Erivedge 150Mg Capsulas Duras
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
▼ Este medicamento está sujeto a un seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas
1. NOMBRE DEL MEDICAMENTO
Erivedge 150 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula dura contiene 150 mg de vismodegib.
Excipiente con efecto conocido:
Cada cápsula dura contiene 71,5 mg de lactosa monohidrato por cápsula. Para consultar la lista completa de excipientes,ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura (cápsula).
Cuerpo de la cápsula de color rosa, opaco, con la inscripción “150 mg” y tapa gris, opaca, con la inscripción “VISMO” en tinta negra. El tamaño de la cápsula es “Tamaño 1” (dimensiones 19,0 x 6,6 mm).
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Erivedge está indicado para el tratamiento de pacientes adultos con:
• carcinoma de células basales metastásico sintomático
• carcinoma de células basales localmente avanzado y no candidatos para cirugía o radioterapia (ver sección 5.1)
4.2 Posología y forma de administración
Erivedge solamente debe prescribirse por un médico especialista con experiencia en el tratamiento de la indicación aprobada.
Posología
La dosis recomendada es una cápsula de 150 mg una vez al día.
Dosis olvidadas
Si se olvida una dosis, el paciente no debe tomar la dosis olvidada, sino que debe reanudar con la siguiente dosis programada.
Duración del tratamiento
En ensayos clínicos, el tratamiento con Erivedge se continuó hasta progresión de la enfermedad o hasta toxicidad inaceptable. Se permitieron interrupciones en el tratamiento de hasta 4 semanas basándose en la tolerancia individual.
Debe evaluarse regularmente el beneficio de continuar el tratamiento, la duración óptima de tratamiento varía para cada paciente.
Poblaciones especiales
Pacientes de edad avanzada
No es necesario un ajuste de la dosis en pacientes >65 años de edad (ver sección 5.2). De un total de 138 pacientes en 4 ensayos clínicos de Erivedge en carcinoma de células basales avanzado, aproximadamente el 40% de los pacientes eran >65 años y no se observaron diferencias generales en la seguridad y eficacia entre estos pacientes y los pacientes más jóvenes.
Insuficiencia renal
No se necesita ajuste de dosis en la insuficiencia renal leve y moderada, ya que no se espera que afecte a la eliminación de vismodegib. Los datos disponibles en pacientes con insuficiencia renal grave son muy limitados. Se debe monitorizar cuidadosamente a los pacientes con insuficiencia renal grave en cuanto a reacciones adversas.
Insuficiencia hepática
No es necesario un ajuste de dosis en pacientes con insuficiencia hepática leve, moderada o grave (ver sección 5.2). La definición del grado de insuficiencia hepática se basa en el criterio del Dysfunction Working Group del National Cancer Institute Organ (NCI-ODWG):
• leve: bilirrubina total (BT) < límite superior de la normalidad (ULN), aspartato aminotransferasa (AST) >ULN o ULN<BT<1,5xULN, cualquier AST
• moderada: 1,5 x ULN < BT< 3 x ULN, cualquier AST
• grave: 3 x ULN < BT < 10 x ULN, cualquier AST (ver sección 5.2)
Población pediátrica
No se ha establecido la seguridad y eficacia de Erivedge en niños y adolescentes menores de 18 años. No se dispone de datos.
Por razones de seguridad (ver secciones 4.4 y 5.3), este medicamento no debe utilizarse en niños y adolescentes menores de 18 años.
Forma de administración
Erivedge se administra por vía oral. Las cápsulas deben tragarse enteras con agua, con o sin alimentos (ver sección 5.2). Las cápsulas no deben abrirse, para evitar la exposición involuntaria a los pacientes y profesionales sanitarios.
4.3 Contraindicaciones
• Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1
• Mujeres que están embarazadas o en periodo de lactancia (ver secciones 4.4 y 4.6)
• Mujeres en edad fértil que no cumplen el Programa de Prevención de Embarazo de Erivedge (ver secciones 4.4 y 4.6)
• La administración concomitante de la hierba de San Juan (Hypericum perforatum) (ver sección 4.5)
4.4 Advertencias y precauciones especiales de empleo
Muerte embriofetal o graves defectos congénitos
Erivedge puede provocar muerte embriofetal o graves defectos congénitos cuando se administra a una mujer embarazada (ver sección 4.6). Se ha demostrado en múltiples especies animales que los inhibidores de la ruta Hedgehog (ver sección 5.1), como vismodegib, son embriotóxicos y/o teratogénicos y pueden provocar graves malformaciones, incluyendo anomalías craneofaciales, defectos de la línea media y defectos en las extremidades (ver sección 5.3). Erivedge no debe usarse durante el embarazo.
Criterios para la mujer en edad fértil
La mujer en edad fértil se define en el Programa de Prevención de Embarazo de Erivedge como:
• Mujer sexualmente madura que
• ha tenido la menstruación en cualquier momento durante los últimos 12 meses consecutivos,
• no haya sido sometida a histerectomía o a ooforectomía bilateral, o que no tenga confirmación médica de insuficiencia ovárica prematura permanente,
• no tenga un genotipo XY, síndrome de Turner, ni agenesia de útero,
• presenta amenorrea después del tratamiento del cáncer, incluyendo el tratamiento con Erivedge.
Asesoramiento
Para la mujer en edad fértil:
Erivedge está contraindicado en la mujer en edad fértil que no cumpla con el Programa de Prevención de Embarazo de Erivedge.
Una mujer en edad fértil debe entender que:
• Erivedge expone a un riesgo teratogénico al feto,
• No debe tomar Erivedge si está embarazada o planea quedarse embarazada,
• Debe haber tenido un test de embarazo negativo, realizado por un profesional sanitario dentro de los 7 días anteriores al comienzo del tratamiento con Erivedge,
• Debe tener un test de embarazo negativo todos los meses durante el tratamiento, incluso si ha estado amenorreica,
• No debe quedarse embarazada mientras toma Erivedge ni durante los 24 meses posteriores a la administración de la última dosis.
• Debe ser capaz de cumplir medidas anticonceptivas eficaces,
• Mientras esté tomando Erivedge, debe utilizar 2 métodos anticonceptivos recomendados (ver la sección “Métodos anticonceptivos” más abajo y la sección 4.6), a menos que se comprometa a no tener relaciones sexuales durante el tratamiento (abstinencia),
• Debe informar a su médico si durante el tratamiento y durante los 24 meses posteriores a la administración de la última dosis ocurre cualquier situación de las siguientes:
• Si se queda embarazada o piensa que por cualquier razón pudiese estar embarazada
• Si tiene alguna falta en su período menstrual
• Si deja de usar métodos anticonceptivos a menos que se comprometa a no tener relaciones sexuales (abstinencia),
• Si necesita cambiar el método anticonceptivo durante el tratamiento,
• No se permite la lactancia durante el tratamiento con Erivedge ni durante los 24 meses posteriores a la administración de la última dosis.
Para hombres
Vismodegib está presente en el semen. Para evitar una exposición potencial al feto durante el embarazo, los pacientes masculinos deben entender que:
• Erivedge expone a un riesgo teratogénico al feto si se mantienen relaciones sexuales sin protección con una mujer embarazada,
• Debe utilizar siempre anticonceptivos recomendados (ver la sección “Métodos anticonceptivos” más abajo y la sección 4.6),
• Debe consultar a su médico si su pareja se queda embarazada mientras esté tomando Erivedge o durante los 2 meses posteriores a la administración de la última dosis.
Para los profesionales sanitarios
Los profesionales sanitarios deben asegurarse que los pacientes entienden y reconocen todas las condiciones del Programa de Prevención de Embarazo de Erivedge.
Métodos anticonceptivos
Mujeres en edad fértil
Las pacientes deben utilizar dos métodos anticonceptivos recomendados, incluyendo uno altamente eficaz y un método de barrera durante el tratamiento con Erivedge y durante los 24 meses posteriores a la administración de la última dosis (ver sección 4.6).
Hombres
Los pacientes masculinos siempre tienen que utilizar preservativo (preferiblemente con espermicida), incluso después de una vasectomía, cuando mantengan relaciones sexuales con una mujer mientras toman Erivedge y durante los 2 meses posteriores a la administración de la última dosis (ver sección 4.6).
Test de embarazo
En mujeres en edad fértil, se debe realizar un test de embarazo realizado por un profesional sanitario y supervisado médicamente, dentro de los 7 días previos al inicio del tratamiento, y una vez al mes durante el tratamiento. Los test de embarazo tienen que tener una sensibilidad mínima de 25 mUI/ml, según disponibilidad local. Las pacientes que presenten amenorrea durante el tratamiento con Erivedge deben continuar realizándose un test de embarazo mensualmente mientras estén en tratamiento.
Restricciones en la prescripción y dispensación para mujeres en edad fértil
La prescripción inicial y la dispensación de Erivedge debe realizarse dentro de los 7 días siguientes a un test de embarazo negativo. Las prescripciones de Erivedge deben limitarse a 28 días de tratamiento, la continuación del tratamiento requiere una nueva prescripción.
Material educacional
Para ayudar a los profesionales sanitarios y a los pacientes a evitar la exposición del embrión y del feto a Erivedge, el titular de la autorización de comercialización proveerá material educacional (Programa de Prevención de Embarazo de Erivedge) para reforzar el manejo de los riesgos potenciales asociados al uso de Erivedge.
Efectos en el desarrollo post-natal
En especies animales, se ha demostrado que vismodegib causa cambios graves e irreversibles en el crecimiento de los dientes (degeneración/necrosis de odontoblastos, formación de quistes llenos de líquido en la pulpa dental, osificación del conducto radicular y hemorragia) y cierre de la placa de crecimiento epifisaria. Estos hallazgos indican un riesgo potencial de baja estatura y deformidades dentales en los lactantes y los niños (ver sección 5.3).
Donación de sangre
Los pacientes no deben donar sangre mientras estén tomando Erivedge ni durante los 24 meses posteriores a la administración de la última dosis.
Donación de semen
Los pacientes varones no deben donar semen mientras estén tomando Erivedge ni durante los 2 meses posteriores a la administración de la última dosis.
Interacciones
Debe evitarse el tratamiento concomitante con inductores potentes de CYP (p.e. rifampicina, carbamazepina, o fenitoína), ya que no se puede excluir el riesgo de una disminución de las concentraciones plasmáticas y una disminución en la eficacia de vismodegib (ver también sección 4.5).
Carcinoma de células escamosas cutáneo (CCEcu)
Los pacientes con carcinoma de células basales avanzado (CCBa) presentan mayor riesgo de desarrollar CCEcu. Se han notificado casos de CCEcu en pacientes con CCB avanzado tratados con Erivedge. No se ha determinado si el CCEcu está relacionado con el tratamiento con Erivedge. Por lo tanto, se debe monitorizar a todos los pacientes de manera rutinaria mientras estén tomando Erivedge, y el CCEcu debe tratarse de acuerdo al estándar de tratamiento.
Precauciones adicionales
Se debe indicar a los pacientes que nunca deben dar este medicamento a otra persona. El paciente debe deshacerse inmediatamente de las cápsulas no utilizadas al final del tratamiento siguiendo la normativa local (si aplica p.ej. devolver las cápsulas a su farmacéutico o médico).
Excipientes
Las cápsulas de Erivedge contienen lactosa monohidrato. Los pacientes con un problema hereditario raro de intolerancia a la galactosa, la deficiencia de lapp lactasa, hipolactasia primaria o malabsorción de glucosa o galactosa no deben tomar este medicamento.
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis, es decir, “esencialmente exento de sodio”.
4.5 Interacción con otros medicamentos y otras formas de interacción
Efectos de medicamentos administrados de forma concomitante con vismodegib
No se esperan interacciones farmacocinéticas (PK) clínicamente significativas entre vismodegib y agentes que aumenten el pH. Los resultados de un estudio clínico demostraron una disminución del 33% de las concentraciones de fármaco libre de vismodegib tras 7 días de tratamiento concomitante con 20 mg de rabeprazol (un inhibidor de la bomba de protones) administrado 2 horas antes de cada administración de vismodegib. No se espera que esta interacción sea clínicamente significativa.
No se esperan interacciones farmacocinéticas clínicamente significativas entre vismodegib e inhibidores del CYP450. Los resultados de un estudio clínico demostraron un aumento del 57% de las concentraciones de fármaco libre de vismodegib en el día 7 tras tratamiento diario con 400 mg de fluconazol (un inhibidor moderado del CYP2C9), sin embargo esta interacción no se espera que sea clínicamente significativo. Itraconazol (un fuerte inhibidor del CYP3A4) a 200 mg diarios no influyó en el AUC 0-24h tras 7 días de tratamiento concomitante en voluntarios sanos.
No se esperan interacciones farmacocinéticas clínicamente significativas entre vismodegib e inhibidores de la P-gp. Los resultados de un estudio clínico en voluntarios sanos demostraron que no hubo interacción farmacocinética clínicamente significativa entre vismodegib e itraconazol (un fuerte inhibidor de la glicoproteína P).
Cuando vismodegib se administra junto con inductores de CYP (rifampicina, carbamazepina, fenitoína, hierba de San Juan), la exposición a vismodegib puede verse reducida (ver secciones 4.3 y 4.4).
Efectos de vismodegib en medicamentos administrados de forma concomitante
Anticonceptivos esteroideos
Los resultados de un estudio de interacción medicamento-medicamento realizado en pacientes con cáncer demostraron que la exposición sistémica a etinil estradiol y noretindrona no se altera cuando se administra de forma concomitante con vismodegib.
Sin embargo, el estudio de interacción fue de solo 7 días de duración y no se puede excluir que vismodegib en tratamientos más largos es un inductor de enzimas que metabolizan esteroides anticonceptivos. La inducción podría conducir a una disminución en la exposición sistémica de los anticonceptivos esteroideos y, por lo tanto, reducir la eficacia anticonceptiva.
Efectos en enzimas específicas y transportadores
Estudios in vitro indican que vismodegib tiene el potencial de actuar como un inhibidor de la proteína de resistencia de cáncer de mama (PRCM). No se dispone de datos de interacción in vivo. No se puede excluir que vismodegib pueda dar lugar a una mayor exposición de los medicamentos transportados por esta proteína, como rosuvastatina, topotecan y sulfasalazina. La administración concomitante debe realizarse con precaución y puede ser necesario un ajuste de dosis.
No se esperan interacciones farmacocinéticas clínicamente significativas entre vismodegib y sustratos del CYP450. In vitro, CYP2C8 fue el isomorfo más sensible para la inhibición del vismodegib. Sin embargo, resultados de un estudio de interacción medicamento-medicamento realizado en pacientes con cáncer demostró que la exposición sistémica de rosiglitazona (un sustrato de CYP2C8) no se alteró cuando se administró de forma concomitante con vismodegib. Por lo tanto, se puede excluir la inhibición in vivo de las enzimas CYP por vismodegib.
In vitro, vismodegib es un inhibidor del OATP1B1. No se puede excluir que vismodegib podria aumentar la exposición a sustratos del OATP1B1, ej. bosentan, ezetimibe, glibenclamida, repaglinida, valsartan y estatinas. En particular, se debe realizar con precaución la administración de vismodegib en combinación con cualquier estatina.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil
Debido al riesgo de muerte embriofetal o graves defectos congénitos provocados por vismodegib, las mujeres que tomen Erivedge no deben estar embarazadas, ni quedarse embarazadas durante el tratamiento, ni durante los 24 meses posteriores a la administración de la última dosis (ver secciones 4.3 y 4.4).
Erivedge está contraindicado en mujeres en edad fértil que no cumplan el Programa de Prevención de Embarazo de Erivedge.
En caso de embarazo o pérdida de período menstrual
Si la paciente se queda embarazada, tiene una falta en su período menstrual o por cualquier razón sospecha que puede estar embarazada debe comunicárselo a su médico inmediatamente.
Se asume que una falta persistente del período menstrual durante el tratamiento con Erivedge indica embarazo hasta evaluación y confirmación médica.
Métodos anticonceptivos en hombres y mujeres
Mujeres en edad fértil
Una mujer en edad fértil debe ser capaz de cumplir medidas anticonceptivas eficaces. Debe usar dos métodos anticonceptivos recomendados, incluyendo un método altamente eficaz y un método de barrera durante el tratamiento con Erivedge y durante los 24 meses posteriores a la administración de la última dosis. Una mujer en edad fértil, cuya menstruación es irregular o se ha interrumpido, debe seguir todas las recomendaciones en cuanto a métodos anticonceptivos eficaces.
Hombres
Vismodegib está presente en el semen. Para evitar una potencial exposición al feto durante el embarazo, los pacientes varones siempre tienen que utilizar preservativo (preferiblemente con espermicida), incluso después de una vasectomía, cuando mantengan relaciones sexuales con una mujer, mientras esté tomando Erivedge y durante los 2 meses posteriores a la administración de la última dosis.
Métodos recomendados altamente eficaces:
• Inyección hormonal depot
• Esterilización tubárica,
• Vasectomía
• Dispositivo intrauterino (DIU).
Métodos de barrera recomendados:
• Cualquier preservativo masculino (preferiblemente con espermicida)
• Diafragma (preferiblemente con espermicida).
Embarazo
Erivedge puede provocar muerte embriofetal o graves defectos congénitos cuando se administra a mujeres embarazadas (ver sección 4.4). Se ha demostrado en múltiples especies animales que los inhibidores de la ruta Hedgehog, como vismodegib (ver sección 5.1), son embriotóxicos y/o teratogénicos y pueden provocar graves malformaciones, incluyendo anomalías craneofaciales, defectos de la línea media y defectos en las extremidades (ver sección 5.3). En caso de que una mujer tratada con Erivedge se quede embarazada, el tratamiento debe interrumpirse inmediatamente.
Lactancia
Se desconoce el grado en el cual vismodegib se excreta en la leche materna. Debido a su potencial de provocar defectos graves en el desarrollo, las mujeres no deben dar el pecho mientras toman Erivedge ni durante los 24 meses posteriores a la administración de la última dosis (ver secciones 4.3 y 5.3)
Fertilidad
La fertilidad femenina humana puede verse comprometida por el tratamiento con Erivedge (ver sección 5.3). Se desconoce si la disfunción de la fertilidad es reversible. Además, en ensayos clínicos se ha observado amenorrea en mujeres en edad fértil (ver sección 4.8). Deben valorarse las estrategias para mantener la capacidad reproductiva en mujeres en edad fértil antes de iniciar el tratamiento con Erivedge.
No se espera que haya disfunción de la fertilidad en varones (ver sección 5.3)
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Erivedge sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas Resumen del perfil de seguridad
Las reacciones adversas a medicamentos más frecuentes (RAM) que ocurren en > 30% de los pacientes, fueron espasmos musculares (74,6 %), alopecia (65,9 %), disgeusia (58,7 %), disminución en el peso (50,0 %), fatiga (47,1 %), náuseas (34,8 %) y diarrea (33,3%).
Lista tabulada de reacciones adversas
Las reacciones adversas (RAM) se presentan a continuación en la tabla 1 utilizando el sistema de clasificación de órganos (SCO) y la frecuencia absoluta.
Las frecuencias se definen como:
Muy frecuentes (> 1/10)
Frecuentes (> 1/100 y < 1/10)
Poco frecuentes (> 1/1.000 y < 1/100)
Raras ( > 1/10.000 y <1/1.000)
Muy raras ( < 1/10.000)
Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Se ha evaluado la seguridad de Erivedge en ensayos clínicos con 138 pacientes tratados de carcinoma de células basales avanzado (CCBa), incluyendo tanto CCB metastasico (CCBm) como CCB localmente avanzado (CCBla). En cuatro ensayos clínicos abiertos fase 1 y 2 los pacientes fueron tratados con al menos una dosis de Erivedge en monoterapia a dosis > 150 mg. En los ensayos clínicos las dosis >150 mg no dieron como resultado un aumento en las concentraciones plasmáticas, los pacientes con dosis >150 mg se incluyeron en los análisis. En general, el perfil de seguridad observado fue consistente en CCBm y CCBla como se describe a continuación.
Tabla 1
RAMs observadas en pacientes tratados con Erivedge en ensayos clínicos
|
MedDRA SCO |
Muy frecuentes |
Frecuentes |
|
Trastornos del metabolismo y de la nutrición |
Disminución del apetito |
Deshidratación Hiponatremia |
|
Trastornos del sistema nervioso |
Disgeusia Ageusia |
Hipogeusia |
|
Trastornos gastrointestinales |
Náusea Diarrea Estreñimiento Vómitos Dispepsia |
Dolor abdominal superior Dolor abdominal |
|
Trastornos de la piel y del tejido subcutáneo |
Alopecia Prurito Erupción |
Madarosis Crecimiento anormal del pelo |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Espasmos musculares Artralgia Dolor en extremidades |
Dolor de espalda Dolor torácico musculoesquelético Mialgia Dolor en flanco Dolor musculoesquelético |
|
Trastornos del aparato reproductor y de la mama |
Amenorrea * | |
|
Trastornos generales y alteraciones en el lugar de administración |
Disminución de peso Fatiga Dolor |
Astenia |
|
Exploraciones complementarias |
Aumento de enzimas hepáticas** | |
|
Todas las notificaciones se basan en las RAMs de todos los grados usando los criterios de terminología común para acontecimientos adversos v3.0 del Instituto Nacional del Cáncer, excepto donde se indique. *De los 138 pacientes con CCB avanzado, 10 eran mujeres en edad fértil. Entre estas mujeres, se observó amenorrea en 3 de ellas (30%). MedDRA = Medical Dictionary for Regulatory Activities (Diccionario Médico para Actividades Regulatorias) **Incluye los términos preferidos: prueba de función hepática anormal, bilirrubina aumentada en sangre, aumento de gamma glutamiltransferasa, aumento de aspartato aminotrasferasa, aumento de fosfatasa alcalina, aumento de las enzimas hepáticas. | ||
Notificación de sospechas de reacciones adversas
Se invita a los sistema nacional
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. profesionales sanitarios a notificar las sospechas de reacciones adversas a través del de notificación incluido en el Apéndice V
4.9 Sobredosis
Erivedge se ha administrado a dosis 3,6 veces mayores que la dosis diaria recomendada de 150 mg. Durante estos ensayos clínicos no se han observado incrementos en plasma de los niveles de vismodegib ni toxicidad.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapeútico: Agentes antineoplásicos, otros agentes antineoplásicos, código ATC: L01XX43.
Mecanismo de acción
Vismodegib es una molécula pequeña, inhibidor de la ruta de Hedgehog, disponible por vía oral. La ruta de señalización de Hedgehog, a través de la proteína transmembrana Smoothened (SMO), conduce a la activación y la localización nuclear de los factores de transcripción de oncogen asociados a glioma (GLI) y la inducción de los genes diana Hedgehog. Muchos de estos genes están involucrados en la proliferación, supervivencia y diferenciación. Vismodegib se une e inhibe la proteína SMO por lo que bloquea la señal de transducción Hedgehog.
Eficacia clínica y seguridad
El ensayo pivotal ERIVANCE BCC (SHH4476g) fue un estudio de dos cohortes, internacional, multicéntrico, con un solo grupo de tratamiento. El CCB metastásico se define como CCB que se ha extendido a través de la piel a otras partes del organismo, incluyendo los nódulos linfáticos, pulmones, huesos y/o órganos internos. Los pacientes con CCB presentan lesiones cutáneas que no son adecuadas para cirugía (inoperable, con múltiples recidivas, donde las resecciones curativas no se estimaron como adecuadas o si la cirugía provocaría una deformidad substancial o morbilidad) y para quienes la radioterapia fracasó, estaba contraindicada o era inapropiada. Previo a la inclusión en el ensayo, el diagnóstico de CCB era confirmado por histología. Los pacientes con síndrome de Gorlin que habían tenido al menos una lesión de CCB avanzado y cumplían los criterios de inclusión fueron elegibles para participar en el ensayo. Los pacientes fueron tratados con una dosis diaria por vía oral de Erivedge de 150 mg.
La mediana de edad de la población sobre la que se evalúa la eficacia fue 62 años (el 46% tenía al menos 65 años), el 61% eran hombres y el 100% de raza blanca. Para la cohorte del CCBm, el 97% de los pacientes tuvo un tratamiento previo, incluyendo cirugía (97%), radioterapia (58%) y tratamientos sistémicos (30%). Para la cohorte de CCBla (n=63), el 94% de los pacientes tuvieron un tratamiento previo incluyendo cirugía (89%), radioterapia (27%) y tratamientos sistémicos/tópicos (11%). La mediana de duración del tratamiento fue 12,9 meses (intervalo de 0,7 a 47,8 meses).
La variable principal fue la tasa de respuesta objetiva (ORR) evaluada por un Comité de Revisión Independiente (CRI) como se recoge en la Tabla 2. La respuesta objetiva se define como la respuesta parcial o completa determinada en dos evaluaciones consecutivas separadas al menos 4 semanas. En la cohorte de CCBm, la respuesta tumoral se evaluó de acuerdo con criterios RECIST (Criterios de Evaluación de la Respuesta en Tumores Sólidos), versión 1.0. En la cohorte de CCBla la respuesta tumoral se evaluó basándose en una evaluación visual del tumor externo y ulceración, escáner del tumor (cuando fue apropiado) y una biopsia del tumor. Un paciente se consideró respondedor en la cohorte de CCBla cuando cumplía al menos uno de los siguientes criterios y el paciente no experimentó progresión: (1) reducción > 30 % en el tamaño de la lesión [suma del diámetro más largo (SDL)], partiendo desde el valor basal en las lesiones diana medidas por radiografía; (2) reducción > 30 % en SDL partiendo desde el valor basal en la dimensión visible externamente de las lesiones diana; (3) resolución completa de la ulceración en todas las lesiones diana. Los datos más relevantes se resumen en la Tabla 2.
Tabla 2 SHH4476g Resultados de eficacia de Erivedge (evaluados a los 21 meses por CRI y a los 39 meses por el investigador tras la inclusión del último paciente): pacientes evaluables para eficacia*’1'
|
Evaluados |
por el CRI |
Evaluados por el investigador | ||
|
CCBm |
CCBla** |
CCBm |
CCBla** | |
|
(n = 33) |
(n = 63) |
(n = 33) |
(n = 63) | |
|
Respondedores |
11 (33,3 %) |
30 (47,6 %) |
16 (48,5 %) |
38 (60,3 %) |
|
IC del 95% para respuesta global |
(19,2%, 51,8 %) |
(35,5 %, 60,6%) |
(30,8%, 66,2 %) |
(47,2 %, 71,7 %) |
|
Respuesta Completa |
0 |
14 (22,2 %) |
0 |
20 (31,7%) |
|
Respuesta Parcial |
11 (33,3 %) |
16 (25,4%) |
16 (48,5 %) |
18 (28,6%) |
|
Enfermedad estable |
20 |
22 |
14 |
15 |
|
Progresión de la enfermedad * |
1 |
8 |
2 |
6 |
|
Duración de la |
7,6 |
9,5 |
14,8 |
26,2 |
|
Respuesta mediana (meses) (IC del 95 %) |
(5,5; 9,4) |
(7,4; 21,4) |
(5,6; 17,0) |
(9,0; 37,6) |
|
Supervivencia Libre de Progresión mediana |
9,5 |
9,5 |
9,3 |
12,9 |
|
(meses) (IC del 95 %) |
(7,4;11,1) |
(7,4; 14,8) |
(7,4; 16,6) |
(10,2; 28,0) |
|
SG mediana, |
33,4 |
NE | ||
|
(meses) (IC del 95 %) |
(18,1; NE) |
(NE; NE) | ||
|
Tasa de supervivencia |
78,7% |
93,2% | ||
|
a 1 año (IC del 95 %) |
(64,7; 92,7) |
(86,8; 99,6) | ||
NE = no estimable
* Población de pacientes evaluables para eficacia se define como todos los pacientes reclutados que recibieron cualquier cantidad de Erivedge y para quienes la interpretación del archivo de tejidos o biopsia basal del patólogo independiente fue consistente con CCB.
f Datos perdidos o no evaluables incluyeron 1 paciente con CCBm y 4 pacientes con CCBla.
{ La progresión en la cohorte de CCBla se define como el cumplimiento de cualquiera de los siguientes criterios: (1) aumento > 20 % en la suma de la dimensión más larga (SDL) desde el punto más bajo en las lesiones diana (ya sea por radiografía o las dimensiones visibles externamente), (2) Nuevas úlceras de las lesiones diana persistentes sin evidencia de curación durante al menos 2 semanas, (3) Nuevas lesiones determinadas por radiografía o exploración física, (4) Progresión de lesiones no diana por criterios RECIST.
** El 54% de los pacientes con CCBla no tenían evidencia histopatológica de CCB a las 24 semanas.
Como se muestra en el gráfico de cascada en las figuras 1 y 2, las cuales muestran la máxima reducción en el tamaño de la(s) lesión(-es) diana para cada paciente, la mayoría de los pacientes en ambas cohortes experimentaron una disminución del tumor, según fue evaluado por el Comité de Revisión Independiente.
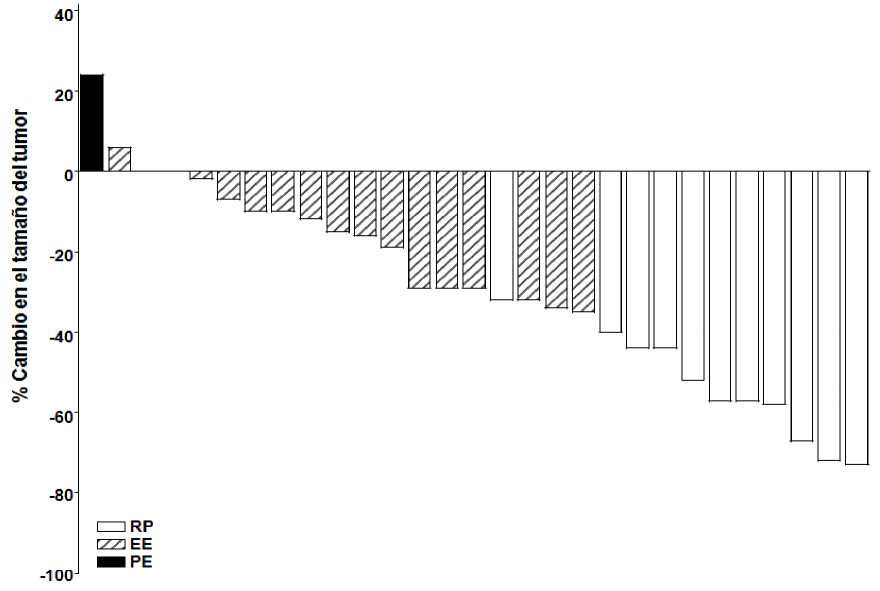
Nota: El tamaño del tumor se basa en la suma de las dimensiones más largas de las lesiones diana. PE= Progresión de la Enfermedad, EE= Enfermedad Estable, RP= Respuesta Parcial. 3 pacientes tuvieron un cambio porcentual mejor en tumor de tamaño 0; éstos están representados por unas barras mínimas positivas en la figura. Cuatro pacientes fueron excluidos de la figura: 3 pacientes con enfermedad estable fueron evaluados sólo por lesiones no diana y 1 paciente no fue evaluable.
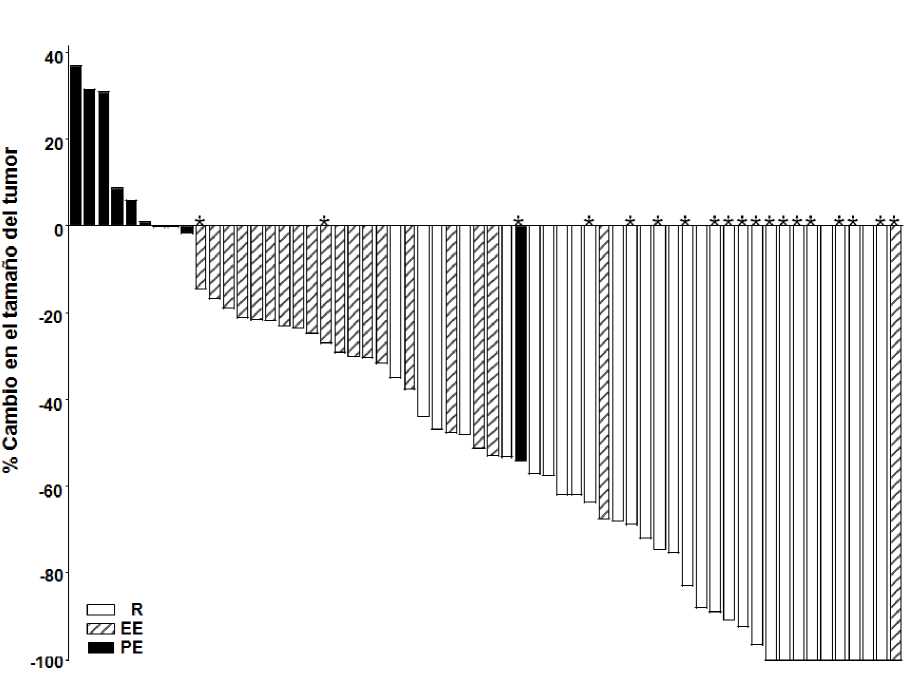
Nota: El tamaño del tumor se basa en la suma de las dimensiones más largas de las lesiones diana.
PE = Progresión de la Enfermedad, EE = Enfermedad Estable, R = Respuesta, *=completa resolución de la(s) úlcera(s). La evaluación de la respuesta se basó en la variable compuesta definida anteriormente. Cuatro pacientes no tuvieron mediciones de lesiones y no se incluyeron en el gráfico.
Tiempo hasta la máxima reducción del tumor
Entre los pacientes que lograron una reducción del tumor, la mediana del tiempo hasta la máxima reducción del tumor fue de 5,6 y 5,5 meses para pacientes con CCBla y para pacientes con CCBm, respectivamente, basándose en la evaluación del Comité de Revisión Independiente (CRI). De acuerdo con la evaluación del investigador, la mediana del tiempo hasta la máxima reducción del tumor fue de 6,7 y 5,5 meses para pacientes con CCBla y para pacientes con CCBm, respectivamente.
Electrofisiología cardiaca
En un ensayo en 60 sujetos sanos donde se estudió el intervalo QTc, no hubo ningún efecto en el intervalo QTc a las dosis terapeúticas de Erivedge.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Erivedge en los diferentes grupos de la población pediátrica con cáncer de células basales (ver sección 4.2 para información sobre el uso en población pediátrica).
Este medicamento se ha autorizado con una «aprobación condicional». Esta modalidad de aprobación significa que se espera obtener más información sobre este medicamento. La Agencia Europea de Medicamentos revisará la información nueva de este medicamento al menos una vez al año y esta Ficha Técnica o Resumen de las Características del Producto (RCP) se actualizará cuando sea necesario.
5.2 Propiedades farmacocinéticas
Absorción
Erivedge es un compuesto altamente permeable con baja solubilidad acuosa (Sistema de Distribución Biofarmacéutica clase 2). La biodisponibilidad absoluta de la media de dosis única (CV %) de Erivedge es 31,8 (14,5)%. La absorción es saturable como se evidencia por la falta de un incremento proporcional a la dosis en la exposición después de una única dosis de 270 mg y 540 mg de Erivedge. Bajo condiciones clínicamente relevantes (estado estacionario), la farmacocinética de vismodegib no se ve afectada por la comida. Por lo tanto, Erivedge se puede administrar con o sin alimentos.
Distribución
El volumen de distribución de vismodegib es bajo, oscila entre 16,4 y 26,6 L. In vitro la unión de vismodegib a las proteínas plasmáticas humanas es alta (97%) a concentraciones clínicamente relevantes. Vismodegib se une a la albúmina sérica humana y a la a-1-glicoproteína ácida. In vitro la unión de la a-1-glicoproteína ácida es saturable a concentraciones clínicamente relevantes. Ex vivo la unión a proteínas plasmáticas en pacientes humanos es > 99 %. Las concentraciones de vismodegib están fuertemente correlacionadas con los niveles de a-1-glicoproteína ácida, mostrando fluctuaciones paralelas con el tiempo de a-1-glicoproteína ácida y vismodegib total y consecuentemente bajos niveles de vismodegib no unido.
Biotransformación
Vismodegib se elimina lentamente por una combinación de metabolismo y excreción del medicamento original. Vismodegib está predominantemente en plasma, con concentraciones representativas mayores del 98 % del total de las concentraciones circulantes (incluidas las asociadas a metabolitos). Las rutas metabólicas de vismodegib en humanos incluyen oxidación, glucuronidación y una escisión poco común del anillo de piridina CYP2C9 parece contribuir en parte al metabolismo de vismodegib in vivo.
Eliminación
Después de una administración oral de una dosis radiomarcada, vismodegib es absorbido y lentamente eliminado mediante una combinación de metabolismo y excreción del medicamento original, la mayoría del cual se recupera en heces (82% de la dosis administrada), con un 4,4% de la dosis administrada recuperada en orina. Vismodegib y los productos metabólicos asociados se eliminan principalmente por vía hepática.
Después de una administración continua de una dosis diaria, la farmacocinética de vismodegib comienza a ser no lineal debido a una absorción saturable y a una unión saturable a proteínas.
Después de una dosis única, vismodegib tiene una semivida terminal de aproximadamente 12 días.
La semivida aparente de vismodegib en estado estacionario se estima que sea de 4 días con una administración diaria continua. Hay una acumulación de concentraciones plasmáticas totales de vismodegib de 3 veces tras una administración diaria continua.
Vismodegib inhibe UGT2B7 in vitro y no se excluye que la inhibición pueda ocurrir in vivo en el intestino.
Poblaciones especiales
Pacientes de edad avanzada
Hay limitados datos en pacientes de edad avanzada. En ensayos clínicos con CCBa aproximadamente el 40% de los pacientes eran de edad avanzada (> 65 años). Los análisis farmacocinéticos poblacionales sugieren que la edad no tuvo un impacto clínicamente significativo en la concentración en estado estacionario de vismodegib.
Sexo
En base al análisis farmacocinético poblacional de los datos combinados de 121 varones y 104 mujeres, el sexo no pareció afectar a la farmacocinética de vismodegib.
Raza
Hay limitados datos en pacientes q no caucasianos. Dado que el número de sujetos que no eran caucasianos fue de < 3% del total de la población (6 de raza negra, 219 caucasianos), la raza no se evaluó como una covariable en el análisis farmacocinético poblacional.
Insuficiencia renal
La excreción renal de vismodegib administrado oralmente es baja. Por lo tanto, es improbable que la insuficiencia renal leve y moderada tenga un efecto clínicamente significativo sobre los parámetros farmacocinéticos de vismodegib. Basándose en los análisis farmacocinéticos poblacionales de pacientes con insuficiencia renal leve (BSA-indexada ClCr 50 a 80 ml/min, n=58) y moderada (BSA-indexada ClCr 30 a 50 ml/min, n=16), la insuficiencia renal leve y moderada no tuvo un efecto clínicamente significativo en los parámetros farmacocinéticos de vismodegib (ver sección 4.2). Se dispone de datos muy limitados en pacientes con insuficiencia renal grave.
Insuficiencia hepática
Las rutas principales de eliminación de vismodegib comprenden metabolismo hepático y secreción biliar/intestinal. En un estudio clínico en pacientes con insuficiencia hepática (grado de insuficiencia basado en los niveles de bilirrubina total y AST de los sujetos), tras múltiples dosis de vismodegib se demostró que el perfil farmacocinético de vismodegib en pacientes con insuficiencia hepática leve (criterios NCI-ODW, n=8), moderada (criterios NCI-ODW, n=8), y grave (criterios NCI-ODW, n=3) fue equiparable al de los sujetos con función hepática normal (n=9) (ver sección 4.2).
Población pediátrica
No hay datos farmacocinéticos suficientes en población pediátrica.
5.3 Datos preclínicos sobre seguridad
El perfil de seguridad preclínico de Erivedge se evaluó en ratones, ratas y perros.
Toxicidad a dosis repetidas
En general, la tolerancia de Erivedge en los estudios de toxicidad a dosis repetida en ratas y perros fue limitada por manifestaciones inespecíficas de toxicidad incluyendo disminución en la ganancia de peso corporal y consumo de alimentos. Hallazgos adicionales a exposiciones clínicamente relevantes incluyeron cambios fecales; espasmos del músculo esquelético o temblores; alopecia; hinchazón; hiperqueratosis folicular; inflamación en las almohadillas de las patas e incremento del colesterol LDL y HDL. En algunos perros se observó un descenso en el hematocrito o en el recuento de plaquetas a exposiciones clínicamente relevantes; sin embargo, no hubo evidencia de un efecto principal en la médula ósea de los animales afectados.
Carcinogenicidad
No se han llevado a cabo ensayos no clínicos dedicados a evaluar la carcinogenicidad de vismodegib. Sin embargo, se observó pilomatricoma (un tumor cutáneo benigno) en la semana 26 en los estudios de toxicidad en ratas. No se ha notificado pilomatricoma en los ensayos clínicos con Erivedge, y, por tanto, la relevancia de este hallazgo en humanos es incierta.
Mutagenicidad
No hay evidencia de genotoxicidad en test in vitro (ensayo de mutación inversa en bacterias [Ames] y ensayo de aberración cromosómica en linfocitos humanos) ni en los test in vivo en micronúcleos de la médula ósea de ratas.
Fertilidad
En el estudio de la fertilidad en ratas con vismodegib a las 26 semanas, se observó un aumento significativo de los pesos absolutos de las vesículas seminales y una reducción de los pesos absolutos de la próstata. Además, la relación peso órgano y peso corporal en el momento del sacrificio fue significativamente mayor para el epidídimo, cola del epidídimo, testículos y vesículas seminales. En el mismo estudio no se encontraron hallazgos histopatológicos en órganos reproductivos masculinos ni tampoco efectos sobre variables de fertilidad masculina, incluyendo porcentaje de espermatozoides móviles, a una dosis de100mg/kg/día al final de la administración o en la fase de recuperación (correspondiente a 1,3 veces el AUC0-24 en el estado estacionario a la dosis recomendada en humanos). Además, en los estudios de toxicidad general de vismodegib hasta 26 semanas en ratas y perros sexualmente maduros, no se observaron efectos en órganos reproductivos masculinos. No se determinó la relación de vismodegib con el aumento del número de células germinales deterioradas e hipospermia en el estudio de toxicidad general, en perros sexualmente inmaduros, a una dosis de > 50 mg/kg/día durante 4 semanas.
En el estudio de fertilidad en ratas con vismodegib a las 26 semanas, se observaron los efectos en órganos reproductivos femeninos relacionados con vismodegib inmediatamente después de la interrupción del tratamiento a una dosis de 100 mg/kg/día, incluyendo disminución de las implantaciones, aumento del porcentaje de pérdidas antes de la implantación y disminución del número de ratas con embriones viables. No se observaron hallazgos similares después del período de recuperación de 16 semanas. No se observaron cambios histopatológicos correlativos. La exposición de ratas hembras a una dosis de 100 mg/kg corresponde a 1,2 veces el AUC0-24h en el estado estacionario en la dosis recomendada en humanos. Además, en el estudio de toxicidad general de vismodegib a una dosis de 100 mg/kg/día a las 26 semanas en ratas, se observó una disminución de cuerpos luteos; el efecto no revirtió al final de un período de recuperación de 8 semanas.
Teratogenicidad
En un estudio de desarrollo embriofetal en el cual se administró vismodegib diariamente a ratas embarazadas durante la organogénesis, vismodegib atravesó la placenta y fue gravemente tóxico para el feto. En fetos de hembras se observaron malformaciones, incluyendo anomalías craneofaciales, perineo abierto y ausencia de dedos y/o dedos fusionados a la dosis que correspondía al 20 % de la exposición a estado estacionario típica en pacientes, y a dosis mayores se observó un 100 % de incidencia de embrioletalidad.
Desarrollo post-natal
No se han llevado a cabo estudios dedicados a evaluar el potencial de vismodegib de afectar al desarrollo post-natal. Sin embargo, defectos irreversibles en el crecimiento de los dientes y un cierre prematuro de la placa epifisaria femoral observados en estudios de toxicidad en ratas a exposiciones clínicamente relevantes representan riesgos para el desarrollo post-natal.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Contenido de la cápsula Celulosa microcristalina Lactosa monohidrato Lauril sulfato sódico Povidona (K29/32)
Almidón glicolato de sodio (Tipo A) Talco
Estearato de magnesio
Cubierta de la cápsula Óxido de hierro negro (E172)
Óxido de hierro rojo (E172)
Dióxido de titanio (E171)
Gelatina
Tinta de impresión Goma laca
Óxido de hierro negro (E172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
3 años.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30 °C.
Mantener el frasco perfectamente cerrado para protegerlo de la humedad.
6.5 Naturaleza y contenido del envase
Frascos de HDPE con un cierre a prueba de niños que contienen 28 cápsulas duras. Cada envase contiene un frasco. El material de la tapa del frasco es de polipropileno. El revestimiento de la tapa es un cartón encerado recubierto de pelicula de aluminio.
6.6 Precauciones especiales de eliminación
El paciente debe deshacerse inmediatamente de las cápsulas no utilizadas al final del tratamiento de acuerdo con la normativa local (si procede p.ej. devolver las cápsulas a su farmacéutico o médico).
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW United Kingdom
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/848/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 12 Julio 2013 Fecha de la última renovación: 27 de Mayo de 2015
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
ANEXO II
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTLIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
E. OBLIGACIÓN ESPECÍFICA DE LLEVAR A CABO MEDIDAS POSTAUTORIZACIÓN EN RELACIÓN CON UNA AUTORIZACIÓN DE COMERCIALIZACIÓN CONDICIONAL
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante responsable de la liberación de los lotes
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes Periódicos de Seguridad
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 quater, apartado 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la fecha de presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
• Medidas adicionales de minimización de riesgos
Antes del lanzamiento en cada Estado Miembro, el Titular de Autorización de Comercialización (TAC) acordará con la Autoridad Nacional Competente lo siguiente:
• La parte nacional de la DHPC
• Metodología para recoger información sobre el uso de Erivedge y el cumplimiento con el programa de prevención de embarazo y su efectividad.
• El formato y contenido del material para profesionales sanitarios y pacientes.
El TAC distribuirá la DHPC en el lanzamiento del medicamento, la cual debe contener lo siguiente:
• Un texto base según lo acordado con el Rapporteur
• Requerimientos específicos nacionales acordados con la Autoridad Competente Nacional en relación a:
• Distribución del producto
• Medidas que aseguren que se han llevado a cabo todas las acciones apropiadas antes de que Erivedge se prescriba y se dispense.
El TAC deberá garantizar continuamente que todos los médicos que prescriban Erivedge cuenten con lo siguiente:
Información del producto
Materiales educacionales para profesionales sanitarios Tarjeta recordatorio para profesionales sanitarios Materiales educacionales para pacientes Tarjeta recordatorio para pacientes
Los materiales educacionales de Erivedge para profesionales sanitarios deben contener los siguientes elementos claves:
• Breve resumen sobre Erivedge, incluyendo su indicación autorizada y posología
• La obligación de informar a los pacientes sobre los riesgos teratogénicos asociados a Erivedge y la necesidad de evitar la exposición fetal
• Descripción del Progama de Prevención de Embarazo y la clasificación de los pacientes en base a sexo y edad fértil
• Información sobre los métodos anticonceptivos recomendados tanto para mujeres como para hombres
• Obligaciones de los profesionales sanitarios en relación con la prescripción de Erivedge
• La necesidad de proveer asesoramiento integral y recomendaciones a los pacientes
• Asegurarse que los pacientes están capacitados para cumplir con los requerimientos para un uso seguro de Erivedge
• La necesidad de proveer a los pacientes con materiales educacionales para pacientes y tarjetas recordatorio para pacientes
• Consejos sobre seguridad para mujeres en edad fértil
• La necesidad de usar métodos anticonceptivos adecuados (incluso si la mujer tiene amenorrea) durante el tratamiento y durante los 24 meses posteriores a la administración de la última dosis de Erivedge.
• Test de embarazo negativo dentro de los 7 días anteriores al comienzo del tratamiento, y test de embarazo supervisados por un médico todos los meses durante el tratamiento
• La necesidad de interrumpir el tratamiento con Erivedge inmediatamente ante sospecha de embarazo
• La necesidad del paciente de notificar una sospecha de embarazo inmediatamente al profesional sanitario que le trate
• Consejos sobre seguridad para hombres
• La necesidad de usar preservativos si su pareja sexual es una mujer embarazada o una mujer en edad fértil (incluso si el hombre se ha sometido a una vasectomía) durante el tratamiento y durante los 2 meses posteriores a la administración de la última dosis de Erivedge
• La necesidad del paciente de notificar inmediatamente al profesional sanitario si su pareja se queda embarazada mientras él esté tomando Erivedge o poco después de que haya interrumpido la toma de Erivedge
• No donar semen durante el tratamiento ni durante los 2 meses posteriores a la administración de la última dosis
• Requisitos en caso de embarazo
• Instrucciones de interrumpir el tratamiento con Erivedge ante sospecha de embarazo
• La necesidad de derivar al paciente a un médico especialista
• Detalles de contacto locales para notificar cualquier sospecha de embarazo
• Formulario de notificación de embarazo
• Informar a los pacientes que no deben donar sangre durante el tratamiento con Erivedge, ni durante los 24 meses posteriores a la administración de la última dosis
• Listado para el profesional sanitario para asegurar que los pacientes reciben el asesoramiento adecuado
• La necesidad de asegurar que todos los pacientes rellenan y firman el formulario de verificación de asesoramiento de Erivedge, que está incluido en el material educacional para profesionales sanitarios
• Formulario de notificación de reacciones adversas
• Información sobre la metodología, de acuerdo con la Autoridad nacional Competente, para recoger información sobre el uso de Erivedge y el cumplimiento con el programa de prevención de embarazo y su efectividad
El material educacional para pacientes de Erivedge debe contener los siguientes elementos claves:
• Información para los pacientes sobre los riesgos teratogénicos asociados a Erivedge y la necesidad de evitar la exposición fetal
• Descripción de la tarjeta recordatorio para pacientes
• La necesidad de una anticoncepción adecuada y definición de anticoncepción adecuada
• Acuerdos específicos nacionales o de otro tipo para una prescripción de Erivedge
• No dar Erivedge a otra persona
• Información relativa a la eliminación de la medicación no deseada
• La necesidad de mantener las cápsulas de Erivedge fuera de la vista y del alcance de los niños
• Que el paciente no debe donar sangre durante el tratamiento ni durante los 24 meses posteriores a la administración de la última dosis
• Que la paciente no debe dar el pecho durante el tratamiento ni durante los 24 meses posteriores a la administración de la última dosis
• Que el paciente debe notificar al profesional sanitario cualquier acontecimiento adverso
• Información para las mujeres en edad fértil
• Descripción del Programa de Prevención de Embarazo
• La necesidad de usar métodos anticonceptivos adecuados durante el tratamiento y durante los 24 meses posteriores a la administración de la última dosis
• Test de embarazo negativo dentro de los 7 días anteriores al comienzo del tratamiento, y test de embarazo supervisados por un médico todos los meses durante el tratamiento
• La necesidad de interrumpir el tratamiento con Erivedge inmediatamente ante sospecha de embarazo
• La necesidad del paciente de notificar una sospecha de embarazo inmediatamente al profesional sanitario que le trate
• Información para hombres
• La necesidad de usar preservativos (incluso si el hombre se ha sometido a una vasectomía) si su pareja sexual es una mujer embarazada o una mujer en edad fértil durante el tratamiento y durante los 2 meses posteriores a la administración de la última dosis de Erivedge
• Que si su pareja se queda embarazada, él debe informar al profesional sanitario que le esté tratando inmediatamente
• No donar semen durante el tratamiento ni durante los 2 meses posteriores a la administración de la última dosis
La tarjeta recordatorio para los profesionales sanitarios debe contener los siguientes elementos clave:
• Información para mujeres en edad fértil
• La necesidad de realizar test de embarazo mensuales incluso si la paciente tiene amenorrea
• La necesidad de usar métodos anticonceptivos adecuados durante el tratamiento y durante los 24 meses posteriores a la administración de la última dosis de Erivedge
• No dar el pecho durante el tratamiento ni durante los 24 meses posteriores a la administración de la última dosis de Erivedge
• Información para los hombres
• La necesidad de usar preservativos cuando mantienen relaciones sexuales con una mujer durante el tratamiento y durante los 2 meses posteriores a la administración de la última dosis de Erivedge
• No donar semen durante el tratamiento ni durante los 2 meses posteriores a la administración de la última dosis
• La necesidad de que los pacientes notifiquen inmediatamente a los profesionales sanitarios que les están tratando si se sospecha de un embarazo en una paciente, o en la pareja de un paciente
• El profesional sanitario debe evaluar el estado de embarazo, asesorar a la paciente sobre el riesgo de teratogenicidad y remitir a la paciente a un médico especialista para recibir asesoramiento
• El profesional sanitario debe notificar los embarazos confirmados al TAC
• Recordar a los pacientes que devuelvan las cápsulas que no han tomado al final del tratamiento (la eliminación dependerá de la normativa local)
Recordar a los pacientes que no pueden donar sangre durante el tratamiento ni durante los 24 meses posteriores a la administración de la última dosis
La tarjeta recordatorio para pacientes debe contener los siguientes elementos clave:
• Información para pacientes de los riesgos teratogénicos asociados a Erivedge y la necesidad de evitar la exposición fetal
• No donar sangre durante el tratamiento ni durante los 24 meses posteriores a la administración de la última dosis
• Información para mujeres en edad fértil
• La necesidad de realizarse test de embarazo mensuales
• La necesidad de usar métodos anticonceptivos adecuados
• La necesidad de contactar con un profesional sanitario inmediatamente si se sospecha de embarazo durante el tratamiento o en los 24 meses siguientes a la finalización del tratamiento
• Información para los hombres
• La necesidad de usar preservativos cuando mantienen relaciones sexuales con una mujer
• No donar semen durante el tratamiento ni durante los 2 meses posteriores a la administración de la última dosis
• La necesidad de contactar con un profesional sanitario si su pareja sospecha que está embarazada mientras el paciente está en tratamiento con Erivedge o durante los 2 meses posteriores al tratamiento
• Devolver las cápsulas que no se han tomado al final del tratamiento (la eliminación dependerá de la normativa local)
• Números de teléfono de contacto de emergencia
E. OBLIGACIÓN ESPECÍFICA DE LLEVAR A CABO MEDIDAS POSTAUTORIZACIÓN EN RELACIÓN CON UNA AUTORIZACIÓN DE COMERCIALIZACIÓN CONDICIONAL
Al ser esta una autorización de comercialización condicional y de según lo que establece el Artículo 14(7) del Reglamento (CE) 726/2004, el TAC deberá llevar a cabo, dentro del plazo establecido, las siguientes medidas:
|
Descripción |
Fecha límite |
|
El solicitante debe proporcionar más datos sobre la seguridad y eficacia en pacientes con CBB metastásico sintomático del análisis final del MO25616 |
1Q.2016 |
ANEXO III
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
Erivedge 150 mg cápsulas duras vismodegib
Cada cápsula contiene 150 mg de vismodegib.
Contiene lactosa.
Para mayor información consultar el prospecto.
28 cápsulas
No rompa, abra, ni mastique la cápsula
Leer el prospecto antes de utilizar este medicamento
Vía oral
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
Riesgo de defectos congénitos graves No usar durante el embarazo ni lactancia
Debe seguir el Programa de Prevención de Embarazo de Erivedge
No conservar a temperatura superior a 30 °C
Mantener el frasco perfectamente cerrado para protegerlo de la humedad
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
Las cápsulas no utilizadas deben ser devueltas al final del tratamiento
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/848/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Erivedge
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC:
SN:
NN:
Erivedge 150 mg cápsulas duras vismodegib
Cada cápsula contiene 150 mg de vismodegib.
Contiene lactosa.
Para mayor información consultar el prospecto
28 cápsulas
No rompa, abra, ni mastique la cápsula
Leer el prospecto antes de utilizar este medicamento
Vía oral
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
Riesgo de defectos congénitos graves No usar durante el embarazo ni lactancia
Debe seguir el Programa de Prevención de Embarazo de Erivedge
No conservar a temperatura superior a 30 °C
Mantener el frasco perfectamente cerrado para protegerlo de la humedad
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
Las cápsulas no utilizadas deben ser devueltas al final del tratamiento
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/13/848/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
B. PROSPECTO
Prospecto: información para el paciente
Erivedge 150 mg cápsulas duras
vismodegib
Erivedge puede causar graves defectos congénitos. Puede provocar la muerte del bebé antes de nacer o poco después de nacer. No debe quedarse embarazada mientras esté tomando este medicamento. Debe seguir los consejos anticonceptivos descritos en este prospecto.
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
• Conserve este prospecto, ya que puede tener que volver a leerlo.
• Si tiene alguna duda, consulte a su médico o farmacéutico.
• Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
• Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4
Contenido del prospecto
1. Qué es Erivedge y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Erivedge
3. Cómo tomar Erivedge
4. Posibles efectos adversos
5. Conservación de Erivedge
6. Contenido del envase e información adicional
1. Qué es Erivedge y para qué se utiliza Qué es Erivedge
Erivedge es un medicamento para el tratamiento del cáncer y contiene el principio activo vismodegib.
Para qué se utiliza Erivedge
Erivedge se utiliza para tratar a adultos con un tipo de cáncer de piel conocido como carcinoma de células basales avanzado. Se utiliza cuando el cáncer:
• Se ha extendido a otras partes del cuerpo (denominado carcinoma de células basales “metastásico”)
• Se ha extendido a las áreas cercanas (denominado carcinoma de células basales “localmente avanzado”) y su médico decide que usted no es candidato al tratamiento con cirugía o radiación.
Cómo funciona Erivedge
El carcinoma de células basales se desarrolla cuando el DNA de las células normales de la piel está dañado y el cuerpo no puede repararlo. Este daño puede cambiar la forma de trabajar de ciertas proteínas en estas células y convertir estas células dañadas en cancerosas empezando a crecer y dividirse. Erivedge es un medicamento para el tratamiento del cáncer que actúa controlando una de las proteínas claves involucrada en el carcinoma de células basales. Esto puede disminuir o parar el crecimiento de las células cancerosas, o puede matarlas. Como resultado, su cáncer de piel puede reducirse.
2. Qué necesita saber antes de empezar a tomar Erivedge
Lea las instrucciones específicas que su médico le ha dado, especialmente sobre los efectos de Erivedge en el feto.
Lea cuidadosamente y siga las instrucciones del prospecto para el paciente y tarjeta de recuerdo que le haya dado su médico.
No tome Erivedge:
• si es alérgico a vismodegib o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
• si está embarazada, piensa que pudiera estarlo, o está planeando quedarse embarazada durante el tratamiento o durante los 24 meses posteriores a la administración de la última dosis de este medicamento. Esto es porque Erivedge puede dañar o provocar la muerte del feto.
• si está dando el pecho o planea dar el pecho durante el tratamiento o durante los 24 meses posteriores a la administración de la última dosis de este medicamento. Esto es porque se desconoce si Erivedge pasa a la leche materna y si puede provocar daño a su bebé.
• si puede quedarse embarazada y no puede o no quiere seguir las medidas necesarias de prevención de embarazo mencionadas en el Programa de Prevención de Embarazo de Erivedge.
• Si está tomando la hierba de San Juan (Hypericum perforatum) - un medicamento a base de plantas usado para la depresión (ver “Toma de Erivedge con otros medicamentos”).
En las secciones “Embarazo, lactancia y fertilidad” y “Métodos anticonceptivos en hombres y mujeres” puede encontrar más información sobre las cuestiones anteriores.
No tome este medicamento si le sucede algo de lo mencionado anteriormente. Si tiene dudas, consulte a su médico o farmacéutico antes de tomar Erivedge.
Advertencias y precauciones
Consulte a su médico o farmacéutico antes de empezar a tomar Erivedge si tiene dudas sobre la información de esta sección:
• No debe donar sangre en ningún momento durante el tratamiento ni durante los 24 meses posteriores a la administración de la última dosis de este medicamento.
• Si es hombre, no debe donar semen en ningún momento durante el tratamiento ni durante 2 meses después de la última dosis.
• Su médico debe revisar su piel regularmente por un tipo de cáncer llamado “carcinoma cutáneo de células escamosas” (CCE). No se sabe si el CCE está relacionado con el tratamiento con Erivedge. Generalmente, este tipo de lesión aparece en pieles dañadas por el sol, es local y se puede curar. Si notara algún cambio en su piel dígaselo a su médico.
• Nunca de este medicamento a nadie más. Debe devolver las cápsulas no utilizadas al final de su tratamiento. Pregunte a su médico o farmacéutico dónde devolverlas.
Niños y adolescentes
No se recomienda el uso de Erivedge en niños y adolescentes menores de 18 años. Esto es porque se desconoce si es seguro o eficaz en este grupo de edad. En estudios en animales con este medicamento se vieron problemas con el crecimiento de los dientes y de los huesos.
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento. Esto también incluye los medicamentos sin receta, vitaminas y plantas medicinales.
Algunos medicamentos pueden influir en el funcionamiento de Erivedge, o hacer que sea más probable que tenga efectos adversos. Erivedge puede influir también en el funcionamiento de otros medicamentos.
En particular, informe a su médico si está tomando alguno de los siguientes medicamentos:
• rifampicina - usado para infecciones bacterianas,
• carbamazepina, fenitoína - usado para la epilepsia,
• ezetimibe y estatinas, tales como atorvastatina, fluvastatina, pravastatina, rosuvastatina, simvastatina - usados para el colesterol alto,
• bosentan, glibenclamida, repaglinida, valsartan,
• topotecan - usado para ciertos tipos de cáncer,
• sulfasalazina - usado para ciertas alteraciones inflamatorias, y especialmente,
• la hierba de San Juan (Hypericum perforatum) - un medicamento a base de plantas usado para la depresión, ya que no debe usarlo a la vez que Erivedge.
Embarazo, lactancia y fertilidad
Embarazo
No tome Erivedge si está embarazada, cree que pudiera estarlo o está planeando quedarse embarazada durante el tratamiento o durante los 24 meses posteriores a la administración de la última dosis de este medicamento.
Debe interrumpir el tratamiento e informar a su médico inmediatamente si: tiene o cree que ha tenido una falta en su período menstrual, o si ha tenido sangrado menstrual anormal o sospecha que está embarazada. Si se queda embarazada durante el tratamiento con Erivedge, debe interrumpir el tratamiento e informar a su médico inmediatamente.
Erivedge puede causar graves defectos congénitos. También puede provocar la muerte del feto. Su médico le dará instrucciones específicas (el Programa de Prevención de Embarazo de Erivedge), especialmente sobre los efectos de Erivedge en los bebés que aún no han nacido.
Lactancia
No debe dar el pecho a su bebé durante el tratamiento ni durante los 24 meses posteriores a la administración de la última dosis de este medicamento. Se desconoce si Erivedge puede pasar a la leche materna y dañar a su bebé.
Fertilidad
Erivedge puede afectar la capacidad de la mujer para tener niños. Algunas mujeres que han tomado Erivedge han dejado de tener la menstruación. Si esto le ocurre, se desconoce si volverá a tener la menstruación. Consulte con su médico si quiere tener hijos en el futuro.
Métodos anticonceptivos - para mujeres y hombres
Para mujeres que tomen Erivedge:
Antes de comenzar el tratamiento, consulte a su médico si puede quedarse embarazada. Incluso si ha dejado de tener la menstruación, es importante consultar a su médico si existe algún riesgo de quedarse embarazada.
Si puede quedarse embarazada:
• debe tomar precauciones para no quedarse embarazada mientras toma Erivedge
• use 2 métodos anticonceptivos, un método altamente eficaz y un método de barrera (por favor, mire los ejemplos más abajo)
• necesita continuar con las medidas anticonceptivas durante los 24 meses posteriores a la administración de la última dosis - porque Erivedge puede permanecer en su cuerpo hasta 24 meses después de la administración de la última dosis.
Métodos anticonceptivos recomendados: Consulte a su médico sobre los dos mejores métodos anticonceptivos para usted.
Use un método altamente eficaz, como:
• inyección anticonceptiva
• dispositivo intrauterino (DIU)
• esterilización quirúrgica.
Debe usar también un método de barrera, como:
• preservativo (preferiblemente con espermicida)
• diafragma (preferiblemente con espermicida)
Su médico se asegurará de realizarle una prueba de embarazo:
• al menos 7 días antes de comenzar su tratamiento, para asegurar que no está embarazada
• cada mes durante el tratamiento.
Debe informar a su médico inmediatamente durante el tiempo de tratamiento o durante los 24 meses después de su última dosis de medicamento si:
• piensa, por cualquier motivo, que ha fallado su método anticonceptivo
• ha dejado de tener la menstruación
• deja de usar métodos de anticoncepción
• necesita cambiar el método de anticoncepción.
Para hombres que tomen Erivedge:
Erivedge puede pasar al semen. Use siempre preservativo (preferiblemente con espermicida), incluso después de una vasectomía, cuando mantenga relaciones sexuales con una mujer. Hágalo durante el tratamiento y durante los 2 meses posteriores a la administración de la última dosis de este medicamento.
No debe donar semen en ningún momento durante el tratamiento ni durante los 2 meses posteriores a la administración de la última dosis de este medicamento.
Conducción y uso de máquinas
No es probable que Erivedge afecte a su capacidad para conducir o para usar herramientas o máquinas. Consulte a su médico si no está seguro.
Erivedge contiene lactosa y sodio
La cápsula de Erivedge contienen un tipo de azúcar llamado lactosa. Si su médico le ha indicado que no puede tolerar o digerir ciertos azúcares, consulte con él antes de tomar este medicamento.
Este medicamento contiene menos de 1 mmol (23 mg) de sodio por cápsula, es decir, está esencialmente “exento de sodio”.
3. Cómo tomar Erivedge
Siga exactamente las instrucciones de administración de Erivedge indicadas por su médico. En caso de duda, consulte de nuevo con su médico o farmacéutico.
La dosis recomendada es una cápsula al día.
• Trague la cápsula entera con un vaso de agua.
• No triture, abra, ni mastique la cápsula, para evitar una exposición involuntaria al contenido de la cápsula.
• Erivedge puede tomarse con o sin comida.
Si toma más Erivedge del que debe
Si toma más Erivedge del que debe consulte a su médico.
Si olvidó tomar Erivedge
No tome una dosis doble para compensar una dosis olvidada, si no que continúe con la siguiente dosis programada.
Si interrumpe el tratamiento con Erivedge
No interrumpa el tratamiento con este medicamento sin consultar antes con su médico, ya que podría hacer que su tratamiento sea menos efectivo.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Erivedge puede provocar graves defectos congénitos. También puede conducir a la muerte de su bebé antes de nacer o poco después de nacer. No debe quedarse embarazada mientras esté tomando este medicamento (ver sección 2 “No tome Erivedge” y “Embarazo, lactancia y fertilidad”).
Otros efectos adversos se presentan ordenados por gravedad y frecuencia:
Muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
• pérdida de la menstruación en mujeres en edad fértil,
• pérdida de apetito y pérdida de peso,
• sensación de cansancio,
• espasmo muscular,
• diarrea,
• pérdida de pelo (alopecia),
• erupción,
• un cambio en el sabor de las cosas o pérdida total del gusto,
• estreñimiento,
• vómitos o sensación de que quiere vomitar (náuseas),
• malestar estomacal o indigestión,
• dolor en articulaciones,
• dolor (en general) o dolor en brazos, piernas,
• picores.
Frecuentes (pueden afectar hasta 1 de cada 10 personas):
• dolor en el pecho, espalda o costado,
• falta de energía o debilidad (astenia),
• pérdida de agua del cuerpo (deshidratación),
• dolor muscular, en tendones, ligamentos, o huesos,
• dolor de estómago,
• pérdida del gusto,
• crecimiento anormal de pelo,
• pérdida de pestañas (madarosis)
• cambios en los análisis de sangre, incluyendo valores aumentados de las pruebas de hígado o valores disminuidos de sodio.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Erivedge
• Mantener este medicamento fuera de la vista y del alcance de los niños.
• No utilice este medicamento después de la fecha de caducidad que aparece en el frasco y en el envase después de CAD. La fecha de caducidad es el último día del mes que se indica.
• No conservar a temperatura superior a 30 ° C.
• Mantener el frasco perfectamente cerrado para protegerlo de la humedad.
• Los medicamentos no se deben tirar por los desagües ni a la basura.
• Cuando termine el tratamiento devuelva todas las cápsulas no usadas. Estas medidas evitarán un mal uso y ayudarán a proteger el medio ambiente. Pregunte a su médico o farmacéutico sobre dónde devolver el medicamento.
6. Contenido del envase e información adicional Composición de Erivedge
• El principio activo es vismodegib. Cada cápsula dura contiene 150 mg de vismodegib.
Los demás componentes son:
• Contenido de la cápsula: celulosa microcristalina, lactosa monohidrato, lauril sulfato sódico, povidona (K29/32), almidón glicolato de sodio (Tipo A), talco y estearato magnésico.
• Cubierta de la cápsula: óxido de hierro rojo (E172), óxido de hierro negro (E172), dióxido de titanio, gelatina,
• Tinta de impresión: goma laca y óxido de hierro negro (E172).
Aspecto de Erivedge y contenido del envase
Las cápsulas tienen un cuerpo coloreado rosa opaco con la inscripción “150 mg” y una tapa gris con la inscripción “VISMO” en tinta comestible negra. Los envases disponibles son frascos con un cierre de rosca a prueba de niños que contienen 28 cápsulas. Cada envase contiene un frasco.
Titular de la autorización de la comercialización
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW United Kingdom
Responsable de fabricación
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Germany
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien N.V. Roche S.A. Tél/Tel: +32 (0) 2 525 82 11 |
Lietuva UAB “Roche Lietuva” Tel: +370 5 2546799 |
|
Etnrapna Pom Etnrapua EOOfl Ten: +359 2 818 44 44 |
Luxembourg/Luxemburg (Voir/siehe Belgique/Belgien) |
|
Ceská republika Roche s. r. o. Tel: +420 -2 20382111 |
Magyarország Roche (Magyarország) Kft. Tel: +36 - 23 446 800 |
|
Danmark Roche a/s Tlf: +45 - 36 39 99 99 |
Malta (See United Kingdom) |
|
Deutschland Roche Pharma AG Tel: +49 (0) 7624 140 |
Nederland Roche Nederland B.V. Tel: +31 (0) 348 438050 |
|
Eesti Roche Eesti OÜ Tel: + 372 -6 177 380 |
Norge Roche Norge AS Tlf: +47 - 22 78 90 00 |
|
EXXáSa Roche (Hellas) A.E. T^: +30 210 61 66 100 |
Osterreich Roche Austria GmbH Tel: +43 (0) 1 27739 |
|
España Roche Farma S.A. Tel: +34 -91 324 81 00 |
Polska Roche Polska Sp.z o.o. Tel: +48 -22 345 18 88 |
|
France Roche Tél: +33 (0) 1 47 61 40 00 |
Portugal Roche Farmacéutica Química, Lda Tel: +351 -21 425 70 00 |
|
Hrvastska Roche d.o.o Tel: +385 1 4722333 |
Romania Roche Romania S.R.L. Tel: +40 21 206 47 01 |
|
Ireland Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700 |
Slovenija Roche farmacevtska druzba d.o.o Tel: +386 - 1 360 26 00 |
|
Ísland Roche a/s c/o Icepharma hf Sími: +354 540 8000 |
Slovenská republika Roche Slovensko, s.r.o. Tel: +421 -2 52638201 |
|
Italia Roche S.p.A. Tel: +39 -039 2471 |
Suomi/Finland Roche Oy Puh/Tel: +358 (0) 10 554 500 |
|
Kúnpoq r.A.Zxa^áxn? & £ra AxS. T^: +357 - 22 76 62 76 |
Sverige Roche AB Tel: +46 (0) 8 726 1200 |
|
Latvija Roche Latvija SIA Tel: +371 -6 7 039831 |
United Kingdom Roche Products Ltd. Tel: +44 (0) 1707 366000 |
Fecha de la última revisión de este prospecto:
Este medicamento se ha autorizado con una «aprobación condicional». Esta modalidad de aprobación significa que se espera obtener más información de este medicamento. La Agencia Europea de Medicamentos revisará la información nueva de este medicamento al menos una vez al año y este prospecto se actualizará cuando sea necesario.
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
En la página web de la Agencia Europea de Medicamentos puede encontrarse este prospecto en todas las lenguas de la Unión Europea/Espacio Económico Europeo.
Como parte del Programa de Prevención de Embarazo de Erivedge, todos los pacientes recibirán:
• Un folleto para el paciente
• Una tarjeta recordatorio para el paciente
Por favor consulte estos documentos para mas información.
39