Fraxiparina 0,8 Ml Solucion Inyectable
FICHA TÉCNICA
1. Nombre del medicamento
Fraxiparina 0,3 ml solución inyectable Fraxiparina 0,4 ml solución inyectable Fraxiparina 0,6 ml solución inyectable Fraxiparina 0,8 ml solución inyectable
2. Composición cualitativa y cuantitativa
1 ml contiene 9.500 UI anti Xa de nadroparina cálcica.
- Fraxiparina 0,3 ml solución inyectable, cada jeringa precargada contiene 2.850 UI anti Xa de nadroparina cálcica.
- Fraxiparina 0,4 ml solución inyectable, cada jeringa precargada contiene 3.800 UI anti Xa de nadroparina cálcica.
- Fraxiparina 0,6 ml solución inyectable, cada jeringa precargada contiene 5.700 UI anti Xa de nadroparina cálcica.
- Fraxiparina 0,8 ml solución inyectable, cada jeringa precargada contiene 7.600 UI anti Xa de nadroparina cálcica.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. Forma farmacéutica
Solución inyectable contenida en jeringas precargadas para inyección subcutánea. En el tratamiento de la angina inestable y el infarto de miocardio sin onda Q se puede administrar además por vía intravenosa.
4. Datos clínicos
4.1. Indicaciones terapéuticas
• Profilaxis de la enfermedad tromboembólica, asociada con cirugía general (Fraxiparina 0,3 ml) y cirugía ortopédica (Fraxiparina 0,3 ml, 0,4 ml y 0,6 ml).
• Profilaxis de la trombosis venosa en pacientes no quirúrgicos inmovilizados, cuya situación pueda definirse como de riesgo moderado o elevado (Fraxiparina 0,3 ml, 0,4 ml y 0,6 ml).
• Prevención de la coagulación en el circuito de circulación extracorpórea (CCE) en hemodiálisis (Fraxiparina 0,3 ml, 0,4 ml y 0,6 ml).
• Tratamiento de la trombosis venosa profunda de las extremidades inferiores con o sin embolia pulmonar (Fraxiparina 0,4 ml, 0,6 ml y 0,8 ml).
• Tratamiento de la angina inestable y el infarto de miocardio sin onda Q (Fraxiparina 0,4 ml, 0,6 ml y 0,8 ml).
4.2. Posología y forma de administración Advertencia:
Las diferentes heparinas de bajo peso molecular (HBPM) no son necesariamente equivalentes. Debe prestarse una especial atención a las instrucciones específicas de posología para cada heparina de bajo peso molecular, debido a la diferente actividad de cada preparado de HBPM.
Las jeringas graduadas facilitan la administración de dosis ajustadas cuando se requiere una adaptación al peso del paciente.
Cuando se administra la inyección por vía subcutánea, la zona de inyección habitual es la pared abdominal antero-lateral, alternativamente en el lado derecho e izquierdo.
La aguja debe ser introducida en toda su longitud, perpendicularmente y no inclinada, en un pliegue cutáneo formado entre el pulgar y el índice, que se mantendrá hasta el final de la inyección.
Deben seguirse recomendaciones específicas en relación al tiempo de administración de nadroparina alrededor de la zona de aplicación de anestesia espinal o epidural o de punción lumbar (ver sección 4.4).
• Adultos
Profilaxis de la enfermedad tromboembólica
- Cirugía general:
Fraxiparina debe administrarse en una dosis única diaria de 0,3 ml (2.850 UI) durante al menos 7 días; en todos los casos la profilaxis debe mantenerse, a criterio del médico, durante el periodo de riesgo, y al menos, hasta la deambulación. En cirugía general, la primera dosis debe ser administrada de 2 a 4 horas antes de la intervención.
- Cirugía ortopédica:
La dosis inicial se administrará 12 horas antes de la intervención y 12 horas después de finalizar la misma. Estas y las siguientes dosis diarias deben ajustarse al peso del paciente según se explica en la tabla siguiente. El tratamiento debe continuarse durante al menos 10 días; en todos los casos, la profilaxis debe mantenerse durante el periodo de riesgo, y al menos, hasta la deambulación.
|
Cirugía ortopédica |
Dosis diaria de Fraxiparina | |
|
Peso corporal |
Desde el preoperatorio |
A partir del 4° día |
|
hasta el 3 er día | ||
|
< 70 kg |
0,3 ml (2.850 UI anti Xa) |
0,4 ml (3.800 UI anti Xa) |
|
> 70 kg |
0,4 ml (3.800 UI anti Xa) |
0,6 ml (5.700 UI anti Xa) |
Ip.
1*4 m
- Pacientes no quirúrgicos:
La posología recomendada para esta indicación se calcula en función del peso corporal y el nivel de riesgo del paciente tal y como se expresa en la siguiente tabla:
|
Prevención en pacientes no quirúrgicos |
Dosis de Fraxiparina cada 24 horas | |
|
Nivel de riesgo |
Peso del paciente (kg) | |
|
Moderado |
0,3 ml (2.850 UI anti Xa) | |
|
51-70 kg |
0,4 ml (3.800 UI anti Xa) | |
|
Alto |
> 70 kg |
0,6 ml (5.700 UI anti Xa) |
La duración del tratamiento coincidirá con la del riesgo tromboembólico.
Prevención de la coagulación en el CCE en hemodiálisis:
Se requiere optimizar la dosis para cada paciente individualmente, teniendo en cuenta las condiciones técnicas de la diálisis. Fraxiparina se administra usualmente en una dosis única en la línea arterial al inicio de cada sesión. Para pacientes sin riesgo hemorrágico, se sugieren las dosis iniciales en función del peso del paciente:
|
Prevención de la coagulación en el CCE en hemodiálisis | |
|
Peso del paciente (kg) |
Dosis de Fraxiparina inyectada al inicio de la hemodiálisis |
|
< 50 |
0,3 ml (2.850 UI anti Xa) |
|
50-69 |
0,4 ml (3.800 UI anti Xa) |
|
> 70 |
0,6 ml (5.700 UI anti Xa) |
En pacientes con riesgo de hemorragia la dosis prevista en función del peso se reducirá a la mitad.
Una pequeña dosis adicional puede ser administrada para sesiones de diálisis que se prolonguen más de 4 horas.
La dosis de sesiones de diálisis posteriores debe ser ajustada de acuerdo al efecto observado en la sesión inicial.
Tratamiento de la trombosis venosa profunda con o sin embolia pulmonar:
Fraxiparina debe administrarse subcutáneamente en dosis de 85,5 UI anti Xa/kg cada 12 horas, con una duración habitual de 10 días ajustando la dosis al peso del paciente según la tabla siguiente:
|
Tratamiento de la enfermedad tromboembólica | |
|
Peso del paciente (kg) |
Dosis de Fraxiparina cada 12 horas |
|
< 50 |
0,4 ml (3.800 UI anti Xa) |
|
50-59 |
0,5 ml (4.750 UI anti Xa) |
|
60-69 |
0,6 ml (5.700 UI anti Xa) |
|
70-79 |
0,7 ml (6.650 UI anti Xa) |
|
> 80 |
0,8 ml (7.600 UI anti Xa) |
Debe iniciarse el tratamiento con anticoagulantes orales tan pronto como sea posible, a menos que estén contraindicados. El tratamiento con Fraxiparina no debe interrumpirse hasta que el valor de la INR esté estabilizado.
Tratamiento de la angina inestable e infarto de miocardio sin onda Q
Fraxiparina debe administrarse subcutáneamente dos veces al día (cada 12 horas) en combinación con ácido acetilsalicílico, hasta un máximo de 325 mg por día. La dosis inicial debe administrarse en bolus intravenoso (IV) con 86 UI anti Xa / Kg seguido de inyecciones subcutáneas de 86 UI anti Xa / Kg. La duración habitual del tratamiento es de 6 días. A título orientativo, la pauta posológica a administrar en función del peso de los pacientes será 0,1 ml/10 kg peso en bolus IV seguido de la misma dosis cada 12 horas. No se debe superar la dosis de 1 ml. Los pacientes con peso inferior a 50 kg se trataran con una dosis de 0,4 ml.
Observaciones generales:
Población pediátrica:
No se recomienda el uso de Fraxiparina en niños y adolescentes al no disponer de datos suficientes de seguridad y eficacia en pacientes menores de 18 años de edad.
Pacientes de edad avanzada:
En los ancianos no se necesita ningún ajuste de la dosis, a menos que la función renal esté alterada. Se recomienda que la función renal se evalúe antes de iniciar el tratamiento con Fraxiparina (ver secciones 4.3 y 5.2).
Insuficiencia renal:
Profilaxis de la enfermedad tromboembólica
No es necesaria una reducción de la dosis en pacientes con insuficiencia renal leve (aclaramiento de creatinina > 50 ml/min). La insuficiencia renal moderada y grave está asociada a una exposición prolongada a nadroparina lo que aumenta el riesgo de tromboembolismo y de hemorragia en los pacientes que la sufren.
Si el médico considera oportuna una reducción de la dosis, al tener en cuenta las características individuales y factores de riesgo de tromboembolismo y hemorragia en pacientes con insuficiencia renal moderada (aclaramiento de creatinina > 30 ml/min y < 50 ml/min), la dosis se debe reducir entre un 25% y un 33% (ver secciones 4.4 y 5.3).
La dosis se debe reducir entre un 25% y un 33% en pacientes con insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min) (ver secciones 4.4 y 5.3).
Tratamientos de la trombosis venosa profunda, angina inestable e infarto de miocardio sin onda Q.
No es necesaria una reducción de la dosis en pacientes con insuficiencia renal leve (aclaramiento de creatinina > 50 ml/min). La insuficiencia renal moderada y grave está asociada a una exposición prolongada a nadroparina lo que aumenta el riesgo de tromboembolismo y de hemorragia en los pacientes que la sufren.
Si el médico considera oportuna una reducción de la dosis, al tener en cuenta las características individuales y factores de riesgo de tromboembolismo y hemorragia en pacientes con insuficiencia renal moderada (aclaramiento de creatinina > 30 ml/min y < 50 ml/min), la dosis se debe reducir entre un 25% y un 33% (ver secciones 4.4 y 5.3).
Fraxiparina con fines de tratamiento está contraindicada en pacientes con insuficiencia renal grave (ver secciones 4.3 y 5.3).
4.3. Contraindicaciones
• Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
• Antecedente de trombocitopenia inducida por nadroparina cálcica (ver sección 4.4).
Al igual que el resto de heparinas de bajo peso molecular, su uso está contraindicado en las siguientes situaciones:
• Hemorragia activa o incremento del riesgo hemorrágico en relación con alteraciones de la hemostasia, excepto las debidas a coagulación intravascular diseminada (CID) no inducida por heparina.
• Lesiones orgánicas susceptibles de sangrar (como úlcera péptica activa).
• Accidente cerebrovascular hemorrágico.
• Endocarditis bacteriana aguda.
• Insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min) en pacientes que reciban Fraxiparina con fines de tratamiento de la trombosis venosa profunda, angina inestable e infarto de miocardio sin onda Q.
• En pacientes que reciban heparina con fines de tratamiento y no de profilaxis, está contraindicada la utilización de anestesia regional en las intervenciones quirúrgicas programadas.
4.4. Advertencias y precauciones especiales de empleo Advertencias
Fraxiparina no debe ser administrada por vía intramuscular.
Debido a la posibilidad de trombocitopenia inducida por heparina, se recomienda un control regular del recuento plaquetario en el curso del tratamiento con nadroparina.
Se han observado casos aislados de trombocitopenia, ocasionalmente severa. Esta puede estar asociada (o no) a trombosis arterial o venosa y el tratamiento debe ser interrumpido. Tal diagnóstico debe ser considerado en caso de:
• trombocitopenia
• cualquier disminución significativa del recuento plaquetario: 30-50 % del valor basal
• empeoramiento de la trombosis inicial durante el tratamiento
• coagulación intravascular diseminada
• trombosis aparecida durante el tratamiento
Estos efectos son probablemente de naturaleza inmunoalérgica y en caso de un primer tratamiento, aparecen principalmente entre el 5° y el 21° día de tratamiento, pero estos pueden manifestarse mucho antes si hay antecedentes de trombocitopenia inducida por heparina (TIH).
Cuando hay antecedentes de trombocitopenia inducida por heparina (estándar o de bajo peso molecular), se puede considerar el tratamiento con nadroparina.
Cuando la trombocitopenia aparece en el curso de un tratamiento con heparina (estándar o de bajo peso molecular), se puede considerar la sustitución por otra clase de antitrombótico, si está disponible. Si no está disponible, puede valorarse la sustitución por otra heparina de bajo peso molecular (HBPM). En tal caso debe realizarse un seguimiento clínico y evaluación del recuento plaquetario al menos una vez al día interrumpiendo el tratamiento tan pronto como sea posible, ya que se han descrito casos en los que se mantiene la trombocitopenia aún después de sustituir la heparina. Los tests de agregación plaquetaria “in vitro” poseen un valor limitado.
Precauciones especiales de empleo
Al igual que el resto de heparinas de bajo peso molecular, se administrará con precaución en los siguientes casos:
• insuficiencia hepática
• hipertensión arterial severa
• antecedentes de úlcera gastroduodenal u otras lesiones orgánicas susceptibles de sangrar
• enfermedad vascular de coroides y retina
• en el periodo postoperatorio tras cirugía cerebral, medular u ocular
• pacientes en tratamiento con anticoagulantes orales
• pacientes en tratamiento con corticosteroides sistémicos
• pacientes en tratamiento con dextranos
Es conocido que la nadroparina se elimina principalmente por vía renal, lo que conlleva un aumento en la exposición a nadroparina en pacientes con insuficiencia renal (ver sección 5.2). Los pacientes con insuficiencia renal tienen un riesgo mayor de sufrir hemorragia y deben ser tratados con precaución.
La decisión sobre si la reducción de la dosis es adecuada en pacientes con aclaramiento de creatinina entre 30 y 50 ml/min debe estar basada en la evaluación que el médico haga del riesgo de hemorragia con respecto al riesgo de tromboembolismo, en cada caso individual.
La administración de heparina puede inhibir la secreción adrenal de aldosterona produciéndose hipercalemia, especialmente en pacientes con niveles plasmáticos de potasio elevados o con riesgo de tenerlos elevados como son pacientes con diabetes mellitus, insuficiencia renal crónica, acidosis metabólica pre-existente o que tomen fármacos que puedan tener este efecto hipercalémico (como los inhibidores del enzima convertidora de la angiotensina (ECA), antiinflamatorios no esteroideos (AINEs). El riesgo de hipercalemia aumenta con la duración del tratamiento y desaparece al retirar el mismo. En pacientes con riesgo, deben monitorizarse los niveles de potasio plasmático.
La prescripción concomitante de anestesia espinal o epidural y de terapia anticoagulante, debe realizarse después de evaluar de forma individual el beneficio frente a los riesgos potenciales.
En pacientes sometidos a anestesia epidural o espinal o a punción lumbar, la administración de heparina con fines profilácticos se ha asociado muy raramente a la aparición de hematomas epidurales o espinales, con el resultado final de parálisis prolongada o permanente. Este riesgo se incrementa por el uso de catéteres epidurales o espinales para anestesia, la administración concomitante de medicamentos con efecto sobre la coagulación como antiinflamatorios no esteroideos (AINEs), antiagregantes plaquetarios o anticoagulantes, y por las punciones traumáticas o repetidas.
En el tratamiento de los desórdenes tromboembólicos venosos el uso concomitante de salicilatos o fármacos antiinflamatorios no esteroideos (AINEs) representa una contraindicación relativa para la administración de nadroparina, así como el uso de antiagregantes plaquetarios, debido a que pueden aumentar el riesgo de hemorragia (ver sección 4.5).
A la hora de decidir el intervalo de tiempo que debe transcurrir entre la administración de heparina a dosis profilácticas y la inserción o retirada de un catéter espinal o epidural, deben tenerse en cuenta las características del paciente y del producto, debiendo transcurrir al menos 12 horas. Una vez insertado o retirado el catéter, deberán transcurrir al menos 12 horas hasta la administración de una nueva dosis de heparina. La siguiente dosis deberá retrasarse hasta que la intervención quirúrgica haya finalizado. En el caso de dosis de tratamiento, el tiempo que debe transcurrir entre la administración de heparina y la inserción o retirada de un catéter espinal o epidural debe ser de al menos 24 horas. En pacientes con insuficiencia renal, se pueden considerar intervalos de tiempo más largos.
Si bajo criterio médico se decide administrar tratamiento anticoagulante durante un procedimiento anestésico espinal o epidural, debe extremarse la vigilancia del paciente para detectar precozmente cualquier signo o síntoma de déficit neurológico, como dolor lumbar, déficit sensorial y motor (entumecimiento y debilidad de extremidades inferiores) y trastornos funcionales del intestino o vejiga. El personal de enfermería debe ser entrenado para detectar tales signos y síntomas. Asimismo, se advertirá a los pacientes que informen inmediatamente al médico o personal de enfermería si experimentan cualquiera de los síntomas antes descritos.
Si se sospecha la aparición de algún signo o síntoma sugestivo de hematoma espinal o epidural, deben realizarse las pruebas diagnósticas con carácter de urgencia e instaurar el tratamiento adecuado, incluyendo la descompresión medular.
Muy raramente se han notificado casos de necrosis cutánea. Ésta es precedida por púrpura o erupción infiltrada, eritematosa, dolorosa, con o sin signos sistémicos. En estos casos, el tratamiento debe ser interrumpido inmediatamente.
El protector de la aguja de la jeringa precargada puede contener látex de caucho natural (goma de látex) que puede producir reacciones alérgicas graves.
4.5. Interacción con otros medicamentos y otras formas de interacción
Debido a que pueden incrementar el riesgo de hemorragia, no se recomienda la terapia combinada de nadroparina con los siguientes fármacos: ácido acetilsalicílico (u otros salicilatos), antiinflamatorios no esteroideos (AINEs) y antiagregantes plaquetarios (ver sección 4.4).
En caso de angina inestable y tratamiento de infarto de miocardio sin onda Q, nadroparina debe combinarse con ácido acetil salicílico, a una dosis máxima de 325 mg/día (ver secciones 4.2 y 4.4).
Combinaciones que requieren precaución:
Anticoagulantes orales, glucocorticoesteroides sistémicos y dextranos. Cuando se vaya a iniciar el tratamiento anticoagulante oral en pacientes en tratamiento con nadroparina, se mantendrá el tratamiento con heparina hasta que el valor de INR esté estabilizado al valor adecuado.
4.6. Fertilidad, embarazo y lactancia Fertilidad
No se han realizado estudios clínicos en relación a los efectos de la nadroparina con la fertilidad.
Embarazo
Estudios en animales no han evidenciado efectos teratogénicos o fetotóxicos con el uso de HBPM. Sin embargo, hay datos clínicos muy limitados en relación con el paso de nadroparina a través de la barrera placentaria en mujeres embarazadas. Por todo ello y como medida de precaución, el uso de nadroparina en el embarazo no es aconsejable a menos que los beneficios derivados del tratamiento sean superiores a los riesgos.
Lactancia
Debido a la limitada información sobre la excreción por leche materna, no es aconsejable el uso de nadroparina durante la lactancia materna.
4.7. Efectos sobre la capacidad para conducir y utilizar máquinas No procede.
4.8. Reacciones adversas
Las reacciones adversas se presentan agrupadas según su frecuencia (muy frecuentes: > 1/10; frecuentes: > 1/100 a < 1/10; poco frecuentes: > 1/1.000 a < 1/100; raras: > 1/10.000 a < 1/1.000; muy raras:< 1/10.000) y según la clasificación de órganos y sistemas.
Trastornos de la sangre y del sistema linfático:
- Muy frecuentes: manifestaciones hemorrágicas en diferentes localizaciones, más frecuentes en pacientes con otros factores de riesgo (ver secciones 4.3 y 4.5).
- Raras: trombocitopenia (incluyendo trombocitopenia inducida por heparina ,ver sección 4.4), trombocitosis.
- Muy raras: eosinofilia, reversible tras la suspensión del tratamiento.
Trastornos del sistema inmunológico:
- Muy raras: reacciones de hipersensibilidad (incluyendo angioedema y reacciones cutáneas), reacción anafilactoide.
Trastornos del metabolismo y de la nutrición:
- Muy raras: hipercalemia reversible relacionada con la inhibición de la aldosterona inducida por heparina, especialmente en pacientes con riesgo (ver sección 4.4).
Trastornos hepatobiliares:
- Frecuentes: elevación de las transaminasas, normalmente de forma transitoria.
Trastornos del aparato reproductor y de la mama:
- Muy raras: priapismo.
Trastornos de la piel y tejidos subcutáneos:
- Raras: erupción cutánea, urticaria, eritema, prurito.
- Muy raras: necrosis cutánea, normalmente localizada en el lugar de inyección (ver sección 4.4).
Trastornos generales y alteraciones en el lugar de la administración:
- Muy frecuentes: pequeños hematomas en el punto de inyección. En algunos casos se puede apreciar la aparición de nódulos firmes que no indican enquistamiento de la heparina y que suelen desaparecer al cabo de unos días.
- Frecuentes: reacción en el lugar de la inyección.
- Raras: calcinosis en el lugar de la inyección.
La calcinosis es más frecuente en pacientes con niveles anormales de fosfato cálcico como ocurre en algunos casos de insuficiencia renal crónica.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite
una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales
sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de medicamentos de Uso Humano: https://www.notificaram.es.
4.9. Sobredosis
El mayor signo clínico de sobredosis subcutánea o intravenosa es la hemorragia. Deben realizarse recuentos plaquetarios y otros parámetros de la coagulación. Las hemorragias menores raramente requieren tratamiento específico y normalmente es suficiente con reducir o espaciar las dosis posteriores.
Debe considerarse el uso de sulfato de protamina únicamente en los casos más severos.
La protamina neutraliza ampliamente el efecto anticoagulante de nadroparina, pero queda remanente una cierta actividad anti-Xa.
0,6 ml de sulfato de protamina neutralizan aproximadamente 950 UI anti-Xa de nadroparina. En el cálculo de la cantidad de protamina a inyectar debe tenerse en cuenta el tiempo transcurrido desde la administración de heparina, pudiendo decidirse una reducción de la dosis.
5. Propiedades farmacológicas
5.1. Propiedades farmacodinámicas
Grupo farmacoterapéutico: nadroparina, código ATC: B01 AB06
Fraxiparina es una heparina cálcica de bajo peso molecular obtenida por despolimerización de la heparina estándar. Está compuesta por glicosaminoglicanos con un peso molecular medio de 4.300 daltons (75-95% alrededor de 2.000-8.000 daltons). Se caracteriza por una elevada actividad anti-Xa (rango 95-130 UI anti-Xa/mg) y una baja actividad anti-IIa (< 40 UI anti-IIa/mg). La relación entre las dos actividades oscila entre 2,5 y 4,0.
Mecanismo de acción
Fraxiparina ejerce su efecto antitrombótico a través de su acción sobre las serinproteasas de la coagulación, principalmente, retardando la generación de trombina y neutralizando la trombina ya formada.
Fraxiparina frente a la heparina estándar tiene mayor actividad fibrinolítica, menor interacción con las plaquetas, y a las dosis habituales no modifica significativamente los tests de la coagulación. Además, su menor unión a las células endoteliales contribuye a prolongar su vida media y la actividad anti-Xa plasmática.
5.2. Propiedades farmacocinéticas
La excreción de Fraxiparina es principalmente por vía renal.
Las propiedades farmacocinéticas han sido determinadas mediante la valoración de la actividad anti Xa en plasma. Tras inyección subcutánea el pico plasmático aparece alrededor de las 3 horas. La vida media de eliminación tras la administración de dosis repetidas es alrededor de 8-10 horas. La actividad anti-Xa (> 0.05 UI/ml) persiste al menos durante 18 horas después de la inyección. La biodisponibilidad es prácticamente completa (aproximadamente 98%).
Biotransformación: los parámetros cinéticos se han evaluado principalmente a través de la desviación de las medidas de la actividad biológica y no por determinación de concentraciones plasmáticas; como consecuencia no se disponen de datos fiables en relación a la biotransformación de Fraxiparina.
Insuficiencia renal: en pacientes con insuficiencia renal puede ser necesaria la reducción de la dosis dado que la eliminación de la actividad anti-Xa es más lenta en estos pacientes (ver sección 4.2).
En un ensayo clínico que ha investigado las propiedades farmacocinéticas de nadroparina administrada por vía intravenosa en pacientes con insuficiencia renal de diverso grado, se encontró una relación entre el aclaramiento de nadroparina y el aclaramiento de creatinina. En pacientes con insuficiencia renal moderada (aclaramiento de creatinina 36-43 ml/min) tanto el valor medio de la AUC como la vida media aumentaron un 52% y un 39%, respectivamente, comparados con voluntarios sanos. En estos pacientes el aclaramiento plasmático de nadroparina disminuyó un 63% respecto de los valores normales. En el ensayo se observó una amplia variabilidad interindividual. En pacientes con insuficiencia renal grave (aclaramiento de creatinina 10-20 ml/min) ambos el valor medio de la AUC y la vida media aumentaron un 95% y un 112%, respectivamente, comparados con los voluntarios sanos. El aclaramiento plasmático en los pacientes con insuficiencia renal grave disminuyó un 50% con respecto al observado en pacientes con valores normales de aclaramiento. En pacientes con insuficiencia renal grave (aclaramiento de creatinina 3-6 ml/min) y en hemodiálisis, tanto el valor medio de la AUC como la vida media aumentaron un 62% y un 65%, respectivamente, comparados con voluntarios sanos. El aclaramiento plasmático en pacientes en hemodiálisis con insuficiencia renal grave disminuyó un 67% respecto de la observada en pacientes con función renal normal (ver secciones 4.2 y 4.4).
Insuficiencia hepática: no se dispone de datos específicos (se administrará con precaución en pacientes afectos de insuficiencia hepática dado que a nivel clínico se ha informado de algunos casos de elevación de transaminasas, generalmente transitorias).
Pacientes de edad avanzada: no se dispone de datos específicos. Sin embargo, se debe resaltar que dada la naturaleza de la patología tratada, la población estudiada en los ensayos clínicos incluía una amplia proporción de sujetos ancianos (ver sección 4.2).
6 . Datos farmacéuticos
6.1. Lista de excipientes
Solución de hidróxido de calcio o ácido clorhídrico diluido (en función del pH alcanzado) para ajustar al pH fisiológico.
Agua para preparaciones inyectables.
6.2. Incompatibilidades
Este medicamento no debe mezclarse con otros preparados.
6.3. Periodo de validez 3 años.
6.4. Precauciones especiales de conservación
No requiere condiciones especiales de conservación.
6.5. Naturaleza y contenido del envase
Cajas que contienen 10 y 50 jeringas precargadas.
Puede que solamente estén comercializados algunos tamaños de envases.
|
Volumen |
Tipo jeringa |
Nadroparina cálcica (UI anti Xa) |
|
0,3 ml |
No graduada (color verde claro) |
2.850 |
|
0,4 ml |
No graduada (color naranja) |
3.800 |
|
0,6 ml |
Graduada (color rojo) |
5.700 |
|
0,8 ml |
Graduada (color verde oscuro) |
7.600 |
Descripción de la jeringa
Cilindro de vidrio tipo I (graduado o no según detalle del cuadro anterior) provisto de una aguja de acero inoxidable.
Protector de aguja de caucho natural (goma de látex).
Junta de pistón de clorobutilo.
6.6. Precauciones especiales de eliminación y otras manipulaciones Ver sección 4.2 Posología y forma de administración.
Instrucciones después del uso:
Este envase contiene una jeringa con un dispositivo de seguridad que sirve para proteger la aguja después de su uso y que se acciona manualmente de la siguiente forma:


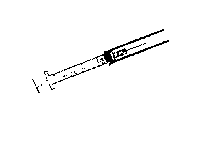
• Antes de la inyección:
• Después de la inyección:
1. Tirar hacia abajo por el punto de sujeción con los dedos (hasta oír el clic).
Simultáneamente y con la otra mano, tirar hacia arriba el dispositivo de seguridad (hasta oír el clic).
2. De esta forma la jeringa se puede desechar, quedando así la aguja completamente protegida.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizara de acuerdo con la normativa local.
7. Titular de la autorización de comercialización
Aspen Pharma Trading Limited 3016 Lake Drive Citywest Business Campus Dublín 24 Irlanda
8. Número(s) de autorización de comercialización
Fraxiparina 0,3 ml solución inyectable: 58496 Fraxiparina 0,4 ml solución inyectable: 58983 Fraxiparina 0,6 ml solución inyectable: 58982 Fraxiparina 0,8 ml solución inyectable: 61783
9. Fecha de la primera autorización/ renovación de la autorización
Fraxiparina 0,3 ml solución inyectable: 27 de Noviembre de 1989 Fraxiparina 0,4 ml solución inyectable: 26 de Junio de 1991 Fraxiparina 0,6 ml solución inyectable: 26 de Junio de 1991 Fraxiparina 0,8 ml solución inyectable: 16 de Abril de 1998
10. Fecha de la revisión del texto
Noviembre 2015
La información detallada y actualizada de este medicamento está disponible en la página Web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) http://www.aemps.gob.es
13 de 13