Fanhdi 100 Ui Fviii/120 Ui Fvw Por Ml Polvo Y Disolvente Para Solucion Inyectable
"I
an
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Fanhdi 25 UI FVIII/30 UI FVW por ml, polvo y disolvente para solución inyectable Fanhdi 50 UI FVIII/60 UI FVW por ml, polvo y disolvente para solución inyectable Fanhdi 100 UI FVIII/120 UI FVW por ml, polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Complejo de factor VIII de coagulación y factor von Willebrand humanos.
Fanhdi se presenta como polvo liofilizado para solución inyectable conteniendo nominalmente 250 UI, 500 UI, 1000 UI o 1500 UI de factor VIII (FVIII) de coagulación humano y 300, 600, 1200 ó 1800 UI de factor von Willebrand (FVW) humano por vial.
Fanhdi 25 UI FVIII/30 UI FVW por ml se reconstituye con 10 ml de agua para inyectables y contiene aproximadamente 25 UI de factor VIII de coagulación humano y 30 UI de factor von Willebrand humano por ml.
Fanhdi 50 UI FVIII/60 UI FVW por ml se reconstituye con 10 ml de agua para inyectables y contiene aproximadamente 50 UI de factor VIII de coagulación humano y 60 UI de factor von Willebrand humano por ml.
Fanhdi 100 UI FVIII/120 UI FVW por ml tiene dos presentaciones que se reconstituyen con 10 o con 15 ml de agua para inyectables. Ambas presentaciones contienen aproximadamente 100 UI de factor VIII de coagulación humano y 120 UI de factor von Willebrand humano por ml.
La potencia de FVIII:C (UI) se determina por el método cromogénico de la Farmacopea Europea. La actividad específica de Fanhdi es como mínimo de 2,5 a 10 UI FVIII:C/mg de proteína dependiendo de la potencia del vial (250, 500, 1000 y 1500 UI).
La potencia de FVW (UI) se determina por el test de la actividad del Cofactor de la Ristocetina (FVW:RCo) en relación con el Estándar Internacional para el concentrado de FVW (OMS).
Para la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Vial conteniendo polvo blanco o amarillo pálido y jeringa con agua para inyectables (disolvente).
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas Hemofilia A
Fanhdi está indicado para el tratamiento y profilaxis de hemorragias en pacientes con hemofilia A (déficit congénito de FVIII).
Este producto puede ser útil en el manejo de deficiencia adquirida de FVIII.
Enfermedad von Willebrand
Fanhdi está indicado para el tratamiento de hemorragias y tratamiento y profilaxis de sangrado quirúrgico en pacientes con la enfermedad de von Willebrand (EVW) cuando el tratamiento solo con desmopresina (DDAVP) no es efectivo o está contraindicado.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de trastornos hemostáticos.
Posología
Hemofilia A
La dosificación y la duración del tratamiento dependen de la gravedad de la deficiencia de FVIII, de la localización y el grado de la hemorragia y del estado clínico del paciente.
El número de unidades de FVIII administradas se expresa en Unidades Internacionales (UI), en relación con el estándar de la Organización Mundial de la Salud (OMS) vigente para concentrados de FVIII. La actividad plasmática de FVIII se expresa como un porcentaje (en relación con el plasma humano normal) o en Unidades Internacionales (en relación con un estándar internacional para FVIII en plasma).
Una Unidad Internacional (UI) de actividad de FVIII equivale a la cantidad de FVIII en un ml de plasma humano normal. El cálculo de la dosis necesaria de FVIII se basa en la observación empírica de que 1 Unidad Internacional (UI) de FVIII por kg de peso corporal aumenta la actividad plasmática de FVIII en un
2,1 ± 0,4% de la actividad normal. La dosis necesaria se determina utilizando la fórmula siguiente:
Unidades requeridas = peso corporal (kg) x aumento deseado de FVIII (%) (UI/dl) x 0,5
La dosis y la frecuencia de administración deben calcularse según la respuesta clínica del paciente.
En el caso de episodios hemorrágicos como los detallados a continuación, la actividad de FVIII no debe ser inferior al nivel plasmático de actividad establecido (en % de plasma normal o UI/dl) en el período correspondiente. Puede emplearse la siguiente tabla como guía de dosificación en episodios hemorrágicos y cirugía:
Grado de la hemorragia/ _Tipo de cirugía
Nivel de FVIII Frecuencia de dosificación (horas)/ requerido (%)(UI/dl) Duración de la terapia (días)_
20 - 40
Hemorragia
Hemartrosis y sangrado muscular u oral menores
Repetir cada 12 - 24 horas. Al menos 1 día, hasta que el episodio hemorrágico manifestado por dolor se detenga o hasta curación.
Hemartrosis y hemorragia muscular o hematoma moderados
30 - 60
Repetir la administración cada 12 - 24 horas durante 3 - 4 días o más hasta que el dolor y la discapacidad aguda desaparezcan.
60 - 100
Hemorragias con peligro para la vida
Repetir la administración cada 8 - 24 horas hasta que el riesgo desaparezca.
Cirugía
Menor 30 - 60
incluyendo extracciones dentales
Cada 24 horas, al menos 1 día hasta curación.
Mayor 80 - 100
(pre- y postoperatorio)
Repetir la administración cada 8 - 24 horas hasta la adecuada cicatrización de la herida, y continuar la terapia durante un mínimo de 7 días para mantener un nivel de actividad de FVIII de 30% a 60% (UI/dl)._
Se recomienda la determinación adecuada de los niveles plasmáticos de FVIII durante todo el tratamiento a fin de calcular la dosis y la frecuencia de las administraciones. Particularmente en las intervenciones de cirugía mayor, es imprescindible una monitorización precisa de la terapia de sustitución por medio de análisis de coagulación (actividad plasmática de FVIII). La respuesta individual de los pacientes a la terapia con FVIII puede variar, alcanzándose diferentes niveles de recuperación in vivo y de semivida.
En la profilaxis a largo plazo para impedir hemorragias en pacientes con hemofilia A grave deben administrarse dosis de 20 a 40 UI de FVIII/kg de peso corporal a intervalos de 2 a 3 días. En algunos casos, especialmente en pacientes jóvenes, puede ser necesario acortar los intervalos de administración o dosis más elevadas.
En los pacientes se debe controlar el desarrollo de inhibidores del FVIII. Si no se obtienen los niveles de actividad plasmática de FVIII esperados, o si el sangrado no se controla con la dosis adecuada, deben realizarse ensayos para determinar la presencia de inhibidores de FVIII. En pacientes con elevados niveles de inhibidor, puede ser que la terapia con FVIII no sea efectiva y deban considerarse otras opciones terapéuticas. Dichas terapias deberán realizarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia.Ver también sección 4.4.
Enfermedad de von Willebrand (EVW)
Generalmente, 1 UI de FVW:RCo/kg eleva el nivel circulante del mismo aproximadamente en un 2%. Deben alcanzarse los niveles de FVW:RCo > 0,6 UI/ml (60%) y de FVIII:C > 0,4 UI/ml (40%).
Normalmente se recomienda 40 - 80 UI/kg de FVW (FVW:RCo) y 20 - 40 UI/kg de FVIII:C para alcanzar la hemostasia.
Se puede necesitar una dosis inicial de 80 UI/kg de FVW, especialmente en pacientes con el tipo 3 de la EVW en que el mantenimiento de niveles adecuados puede necesitar dosis más elevadas que en otros tipos de la EVW.
Se debe readministrar una dosis apropiada cada 12 - 24 horas. La dosis y la duración del tratamiento depende del estado clínico del paciente, del tipo y severidad de la hemorragia, y de los niveles de FVW:RCo y FVIII:C.
En el uso de un preparado de FVW que contenga FVIII, el médico que realiza el tratamiento debe tener en cuenta que el tratamiento continuado puede causar un aumento excesivo de FVIII:C. Después de 24 - 48 h de tratamiento, y para evitar un aumento excesivo de FVIII:C, debe considerarse la reducción de la dosis
aftPs
y/o prolongación del intervalo en la administración de la dosis, o bien se debe considerar el uso de productos con FVW que contengan un bajo nivel de FVIII.
No se dispone de datos suficientes procedentes de ensayos clínicos para recomendar el uso de Fanhdi en niños menores de 6 años para las indicaciones autorizadas.
Forma de administración
Reconstituir el producto como se describe en sección 6.6. El producto debe administrarse por vía intravenosa. La velocidad de administración no debe sobrepasar los 10 ml/min.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes (ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
Al igual que con cualquier producto proteico para administración intravenosa, es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico. El producto contiene trazas de otras proteínas humanas, además de FVIII. Los pacientes deben ser informados acerca de los signos iniciales de las reacciones de hipersensibilidad, que incluyen erupciones cutáneas que pueden llegar a urticaria generalizada, opresión torácica, dificultad al respirar, hipotensión y anafilaxia. Si se producen reacciones de este tipo, se recomienda interrumpir la administración del preparado y contactar inmediatamente con el médico.
En caso de shock, se seguirán las recomendaciones vigentes para tratamiento del shock.
Las medidas estándar para prevenir infecciones resultantes del uso de medicamentos preparados a partir de sangre o plasma humano incluyen la selección de los donantes, la realización de pruebas de detección para las donaciones individuales y de las mezclas de plasma para marcadores específicos de infección, y la inclusión de etapas de fabricación eficaces para la inactivación o eliminación de virus. A pesar de estas medidas, si se administran medicamentos preparados a partir de sangre o plasma humano, no se puede excluir totalmente la posibilidad de transmisión de agentes infecciosos. Esto es aplicable también a los virus desconocidos o emergentes y a otros patógenos.
Las medidas adoptadas se consideran eficaces para los virus envueltos como el VIH, VHB y VHC, y para el virus no envuelto de la hepatitis A. Las medidas tomadas pueden tener un valor limitado para virus no envueltos tales como el parvovirus B19.
La infección por parvovirus B19 puede ser grave para una mujer embarazada (infección fetal) y para sujetos con inmunodeficiencia o con una producción aumentada de hematíes (ej. con anemia hemolítica).
Hemofilia A
La formación de anticuerpos neutralizantes (inhibidores) es bien conocida en el tratamiento de pacientes con hemofilia A. Estos inhibidores son generalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del FVIII. Esta actividad se cuantifica en unidades Bethesda (UB) por ml de plasma utilizando el método modificado de Nijmegen. El riesgo de desarrollar inhibidores se correlaciona con la exposición a FVIII antihemofílico, siendo este riesgo más alto los primeros 20 días de exposición. Raramente, pueden desarrollarse inhibidores tras los primeros 100 días de exposición. En los pacientes tratados con el FVIII humano de coagulación se debe controlar el posible desarrollo de inhibidores mediante observación clínica y pruebas de laboratorio adecuadas. Ver también sección 4.8.
Enfermedad de von Willebrand
En el uso de un preparado de FVW que contenga FVIII, el médico que realiza el tratamiento debe tener en cuenta que el tratamiento continuado puede causar un aumento excesivo de FVIII. En pacientes que reciban un preparado de FVW que contenga FVIII, deberán monitorizarse los niveles de FVIII:C para evitar niveles
4 de 1 1 MINISTRO DE
SANIDAD, POLITICA SOCIAL E IGUALDAD Agencia españoiaóe medie amentos y proouctos san-tanos
ÍTTI
excesivos sostenidos de FVIII:C en plasma, lo cual podría incrementar el riesgo de complicaciones tromboembólicas.
En el uso de un preparado de FVW que contenga FVIII en pacientes con la EVW existe riesgo de aparición de efectos trombóticos, particularmente en pacientes con riesgos clínicos o de laboratorio conocidos. Así pues, los pacientes con riesgo deben ser monitorizados ante la aparición de los signos iniciales de trombosis. Debe iniciarse profilaxis contra tromboembolismo vascular, según las recomendaciones vigentes.
Los pacientes con EVW, especialmente aquellos pacientes con el tipo 3, pueden desarrollar anticuerpos neutralizantes (inhibidores) al FVW. Si no se alcanzan los niveles esperados de actividad de FVW:RCo en plasma, o si la hemorragia no se controla con la dosis apropiada, deberá realizarse un ensayo para determinar la presencia de inhibidor de FVW. La terapia con FVW puede no ser efectiva en aquellos pacientes con altos niveles de inhibidor, por lo que deberán considerarse otras opciones terapéuticas.
Complicaciones relacionadas con el catéter
En el caso que se necesite usar un dispositivo de acceso venoso central (DAVC) debe tenerse en cuenta el riesgo de complicaciones relacionadas con DAVC incluyendo infecciones locales, bacteriemia y trombosis en el lugar de inserción del catéter.
Se recomienda la vacunación apropiada (hepatitis A y B) para los pacientes que reciban concentrados de FVIII.
Cada vez que se administra Fanhdi a un paciente, se recomienda indicar el nombre y el número de lote del medicamento para mantener la trazabilidad del producto.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han observado interacciones del complejo FVIII/FVW humano con otros medicamentos.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción en animales con el complejo FVIII/FVW. Por tanto, durante el embarazo y la lactancia puede utilizarse el complejo FVIII/FVW únicamente si está claramente indicado.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Fanhdi sobre la capacidad de conducir o utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Rara vez se han observado reacciones alérgicas o de hipersensibilidad (que pueden incluir angioedema, sensación de ardor y picor en el lugar de inyección, escalofríos, enrojecimiento, erupciones cutáneas que pueden llegar a urticaria generalizada, cefalea, hipotensión, somnolencia, náuseas, inquietud, taquicardia, opresión torácica, hormigueo, vómitos, dificultad al respirar) en pacientes tratados con productos que contienen FVIII. En ciertos casos, estas reacciones han progresado hasta anafilaxia grave (incluyendo shock).
En raras ocasiones se ha observado fiebre.
Algunos pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes contra el FVIII (inhibidores), lo que ocasiona una respuesta clínica insuficiente al tratamiento. En tales casos, se recomienda contactar con un centro especializado en hemofilia.
Los pacientes con la EVW, especialmente aquellos pacientes de tipo 3, pueden desarrollar anticuerpos neutralizantes (inhibidores) al FVW en muy raras ocasiones. Si dichos inhibidores aparecen, esta condición
ÍTTI
se manifestará en forma de una respuesta clínica inadecuada. Dichos anticuerpos pueden aparecer asociados a reacciones anafilácticas. Así pues, en aquellos pacientes que experimenten reacciones anafilácticas deberá evaluarse la presencia de inhibidores. En tales casos, se recomienda se contacte un centro de hemofilia especializado.
En aquellos pacientes que reciban un preparado de FVW que contenga FVIII, niveles excesivos de FVIII:C de forma sostenida podrían incrementar el riesgo de trastornos trombóticos.
En el uso de un preparado de FVW que contenga FVIII en pacientes con la EVW, existe el riesgo de que se produzcan trastornos trombóticos, particularmente en pacientes con riesgos clínicos o de laboratorio conocidos. Así pues, los pacientes con riesgo deben ser monitorizados ante la aparición de signos iniciales de trombosis. Debe iniciarse profilaxis contra tromboembolismo vascular, según recomendaciones vigentes.
Para la seguridad con respecto a agentes transmisibles, ver sección 4.4.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaRAM.es
4.9 Sobredosis
No se han notificado casos de sobredosificación con factor VIII de coagulación humano.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo Farmaterapéutico: Antihemorrágico: factores de coagulación sanguínea: factor von Willebrand y factor VIII de coagulación sanguínea en combinación. Código ATC: B02BD06.
En Fanhdi, el FVIII se presenta en forma de complejo con el FVW.
El complejo FVIII/FVW esta compuesto por dos moléculas (FVIII y FVW) con funciones fisiológicas diferentes.
Hemofilia A
Cuando se infunde en un paciente hemofílico, el FVIII se liga al FVW en la circulación del paciente.
El FVIII activado actúa como cofactor del factor IX activado, acelerando la conversión del factor X a factor X activado. El factor X activado transforma la protrombina en trombina, la cual a su vez convierte el fibrinógeno en fibrina, de manera que puede formarse el coágulo.
La hemofilia A es un trastorno hereditario de la coagulación sanguínea ligado al sexo, debido a la disminución del nivel de FVIII, lo que produce hemorragias graves en las articulaciones, músculos u órganos internos, bien de forma espontánea o como consecuencia de un accidente o trauma quirúrgico. Mediante una terapia de sustitución aumenta el nivel plasmático de FVIII y por tanto se corrige temporalmente la deficiencia de actividad de dicho factor y se controlan y previenen las hemorragias.
Enfermedad de von Willebrand (EVW)
Fanhdi se comporta de la misma forma que el FVW endógeno.
La administración de FVW permite corregir las anomalías hemostáticas mostradas por pacientes con deficiencia de FVW (EVW) a dos niveles:
- el FVW restablece la adhesión plaquetaria al subendotelio vascular en el lugar del daño vascular (ya que se une al subendotelio vascular y a la membrana plaquetaria), facilitando la hemostasia primaria tal y como se muestra en la reducción del tiempo de hemorragia. Este efecto se produce inmediatamente y se sabe que depende en gran medida del nivel de polimerización de la proteína.
- el FVW produce una corrección retardada de la deficiencia asociada al FVIII. Administrado de forma intravenosa, el FVW se une al FVIII endógeno (producido normalmente por el paciente), y mediante la estabilización de dicho factor, evita su rápida degradación. Debido a esto, la administración de FVW puro (producto FVW con un bajo nivel de FVIII) restablece el nivel de FVIIEC a la normalidad como un efecto secundario tras la primera administración.
La administración de un preparado de FVW que contenga FVIII restablece el nivel normal de FVIIEC de forma inmediata después de la primera administración.
No hay experiencia procedente de ensayos clínicos en niños menores de 6 años para las indicaciones autorizadas.
Experiencia en inmunotolerancia
Se han recogido datos en Inducción a la Inmunotolerancia (IIT) de pacientes pediátricos y adultos con hemofilia A que presentaban inhibidores contra el FVIII. Entre los 57 pacientes procedentes de un estudio retrospectivo y los 14 procedentes de estudios prospectivos se incluye un amplio espectro de pacientes con tratamiento primario y de rescate, con factores prognosticos variados para la obtención de la inmunotolerancia. Los datos indican que Fanhdi se usa para inducir inmunotolerancia. En aquellos pacientes en los que se logró tolerancia, los sangrados se pudieron prevenir o controlar mediante tratamiento profiláctico o a demanda usando concentrados de FVIII.
5.2 Propiedades farmacocinéticas
La actividad plasmática de FVIII disminuye según una degradación exponencial bifásica.
La semivida del FVIII humano obtenida en el ensayo clínico realizado con Fanhdi es de 14,18 ± 2,55 horas, con una recuperación in vivo de 105,5 ± 18,5% equivalente aproximadamente a 2,1 ± 0,4 UI/dl por UI/kg administrada (determinaciones realizadas por el método cromogénico). A continuación se detallan los valores de los siguientes parámetros: MRT 20,6 ± 4,8 h, AUC 19,3 ± 3,7 UI h/ml y aclaramiento 2,6 ± 0,5 ml/h/Kg.
La semivida del FVIII y FVW humanos obtenida en el ensayo clínico realizado con Fanhdi en pacientes con la EVW, es de 14,4 ± 10,5 horas para el FVW y de 29,5 ± 16 horas para el FVIII, con una recuperación in vivo de 1,9 ± 0,6 UI/dl por UI/Kg administrada, para el FVW y 2,5 ± 0,5 UI/dl por UI/Kg administrada para el FVIII. Las determinaciones del FVW se han realizado como medida del Cofactor de Ristocetina y las de FVIII como FVIII:C, utilizando el método cromogénico. A continuación se detallan los valores de los siguientes parámetros: AUC para FVW:RCo 15,29 ± 10,03; AUC para FVIII:C 36,60 ± 32,73 UI h/ml; aclaramiento para FVW:RCo 5,6 ± 3,3 y para FVIII:C 1,8 ± 1,0 ml/h/kg.
Los máximos niveles de FVW en plasma aparecen normalmente antes de 30 minutos después de la inyección.
ÍTTI
5.3 Datos preclínicos sobre seguridad
El FVIII de coagulación y el FVW humano (principios activos de Fanhdi) son componentes normales del plasma humano y actúan de la misma forma que las correspondientes proteínas endógenas. Las pruebas de toxicidad a dosis única no son relevantes ya que altas dosis del producto dan lugar a sobrecarga.
Las pruebas de toxicidad a dosis repetidas en animales son impracticables debido a las interferencias con los anticuerpos desarrollados contra las proteínas heterólogas.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
- Histidina
- Albúmina (humana)
- Arginina
- Agua para inyectables (disolvente)
6.2 Incompatibilidades
Fanhdi no debe mezclarse con otros medicamentos.
Únicamente debe utilizarse el equipo para inyección que se suministra para evitar un posible error en el tratamiento como consecuencia de la adsorción del complejo FVIII/FVW a la superficie interna de cualquier otro equipo para inyección.
6.3 Periodo de validez
El período de validez de Fanhdi es de 3 años conservado a temperatura no superior a 30 °C.
Tras la reconstitución el producto es estable química y físicamente durante 12 horas a 25 °C. Desde un punto de vista microbiológico, el producto debe utilizarse inmediatamente. Si no se utiliza inmediatamente, el tiempo y las condiciones de conservación antes de su uso son responsabilidad del usuario y, normalmente no serán más de 24 horas a 2 °C - 8 °C a menos que la reconstitución se haya realizado en condiciones asépticas controladas y validadas.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30 °C. No congelar.
Para las condiciones de conservación del medicamento reconstituido, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Fanhdi se presenta en viales de vidrio tipo I/II que contienen 250, 500, 1000 ó 1500 UI de FVIII (liofilizado) y jeringas de vidrio tipo I con 10 ml para las presentaciones de 250, 500 y 1000 UI ó 15 ml para la presentación de 1500 UI de agua para inyectables (disolvente).
Cada vial de Fanhdi va acompañado de los accesorios necesarios para su reconstitución y administración (microfiltro, 2 toallitas con alcohol, adaptador de vial y equipo para inyección).
Puede que solamente estén comercializados algunos tamaños de envases.
ÍTTI
Contenido de la caja: 1 vial de liofilizado, 1 jeringa precargada de disolvente y accesorios.
6.6 Precauciones especiales de eliminación y otras manipulaciones
No debe utilizarse después de la fecha de caducidad indicada en la etiqueta.
En ningún caso se aprovechará la fracción que no se haya utilizado, ni guardándola en nevera.
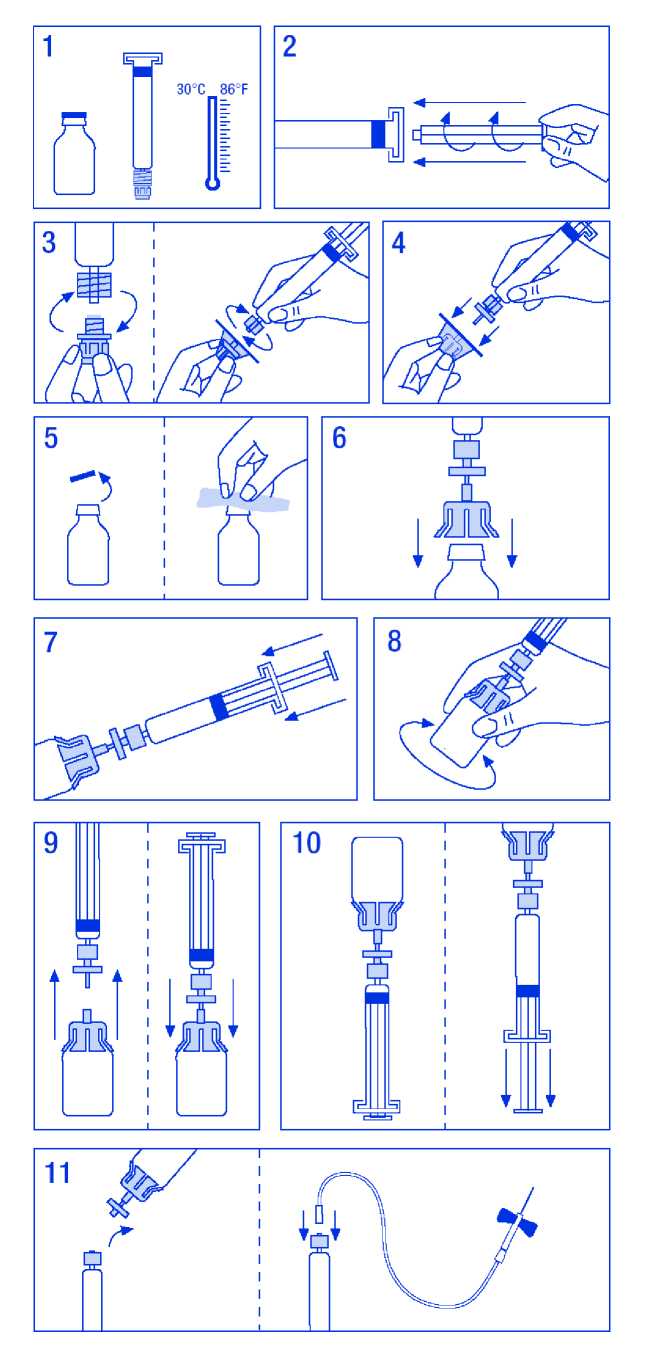
Preparación de la solución:
1. Atemperar el vial y la jeringa del disolvente sin sobrepasar los 30 °C.
2. Acoplar el émbolo a la j eringa del disolvente.
3. Desprecintar el filtro. Separar el tapón del cono de la jeringa del disolvente y acoplarla al filtro.
4. Desprecintar el adaptador de vial y acoplarlo al conjunto filtro-jeringa.
5. Desprecintar el vial, desinfectando el tapón con una de las toallitas con alcohol.
6. Introducir la espina del adaptador en el vial.
7. Trasvasar todo el disolvente de la jeringa al vial.
8. Girar suavemente el vial procurando no producir espuma hasta la total disolución. No agitar. Como todos los productos de administración parenteral, no utilizar si la disolución es incompleta o presenta partículas.
9. Separar el conjunto filtro-jeringa del resto para facilitar la aspiración posterior de la solución y acoplar inmediatamente de nuevo el conjunto filtro-jeringa al vial.
10. Invertir el vial y aspirar el contenido en la jeringa.
11. Preparar la zona de inyección del paciente, separar la jeringa del resto e inyectar el producto utilizando la aguja mariposa suministrada a una velocidad de 3 ml/min por vía intravenosa. La velocidad de administración no debe sobrepasar los 10 ml/min para evitar reacciones vasomotoras.
No deben reutilizarse los equipos de administración.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Generalmente la solución es clara o ligeramente opalescente. No utilizar soluciones que presenten turbidez o sedimentos.
Una vez reconstituida, la solución debe desecharse si se observan partículas en su interior o algún tipo de decoloración.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Instituto Grifols, S.A.
C/ Can Guasch, 2 - Parets del Valles 08150 Barcelona - ESPAÑA
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
Fanhdi 25 UI FVIII/30 UI FVW por ml No. Reg. 60601 Fanhdi 50 UI FVIII/60 UI FVW por ml No. Reg. 60600
Fanhdi 100 UI FVIII/120 UI FVW por ml (1000 UI FVIII/1200 UI FVW por 10 ml, 1500 UI FVIII/1800 UI FVW por 15 ml) No. Reg. 60602
9 de 1 1 MINISTWIODE
SANIDAD, POLITICA SOCIAL E IGUALDAD Agencia españoiade medie ámenlos y prooucios sabíanos
ÍTTI
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Autorización 23/12/1994. Última renovación 23/12/2009
10. FECHA DE LA REVISIÓN DEL TEXTO
Febrero 2015.
La información detallada y actualizada de este medicamento está disponible en la página Web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) http://www.aemps.es/
11 de 11