Fragmin 10.000 Ui/0,4 Ml Solucion Inyectable En Jeringas Precargadas
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
FRAGMIN 2.500 UI/0,2 ml, solución inyectable en jeringas precargadas. FRAGMIN 5.000 UI/0,2 ml, solución inyectable en jeringas precargadas. FRAGMIN 7.500 UI/0,3 ml, solución inyectable en jeringas precargadas. FRAGMIN 10.000 UI/0,4 ml, solución inyectable en jeringas precargadas. FRAGMIN 12.500 UI/0,5 ml, solución inyectable en jeringas precargadas. FRAGMIN 15.000 UI/0,6 ml, solución inyectable en jeringas precargadas. FRAGMIN 18.000 UI/0,72 ml, solución inyectable en jeringas precargadas. FRAGMIN 10.000 UI/ml, solución inyectable en ampollas.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Composición cualitativa y cuantitativa
|
Presentación |
Principio activo/ml de solución |
Contenido total |
|
Fragmin 2.500 UI/0,2 ml jeringas precargadas |
Dalteparina sódica (D.C.I.) 12.500 UI (anti-Xa) |
2.500 UI (anti-Xa)* |
|
Fragmin 5.000 UI/0,2 ml jeringas precargadas |
Dalteparina sódica (D.C.I.) 25.000 UI (anti-Xa) |
5.000 UI (anti-Xa)* |
|
Fragmin 7.500 UI/0,3 ml jeringas precargadas |
Dalteparina sódica (D.C.I.) 25.000 UI (anti-Xa) |
7.500 UI (anti-Xa)* |
|
Fragmin 10.000 UI/0,4 ml jeringas precargadas |
Dalteparina sódica (D.C.I.) 25.000 UI (anti-Xa) |
10.000 UI (anti-Xa)* |
|
Fragmin 12.500 UI/0,5 ml jeringas precargadas |
Dalteparina sódica (D.C.I.) 25.000 UI (anti-Xa) |
12.500 UI (anti-Xa)* |
|
Fragmin 15.000 UI/0,6 ml jeringas precargadas |
Dalteparina sódica (D.C.I.) 25.000 UI (anti-Xa) |
15.000 UI (anti-Xa)* |
|
Fragmin 18.000 UI/0,72ml jeringas precargadas |
Dalteparina sódica (D.C.I.) 25.000 UI (anti-Xa) |
18.000 UI (anti-Xa)* |
|
Fragmin 10.000 UI/ml solución inyectable en ampollas |
Dalteparina sódica (D.C.I.) 10.000 UI (anti-Xa) |
10.000 UI (anti-Xa)* |
* Una unidad internacional (anti factor Xa) de dalteparina sódica, corresponde a la actividad de una unidad del primer estándar internacional OMS para heparina de bajo peso molecular respecto a la
inhibición del Factor Xa de la coagulación, utilizando el sustrato cromogénico Kabi Pharmacia Diagnostica, Suecia (método amidolítico).
Excipiente(s) con efecto conocido
Cada jeringa precargada de Fragmin 2.500 UI/0,2 ml contiene hasta un máximo de 0,59 mg de sodio. Cada ampolla de Fragmin 10.000 UI/ ml contiene hasta un máximo de 3,10 mg de sodio.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable.
- Solución inyectable de administración intravenosa o subcutánea con 10.000 UI (anti-Xa)/ml en ampollas de 1 ml.
- Solución inyectable de administración subcutánea en jeringas precargadas con 2.500 UI (anti-Xa)/0,2 ml,
5.000 UI (anti-Xa)/0,2 ml, 7.500 UI/0,3 ml, 10.000 UI/0,4 ml, 12.500 UI/0,5 ml, 15.000 UI/0,6 ml ó 18.000 UI/0,72 ml.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento de la trombosis venosa profunda con o sin embolia pulmonar (Fragmin 5.000 UI/0,2 ml, Fragmin 7.500 UI/0,3 ml, Fragmin 10.000 UI/0,4 ml, Fragmin 12.500 UI/0,5 ml, Fragmin 15.000 UI/0,6 ml y Fragmin 18.000 UI/0,72 ml jeringas precargadas, y Fragmin 10.000 UI/ml solución inyectable en ampollas).
Prevención de los coágulos del sistema extracorpóreo durante la hemodiálisis y hemofiltración en los enfermos con insuficiencia renal crónica (Fragmin 5.000 UI/0,2 ml jeringas precargadas y Fragmin 10.000 UI/ml solución inyectable en ampollas).
Profilaxis de la enfermedad tromboembólica en cirugía (Fragmin 2.500 UI/0,2 ml y Fragmin 5.000 UI/0,2 ml j eringas precargadas).
Tratamiento de la angina inestable e infarto de miocardio sin onda Q (Fragmin 10.000 UI/ml ampollas 1 ml y Fragmin 7.500 UI/0,3 ml jeringas precargadas).
Profilaxis de la enfermedad tromboembólica en pacientes no quirúrgicos inmovilizados, cuya situación pueda definirse como de riesgo moderado o elevado (Fragmin 2.500 UI/0,2 ml y Fragmin 5.000 UI/0,2 ml jeringas precargadas).
Prevención secundaria de la enfermedad tromboembólica venosa (ETV) en pacientes oncológicos con trombosis venosa profunda y/o embolismo pulmonar.
4.2 Posología y forma de administración
Posología
ADVERTENCIA:
Las diferentes heparinas de bajo peso molecular no son necesariamente equivalentes. En consecuencia, se debe respetar la dosificación y el modo de empleo específico de cada una de estas especialidades farmacéuticas.
1) Tratamiento de la trombosis venosa profunda con o sin embolia pulmonar:
Fragmin se puede administrar por vía subcutánea en una sola dosis al día o en dos dosis diarias. Por regla general, debe iniciarse al mismo tiempo tratamiento con antagonistas de la vitamina K.
En general, no es necesaria la monitorización de la actividad anticoagulante.
Si se considera necesario, la actividad de Fragmin puede monitorizarse mediante la determinación de antiXa por un método funcional. Los niveles máximos en plasma se obtienen a las 3-4 horas después de la inyección subcutánea.
El tratamiento con Fragmin debe iniciarse lo antes posible y continuarse durante al menos 5 días o hasta que los niveles del complejo protrombínico (factores II, VII, IX, X) hayan descendido hasta el rango terapéutico.
Administración de una dosis diaria:
La dosis es de 200 UI/kg/día por vía subcutánea.
Esta dosis/día no debe exceder de las 18.000 UI.
Administración de dos dosis diarias:
La dosis es de 100 UI/kg/12 horas por vía subcutánea.
Esta pauta es recomendable en pacientes que requieran > 18.000 UI o que tengan factores de riesgo de sangrado, o en los que sea necesario monitorizar la actividad anti-Xa.
Los niveles de anti-Xa recomendados obtenidos a las 3-4 horas postadministración deben estar entre 0,51,0 UI/ml. 1 2
Cirugía general (riesgo moderado de trombosis):
El día de la intervención, administración de 2.500 UI (anti-Xa) por vía subcutánea, 2 a 4 horas antes de la misma. Los días siguientes, se administrarán 2.500 UI (anti-Xa) por vía subcutánea, una vez al día.
Cirugía oncológica y ortopédica (riesgo elevado de trombosis):
El día de la intervención, administración de 2.500 UI (anti-Xa) por vía subcutánea, 2 a 4 horas antes de la intervención. Doce horas después de la operación, administrar nuevamente 2.500 UI (anti-Xa) por vía subcutánea.
Los días siguientes, administrar 5.000 UI (anti-Xa) por vía subcutánea una vez al día o dos dosis de 2.500 UI al día.
En ambos casos, el tratamiento debe seguirse durante el periodo de riesgo o hasta la deambulación del paciente.
4) Profilaxis de la enfermedad tromboembólica en pacientes no quirúrgicos:
En pacientes de riesgo moderado, la posología será de 2.500 UI por vía subcutánea administradas una vez al día, y en pacientes de riesgo elevado, la posología será de 5.000 UI/día.
La profilaxis debe mantenerse mientras persista la situación de riesgo tromboembólico venoso o hasta la deambulación del paciente, siempre según criterio médico.
5) Enfermedad coronaria inestable, es decir, angina inestable e infarto de miocardio sin onda Q:
La dosis es de 120 UI/kg de peso corporal, administrada dos veces al día en inyección subcutánea, con una dosis máxima de 10.000 UI/12 horas.
La duración del tratamiento recomendado es de 6 a 8 días.
Se recomienda el tratamiento concomitante con dosis bajas de ácido acetilsalicílico.
6) Prevención secundaria de la enfermedad tromboembólica venosa (ETV) en pacientes oncológicos con trombosis venosa profunda y/o embolismo pulmonar:
Mes 1°:
Administrar subcutáneamente (SC) 200 UI/kg de peso corporal de dalteparina una vez al día durante los primeros 30 días de tratamiento. La dosis total diaria no debe superar las 18.000 UI.
Meses 2-6:
Debe administrarse subcutáneamente una dosis de aproximadamente 150 UI/kg de dalteparina, una vez al día según la tabla siguiente:
|
Peso corporal (Kg) |
Dosis de Dalteparina (UI) |
|
<56 |
7.500 |
|
57 a 68 |
10.000 |
|
69 a 82 |
12.500 |
|
83 a 98 |
15.000 |
|
>99 |
18.000 |
Reducción de la dosis debido a trombocitopenia inducida por quimioterapia:
Trombocitopenia: En el caso de trombocitopenia inducida por quimioterapia con recuentos plaquetarios <50.000/mm2, debe interrumpirse el tratamiento con dalteparina hasta que se recupere el recuento plaquetario por encima de 50.000/mm2.
Con recuentos plaquetarios entre 50.000 y 100.000/mm3, debe reducirse la dosis de dalteparina de un 17 a un 33% de la dosis inicial, dependiendo del peso del paciente (Tabla 1). Una vez se recupere el recuento plaquetario a >100.000/mm3, debe reiniciarse el tratamiento con la dosis completa.
Tabla 1. Reducción de la Dosis de Dalteparina con Trombocitopenia de 50.000-
100.000/mm3
|
Peso Corporal (kg) |
Dosis de Dalteparina Programada (UI) |
Dosis de Dalteparina Reducida (UI) |
Reducción Media de la Dosis (%) |
|
<56 |
7.500 |
5.000 |
33 |
|
57 to 68 |
10.000 |
7.500 |
25 |
|
69 to 82 |
12.500 |
10.000 |
20 |
|
83 to 98 |
15.000 |
12.500 |
17 |
|
>99 |
18.000 |
15.000 |
17 |
Abreviaturas: UI = Unidad Internacional
Insuficiencia renal: En caso de insuficiencia renal significativa, definida como un nivel de creatinina de >3 x LSN, debe ajustarse la dosis de dalteparina para mantener el nivel terapéutico anti-Xa de 1 UI/mL (intervalo de 0,5-1,5 UI/mL) medido a las 4-6 horas tras la inyección de dalteparina. Si el nivel anti-Xa está por debajo o por encima del rango terapéutico, debe aumentarse o reducirse la dosis de dalteparina, respectivamente, utilizando jeringas con otra dosis y debe repetirse la determinación anti-Xa tras administrar 3-4 nuevas dosis. Este ajuste de dosis debe repetirse hasta que se obtenga el nivel anti-Xa terapéutico.
La duración de la profilaxis que ha demostrado ser eficaz en la prevención secundaria de la ETV en pacientes oncológicos es de 6 meses.
Población pediátrica;
No se ha establecido la seguridad y eficacia de dalteparina sódica en niños. Los datos actualmente disponibles se describen en las secciones 5.1 y 5.2, pero no puede hacerse ninguna recomendación posológica.
Control de los niveles de anti-Xa en niños
En ciertas poblaciones especiales tratadas Fragmin, como los niños, debe considerarse la determinación de los niveles máximos de anti-Xa aproximadamente a las 4 horas de la administración. Para el tratamiento terapéutico con dosis administradas una vez al día, los niveles máximos de anti-Xa generalmente se deben mantener entre 0,5 y 1,0 IU/mL medidos a las 4 horas de la administración. En caso de una función renal fisiológica baja y cambiante, como en los recién nacidos, es necesario un control cuidadoso de los niveles de anti-Xa. Para el tratamiento profiláctico los niveles de anti-Xa deben mantenerse generalmente entre 0,2-0,4 IU/mL.
Como ocurre con todos los agentes antitrombóticos, existe riesgo de hemorragia sistémica con la administración de Fragmin. Debe tenerse precaución en el tratamiento con dosis altas de Fragmin en pacientes recién operados. Después de iniciado el tratamiento se deben vigilar cuidadosamente las complicaciones hemorrágicas. Esto puede hacerse mediante exámenes físicos regulares de los pacientes, estrecha observación del drenaje quirúrgico y determinaciones periódicas de hemoglobina y de anti-Xa.
Forma de administración
Técnica de la inyección subcutánea: La inyección debe ser realizada en el tejido celular subcutáneo de la cintura abdominal anterolateral y posterolateral, alternativamente en el lado derecho e izquierdo. La aguja debe ser introducida de forma completa, perpendicularmente y no tangencialmente en el espesor de un pliegue cutáneo formado entre el pulgar y el índice, y que debe ser mantenido durante toda la inyección.
4.3 Contraindicaciones
Fragmin está contraindicado en pacientes con:
• Hipersensibilidad al principio activo o a alguno de los excipientes o a otras heparinas de bajo peso molecular, otras heparinas, o a productos derivados del cerdo.
• Historia confirmada o sospechosa de trombocitopenia de origen inmunológico inducida por heparinas.
• Sangrados activos clínicamente significativos (tales como ulceración o sangrado gastrointestinal, o hemorragia cerebral).
• Alteraciones graves de la coagulación.
• Endocarditis séptica aguda o subaguda.
• Reciente traumatismo o cirugía del sistema nervioso central, ojos y oídos.
Está contraindicada la administración de dosis altas de dalteparina (usadas, por ejemplo, para tratar trombosis venosa profunda, embolismo pulmonar o enfermedad coronaria inestable) en pacientes que deban recibir anestesia epidural o que se sometan a procesos que requieran punción espinal (ver sección 4.4), debido a un mayor riesgo de sangrado (ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
No administrar por vía intramuscular. Debido al riesgo de hematomas, debe evitarse la inyección intramuscular de otros fármacos cuando la dosis de dalteparina supere las 5.000 UI en 24 horas.
Riesgo de hemorragia
En pacientes con un mayor riesgo de hemorragia como aquellos con trombocitopenia, defectos plaquetarios, insuficiencia renal o hepática grave, hipertensión no controlada, retinopatía hipertensiva o diabética, dalteparina debe utilizarse con precaución. Asimismo, altas dosis de dalteparina deben administrarse con cautela en pacientes que hayan sido sometidos recientemente a un proceso quirúrgico.
Si ocurre un infarto de miocardio transmural en un paciente con enfermedad coronaria inestable, es decir, con angina inestable o con infarto de miocardio sin onda Q, en el que esté indicado el tratamiento trombolítico, no es necesario suspender el tratamiento con Fragmin aunque éste puede incrementar el riesgo de hemorragia.
Monitorización de los niveles Anti-Xa
Normalmente no es necesario, pero en ciertos casos como en niños, pacientes con insuficiencia renal, o pacientes extremadamente delgados o con obesidad mórbida, embarazadas o que estén en riesgo de sufrir hemorragia o repetir la trombosis, debe considerarse la monitorización el efecto anticoagulante de dalteparina. Las pruebas de laboratorio que utilizan un sustrato cromogénico se consideran los métodos de elección para determinar los niveles anti-Xa. Se recomienda no utilizar el tiempo de tromboplastina parcial activado (TTPA) o el tiempo de protrombina (TP) como método para medir los niveles de anti-Xa, ya que son pruebas relativamente insensibles a la actividad de dalteparina. El aumento de la dosis de dalteparina con objeto de prolongar TTPA, puede provocar una sobredosificación y hemorragia (ver sección 4.9).
Por lo general los pacientes sometidos a hemodiálisis crónica en tratamiento con dalteparina, requieren menos ajustes de dosis y en consecuencia menos controles de los niveles anti-Xa. Los pacientes sometidos a hemodiálisis aguda pueden ser más inestables y precisarán una monitorización más completa de los niveles anti-Xa.
Hiperpotasemia
La heparina y las heparinas de bajo peso molecular pueden inhibir la secreción adrenal de aldosterona dando lugar a hiperpotasemia, en especial en pacientes con diabetes mellitus, insuficiencia renal crónica, acidosis metabólica preexistente, niveles de potasio en plasma elevados o que reciben tratamiento con fármacos ahorradores de potasio.
El riesgo de hiperpotasemia parece aumentar normalmente a medida que se prolonga el tratamiento, pero normalmente es reversible al interrumpirlo. Deben vigilarse los niveles plasmáticos de potasio en aquellos pacientes con riesgo de sufrir hiperpotasemia, antes de iniciar el tratamiento con heparinas y después controlarlo regularmente especialmente si el tratamiento se prolonga más de 7 días.
Anestesia epidural o espinal
En pacientes sometidos a anestesia epidural o espinal o a punción raquídea, la administración de heparina con fines profilácticos se ha asociado muy raramente a la aparición de hematomas epidurales o espinales, con el resultado final de parálisis prolongada o permanente. Este riesgo se incrementa por el uso de catéteres epidurales o espinales para anestesia, la administración concomitante de medicamentos con efecto sobre la coagulación como antiinflamatorios no esteroideos (AINES), antiagregantes plaquetarios o anticoagulantes, y por las punciones traumáticas o repetidas.
A la hora de decidir el intervalo de tiempo que debe transcurrir entre la administración de heparina a dosis profilácticas y la inserción o retirada de un catéter espinal o epidural, deben tenerse en cuenta las características del paciente y del producto, debiendo transcurrir al menos de 10 a 12 horas para las heparinas de bajo peso molecular. Una vez insertado o retirado el catéter, deberán transcurrir al menos cuatro horas hasta la administración de una nueva dosis de heparina. La siguiente dosis deberá retrasarse hasta que la intervención quirúrgica haya finalizado. En aquellos pacientes que reciben dosis terapéuticas de dalteparina más altas (por ejemplo, de 100 UI/kg a 120 UI/kg cada 12 horas o 200 UI/kg una vez al día), el intervalo debe ser como mínimo de 24 horas.
Si bajo criterio médico se decide administrar tratamiento anticoagulante durante un procedimiento anestésico espinal o epidural, debe extremarse la vigilancia del paciente para detectar precozmente cualquier signo o síntoma de déficit neurológico, como dolor lumbar, déficit sensorial y motor (entumecimiento y debilidad de extremidades inferiores) y trastornos funcionales del intestino o vejiga. El personal de enfermería debe ser entrenado para detectar tales signos y síntomas. Asimismo, se advertirá a los pacientes que informen inmediatamente al médico o personal de enfermería si experimentan cualquiera de los síntomas antes descritos.
Si se sospecha la aparición de algún signo o síntoma sugestivo de hematoma espinal o epidural, deben realizarse las pruebas diagnósticas con carácter de urgencia e instaurar el tratamiento adecuado, incluyendo la descompresión medular.
Enfermedad cardiaca
No se han realizado estudios adecuados para determinar la seguridad y el uso efectivo de Fragmin en la prevención de la trombosis valvular en pacientes con prótesis valvular cardiaca. Las dosis profilácticas de Fragmin no son suficientes para prevenir la trombosis valvular en pacientes con prótesis valvular cardiaca. No se puede recomendar el uso de Fragmin para este propósito.
En tratamientos prolongados de enfermedad arterial coronaria inestable como, por ejemplo, antes de una revascularización, se debe considerar la reducción de la dosis cuando existe una insuficiencia renal (S-creatinina > 150 pmol/l).
Trombocitopenia
Antes de iniciar el tratamiento con dalteparina y de forma regular durante el mismo, se recomienda realizar recuentos de plaquetas. Es necesaria una precaución especial si se desarrolla trombocitopenia rápidamente o en un grado significativo (menos de 100.000/pl o mm3) durante el tratamiento con dalteparina. En cualquier caso, se recomienda un estudio in vitro de anticuerpos antiplaquetarios en presencia de heparinas o heparinas de bajo peso molecular. Debe interrumpirse el tratamiento con heparina si el resultado de este estudio in vitro es positivo o no concluyente, o no se realiza el estudio.
Intercambiabilidad con otros anticoagulantes:
No se puede intercambiar dalteparina (unidad por unidad) con heparinas no fraccionadas, otras heparinas de bajo peso molecular, o polisacáridos sintéticos. Cada uno de estos medicamentos difieren en sus materias primas, proceso de fabricación, propiedades fisico-químicas, biológicas y clínicas, que conducen a diferencias en la identidad bioquímica, dosis, y posiblemente en la eficacia clínica y seguridad. Cada uno de estos medicamentos es único y tiene sus propias instrucciones de uso.
Población pediátrica
Se dispone de información limitada sobre la seguridad y eficacia de dalteparina en pacientes pediátricos. Si se utiliza dalteparina en estos pacientes, deben monitorizarse los niveles de anti-Xa.
Pacientes de edad avanzada
Dentro del intervalo de dosis terapéuticas, los pacientes de edad avanzada (especialmente a partir de los 80 años) pueden tener mayor riesgo de complicaciones hemorrágicas. Se recomienda una cuidadosa vigilancia de estos pacientes.
Advertencias sobre excipientes
Fragmin 2.500 UI/0.2 ml contiene menos de 23 mg (1 mmol) de sodio por 0,2 ml, por lo que se considera esencialmente “exento de sodio”.
Fragmin 10.000 Ul/ml, contiene menos de 23 mg (1 mmol) de sodio por 1 ml, por lo que se considera esencialmente “exento de sodio”.
4.5 Interacción con otros medicamentos y otras formas de interacción
La administración concomitante de fármacos que actúan sobre la hemostasia como los fármacos trombolíticos, otros anticoagulantes, inhibidores plaquetarios, ácido acetilsalicílico (AAS), antiinflamatorios no esteroideos (AINES), antagonistas de la vitamina K y dextrano, puede potenciar el efecto anticoagulante de Fragmin.
Sin embargo, a menos que exista una contraindicación concreta, los pacientes con enfermedad coronaria inestable, es decir, con angina inestable o infarto de miocardio sin onda Q, deben recibir ácido acetilsalicílico en dosis bajas por vía oral.
Dado que las dosis analgésicas/antiinflamatorias de los AINEs y AAS reducen la producción de prostaglandinas vasodilatadoras y en consecuencia el flujo sanguíneo renal y la excreción renal, debe tenerse especial precaución cuando se administre dalteparina junto con AINEs o dosis elevadas de AAS en pacientes con insuficiencia renal.
Como la heparina ha demostrado que interacciona con la nitroglicerina intravenosa, dosis elevadas de penicilina, sulfinpirazona, probenecid y ácido etacrínico, citostáticos, quinidina, antihistamínicos, digitálicos, tetraciclinas, tabaco y ácido ascórbico, no pueden descartarse estas interacciones para dalteparina.
4.6 Fertilidad, embarazo y lactancia
Embarazo
La dalteparina no atraviesa la placenta. Existen un elevado número de datos en mujeres embarazadas (datos en más de 1.000 embarazos) que indican que no produce malformaciones ni toxicidad fetal/neonatal. Se puede utilizar Fragmin durante el embarazo si es necesario desde el punto de vista clínico.
Se han publicado más de 2.000 casos (estudios, series de casos e informes de casos) sobre la administración de dalteparina durante el embarazo. En comparación con la heparina no fraccionada, se notificó una diátesis hemorrágica menor y una reducción del riesgo de fractura osteoporótica. En el estudio prospectivo más amplio que se ha realizado, “Efficacy of Thromboprophylaxis as an Intervention during Gravidity” (EThIG), participaron 810 mujeres embarazadas y se investigó un esquema específico del embarazo para la estratificación del riesgo (riesgo bajo, alto y muy alto de tromboembolismo venoso) con dosis diarias de dalteparina, de entre 50 y 150 UI/kg de peso corporal (en casos específicos de hasta 200 UI/kg de peso corporal). No obstante, solo existe un número limitado de estudios controlados aleatorios sobre el uso de heparinas de bajo peso molecular durante el embarazo.
Los experimentos realizados con animales no han demostrado ninguna propiedad teratogénica o fetotóxica de la dalteparina (ver sección 5.3).
La anestesia epidural durante el parto está totalmente contraindicada en mujeres que reciben dosis elevadas de anticoagulantes (ver sección 4.3). Se recomienda precaución en pacientes con mayor riesgo de hemorragia, como las mujeres perinatales (ver sección 4.4). En mujeres embarazadas, durante el primer trimestre de embarazo, se determinaron semividas de la actividad anti-Xa de dalteparina de 4 a 5 horas.
Se han notificado fracasos terapéuticos en mujeres embarazadas con prótesis valvular cardiaca y en tratamiento con dosis anticoagulantes completas de heparina de bajo peso molecular. No se ha estudiado de manera adecuada el uso de Fragmin en mujeres embarazadas con prótesis valvular cardiaca.
Lactancia
Dalteparina pasa en pequeñas cantidades a la leche materna. Los estudios han revelado niveles anti-Xa del 2 al 8% de los niveles plasmáticos en leche materna (15 mujeres, del tercer al quinto día de lactancia, 2-3 horas tras la administración subcutánea). Parece poco probable un efecto anticoagulante en el niño.
No se puede descartar un posible riesgo para el lactante. A la hora de decidir si continuar o interrumpir la lactancia materna o el tratamiento con Fragmin se deberá tener en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento con Fragmin para la madre.
Fertilidad
Los datos clínicos actuales no demuestran que la dalteparina sódica afecte a la fertilidad. No ha detectado ningún efecto de la dalteparina sódica en la fertilidad, el coito o el desarrollo perinatal y posnatal en los ensayos realizados con animales.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Fragmin sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Aproximadamente un 3% de los pacientes que han sido tratados de forma profiláctica han notificado efectos adversos.
En la tabla siguiente se listan las reacciones adversas que pueden estar asociadas con dalteparina agrupadas por grupos de órganos y frecuencias: frecuente (>1/100, < 1/10), poco frecuente (>1/1.000, <1/100); rara (>1/10.000, <1/1.000) y frecuencia no conocida (reacciones adversas notificadas durante la experiencia post-comercialización).
|
Clasificación de Órganos del Sistema |
Frecuencia |
Reacción adversa |
|
Trastornos de la sangre y del sistema linfático: |
Frecuente |
Trombocitopenia reversible no inmune (tipo 1) |
|
No conocida* |
Trombocitopenia inmune inducida por heparina (tipo 2, con o sin complicaciones tromboembólicas asociadas) | |
|
Trastornos del sistema inmunológico |
No conocida* |
Reacciones anafilácticas |
|
Trastornos del sistema nervioso |
No conocida* |
Sangrado intracraneal, que en algunos casos ha sido mortal |
|
Trastornos vasculares |
Frecuente |
Hemorragia (sangrado en cualquier punto), que en algunos casos ha sido mortal |
|
Trastornos gastrointestinales |
No conocida* |
Sangrado retroperitoneal, que en algunos casos ha sido mortal |
|
Trastornos hepatobiliares |
Frecuente |
Elevación transitoria de las transaminasas hepáticas (Aspartato aminotransferasa, Alanina aminotransferasa) |
|
Trastornos de la piel y del tejido subcutáneo: |
Rara |
Necrosis cutánea, alopecia transitoria, |
|
No conocida* |
Erupción | |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
No conocida |
Hematoma espinal o epidural (ver secciones 4.3 y 4.4) |
|
Trastornos generales y alteraciones en el lugar de administración |
Frecuente |
Hematoma subcutáneo en el lugar de la inyección, dolor en el lugar de la inyección |
N(no se puede establecer con los datos disponibles)
Se han notificado los siguientes efectos adversos durante la experiencia post-comercialización:
El riesgo de hemorragia es dosis dependiente. La mayoría de las hemorragias son leves. Se han notificado casos de hemorragias graves que en algunos casos han sido mortales.
La heparina puede causar hipoaldosteronismo que puede producir un aumento de los niveles de potasio en plasma. Raramente puede producirse una hiperpotasemia clínicamente significativa, especialmente en pacientes con insuficiencia renal crónica y diabetes mellitus (ver sección 4.4).
El tratamiento a largo plazo con heparina se ha asociado con riesgo de osteoporosis. Aunque esto no se ha observado con dalteparina no puede excluirse el riesgo de osteoporosis.
Población pediátrica:
Se espera que la frecuencia, tipo y gravedad de reacciones adversas en los niños que sean las mismas que en adultos. No se ha establecido la seguridad de la administración de dalteparina a largo plazo.
Notificación de sospechas de reacciones adversas
Es importante notificar las sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaram.es.
4.9 Sobredosis
El efecto anticoagulante inducido por la dalteparina sódica puede ser inhibido con la protamina. La prolongación del tiempo de coagulación se neutraliza completamente, pero la actividad anti-Xa de dalteparina se mantiene en un 25-50%. Cada mg de protamina neutraliza parcialmente el efecto de 100 unidades (anti-Xa) de dalteparina sódica. La propia protamina inhibe la hemostasia primaria y sólo debe utilizarse como medida de emergencia.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agentes antitrómbicos, código ATC: B01AB04
Fragmin es un producto antitrombótico que contiene una heparina de bajo peso molecular denominada dalteparina sódica. La dalteparina sódica es una sal sódica de heparina despolimerizada, obtenida por degradación de heparina de la mucosa intestinal porcina con ácido nitroso. La masa molecular media es de 6.000. El porcentaje de fragmentos con una masa molecular superior a 8.000 está entre 15-25 %, y el porcentaje de fragmentos con una masa molecular inferior a 3.000 está entre 5-13 %.
Los límites de actividad anti Xa UI/ml y anti IIa UI/ml para las diferentes presentaciones de Fragmin son los siguientes:
|
Anti Xa UI/ml |
Anti IIa UI/ml | |
|
Fragmin 2.500 UI/0,2 ml |
11250 - 13750 |
3850 - 5950 |
|
Fragmin 5.000 UI/0,2 ml |
|
Fragmin 7.500 UI/0,3 ml Fragmin 10.000 UI/0,4 ml Fragmin 12.500 UI/0,5 ml Fragmin 15.000 UI/0,6 ml Fragmin 18.000 UI/0,72 ml |
22500 - 27500 |
7700 - 11900 |
|
Fragmin 10.000 UI/ml |
9000 - 11000 |
3080 - 4760 |
El efecto antitrombótico de la dalteparina sódica depende de su capacidad para potenciar la inhibición del factor Xa y de la trombina por la antitrombina (AT). La dalteparina sódica muestra una capacidad relativamente mayor para potenciar la inhibición del factor Xa que para prolongar el tiempo de coagulación del plasma (TTPA). La dalteparina sódica posee un menor efecto sobre la función y adherencia plaquetaria que la heparina y en consecuencia, un efecto mínimo sobre la hemostasia primaria. Se piensa, sin embargo, que algunas de las propiedades antitrombóticas de la dalteparina sódica son mediadas a través de su efecto sobre la pared vascular o el sistema fibrinolítico.
Población pediátrica:
Se dispone de limitada información sobre la seguridad y eficacia de dalteparina en pacientes pediátricos. Si se utiliza dalteparina en estos pacientes, deben vigilarse los niveles anti-Xa.
El mayor estudio prospectivo investigó la eficacia, seguridad y la relación de las dosis con la actividad antiXa plasmática de dalteparina en la profilaxis y tratamiento de trombosis arterial y venosa en 48 pacientes pediátricos (Nohe et al, 1999).
Nohe et al (1999) Demografía y diseño del estudio
|
Diseño del estudio |
Pacientes |
Diagnóstico |
Indicación, dosis de Fragmin, anti-Xa objetivo, Duración | ||
|
Monocéntri |
Edad: |
Trombosis |
Profiláxis: |
Tratamiento |
Tratamiento |
|
co, abierto; |
31 semanas |
arterial o |
primario: |
secundario: | |
|
(n = 48) |
pretérmino a |
venosa; |
(n = 10) |
(n = 25) |
(n = 13) |
|
18 años |
EVOP; | ||||
|
HPP* |
95 ± 52 anti- |
129 ± 43 anti- |
129 ± 43 anti- | ||
|
Sexo: |
Xa IU/kg sc |
Xa |
Xa | ||
|
32 varones, |
una vez al |
IU/kg sc una vez |
IU/kg sc una vez | ||
|
16 mujeres |
día; |
al día; |
al día; | ||
|
0,2 a 0,4 |
0,4 a 1,0 IU/mL |
0,4 a 1,0 IU/mL | |||
|
IU/mL | |||||
|
3-6 meses |
3-6 meses |
3-6 meses | |||
* Enfermedad veno-oclusiva pulmonar (EVOP), Hipertensión pulmonar primaria (HPP)
En este estudio, no se produjeron eventos tromboembólicos en los 10 pacientes que recibieron dalteparina para tromboprofilaxis. En los 23 pacientes que recibieron dalteparina para tratamiento antitrombótico primario de trombosis arterial o venosa, se observó una recanalización completa en 7/23 (30%), recanalización parcial en 7/23 (30%) y no recanalización en 9/23 (40%). En 8 pacientes que recibieron dalteparina para el tratamiento antitrombótico secundario tras trombolisis exitosa, se mantuvo o mejoró la
recanalización. No se observó recanalización en los 5 pacientes que recibieron dalteparina como tratamiento secundario tras el fracaso de la trombólisis. Se notificó un sangrado menor en 2/48 niños (4%), que se resolvió tras la reducción de la dosis. Los recuentos plaquetarios en los pacientes oscilaron entre 37.000/pl a 574.000/pl. Los autores atribuyen los recuentos plaquetarios por debajo de lo normal (150.000/pl) al tratamiento inmunosupresor. No se observó una reducción del recuento plaquetario > 50% del valor inicial, una señal del tipo de trombocitopenia tipo 2 (HIT 2) inducida por heparina, en ningún paciente. En los grupos tanto de terapia como de profilaxis, la dosis de dalteparina (UI anti-Xa/kg) necesaria para alcanzar la actividad anti-Xa objetivo (IU/ml) fué inversamente proporcional a la edad (r2 = 0,64, P = 0,017; r2 = 0,13, P = 0,013). La predictibilidad del efecto anticoagulante con la dosis ajustada por el peso parece reducirse en los niños, en comparación con los adultos, posiblemente debido a una alteración en la unión en plasma (ver sección 5.2).
5.2 Propiedades farmacocinéticas
La vida media de la dalteparina tras su inyección i.v. es de 2 horas, y tras la administración s.c., de 3-4 horas. La biodisponibilidad del principio activo tras su inyección s.c. es de aproximadamente el 90%, y la farmacocinética apenas depende de la dosis. La vida media se prolonga en los pacientes urémicos. La dalteparina sódica se elimina fundamentalmente por el riñón.
Población pediátrica:
Los bebés menores de aproximadamente 2 a 3 meses de edad o < 5 kg requieren mayores dosis HBPM por kg, posiblemente debido a su mayor volumen de distribución. Explicaciones alternativas para este aumento de las dosis de HBPM por peso corporal en niños pequeños incluyen una farmacocinética de la heparina alterada y/o una disminución en la expresión de la actividad anticoagulante de la heparina en los niños debido a las reducidas concentraciones en plasma de antitrombina.
5.3 Datos preclínicos sobre seguridad
La toxicidad aguda de la dalteparina sódica es considerablemente menor que la de la heparina. El único dato significativo que se ha observado en todos los estudios de toxicidad tras la administración subcutánea de altas dosis, es una hemorragia en el lugar de inyección cuya incidencia y gravedad se relacionan con la dosis. No se ha detectado ningún efecto acumulativo de las hemorragias en los lugares de inyección.
La reacción hemorrágica se tradujo en cambios del efecto anticoagulante, dependientes de la dosis, a juzgar por TTPA y la actividad anti-Xa.
No se pudo demostrar que la dalteparina sódica ejerciera un mayor efecto osteopénico que la heparina, ya que la actividad osteopénica de dosis equivalentes resultó similar.
Los resultados obtenidos no revelaron toxicidad orgánica, con independencia de la vía de administración, posología o duración del tratamiento. Tampoco se hallaron efectos mutagénicos. No se ha descrito ninguna acción embriotóxica o teratógena, ni tampoco se han observado anomalías de la capacidad reproductora o desarrollo peri o postnatal.
6 . DATOS FARMACÉUTICOS 6.1 Lista de excipientes
Hidróxido de sodio (F. Eur.) y ácido clorhídrico (F. Eur.) para ajuste de pH.
Cloruro de sodio (F. Eur.): sólo en las presentaciones de Fragmin 2.500 UI/0,2 ml y Fragmin 10.000 Ul/ml. Agua para preparaciones inyectables (F. Eur.).
6.2 Incompatibilidades
Fragmin, solución inyectable, es compatible con soluciones de cloruro sódico isotónicas (9 mg/ml) o de glucosa (50 mg/ml), tanto en frascos de vidrio como en envases de plástico. No se ha investigado la compatibilidad entre FRAGMIN y otros productos.
6.3 Periodo de validez
Fragmin, solución inyectable, se mantiene estable durante 3 años.
6.4 Precauciones especiales de conservación
Conservar por debajo de 25° C: Fragmin 10.000 UI/0,4 ml, Fragmin 12.500 UI/0,5 ml, Fragmin 15.000 UI/0,6 ml y Fragmin 18.000 UI/0,72 ml jeringas precargadas.
Conservar por debajo de 30° C: Fragmin 2.500 UI/0,2 ml, Fragmin 5.000 UI/0,2 ml, Fragmin 7.500 UI/0,3 ml jeringas precargadas y Fragmin 10.000 UI/ml solución inyectable en ampollas.
6.5 Naturaleza y contenido del envase
Solución inyectable en ampollas:
Fragmin 10.000 UI/ml solución inyectable, se presenta en ampollas de vidrio claro (vidrio Tipo I) de 1 ml (10.000 UI/ml) en envase con 10 ampollas.
Jeringas precargadas con dispositivo de protección para la aguja:
Fragmin 2.500 UI/0,2 ml y Fragmin 5.000 UI/0,2 ml solución inyectable en jeringas precargadas: Solución inyectable en jeringas precargadas (vidrio Tipo I), con capuchón para la aguja (exento de látex), tapón del émbolo, émbolo y un dispositivo de protección para la aguja en envases con 2, 10 y 25 jeringas precargadas y envases clínicos con 100 jeringas precargadas.
Fragmin 7.500 UI/0,3 ml solución inyectable en jeringas precargadas: Solución inyectable en jeringas precargadas (vidrio Tipo I), con capuchón para la aguja (exento de látex), tapón del émbolo, émbolo y un dispositivo de protección para la aguja en envase con 10 jeringas precargadas.
Fragmin 10.000 UI/0,4 ml, Fragmin 12.500 UI/0,5 ml Fragmin 15.000 UI/0,6 ml y Fragmin 18.000 UI/0,72 ml, solución inyectable en jeringas precargadas: Solución inyectable en jeringas precargadas (vidrio Tipo I), con capuchón para la aguja (exento de látex), tapón del émbolo, émbolo y un dispositivo de protección para la aguja en envase con 5 jeringas precargadas.
6.6 Precauciones especiales de eliminación
Los medicamentos para administración parenteral deben inspeccionarse visualmente para descartar la existencia de partículas y/o decoloración antes de su administración. Desechar el medicamento, si presenta partículas o si aparece decoloración. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
Utilización de las ampollas:
Administrar siguiendo el protocolo estándar.
Utilización de las jeringas precargadas con dispositivo de protección para la aguja:
Administrar siguiendo el protocolo estándar.
El dispositivo de protección para la aguja consiste en un bloqueante de plástico sujeto a la etiqueta de la jeringa. El bloqueante de plástico llega hasta la punta de la aguja y está alineado en paralelo con el capuchón de la misma. El dispositivo se ha diseñado específicamente para prevenir los pinchazos accidentales tras la administración del medicamento.
El dispositivo de protección debe ser activado por el usuario, lo que hará que la aguja sea inofensiva una vez administrada la inyección.
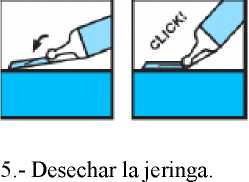
1.- Separar la punta del bloqueante de plástico del capuchón de la aguja.

2.- Retirar el capuchón de la aguja.

3.- Administrar la inyección normalmente.
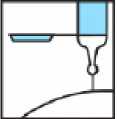
4.- Al terminar la inyección, activar el dispositivo de protección apoyando el bloqueante de plástico sobre una superficie dura y estable, doblando el cuerpo de la jeringa hacia arriba para forzar que la aguja entre en el bloqueante y quede fija (se oye un clic cuando la aguja queda atrapada en el bloqueante). Doblar la aguja hasta superar un ángulo de 45 grados con la superficie plana hasta que quede totalmente inservible.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer, S.L.
Avda. de Europa 20-B Parque Empresarial La Moraleja 28108 Alcobendas (Madrid)
Tel.: 91. 490.99.00 Fax: 91. 490.97.00
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
Fragmin 2.500 UI/0,2 ml, solución inyectable en jeringas precargadas: 58.536 Fragmin 5.000 UI/0,2 ml, solución inyectable en jeringas precargadas: 58.537 Fragmin 7.500 UI/0,3 ml, solución inyectable en jeringas precargadas: 62.567 Fragmin 10.000 UI/0,4 ml, solución inyectable en jeringas precargadas: 62.155 Fragmin 12.500 UI/0,5 ml, solución inyectable en jeringas precargadas: 62.156 Fragmin 15.000 UI/0,6 ml, solución inyectable en jeringas precargadas: 62.157 Fragmin 18.000 UI/0,72 ml, solución inyectable en jeringas precargadas: 62.158 Fragmin 10.000 UI/ml, solución inyectable en ampollas: 61.523
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Fragmin 2.500 UI/0,2 ml y Fragmin 5.000 UI/0,2 ml, solución inyectable en jeringas precargadas: Noviembre de 1989/noviembre 2002.
Fragmin 7.500 UI/0,3 ml, solución inyectable en jeringas precargadas: Agosto 1999/ agosto 2004. Fragmin 10.000 UI/0,4 ml, Fragmin 12.500 UI/0,5 ml, Fragmin 15.000 UI/0,6 ml y Fragmin 18.000 UI/0,72 ml, solución inyectable en jeringas precargadas: Febrero de 1999/febrero 2004.
Fragmin 10.000 UI/ml, solución inyectable en ampollas: Septiembre de 1997/septiembre 2002.
10. FECHA DE LA REVISIÓN DEL TEXTO
05/2015
La información detallada y actualizada de este medicamento está disponible en la página Web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) http://www.aemps.gob.es
16 de 16
Prevención de los coágulos durante la hemodiálisis y hemofiltración:
Pacientes con insuficiencia renal crónica sin diátesis hemorrágica:
Hemodiálisis y hemofiltración durante, como máximo, 4 horas:
Se administra la misma posología que se indica más adelante o únicamente una inyección en bolo intravenoso de 5.000 UI.
Hemodiálisis y hemofiltración durante más de 4 horas:
Se administra una inyección en bolo intravenoso de 30-40 UI/kg de peso corporal seguido de la infusión intravenosa de 10-15 UI/kg de peso corporal y hora.
En ambos casos se recomienda mantener los niveles de actividad anti-Xa en el intervalo de 0,5-1,0 UI anti-Xa/ml.
Profilaxis de la enfermedad tromboembólica en cirugía: