Hibor 10.000 Ui Anti-Xa/0,4 Ml Solución Inyectable En Jeringas Precargadas
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
HIBOR 5.000 UI anti -Xa/0,2 ml solución inyectable en jeringas precargadas HIBOR 7.500 UI anti -Xa/0,3 ml solución inyectable en jeringas precargadas HIBOR 10.000 UI anti -Xa/0,4 ml solución inyectable en jeringas precargadas HIBOR 12.500 UI anti -Xa/0,5 ml solución inyectable en jeringas precargadas
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
|
Presentación |
Principio activo/ ml de solución |
Contenido total |
|
HIBOR 5.000 UI/ 0,2 ml jeringas precargadas |
Bemiparina sódica (DCI) 25.000 UI (anti-Xa) |
5.000 UI (anti-Xa)* |
|
HIBOR 7.500 UI/ 0,3 ml jeringas precargadas |
Bemiparina sódica (DCI) 25.000 UI (anti-Xa) |
7.500 UI (anti-Xa)* |
|
HIBOR 10.000 UI/ 0,4 ml jeringas precargadas |
Bemiparina sódica (DCI) 25.000 UI (anti-Xa) |
10.000 UI (anti-Xa)* |
|
HIBOR 12.500 UI/ 0,5 ml jeringas precargadas |
Bemiparina sódica (DCI) 25.000 UI (anti-Xa) |
12.500 UI (anti-Xa)* |
* Actividad aproximada anti Factor Xa en unidades internacionales (UI) valorada frente al primer estándar internacional de la OMS para heparinas de bajo peso molecular con el método anti-Xa amidolítico con sustratos específicos y utilizando el patrón internacional LMWHs (NIBSC).
Bemiparina sódica se obtiene a partir de la mucosa intestinal del cerdo.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable en jeringa precargada.
Solución incolora o ligeramente amarillenta, transparente, exenta de partículas visibles.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento de la trombosis venosa profunda establecida, con o sin embolismo pulmonar.
4.2 Posología y forma de administración
ADVERTENCIA: Las diferentes heparinas de bajo peso molecular no son necesariamente equivalentes. En consecuencia, se debe respetar la dosificación y el modo de empleo específico de cada uno de estos medicamentos.
Posología
Adultos
Tratamiento de la trombosis venosa profunda:
HIBOR debe administrarse a la dosis fija curativa de 115 UI anti-Xa/kg peso/día, por vía subcutánea, durante 7±2 días como norma general. Esta pauta corresponde, aproximadamente, según el peso corporal, a los rangos: <50 kg, 0,2 ml (5.000 UI anti-Xa); 50-70 kg, 0,3 ml (7.500 UI anti-Xa), 70-100 kg, 0,4 ml (10.000 UI anti-Xa) y 100-120 kg, 0,5 ml (12.500 UI anti-Xa). En pacientes de > 120 kg de peso, la dosis a administrar debe ajustarse al peso, a razón de 115 UI anti-Xa/kg/día, considerando la concentración de 25.000 UI/ml.
Salvo contraindicación se iniciará tratamiento anticoagulante oral entre los días 3-5 después de comenzar la administración de HIBOR, en dosis ajustadas para mantener el INR de 2 a 3 sobre el valor control. La administración de bemiparina puede interrumpirse una vez alcanzado el citado valor de INR. La anticoagulación oral debería continuarse durante un mínimo de 3 meses.
En pacientes con trombosis venosa profunda y factores de riesgo transitorios, como alternativa terapéutica a la administración de anticoagulantes orales o en casos de contraindicación de su uso, se podrá administrar HIBOR a la dosis fija de 3.500 UI una vez al día hasta un máximo de tres meses.
Población pediátrica
HIBOR no está recomendado para uso en niños menores de 18 años debido a la ausencia de datos sobre seguridad y eficacia.
Ancianos
No se requiere ajuste de dosis, a menos que la función renal esté alterada (Ver secciones: 4.2 Posología y forma de administración, Insuficiencia renal; 4.4 Advertencias y precauciones especiales de empleo; 5.2 Propiedades farmacocinéticas).
Insuficiencia renal
(Ver secciones 4.4 Advertencias y precauciones especiales de empleo y 5.2 Propiedades farmacocinéticas)
- Insuficiencia renal leve o moderada (aclaramiento de creatinina 30-80 ml/min): no es necesario ajustar la dosis. Sin embargo, se recomienda un seguimiento clínico cuidadoso.
- Insuficiencia renal grave (aclaramiento de creatinina <30 ml/min): la farmacocinética de bemiparina puede verse afectada. Después de una cuidadosa valoración del riesgo de hemorragias y trombosis en pacientes con insuficiencia renal grave (especialmente si presentan embolismo pulmonar), puede ser necesario ajustar la dosis. De acuerdo a los datos farmacocinéticos en pacientes con insuficiencia renal grave se recomienda ajustar la dosis al 75% (aproximadamente 85 UI anti-Xa/kg una vez al día) para el tratamiento de la trombosis venosa profunda establecida, durante la fase aguda. Se
¿ijiZ
recomienda un seguimiento clínico cuidadoso. Debe considerarse realizar una medida de los niveles anti-Xa sobre las 4 horas de la administración de una dosis.
Insuficiencia hepática
No hay datos suficientes para recomendar un ajuste de la dosis de bemiparina en este grupo de pacientes.
Forma de administración
Técnica de la inyección subcutánea:
Debe seguir estos pasos:
- Lávese bien las manos. El paciente debe estar sentado o tumbado en una posición cómoda en el momento de la administración de Hibor.
- La administración de HIBOR por vía subcutánea se realiza inyectando la jeringa en el tejido celular subcutáneo de la cintura abdominal anterolateral o posterolateral, a 5 centímetros del ombligo y de cualquier cicatriz o moratón. Limpie bien la piel de esa zona.
- Utilice cada día sitios diferentes para la inyección, por ejemplo, primero en el lado izquierdo y la próxima vez en el derecho.
- Quite el capuchón que tapa la aguja de la jeringa de HIBOR.
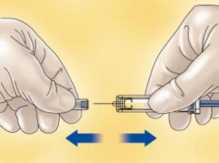
Para mantener la aguja estéril, asegúrese de que no toca nada.
La jeringa precargada ya está lista para su uso.
Antes de la inyección, las jeringas no deben ser purgadas, porque puede perder medicamento.
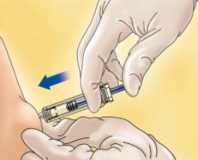
Coja la jeringa con una mano y con la otra, usando los dedos índice y pulgar, coja un pellizco de la zona de piel que había limpiado para formar un pliegue.
Introduzca la aguja entera en el pliegue de piel manteniendo la jeringa lo más erguida posible sobre la superficie del cuerpo, en un ángulo de 90°.
Empuje el vástago asegurándose de que mantiene el pliegue de piel en la misma posición hasta que el vástago esté abajo del todo.
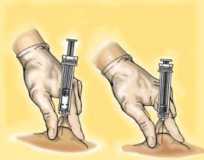
Retire la jeringa del lugar de la inyección manteniendo el dedo sobre el vástago del émbolo y la jeringa erguida. Suelte el pliegue de piel.
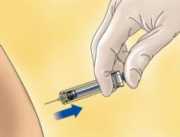
Para jeringas con dispositivo de seguridad: Oriente la aguja lejos de usted y de cualquiera que se encuentre presente, active el sistema de seguridad presionando firmemente sobre el vástago del émbolo. La funda protectora cubrirá automáticamente la aguja y se percibirá un clic audible que confirmará la activación del protector.
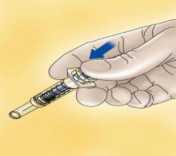
Deseche inmediatamente la jeringa arrojándola al contenedor de objetos punzantes más cercano (la aguja hacia dentro), cierre bien el contenedor con la tapa y póngalo fuera del alcance de los niños.
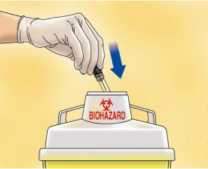
Advertencias:
- El sistema de seguridad sólo puede activarse una vez que se ha vaciado la jeringa.
- La activación del sistema de seguridad sólo debe efectuarse tras retirar la aguja de la piel del paciente.
- No reutilice la protección de la aguja tras la inyección.
- La activación del sistema de seguridad puede salpicar una mínima cantidad de líquido. Para su máxima seguridad, active el sistema de seguridad orientándolo hacia abajo y lejos de usted y de cualquiera que esté presente.
No frote la piel donde se ha puesto la inyección. Esto ayudará a evitar que salgan moratones.
4.2.1 Posología
4.2.2 Forma de administración
4.3 Contraindicaciones
Hipersensibilidad a bemiparina sódica, heparina , sustancias de origen porcino o a alguno de los excipientes incluidos en la sección 6.1.
Antecedentes o sospecha de trombocitopenia inducida por heparina mediada inmunológicamente (TIH) (ver sección 4.4).
Hemorragia activa o incremento del riesgo de sangrado debido a alteraciones de la hemostasia.
Trastorno grave de la función hepática o pancreática.
Daños o intervenciones quirúrgicas en el sistema nervioso central, ojos y oídos que hayan tenido lugar en los últimos 2 meses.
Coagulación Intravascular Diseminada (CID) atribuible a una trombocitopenia inducida por heparina. Endocarditis bacteriana aguda y endocarditis lenta.
Lesiones orgánicas susceptibles de sangrar (ej.: úlcera péptica activa, accidente cerebrovascular hemorrágico, aneurismas o neoplasias cerebrales).
En pacientes que reciban heparina con fines de tratamiento y no de profilaxis, está contraindicada la utilización de anestesia regional en las intervenciones quirúrgicas programadas.
4.4 Advertencias y precauciones especiales de empleo
No administrar por vía intramuscular.
Debido al riesgo de hematoma durante la administración de bemiparina, debería evitarse la inyección intramuscular de otros agentes.
En pacientes con insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min) puede verse afectada la farmacocinética de bemiparina. Se recomienda un control clínico regular en esta población especialmente si se le administran dosis terapéuticas. Tras la valoración individual del riesgo trombótico y hemorrágico, podría ser necesario un ajuste de dosis. En pacientes con insuficiencia renal leve o moderada (aclaramiento de creatinina 30-80 ml/min) no se requiere ajuste de dosis aunque se recomienda un seguimiento clínico cuidadoso (ver secciones 4.2 Posología y forma de administración y 5.2 Propiedades farmacocinéticas).
Se recomienda tener precaución en los casos de insuficiencia hepática, hipertensión arterial no controlada, antecedentes de úlcera gastroduodenal, trombocitopenia, nefrolitiasis y/o uretrolitiasis, enfermedad vascular de coroides y retina, o cualquier otra lesión orgánica susceptible de sangrar, o en pacientes sometidos a anestesia espinal o epidural y/o punción lumbar.
Bemiparina, al igual que otras HBPM, puede suprimir la secreción suprarrenal de la aldosterona ocasionando una hiperpotasemia, especialmente en pacientes con diabetes mellitus, insuficiencia renal crónica, antecedentes de acidosis metabólica, niveles elevados de potasio en plasma o aquellos que estén recibiendo fármacos ahorradores de potasio. El riesgo de hiperpotasemia parece aumentar con la duración de la terapia pero es normalmente reversible (ver sección 4.8). Deben medirse los electrolitos séricos en pacientes de riesgo antes de comenzar la terapia con bemiparina y controlarlos regularmente a partir de ese momento especialmente si el tratamiento se prolonga más de 7 días.
Se han comunicado casos de trombocitopenia transitoria leve (tipo I) al inicio del tratamiento con heparina con recuento de plaquetas entre 100.000/mm3 y 150.000/mm3 debido a una activación plaquetaria temporal (ver sección 4.8). Por regla general no se producen complicaciones y el tratamiento puede continuar.
En raras ocasiones se han observado casos de trombocitopenia grave mediada por anticuerpos (tipo II) con recuentos de plaquetas claramente inferiores a 100.000/ mm3(ver sección 4.8). Estos efectos suelen aparecer entre el 5° y el 21° día de tratamiento, aunque pueden manifestarse mucho antes si hay antecedentes de trombocitopenia inducida por heparina.
Por ello, antes de comenzar la administración de bemiparina, se recomienda efectuar un recuento de plaquetas en el primer día de tratamiento y posteriormente de forma regular cada 3 o 4 días, y al final del
tratamiento. En la práctica, el tratamiento deberá interrumpirse de forma inmediata y se iniciará una terapia alternativa, si se observa una reducción significativa de las plaquetas (30-50%) asociada con resultados positivos o desconocidos del test in-vitro de anticuerpos plaquetarios en presencia de bemiparina, otras HBPM y/o heparinas.
Se han descrito con bemiparina, al igual que con otras heparinas, algunos casos de necrosis cutánea, precedida, a veces, por púrpura o lesiones eritematosas dolorosas (ver sección 4.8). En tales casos se aconseja suspender inmediatamente el tratamiento.
En pacientes sometidos a anestesia epidural o espinal o a punción lumbar, la administración de heparina con fines profilácticos se ha asociado muy raramente a la aparición de hematomas epidurales o espinales, con el resultado final de parálisis prolongada o permanente (ver sección 4.8). Este riesgo se incrementa por el uso de catéteres epidurales o espinales para anestesia, la administración concomitante de medicamentos con acción sobre la coagulación como antiinflamatorios no esteroideos (AINES), antiagregantes plaquetarios o anticoagulantes (ver sección 4.5), y por las punciones traumáticas o repetidas.
A la hora de decidir el intervalo de tiempo que debe transcurrir entre la administración de heparina a dosis profilácticas y la inserción o retirada de un catéter espinal o epidural, deben tenerse en cuenta las características del paciente y del producto. La siguiente dosis de bemiparina deberá ser administrada al menos 4 horas después de la extracción del catéter. La siguiente dosis deberá retrasarse hasta que la intervención quirúrgica haya finalizado. Si el paciente recibiera dosis de tratamiento de bemiparina sódica (115 UI/Kg una vez al día) sería necesario aumentar el tiempo de espera (24 horas).
Si bajo criterio médico se decide administrar tratamiento anticoagulante durante un procedimiento anestésico espinal o epidural debe extremarse la vigilancia del paciente y realizar controles frecuentes, para detectar precozmente cualquier signo o síntoma de déficit neurológico, como dolor de espalda, déficit sensorial y motor (entumecimiento y debilidad de extremidades inferiores) y trastornos funcionales del intestino o vejiga. El personal de enfermería debe ser entrenado para detectar tales signos y síntomas. Así mismo, se advertirá a los pacientes de que informen inmediatamente al médico o personal de enfermería si experimentan cualquiera de los síntomas antes descritos.
Si se sospecha la aparición de algún signo o síntoma sugestivo de hematoma espinal o epidural, deben realizarse las pruebas diagnósticas con carácter de urgencia e iniciar un tratamiento urgente, incluyendo la descompresión medular.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones de bemiparina sódica con otros fármacos, por lo que la información de este apartado se deriva de los datos disponibles para otras heparinas de bajo peso molecular.
No se recomienda la administración concomitante de bemiparina con los siguientes fármacos:
Antagonistas de la vitamina K, salvo en la fase aguda del tratamiento de pacientes con enfermedad trombovenosa.
Otros anticoagulantes, ácido acetilsalicílico, otros salicilatos y antiinflamatorios no esteroideos, ticlopidina, clopidogrel y otros agentes antiagregantes plaquetarios, glucocorticoides sistémicos y dextrano.
Todos estos fármacos potencian el efecto farmacológico de bemiparina, ya que interfieren con los mecanismos de la coagulación y/o la función plaquetar, con el consiguiente incremento del riesgo de sangrado. Cuando sea imprescindible dicha asociación, deberá realizarse un cuidadoso control analítico y clínico.
Los fármacos que incrementan la concentración de potasio sérico sólo se deberían tomar bajo supervisión médica especial.
La interacción de la heparina con la nitroglicerina intravenosa (que puede resultar en un descenso de su eficacia), no debe descartarse en el caso de la bemiparina.
4.6 Fertilidad, embarazo y lactancia
4.6.1 Embarazo
Los estudios en animales no han mostrado evidencia de efectos teratogénicos con el uso de bemiparina (ver sección 5.3). Los datos clínicos relativos al uso de bemiparina en mujeres embarazadas son escasos. Por lo tanto, deberá administrarse con cuidado en este tipo de pacientes.
Se desconoce si bemiparina atraviesa la barrera placentaria.
4.6.2 Lactancia
No se dispone de información suficiente relativa a la excreción de bemiparina en la leche materna. La absorción oral de bemiparina es improbable. Sin embargo, cuando sea necesario administrar HIBOR a mujeres lactantes, por precaución se les recomendará que eviten la lactancia.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de HIBOR sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
La reacción adversa más frecuente es hematoma y/o equimosis en el lugar de la inyección, que ocurre aproximadamente en el 15% de los pacientes que reciben HIBOR.
La aparición de osteoporosis se ha asociado con tratamientos a largo plazo con heparinas.
Las reacciones adversas se indican por clasificación de órganos y sistemas y frecuencia:
- Muy frecuentes (> 1/10)
- Frecuentes (> 1/100 a < 1/10)
- Poco frecuentes (> 1/1.000 a < 1/100)
- Raras (> 1/10.000 a < 1/1.000)
- Muy raras (< 1/10.000)
- Frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
La frecuencia de reacciones adversas comunicadas con bemiparina es similar a las comunicadas con otras HBPMs y se cita a continuación.
|
Clasificación por órganos y sistemas |
Muy frecuentes (> 1/10) |
Frecuentes (>1/100 a <1/10) |
Poco frecuentes (>1/1.000 a <1/100) |
Raras (>1/10.000 a <1/1.000) |
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles) |
|
Trastornos de la sangre y del sistema linfático |
Complicaciones hemorrágicas (piel, mucosas, heridas, tracto gastrointestinal y urogenital) |
Trombocitopenia transitoria leve (tipo I) (ver sección 4.4) |
Trombocitopenia grave (tipo II) (ver sección 4.4) | ||
|
Trastornos del |
Reacciones |
Reacciones |
|
sistema inmunológico |
alérgicas cutáneas (urticaria, prurito). |
anafilácticas (nauseas, vómitos, fiebre, disnea, broncoespasmo, edema de glotis, hipotensión, urticaria, prurito) | |||
|
Trastornos del metabolismo y de la nutrición |
Hiperpotasemia (ver sección 4.4) | ||||
|
Trastornos hepatobiliares |
Elevación moderada y transitoria de los niveles de transaminasas (Aspartato aminotransferasa: AST, Alanino aminotransferasa: ALT) y gamma-glutamil transpeptidasa (GGT) | ||||
|
Trastornos de la piel y del tejido subcutáneo |
Necrosis cutánea en el lugar de la inyección (ver sección 4.4) | ||||
|
Trastornos generales y alteraciones en el lugar de administración |
Equimosis, hematoma, y dolor en el lugar de la inyección |
Hematomas espinales y epidurales tras anestesia epidural o espinal y punción lumbar. Estos hematomas han causado diferentes grados de déficit neurológico, incluyendo parálisis prolongada o permanente (ver sección 4.4) |
Notificación de sospechas de reacciones adversas:
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de medicamentos de Uso Humano: https://www.notificaram.es.
4.9 Sobredosis
El síntoma clínico principal de sobredosificación es la hemorragia. Si se produce hemorragia debe interrumpirse el tratamiento con bemiparina, dependiendo de la gravedad de la hemorragia y del riesgo de trombosis.
Las hemorragias menores rara vez requieren tratamiento específico. En casos de hemorragia grave puede ser necesaria la utilización de sulfato de protamina.
La neutralización de bemiparina con sulfato de protamina se ha estudiado en un sistema in-vitro e in-vivo, con el objeto de observar la reducción de la actividad anti-Xa y su efecto sobre el tiempo parcial de tromboplastina activada (TPTA). El sulfato de protamina produce un descenso parcial de la actividad antiXa durante las 2 horas siguientes a su administración intravenosa, a una dosis de 1,4 mg de sulfato de protamina por cada 100 UI anti-Xa administradas.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agente Antitrombótico, Grupo Heparinas. Código ATC: B01AB12.
Bemiparina sódica es una HBPM obtenida por despolimerización de heparina sódica de mucosa intestinal porcina. Su peso molecular (Pm) medio aproximado es de 3.600 daltons. El porcentaje de cadenas de Pm inferior a 2.000 daltons es menor del 35%, el porcentaje de cadenas de Pm entre 2.000 y 6.000 daltons está comprendido entre el 50% y el 75%, y el porcentaje de cadenas de Pm superior a 6.000 daltons es menor del 15%.
Su actividad anti-Xa está comprendida entre 80 y 120 UI anti-Xa por miligramo y su actividad anti-IIa está comprendida entre 5 y 20 UI anti-IIa por miligramo, calculadas en relación a la sustancia seca. La relación entre las actividades anti-Xa y anti-IIa es aproximadamente de 8.
En modelos de experimentación animal, bemiparina ha mostrado actividad antitrombótica y un moderado efecto hemorrágico.
En humanos, bemiparina confirma su eficacia antitrombótica y no produce, a las dosis recomendadas, prolongación significativa de los tests globales de coagulación.
5.2 Propiedades farmacocinéticas
Los parámetros farmacocinéticos de bemiparina han sido estudiados a partir de la evolución de la actividad anti-Xa plasmática. La determinación se efectúa por método amidolítico, de acuerdo al primer estándar internacional para heparinas de bajo peso molecular LMWH (NIBSC).
Los procesos de absorción y eliminación siguen una cinética lineal, de orden 1.
5.2.1 Absorción
Tras la inyección por vía sc, la absorción es rápida y se estima que la biodisponibilidad es del 96%. El efecto máximo anti-Xa a dosis profilácticas de 2.500 UI y 3.500 UI se observó entre 2 y 3 horas después de la inyección por vía sc de bemiparina, alcanzando valores de 0,34 ± (0,08) y 0,45 ± (0,07) UI anti-Xa/ml respectivamente, sin que se detectase actividad anti-IIa. El efecto máximo anti-Xa a dosis de tratamiento de 5.000 UI, 7.500 UI, 10.000 UI y 12.500 UI se observó entre 3 y 4 horas después de la inyección subcutánea de bemiparina, alcanzando valores de 0,54 ± (0,06), 1,22 ± (0,27), 1,42 ± (0,19) y 2,03 ± (0,25) UI anti-Xa/ml respectivamente, detectándose una actividad anti-IIa de 0,01 UI/ml a las dosis de 7.500 UI, 10.000 UI y 12.500 UI.
5.2.4 Eliminación
Bemiparina en el rango de dosis de 2.500 UI a 12.500 UI tiene una semivida aproximada entre 5 y 6 horas, lo que justifica su administración una vez al día.
Hasta la fecha no hay datos sobre la unión a proteínas plasmáticas, metabolismo y excreción de bemiparina en humanos.
Ancianos: los resultados de un análisis farmacocinético de un ensayo clínico realizado en voluntarios sanos jóvenes y ancianos (>65 años) muestran que no existen diferencias significativas en el perfil cinético de bemiparina entre jóvenes y ancianos cuando la función renal es normal.
Insuficiencia renal: (ver secciones 4.2 Posología y forma de administración y 4.4 Advertencias y precauciones de empleo) los resultados de un análisis farmacocinético de un ensayo clínico realizado en jóvenes, ancianos y sujetos con diferentes grados de insuficiencia renal (aclaramiento de creatinina <80 ml/min), en el que se utilizaron dosis profilácticas múltiples (3.500 UI/24 h) y terapéuticas únicas (115 UI/kg) de bemiparina, mostró una correlación entre el aclaramiento de creatinina y la mayoría de los parámetros farmacocinéticos de actividad anti-Xa. Asimismo se evidenció que la exposición a bemiparina (basada en el AUC de la actividad anti-Xa) se encontró significativamente elevada en el grupo de voluntarios con insuficiencia renal grave (aclaramiento de creatinina <30 ml/min) con respecto al resto de grupos de voluntarios.
Por otro lado, se realizaron simulaciones farmacocinéticas para evaluar el perfil de bemiparina tras la administración de diez dosis diarias consecutivas. El promedio de la actividad anti-Xa máxima (Amax) simulada tras 10 dosis profilácticas (3.500 UI/24 h) se encontraba en todos los grupos entre 0,35 y 0,60 UI anti-Xa/ml; no obstante, en el grupo de insuficiencia renal grave (aclaramiento de creatinina <30 ml/min) un sujeto presentó un valor de Amax de 0,81 UI anti-Xa/ml tras la décima dosis. Al simular una reducción de dosis a 2.500 UI/24 h, el modelo predijo valores de Amax menores de 0,60 UI anti-Xa/ml (valor medio
de Amax= 0,42 UI anti-Xa /ml) para todos los voluntarios del grupo de insuficiencia renal grave. Por otra parte, las medias predichas de Amax tras 10 dosis terapéuticas (115 UI/kg/24 h) se situaron entre 0,89 y 1,22 UI anti-Xa/ml en todos los grupos; igualmente, un voluntario del grupo de insuficiencia renal grave presentó un valor de Amax superior a 2,09 UI anti-Xa/ml tras la última administración. Cuando se simuló el ajuste a un 75% de la dosis terapéutica (86,25 UI/kg/24 h) se predijo una Amax para dicho voluntario de 1,60 UI anti-Xa/ml, y al mismo tiempo la Amax media (0,91 UI anti-Xa/ml) del grupo de insuficiencia renal grave se mantuvo dentro del rango observado para el resto de los grupos sin ajuste de dosis.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos de bemiparina no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis únicas y repetidas, genotoxicidad y toxicidad para la reproducción.
Los estudios de toxicidad aguda y a dosis repetidas tras la administración subcutánea de bemiparina en animales revelan alteraciones que consisten esencialmente en lesiones hemorrágicas reversibles y dosis-dependientes en las áreas de inyección. Éstos se consideraron resultado de una actividad farmacológica exacerbada.
Estudios de toxicidad en la reproducción, realizados con bemiparina en ratas y conejos gestantes, entre los días 6 y 18 de la gestación, no registraron muertes entre las hembras tratadas con bemiparina. Los principales signos clínicos registrados fueron hematomas subcutáneos que fueron atribuíbles a los efectos farmacológicos del ensayo. En el examen de los fetos no se registraron efectos embriotóxicos relacionados con el tratamiento, ni alteraciones externas esqueléticas y/o viscerales.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes Agua para inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
Una vez abierto HIBOR debe utilizarse inmediatamente.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 25° C. No congelar.
6.5 Naturaleza y contenido del envase
Jeringas precargadas desechables (vidrio Tipo I) con vástago de polipropileno, émbolo-tapón de elastómero de clorobutilo y aguja de acero inoxidable, con 0,2 ml, 0,3 ml, 0,4 ml y 0,5 ml de solución inyectable. Envases de 2, 10, 30 y 50 jeringas.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Envase para un sólo uso. Desechar cualquier fracción no utilizada del producto. No administrar si el envase protector está dañado o abierto. Sólo se utilizará si la solución se presenta transparente e incolora o ligeramente amarillenta y exenta de partículas visibles.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
LABORATORIOS FARMACÉUTICOS ROVI, S.A.
Julián Camarillo, 35-28037 MADRID
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
|
HIBOR 5.000 UI |
Número de registro: 64.164 |
|
HIBOR 7.500 UI |
Número de registro: 64.165 |
|
HIBOR 10.000 UI |
Número de registro: 64.166 |
|
HIBOR 12.500 UI |
Número de registro: 74.793 |
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Octubre 2011
10. FECHA DE LA REVISIÓN DEL TEXTO
12/2015
La información detallada de este medicamento está disponible en la página web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) http://www.aemps.gob.es/
12 de 12