Fuzeon 90 Mg/Ml Polvo Y Disolvente Para Solucion Inyectable
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Fuzeon 90 mg/ml polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene 108 mg de enfuvirtida.
Cada ml de solución reconstituida contiene 90 mg de enfuvirtida.
Excipientes con efecto conocido: sodio. Contiene menos de 1 mmol de sodio (23 mg) por dosis; esto es, esencialmente “exento de sodio”.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable Polvo liofilizado de color blanco a blanquecino.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Fuzeon está indicado en combinación con otros medicamentos antirretrovirales en el tratamiento de pacientes infectados por el VIH-1 que han recibido tratamiento, y a los que les han fallado los tratamientos con al menos un medicamento de cada una de las siguientes clases antirretrovirales: inhibidores de la proteasa, inhibidores de la transcriptasa inversa no análogos de nucleósidos e inhibidores de la transcriptasa inversa análogos de nucleósidos, o que tienen intolerancia a tratamientos antirretrovirales previos (ver la sección 5.1).
Para decidir el nuevo tratamiento en los pacientes que hayan fallado a un tratamiento antirretroviral, hay que prestar atención especial a la historia terapéutica de cada paciente y a los patrones de mutaciones asociados con los distintos medicamentos. Se deberían realizar tests de resistencias siempre que fuera posible. (ver las secciones 4.4 y 5.1)
4.2 Posología y forma de administración
Fuzeon debe ser prescrito por médicos con experiencia en el tratamiento de la infección por VIH. Posología
Adultos y adolescentes >16 años: La dosis recomendada de Fuzeon es de 90 mg, dos veces al día, en inyección subcutánea en el brazo, la cara anterior del muslo o el abdomen.
En caso de que se olvide una dosis de Fuzeon, se debe instruir a los pacientes a administrarse la dosis lo antes posible, si quedan menos de 6 horas antes de la siguiente dosis habitual, se debe omitir la dosis olvidada.
Pacientes ancianos: No hay experiencia en pacientes mayores de 65 años.
Niños > 6 años y adolescentes: La experiencia en niños es limitada (ver la sección 5.2). La pauta posológica que se utilizó en los ensayos clínicos es la que se indica en la Tabla 1:
Tabla 1: Posología en pediatría
|
Peso (kg) |
Dosis para una inyección dos veces al día (mg/dosis) |
Volumen de inyección (90 mg de enfuvirtida por ml) |
|
de 11,0 a 15,5 |
27 |
0,3 ml |
|
de 15,6 a 20,0 |
36 |
0,4 ml |
|
de 20,1 a 24,5 |
45 |
0,5 ml |
|
de 24,6 a 29,0 |
54 |
0,6 ml |
|
de 29,1 a 33,5 |
63 |
0,7 ml |
|
de 33,6 a 38,0 |
72 |
0,8 ml |
|
de 38,1 a 42,5 |
81 |
0,9 ml |
|
> 42,6 |
90 |
1,0 ml |
Fuzeon no está recomendado para su uso en niños menores de 6 años debido a que los datos sobre seguridad y eficacia disponibles no son suficientes (ver sección 5.2).
Insuficiencia renal: No es necesario ajustar la dosis de los pacientes con insuficiencia renal incluyendo aquellos tratados con diálisis. (ver las secciones 4.4 y 5.2).
Insuficiencia hepática: No se dispone de datos para establecer recomendaciones posológicas para los pacientes con insuficiencia hepática. (ver las secciones 4.4 y 5.2).
Forma de administración
Fuzeon sólo debe administrarse en inyección subcutánea. Para consultar las instrucciones de reconstitución antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Fuzeon debe administrarse como parte de un tratamiento combinado. Se debe consultar también los resúmenes de las características del producto de los otros medicamentos antirretrovirales que se empleen en la combinación. Al igual que otros antirretrovirales, la enfuvirtida debe combinarse lo más adecuadamente posible con otros antirretrovirales a los cuales el virus del paciente sea sensible. (ver la sección 5.1)
Se debe informar a los pacientes que Fuzeon no cura la infección por el VIH-1. A pesar de que se ha probado que la supresión viral con tratamiento antirretroviral eficaz reduce sustancialmente el riesgo de transmisión sexual, no se puede excluir un riesgo residual. Se deben tomar precauciones, conforme a las directrices nacionales, para prevenir la transmisión.
Los estudios en animales han demostrado que enfuvirtida puede afectar a algunas de las funciones inmunes (ver sección 5.3). En ensayos clínicos se ha observado un aumento de la incidencia de algunas infecciones bacterianas, de manera más notable una mayor incidencia de neumonía, en pacientes tratados con Fuzeon; sin embargo, un aumento del riesgo de neumonía bacteriana relacionado con el uso de Fuzeon no ha sido confirmado mediante datos epidemiológicos posteriores.
El tratamiento con enfuvirtida se ha asociado ocasionalmente con reacciones de hipersensibilidad y en raras ocasiones, las reacciones de hipersensibilidad se han repetido con la reexposición. Las complicaciones comprenden rash, fiebre, náuseas y vómitos, escalofríos, rigidez, tensión sanguínea baja y transaminasas hepáticas elevadas en suero en varias combinaciones, y posible reacción primaria del complejo inmune, dificultad respiratoria y glomerulonefritis. Los pacientes que presenten signos o síntomas de una reacción de hipersensibilidad sistémica suspenderán el tratamiento con enfuvirtida y acudirán al médico para su evaluación de inmediato. El tratamiento con enfuvirtida no se debe reiniciar tras la aparición de signos y síntomas sistémicos consistentes con una reacción de hipersensibilidad que se considere como relacionada con enfuvirtida. No se han identificado los factores de riesgo que predicen la aparición o la gravedad de la hipersensibilidad a la enfuvirtida.
Enfermedad hepática: La seguridad y la eficacia de la enfuvirtida no se han estudiado de manera específica en pacientes con trastornos hepáticos subyacentes significativos. Los pacientes con hepatitis crónica B y C, en tratamiento con antirretrovirales tienen mayor riesgo de experimentar acontecimientos adversos hepáticos graves o potencialmente mortales. Algunos pacientes que participaron en los ensayos en fase III estaban co-infectados por la hepatitis B/C. En ellos la adición de Fuzeon no aumentó la incidencia de acontecimientos hepáticos. En caso de tratamiento antirretroviral concomitante para la hepatitis B o C, se deberá consultar también la información del producto relevante para estos medicamentos.
La administración de Fuzeon a sujetos no infectados por el VIH-1 puede provocar la formación de anticuerpos antienfuvirtida que reaccionen de forma cruzada con el antígeno gp41 del VIH. Esto podría ocasionar un resultado falso positivo del test de ELISA de anticuerpos anti-VIH.
No hay experiencia en pacientes con la función hepática disminuida. Los datos son limitados en pacientes con insuficiencia renal de moderada a grave y en pacientes mantenidos con diálisis. Fuzeon se debe emplear con precaución en estas poblaciones (ver las secciones 4.2 y 5.2).
Síndrome de Reconstitución Inmune: Cuando se instaura una terapia antirretroviral combinada en pacientes infectados por VIH con deficiencia inmune grave puede aparecer una respuesta inflamatoria frente a patógenos oportunistas latentes o asintomáticos y provocar situaciones clínicas graves, o un empeoramiento de los síntomas. Normalmente estas reacciones se han observado en las primeras semanas o meses después del inicio de la terapia antirretroviral combinada. Algunos ejemplos relevantes de estas reacciones son: retinitis por citomegalovirus, infecciones micobacterianas generalizadas y/o localizadas y neumonía por Pneumocystis carinii. Se debe evaluar cualquier síntoma inflamatorio y establecer un tratamiento cuando sea necesario.
También se ha notificado la aparición de trastornos autoinmunitarios (como por ejemplo la enfermedad de Graves) durante la reconstitución inmune; sin embargo, el tiempo notificado hasta su aparición es más variable y estos acontecimientos pueden suceder muchos meses después del inicio del tratamiento.
Osteonecrosis:
Se han notificado casos de osteonecrosis, especialmente en pacientes con infección avanzada por VIH y/o exposición prolongada al tratamiento antirretroviral combinado (TARC), aunque se considera que la etiología es multifactorial (incluyendo uso de corticosteroides, consumo de alcohol, inmunodepresión grave, índice de masa corporal elevado). Se debe aconsejar a los pacientes que consulten al médico si experimentan molestias o dolor articular, rigidez articular o dificultad para moverse.
4.5 Interacción con otros medicamentos y otras formas de interacción
Los estudios de interacciones se han realizado sólo en adultos.
No se esperan interacciones farmacocinéticas con repercusión clínica entre la enfuvirtida y los otros medicamentos que se administran de forma simultánea y se metabolizan por las enzimas del citocromo P450.
Efecto de la enfuvirtida sobre el metabolismo de medicamentos concomitantes: Según un estudio in vivo en el metabolismo humano, la enfuvirtida, en las dosis recomendadas de 90 mg dos veces al día, no inhibe el metabolismo de los sustratos de las isoenzimas CYP3A4 (dapsona), la CYP2D6 (debrisoquina), la CYP1A2 (cafeína), la CYP2C19 (mefenitoína) y la CYP2E1 (clorzoxazona).
Efecto de medicamentos concomitantes sobre el metabolismo de la enfuvirtida: Según estudios independientes de interacción farmacocinética, la co-administración de ritonavir (inhibidor potente de la CYP3A4) o saquinavir en combinación con una dosis potenciada de ritonavir o rifampicina (inductor potente de la CYP3A4) no produjo cambios clínicos significativos de la farmacocinética de la enfuvirtida.
4.6 Fertilidad, embarazo y lactancia
Embarazo: No se han realizado estudios adecuados y bien controlados en mujeres embarazadas. Los estudios en animales no muestran efectos dañinos sobre el desarrollo fetal. La enfuvirtida debe utilizarse durante el embarazo solo si el beneficio potencial justifica el riesgo potencial para el feto.
Lactancia: No se sabe si la enfuvirtida se excreta por la leche humana. Se debe informar a las madres que no deben dar el pecho si están recibiendo enfuvirtida debido a la posibilidad de transmisión del VIH y de cualquier posible efecto adverso en los lactantes.
4.7 Efectos sobre la capacidad para conducir o utilizar máquinas
No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas. No existen pruebas de que la enfuvirtida pueda alterar la capacidad del paciente para conducir o utilizar máquinas; no obstante, hay que tener en cuenta el perfil de reacciones adversas de la enfuvirtida. (Ver la sección 4.8)
4.8 Reacciones adversas
a. Resumen del perfil de seguridad
Los datos de seguridad se refieren principalmente a los datos de 48 semanas de los ensayos TORO 1 y TORO 2 combinados (ver la sección 5.1). Los resultados de seguridad se expresan como el número de pacientes con una reacción adversa por cada 100 pacientes-año de exposición (a excepción de las reacciones en el lugar de inyección).
Los acontecimientos notificados más frecuentemente fueron reacciones en el lugar de inyección, diarrea y náuseas. La adición de Fuzeon a la terapia antirretroviral previa generalmente no aumenta la frecuencia o la gravedad de la mayoría de las reacciones adversas.
b. Tabla de reacciones adversas
La Tabla 2 muestra los acontecimientos observados en una proporción mayor entre pacientes tratados con Fuzeon + OB que entre pacientes tratados solo con régimen OB; este incremento, ajustado por la exposición, fue de al menos 2 pacientes con acontecimiento por cada 100 pacientes-año. Se observó un incremento estadísticamente significativo para neumonía y linfoadenopatía. La mayoría de las reacciones adversas fueron de intensidad leve o moderada. Las reacciones adversas se ordenan según el sistema de clasificación de órganos de MeDRA y por categoría de frecuencia. Las categorías de frecuencia se definen utilizando la siguiente convención: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 2: Reacciones adversas atribuidas al tratamiento con Fuzeon en los ensayos TORO 1 y TORO 2 combinados
|
Sistema de clasificación de órganos Frecuencia |
Reacción adversa |
|
Infecciones e infestaciones Frecuentes |
Sinusitis, papiloma cutáneo, gripe, neumonia, infección de oído |
|
Trastornos de la sangre y del sistema linfático Frecuentes |
Linfadenopatía |
|
Trastornos del metabolismo y de la nutrición Frecuentes |
Disminución del apetito, anorexia, hipertrigliceridemia, incremento de triglicéridos en sangre, diabetes mellitus |
|
Trastornos psiquiátricos Frecuentes |
Ansiedad, pesadillas, irritabilidad |
|
Trastornos del sistema nervioso Muy frecuentes Frecuentes |
Neuropatía periférica Hipoestesia, problemas de atención, temblores |
|
Trastornos oculares Frecuentes |
Conjuntivitis |
|
Trastornos del oído y del laberinto Frecuentes |
Vértigo |
|
Trastornos respiratorios, torácicos y mediastínicos Frecuentes |
Congestión nasal |
|
Trastornos gastrointestinales Frecuentes |
Pancreatitis, reflujo gastroesofágico |
|
Trastornos de la piel y del tejido subcutáneo Frecuentes |
Sequedad de piel, eczema seborreico, eritema, acné |
|
Trastornos musculoesqueléticos, del tejido conjuntivo y óseos Frecuentes |
Mialgia |
|
Trastornos renales y urinarios Frecuentes |
Nefrolitiasis, hematuria |
|
Trastornos generales y alteraciones en el lugar de administración Muy frecuentes Frecuentes |
Pérdida de peso Enfermedad pseudogripal, debilidad |
c. Descripción de reacciones adversas seleccionadas Reacciones en el lugar de inyección
Las reacciones en el lugar de inyección (RLIs) fueron las reacciones adversas más frecuentes y ocurrieron en el 98% de los pacientes (Tabla 3). La inmensa mayoría de las RLIs ocurrieron en la primera semana de tratamiento con Fuzeon, y se asociaron con un dolor o molestia de carácter leve a moderado en el lugar de inyección, que no limitaron las actividades habituales. La gravedad del dolor y de las molestias no aumentaron con la duración del tratamiento. Por lo general, la duración de los signos y los síntomas fue igual o menor de 7 días. Las infecciones en el lugar de inyección (incluidos los abscesos y la celulitis) afectaron al 1,5% de los pacientes.
Tabla 3: Resumen de los signos y síntomas individuales característicos de las reacciones
locales en el lugar de inyección en los ensayos TORO 1 y TORO 2 combinados (% de pacientes)__
|
n=663 | |||
|
Tasa de abandonos por RLI |
4% | ||
|
Categoría de la reacción |
Fuzeon + Tratamiento Optimizado11 |
% de acontecimientos con reacciones de grado 3 |
% de acontecimientos con reacciones de grado 4 |
|
Dolor / molestias |
96,1% |
11,0%b |
0% |
|
Eritema |
90,8% |
23,8%c |
10,5%c |
|
Induración |
90,2% |
43,5%d |
19,4%d |
|
Nódulos y quistes |
80,4% |
29,1%e |
0,2%e |
|
Prurito |
65,2% |
3,9%f |
No procede |
|
Equimosis |
51,9% |
8,7%g |
4,7%g |
“Cualquier grado de intensidad.
bGrado 3 = dolor intenso con necesidad de analgésicos (o administración de analgésicos narcóticos durante < 72 horas) y/o con limitación de las actividades habituales; Grado 4 = dolor intenso con necesidad de hospitalización o prolongación de la estancia hospitalaria, causante de muerte, de discapacidad/incapacidad persistente o graves, potencialmente mortal, o de importancia médica.
cGrado 3 = diámetro medio > 50 mm pero < 85 mm; Grado 4 = diámetro medio > 85 mm. dGrado 3 = diámetro medio > 25 mm pero < 50 mm; Grado 4 = diámetro medio > 50 mm. eGrado 3 = > 3 cm; Grado 4 = drenaje.
fGrado 3 = resistente al tratamiento por vía tópica o con necesidad de tratamiento oral o parenteral; Grado 4 = no definido. gGrado 3 = > 3 cm pero < 5 cm; Grado 4= > 5 cm.
Además, se observó un pequeño número de reacciones de hipersensibilidad, atribuidas a la enfuvirtida y, en algunos casos recidivaron tras la reexposición (ver la sección 4.4).
Otras reacciones adversas
Al inicio de la terapia antirretroviral combinada, en los pacientes infectados por VIH con deficiencia inmune grave, puede aparecer una respuesta inflamatoria frente a infecciones oportunistas latentes o asintomáticas. También se han notificado trastornos autoinmunitarios (como por ejemplo la enfermedad de Graves); sin embargo, el tiempo notificado hasta su aparición es más variable y estos acontecimientos pueden suceder muchos meses después del inicio del tratamiento (ver sección 4.4).
Se han notificado casos de osteonecrosis, especialmente en pacientes con factores de riesgo generalmente reconocidos, enfermedad avanzada por VIH o exposición prolongada al tratamiento antirretroviral combinado (TARC). Se desconoce la frecuencia de esta reacción adversa (ver sección 4.4).
Enfuvirtida, como péptido, puede causar amiloidosis cutánea en el lugar de inyección.
Anomalías de laboratorio
La mayoría de los pacientes no experimentó cambios en el grado de toxicidad de ninguno de los parámetros de laboratorio a lo largo del estudio, a excepción de los relacionados en la Tabla 4. Durante la semana 48, la eosinofilia [recuento mayor al Límite Superior de Normalidad (ULN)
> 0,7 x 109/l ] se dio en mayor porcentaje entre el grupo de los pacientes tratados con Fuzeon (12,4 pacientes con acontecimiento por cada 100 pacientes-año) que en los tratados solamente con OB (5,6 pacientes con acontecimiento por cada 100 pacientes-año). Cuando se usa un umbral más alto para la eosinofilia (> 1,4 x 109/l), la proporción de eosinofilia ajustada por la exposición del paciente es igual en ambos grupos (1,8 pacientes con acontecimiento por cada 100 pacientes-año).
Tabla 4: Exposición ajustada de anomalías de laboratorio Grado 3 y 4 entre pacientes
tratados con Fuzeon + OB y solo con OB, notificados en más de 2 pacientes con acontecimiento por cada 100 pacientes-año__
|
Parámetros de laboratorio |
Régimen de Fuzeon + OB |
Régimen de OB solo |
|
Grado |
Por cada 100 pacientes-año |
Por cada 100 pacientes-año |
|
n |
663 |
334 |
|
(Exposición Total de los pacientes, por año de tratamiento) |
(557,0) |
(162,1) |
|
ALAT | ||
|
Gr. 3 (> 5-10 x ULN) |
4,8 |
4,3 |
|
Gr. 4 (> 10 x ULN) |
1,4 |
1,2 |
|
Hemoglobina | ||
|
Gr. 3 (6,5-7,9 g/dl) |
2,0 |
1,9 |
|
Gr. 4 (< 6,5 g/dl) |
0,7 |
1,2 |
|
Creatinina Fosfoquinasa | ||
|
Gr. 3 (> 5-10 x ULN) |
8,3 |
8,0 |
|
Gr. 4 (> 10 x ULN) |
3,1 |
8,6 |
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se han notificado casos de sobredosis. La dosis máxima administrada a 12 pacientes fue de 180 mg en inyección subcutánea única durante un ensayo clínico. Estos pacientes no experimentaron ninguna reacción adversa que no se observara ya con las dosis recomendadas. En un estudio correspondiente al Programa de Acceso Precoz, se administró en una ocasión 180 mg de Fuzeon a un paciente como dosis única. El paciente no experimentó ninguna reacción adversa como resultado.
No existe ningún antídoto específico para tratar la sobredosis de enfuvirtida. El tratamiento de la sobredosis consistiría en las medidas generales de apoyo.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico:Otros antivirales. Código ATC: J05A X07
Mecanismo de acción: La enfuvirtida es un miembro de la clase terapéutica denominada inhibidores de la fusión. Se trata de un inhibidor de la reordenación estructural de la gp41 de VIH-1 y actúa uniéndose extracelularmente a esta proteína del virus de manera específica, bloqueando la fusión entre la membrana del virus y la membrana de la célula diana, previniendo la entrada del ARN viral en dicha célula.
Actividad antiviral in vitro: La susceptibilidad basal a la enfuvirtida, medida en 612 VIH recombinantes que contenían genes env, procedentes de muestras de ARN del VIH de pacientes de ensayos en fase III, resultó en una media geométrica EC50 de 0,259 microgramo/ml (media geométrica + 2DE = 1,96 microgramo/ml) en un ensayo de entrada de recombinación de genotipos VIH. Además, la enfuvirtida inhibió la fusión entre células mediada por la cubierta del VIH-1. Los estudios de combinación de enfuvirtida con miembros representativos de las distintas clases antirretrovirales mostraron una actividad antiviral de aditiva a sinérgica y la ausencia de antagonismo. No se ha establecido ninguna relación entre la sensibilidad in vitro del VIH-1 a la enfuvirtida y la inhibición de la replicación de VIH-1 en la especie humana.
Resistencia a los antirretrovirales: La supresión viral incompleta puede llevar al desarrollo de resistencia farmacológica a uno o más componentes del tratamiento.
Resistencia in vitro a la enfuvirtida: Se han seleccionado cepas de VIH-1 in vitro con menor sensibilidad a la enfuvirtida que contienen sustituciones en los aminoácidos (aa) 36-38 del ectodominio gp41. Estos cambios se correlacionan con varios niveles de disminución en la sensibilidad a la enfuvirtida de cepas de VIH con mutación específica de sitio.
Resistencia in vivo a la enfuvirtida: Se observó una reducción de la susceptibilidad a la enfuvirtida de más de 4 veces al comparar los recombinantes VIH que contenían genes env, obtenidos a partir de muestras de ARN del VIH tomadas antes de la semana 24, y procedentes de 187 pacientes participantes en los ensayos clínicos en fase III, frente a las muestras pretratamiento correspondientes. De estos, 185 (98,9%) genes env portaban sustituciones específicas en la región del aa 36-45 de la gp41. Las sustituciones observadas, en orden decreciente de frecuencia, se localizaron en los aminoácidos de las posiciones 38, 43, 36, 40, 42 y 45. Cada sustitución única específica en estos residuos de la gp41 originó una disminución gradual en la susceptibilidad viral recombinante a la enfuvirtida en comparación con las condiciones iniciales. Los cambios de la media geométrica oscilaban entre 15,2 veces para V38M y 41,6 veces para V38A. Los ejemplos de sustituciones múltiples fueron insuficientes para determinar un patrón de sustituciones consistente o su efecto en la susceptibilidad viral a la enfuvirtida. No se ha establecido ninguna relación entre estas sustituciones y la eficacia in vivo del tratamiento con enfuvirtida. La disminución de la sensibilidad viral estaba correlacionada con el grado de resistencia pretratamiento a la terapia optimizada. (ver Tabla 6)
Resistencia cruzada: Como consecuencia de la nueva diana viral, la enfuvirtida se muestra igual de eficaz in vitro frente a las cepas salvajes de laboratorio y clínicas que frente a aquellas con resistencia a 1, 2 ó 3 clases de antirretrovirales diferentes (inhibidores de la transcriptasa inversa análogos de nucleósidos, inhibidores de la transcriptasa inversa no análogos de nucleósidos e inhibidores de la proteasa). A la inversa, las mutaciones de los aminoácidos 36-45 de la gp41, que confieren resistencia a la enfuvirtida, no deberían dar resistencia cruzada a otras clases de antirretrovirales.
Datos clínicos farmacodinámicos
Ensayos en Pacientes Pretratados con Antirretrovirales: La actividad clínica de Fuzeon (en combinación con otros antirretrovirales) sobre los valores plasmáticos del ARN del VIH y recuento de linfocitos CD4 se ha investigado en dos ensayos aleatorizados, multicéntricos y controlados (TORO 1 y TORO 2) de 48 semanas de duración. La población por intención de tratar comprendía 995 pacientes. Los datos demográficos de los pacientes de cada grupo, Fuzeon + OB y OB, fueron una mediana basal de ARN del VIH-1 de 5,2 logi0 copias/ml y 5,1 logi0 copias/ml y una mediana basal del recuento de células CD4 de 88 células/mm3 y 97 células/mm3, respectivamente. Los pacientes tuvieron una exposición previa a una mediana de 12 antirretrovirales durante una mediana de 7 años. Todos los pacientes recibieron un tratamiento optimizado (OB) que contenía de 3 a 5 agentes antirretrovirales seleccionados de acuerdo con la historia terapéutica previa de cada paciente, así como con los resultados basales de resistencias virales genotípicas y fenotípicas.
La proporción de pacientes que alcanzaron una carga viral de < 400 copias/ml en la semana 48 fue del 30,4% entre los pacientes en el régimen de Fuzeon + OB comparado con el 12% entre los pacientes que recibieron el régimen de OB sólo. El principal aumento de recuento de células CD4 fue mayor en los pacientes en el régimen de Fuzeon + OB que en los pacientes en el régimen OB sólo (Ver Tabla 5).
Tabla 5 Resultados a las 48 semanas del tratamiento randomizado (análisis combinado de
los ensayos TORO 1 y TORO 2, ITT)
|
Resultados |
Fuzeon + OB 90 mg bid (dos veces al día) (N=661) |
OB (N=334) |
Diferencia del Tratamiento |
Intervalo de Confianza 95% |
Valor-p |
|
ARN del VIH-1 Cambio Logarítmico desde el inicio (log10 copias/ml)* |
-1,48 |
-0,63 |
LSM -0,85 |
-1,073, -0,628 |
<,0001 |
|
Recuento de células CD4+ Cambio desde el inicio (células/mm3)# |
+91 |
+45 |
LSM 46,4 |
25,1, 67,8 |
<,0001 |
|
ARN del VIH >1 log por debajo de los valores basales** |
247 (37,4%) |
57 (17,1%) |
Odds ratio 3,02 |
2,16, 4,20 |
<,0001 |
|
ARN del VIH < 400 copias/ml |
201 (30,4%) |
40 (12,0%) |
Odds Ratio 3,45 |
2,36, 5,06 |
<,0001 |
|
ARN del VIH < 50 copias/ml |
121 (18,3%) |
26 (7,8%) |
Odds Ratio 2,77 |
1,76, 4,37 |
<,0001 |
|
Interrupciones debidas a reacciones adversas/enfermedades recurrentes/parámetros de t laboratorio |
9% |
11% | |||
|
Interrupciones debidas a reacciones en el lugar de t inyección |
4% |
N/A | |||
|
Interrupciones por otros • t$§ motivos |
13% |
25% |
*
Basado en los resultados obtenidos del análisis combinado de la población por intención de tratar en los ensayos TORO 1 y TORO 2. La carga viral de la semana 48 en sujetos con pérdida de seguimiento, que interrumpieron la terapia, o tuvieron fallo virológico fue reemplazada por la última observación (LOCF).
Último valor arrastrado.
Ensayo M-H: Interrupciones o fallo virológico considerados como fallos.
Porcentajes basados en la población análizada para la obtención de datos de seguridad, Fuzeon+tratamiento optimizado (N=663) y tratamiento optimizado (N=334). Denominador para pacientes sin cambio de brazo de tratamiento: N=112.
Según el criterio del investigador.
Incluye interrupciones por pérdida de seguimiento, rechazo del tratamiento y otras razones.
#
**
t $
§
La terapia de Fuzeon + OB se asoció con una mayor proporción de pacientes que alcanzaron
< 400 copias/ml (o < 50 copias/ml) en todos los subgrupos establecidos según los CD4 basales, el ARN del VIH-1 basal, según el número previo de antirretrovirales (ARVs) o según el número de ARVs activos en el régimen OB. Sin embargo, los pacientes con valores iniciales de CD4
> 100 células/mm3, valores iniciales de ARN del VIH-1 < 5,0 logio copias/ml, < 10 ARVs previos, y/u otros ARVs activos en su régimen OB tuvieron más probabilidades de alcanzar un ARN del VIH-1 de
< 400 copias/ml (o < 50 copias/ml) en cualquiera de los tratamientos. (ver Tabla 6)
Tabla 6 Proporción de Pacientes que alcanzan < 400 copias/ml y < 50 copias/ml a las 48 _semanas por subgrupos (análisis combinado de TORO 1 y TORO 2, ITT)_
|
Subgrupos |
ARN del VIH-1 < 400 copias/ml |
ARN del VIH-1 < 50 copias/ml | ||
|
Fuzeon + OB 90 mg bid (N=661) |
OB (N=334) |
Fuzeon + OB 90 mg bid (N=661) |
OB (N=334) | |
|
ARN del VIH-1 basal < 5,0 log101 copias/ml |
118/269 (43,9%) |
26/144 (18,1%) |
77/269 (28,6%) |
18/144 (12,5%) |
|
ARN del VIH-1 basal > 5,0 log^1 copias/ml |
83/392 (21,2%) |
14/190 (7,4%) |
44/392 (11,2%) |
8/190 (4,2%) |
|
ARVs previos totales < 101 |
100/215 (46,5%) |
29/120 (24,2%) |
64/215 (29,8%) |
19/120 (15,8%) |
|
ARVs previos totales > 101 |
101/446 (22,6%) |
11/214 (5,1%) |
57/446 (12,8%) |
7/214 (3,3%) |
|
0 ARVs Activos en el tratamiento optimizado1,2 |
9/112 (8,0%) |
0/53 (0%) |
4/112 (3,5%) |
0/53 (0%) |
|
1 ARV Activo en el tratamiento optimizado 1,2 |
56/194 (28,9%) |
7/95 (7,4%) |
34/194 (17,5%) |
3/95 (3,2%) |
|
> 2 ARVs Activos en el tratamiento optimizado 1,2 |
130/344 (37,8%) |
32/183 (17,5%) |
77/334 (22,4%) |
22/183 (12,0%) |
'Interrupciones o fallos virológicos considerados como fallos. 2Basados en los resultados de los ensayos de resistencia genotípica.
5.2 Propiedades farmacocinéticas
Las propiedades farmacocinéticas de la enfuvirtida se han examinado en adultos y niños infectados por el VIH-1.
Absorción: La biodisponibilidad absoluta tras la administración subcutánea de 90 mg de enfuvirtida en el abdomen resultó de 84,3 ± 15,5%. La Cmax media (± DE) fue de 4,59 ± 1,5 microgramo/ml, y el AUC fue de 55,8 ± 12,1 microgramo*h/ml. La absorción subcutánea de la enfuvirtida es proporcional a la dosis administrada en el intervalo de 45 a 180 mg. La absorción subcutánea de la dosis de 90 mg es comparable cuando se inyecta en el abdomen, el muslo o el brazo. El valor medio de concentración plasmática mínima en estado estacionario varió entre 2,6 y 3,4 microgramo/ml en cuatro ensayos diferentes (N= 9 a 12).
Distribución: El volumen de distribución en el estado estacionario tras la administración intravenosa de una dosis de 90 mg de enfuvirtida fue de 5,5 ± 1,1 l. La enfuvirtida se une en un 92% a las proteínas del plasma infectado por el VIH, en un intervalo de concentraciones plasmáticas de 2 a 10 microgramo/ml. La enfuvirtida se une sobre todo a la albúmina y, en menor medida, a la glicoproteína ácida a-1. En los estudios in vitro, la enfuvirtida no fue desplazada de su lugar de unión por otros fármacos, ni tampoco desplazaba a otros fármacos de sus lugares de unión. Se han notificado niveles insignificantes de enfuvirtida en el líquido cefalorraquídeo en pacientes con VIH.
Biotransformación: La enfuvirtida, como péptido que es, se cataboliza en sus aminoácidos constituyentes; luego, los aminoácidos se reciclan dentro del organismo. Los estudios in vitro con microsomas humanos y los estudios in vivo señalan que la enfuvirtida no inhibe las enzimas del citocromo P450. En estudios in vitro con microsomas y hepatocitos humanos, la hidrólisis del grupo amida de la fenilalanina carboxiterminal determina un metabolito desamidado, cuya formación no depende del NADPH. Este metabolito se detecta en el plasma humano después de administrar la enfuvirtida y con un valor del AUC que varía entre el 2,4 y el 15% del AUC de la enfuvirtida.
Eliminación: El aclaramiento de la enfuvirtida tras la administración intravenosa de 90 mg fue de 1,4 ± 0,28 l/h y la semivida de eliminación fue de 3,2 ± 0,42 h. Después de administrar una dosis subcutánea de 90 mg de enfuvirtida, la semivida de eliminación (± DE) es de 3,8 ± 0,6 h. No se han realizado estudios de balance de masa en humanos para determinar la(s) ruta(s) de eliminación de la enfuvirtida.
Insuficiencia Hepática: No se ha investigado la farmacocinética de la enfuvirtida en pacientes con alteración de la función hepática.
Insuficiencia Renal: El análisis de los datos de la concentración plasmática de los pacientes que intervinieron en los ensayos clínicos revela que el aclaramiento de la enfuvirtida no se ve afectado con ningún efecto clínico relevante en pacientes con insuficiencia renal de leve a moderada. En un ensayo en pacientes con insuficiencia renal, el AUC de enfuvirtida aumentó por término medio un 43-62 % en los pacientes con insuficiencia renal grave y en los pacientes en el estado final de la enfermedad renal, en comparación con los pacientes con la función renal normal. La hemodiálisis no modifica considerablemente el aclaramiento de enfuvirtida. Se eliminó menos de un 13 % de la dosis durante la hemodiálisis. No se requiere ajuste de dosis en los pacientes con la función renal afectada.
Pacientes de edad avanzada: La farmacocinética de la enfuvirtida tampoco se ha investigado de manera formal entre personas de 65 ó más años.
Sexo y peso: El análisis de los datos de la concentración plasmática de los pacientes de los ensayos clínicos puso de relieve que el aclaramiento de la enfuvirtida es un 20% inferior en el sexo femenino que en el sexo masculino, con independencia del peso, y aumenta según lo hace el peso corporal, con independencia del sexo (20% superior para personas de 100 kg y 20% inferior para pacientes de 40 kg, en relación con un paciente prototipo de 70 kg). Sin embargo, estas variaciones carecen de significado clínico y no se precisa ningún ajuste posológico.
Raza: El análisis de los datos de la concentración plasmática de los pacientes de los ensayos clínicos indica que el aclaramiento de la enfuvirtida no difiere entre afroamericanos comparado con caucásicos. Los demás estudios farmacocinéticos tampoco señalan diferencias entre los asiáticos y los caucásicos, una vez ajustada la exposición según el peso corporal.
Población pediátrica: Se ha investigado la farmacocinética de la enfuvirtida en 37 niños. La dosis de 2 mg/kg aplicada dos veces al día (con un máximo de 90 mg, dos veces al día) proporcionó concentraciones plasmáticas de enfuvirtida similares a las de los pacientes adultos tratados con 90 mg, dos veces al día. Los valores obtenidos entre 25 niños de 5 a 16 años tratados con una dosis de 2 mg/kg, dos veces al día, inyectada en el brazo, cara anterior del muslo o abdomen fueron estos: Mediana del AUC en el estado estacionario de 54,3 ± 23,5 microgramo*h/ml, Cmax de 6,14 ± 2,48 microgramo/ml, y Cmin de 2,93 ± 1,55 microgramo/ml.
5.3 Datos preclínicos sobre seguridad
Los datos en los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas, genotoxicidad y desarrollo embrionario tardío. No se han realizado estudios de carcinogenicidad a largo plazo en animales.
En los estudios con cobayas se observó que la enfuvirtida induce una hipersensibilidad tardía por contacto. En un modelo con ratas para estudiar la resistencia a la infección por influenza, se observó una alteración en la producción de IFN-y (Interferon-y). La resistencia a la infección por influenza y estreptococo en ratas tan solo estuvo débilmente comprometida. Se desconoce la relevancia clínica de estos hallazgos.
DATOS FARMACÉUTICOS
6.
6.1 Lista de excipientes
Polvo
Carbonato de sodio Manitol
Hidróxido de sodio Ácido clorhídrico
Disolvente
Agua para preparaciones inyectables.
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros excepto con los mencionados en la sección 6.6.
6.3 Periodo de validez
Polvo 4 años
Disolvente 4 años
Periodo de Validez tras la reconstitución
Después de su reconstitución: Conservar en nevera (entre 2°C y 8°C).
En la práctica diaria se ha demostrado que la solución reconstituida es estable química y físicamente durante 48 horas a 5 °C si se protege de la luz.
Desde el punto de vista microbiológico, el producto se debe emplear inmediatamente. Si no se utiliza inmediatamente, los tiempos de conservación y las condiciones previas a su utilización son responsabilidad del usuario y normalmente no deberían ser superiores a las 24 horas a 2 °C - 8 °C en la práctica diaria, a menos que la reconstitución se haya realizado en condiciones asépticas controladas y validadas.
6.4 Precauciones especiales de conservación
Polvo
Conservar el vial en el embalaje exterior para protegerlo de la luz. Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
Disolvente
No requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del recipiente
vial de 3 ml, incoloro, de vidrio de tipo 1 tapón de liofilizados, de goma (sin látex) cierre de aluminio con cápsula de fácil apertura
Polvo Vial:
Tapón Cierre
Disolvente
Vial: vial de 2 ml, incoloro, de vidrio de tipo 1
Tapón: tapón de goma (sin látex)
Cierre: cierre de aluminio con cápsula de fácil apertura
Tamaños de envase
60 viales del polvo para solución inyectable 60 viales del disolvente 60 jeringas de 3 ml 60 jeringas de 1 ml 180 toallitas de alcohol
6.6 Precauciones especiales de eliminación y reconstitución
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
Los pacientes deben recibir instrucciones de los profesionales sanitarios sobre el uso y la administración de Fuzeon antes de utilizarlo por primera vez.
Fuzeon sólo debe reconstituirse con 1,1 ml de Agua para Preparaciones Inyectables. Los pacientes deben aprender a añadir el agua para preparación inyectable y golpear el vial suavemente con la yema de los dedos hasta que el polvo empiece a disolverse. Nunca se debe agitar el vial ni invertirlo para mezclarlo pues esto provocará que se produzca demasiada espuma. Una vez que el polvo empiece a disolverse se puede dejar reposar el vial para permitir la completa disolución. El polvo puede tardar hasta 45 minutos en disolverse. El paciente puede hacer rodar el vial suavemente entre sus manos después de añadir el agua para preparación inyectable hasta que el polvo esté completamente disuelto, lo que puede reducir el tiempo que tarda éste en disolverse. Antes de retirar la solución para su administración, el paciente debe realizar una inspección visual del vial para verificar que se ha disuelto todo el contenido, que la solución es transparente y que no presenta burbujas ni partículas. Si se observan partículas en suspensión, no debe utilizarse el vial, sino que se debe desechar o devolver a la farmacia.
Los viales de disolvente contienen 2 ml de agua para preparación inyectable, de los cuales 1,1 ml deben extraerse para la reconstitución del polvo. Se debe informar a los pacientes de que desechen el volumen restante de los viales de disolvente.
Fuzeon no contiene conservantes. Una vez reconstituido, debe inyectarse de inmediato. Si no se puede inyectar enseguida la solución reconstituida, debe mantenerse en el frigorífico y utilizarse antes de 24 horas. La solución reconstituida y refrigerada debe llevarse a la temperatura ambiente antes de su inyección.
1 ml de solución reconstituida se inyecta por vía subcutánea en el brazo, abdomen o cara anterior del muslo. La inyección debe realizarse en un sitio diferente al de la inyección previa y donde no se haya producido una reacción en el lugar de inyección. Cada vial sirve para un sólo uso; se deben desechar las porciones no utilizadas.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/252/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 27 de Mayo de 2003
Fecha de la última renovación: 27 de Mayo de 2008
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamento: http://www.ema.europa.eu/.
A. FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
Nombre o dirección del fabricante responsable de la liberación de los lotes Roche Pharma AG, Emil-Barrel-Str. 1, D-79639 Grenzach-Wyhlen, Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
No procede.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
Fuzeon 90 mg/ml polvo y disolvente para solución inyectable Enfuvirtida
2. PRINCIPIO(S) ACTIVO(S)
Cada vial contiene 108 mg de enfuvirtida.
1 ml de solución reconstituida contiene 90 mg de enfuvirtida.
3. LISTA DE EXCIPIENTES
Cada vial de polvo también contiene carbonato de sodio (anhidro), manitol, hidróxido de sodio y ácido clorhídrico.
Cada vial de disolvente contiene 2 ml de Agua para Preparaciones Inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable Este envase contiene:
60 viales con polvo para solución inyectable 60 viales de disolvente 60 jeringas de 3 ml 60 jeringas de 1 ml 180 toallitas de alcohol
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía subcutánea
Leer el prospecto antes de utilizar
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar el vial en el embalaje exterior para protegerlo de la luz Conservar en nevera después de su reconstitución
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Tras la extracción de los 1,1 ml necesarios para la reconstitución, debe desecharse el agua para preparaciones inyectables que quede en el vial del disolvente
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/252/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único.
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC:
SN:
NN:
1. NOMBRE DEL MEDICAMENTO
Fuzeon 90 mg/ml polvo para solución inyectable Enfuvirtida
2. PRINCIPIO(S) ACTIVO(S)
Cada vial contiene 108 mg de enfuvirtida.
1 ml de solución reconstituida contiene 90 mg de enfuvirtida.
3. LISTA DE EXCIPIENTES
Cada vial también contiene carbonato de sodio (anhidro), manitol, hidróxido de sodio y ácido clorhídrico.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo para solución inyectable
60 viales con polvo para solución inyectable
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía subcutánea
Leer el prospecto antes de utilizar
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar el vial en el embalaje exterior para protegerlo de la luz Conservar en nevera después de su reconstitución
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/252/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único.
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC:
SN:
NN:
ETIQUETA DEL VIAL DE FUZEON
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Fuzeon 90 mg/ml polvo para solución inyectable
Enfuvirtida
Vía subcutánea
2. FORMA DE ADMINISTRACIÓN
Consultar el prospecto antes de usar
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
108 mg de enfuvirtida
6. OTROS
INFORMACIÓN QUE DEBE FIGURAR EN EL ACONDICIONAMIENTO PRIMARIO ESTUCHE DE LOS VIALES DE AGUA PARA PREPARACIONES INYECTABLES
1. NOMBRE DEL MEDICAMENTO
Disolvente para solución
Agua para Preparaciones Inyectables
2. PRINCIPIO(S) ACTIVO(S)
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Disolvente para uso parenteral
Este envase contiene 60 viales con 2 ml de agua para preparaciones inyectables
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
El agua para preparaciones inyectables se debe emplear para la reconstitución de Fuzeon 90 mg/ml polvo para solución inyectable para obtener una solución para su uso por vía subcutánea Leer el prospecto antes de utilizar
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
Tras la extracción de los 1,1 ml necesarios para la reconstitución, debe desecharse el agua para preparaciones inyectables que quede en el vial del disolvente
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/252/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único.
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC:
SN:
NN:
ETIQUETA DEL VIAL DE AGUA PARA PREPARACIONES INYECTABLES
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Disolvente para solución
Agua para Preparaciones Inyectables
Vía subcutánea
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
2 ml
6. OTROS
B. PROSPECTO
Prospecto: información para el paciente
Fuzeon 90 mg/ml polvo y disolvente para solución inyectable
Enfuvirtida
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
• Conserve este prospecto, ya que puede tener que volver a leerlo.
• Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
• Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
• Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfernero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Fuzeon y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Fuzeon
3. Cómo usar Fuzeon
4. Posibles efectos adversos
5. Conservación de Fuzeon
6. Contenido del envase e información adicional
7. Guía paso a paso sobre cómo inyectarse Fuzeon
1. Qué es Fuzeon y para qué se utiliza Qué es Fuzeon
Fuzeon contiene la sustancia activa “enfuvirtida” y pertenece al grupo de medicamentos denominado “antirretrovirales”.
Para qué se utiliza Fuzeon
Fuzeon se utiliza para el tratamiento del Virus de la Inmunodeficiencia Humana (VIH) en combinación con otros medicamentos antirretrovirales en pacientes infectados por el VIH.
• Su médcio le ha prescrito Fuzeon para ayudar a controlar su infección por VIH.
• Fuzeon no cura la infección por VIH.
Cómo funciona Fuzeon
El VIH ataca células de su sangre conocidas como linfocitos CD4 o T. El virus necesita entrar en contacto con ellas, y consigue entrar en estas células para que el virus se multiplique. Fuzeon ayuda a prevenir esto.
2. Qué necesita saber antes de empezar a usar Fuzeon No use Fuzeon si
• es alérgico a la enfuvirtida o a cualquiera de los demás componentes de este medicamento. (incluidos en la sección 6).
Si no está seguro, consulte a su médico, farmacéutico o enfermero antes de usar Fuzeon. Advertencias y precauciones
Consulte a su médico, farmacéutico o enfermero antes de empezar a usar Fuzeon si:
• ha padecido alguna vez problemas pulmonares
• si ha tenido alguna vez problemas de riñón.
• si tiene hepatitis crónica B o C u otra enfermedad hepática -es más probable que padezca problemas graves del hígado mientras toma este medicamento
Signos de infecciones previas
En algunos pacientes con infección por VIH (SIDA) avanzada y antecedentes de infecciones oportunistas pueden aparecer signos y síntomas de inflamación de infecciones previas poco después de iniciar el tratamiento anti-VIH. Se cree que estos síntomas se deben a una recuperación del sistema inmune del organismo. Esta mejoría permite al organismo combatir infecciones que estaban presentes sin ningún síntoma aparente. Si observa cualquier síntoma de infección, por favor informe a su médico inmediatamente.
Signos de alteraciones autoinmunes
Además de las infecciones oportunistas, también pueden aparecer trastornos autoinmunitarios (una afección que ocurre cuando el sistema inmunitario ataca tejido corporal sano) después de que usted haya empezado a tomar medicamentos para el tratamiento de su infección por VIH. Los trastornos autoinmunitarios pueden aparecer muchos meses después del inicio del tratamiento. Si observa cualquier síntoma de infección u otros síntomas como por ejemplo debilidad muscular, debilidad que empieza en las manos y pies y que asciende hacia el tronco del cuerpo, palpitaciones, temblor o hiperactividad, informe a su médico inmediatamente para recibir el tratamiento necesario.
Pacientes con enfermedad hepática
Los pacientes con hepatitis crónica B ó C y que estén en tratamiento con terapia anti-VIH presentan un mayor riesgo de experimentar problemas graves del hígado. Consulte con su médico si tiene antecedentes de enfermedad hepática.
Enfermedad ósea (osteonecrosis)
Algunos pacientes que reciben medicamentos anti- VIH combinado pueden desarrollar una enfermedad de los huesos llamada osteonecrosis. El tejido óseo muere porque se ha perdido el suministro de sangre (muerte de tejido óseo provocada por la pérdida de aporte de sangre al hueso).
• Los signos de osteonecrosis son rigidez en las articulaciones, dolor y molestias (especialmente en cadera, rodilla y hombro) y dificultad de movimiento. Si nota cualquiera de estos síntomas, comuníqueselo a su médico.
• Los factores de riesgo para desarrollar esta enfermedad incluyen: cuánto tiempo ha estado tomando medicamentos anti VIH, si toma corticoesteroides, cuánto alcohol bebe, cómo funciona su sistema inmune y tener sobrepeso.
Transmisión de VIH a otras personas
Mientras esté tomando este medicamento aún puede transmitir el VIH a los demás, por lo que aunque el tratamiento antiviral eficaz reduzca el riesgo. Consulte a su médico sobre qué precauciones son necesarias para no infectar a otras personas.
Uso de Fuzeon con otros medicamentos
Informe a su médico, farmacéutico o enfermero si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento. Esto incluye medicamentos sin receta médica y medicamentos a base de plantas. Fuzeon no ha mostrado interacciones con los otros medicamentos que forman parte de su tratamiento anti-VIH ni con la rifampicina (un antibiótico).
Uso de Fuzeon con alimentos y bebidas
Puede usar Fuzeon con o sin alimentos. Sin embargo, debe seguir las instrucciones indicadas en los prospectos de los otros medicamentos que esté usando.
Embarazo y lactancia
• Si está embarazada, piensa que podría estar embarazada o está planeando tener un bebé, consulte con su médico antes de tomar este medicamento. No debe usar Fuzeon a menos que se lo diga específicamente su médico.
• Si tiene el VIH, no debe dar el pecho a su bebé, porque puede pasarle el VIH.
Conducción y uso de máquinas
No se han examinado los efectos de Fuzeon sobre la capacidad para conducir vehículos o utilizar máquinas. No deberá conducir ni utilizar máquinas si mientras usa Fuzeon se siente mareado.
Fuzeon contiene sodio
Fuzeon contiene menos de 1 mmol de sodio (23 mg), por dosis; esto es, esencialmente “exento de sodio”.
3. Cómo usar Fuzeon
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Cómo preparar e inyectar Fuzeon
Fuzeon debe administrarse como inyección por debajo de la piel - denominada “inyección subcutánea”. En la sección 7 se indica cómo preparar Fuzeon y cómo autoadministrarse una inyección.
Cuánto usar
• La dosis recomendada es de 90 mg, dos veces al día, para adultos y adolescentes (16 años y mayores).
• Se administra como una inyección debajo de la piel de 1 ml.
• Es mejor usar Fuzeon a la misma hora cada día.
• Trate de espaciar las dosis convenientemente separadas en momentos que sean buenos para usted - por ejemplo, a primera hora de la mañana y por la tarde.
Consulte al final de este prospecto la información adicional sobre como usar Fuzeon (ver sección 7). Allí encontrará instrucciones sobre como preparar Fuzeon y como administrarse usted mismo una inyección.
Si usa más Fuzeon del que debe
Si usa más Fuzeon del que debe, consulte con su médico o vaya al hospital inmediatamente. Lleve el envase del medicamento.
Si olvidó usar Fuzeon
• Si olvida usar una dosis, inyéctese la dosis en cuanto se acuerde. Sin embargo, si quedan menos de 6 horas antes de la siguiente dosis habitual, no tome la dosis olvidada.
• No debe inyectarse una dosis doble para compensar la dosis olvidada.
Si interrumpe el tratamiento con Fuzeon
• Continúe usando su medicamento hasta que su médico le diga lo contrario. Si para y hay una interrupción en su tratamiento puede acelerar la posibilidad de que el VIH en su sangre llegue a ser resistente a Fuzeon. Esto es menos probable si lo usa regularmente y sin interrupciones en su tratamiento.
• Con el tiempo el virus VIH en su sangre puede llegar a ser resistente a Fuzeon. Si esto ocure, sus niveles de virus en sangre empezarán a aumentar. Su médico puede decidir no continuar tratándole con Fuzeon. Su médico lo comentará con usted en ese momento.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Interrumpa el tratamiento con Fuzeon y acuda a su médico inmediatamente si nota cualquiera de los siguientes efectos adversos graves porque puede necesitar tratamiento médico urgente:
• Reacción alérgica (hipersensibilidad) - los signos pueden incluir: erupción, alta temperatura o escalofríos, sensación de cansancio o malestar, sudores o temblores.
Este efecto adverso es raro (afecta a menos de 1 de cada 1.000 personas). Estos signos no significan que definitivamente sea alérgico a este medicamento.
Consulte con su médico si presenta efectos adversos en el lugar de la inyección
Los efectos adversos más freceuntes (afectan a más de 1 de cada 10 personas) son problemas en el lugar del cuerpo donde se administra la inyección. Es muy probable que tenga una o más de las siguientes reacciones, de carácter leve a moderado:
• enrojecimiento^ hinchazón
• sensación de picor^ cardenales
• endurecimiento de la piel o bultos
• dolor, sensación de dolor o molestia
Estas reacciones pueden aparecer en la primera semana de tratamiento y normalmente sólo duran hasta 7 días. Generalmente no empeoran después de este tiempo. Si tiene alguna de estas reacciones no interrumpa el tratamiento con Fuzeon pero consúltelo con su médico.
Estas reacciones pueden empeorar cuando se repite la inyección en el mismo sitio del cuerpo. También pueden empeorar cuando la inyección se administra más profundamente de lo que se pretende (por ejemplo, dentro del músculo). Raramente, puede tener infección en un lugar donde se administró una inyección. Para disminuir el riesgo de infección es importante que siga las Instrucciones que se presentan en la Sección 7.
Fuzeon puede causar una acumulación de un tipo de proteína que se llama amiloide debajo de la piel en el lugar de inyección. Usted puede notar bultos debajo de la piel. Por favor póngase en contacto con su médico si esto le ocurre.
Otros posibles efectos adversos
Muy frecuentes (afectan a más de 1 de cada 10 personas)
• diarrea
• malestar
• pérdida de peso
• dolor y sensación de entumecimiento de manos, pies o piernas.
Frecuentes (afectan hasta 1 de cada 10 personas)
• neumonía
• infección de oídos
• aumento del tamaño de los ganglios linfáticos (nódulos linfáticos)
• ojos inflamados (conjuntivitis)
• gripe o síntomas seudogripales
• senos inflamados
• congestión nasal
• anorexia
• acidez
• inflamación del páncreas
• disminución del apetito
• diabetes,
• pesadillas
• sensación de mareo
• sacudidas (temblor)
• sentimiento de ansiedad o irritabilidad
• no ser capaz de concentrarse
• disminución de la sensibilidad
• acné
• enrojecimiento de la piel
• eczema
• sequedad de piel
• verrugas
• dolor muscular
• piedras en el riñón
• sensación de debilidad
• sangre en la orina
• cambios mostrados en los análisis de sangre (aumento de grasas en la sangre)
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V*. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Fuzeon
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece o bien en la etiqueta de los viales de Fuzeon o en la de los viales de Agua para Preparaciones Inyectables después de “CAD”. La fecha de caducidad es el último día del mes que se indica.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Una vez preparada la solución, la inyección debe efectuarse de inmediato. Si no se inyecta de inmediato el medicamento, consérvelo en nevera (entre 2 °C y 8 °C) y utilícelo antes de 24 horas.
No utilice este medicamento si observa alguna partícula en el polvo o en la solución después de agregar el agua para preparaciones inyectables. Tampoco utilice el agua para preparaciones inyectables si observa partículas en el interior del vial o si el agua está turbia.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente
Contenido del envase e información adicional
6.
Composición de Fuzeon
• El principio activo es la enfuvirtida. Cada vial contiene 108 mg de enfuvirtida. Después de la reconstitución con el disolvente que se incluye en el envase, 1 ml de solución reconstituida contiene 90 mg de enfuvirtida.
• Los demás componentes son:
Polvo
Carbonato de sodio anhidro Manitol
Hidróxido de sodio Ácido clorhídrico
Disolvente
Agua para preparaciones inyectables
Aspecto del producto y contenido del envase
Fuzeon, polvo y disolvente para solución inyectable, consta de un envase que contiene:
60 viales de Fuzeon
60 viales de Agua para preparaciones inyectables que se usan para reconstituir el polvo de Fuzeon 60 jeringas de 3 ml 60 jeringas de 1 ml 180 toallitas de alcohol
Este envase trae todo lo que necesita para preparar e inyectarse Fuzeon durante 30 días de tratamiento.
Titular de la autorización de comercialización
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
El fabricante responsable de la liberación de los lotes es
Roche Pharma AG Emil-Barell-Str. 1,
D-79639 Grenzach-Wyhlen Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien N.V. Roche S.A. Tél/Tel: +32 (0) 2 525 82 11 |
Luxembourg/Luxemburg (Voir/siehe Belgique/Belgien) |
|
Etarapun Pom Etarapna EOOfl Tea: +359 2 818 44 44 |
Magyarország Roche (Magyarország) Kft. Tel: +36 - 23 446 800 |
|
Ceská republika Roche s. r. o. Tel: +420 - 2 20382111 |
Malta (See United Kingdom) |
|
Danmark Roche a/s Tlf: +45 - 36 39 99 99 |
Nederland Roche Nederland B.V. Tel: +31 (0) 348 438050 |
|
Deutschland Roche Pharma AG Tel: +49 (0) 7624 140 |
Norge Roche Norge AS Tlf: +47 - 22 78 90 00 |
|
Eesti Roche Eesti OÜ Tel: + 372 6 177 380 |
Osterreich Roche Austria GmbH Tel: +43 (0) 1 27739 |
|
EkXába Roche (Hellas) A.E. Tr|U +30 210 61 66 100 |
Polska Roche Polska Sp.z o.o. Tel: +48 - 22 345 18 88 |
|
España Roche Farma S.A. Tel: +34 - 91 324 81 00 |
Portugal Roche Farmacéutica Química, Lda Tel: +351 - 21 425 70 00 |
|
Hrvatska Roche d.o.o. Tel: + 385 1 47 22 333 France Roche Tél: +33 (0) 1 47 61 40 00 |
Romania Roche Romania S.R.L. Tel: +40 21 206 47 01 |
|
Ireland Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700 |
Slovenija Roche farmacevtska druzba d.o.o. Tel: +386 - 1 360 26 00 |
|
Ísland Roche a/s c/o Icepharma hf Sími: +354 540 8000 |
Slovenská republika Roche Slovensko, s.r.o. Tel: +421 - 2 52638201 |
|
Italia Roche S.p.A. Tel: +39 - 039 2471 |
Suomi/Finland Roche Oy Puh/Tel: +358 (0) 10 554 500 |
Kúrcpog
UA.Exapáxn? & Sia AxS. Tn^: +357 - 22 76 62 76
Sverige
Roche AB
Tel: +46 (0) 8 726 1200
Latvija
Roche Latvija SIA
Tel: +371 - 6 7039831
United Kingdom
Roche Products Ltd.
Tel: +44 (0) 1707 366000
Fecha de la última revisión de este prospecto:
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu/.
7. GUÍA PASO A PASO SOBRE CÓMO INYECTARSE FUZEON
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero.
Qué debe hacer si es zurdo
Los dibujos de este prospecto muestran personas diestras. Si es zurdo actúe con naturalidad. Posiblemente, le resulte más cómodo:
• sujetar la jeringa con la mano izquierda y
• tomar el vial entre el pulgar y el índice de la mano derecha.
Cuándo solicitar la ayuda al personal de apoyo
Al principio, puede resultar difícil la inyección en algunos lugares, como el brazo. Pida ayuda si la necesita, a su pareja, a un amigo o a un familiar. Es posible que quiera pedirle a alguien que le acompañe a una sesión de entrenamiento en la técnica de la inyección con su médico o enfermero.
Las jeringas
Las jeringas que se suminstran con este medicamento cuentan con un protector coloreado de la aguja. Éste está unido a la aguja y cubre la aguja después de su uso para reducir el riesgo de que accidentalmente otra persona se pinche con la aguja. Aunque estas jeringas tienen esta medida de seguridad, es importante que una vez usadas las deseche convenientemente. Siga las instrucciones que le haya dado su médico, farmacéutico o enfermero.
Consejos de Seguridad
• Lávese bien las manos. Esto reducirá el riesgo de infecciones bacterianas.
• Una vez que se haya lavado las manos, no toque nada, salvo el medicamento y el material suministrado para la inyección.
• Cuando manipule la jeringa, no toque la aguja.
• No toque los tapones de los viales, una vez que los haya limpiado con las toallitas de alcohol.
• No utilice nunca materiales abiertos. Antes de usarlo, compruebe que todos los elementos del envase están cerrados.
• No utilice ni comparta nunca agujas usadas.
• No utilice nunca una aguja doblada o dañada.
• No mezcle nunca el medicamento con el agua del grifo.
• No se inyecte nunca el medicamento con otros medicamentos inyectables.
• Sólo inyecte Fuzeon debajo de la piel (“vía subcutánea”).
• No se inyecte Fuzeon en la vena (“vía intravenosa”), ni en el músculo (“vía intramuscular”).
• Elimine todo el material utilizado en el recipiente con tapadera para los materiales desechables. Haga esto incluso si los viales contienen cantidades no utilizadas de medicamento o agua para preparaciones inyectables ya que éstos son para un solo uso. Consulte a su médico, farmacéutico o enfermero si tiene alguna duda acerca del vertido seguro de este material.
La siguiente es una guía básica, paso a paso, para inyectar el medicamento.
Paso A: Para empezar
1. Reúna los siguientes materiales:
• Un vial de Fuzeon (recipiente de vidrio con polvo blanco en su interior)
• Un vial de agua para preparaciones inyectables (recipiente de vidrio con líquido transparente e
incoloro en su interior)
• Una jeringa de 3 ml (jeringa grande) con una aguja de 25 mm
• Una jeringa de 1 ml (jeringa pequeña) con una aguja de 13 mm
• Tres toallitas de alcohol
• Recipiente con tapadera para un vertido seguro de los materiales desechables
2. Abra los envases de las jeringas y retire las cápsulas de cierre del vial.
• Deposite los envases y las cápsulas del vial en el recipiente con tapadera para los materiales
dsechables.
• Coloque las jeringas y los viales sobre una superficie limpia.
3. Lávese minuciosamente las manos.
• Después de lavarse las manos, no toque nada, salvo el material para la inyección y el lugar donde se vaya a administrar la inyección.
4. Limpie los tapones de los viales.
• Limpie cada tapón de los viales con una toallita de alcohol limpia. Deje que se seque el tapón al aire.
• Asegúrese de que no toca los tapones de goma una vez limpios. Si los toca, asegúrese de limpiarlos de nuevo.
Extraiga el Agua para Preparaciones Inyectables
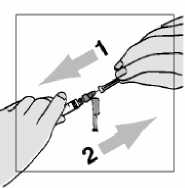
1. Tome la jeringa grande de 3 ml. Utilice el dedo índice para mover el protector coloreado de la aguja y separarlo de la aguja.

2. Para asegurarse de que la aguja está firmemente colocada en la jeringa:
• sujete el capuchón de plástico por debajo del protector de la aguja
• apriete la aguja y el capuchón con un suave giro, en el sentido de las agujas del reloj. No ejerza demasiada fuerza pues la aguja podría aflojarse.
3. Para retirar el capuchón de plástico transparente:
• agarre la jeringa y tire del capuchón.
4. Aspire 1,1 ml de aire
5. Introduzca la aguja de la jeringa en el tapón de goma del vial con el agua para preparaciones inyectables y presione el émbolo. Éste inyecta el aire.

6. Invierta suavemente el vial. Asegúrese de que la punta de la aguja queda en todo momento por debajo de la superficie del agua para preparaciones inyectables para que no entre ninguna burbuja de aire en la jeringa.
7. Extraiga lentamente el agua tirando del émbolo hasta la marca de 1,1 ml. Por favor, tenga en cuenta que el vial contiene más líquido del que necesita (2 ml); sólo tiene que extraer 1,1 ml para preparar su inyección adecuadamente.
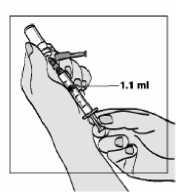
8. Golpee suavemente la jeringa para que las burbujas de aire suban.
• Si ha entrado demasiado aire en la jeringa, empuje suavemente el émbolo para reintroducir las burbujas de aire en el vial.
• Luego extraiga de nuevo el agua,
• Asegúrese de que cuenta con 1,1 ml de agua para preparaciones inyectables en la jeringa.
• Este paso puede repetirse hasta conseguir la cantidad correcta de agua para preparaciones inyectables en la jeringa.
9. Retire la aguja del vial. Asegúrese de que no toca la aguja con los dedos ni con nada más en ningún momento.
10. Deseche el vial y el agua para preparaciones inyectables en el recipiente con tapadera para los materiales desechables- este vial es para un solo uso.
Inyecte el Agua para Preparaciones Inyectables en el Vial de Fuzeon
1. Golpee suavemente el vial para dispersar el polvo.
2. Sujete la parte principal de la jeringa que contiene el agua, e introduzca la aguja a través del tapón de goma del vial con una ligera inclinación.
3. Presione lentamente el émbolo de la jeringa.
• Dejando que el agua resbale por las paredes internas del vial.
• Procure no inyectar bruscamente el agua sobre el polvo, porque podría formarse espuma.
• Si se formara espuma, el polvo tardaría más en disolverse por completo.

4. Una vez que haya inyectado todo el agua para preparaciones inyectables al vial de Fuzeon, retire la jeringa del vial.
5. Sujete la jeringa por la parte principal con una mano y, sobre una superficie plana, presione con suavidad hacia abajo el protector coloreado de la aguja, hasta que ésta quede cubierta por el protector.
- Oirá un clic. No utilice la mano libre para presionar el protector sobre la aguja.

6. Deseche la jeringa en un recipiente con tapadera para materiales desechables.
Mezcla del Agua para Preparaciones Inyectables con el Polvo de Fuzeon
1. Golpee suavemente el vial con la yema de los dedos hasta que empiece a disolverse el polvo. No agite nunca el vial ni lo invierta para mezclarlo porque podría formarse mucha espuma.
2. Cuando empiece a disolverse el polvo, aparte el vial a un lado y deje que se disuelva por completo.
• El polvo puede tardar hasta 45 minutos en disolverse.
• El paciente puede hacer rodar el vial suavemente entre sus manos después de añadir el agua para preparaciones inyectables hasta que el polvo esté completamente disuelto
• Esto puede reducir el tiempo que tarda en disolverse.
3. Después de que el polvo se haya disuelto por completo
• Deje que las posibles burbujas que se hayan formado sedimenten.
• Si todavía quedan burbujas, golpee suavemente los lados del vial para que sedimenten.
4. Es importante comprobar si el líquido contiene fragmentos (partículas).
• Si observa algún fragmento en el líquido, no lo utilice.
• Deseche el vial en el recipiente con tapadera para los materiales desechables o devuélvalo a la farmacia. Comience otra vez con un vial nuevo de polvo de Fuzeon.
5. Si, por accidente, toca el tapón de goma, asegúrese de limpiarlo otra vez con una toallita de alcohol nueva.
6. Una vez mezclada la dosis con el agua para preparaciones inyectables, debe utilizarla de inmediato. Si no la usa, puede conservarla en el frigorífico y usarla antes de 24 horas.
• Espere a que el líquido alcance la temperatura ambiente antes de utilizarlo.
7. Si está preparando las dos dosis diarias a la vez, asegúrese de utilizar jeringas nuevas, agua para preparaciones inyectables nueva y un nuevo vial de Fuzeon para cada dosis.
Extraiga Fuzeon con la jeringa de 1 ml
1. Limpie otra vez el tapón del vial de Fuzeon con una toallita de alcohol nueva.
2. Tome la jeringa pequeña de 1 ml. Utilice el dedo índice para mover hacia atrás el protector coloreado de la aguja y separarlo de la aguja.
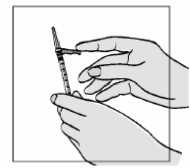
3. Para asegurarse de que la aguja está firmemente colocada en la jeringa:
• sujete el capuchón de plástico bajo el protector de la aguja
• apriete la aguja y el capuchón girando ligeramente y empujándola hacia la jeringa.
4. Para retirar el capuchón de plástico transparente:
• sujete la jeringa y tire del capuchón.
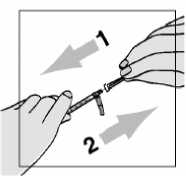
5. Aspire 1 ml de aire.
• Tenga cuidado de no tirar del émbolo demasiado deprisa- podría rebasar la marca de 1 ml o sacar la jeringa.
6. Introduzca la aguja de la jeringa en el tapón de goma del vial de Fuzeon y presione el émbolo. Esto inyecta el aire.
7. Invierta el vial con suavidad varias veces.
Procure que la punta de la aguja quede siempre debajo de la superficie de la solución para que no entren burbujas de aire en la jeringa.
8. Tire lentamente del émbolo hasta que la solución alcance la marca de 1,0 ml.
• Tenga cuidado de no tirar del émbolo demasiado deprisa- podría rebasar la marca de 1 ml o sacar la jeringa.
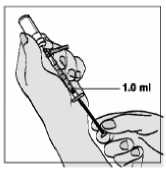
9. Golpee con suavidad la jeringa para que asciendan las posibles burbujas de aire.
• Si entra demasiado aire en la jeringa, empuje con suavidad el émbolo para que el aire regrese al vial.
• Luego retire de nuevo el líquido.
• Asegúrese de que haya 1,0 ml de líquido en la jeringa (o la cantidad correspondiente que le haya prescrito el médico, si fuera diferente).
• Este paso puede repetirse hasta que entre la cantidad correcta de la solución dentro de la jeringa.
10. Retire la jeringa del vial.
Paso D: Inyección de Fuzeon
Consejo: Su médico o enfermero le sugerirá técnicas de inyección diferentes que le vayan mejor en
su caso.
Dónde inyectar
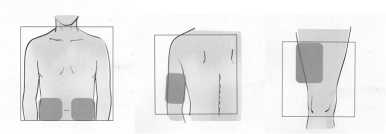
• Fuzeon se administra en una inyección de 1 ml por debajo de la piel - conocida como “inyección subcutánea”.
• Puede inyectarlo en los brazos, la cara anterior del muslo o área del estómago (abdomen).
• Elija una zona diferente a la de la última inyección que se haya administrado.
• No se inyecte el medicamento en un sitio donde todavía haya una reacción de la anterior dosis. Compruebe los lugares donde puede haber una reacción presionando la piel para ver si hay bultos duros.
• No se inyecte el medicamento en zonas que podrían irritarse por el cinturón o por el roce de la ropa.
• No se inyecte el medicamento en lunares, cicatrices, cardenales o en el ombligo.
Limpie el Lugar de Inyección
Limpie la zona de inyección con una toallita de alcohol en un movimiento circular hacia afuera. Deje que la zona se seque por completo.
Introduzca la aguja y póngase la inyección 1. Pellízquese la piel formando un pliegue tan grande como sea posible- sin hacerse daño.
2. Introduzca la aguja en la piel con un ángulo de 45 grados.
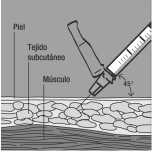
3. Cuando se introduzca la aguja:
• suelte la piel
• con la mano que tiene libre, sujete la parte principal de la jeringa- esto ayudará a mantenerla recta y evitar que se mueva.
4. Con el pulgar de su otra mano, empuje el émbolo para inyectar el líquido.
• Una vez inyectada toda la dosis, retire la aguja de la piel.
Después de retirar la aguja
1. Sujete la parte principal de la jeringa con una mano
• luego sobre una superficie plana, presione con suavidad hacia abajo el protector coloreado de la aguja, hasta que ésta quede cubierta por el protector.
• Oirá un clic.
No utilice la mano libre para presionar el protector sobre la aguja.

2. Deseche la jeringa en un recipiente con tapadera para los materiales desechables.
3. Si hay algo de sangre donde se ha puesto la inyección, cubra la piel con un apósito.
Paso E: Cómo desechar el material utilizado
• Deseche directamente todo el material utilizado en el recipiente con tapadera para los materiales desechables. Haga esto incluso si los viales contienen restos del medicamento o de agua para preparaciones inyectables ya que son para un solo uso.
• Mantenga siempre cerrada la tapa del recipiente y colóquelo fuera del alcance de los niños.
• Pregunte a su médico, farmacéutico o enfermero cómo desechar convenientemente el recipiente.
• Si tiene alguna duda o le preocupa la eliminación segura del material, consulte con su médico, farmacéutico o enfermero.
ANEXO IV
CONCLUSIONES CIENTÍFICAS Y MOTIVOS POR LOS QUE SE RECOMIENDA LA MODIFICACIÓN DE LAS CONDICIONES DE LAS AUTORIZACIONES DE
COMERCIALIZACIÓN
Conclusiones científicas
Teniendo en cuenta lo dispuesto en el Informe de Evaluación del Comité para la Evaluación de Riesgos en Farmacovigilancia (PRAC) sobre los informes periódicos de seguridad (IPSs) para enfuvirtida, las conclusiones científicas del Comité de Medicamentos de Uso Humano (CHMP) son las siguientes:
Durante las actividades rutinarias de detección de señales, se revisaron, 5 casos notificados de amiloidosis de alto nivel relacionados con enfuvirtida en Eudravigilance. La señal se consideró potencialmente grave y el titular de la autorización de comercialización llevó a cabo una revisión acumulada de amiloidosis de alto nivel. La información proporcionada con respecto a la señal de amiloidosis local, evidencia, en base a 2 casos bien documentados, que enfuvirtida como péptido puede causar amiloidosis cutánea en el lugar de la inyección. A la vista de lo anterior, se consideró que en la información del producto para Fuzeon debía constar la amiloidosis cutánea en el lugar de la inyección como reacción adversa.
Por lo tanto, en vista de los datos disponibles sobre Fuzeon, el PRAC consideró que estaba justificado modificar la información del producto.
El CHMP está de acuerdo con las conclusiones científicas realizadas por el PRAC.
Motivos por los que se recomienda la modificación de las condiciones de la(s) Autorización(es) de Comercialización
De acuerdo con conclusiones científicas para enfuvirtida, el CHMP considera que el balance beneficio-riesgo del medicamento o medicamentos que contiene(n) enfuvirtida es favorable sujeto a los cambios propuestos a la información del producto.
El CHMP recomienda que se modifiquen las condiciones de la(s) Autorización(es) de Comercialización.
46