Relacionada con la salud y la calidad de vida los resultados de los estudios G2305 y G2301 El tratamiento con Ilaris mejoró clínicamente de manera significativa la función física de los pacientes y la calidad de vida. En el estudio G2305, la mejoría media del Cuestionario de Evaluación de Salud Infantil de Mínimos Cuadrados fue de 0,69 para Ilaris vs placebo, lo que representa una diferencia mínima clínicamente importante de 3,6 veces de 0,19 (p=0,0002). La mejoría media al final de la primera parte del estudio G2301 fue de 0,88 (79%). Se notificaron mejorías estadísticamente significativas en el Cuestionario de Salud Infantil - en los resultados PF50 de Ilaris vs placebo en el estudio G2305 (p=0,0012 físicamente, p=0,0017 estado psicosocial).
Análisis de Eficacia
Para evaluar el mantenimiento de la eficacia se combinaron los datos de los estudios G2305, G2301 y el estudio de extensión de las primeras 12 semanas de tratamiento con Ilaris. Estos datos mostraron mejoras similares desde el inicio hasta la semana 12 en las respuestas ACR adaptado pediátrico y de sus manifestaciones a los observados en el estudio controlado con placebo (G2305). En la semana 12, las respuestas ACR adaptado pediátrico 30, 50, 70, 90 y 100 fueron del 70%, 69%, 61%, 49% y 30%, respectivamente, y el 28% de los pacientes tenían enfermedad inactiva (N=178).
La eficacia observada en los estudios G2305 y G2301 se mantuvo en el estudio abierto de extensión a largo plazo en marcha (datos disponibles a través de la mediana de 49 semanas de seguimiento). En este estudio, 25 pacientes que presentaron una respuesta importante de ACR durante un mínimo de
5 meses, se les redujo su dosis a 2 mg/kg de Ilaris cada 4 semanas y se le dio la dosis reducida (media de 32 semanas, 8-124 semanas) los que mantuvieron una respuesta ACR pediátrico 100 a lo largo del tiempo.
Existe evidencia, aunque limitada, de los ensayos clínicos que indica que los pacientes que no responden a tocilizumab o anakinra, pueden responder a canakinumab.
AIJS en adultos jóvenes y ESA
La eficacia de Ilaris en un subgrupo de pacientes con AIJS adultos jóvenes de 16 a 20 años de edad fue consistente con la eficacia observada en pacientes menores de 16 años de edad. Se espera que el perfil de eficacia en pacientes con ESA sea similar al de pacientes con AIJS de acuerdo con los informes de la literatura.
Gota artrítica
La eficacia de Ilaris para el tratamiento de los ataques agudos de gota artrítica se demostró en dos estudios multicéntricos, controlados con activo, doble ciego, aleatorizados, en pacientes con gota artrítica frecuente (> 3 ataques en los 12 meses previos) que no podían utilizar AINEs o colchicina (debido a contraindicación, intolerancia o falta de eficacia). Estos estudios eran de 12 semanas seguidos de una extensión de 12 semanas doble ciego. Un total 225 pacientes fueron tratados con 150 mg de Ilaris subcutáneo y 229 pacientes fueron tratados con 40 mg de acetónido de triamcinolona (TA) al iniciar el tratamiento y después de experimentar un nuevo ataque. El número medio de ataques de gota artrítica en los 12 meses previos era 6,5. Más del 85% de los pacientes tuvieron comorbilidad, incluyendo hipertensión (60%), diabetes (15%), cardiopatía isquémica (12%), y enfermedad renal crónica en fase > 3 (25%). Aproximadamente un tercio de los pacientes incluidos (76 [33,8%] en el grupo Ilaris y 84 [36,7%] en el grupo acetónido de triamcinolona) tenían incapacidad documentada (intolerancia, contraindicación o ausencia de respuesta) para utilizar tanto AINEs como colchicina. Al inicio del estudio se notificó tratamiento concomitante con ULTs en el 42% de los pacientes.
Las dos variables principales de evaluación fueron: (i) intensidad del dolor de la gota artrítica (escala analógica visual, EAV) a las 72 horas post-dosis, y (ii) tiempo hasta un nuevo ataque de gota artrítica.
Para la población general del estudio, la intensidad del dolor fue menor estadísticamente significativa para Ilaris 150 mg comparado con acetónido de triamcinolona a las 72 horas. Ilaris también disminuyó el riesgo de ataques posteriores (ver Tabla 4).
Los resultados de eficacia en pacientes que no pueden utilizar ni AINEs ni colchicina y que no estaban en tratamiento con ULT, no respondieron al tratamiento con ULT o estaban contraindicados de ULT (N=101) fueron consistentes en la población general del estudio con una diferencia estadísticamente significativa comparado a acetónido de triamcinolona en la intensidad del dolor a las 72 horas (-10,2 mm, p=0,0208) y en la disminución del riesgo de ataques posteriores(Cociente del riesgo 0,39, p=0,0047 a las 24 semanas).
En la Tabla 4 se presentan los resultados de eficacia para un subgrupo más restringido limitado a los pacientes que están utilizando ULT (N=62). El tratamiento con Ilaris produjo una reducción del dolor y disminuyó el riesgo de ataques posteriores en pacientes que estaban utilizando ULT y que no podían utilizar ni AINEs ni colchicina, a pesar de que la diferencia observada durante el tratamiento comparado con acetónido de triamcinolona fue menos pronunciada que con la población de estudio general.
Tabla 4 Eficacia de la población general y en un subgrupo de pacientes que utilizan ULT y que no pueden utilizar ni AINEs ni colchicina
Variable de eficacia Población general del Incapacidad de utilizar tanto
estudio; AINEs como colchicina con
N=454 ULT
_N=62_
Tratamiento de los ataques de gota artrítica medidos por la intensidad del dolor (EAV) a las
Diferencia estimada del promedio de -10,7 -3,8
Mínimos Cuadrados a acetónido de
triamcinolona
IC (-15,4, -6,0) (-16,7, 9,1)
Valor p, 1 cola p < 0,0001* p=0,2798
Reducción del riesgo de ataques posteriores de gota artrítica medida por el tiempo hasta un
nuevo ataque (24 semanas)
Cociente de riesgo a acetónido de 0,44 0,71
triamcinolona
IC (0,32, 0,60) (0,29, 1,77)
Valor p, 1 cola p < 0,0001* p=0,2337
* Representa un valor-p significativamente < 0,025_
Los resultados de eficacia mostraron una incidencia mayor de efectos adversos para canakinumab comparado con acetónido de triamcinolona con el 66% vs el 53% de los pacientes notificando algún efecto adverso y el 20% vs el 10% de los pacientes notificando una infección como reacción adversa después de las 24 semanas.
Pacientes de edad avanzada
En global, el perfil de eficacia, seguridad y tolerabilidad de Ilaris en pacientes de edad avanzada de > 65 años de edad fue comparable a los pacientes < 65 años de edad.
Pacientes en tratamiento con reductores de uratos (ULT)
En los ensayos clínicos, Ilaris se ha administrado de forma segura con ULT. En el conjunto de la población de los estudios, los pacientes con ULT tuvieron una diferencia menos pronunciada durante el tratamiento tanto en la reducción del dolor como en la reducción del riesgo de ataques posteriores de gota artrítica comparado con pacientes que no estaban en tratamiento con ULT.
Inmugenecidad
En aproximadamente 1,5%, 3% y 2% de los pacientes tratados con Ilaris para CAPS, AIJS y gota artrítica respectivamente, se observaron anticuerpos contra Ilaris. No se detectaron anticuerpos neutralizantes. No se observó una correlación aparente entre el desarrollo de anticuerpos y la respuesta clínica o efectos adversos.
Este medicamento se ha autorizado para CAPS en «circunstancias excepcionales». Esta modalidad de aprobación significa que debido a la rareza de la enfermedad no ha sido posible obtener información completa de este medicamento. La Agencia Europea de Medicamentos revisará anualmente la información nueva de este medicamento que pueda estar disponible, y esta Ficha Técnica o Resumen de las Características del Producto (RCP) se actualizará cuando sea necesario.
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Ilaris en uno o más grupos de la población pediátrica con Síndromes Periódicos Asociados a la Criopirina (CAPS) y Artritis idiopática juvenil (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Ilaris en los diferentes grupos de la población pediátrica en gota artrítica (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 Propiedades farmacocinéticas
CAPS
Absorción
La concentración sérica máxima (Cmax) de canakinumab se produjo aproximadamente a los 7 días tras una administración única de 150 mg por vía subcutánea a pacientes de CAPS adultos. La semivida terminal media fue de 26 días. Los valores medios de Cmax y AUCinf después de una única dosis subcutánea de 150 mg en un paciente adulto común de CAPS (70 kg) fueron 15,9 pg/ml y 708 pg*d/ml. Se estimó que la biodisponibilidad absoluta de canakinumab administrado por vía subcutánea era del 66%. Los parámetros relativos a la exposición (tales como AUC y Cmáx) se incrementaron de forma proporcional a la dosis en el intervalo de dosis comprendidas entre 0,30 a 10,0 mg/kg por perfusión intravenosa y entre 150 a 600 mg por inyección subcutánea. Los valores previstos de exposición en estado estacionario (Cmin,ss, Cmaxss, AUC,ss,8w) tras la administración de 150 mg subcutánea (ó 2 mg/kg, respectivamente) fueron ligeramente superiores en la categoría de peso 40-70 kg (6,6 pg/ml, 24,3 pg/ml, 767 pg*d/ml) en comparación con las categorías de peso < 40 kg (4,0 pg/ml, 19,9 pg/ml, 566 pg*d/ml) y > 70 kg (4,6 pg/ml, 17,8 pg/ml, 545 pg*d/ml). El coeficiente de acumulación esperado fue de 1,3 veces tras 6 meses de administración subcutánea de 150 mg de canakinumab cada 8 semanas.
Distribución
Canakinumab se une a la IL-1 beta sérica. El volumen de distribución (Vss) de canakinumab varió según el peso corporal. Se estimó que es de 6,2 litros en un paciente de CAPS de 70 kg de peso.
Eliminación
El aclaramiento aparente (CL/F) de canakinumab se incrementa con el peso corporal. Se estimó que era 0,17 l/d en un paciente CAPS de 70 kg de peso y 0,11 l/d en un paciente AIJS de 33 kg de peso. Tras tener en cuenta las diferencias de peso corporal, no se observaron diferencias clínicamente significativas en las propiedades farmacocinéticas de canakinumab entre los pacientes CAPS y los AIJS.
No se observó aclaramiento acelerado o modificación con el tiempo de las propiedades farmacocinéticas de canakinumab tras la administración de dosis repetidas. Tampoco se observaron diferencias por edad y sexo en los parámetros farmacocinéticos una vez efectuada la corrección en función del peso.
Enfermedad de Still (ESA y AIJS)
La biodisponibilidad en pacientes con AIJS no se ha determinado de forma independiente. El aclaramiento aparente por kg de peso corporal (CL/F por kg) fue comparable entre la población de AIJS y la de CAPS (0,004 l/d por kg). El volumen aparente de distribución por kg (V/F por kg) fue de 0,14 l/kg.
El coeficiente de acumulación de canakinumab fue de 1,6 veces tras la administración subcutánea de 4 mg/kg de canakinumab cada 4 semanas en pacientes con AIJS. El estado estacionario se alcanzó a los 110 días. La media prevista total (±SD) para Cminss, Cmaxss y AUC,ss4w fue 14,7±8,8 pg/ml, 36,5±14,9 pg/ml y 696,1±326,5 pg*d/ml, respectivamente.
Las AUCss4w en cada grupo de edad fueron de 692, 615, 707 y 742 pg*d/ml de 2-3, 4-5, 6-11, y 12-19 años respectivamente. Cuando se estratificó por peso, se observó un menor exposición media (30-40%) para Cminss (11,4 vs 19 pg/ml) y AUCss (594 vs 880 pg*d/ml) para la categoría de menor peso (< 40 kg) vs la categoría de más peso (> 40 kg).
De acuerdo con el análisis del modelo farmacocinético poblacional, la farmacocinética de canakinumab en pacientes con AIJS adultos jóvenes de 16 a 20 años de edad fue similar a la de pacientes menores de 16 años de edad. Las exposiciones predecibles de canakinumab en estado estacionario al nivel de dosis de 4 mg/kg (máximo 300 mg) en pacientes mayores de 20 años de edad fueron comparables a las observadas en pacientes con AIJS menores de 20 años de edad.
Pacientes con gota artrítica
La biodisponibilidad en pacientes con gota artrítica no ha sido determinada independientemente. El aclaramiento aparente por kg de peso corporal (CL/F por kg) fue comparable entre la población de gota y de CAPS (0,004 l/d/kg). La exposición media en un paciente común de gota artrítica (93 kg) después de una única dosis subcutánea de 150 mg (Cmax: 10,8 pg/ml y AUCinf: 495 pg*d/ml) fue menor que en un paciente común de CAPS de 70 kg (15,9 pg/ml y 708 pg*d/ml). Esto es consistente con el incremento observado de CL/F con el peso corporal.
El coeficiente de acumulación de canakinumab fue de 1,1 veces tras la administración subcutánea de 150 mg de canakinumab cada 12 semanas.
Población pediátrica
Las concentraciones máximas de canakinumab se alcanzaron entre los 2 y los 7 días (Tmax) después de una administración subcutánea única de 150 mg ó 2 mg/kg de canakinumab en pacientes pediátricos a partir de 4 años de edad. La semivida terminal osciló entre 22,9 y 25,7 días, similar a las propiedades farmacocinéticas observadas en adultos. En base al análisis de los modelos de población farmacocinético, la farmacocinética de canakinumab en niños de 2 a < 4 años fue similar a la de los pacientes de 4 años de edad o mayores. El coeficiente de absorción subcutánea se estima que disminuye con la edad y aparentemente es más rápido en pacientes más jóvenes. En consecuencia, Tmax más corta (3,6 días) en pacientes jóvenes con AIJS (2-3 años) en comparación con pacientes mayores de AISJ (12-19 años; Tmax 6 días). No afecta la biodisponibilidad (AUCss).
Un análisis farmacocinético adicional mostró que la farmacocinética de canakinumab en 6 pacientes pediátricos con CAPS, menores de 2 años de edad, era similar a la farmacocinética en pacientes pediátricos entre 2-4 años de edad. De acuerdo al análisis de los modelos de población farmacocinético, la exposición esperada después de una dosis de 2 mg/kg fue comparable entre los grupos de edad pediátrica con CAPS, pero fue aproximadamente el 40% inferior en pacientes pediátricos de muy bajo peso corporal (p.ej., 10 kg) comparado con pacientes adultos (dosis de 150 mg). Esto es consistente con las observaciones de exposiciones más elevadas en grupos de peso corporal más elevado en pacientes con CAPS.
Las propiedades farmacocinéticas de poblaciones pediátricas CAPS y AIJS son similares.
Población en edad avanzada
No se observaron cambios en los parámetros farmacocinéticos en base al aclaramiento o al volumen de distribución entre pacientes de edad avanzada y pacientes adultos < 65 años de edad.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios de reactividad cruzada, toxicidad a dosis repetidas, inmunotoxicidad, toxicidad para la reproducción y estudios de toxicidad juvenil realizados con canakinumab o con un anticuerpo murino anti IL-1 beta murina.
Como canakinumbab se une a la IL-1 beta del hombre y monos babuinos (C. jacchus) con afinidad similar, la seguridad de canakinumab se ha investigado en estos animales. No se observaron efectos adversos de canakinumab después de la administración dos veces por semana durante 26 semanas a monos babuinos o en el estudio de toxicidad de desarrollo embriofetal en monas embarazadas. Las concentraciones plasmáticas que están bien toleradas en animales exceden como mínimo 42 veces (Cmax) y 78 veces (Cavg) las concentraciones plasmáticas en pacientes pediátricos con CAPS (peso corporal 10 kg) tratados con dosis clínicas de canakinumab hasta 8 mg/kg por vía subcutánea cada 8 semanas. Las concentraciones plasmáticas que se toleran bien en animales son 62 veces superiores (Cmáx) y 104 veces (Cavg) a las concentraciones plasmáticas en los pacientes pediátricos con SJIA tratados con hasta 4 mg / kg por vía subcutánea cada 4 semanas. Además, no se detectaron anticuerpos frente a canakinumab en estos estudios. No se demostró que existiera reactividad cruzada inespecífica cuando se aplicó canakinumab a tejidos humanos normales.
No se han realizado estudios de carcinogénesis formales con canakinumab.
En el estudio de desarrollo embrionario en monos babuinos, canakinumab no mostró toxicidad materna, embrionaria o potencial teratogénico cuando se administró durante la organogénesis.
No se apreciaron efectos no deseados de un anticuerpo murino anti IL-1 beta murina en el conjunto global de estudios de reproducción y en animales jóvenes realizados en ratones. El anticuerpo anti-murino IL-1 beta no provocó acontecimientos adversos en el crecimiento fetal o neonatal al administrarse durante la parte final de la gestación, parto y lactancia (ver sección 4.6). La dosis alta utilizada para estos estudios excedió la dosis máxima efectiva en términos de supresión y de la actvidad de IL-1 beta.
En un estudio inmunotoxicológico en ratones con un anticuerpo murino anti 1L-1 beta murino mostró que la neutralización de IL-1 beta no tiene efectos sobre los parámetros inmunes y no deteriora la función inmune de los ratones.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo para solución inyectable:
Sacarosa
Histidina
Histidina hidrocloruro monohidrato Polisorbato 80
Disolvente:
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
3 años.
Desde un punto de vista microbiológico, el producto debería utilizarse inmediatamente tras la reconstitución. Si no se usa inmediatamente, el tiempo y las condiciones antes de uso estarán bajo responsabilidad exclusiva del usuario y no deberían superar 24 horas a 2°C - 8°C.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Polvo: 150 mg de polvo para solución inyectable en un vial de 6 ml (vidrio tipo I) con un tapón (recubierto de goma de clorobutilo) y una cápsula flip-off (aluminio/cápsula de plástico).
Disolvente: 5 ml de agua para preparaciones inyectables en un vial de 6 ml (vidrio tipo I) con un tapón (de goma de clorobutilo recubierto de fluoropolímero) y una cápsula flip-off (aluminio/cápsula de plástico).
Un kit para la inyección de Ilaris contiene 1 vial de polvo para solución inyectable, 1 vial de disolvente, jeringas de 1 ml para la inyección, 1 aguja de seguridad, 2 adaptadores para el vial y
4 toallitas limpiadoras.
6.6 Precauciones especialies de eliminación y otras manipulaciones
Ilaris 150 mg polvo y disolvente para solución inyectable se suministra en un vial de un solo uso para uso individual. Utilice solo el contenido incluido en el kit para inyectar Ilaris. La eliminación del medicamento no utilizado y de todos los materiales o jeringas que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Instrucciones para la reconstitución
Con técnica aséptica, acoplar los adaptadores para el vial con los viales de Ilaris y de disolvente. Transferir 1,0 ml de aire al vial de disolvente, a continuación retirar 1,0 ml de agua para preparaciones inyectables del vial de disolvente utilizando la jeringa que se suministra en el kit. Reconstituir el contenido del vial de Ilaris a temperatura ambiente (normalmente entre 15°C y 25°C) mediante la inyección lenta de 1,0 ml de agua para preparaciones inyectables del vial de disolvente. Remover lentamente el vial con un ángulo de 45° aproximadamente durante 1 minuto y dejar reposar durante
5 minutos. A continuación, girar suavemente el vial de abajo a arriba unas 10 veces. Dejar reposar alrededor de 15 minutos a temperatura ambiente. No agitar. No utilizar si se observan partículas en la solución.
Dar ligeros golpecitos en las paredes laterales del vial para eliminar cualquier posible residuo de líquido del tapón. La solución debería estar libre de partículas visibles y no turbia. La solución puede ser incolora o presentar un ligero color pardo amarillo. Si la solución presenta un color pardo evidente, no debe utilizarse. Si no se utiliza inmediatamente después de la reconstitución, la solución debería mantenerse entre 2°C y 8°C y utilizarse en las próximas 24 horas.
Instrucciones para la administración
Extraer con cuidado la cantidad de volumen requerido en función de la dosis que deba administrarse (0,2 ml a 1,0 ml) e inyectar por vía subcutánea con una aguja de seguridad suministrada en el kit.
Se recomiendan los siguientes lugares de inyección: muslo superior, abdomen, brazo superior o glúteos. Escoja un lugar de inyección diferente cada vez que se inyecte para evitar que le duela. Deben evitarse las áreas de piel lesionada, con hematomas o con erupciones. Debe evitarse inyectar en el tejido cicatricial puesto que podría resultar en una exposición insuficiente a Ilaris.
Eliminación
Después de la inyección el volumen restante se debe desechar innediatamente. Los pacientes y el personal sanitario al cuidado deben ser formados en relación a la eliminación de los viales, jeringas y agujas de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/09/564/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 23 de octubre de 2009 Fecha de la última renovación: 19 de junio de 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
E. OBLIGACIÓN ESPECÍFICA DE LLEVAR A CABO MEDIDAS POSAUTORIZACIÓN EN RELACIÓN CON UNA AUTORIZACIÓN DE COMERCIALIZACIÓN EN CIRCUNSTANCIAS EXCEPCIONALES
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y
FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante del principio activo biológico
Novartis Pharma S.A.S.
Centre de Biotechnologie 8, rue de l’Industrie 68330 Huningue Francia
Nombre y dirección del fabricante responsable de la liberación de los lotes
Novartis Pharma GmbH Roonstrasse 25 D-90429 Nürnberg Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y posteriores actualizaciones publicadas en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado
en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en
cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
• Medidas adicionales de minimización de riesgos
El Titular de la Autorización de Comercialización (TAC) debe asegurar que, antes del lanzamiento, todos los médicos que se espera que receten/usen Ilaris, reciben un paquete informativo que contenga lo siguiente:
• El Resumen de las Características del Producto
• Información para el médico
• Tarjeta de Advertencia para el Paciente
La información para el médico debe contener los siguientes mensajes clave:
• El riesgo de infecciones graves, incluyendo infecciones oportunistas bacterianas, víricas y fúngicas, en pacientes tratados con Ilaris;
• El riesgo de reacciones agudas relacionadas con la inyección;
• Para los pacientes con CAPS: la necesidad de instruir a los pacientes sobre las técnicas adecuadas para la auto-administración cuando el paciente lo desee y sea capaz de hacerlo, e instrucciones para los Profesionales Sanitarios sobre cómo notificar los errores de administración;
• El riesgo de inmunogenicidad identificado o potencial que podría comportar síntomas mediados por el sistema inmune. Para pacientes con gota artrítica: resaltar que el tratamiento intermitente o la reexposicion después de un intervalo largo sin tratamiento puede estar asociado a una respuesta inmune aumentada (o pérdida de tolerancia inmune) a Ilaris y por ello se debe considerar que los pacientes que han vuelto a ser tratados están en riesgo de reacciones de hipersensibilidad;
• Para el tratamiento crónico en CAPS: la necesidad de los Profesionales Sanitarios de realizar una valoración clínica anual de pacientes respecto al aumento potencial de riesgo de aparición de procesos malignos;
• La necesidad de controlar el recuento de neutrófilos antes de iniciar el tratamiento y de nuevo, después de 1 ó 2 meses, puesto que no debe iniciarse el tratamiento con Ilaris en pacientes con neutropenia. Para el tratamiento crónico en pacientes con CAPS o tratamiento repetido en pacientes con gota artrítica, se recomienda controlar el recuento de neutrófilos periódicamente durante el tratamiento;
• Para los pacientes con AIJS, la necesidad de los Profesionales Sanitarios de estar atentos a los síntomas de la infección o empeoramiento de AIJS, ya que éstos se sabe que desencadenan el síndrome de activación macrofágica (SAM), que es un conocido trastorno potencialmente mortal que puede desarrollarse en pacientes con enfermedades reumáticas, especialmente los pacientes con AIJS. Si se produce SAM, o se sospecha, debe evaluarse y tratarse lo antes posible;
• La necesidad de controlar los cambios en el perfil lipídico de los pacientes;
• El desconocimiento de la seguridad de Ilaris en mujeres embarazadas o en periodo de lactancia, por tanto la necesidad por parte de los médicos de comentar con las pacientes los riesgos en caso de quedarse embarazadas o si desean quedarse embarazadas;
• La gestión adecuada del paciente respecto a la interacción con las vacunas;
• La posibilidad de incluir pacientes en el estudio de registro para facilitar la recogida de datos de eficacia y seguridad a largo plazo;
• El papel y el uso de la tarjeta de advertencia para el paciente.
E. OBLIGACIÓN ESPECÍFICA DE LLEVAR A CABO MEDIDAS POSAUTORIZACIÓN EN RELACIÓN CON UNA AUTORIZACIÓN DE COMERCIALIZACIÓN EN CIRCUNSTANCIAS EXCEPCIONALES
Al ser esta una autorización de comercialización en circustancias excepcionales y según lo que establece el artículo 14(8) del Reglamento (CE) 726/2004, el TAC deberá llevar a cabo, dentro del plazo establecido, las siguientes medidas:
|
Área |
Descripción |
Fecha límite |
|
Clínica OE 1 |
Se requiere al solicitante que aporte informes del registro B-Confident (CACZ885D2401), que fue diseñado para aportar datos sobre la seguridad a largo plazo y eficacia del tratamiento de Ilaris en la práctica clínica habitual en los pacientes de CAPS adultos y pediátricos. En estos informes se requiere que el TAC evalúe casos para los cuales hay una pérdida de eficacia (pacientes para los que se ha notificado la discontinuación de Ilaris por ausencia de respuesta en el tratamiento) para determinar si esto es debido a cambios en la PK/PD que aparecen con el tiempo o al desarrollo de anticuerpos (cuando estén disponibles los datos) o en los cuales un ajuste de dosis ha producido una respuesta terapéutica aumentada (pacientes con un escalado de dosis sin discontinuación en ausencia de respuesta terapéutica). |
Anualmente con la reevaluación anual |
|
Se requiere al TAC que aporte, anualmente con la reevaluación anual, actualizaciones sobre las tasas de reclutamiento y de cualquier resultado intermedio. | ||
|
Se deberán incluir pacientes en el Registro hasta que se cumplan las dos condiciones siguientes: un periodo de reclutamiento de 5 años y la inclusión de 200 pacientes. |
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
Ilaris 150 mg polvo para solución inyectable Canakinumab
2. PRINCIPIO(S) ACTIVO(S)
Un vial contiene 150 mg de canakinumab.
3. LISTA DE EXCIPIENTES
Además contiene: sacarosa, histidina, histidina hidrocloruro monohidrato, polisorbato 80.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo para solución inyectable. 1 vial de polvo.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía subcutánea.
Para un único uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/09/564/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Ilaris 150 mg
1. NOMBRE DEL MEDICAMENTO
Ilaris 150 mg polvo para solución inyectable Canakinumab
2. PRINCIPIO(S) ACTIVO(S)
Un vial contiene 150 mg de canakinumab.
3. LISTA DE EXCIPIENTES
Además contiene: sacarosa, histidina, histidina hidrocloruro monohidrato, polisorbato 80.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo para solución inyectable.
Envase múltiple que contiene 4 envases intermedios cada uno conteniendo 1 vial de polvo.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía subcutánea.
Para un único uso.
Leer el prospecto antes de utilizar el medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/09/564/002
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Ilaris 150 mg
1. NOMBRE DEL MEDICAMENTO
Ilaris 150 mg polvo para solución inyectable Canakinumab
2. PRINCIPIO(S) ACTIVO(S)
Un vial contiene 150 mg de canakinumab.
3. LISTA DE EXCIPIENTES
Además contiene: sacarosa, histidina, histidina hidrocloruro monohidrato, polisorbato 80.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo para solución inyectable.
Componente del envase múltiple que contiene 4 envases intermedios cada uno conteniendo 1 vial.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía subcutánea.
Para un único uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/09/564/002
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Ilaris 150 mg
ETIQUETA DEL VIAL_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Ilaris 150 mg polvo para solución inyectable Canakinumab
Vía subcutánea después de la reconstitución
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
150 mg
6. OTROS
1. NOMBRE DEL MEDICAMENTO
Ilaris 150 mg polvo y disolvente para solución inyectable Canakinumab
2. PRINCIPIO(S) ACTIVO(S)
Un vial contiene 150 mg de canakinumab.
3. LISTA DE EXCIPIENTES
Polvo: sacarosa, histidina, histidina hidrocloruro monohidrato, polisorbato 80. Disolvente: agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable.
1 vial de polvo + 1 vial de disolvente + jeringa de 1 ml para la inyección + 1 aguja de seguridad +
2 adaptadores para el vial + 4 toallitas limpiadoras.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía subcutánea.
Para un único uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/09/564/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Ilaris 150 mg
1. OTROS
Contenido

1 vial de polvo
1 vial de disolvente
1 jeringa de seguridad
2 adaptadores para el vial
4 toallitas limpiadoras
Jeringa de 1 ml para la inyección
ETIQUETA DEL VIAL - POLVO_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Ilaris 150 mg polvo para solución inyectable Canakinumab
Vía subcutánea después de la reconstitución
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
150 mg
6. OTROS
ETIQUETA DEL VIAL - DISOLVENTE
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Disolvente para Ilaris
Agua para preparaciones inyectables
|
2. |
FORMA DE ADMINISTRACIÓN |
|
3. |
FECHA DE CADUCIDAD |
|
EXP |
4. NÚMERO DE LOTE
|
Lot | |
|
5. |
CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES |
|
5 ml | |
|
6. |
OTROS |
B. PROSPECTO
Prospecto: información para el usuario
Ilaris 150 mg polvo para solución inyectable
Canakinumab
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Ilaris y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Ilaris
3. Cómo usar Ilaris
4. Posibles efectos adversos
5. Conservación de Ilaris
6. Contenido del envase e información adicional
1. Qué es Ilaris y para qué se utiliza Qué es Ilaris
Ilaris contiene el principio activo canakinumab, un anticuerpo monoclonal que pertenece al grupo farmacoterapéutico de los inhibidores de interleucinas. En el organismo bloquea la actividad de una sustancia llamada interleucina-1 beta (IL-1 beta), que se encuentra a niveles elevados en las enfermedades inflamatorias tales como los Síndromes Periódicos Asociados a la Criopirina (CAPS), la enfermedad de Still incluyendo la Enfermedad de Still del Adulto (EAS) y la Artritis Idiopática Juvenil Sistémica (AIJS) y gota artrítica.
Ilaris se utiliza para el tratamiento de los Síndromes Periódicos Asociados a Criopirina (CAPS), la enfermedad de Still incluyendo la Enfermedad de Still del Adulto (EAS) y la Artritis Idiopática Juvenil Sistémica (AIJS) y gota artrítica.
Para qué se utiliza Ilaris
Síndromes Periódicos Asociados a la Criopirina
Ilaris se utiliza en adultos, adolescentes y niños a partir de 2 años y con un peso de 7,5 kg o superior, para tratar las siguientes enfermedades autoinflamatorias que se conocen en conjunto con el nombre de Síndromes Periódicos Asociados a la Criopirina (CAPS):
- Síndrome de Muckle-Wells (MWS),
- Enfermedad Neonatal Multisistémica Inflamatoria (NOMID), también conocida como Síndrome Infantil Neurológico, Cutáneo y Articular Crónico (CINCA),
- Manifestaciones graves del Síndrome Autoinflamatorio Familiar inducido por el Frío (FCAS) / Urticaria Familiar Fría (FCU) que presente signos y síntomas más allá de la erupción de la piel de tipo urticaria inducido por el frío.
En pacientes con CAPS, el organismo produce una cantidad excesiva de IL-1 beta. Esto puede ocasionar síntomas tales como fiebre, dolor de cabeza, fatiga, erupción cutánea o dolores en las articulaciones y músculos. Mediante el bloqueo de la actividad de IL-1 beta, Ilaris puedefacilitar la mejoría de estos síntomas.
Enfermedad de Still
Ilaris se utiliza en pacientes adultos, adolescentes y niños a partir de los 2 años de edad para el tratamiento de la enfermedad de Still activa incluyendo la Enfermedad de Still del Adulto (ESA) y la Artritis Idiopática Juvenil Sistémica (AIJS) cuando los otros tratamientos no hayan funcionado bien. Ilaris puede utilizarse sólo o en combinación con metrotrexato.
La enfermedad de Still que incluye EAS y AIJS es una enfermedad inflamatoria que puede provocar dolor, hinchazón e inflamación de una o más articulaciones, así como sarpullido y fiebre. La proteina pro-inflamatoria IL-1beta juega un importante papel en el proceso inflamatorio de la enfermedad de Still y Ilaris puede mejorar los signos y síntomas de la enfermedad de Still bloqueando la actividad de IL-1beta.
Gota artrítica
Ilaris se utiliza en adultos para tratar los síntomas de ataques frecuentes de gota artrítica si otros tratamientos no han funcionado suficientemente bien.
La gota artrítica está causada por el depósito en el organismo de unas sustancias químicas llamadas cristales de urato. Estos cristales de urato causan una producción excesiva de IL-1 beta, que a su vez pueden provocar un repentino dolor grave, enrojecimiento, calor, e hinchazón de las articulaciones (conocido como ataque de gota). Mediante el bloqueo de IL-1 beta, Ilaris puede conseguir mejorar estos síntomas.
Si tiene dudas sobre cómo actúa Ilaris o por qué le han recetado este medicamento, consulte a su médico, farmacéutico o enfermero.
2. Qué necesita saber antes de empezar a usar Ilaris
No use Ilaris
- si es alérgico al canakinumab o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
- si tiene, o sospecha que tiene, una infección grave activa.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar Ilaris, si se encuentra en alguna de las siguientes
situaciones:
- si padece alguna infección en la actualidad o si ha tenido infecciones repetidas o sufre alguna enfermedad tal como nivel bajo de góbulos blancos que le hace más vulnerable a tener infecciones.
- si tiene o alguna vez ha tenido tuberculosis o contacto directo con una persona con una infección activa de tuberculosis. Su médico también puede comprobar si tiene tuberculosis utilizando una prueba específica.
- si tiene signos de reacciones alérgicas tales como dificultad para respirar o tragar, náuseas, mareo, erupción en la piel, picor, urticaria, palpitaciones y tensión arterial baja.
- si tiene signos de un trastorno del hígado tales como coloración amarilla en la piel y los ojos, náuseas, pérdida del apetito, color oscuro en la orina y heces blanquecinas.
- si tiene que ponerse alguna vacuna. Se recomienda evitar ser vacunado con un tipo de vacuna (también conocida como vacuna atenuada) mientras está usando Ilaris (ver también «Uso de Ilaris con otros medicamentos (incluyendo vacunas)»).
Enfermedad de Still
- Los pacientes con enfermedad de Still pueden desarrollar una enfermedad llamada síndrome de activación macrofágica (SAM) que puede causar la muerte. Su médico le hará un seguimiento de los posibles factores desencadenantes de SAM, que incluyen infecciones y re-activación de la enfermedad de Still (empeoramiento).
Niños y adolescentes
- CAPS y AIJS: Ilaris puede utilizarse en niños a partir de de 2 años de edad.
- Gota artrítica: Ilaris no está recomendado para niños o adolescentes menores de 18 años de edad.
Uso de Ilaris con otros medicamentos (incluyendo vacunas)
Informe a su médico, farmacéutico o enfermero si está utilizando, ha utilizado recientemente o pudiera tener que utilizar cualquier otro medicamento.
- Vacunas atenuadas: Se recomienda evitar ser vacunado con un tipo de vacuna conocida como vacuna atenuada mientras está en tratamiento con Ilaris. Puede que su médico necesite comprobar su historial de vacunaciones y darle aquellas vacunas que no ha recibido antes de iniciar el tratamiento con Ilaris. Si es necesario que le administren una vacuna atenuada después de iniciar el tratamiento con Ilaris, es aconsejable que se espere durante al menos 3 meses después de la última inyección de Ilaris y antes de la próxima inyección.
- Medicamentos conocidos como los inhibidores del factor de necrosis tumoral (TNF), tales como etanercept, adalimumab o infliximab. Éstos se utilizan principalmente en enfermedades reumáticas y autoinmunes. No deben utilizarse con Ilaris porque puede aumentar el riesgo de infecciones.
Embarazo y lactancia
- No se ha investigado Ilaris en mujeres embarazadas. Debe evitar quedar embarazada y se recomienda utilizar medidas anticonceptivas adecuadas mientras esté tomando Ilaris y durante al menos tres meses después de la última administración. Es importante que informe a su médico si está embarazada, si puede estar embarazada o si está planeando tener un bebé. Su médico le informará acerca del riesgo potencial de utilizar Ilaris durante el embarazo.
- Si recibe canakinumab mientras está embarazada, es importante que informe al pediatra o enfermero antes de que le administren alguna vacuna a su bebé. Su bebé no debe recibir vacunas vivas hasta al menos 16 semanas después de que usted haya recibido su última dosis de canakinumab antes del parto.
- Se desconoce si Ilaris pasa a la leche materna. Su médico le informará acerca de los riesgos potenciales de utilizar Ilaris antes de dar el pecho.
Conducción y uso de máquinas
El tratamiento con Ilaris puede producirle una sensación de que todo da vueltas (mareo/vértigo) o cansancio intenso (astenia). Esto puede afectar a la capacidad de conducir o usar herramientas o máquinas. Si nota la sensación de que todo le da vueltas o se siente cansado, no conduzca ni use herramientas o máquinas hasta que se sienta de nuevo bien.
3. Cómo usar Ilaris
Si tiene CAPS o enfermedad de Still (ESA o AIJS), después de una formación adecuada, usted mismo o su cuidador podría inyectar Ilaris.
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero.
Si tiene gota artrítica, un médico con formación especializada supervisará su tratamiento. Ilaris solo debe ser inyectado por un profesional sanitario.
Mantenga a su médico informado de su enfermedad y de cualquier síntoma antes de utilizar o que se le administre Ilaris (ver sección 2). Su médico puede decidir postponer o interrumpir su tratamiento, solo cuando sea necesario.
Ilaris debe utilizarse por vía subcutánea. Esto significa que se inyecta con una aguja corta dentro del tejipo adiposo por debajo de la piel.
Cuánto Ilaris debe utilizar
Síndromes Periódicos Asociados a la Criopirina La dosis inicial recomendada de Ilaris es:
Adultos, adolescentes y niños de 4 años o más
- 150 mg para pacientes cuyo peso corporal sea más de 40 kg
- 2 mg/kg para pacientes cuyo peso corporal sea de 15 kg o más, hasta un máximo de 40 kg
- 4 mg/kg para pacientes cuyo peso corporal sea de7,5 kg o más, pero menos de 15 kg
Niños de 2 o 3 años
- 4 mg/kg para pacientes cuyo peso corporal sea de 7,5 kg o más Ilaris se utiliza cada 8 semanas como una inyección única.
Si no responde suficientemente al tratamiento a los 7 días, su médico puede considerar darle una dosis repetida de 150 mg ó 2 mg/kg. Si responde suficientemente a ésta, su tratamiento continuará con esta dosis más alta de 300 mg o 4 mg/kg cada 8 semanas. Si no responde suficientemente a la dosis de repetición, se puede considerar una tercera dosis de Ilaris a 300 mg o 4 mg/kg, y si responde suficientemente a ésta, su tratamiento puede continuar con 600 mg y 8 mg/kg cada 8 semanas.
En pacientes que han iniciado con una dosis inicial de 4 mg/kg y que no han respondido suficientemente después de 7 días, se puede considerar una segunda dosis de 4 mg/kg. Si el paciente responde suficientemente a ésta, se puede continuar el tratamiento con 8 mg/kg cada 8 semanas.
Enfermedad de Still (ESA y AIJS)
La dosis recomendada de Ilaris para pacientes con enfermedad de Still (ESA o AIJS) cuyo peso corporal sea de 7,5 kg o más es de 4 mg/kg (hasta un máximo de 300 mg). Ilaris se inyecta bajo la piel cada 4 semanas.
Gota artrítica
Su médico comentará con usted la necesidad de empezar o ajustar un tratamiento con reductores de urato para disminuir el nivel de ácido úrico de su sangre.
La dosis recomendada para Ilaris en pacientes adultos con gota es 150 mg administrada como una única dosis durante un ataque de gota artrítica.
Si necesita otro tratamiento con Ilaris, y con la última dosis obtuvo alivio, debe esperar un mínimo de 12 semanas antes de una siguiente dosis.
Autoinyección de Ilaris o inyección de Ilaris en un niño
Si tiene CAPS o enfermedad de Still (ESA o AIJS), los pacientes o los cuidadores de pacientes de niños pueden poner ellos mismos las inyecciones después de recibir una formación adecuada en relación con la técnica de inyección.
- El paciente o cuidador y el médico decidirán quién pondrá las inyecciones de Ilaris.
- Su médico o enfermero le enseñarán cómo poner las inyecciones de Ilaris.
- No debe tratar de ponerse una inyección si no ha recibido la formación necesaria o no está seguro de cómo hacerlo.
- Ilaris 150 mg polvo para solución inyectable se suministra en un vial de un solo uso para uso individual.
- No reutilice la solución sobrante.
Para tener más información sobre cómo poner las inyecciones de Ilaris, consulte la sección «Instrucciones de uso» al final de este prospecto. Si tiene dudas, hable con su médico, farmacéutico o enfermero.
Duración del tratamiento con Ilaris
- Si tiene CAPS o enfermedad de Still (ESA o AIJS), debe continuar utilizando Ilaris durante el tiempo que su médico le aconseje.
- Si tiene un ataque de gota artrítica, le administrarán una única dosis de Ilaris. Si experimenta un nuevo ataque, su médico puede considerar administrarle una nueva dosis de Ilaris pero no antes de las 12 semanas de haberle administrado la dosis previa.
Si usa más Ilaris del que debe
Si se inyecta accidentalmente más Ilaris del de la dosis recomendada, no es probable que sea grave pero debe informar a su médico, farmacéutico o enfermero lo antes posible.
Si se pone Ilaris antes de lo que debe
- En CAPS, no debe inyectarse Ilaris antes de transcurridas las 8 semanas de la última dosis, a menos que su médico se lo diga.
- En enfermedad de Still (ESA o AIJS) no debe inyectarse Ilaris antes de transcurridas las 4 semanas de la última dosis.
Si se inyecta Ilaris acccidentalmente antes del plazo recomendado, lo debe decir a su médico, farmacéutico o enfermero lo antes posible.
Si olvidó usar Ilaris
Si tiene CAPS o enfermedad de Still (ESA o AIJS) y ha olvidado inyectarse una dosis de Ilaris, la dosis siguiente debe inyectarse tan pronto como lo recuerde. Entonces hable con el médico para acordar cuándo debe inyectare la dosis siguiente. A continuación, debe seguirse con la inyección a los intervalos recomendados como antes.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. La mayoría de efectos adversos son de carácter leve o moderado y desaparecen generalmente a los pocos días o a las pocas semanas de la administración.
Algunos efectos adversos pueden ser graves. Informe a su médico inmediatamente, si experimenta cualquiera de los siguientes efectos adversos:
- Fiebre que se prolongue más de 3 días o cualquier síntoma que puede deberse a una infección. Esto incluye temblores, escalofríos, malestar general, pérdida de apetito, dolores en el cuerpo, por lo general relacionado con la aparición repentina de la enfermedad, dolor de garganta o úlceras en la boca, tos, flema, dolor en el pecho, dificultad para respirar, dolor de oído, dolor de cabeza prolongado y enrojecimiento, calor e hinchazón localizadas en la piel o inflamación del tejido conectivo (celulitis). Estos síntomas pueden deberse a una infección causada por los bajos niveles de glóbulos blancos (denominado leucopenia y neutropenia). Si lo considera necesario su médico puede hacerle análisis de sangre de forma regular.
- Reacciones alérgicas con sarpullido y picor y posiblemente también urticaria, dificultad para respirar o tragar, mareo, conciencia inusual del latido del corazón (palpitaciones) y tensión arterial baja.
Otros efectos adversos de Ilaris incluyen:
Muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
- Inflamación de garganta con secreción nasal, nariz taponada, estornudos, sensación de presión o dolor en las mejillas y/o en la frente con o sin fiebre (nasofaringitis, faringitis, rinitis, sinusitis).
- Combinación de inflamación de garganta, fiebre, hinchazón o enrojecimiento de las amígdalas, tos, dificultad para tragar y dolor de cabeza (amigdalistis), comunicado con menor frecuencia en los pacientes con gota artrítica.
- Dolor y necesidad de orinar frecuentemente con o sin fiebre (infección del tracto urinario).
- Descenso de los niveles de plaquetas en sangre (denominado trombocitopenia).
- Dolor de estómago y sensación de mareo (gastroenteritis).
- Dolor abdominal.
- Dolor en los músculos, huesos y articulaciones.
- Resultados anormales de la función renal (disminución del aclaramiento renal, proteinuria).
- Reacciones en el lugar de inyección (tales como enrojecimiento, hinchazón, calor y picor).
Frecuentes (pueden afectar hasta 1 de cada 10 personas):
- Niveles anómalos de triglicéridos en su sangre (trastorno del metabolismo lipídico).
- Resultados de la prueba de la función del hígado anormal (transaminasas aumentadas).
- Nivel alto de bilirrubina en la sangre, con o sin amarillamiento de la piel y ojos
(hiperbilirrubinemia).
- Sentirse mareado, sensación que todo le da vueltas (vértigo).
- Sensación de debilidad o muy cansado (astenia).
- Dolor de espalda.
Poco frecuentes (pueden afectar hasta 1 de cada 100 personas):
- Ardor (reflujo gastroesofágico).
No conocida (frecuencia no puede estimarse a partir de los datos disponibles):
- Estar mareado (vómitos).
- Se ha observado en estudios con pacientes con brotes de gota artrítica un aumento de los niveles de ácido úrico.
- Fiebre prolongada (fiebre que dura más de tres días) o cualquier síntoma que pueda estar relacionado con una infeccción, como tos persistente, flema, dolor en el pecho, sangre en el esputo (saliva y flema que se escupe), dificultad para respirar, dolor de oído, dolor de cabeza prolongado o zona de la piel con enrojecimiento localizado, calor o hinchazón. Éstos pueden ser síntomas de una típica infección o de una que puede ser más grave (infección oportunista).
Informe inmediatamente a su médico o al médico de su hijo si nota alguno de estos síntomas.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en las etiquetas y la caja. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar en el embalaje original para protegerlo de la luz.
Desde el punto de vista de la conservación microbiológica, después de la reconstitución el producto se debe utilizar inmediatamente. Sin embargo, se ha demostrado una estabilidad química y física en uso de 24 horas entre 2°C y 8°C.
No utilice Ilaris si observa que la solución no es de transparente a opalescente o contiene partículas.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Ilaris
- El principio activo es canakinumab. Un vial de polvo contiene 150 mg de canakinumab. Tras la reconstitución, cada ml de solución contiene 150 mg de canakinumab.
- Los demás componentes son: sacarosa, histidina,histidina hidrocloruro monohidrato, polisorbato 80.
Aspecto del producto y contenido del envase
- Ilaris se presenta en forma de polvo para solución inyectable dentro de un vial de vidrio de 150 mg.
- El polvo es blanco.
- Ilaris se encuentra disponible en cajas que contienen un vial o en cajas multienvase conteniendo 4 cajas intermedias, cada una de las cuales contiene un vial. Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH Roonstrasse 25 D-90429 Nürnberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Etarapun Novartis Pharma Services Inc. Tea.: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
|
Ceská republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 111 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EXláda Novartis (Hellas) A.E.B.E. Tpk: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
Kúrcpog
Novartis Pharma Services Inc. Tr(k: +357 22 690 690
Latvija
Novartis Pharma Services Inc. Tel: +371 67 887 070
Sverige
Novartis Sverige AB Tel: +46 8 732 32 00
United Kingdom
Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370
Fecha de la última revisión de este prospecto:
Este medicamento se ha autorizado en «circunstancias excepcionales». Esta modalidad de aprobación significa que debido a la rareza de su enfermedad no ha sido posible obtener información completa de este medicamento. La Agencia Europea de Medicamentos revisará anualmente la información nueva de este medicamento que pueda estar disponible y este prospecto se actualizará cuando sea necesario.
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
INSTRUCCIONES DE USO DE ILARIS POLVO PARA SOLUCIÓN INYECTABLE
Por favor, tenga en cuenta que la preparación de la inyección a temperatura ambiente puede durar unos 30 minutos.
Consulte asimismo la sección 3, «Autoinyección de Ilaris o inyección de Ilaris en un niño».
Lea estas instrucciones por completo antes de empezar.
Preparativos esenciales:
- Encuentre un sitio limpio en el que preparar y administrar la inyección.
- Lávese las manos con agua y jabón.
- Compruebe la fecha de caducidad del vial y de la jeringa. No los utilice después de la fecha de caducidad que aparece en la etiqueta y el envase. La fecha de caducidad es el último día del mes que se indica.
- Utilice siempre jeringas y agujas nuevas que estén en envases cerrados. Evite el contacto con las agujas y el tapón del vial.
Reúna todos los elementos que precisa:
Incluidos en el envase
Un vial de Ilaris polvo para solución inyectable (mantener en la nevera).
No incluidos en el envase
- un vial (o ampolla) de agua estéril para inyección («agua») (no conservar en nevera)
- una jeringa de 1,0 ml
- una aguja de 18 G x 2 pulgadas (50 mm) para reconstituir el polvo («aguja para la transferencia»)
- una aguja de 27 G x 0,5 pulgadas (13 mm) para la inyección («aguja de inyección»)
- toallitas con alcohol
- algodón seco y limpio
- una tirita adhesiva
- un contenedor apropiado para depositar las agujas, jeringa y el vial utilizados (contenedor de material de desecho)
Preparación de la mezcla de Ilaris

1. Sacar el protector de los viales de Ilaris y agua. No tocar los tapones de goma de los viales. Limpiar los tapones con una toallita con alcohol.
2. Abrir las bolsas que contienen la jeringa y la aguja para la transferencia (la mayor) y acoplar ésta a la jeringa.
3. Con cuidado, quitar el capuchón de la aguja para la transferencia. Tirar del émbolo hacia abajo hasta alcanzar la señal de 1,0 ml, llenando la jeringa con aire. Introducir la aguja en el vial del agua por el centro del tapón de goma (Fig. 1).
4. Empujar lentamente el émbolo hacia abajo de tal modo que el aire existente en la jeringa pase al vial.

13.
Invertir el conjunto de vial y jeringa y situarlo a la altura de los ojos (Fig. 2).
Asegurar que el extremo de la aguja de transferencia está cubierto por el agua y tirar lentamente del émbolo hasta sobrepasar ligeramente la señal de 1,0 ml. Si se observan burbujas en la jeringa, éstas deben eliminarse de acuerdo con las instrucciones recibidas por su enfermero o farmacéutico.
Asegurar que la jeringa contiene 1,0 ml de agua; a continuación, retirar la aguja del vial. (Debe quedar agua en el vial.)_
Insertar la aguja de transferencia por el centro del tapón del vial de polvo de Ilaris, teniendo cuidado de no tocar la aguja o el tapón. Inyectar lentamente 1,0 ml de agua en el vial de polvo (Fig. 3).
Sacar con cuidado la jeringa y la aguja para la transferencia del vial y tapar la aguja con su capuchón tal y como le ha mostrado su enfermero o farmacéutico.
Sin tocar el tapón, remover (no agitar) el vial lentamente con un ángulo de 45 grados durante aproximadamente 1 minuto (Fig. 4a). Dejar reposar durante 5 minutos.
A continuación, girar suavemente el vial de arriba a abajo unas 10 veces, tomando precauciones para no tocar el tapón de goma del vial (Fig. 4b).
Dejar reposar durante unos 15 minutos a temperatura ambiente para obtener una solución de transparente a opalescente. No agitar. No utilizar si se observan partículas en la solución.
Comprobar que toda la solución está en el fondo del vial. En caso de observar gotas en el tapón, dar ligeros golpes en las paredes del vial para eliminarlas. La solución debe ser de transparente a opalescente y no debe presentar partículas. La solución debe ser incolora o puede tener una ligera coloración amarillo-pardusco.
- Si la solución no se utiliza inmediatamente después de la reconstitución, ésta debe conservarse en la nevera (2°C a 8°C) y _usarse durante las próximas 24 hours._

15.
16.
17.
18.
19.
20.
Limpiar el tapón de goma del vial que contiene la solución de Ilaris con una nueva toallita de algodón.
Quitar el capuchón de la aguja para la transferencia. Tirar del émbolo de la jeringa hacia abajo hasta alcanzar la señal de 1,0 ml, llenando la jeringa con aire. Introducir la aguja de la jeringa en el vial de Ilaris en el centro del tapón de goma (Fig. 5). Lentamente, empujar el émbolo hacia abajo hasta que se inyecte el aire en el vial. No debe inyectarse aire en el medicamento._
No debe invertirse el conjunto de la jeringa y el vial (Fig. 6a). Introducir la aguja hasta el fondo del vial.
Inclinar el vial con el fin de garantizar que se pueda extraer una cantidad adecuada de solución del vial (Fig. 6b).
NOTA: La cantidad necesaria depende de la dosis que deba administrarse (0,2 ml a 1,0 ml). Su médico le mostrará la cantidad que ha de inyectarse.
Lentamente, tirar del émbolo hasta situarlo en la señal adecuada (0,2 ml a 1,0 ml), llenando la jeringa con la solución de Ilaris. Si aparecen burbujas de aire en la jeringa, eliminarlas de acuerdo con las instrucciones recibidas del profesional sanitario. Comprobar que en la jeringa se encuentra la cantidad de solución adecuada. Sacar la jeringa con la aguja del vial. (Puede quedar solución en el vial.) Tapar de nuevo la aguja tal y como le ha enseñado su enfermero o farmacéutico. Separar la aguja de transferencia de la jeringa. Tirar la aguja de transferencia en el contenedor de instrumentos afilados.
Abrir el envoltorio que protege la aguja para inyección y acoplar ésta a la jeringa. Dejar la jeringa a un lado._

21.
22.
23.
24.

8
Elegir el área de inyección en la parte superior del muslo, abdomen, parte superior del brazo o glúteos. No debe elegirse una zona con erupción o lesión en la piel, o que presente un morado o hinchado. No inyectar en cicatrices ya que puede disminuir la exposición al medicamento. No debe inyectarse en vena.
Limpiar el lugar de inyección con una nueva toallita de algodón. Permitir que se seque. Destapar la aguja.
Pellizcar y elevar suavemente la piel del área de inyección. Sostener la jeringa con un ángulo de 90 grados y de forma suave con un único movimiento, empujar la aguja hacia abajo para
introducirla en la piel (Fig. 7)._
Mantener la aguja en la piel mientras se empuja lentamente el émbolo hasta que la jeringa esté vacía (Fig. 8). Liberar la zona de inyección y extraer la aguja. Tirar la aguja y la jeringa en el contenedor de instrumentos afilados sin necesidad de protegerla de nuevo o separar la aguja.

9


25. No frotar la zona de inyección. Si hay sangrado, aplicar un algodón seco sobre el área y presionar suavemente durante 1 ó 2 minutos, o hasta que se detenga la hemorragia (Fig. 9). A continuación, proteger con una tirita adhesiva.
26.
27.
Depositar con cuidado las agujas y las jeringas en los contenedores provistos a tal efecto de acuerdo con las instrucciones dadas por el profesional sanitario o farmacéutico (Fig. 10). Las jeringas y las agujas no deben reutilizarse.
Eliminar los viales con agua y solución sobrante de Ilaris tal y como el profesional sanitario o farmacéutico le haya indicado. Todo producto no utilizado o de deshecho debe eliminarse de acuerdo con las normas locales.
Mantener el contenedor de material de desecho fuera del alcance de los niños. Eliminarlo de acuerdo con las instrucciones recibidas del profesional sanitario o farmacéutico.
Prospecto: información para el usuario
Ilaris 150 mg polvo y disolvente para solución inyectable
Canakinumab
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Ilaris y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Ilaris
3. Cómo usar Ilaris
4. Posibles efectos adversos
5. Conservación de Ilaris
6. Contenido del envase e información adicional
1. Qué es Ilaris y para qué se utiliza Qué es Ilaris
Ilaris contiene el principio activo canakinumab, un anticuerpo monoclonal que pertenece al grupo farmacoterapéutico de los inhibidores de interleucinas. En el organismo bloquea la actividad de una sustancia llamada interleucina-1 beta (IL-1 beta), que se encuentra a niveles elevados en las enfermedades inflamatorias tales como los Síndromes Periódicos Asociados a la Criopirina (CAPS), la enfermedad de Still incluyendo la Enfermedad de Still del Adulto (EAS) y la Artritis Idiopática Juvenil Sistémica (AIJS) y gota artrítica.
Ilaris se utiliza para el tratamiento de los Síndromes Periódicos Asociados a Criopirina (CAPS), la enfermedad de Still incluyendo la Enfermedad de Still del Adulto (EAS) y la Artritis Idiopática Juvenil Sistémica (AIJS) y gota artrítica.
Para qué se utiliza Ilaris
Síndromes Periódicos Asociados a la Criopirina
Ilaris se utiliza en adultos, adolescentes y niños a partir de 2 años y con un peso de 7,5 kg o superior, para tratar las siguientes enfermedades autoinflamatorias que se conocen en conjunto con el nombre de Síndromes Periódicos Asociados a la Criopirina (CAPS):
- Síndrome de Muckle-Wells (MWS),
- Enfermedad Neonatal Multisistémica Inflamatoria (NOMID), también conocida como Síndrome Infantil Neurológico, Cutáneo y Articular Crónico (CINCA),
- Manifestaciones graves del Síndrome Autoinflamatorio Familiar inducido por el Frío (FCAS) / Urticaria Familiar Fría (FCU) que presente signos y síntomas más allá de la erupción de la piel de tipo urticaria inducido por el frío.
En pacientes con CAPS, el organismo produce una cantidad excesiva de IL-1 beta. Esto puede ocasionar síntomas tales como fiebre, dolor de cabeza, fatiga, erupción cutánea o dolores en las articulaciones y músculos. Mediante el bloqueo de la actividad de IL-1 beta, Ilaris puedefacilitar la mejoría de estos síntomas.
Enfermedad de Still
Ilaris se utiliza en pacientes adultos, adolescentes y niños a partir de los 2 años de edad para el tratamiento de la enfermedad de Still activa incluyendo la Enfermedad de Still del Adulto (ESA) y la Artritis Idiopática Juvenil Sistémica (AIJS) cuando los otros tratamientos no hayan funcionado bien. Ilaris puede utilizarse sólo o en combinación con metrotrexato.
La enfermedad de Still que incluye EAS y AIJS es una enfermedad inflamatoria que puede provocar dolor, hinchazón e inflamación de una o más articulaciones, así como sarpullido y fiebre. La proteina pro-inflamatoria IL-1beta juega un importante papel en el proceso inflamatorio de la enfermedad de Still y Ilaris puede mejorar los signos y síntomas de la enfermedad de Still bloqueando la actividad de IL-1beta.
Gota artrítica
Ilaris se utiliza en adultos para tratar los síntomas de ataques frecuentes de gota artrítica si otros tratamientos no han funcionado suficientemente bien.
La gota artrítica está causada por el depósito en el organismo de unas sustancias químicas llamadas cristales de urato. Estos cristales de urato causan una producción excesiva de IL-1 beta, que a su vez pueden provocar un repentino dolor grave, enrojecimiento, calor, e hinchazón de las articulaciones (conocido como ataque de gota). Mediante el bloqueo de IL-1 beta, Ilaris puede conseguir mejorar estos síntomas.
Si tiene dudas sobre cómo actúa Ilaris o por qué le han recetado este medicamento, consulte a su médico, farmacéutico o enfermero.
2. Qué necesita saber antes de empezar a usar Ilaris
No use Ilaris
- si es alérgico al canakinumab o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
- si tiene, o sospecha que tiene, una infección grave activa.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar Ilaris, si se encuentra en alguna de las siguientes
situaciones:
- si padece alguna infección en la actualidad o si ha tenido infecciones repetidas o sufre alguna enfermedad tal como nivel bajo de góbulos blancos que le hace más vulnerable a tener infecciones.
- si tiene o alguna vez ha tenido tuberculosis o contacto directo con una persona con una infección activa de tuberculosis. Su médico también puede comprobar si tiene tuberculosis utilizando una prueba específica.
- si tiene signos de reacciones alérgicas tales como dificultad para respirar o tragar, náuseas, mareo, erupción en la piel, picor, urticaria, palpitaciones y tensión arterial baja.
- si tiene signos de un trastorno del hígado tales como coloración amarilla en la piel y los ojos, náuseas, pérdida del apetito, color oscuro en la orina y heces blanquecinas.
- si tiene que ponerse alguna vacuna. Se recomienda evitar ser vacunado con un tipo de vacuna (también conocida como vacuna atenuada) mientras está usando Ilaris (ver también «Uso de Ilaris con otros medicamentos (incluyendo vacunas)»).
Enfermedad de Still
- Los pacientes con enfermedad de Still pueden desarrollar una enfermedad llamada síndrome de activación macrofágica (SAM) que puede causar la muerte. Su médico le hará un seguimiento de los posibles factores desencadenantes de SAM, que incluyen infecciones y re-activación de la enfermedad de Still (empeoramiento).
Niños y adolescentes
- CAPS y AIJS: Ilaris puede utilizarse en niños a partir de de 2 años de edad.
- Gota artrítica: Ilaris no está recomendado para niños o adolescentes menores de 18 años de edad.
Uso de Ilaris con otros medicamentos (incluyendo vacunas)
Informe a su médico, farmacéutico o enfermero si está utilizando, ha utilizado recientemente o pudiera tener que utilizar cualquier otro medicamento.
- Vacunas atenuadas: Se recomienda evitar ser vacunado con un tipo de vacuna conocida como vacuna atenuada mientras está en tratamiento con Ilaris. Puede que su médico necesite comprobar su historial de vacunaciones y darle aquellas vacunas que no ha recibido antes de iniciar el tratamiento con Ilaris. Si es necesario que le administren una vacuna atenuada después de iniciar el tratamiento con Ilaris, es aconsejable que se espere durante al menos 3 meses después de la última inyección de Ilaris y antes de la próxima inyección.
- Medicamentos conocidos como los inhibidores del factor de necrosis tumoral (TNF), tales como etanercept, adalimumab o infliximab. Éstos se utilizan principalmente en enfermedades reumáticas y autoinmunes. No deben utilizarse con Ilaris porque puede aumentar el riesgo de infecciones.
Embarazo y lactancia
- No se ha investigado Ilaris en mujeres embarazadas. Debe evitar quedar embarazada y se recomienda utilizar medidas anticonceptivas adecuadas mientras esté tomando Ilaris y durante al menos tres meses después de la última administración. Es importante que informe a su médico si está embarazada, si puede estar embarazada o si está planeando tener un bebé. Su médico le informará acerca del riesgo potencial de utilizar Ilaris durante el embarazo.
- Si recibe canakinumab mientras está embarazada, es importante que informe al pediatra o enfermero antes de que le administren alguna vacuna a su bebé. Su bebé no debe recibir vacunas vivas hasta al menos 16 semanas después de que usted haya recibido su última dosis de canakinumab antes del parto.
- Se desconoce si Ilaris pasa a la leche materna. Su médico le informará acerca de los riesgos potenciales de utilizar Ilaris antes de dar el pecho.
Conducción y uso de máquinas
El tratamiento con Ilaris puede producirle una sensación de que todo da vueltas (mareo/vértigo) o cansancio intenso (astenia). Esto puede afectar a la capacidad de conducir o usar herramientas o máquinas. Si nota la sensación de que todo le da vueltas o se siente cansado, no conduzca ni use herramientas o máquinas hasta que se sienta de nuevo bien.
3. Cómo usar Ilaris
Si tiene CAPS o enfermedad de Still (ESA o AIJS), después de una formación adecuada, usted mismo o su cuidador podría inyectar Ilaris.
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico farmacéutico o enfermero.
Si tiene gota artrítica, un médico con formación especializada supervisará su tratamiento. Ilaris solo debe ser inyectado por un profesional sanitario.
Mantenga a su médico informado de su enfermedad y de cualquier síntoma antes de utilizar o que se le administre Ilaris (ver sección 2). Su médico puede decidir postponer o interrumpir su tratamiento, solo cuando sea necesario.
Ilaris debe utilizarse por vía subcutánea. Esto significa que se inyecta con una aguja corta dentro del tejipo adiposo por debajo de la piel.
Cuánto Ilaris debe utilizar
Síndromes Periódicos Asociados a la Criopirina La dosis inicial recomendada de Ilaris es:
Adultos, adolescentes y niños de 4 años o más
- 150 mg para pacientes cuyo peso corporal sea más de 40 kg
- 2 mg/kg para pacientes cuyo peso corporal sea de 15 kg o más, hasta un máximo de 40 kg
- 4 mg/kg para pacientes cuyo peso corporal sea de7,5 kg o más, pero menos de 15 kg
Niños de 2 o 3 años
- 4 mg/kg para pacientes cuyo peso corporal sea de 7,5 kg o más Ilaris se utiliza cada 8 semanas como una inyección única.
Si no responde suficientemente al tratamiento a los 7 días, su médico puede considerar darle una dosis repetida de 150 mg ó 2 mg/kg. Si responde suficientemente a ésta, su tratamiento continuará con esta dosis más alta de 300 mg o 4 mg/kg cada 8 semanas. Si no responde suficientemente a la dosis de repetición, se puede considerar una tercera dosis de Ilaris a 300 mg o 4 mg/kg, y si responde suficientemente a ésta, su tratamiento puede continuar con 600 mg y 8 mg/kg cada 8 semanas.
En pacientes que han iniciado con una dosis inicial de 4 mg/kg y que no han respondido suficientemente después de 7 días, se puede considerar una segunda dosis de 4 mg/kg. Si el paciente responde suficientemente a ésta, se puede continuar el tratamiento con 8 mg/kg cada 8 semanas.
Enfermedad de Still (ESA y AIJS)
La dosis recomendada de Ilaris para pacientes con enfermedad de Still (ESA o AIJS) cuyo peso corporal sea de 7,5 kg o más es de 4 mg/kg (hasta un máximo de 300 mg). Ilaris se inyecta bajo la piel cada 4 semanas.
Gota artrítica
Su médico comentará con usted la necesidad de empezar o ajustar un tratamiento con reductores de urato para disminuir el nivel de ácido úrico de su sangre.
La dosis recomendada para Ilaris en pacientes adultos con gota es 150 mg administrada como una única dosis durante un ataque de gota artrítica.
Si necesita otro tratamiento con Ilaris, y con la última dosis obtuvo alivio, debe esperar un mínimo de 12 semanas antes de una siguiente dosis.
Autoinyección de Ilaris o inyección de Ilaris en un niño
Si tiene CAPS o enfermedad de Still (ESA o AIJS), los pacientes o los cuidadores de pacientes de niños pueden poner ellos mismos las inyecciones después de recibir una formación adecuada en relación con la técnica de inyección.
- El paciente o cuidador y el médico decidirán quién pondrá las inyecciones de Ilaris.
- Su médico o enfermero le enseñarán cómo poner las inyecciones de Ilaris.
- No debe tratar de ponerse una inyección si no ha recibido la formación necesaria o no está seguro de cómo hacerlo.
- Los viales de Ilaris son de un solo uso, solo para uso individual. Utilice solo el contenido incluido en el kit para inyectar Ilaris.
- No reutilice la solución sobrante o cualquier parte del kit.
Para tener más información sobre cómo poner las inyecciones de Ilaris, consulte la sección «Instrucciones de uso» al final de este prospecto. Si tiene dudas, hable con su médico, farmacéutico o enfermero.
Duración del tratamiento con Ilaris
- Si tiene CAPS o enfermedad de Still (ESA o AIJS), debe continuar utilizando Ilaris durante el tiempo que su médico le aconseje.
- Si tiene un ataque de gota artrítica, le administrarán una única dosis de Ilaris. Si experimenta un nuevo ataque, su médico puede considerar administrarle una nueva dosis de Ilaris pero no antes de las 12 semanas de haberle administrado la dosis previa.
Si usa más Ilaris del que debe
Si se inyecta accidentalmente más Ilaris del de la dosis recomendada, no es probable que sea grave pero debe informar a su médico, farmacéutico o enfermero lo antes posible.
Si se pone Ilaris antes de lo que debe
- En CAPS, no debe inyectarse Ilaris antes de transcurridas las 8 semanas de la última dosis, a menos que su médico se lo diga.
- En enfermedad de Still (ESA o AIJS) no debe inyectarse Ilaris antes de transcurridas las 4 semanas de la última dosis.
Si se inyecta Ilaris acccidentalmente antes del plazo recomendado, lo debe decir a su médico, farmacéutico o enfermero lo antes posible.
Si olvidó usar Ilaris
Si tiene CAPS o enfermedad de Still (ESA o AIJS) y ha olvidado inyectarse una dosis de Ilaris, la dosis siguiente debe inyectarse tan pronto como lo recuerde. Entonces hable con el médico para acordar cuándo debe inyectarse la dosis siguiente. A continuación, debe seguirse con la inyección a los intervalos recomendados como antes.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. La mayoría de efectos adversos son de carácter leve o moderado y desaparecen generalmente a los pocos días o a las pocas semanas de la administración.
Algunos efectos adversos pueden ser graves. Informe a su médico inmediatamente, si experimenta cualquiera de los siguientes efectos adversos:
- Fiebre que se prolongue más de 3 días o cualquier síntoma que puede deberse a una infección. Esto incluye temblores, escalofríos, malestar general, pérdida de apetito, dolores en el cuerpo, por lo general relacionado con la aparición repentina de la enfermedad, dolor de garganta o úlceras en la boca, tos, flema, dolor en el pecho, dificultad para respirar, dolor de oído, dolor de cabeza prolongado y enrojecimiento, calor e hinchazón localizadas en la piel o inflamación del tejido conectivo (celulitis). Estos síntomas pueden deberse a una infección causada por los bajos niveles de glóbulos blancos (denominado leucopenia y neutropenia). Si lo considera necesario su médico puede hacerle análisis de sangre de forma regular.
- Reacciones alérgicas con sarpullido y picor y posiblemente también urticaria, dificultad para respirar o tragar, mareo, conciencia inusual del latido del corazón (palpitaciones) y tensión arterial baja.
Otros efectos adversos de Ilaris incluyen:
Muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
- Inflamación de garganta con secreción nasal, nariz taponada, estornudos, sensación de presión o dolor en las mejillas y/o en la frente con o sin fiebre (nasofaringitis, faringitis, rinitis, sinusitis).Dolor y necesidad de orinar frecuentemente con o sin fiebre (infección del tracto urinario).
- Combinación de inflamación de garganta, fiebre, hinchazón o enrojecimiento de las amígdalas, tos, dificultad para tragar y dolor de cabeza (amigdalistis), comunicado con menor frecuencia en los pacientes con gota artrítica.
- Descenso de los niveles de plaquetas en sangre (denominado trombocitopenia).
- Dolor de estómago y sensación de mareo (gastroenteritis).
- Dolor abdominal.
- Dolor en los músculos, huesos y articulaciones.
- Resultados anormales de la función renal (disminución del aclaramiento renal, proteinuria).
- Reacciones en el lugar de inyección (tales como enrojecimiento, hinchazón, calor y picor).
Frecuentes (pueden afectar hasta 1 de cada 10 personas):
- Niveles anómalos de triglicéridos en su sangre (trastorno del metabolismo lipídico).
- Resultados de la prueba de la función del hígado anormal (transaminasas aumentadas).
- Nivel alto de bilirrubina en la sangre, con o sin amarillamiento de la piel y ojos
(hiperbilirrubinemia).
- Sentirse mareado, sensación que todo le da vueltas (vértigo).
- Sensación de debilidad o muy cansado (astenia).
- Dolor de espalda.
Poco frecuentes (pueden afectar hasta 1 de cada 100 personas):
- Ardor (reflujo gastroesofágico).
No conocida (frecuencia no puede estimarse a partir de los datos disponibles):
- Estar mareado (vómitos).
- Se ha observado en estudios con pacientes con brotes de gota artrítica un aumento de los niveles de ácido úrico.
- Fiebre prolongada (fiebre que dura más de tres días) o cualquier síntoma que pueda estar relacionado con una infeccción, como tos persistente, flema, dolor en el pecho, sangre en el esputo (saliva y flema que se escupe), dificultad para respirar, dolor de oído, dolor de cabeza prolongado o zona de la piel con enrojecimiento localizado, calor o hinchazón. Éstos pueden ser síntomas de una típica infección o de una que puede ser más grave (infección oportunista).
Informe inmediatamente a su médico o al médico de su hijo si nota alguno de estos síntomas.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Ilaris
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta y la caja. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2°C y 8°C). No congelar.
Conservar en el embalaje original para protegerlo de la luz.
Desde el punto de vista de la conservación microbiológica, después de la reconstitución el producto se debe utilizar inmediatamente. Sin embargo, se ha demostrado una estabilidad química y física en uso de 24 horas entre 2°C y 8°C.
No utilice Ilaris si observa que la solución no es de transparente a opalescente o contiene partículas.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Ilaris
- El principio activo es canakinumab. Un vial de polvo contiene 150 mg de canakinumab. Tras la reconstitución, cada ml de solución contiene 150 mg de canakinumab.
- Los demás componentes son:
Polvo: sacarosa, histidina,histidina hidrocloruro monohidrato, polisorbato 80.
Disolvente: agua para preparaciones inyectables.
Aspecto del producto y contenido del envase
- Ilaris polvo y disolvente para solución inyectable consiste en un polvo blanco (150 mg en un vial de vidrio de 6 ml) y un disolvente transparente e incoloro (5 ml en un vial de vidrio independiente).
- Ilaris polvo y disolvente para solución inyectable se encuentra disponible en un kit para la inyección conteniendo 1 vial de polvo para solución inyectable, 1 vial de disolvente, 1 jeringa para la inyección, 1 aguja de seguridad, 2 adaptadores para el vial y 4 toallitas limpiadoras.
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Novartis Pharma GmbH Roonstrasse 25 D-90429 Nürnberg Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Etarapun Novartis Pharma Services Inc. Tea.: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
|
Ceská republika Novartis s.r.o. Tel: +420 225 775 111 Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 111 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EXláda Novartis (Hellas) A.E.B.E. Tpk: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
Kúrcpog
Novartis Pharma Services Inc.
Tel: +46 8 732 32 00
Tr(k: +357 22 690 690
Fecha de la última revisión de este prospecto:
Este medicamento se ha autorizado en «circunstancias excepcionales». Esta modalidad de aprobación significa que debido a la rareza de su enfermedad no ha sido posible obtener información completa de este medicamento. La Agencia Europea de Medicamentos revisará anualmente la información nueva de este medicamento que pueda estar disponible y este prospecto se actualizará cuando sea necesario.
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu
INSTRUCCIONES DE USO DE ILARIS POLVO Y DISOLVENTE PARA SOLUCIÓN INYECTABLE (KIT PARA LA INYECCIÓN)
Lea estas instrucciones por completo antes de empezar a preparar su medicación.
• Su médico o enfermero le enseñarán cómo inyectarse usted mismo (ver Sección 3. «Autoinyección de Ilaris o inyección de Ilaris en un niño»).
• La preparación de la inyección dura unos 30 minutos.
• En esta sección, denominamos «kit» al kit para la inyección.
Antes de empezar:
1. Saque el kit de la nevera.
• Compruebe la fecha de caducidad del envase.
• No lo utilice después de la fecha de caducidad que aparece en la etiqueta y el envase.
• La fecha de caducidad es el último día del mes que se indica.
2. Mantenga el kit sin abrir durante 15 minutos.
• Esto hará que el contenido llegue a temperatura ambiente.
• No intente calentar el kit - deje que se caliente por sí solo.
3. Encuentre un sitio limpio en el que preparar y administrarse usted mismo la inyección. Lávese las manos con agua y jabón, y entonces, séqueselas con una toalla limpia.
4. Abra el envase y saque todo su contenido.
• Compruebe que tiene todos los elementos que se muestran en el esquema inferior.
• Si falta cualquier parte del kit o está dañado, devuelva todo el envase a su médico o farmacéutico.
• Utilice solo los elementos incluidos en el kit- no utilice nada más.
5. No toque las agujas o la parte superior de los viales.
QUÉ HAY EN EL KIT:
a.

b.

a. Vial con polvo
b. Vial con disolvente
c.
d.

e.

f.

c. 1 jeringa de 1 ml para la inyección
d. Aguja de seguridad
e. 2 adaptadores para el vial
f. 4 toallitas limpiadoras
Saque la cápsula flip-off del vial con polvo.
Paso 1: Preparación del vial con polvo

Utilice una toallita limpiadora limpia para limpiar el tapón de goma.
No toque el tapón de goma después de haberlo limpiado.
Paso 2: Acoplamiento del adaptador para el vial al vial con polvo

• Coja uno de los capuchones protectores que contienen los adaptadores para el vial.
• Sujete el capuchón firmemente.
• Saque la lámina protectora.
• No saque el adaptador del vial del capuchón protector.
No toque el adaptador para el vial en ningún momento.



Ponga el vial con polvo sobre una superficie plana.
• Sujete el capuchón protector y ponga el adaptador para el vial en la parte superior del vial con polvo.
• Apriete completamente el capuchón protector hacia abajo. Oirá el ruido que hace el adaptador para el vial al acoplarse.
Sujete el capuchón protector por la parte superior.
• Levante el capuchón protector hacia arriba para que se desprenda del adaptador del vial.
Compruebe que el adaptador para el vial está en la posición correcta. Si no lo está:
No toque el adaptador para el vial.
Vuelva a poner el capuchón protector sobre el adaptador para el vial.
Entonces cambie la posición del adaptador para el vial.
Paso 3: Acoplamiento del adaptador para
el
vial al vial con disolvente.
Repita los pasos 1 y 2 para poner el adaptador para el vial sobre el vial con disolvente.
• El disolvente contiene agua para preparaciones inyectables.
Ahora los dos viales tienen su adaptador para el vial acoplado. Están preparados para su uso.
Paso 4: Introducción de 1,0 ml de aire a la jeringa

Quitar la bolsa que contiene la jeringa y sacar la jeringa.
• No tocar la punta de la jeringa.
• Sitúe la jeringa a la altura de los ojos, para poder medir la dosis correctamente.
• Tire el émbolo hacia abajo hasta que la señal de 1,0 ml está en línea con el límite superior del émbolo. Esto se muestra en el esquema.
El transferir 1,0 ml de aire en la jeringa facilita introducir el agua para preparaciones inyectables en la jeringa. Esto también ayuda a evitar que las burbujas de aire entren en la jeringa.
Paso 5: Acoplamiento de la jeringa al vial de disolvente

No toque la punta de la jeringa o del adaptador para el vial cuando realice esto.
• Sujete el vial con disolvente.
• Enrosque cuidadosamente la jeringa en el adaptador para el vial.
• No utilice la fuerza.
En esta fase no necesita ninguna aguja.
Paso 6: Trasvase de 1,0 ml de aire en el vial con disolvente

Empuje lentamente el émbolo hacia abajo completamente.
• Esto empujará el aire que acaba de introducir en la jeringa al vial con disolvente.
• Mantenga el émbolo hacia abajo.
Paso 7: Introducción de 1,0 ml de agua en la jeringa

Gire la jeringa hacia arriba - esto significa que el vial con disolvente ahora está hacia abajo.
• Situé la jeringa a la altura de los ojos, para poder medir la dosis correctamente.
• Tire lentamente el émbolo hacia abajo hasta que la señal de 1.0 ml está en línea con el límite superior del émbolo. Esto se muestra en el esquema.
Mantenga el vial hacia abajo.
• Comprueba que la jeringa no tiene burbujas grandes de aire.
Paso 8: Eliminación de las burbujas de aire de la jeringa

Si en la jeringa hay burbujas grandes de aire, tiene de eliminarlas.
• Dé ligeros golpecitos con cuidado para que las burbujas grandes de aire suban hacia arriba.
• Empuje con cuidado el émbolo hacia arriba. Esto hace que cualquier burbuja grande de aire entre en el vial.
• Entonces tire lentamente el émbolo hacia abajo, de nuevo hasta la señal de 1,0 ml.
• Repita estos pasos hasta que se hayan eliminado todas las burbujas grandes.
• Después de esto, compruebe que la jeringa aún contiene 1,0 ml de agua.
Gire la jeringa y el vial hacia el otro lado. Esto significa que el vial ahora está hacia arriba.
• Ponga el vial sobre una superficie limpia y plana.
• El utilizar una superficie plana le ayudará a impedir que el agua se derrame.
• Sujete el vial con una mano.
• Con la otra mano, sujete el émbolo y desenrosque la jeringa del vial.
Aún habrá un poco de agua en el vial con disolvente.
Paso 9: Enroscamiento de la jeringa con el vial con polvo

No toque la punta de la jeringa o del vial.
• Mantenga el vial con polvo en la superficie limpia y plana.
• Enrosque la jeringa conteniendo 1,0 ml de agua con el adaptador para el vial.
• No lo fuerce.
Paso 10: Trasvase de 1,0 ml de agua en el vial con polvo

agua
Apriete lentamente el émbolo hasta el final.
• Esto hará trasvasar el agua de la jeringa al vial con polvo.
• No saque todavía la jeringa.

• Mantenga la jeringa en un ángulo de unos 45 grados.
• Mueva cuidadosamente en círculo la jeringa y vial durante un mínimo de 1 minuto.
• Hágalo cuidadosamente - no agite la jeringa y el vial.
Paso 12: Dejar la jeringa y el vial en reposo
• Ponga la jeringa y el vial sobre una superficie plana.

• Déjelos reposar durante 5 minutos.
No se preocupe si el émbolo se mueve hace arriba. Esto puede ocurrir si hay demasiada presión en el vial.
Paso 13: Nueva mezcla del polvo y el agua

• Transcurridos 5 minutos, apriete hacia abajo el émbolo hasta el final, introduciéndolo en la jeringa.
• Mueva cuidadosamente la jeringa y el vial hacia arriba de manera que el conjunto se haya invertido. Entonces gire de nuevo la jeringa y el vial de manera que vuelvan a la posición que estaban.
• Realice este paso diez veces.
• Hágalo con cuidado - no agite la jeringa y el vial. Paso 14: Comprobación de la medicación

• Deje reposar el vial y la jeringa durante 15 minutos.
Después de 15 minutos, compruebe que el medicamento no
está turbio y no tiene partículas.
• No agite el vial y la jeringa.
• Puede que haya espuma en la parte superior de la medicación. No tiene de preocuparse por ello.
Si todavía hay partículas en la medicación, repita el
paso 13.
• Entonces, deje reposar el vial y la jeringa durante otros 5 minutos.
• Compruebe de nuevo que la medicación no tiene partículas. Si la medicación no está turbia y no tiene partículas dentro, está lista para su uso.
Si no utiliza su medicación inmediatamente después de haberla preparado:
• Guarde la medicación en una nevera a una temperatura entre 2°C y 8°C.
• Utilice su medicación dentro de las 24 horas después de haberla preparado.
Paso 15: Llenado de la jeringa con 1,0 ml de la medicación preparada
• Gire la jeringa y el vial hacia abajo.
Tire cuidadosamente hacia abajo el émbolo hasta que la señal de 1,0 ml este en línea con el límite superior del émbolo. Eso se muestra en el esquema.


No saque todavía la jeringa.
• Empuje lentamente el émbolo hasta el final. Esto hará retornar la medicación al vial.
Apretando el émbolo hasta el final asegura que la medicación esté completamente mezclada. Además ayuda a evitar las burbujas de aire.
Paso 16: Medición de la dosis de medicación preparada

Su médico le informará cuál es la dosis de medicamento que se debe administrar.
• Sitúe la jeringa a la altura de los ojos, para poder medir la dosis correctamente.
• La dosis habitual está entre 0,2 y 1,0 ml.
• Tire lentamente del émbolo hacia abajo otra vez hasta que el límite superior del émbolo esté en línea con la señal que corresponde a su dosis de medicamento. Esto se muestra en el esquema.
Mantenga el vial hacia abajo.
• Compruebe que su medicación no tiene burbujas grandes de aire.
• Puede que aún quede algo de medicación en el vial.
Paso 17: Eliminación de burbujas grandes de aire de la jeringa

Elimine las burbujas grandes de aire de la solución del siguiente modo:
• Dé ligeros golpecitos con cuidado para que las burbujas grandes de aire suban hacia arriba.
• Empuje con cuidado el émbolo hacia arriba. Esto hace que cualquier burbuja grande de aire entre en el vial.
• Tire lentamente el émbolo hacia abajo hasta la dosis prescrita por su médico.
• Repita estos pasos hasta que se hayan eliminado todas las burbujas grandes.
• Compruebe que la jeringa contiene la dosis prescrita por su médico.
Ponga la jeringa preparada sobre una superficie limpia y plana.
• Utilizando una superficie plana le ayudará a evitar que el medicamento se pueda derramar.
• Sujete el émbolo y desenrosque la jeringa.
• No toque la punta de la jeringa.
Paso 18: Acoplamiento de la jeringa

No toque el extremo de la aguja o la punta de la jeringa.
• Saque la aguja de seguridad de su envoltorio protector.
• Enrosque la aguja de seguridad con la jeringa preparada.
• Mueva la parte articulada del protector de seguridad hacia la jeringa- como se muestra en la figura.
Ponga la jeringa sobre una superficie plana. Ahora debe escoger el lugar de su cuerpo donde usted mismo se inyectará la inyección.
DONDE INYECTARSE LA INYECCIÓN

Las zonas sombreadas en este esquema muestran los lugares donde puede inyectarse la inyección. Elija uno de estos lugares:
• parte inferior derecha de su abdomen o
• parte inferior izquierda de su abdomen o
• parte superior de su muslo derecho o
• parte superior de su muslo izquierdo.
También se puede administrar la inyección en la parte superior del brazo o glúteos.
Elija un lugar diferente cada vez que se inyecte. Esto le ayudará a evitar el dolor.
• Nunca se inyecte en un área donde la piel esté dañada o inflamada.
Paso 19: Preparación de la piel y de la aguja

Limpie el área de la piel donde va administrase la inyección con una toallita limpiadora limpia. Saque el tapón protector de la aguja. No toque o doble la jeringa.

Pellizque cuidadosamente un pliegue de la piel en el lugar que ha elegido para autoadministrarse la inyección. Esta área tiene de ser una capa grasa de la piel.
Sujete la aguja como se muestra en el esquema. Ponga la aguja entera en el pliegue de la piel en un ángulo recto.
Presione cuidadosamente en el émbolo hasta el fondo. Así se inyectará toda la dosis.
Entonces espere 10 segundos y extraiga la aguja fuera de su piel.
Si empieza a sangrar:
• No frote el área de la inyección.
• Ponga un algodón limpio y seco sobre el área.
• Presione suavemente durante 1 ó 2 minutos. A continuación póngase una tirita adhesiva.
DESPUÉS DE LA INYECCIÓN
Paso 21: Poner el protector de seguridad en la aguja

Paso 22: Eliminación

Inmediatamente ponga el protector de seguridad en la
aguja:
• Mantenga sus dedos alejados de la punta de la aguja y del protector de seguridad.
• Presione el protector de seguridad de la aguja sobre una superficie dura, como por ejemplo una mesa. No utilice los dedos para apretar el protector de seguridad.
• Cuando el protector de seguridad se haya colocado correctamente en la aguja, oirá un clic.
Inmediatamente elimine la jeringa, la aguja y los viales utilizados, incluyendo cualquier solución sobrante.
• Póngalos en el contenedor de material de desecho o sigue las instrucciones recibidas de su médico, enfermero o farmacéutico si difieren de éstas.
• Mantener el contenedor de material de desecho fuera del alcance de los niños.
Nunca intente reutilizar cualquier parte del kit o solución sobrante.
93