Emend 125 Mg Polvo Para Suspension Oral
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
EMEND 40 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula contiene 40 mg de aprepitant.
Excipiente con efecto conocido
Cada cápsula contiene 40 mg de sacarosa.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura.
Las cápsulas son opacas con cuerpo blanco y tapa amarillo mostaza, con “464” y “40 mg” impreso en forma radial en tinta negra en el cuerpo.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
EMEND 40 mg está indicado para la prevención de náuseas y vómitos posquirúrgicos (NVPQ) en adultos.
4.2 Posología y forma de administración
Posología
Se deben considerar las directrices de tratamiento clínico en cuanto a la necesidad de tratamiento preventivo frente a las náuseas y vómitos posquirúrgicos (NVPQ).
La dosis oral recomendada de EMEND es una dosis única de 40 mg en el transcurso de las 3 horas anteriores a la inducción de la anestesia.
Poblaciones especiales
Pacientes de edad avanzada (> 65 años)
No es necesario ajustar la dosis en los pacientes de edad avanzada (ver sección 5.2).
Sexo
No es necesario ajustar la dosis según el sexo (ver sección 5.2).
Insuficiencia renal
No es necesario ajustar la dosis en los pacientes con insuficiencia renal ni en los pacientes con nefropatía terminal sometidos a hemodiálisis (ver sección 5.2).
Insuficiencia hepática
No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. Existen datos limitados en pacientes con insuficiencia hepática moderada y no existen datos en pacientes con insuficiencia hepática grave. Aprepitant se debe usar con precaución en estos pacientes (ver las secciones 4.4 y 5.2).
Población pediátrica
No se ha establecido la seguridad y eficacia de EMEND 40 mg en niños y adolescentes menores de 18 años de edad. Los datos actualmente disponibles están descritos en las secciones 5.1 y 5.2, sin embargo no se puede hacer una recomendación posológica.
Forma de administración
Las cápsulas duras se deben tragar enteras.
EMEND puede tomarse con o sin alimentos.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Pacientes con insuficiencia hepática moderada a grave
Existen datos limitados en pacientes con insuficiencia hepática moderada y no existen datos en pacientes con insuficiencia hepática grave. EMEND se debe usar con precaución en estos pacientes (ver sección 5.2).
Interacciones con el CYP3A4
EMEND (40 mg) se debe usar con precaución en pacientes que estén recibiendo de forma concomitante la administración de pimozida, terfenadina, astemizol, cisaprida o derivados de los alcaloides del ergot. La inhibición de la isoenzima 3A4 del citocromo P450 (CYP3A4) por aprepitant podría dar lugar a elevaciones de las concentraciones plasmáticas de estos principios activos, lo que podría provocar reacciones adversas graves (ver sección 4.5).
Administración conjunta con anticonceptivos hormonales
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de la administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo (ver sección 4.5).
Para más información sobre la posible interacción de aprepitant a dosis elevadas y múltiples, consulte la Ficha Técnica o Resumen de las Características del Producto de EMEND 80 mg cápsulas duras y EMEND 125 mg cápsulas duras.
Excipientes
EMEND cápsulas contiene sacarosa. Los pacientes con intolerancia hereditaria a la fructosa, problemas de absorción de glucosa o galactosa, o insuficiencia de sacarasa-isomaltasa, no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Aprepitant es un sustrato, y un inhibidor dependiente de la dosis, y un inductor de CYP3A4.
Aprepitant es también un inductor de CYP2C9. Durante el tratamiento, la dosis única de 40 mg de aprepitant recomendada para NVPQ da lugar a una inhibición débil de CYP3A4. Después del tratamiento, EMEND causa una inducción transitoria suave de CYP2C9, CYP3A4 y glucuronidación. Aprepitant se ha estudiado a dosis más elevadas. Durante el tratamiento de las náuseas y los vómitos inducidos por la quimioterapia (NVIQ), el tratamiento de 3 días con un régimen de 125 mg/80 mg de aprepitant da lugar a una inhibición moderada de CYP3A4. Aprepitant no parece que interaccione con el transportador de la P-glucoproteína, como sugiere la falta de interacción de aprepitant con digoxina.
Efecto de aprepitant sobre la farmacocinética de otros principios activos
Inhibición de CYP3A4
Como inhibidor débil de CYP3A4, aprepitant (40 mg) puede aumentar las concentraciones plasmáticas de los principios activos coadministrados por vía oral, que se metabolizan a través de
CYP3A4. La exposición total de los sustratos de CYP3A4 que se administran por vía oral puede aumentar hasta aproximadamente 1,5 veces después de una dosis única de 40 mg de aprepitant; se estima que el efecto de aprepitant sobre las concentraciones plasmáticas de los sustratos de CYP3A4 que se administran por vía intravenosa sea menor.
EMEND 40 mg se debe usar con precaución en pacientes que estén recibiendo pimozida, terfenadina, astemizol, cisaprida o derivados de los alcaloides del cornezuelo. La inhibición de CYP3A4 por aprepitant podría dar lugar a elevaciones de las concentraciones plasmáticas de estos principios activos, lo que podría provocar reacciones graves.
Corticoesteroides
Dexametasona: Una dosis única de 40 mg de aprepitant, cuando se administra conjuntamente con una dosis única oral de 20 mg de dexametasona, aumenta el AUC de dexametasona en 1,45 veces. No se recomienda el ajuste de dosis.
Metilprednisolona: Aunque no se ha estudiado la administración concomitante de metilprednisolona con una dosis única de 40 mg de aprepitant, una dosis única de 40 mg de aprepitant produce una inhibición débil de CYP3A4 y no se espera que altere las concentraciones plasmáticas de metilprednisolona a un nivel clínicamente significativo. Por tanto, no se recomienda el ajuste de dosis.
Midazolam
El AUC de midazolam aumentó en 1,2 veces cuando una dosis única de 40 mg de aprepitant se administró conjuntamente con una dosis oral única de 2 mg de midazolam; este efecto no se consideró clínicamente importante.
Inducción
Como inductor suave de CYP2C9, CYP3A4 y glucuronidación, aprepitant puede disminuir las concentraciones plasmáticas de sustratos eliminados por estas vías durante las dos semanas posteriores al inicio del tratamiento. Para los sustratos CYP2C9 y CYP3A4 la inducción es transitoria con un efecto máximo alcanzado después de 3-5 días. El efecto se puede mantener durante unos pocos días y se espera que sea clínicamente insignificante a las 2 semanas después de terminar el tratamiento con EMEND. No existen datos relativos a los efectos sobre CYP2C8 y CYP2C19. La administración conjunta de EMEND con principios activos que se sabe que son metabolizados por CYP2C9 (por ej. fenitoína, warfarina) puede dar lugar a concentraciones plasmásticas más bajas de estos principios activos. En base a los ensayos de interacciones con tolbutamida y anticonceptivos orales, la exposición total de principios activos metabolizados por CYP2C9 o CYP3A4 administrados conjuntamente puede reducirse hasta el 15-30%.
Anticonceptivos hormonales
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de la administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo.
Antagonistas 5-HT3
En ensayos clínicos de interacción, aprepitant no tuvo efectos clínicamente importantes sobre la farmacocinética de ondansetrón, granisetrón ni hidrodolasetrón (el metabolito activo de dolasetrón).
Efecto de otros medicamentos sobre la farmacocinética de aprepitant
La administración concomitante de EMEND con principios activos que inhiben la actividad de CYP3A4 (por ejemplo, ketoconazol, itraconazol, voriconazol, posaconazol, claritromicina, telitromicina, nefazodona e inhibidores de la proteasa) se debe abordar con precaución, ya que la combinación se espera que provoque una elevación de varias veces las concentraciones plasmáticas de aprepitant (ver sección 4.4).
La administración concomitante de EMEND con principios activos que inducen fuertemente la actividad de CYP3A4 (por ejemplo, rifampicina, fenitoína, carbamazepina, fenobarbital) se debe
evitar ya que la combinación provoca reducciones en las concentraciones plasmáticas de aprepitant lo que podría provocar una disminución de la eficacia de EMEND. No se recomienda la administración concomitante de EMEND con plantas medicinales que contienen hipérico (Hypericum perforatum).
Ketoconazol
Al administrarse una dosis única de 125 mg de aprepitant el día 5 de un régimen de 10 días de 400 mg/día de ketoconazol, un potente inhibidor de CYP3A4, el AUC de aprepitant aumentó aproximadamente 5 veces y la semivida terminal media de aprepitant aumentó aproximadamente 3 veces.
Rifampicina
Al administrarse una dosis única de 375 mg de aprepitant el día 9 de un régimen de 14 días de 600 mg/día de rifampicina, un potente inductor de CYP3A4, el AUC de aprepitant disminuyó un 91% y la semivida terminal media disminuyó un 68%.
4.6 Fertilidad, embarazo y lactancia
Anticoncepción en varones y mujeres
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de su administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo (ver las secciones 4.4 y 4.5).
Embarazo
No hay datos clínicos disponibles sobre la exposición a aprepitant durante el embarazo. En estudios en animales, no hubo indicios de efectos perjudiciales directos ni indirectos en términos del embarazo, el desarrollo embrionario/fetal, el parto o el desarrollo posnatal (ver sección 5.3). Se desconocen los posibles efectos sobre la reproducción de alteraciones en la regulación de la neurocinina. EMEND no se debe utilizar durante el embarazo excepto si fuese claramente necesario.
Lactancia
Aprepitant se excreta en la leche de ratas lactantes. Se desconoce si aprepitant se excreta en la leche materna; por consiguiente, no se recomienda la lactancia durante el tratamiento con EMEND.
Fertilidad
Los estudios de fertilidad no sugirieron efectos perjudiciales directos ni indirectos en términos del estado de apareamiento, fertilidad, desarrollo embrionario/fetal, o recuento de esperma y movilidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de EMEND sobre la capacidad para conducir y utilizar máquinas es pequeña. Se pueden producir mareos y fatiga después de la administración de EMEND (ver sección 4.8).
4.8 Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad de aprepitant se evaluó en aproximadamente 6.500 adultos.
Las reacciones adversas se notificaron en aproximadamente un 4% de los adultos tratados con 40 mg de aprepitant en comparación con aproximadamente un 6% de los pacientes que recibieron 4 mg de ondansetrón vía intravenosa. En ensayos clínicos controlados en adultos recibiendo anestesia general, se administraron 40 mg de aprepitant por vía oral a 564 pacientes y se administraron 4 mg de ondansetrón vía intravenosa a 538 pacientes. La mayoría de las reacciones adversas en estos ensayos clínicos fueron descritas con intensidad de leve a moderada.
La reacción adversa más frecuente, notificada con mayor incidencia en adultos tratados con 40 mg de aprepitant (1,1 %) que con ondansetrón (1,0 %) fue ALT elevada.
Tabla de reacciones adversas
Las reacciones adversas siguientes se observaron en ensayos en NVPQ en adultos tratados con aprepitant con una incidencia mayor que con ondansetrón o en el uso después de la comercialización:
Las frecuencias se definen como: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000) y muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
|
Sistema de clasificación de órganos |
Reacción adversa |
Frecuencia |
|
Trastornos del sistema inmunológico |
reacciones de hipersensibilidad incluyendo reacciones anafilácticas |
no conocida |
|
Trastornos psiquiátricos |
insomnio |
poco frecuentes |
|
Trastornos del sistema nervioso |
disartria, hipoestesia, alteración sensitiva |
poco frecuentes |
|
Trastornos oculares |
miosis, agudeza visual disminuida |
poco frecuentes |
|
Trastornos cardiacos |
bradicardia |
poco frecuentes |
|
Trastornos respiratorios, torácicos y mediastínicos |
disnea, sibilancia |
poco frecuentes |
|
Trastornos gastrointestinales |
dolor abdominal alto, ruidos intestinales anormales, boca seca, náuseas, molestias en el estómago, estreñimiento*, subíleo* |
poco frecuentes |
|
Trastornos de la piel y del tejido subcutáneo |
prurito, erupción, urticaria, síndrome de Stevens-Johnson/necrolisis epidérmica tóxica |
no conocida |
|
Exploraciones complementarias |
ALT elevada |
frecuentes |
*Notificadas en pacientes que tomaban una dosis más alta de aprepitant.
Descripción de reacciones adversas seleccionadas
Se observaron otras reacciones adversas en adultos tratados con el régimen de aprepitant (125 mg/80 mg) para náuseas y vómitos inducidos por quimioterapia (NVIQ), con una incidencia mayor que con el tratamiento estándar: distensión abdominal, dolor abdominal, acné, anemia, ansiedad, AST elevada, astenia, fosfatasa alcalina aumentada, sodio disminuido en sangre, candidiasis, trastorno cardiovascular, malestar torácico, trastorno cognoscitivo, conjuntivitis, tos, apetito disminuido, desorientación, mareo, perforación de úlcera duodenal, disgeusia, dispepsia, disuria, eructos, estado de ánimo eufórico, heces duras, fatiga, neutropenia febril, flatulencia, alteración de la marcha, enfermedad por reflujo gastroesofágico, presencia de glucosuria, hipo, acaloramiento, hiperhidrosis, letargia, malestar general, espasmos musculares, debilidad muscular, náuseas*, colitis neutropénica, recuento disminuido de neutrófilos, edema, dolor orofaríngeo, palpitaciones, reacción de fotosensibilidad, polaquiuria, polidipsia, goteo postnasal, erupción prurítica, hematíes en orina positivos, seborrea, lesión de la piel, estornudos, somnolencia, infección estafilocócica, estomatitis, irritación de garganta, acúfenos, excreción urinaria aumentada, vómitos*, peso disminuido.
*Náuseas y vómitos fueron parámetros de eficacia en los 5 primeros días de tratamiento post-quimioterapia y sólo después se notificaron como reacciones adversas.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
En caso de sobredosis, se debe suspender la administración de EMEND y proporcionar tratamiento de apoyo general y vigilancia. Debido a la actividad antiemética de aprepitant, es posible que la emesis inducida por un medicamento no resulte eficaz.
Aprepitant no puede eliminarse mediante hemodiálisis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antieméticos y antinauseosos, código ATC: A04AD12
Aprepitant es un antagonista selectivo de alta afinidad por los receptores de la sustancia P neurocinina 1 (NKi) humana.
En 2 ensayos clínicos en adultos, en fase III de grupos paralelos, multicéntricos, aleatorizados, doble ciego, controlados con un comparador activo, se comparó aprepitant con ondansetrón en la prevención de NVPQ en 1.658 pacientes sometidos a cirugía abdominal abierta. La mayoría de los adultos eran mujeres (>90%), principalmente sometidas a cirugía ginecológica. Los pacientes fueron aleatorizados a recibir 40 mg de aprepitant, 125 mg de aprepitant o 4 mg de ondansetrón. Se administró aprepitant por vía oral con 50 ml de agua de 1 a 3 horas antes de la anestesia. Ondansetrón se administró por vía intravenosa inmediatamente antes de la inducción de la anestesia. Se evaluó la actividad entiemética de aprepitant durante el periodo de 0 a 48 horas después de finalizar la operación.
Los resultados muestran que un porcentaje mayor de adultos posquirúrgicos experimentaron una respuesta completa (sin emesis y sin utilizar medicación de rescate) con 40 mg de aprepitant que con 4 mg de ondansetrón (el límite inferior del IC es 0,0, lo que indica significación limítrofe), como se describe en la Tabla 1:
Tabla 1
Porcentaje de adultos posquirúrgicos que respondieron por grupo de tratamiento Resultados combinados de 2 ensayos en fase III
|
Aprepitant 40 mg, vía oral (N=541) |
Ondansetrón 4 mg, vía intravenosa (N=526) |
Diferencia de puntos de porcentaje (%) § e IC del 95% # | ||||
|
n/m |
(%) |
n/m |
(%) |
% |
IC del 95% | |
|
Respuesta completa (0-24 horas)f |
298/541 |
(55,1) |
258/526 |
(49,0) |
5,9 |
(0,0, 11,8) |
f Respuesta completa: sin emesis y sin medicación de rescate
§ Diferencia (%) calculada como aprepitant 40 mg menos ondansetrón 4 mg
# Diferencia (%) e IC del 95% calculado usando el método estratificado de Miettinen-Nurminen,
usando los pesos de Cochran-Mantel-Haenszel
La reducción del riesgo de un episodio de vómitos en el periodo de 0 a 24 horas, con 40 mg de aprepitant en relación con 4 mg de ondansetrón fue del 53,3 % (IC del 95%: 35,3 a 66,3), en un análisis que no tuvo en cuenta a los pacientes en el momento de usar la medicación de rescate.
Población pediátrica
La seguridad y la eficacia exploratoria de aprepitant se evaluaron en un ensayo clínico de fase I (n=50) usando polvo para suspensión oral de 40 mg. El porcentaje de sujetos que no notificó vómitos durante las primeras 24 horas tras la cirugía fue similar entre los sujetos que recibieron aprepitant y los tratados con ondansetrón. No se identificaron nuevos problemas de seguridad. No obstante, los datos de este pequeño estudio no respaldan una conclusión acerca de la pauta posológica óptima. Están en curso ensayos clínicos adicionales para evaluar el uso de aprepitant en pacientes pediátricos (ver sección 4.2 para la información sobre el uso pediátrico).
5.2 Propiedades farmacocinéticas
Aprepitant muestra una farmacocinética no lineal. Tanto el aclaramiento como la biodisponibilidad absoluta disminuyen al aumentar la dosis.
Absorción
La biodisponibilidad oral absoluta media de aprepitant es de 67% para la cápsula de 80 mg y de 59% para la cápsula de 125 mg. La concentración plasmática máxima media (Cmax) de aprepitant se alcanzó aproximadamente a las 4 horas (tmax).
Tras la administración oral de una dosis única de 40 mg de EMEND en ayunas, el AUC0-a,
(media ± DE) fue 8,0 ±2,1 ^g»h/ml y la Cmax fue de 0,7 ± 0,24 ^g/ml. La tmax media fue de 3,0 h.
La ingesta concomitante de una dosis de 40 mg con un desayuno estándar, sólo disminuyó la Cmax de aprepitant en un 18 % pero no afectó al AUC. Esto no se consideró clínicamente importante.
Distribución
Aprepitant se une fuertemente a proteínas, con una media del 97%. La media geométrica del volumen aparente de distribución en el estado equilibrio (Vdee) es aproximadamente de 66 litros en el ser humano.
Biotransformación
Aprepitant se metaboliza extensamente. En adultos jóvenes sanos, aprepitant representa aproximadamente el 19% de la radiactividad plasmática durante 72 horas después de una dosis única intravenosa de 100 mg de [C14]-fosaprepitant, un profármaco de aprepitant, lo que indica una importante presencia de metabolitos en el plasma. En el plasma humano se han identificado doce metabolitos de aprepitant. El metabolismo de aprepitant se produce en gran medida por oxidación en el anillo de morfolina y sus cadenas laterales y los metabolitos resultantes sólo fueron débilmente activos. Estudios in vitro en los que se usaron microsomas hepáticos humanos indicaron que aprepitant se metaboliza principalmente a través de CYP3A4 y posiblemente con una contribución menor a través de CYP1A2 y CYP2C19.
Eliminación
Aprepitant no se elimina inalterado en la orina. Los metabolitos se eliminan en la orina y a través de excreción biliar en las heces. Después de una dosis única intravenosa de 100 mg de [C14]-fosaprepitant, un profármaco de aprepitant a sujetos sanos, el 57% de la radiactividad se recuperó en la orina y el 45% en las heces.
El aclaramiento plasmático de aprepitant es dependiente de la dosis, disminuyendo al aumentar la dosis y oscilando aproximadamente entre 60 a 72 ml/min en el intervalo de la dosis terapéutica. La semivida terminal es de aproximadamente 9 horas después de la administración de una dosis única de 40 mg.
Farmacocinética en poblaciones especiales
Pacientes de edad avanzada: Tras la administración oral de una dosis única de 125 mg de aprepitant el día 1 y 80 mg una vez al día los días 2 a 5, el AUC0-24h de aprepitant fue un 21% superior el día 1 y un 36% superior el día 5 en pacientes de edad avanzada (>65 años) respecto de los adultos más jóvenes. La Cmax fue un 10% superior el día 1 y un 24% superior el día 5 en pacientes de edad avanzada respecto de los adultos más jóvenes. Estas diferencias no se consideraron clínicamente significativas. EMEND no requiere ajuste de dosis en los pacientes de edad avanzada.
Sexo: Tras la administración oral de una dosis única de 125 mg de aprepitant, la Cmax de aprepitant es un 16% superior en las mujeres en comparación con los varones. La semivida de aprepitant es un 25% inferior en las mujeres en comparación con los varones y su tmax se produce en aproximadamente el mismo tiempo. Estas diferencias no se consideraron clínicamente significativas. EMEND no requiere ajuste de dosis en función del sexo.
Insuficiencia hepática: La insuficiencia hepática leve (Child-Pugh clase A) no afecta a la farmacocinética de aprepitant en un grado clínicamente relevante. No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. De los datos disponibles no pueden extraerse conclusiones relativas a la influencia de la insuficiencia hepática moderada (Child-Pugh clase B) sobre la farmacocinética de aprepitant. No existen datos clínicos ni farmacocinéticos de pacientes con insuficiencia hepática grave (Child-Pugh clase C).
Insuficiencia renal: Se administró una dosis única de 240 mg de aprepitant a pacientes con insuficiencia renal grave (CrCl<30 ml/min) y a pacientes con nefropatía terminal que requería hemodiálisis.
En los pacientes con insuficiencia renal grave, el AUC0_<» de aprepitant total (no unido y unido a proteínas) disminuyó en un 21 % y la Cmax disminuyó en un 32 %, respecto de los sujetos sanos. En los pacientes con nefropatía terminal sometidos a hemodiálisis, el AUC0_<» de aprepitant total disminuyó en un 42 % y la Cmax disminuyó en un 32 %. Debido a los modestos descensos en la unión a proteínas de aprepitant en los pacientes con enfermedad renal, el AUC de aprepitant no unido farmacológicamente activo no se vio significativamente afectado en los pacientes con insuficiencia renal en comparación con los sujetos sanos. La hemodiálisis realizada 4 ó 48 horas después de la administración no tuvo efectos significativos sobre la farmacocinética de aprepitant; en el dializado se recuperó menos de 0,2 % de la dosis.
En pacientes con insuficiencia renal o en pacientes con nefropatía terminal sometidos a hemodiálisis no es necesario ajustar la dosis de EMEND.
Población pediátrica: En un ensayo en el que se utilizó una formulación de polvo para suspensión oral, la administración de una única dosis de 40 mg de aprepitant a pacientes adolescentes (con edades de entre 12 y 17 años) dio lugar a una media del AUC0-48h de 6 ^g/ml, alcanzándose una concentración plasmática máxima media (Cmax) de 0,5 ^g/ml aproximadamente a las 4 horas. La administración de dosis ajustadas en función de la superficie corporal a pacientes de entre 6 meses y menos de 12 años de edad logró una media del AUC0-48h superior a 4 ^g/ml, alcanzándose una Cmax media superior a 0,5 (rg/ml aproximadamente a las 3 horas.
Relación entre concentración y efecto
Usando un trazador altamente específico del receptor NKj, los estudios de tomografía por emisión de positrones (PET) en varones jóvenes sanos han demostrado que aprepitant penetra en el cerebro y ocupa los receptores NK de forma dependiente de la dosis y de la concentración plasmática. Se predice que las concentraciones plasmáticas de aprepitant alcanzadas con el régimen de 3 días de EMEND proporcionarán una ocupación superior al 95 % de los receptores NK cerebrales.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de toxicidad, genotoxicidad, potencial carcinogénico, toxicidad para la reproducción y el desarrollo, a dosis únicas y repetidas. Se deberá tener en cuenta que la exposición sistémica en ratas macho adultas fue inferior a la exposición terapéutica en seres humanos a la dosis de 40 mg. Consecuentemente, no se puede hacer una evaluación adecuada de los posibles efectos sobre la fertilidad en ratas macho. Sin embargo, en un estudio de 9 meses en perros, no se observaron cambios en el peso de los órganos, ni hallazgos macroscópicos ni histomorfológicos en órganos reproductores masculinos a niveles de exposición sistémica 35 veces superior a la exposición terapéutica en seres humanos a la dosis de 40 mg. Aunque no se observaron efectos adversos en los estudios sobre la reproducción cuando animales hembra adultos se expusieron a 3,5 a 4 veces por encima de la exposición terapéutica en seres humanos con 40 mg, se desconocen los posibles efectos sobre la reproducción de alteraciones en la regulación de la neurocinina.
En un estudio de toxicidad juvenil en ratas tratadas desde el día 10 después del nacimiento hasta el día 63, aprepitant ocasionó en las hembras una apertura vaginal prematura a partir de 250 mg/kg dos veces al día y en los machos un retraso en la separación del prepucio a partir de 10 mg/kg dos veces al día. No hubo márgenes de exposición clínicamente relevantes. No se observaron efectos sobre el apareamiento, la fertilidad o la supervivencia embrionaria o fetal ni cambios patológicos en los órganos reproductores relacionados con el tratamiento. En un estudio de toxicidad juvenil en perros tratados desde el día 14 después del nacimiento hasta el día 42, se detectó una disminución del peso testicular y del tamaño de las células de Leydig en los machos con 6 mg/kg/día y un aumento del peso del útero, hipertrofia del útero y del cuello del útero y edema de los tejidos vaginales en las hembras a partir de 4 mg/kg/día. No hubo márgenes de exposición clínicamente relevantes de aprepitant. En el tratamiento a corto plazo de acuerdo con la pauta posológica recomendada, se considera poco probable que estos resultados sean clínicamente relevantes.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Contenido de la cápsula Sacarosa
Celulosa microcristalina (E-460)
Hidroxipropilcelulosa (E-463)
Lauril sulfato de sodio
Cubierta de la cápsula Gelatina
Dióxido de titanio (E-171)
Óxido de hierro amarillo (E-172)
Tinta para impresión Laca
Hidróxido de potasio Óxido de hierro negro (E-172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
4 años.
6.4 Precauciones especiales de conservación
Conservar en el embalaje original para protegerlo de la humedad.
6.5 Naturaleza y contenido del envase
Están disponibles diferentes tamaños de envase:
Blister de aluminio que contiene una cápsula de 40 mg.
5 blisters de aluminio que contienen una cápsula de 40 mg cada uno.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
Ninguna especial para su eliminación.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/262/007
EU/1/03/262/008
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 11/noviembre/2003 Fecha de la última renovación: 22/septiembre/2008
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
NOMBRE DEL MEDICAMENTO
1.
EMEND 165 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula contiene 165 mg de aprepitant.
Excipiente con efecto conocido
Cada cápsula contiene165 mg de sacarosa.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura.
Las cápsulas son opacas con tapa azul claro y cuerpo blanco con “466” y “165 mg” impreso en forma radial en tinta negra en una parte el cuerpo y el logo de Merck en la otra parte.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Prevención de las náuseas y los vómitos agudos y diferidos que se asocian con la quimioterapia antineoplásica altamente emetógena basada en el cisplatino en adultos.
Prevención de las náuseas y los vómitos que se asocian con la quimioterapia antineoplásica moderadamente emetógena en adultos.
EMEND 165 mg se administra como parte de un tratamiento de combinación (ver sección 4.2).
4.2 Posología y forma de administración Posología
EMEND 165 mg se administra sólo el día 1, aproximadamente una hora antes de empezar la quimioterapia como parte de un régimen de tratamiento que incluye un corticosteroide y un antagonista 5-HT3.
Se recomiendan los siguientes regímenes de administración en adultos para la prevención de las náus eas y los vómitos asociados con la quimioterapia antineoplásica emetógena:
Régimen de quimioterapia altamente emetógena
|
Día 1 |
Día 2 |
Día 3 |
Día 4 | |
|
EMEND |
165 mg vía oral |
nada |
nada |
nada |
|
Dexametasona |
12 mg vía oral |
8 mg vía oral |
8 mg vía oral 2 veces al día |
8 mg vía oral 2 veces al día |
|
Antagonistas 5-HT3 |
Dosis habituales de los antagonistas 5-HT3. Ver la información de producto del antagonista 5-HT3 escogido para obtener información sobre la dosis adecuada |
nada |
nada |
nada |
Dexametasona se debe administrar 30 minutos antes de la quimioterapia el día 1 y por la mañana los días 2 a 4. Dexametasona también se debe administrar por las noches los días 3 y 4. La dosis de dexametasona es responsable de las interacciones del principio activo.
Régimen de quimioterapia moderadamente emetógena
|
Día 1 | |
|
EMEND |
165 mg vía oral |
|
Dexametasona |
12 mg vía oral |
|
Antagonistas 5-HT3 |
Dosis habituales de los antagonistas 5-HT3. Ver la información de producto del antagonista 5-HT3 escogido para obtener información sobre la dosis adecuada |
Dexametasona se debe administrar 30 minutos antes de la quimioterapia el día 1. La dosis de dexametasona es responsable de las interacciones del principio activo.
Los datos de eficacia en combinación con otros corticoesteroides y antagonistas 5-HT3 son limitados. Para información adicional sobre la co-administración con corticoesteroides, ver sección 4.5. Consultar la Ficha Técnica de los medicamentos antagonistas 5-HT3 coadministrados.
Fosaprepitant 150 mg, un profármaco liofilizado de aprepitant para administración intravenosa, está también disponible en dosis única y se puede usar como una alternativa a EMEND 165 mg vía oral.
Poblaciones especiales
Pacientes de edad avanzada (> 65 años)
No es necesario ajustar la dosis en los pacientes de edad avanzada (ver sección 5.2).
Sexo
No es necesario ajustar la dosis según el sexo (ver sección 5.2).
Insuficiencia renal
No e s necesario ajustar la dosis en los pacientes con insuficiencia renal ni en los pacientes con nefropatía terminal sometidos a hemodiálisis (ver sección 5.2).
Insuficiencia hepática
No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. Existen datos limitados en pacientes con insuficiencia hepática moderada y no existen datos en pacientes con insuficiencia hepática grave. Aprepitant se debe usar con precaución en estos pacientes (ver las secciones 4.4 y 5.2).
Población pediátrica
No se ha establecido la seguridad y eficacia de EMEND 165 mg en niños y adolescentes menores de 18 años. No se dispone de datos. Otras formas farmacéuticas/dosis pueden ser más apropiadas para la administración a esta población.
Forma de administración
Las cápsulas duras se deben tragar enteras.
EMEND puede tomarse con o sin alimentos.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Co-administración con pimozida, terfenadina, astemizol o cisaprida (ver sección 4.5).
4.4 Advertencias y precauciones especiales de empleo Pacientes con insuficiencia hepática moderada a grave
Existen datos limitados en pacientes con insuficiencia hepática moderada y no existen datos en pacientes con insuficiencia hepática grave. EMEND se debe usar con precaución en estos pacientes (ver sección 5.2).
Interacciones con el CYP3A4
EMEND se debe usar con precaución en pacientes que estén recibiendo de forma concomitante principios activos metabolizados principalmente a través de CYP3A4 y con un rango terapéutico estrecho, tales como ciclosporina, tacrolimus, sirolimus, everolimus, alfentanilo, alcaloides derivados del ergot, fentanilo y quinidina (ver sección 4.5). Adicionalmente, la administración concomitante con irinotecán se debe abordar con especial prudencia ya que esta combinación puede provocar un aumento de la toxicidad.
Administración conjunta con warfarina (un sustrato CYP2C9)
En pacientes en tratamiento crónico con warfarina, el coeficiente internacional normalizado (INR) se debe vigilar estrechamente durante el tratamiento con EMEND y durante 14 días después del uso de EMEND (ver sección 4.5).
Administración conjunta con anticonceptivos hormonales
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de la administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo (ver sección 4.5).
Excipientes
EMEND cápsulas contiene sacarosa. Los pacientes con intolerancia hereditaria a la fructosa, malabsorción de glucosa o galactosa, o insuficiencia de sacarasa-isomaltasa, no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Aprepitant es un sustrato, un inhibidor moderado, y un inductor de CYP3A4. Aprepitant es también un inductor de CYP2C9. Durante el tratamiento con EMEND, CYP3A4 se inhibe hasta 4 días. EMEND produce una inducción transitoria suave de CYP2C9, de CYP3A4 y glucuronidación, aproximadamente una semana después del tratamiento. Aprepitant no parece que interaccione con el transportador de la P-glucoproteína, como sugiere la falta de interacción de aprepitant con digoxina.
Efecto de aprepitant sobre la farmacocinética de otros principios activos
Inhibición CYP3A4
Como inhibidor moderado de CYP3A4, aprepitant puede aumentar las concentraciones plasmáticas de los principios activos que se metabolizan a través de CYP3A4 cuando se administran conjuntamente. La exposición total de los sustratos de CYP3A4 que se administran por vía oral puede aumentar hasta aproximadamente 3 veces durante 2 días tras el tratamiento con una dosis única de EMEND 165 mg y disminuir hasta la línea basal aproximadamente 4 días después de la dosis de EMEND 165 mg. Se estima que el efecto de aprepitant sobre las concentraciones plasmáticas de los sustratos de CYP3A4 que se administran por vía intravenosa sea menor. EMEND no se debe usar simultáneamente con pimozida, terfenadina, astemizol o cisaprida (ver sección 4.3). La inhibición de CYP3A4 por aprepitant podría dar lugar a elevaciones de las concentraciones plasmáticas de estos principios activos, lo que podría provocar reacciones graves o potencialmente mortales. Se aconseja precaución durante la administración concomitante de EMEND y principios activos metabolizados principalmente a través de CYP3A4 y con un rango terapéutico estrecho, tales como ciclosporina, tacrolimus, sirolimus, everolimus, alfentanilo, diergotamina, ergotamina, fentanilo y quinidina (ver sección 4.4).
Corticoesteroides
Dexametasona: No se han llevado a cabo ensayos de interacción con aprepitant 165 mg y dexametasona; sin embargo, se debe tener en cuenta el siguiente ensayo con 200 mg de aprepitant cuando se use EMEND 165 mg con dexametasona. Aprepitant, administrado como una dosis única de 200 mg en estado alimentado (desayuno ligero estándar) el día 1 con dexametasona oral, administrada concomitantemente vía oral 12 mg el día 1 y 8 mg los días 2 a 4, aumentó el AUC0_24h de dexametasona 2,1 y 2,3 veces los días 1 y 2, en una menor cantidad (aumento de 1,4 veces) el día 3 y sin efecto el día 4 (1,1 veces). La dosis diaria de dexametasona los días 1 y 2 se debe reducir aproximadamente un 50 % cuando se administra conjuntamente con EMEND 165 mg el día 1 para alcanzar exposiciones de dexametasona similares a las obtenidas cuando se administra sin EMEND 165 mg.
Metilprednisolona: No se han llevado a cabo ensayos de interacción con aprepitant 165 mg y metilprednisolona; sin embargo, se debe tener en cuenta el siguiente ensayo con aprepitant 125 mg/80 mg cuando se use EMEND 165 mg con metilprednisolona. EMEND, administrado en un régimen de 125 mg el día 1 y 80 mg/día los días 2 y 3, aumentó el AUC de metilprednisolona, un sustrato de CYP3A4, 1,3 veces el día 1 y 2,5 veces el día 3, al administrarse conjuntamente metilprednisolona por vía intravenosa, 125 mg el día 1, y por vía oral, 40 mg los días 2 y 3.
Durante el tratamiento continuo con metilprednisolona, el AUC de metilprednisolona puede disminuir en puntos de tiempo posteriores en el transcurso de los 14 días siguientes al inicio de la administración de EMEND, debido al efecto inductor de aprepitant sobre CYP3A4. Puede ser que este efecto sea más pronunciado para metilprednisolona administrada oralmente.
Antineoplásicos
No se han llevado a cabo ensayos de interacción con aprepitant 165 mg y antineoplásicos; sin embargo, en base a ensayos con el régimen de 3 días de tratamiento de aprepitant oral y docetaxel y vinorelbina, no se espera que EMEND 165 mg tenga interacciones clínicamente significativas con la administración por vía intravenosa de docetaxel y vinorelbina. En ensayos farmacocinéticos, EMEND, administrado en un régimen de 125 mg el día 1 y 80 mg/día los días 2 y 3, no influyó en la farmacocinética de docetaxel administrado por vía intravenosa el día 1 ni en la de vinorelbina administrada por vía intravenosa el día 1 o el día 8. Debido a que el efecto de EMEND sobre la farmacocinética de sustratos de CYP3A4 administrados por vía oral es mayor que el efecto de
EMEND sobre la farmacocinética de sustratos de CYP3A4 administrados por vía intravenosa, no puede excluirse una interacción con antineoplásicos administrados por vía oral que se metabolizan principal o parcialmente a través de CYP3A4 (p. ej. etopósido, vinorelbina). En pacientes que reciben medicamentos que se metabolizan principal o parcialmente a través de CYP3A4, se aconseja precaución y puede ser conveniente una vigilancia adicional (ver sección 4.4). Se han notificado acontecimientos adversos de neurotoxicidad postcomercialización, una reacción adversa potencial de ifosfamida, tras la administración simultánea de aprepitant e ifosfamida.
Inmunosupresores
Después de una dosis única de aprepitant 165 mg, se espera un incremento moderado transitorio durante dos días seguido de una leve disminución en la exposición de los inmunosupresores metabolizados por CYP3A4 (por ej. ciclosporina, tacrolimus, everolimus y sirolimus). Dada la corta duración de la exposición aumentada, no se recomienda una reducción de dosis de los inmunosupresores en base a la monitorización de la dosis terapéutica en el mismo día y el día después de la administración de EMEND 165 mg.
Midazolam
No se han llevado a cabo ensayos de interacción con aprepitant 165 mg y midazolam; sin embargo, se debe tener en cuenta el siguiente ensayo con 200 mg de aprepitant cuando se use EMEND 165 mg con medicamentos metabolizados vía CYP3A4. En un ensayo con 2 mg de midazolam vía oral administrado conjuntamente con 200 mg de aprepitant en estado alimentado (desayuno ligero estándar), el AUC0-<X) de midazolam, un sustrato CYP3A4 sensible, aumentó 3,2 veces el día 1. No hubo un efecto clínicamente importante el día 4 (aumento del AUC0-<X) de midazolam 1,2 veces) y se observó un cambio ligero del AUC0-a, de midazolam el día 8 (disminución del 35%).
Los posibles efectos de aumentos en las concentraciones plasmáticas de midazolam u otras benzodiazepinas metabolizadas a través de CYP3A4 (alprazolam, triazolam) se deben tener en cuenta al administrar estos medicamentos conjuntamente con EMEND 165 mg.
Inducción
Como inductor suave de CYP2C9, de CYP3A4 y de la glucuronidación, aprepitant puede disminuir las concentraciones plasmáticas de los sustratos eliminados por estas vías. Este efecto puede hacerse evidente aproximadamente 7 días después de la administración de una dosis única de EMEND 165 mg. El efecto se mantiene durante unos pocos días, después desciende lentamente y es clínicamente insignificante a los 14 días después de finalizar el tratamiento con EMEND. Una dosis única de aprepitant 200 mg el día 1 administrada junto con midazolam, un sustrato sensible de CYP3A4, los días 1, 4 y 8 produjo un 35% de reducción del AUC0-m de midazolam el día 8. Se espera que EMEND 165 mg cause una inducción de CYP2C9, CYP3A4 y de la glucuronidación similar a la causada por la administración de un régimen de tratamiento de 3 días de aprepitant oral, para el cual se ha observado una inducción transitoria, con su máximo efecto lo días 6 a 8 después de la primera dosis de aprepitant. El régimen de tratamiento de 3 días con aprepitant oral produjo una reducción de aproximadamente el 30-35% en el AUC de sustratos de CYP2C9 y una disminución de hasta un 64% en las concentraciones mínimas de etinil estradiol. Se carece de datos relativos a los efectos sobre CYP2C8 y CYP2C19. Se aconseja precaución al administrar EMEND 165 mg con warfarina, acenocumarol, tolbutamida, fenitoína u otros principios activos que se sabe que son metabolizados por CYP2C9.
Warfarina
En pacientes en tratamiento crónico con warfarina, el tiempo de protrombina (INR) se debe vigilar estrechamente durante el tratamiento con EMEND 165 mg y durante 14 días después del uso de EMEND 165 mg para las náuseas y vómitos inducidos por quimioterapia (ver sección 4.4). Al administrarse una dosis única de 125 mg de EMEND el día 1 y 80 mg/día los días 2 y 3 a sujetos sanos estabilizados en un tratamiento crónico con warfarina, no se observó ningún efecto de EMEND sobre el AUC plasmático de R(+) o S(-) warfarina determinado el día 3; sin embargo, se observó un descenso del 34% en la concentración mínima de S(-) warfarina (un sustrato de CYP2C9) acompañado de un descenso del 14% en el INR 5 días después de finalizar la administración de EMEND.
Tolbutamida
EMEND, administrado como 125 mg el día 1 y 80 mg/día los días 2 y 3, disminuyó el AUC de tolbutamida (un sustrato de CYP2C9) en un 23% el día 4, un 28% el día 8 y un 15% el día 15, al administrarse una dosis única de tolbutamida 500 mg por vía oral antes de la administración de un régimen de 3 días de EMEND y en los días 4, 8 y 15.
Anticonceptivos hormonales
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de la administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo.
En un ensayo clínico, dosis únicas de un anticonceptivo oral que contenía etinil estradiol y noretindrona se administraron en los días 1 hasta el 21 con EMEND, administrado con una pauta posológica de 125 mg en el día 8 y 80 mg/día en los días 9 y 10 con ondansetrón 32 mg vía intravenosa en el día 8 y dexametasona oral administrada como 12 mg en el día 8 y 8 mg/día los días 9, 10 y 11. Durante los días 9 hasta el 21 en este ensayo, hubo un descenso hasta del 64% en las concentraciones mínimas de etinil estradiol y hasta del 60% en las concentraciones mínimas de noretindrona.
Antagonistas 5-HT3
En ensayos clínicos de interacción, aprepitant, administrado como 125 mg el día 1 y 80 mg los días 2 y 3, no tuvo efectos clínicamente importantes sobre la farmacocinética de ondansetrón, granisetrón ni hidrodolasetrón (el metabolito activo de dolasetrón).
Efecto de otros medicamentos sobre la farmacocinética de aprepitant
La administración concomitante de EMEND con principios activos que inhiben la actividad de CYP3A4 (por ejemplo, ketoconazol, itraconazol, voriconazol, posaconazol, claritromicina, telitromicina, nefazodona e inhibidores de la proteasa) se debe abordar con precaución, ya que la combinación se espera que provoque una elevación de varias veces las concentraciones plasmáticas de aprepitant (ver sección 4.4).
La administración concomitante de EMEND con principios activos que inducen fuertemente la actividad de CYP3A4 (por ejemplo, rifampicina, fenitoína, carbamazepina, fenobarbital) se debe evitar ya que la combinación provoca reducciones en las concentraciones plasmáticas de aprepitant, lo que podría provocar una disminución de la eficacia de EMEND. No se recomienda la administración concomitante de EMEND con plantas medicinales que contienen hipérico (Hypericum perforatum).
Ketoconazol
Al administrarse una dosis única de 125 mg de aprepitant el día 5 de un régimen de 10 días de 400 mg/día de ketoconazol, un potente inhibidor de CYP3A4, el AUC de aprepitant aumentó aproximadamente 5 veces y la semivida terminal media de aprepitant aumentó aproximadamente 3 veces.
Rifampicina
Al administrarse una dosis única de 375 mg de aprepitant el día 9 de un régimen de 14 días de 600 mg/día de rifampicina, un potente inductor de CYP3A4, el AUC de aprepitant disminuyó un 91% y la semivida terminal media disminuyó un 68%.
4.6 Fertilidad, embarazo y lactancia
Anticoncepción en varones y mujeres
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de su administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo (ver las secciones 4.4 y 4.5).
Embarazo
No hay datos clínicos disponibles sobre la exposición a aprepitant en embarazos. La capacidad de aprepitant para provocar toxicidad sobre la reproducción no se ha caracterizado completamente, ya que en los estudios en animales no se pudieron alcanzar niveles de exposición por encima de la exposición terapéutica en seres humanos a la dosis de 125 mg/80 mg y 165 mg. Estos estudios no sugirieron efectos perjudiciales directos ni indirectos en términos del embarazo, el desarrollo embrionario/fetal, el parto o el desarrollo posnatal (ver sección 5.3). Se desconocen los posibles efectos sobre la reproducción de alteraciones en la regulación de la neurocinina. EMEND no se debe utilizar durante el embarazo excepto si fuese claramente necesario.
Lactancia
Aprepitant se excreta en la leche de ratas lactantes. Se desconoce si aprepitant se excreta en la leche materna; por consiguiente, no se recomienda la lactancia durante el tratamiento con EMEND.
Fertilidad
El potencial efecto de aprepitant sobre la fertilidad no se ha caracterizado completamente, ya que en los estudios en animales no se pudieron alcanzar niveles de exposición por encima de la exposición terapéutica en seres humanos. Estos estudios de fertilidad no sugirieron efectos perjudiciales directos ni indirectos en términos del estado de apareamiento, fertilidad, desarrollo embrionario/fetal, o recuento de esperma y movilidad (ver sección 5.3)
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de EMEND sobre la capacidad para conducir y utilizar máquinas es pequeña. Se pueden producir mareos y fatiga después de la administración de EMEND (ver sección 4.8).
4.8 Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad de aprepitant se evaluó en aproximadamente 6.500 adultos.
En base a un perfil comparativo farmacocinético/farmacodinámico, el régimen de tratamiento oral de 1 día de EMEND 165 mg se espera que tenga un perfil de seguridad y tolerabilidad similar al del régimen de tratamiento de 1 día de fosaprepitant 150 mg y al del régimen oral de 3 días de aprepitant en pacientes con quimioterapia (ver sección 5.2).
Las reacciones adversas más frecuentes que se notificaron con una mayor incidencia en adultos tratados con el régimen de 3 días de aprepitant oral que con el tratamiento estándar en pacientes que estaban recibiendo quimioterapia altamente emetógena (HEC) fueron: hipo (4,6 % versus 2,9 %), alanina aminotransferasa elevada (ALT) (2,8 % versus 1,1 %), dispepsia (2,6 % versus 2,0 %), estreñimiento (2,4 % versus 2,0 %), cefalea (2,0 % versus 1,8 %) y apetito disminuido (2,0 % versus 0,5 %). La reacción adversa más frecuente notificada con una mayor incidencia en pacientes tratados con el régimen de 3 días de aprepitant oral que con el tratamiento estándar en pacientes que estaban recibiendo quimioterapia moderadamente emetógena (MEC) fue fatiga (1,4 % versus 0,9 %).
Tabla de reacciones adversas
Las reacciones adversas siguientes se observaron en un análisis combinado de los ensayos en HEC y en MEC con una incidencia mayor con aprepitant que con el tratamiento estándar en adultos o en el uso después de la comercialización:
Las frecuencias se definen como: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000) y muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
|
Sistema de clasificación de órganos |
Reacción adversa |
Frecuencia |
|
Infecciones e infestaciones |
candidiasis, infección estafilocócica |
raras |
|
Trastornos de la sangre y del sistema linfático |
neutropenia febril, anemia |
poco frecuentes |
|
Trastornos del sistema inmunológico |
reacciones de hipersensibilidad incluyendo reacciones anafilácticas |
no conocida |
|
Trastornos del metabolismo y de la nutrición |
apetito disminuido |
frecuentes |
|
polidipsia |
raras | |
|
Trastornos psiquiátricos |
ansiedad |
poco frecuentes |
|
desorientación, estado de ánimo eufórico |
raras | |
|
Trastornos del sistema nervioso |
cefalea |
frecuentes |
|
mareo, somnolencia |
poco frecuentes | |
|
trastorno cognoscitivo, letargia, disgeusia |
raras | |
|
Trastornos oculares |
conjuntivitis |
raras |
|
Trastornos del oído y del laberinto |
acúfenos |
raras |
|
Trastornos cardiacos |
palpitaciones |
poco frecuentes |
|
bradicardia, trastorno cardiovascular |
raras | |
|
Trastornos vasculares |
acaloramiento |
poco frecuentes |
|
Trastornos respiratorios, torácicos y mediastínicos |
hipo |
frecuentes |
|
dolor orofaríngeo, estornudos, tos, goteo postnasal, irritación de garganta |
raras | |
|
Trastornos gastrointestinales |
estreñimiento, dispepsia |
frecuentes |
|
eructos, náuseas*, vómitos*, enfermedad por reflujo gastroesofágico, dolor abdominal, boca seca, flatulencia |
poco frecuentes | |
|
perforación de úlcera de duodeno, estomatitis, distensión abdominal, heces duras, colitis neutropénica |
raras | |
|
Trastornos de la piel y del tejido subcutáneo |
erupción, acné |
poco frecuentes |
|
reacción de fotosensibilidad, hiperhidrosis, seborrea, lesión de la piel, erupción prurítica, síndrome de Stevens-Johnson/necrolisis epidérmica tóxica |
raras | |
|
prurito, urticaria |
no conocida | |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
debilidad muscular, espasmos musculares |
raras |
|
Trastornos renales y urinarios |
disuria |
poco frecuentes |
|
polaquiuria |
raras | |
|
Trastornos generales y alteraciones en el lugar de administración |
fatiga |
frecuentes |
|
astenia, malestar general |
poco frecuentes | |
|
edema, malestar torácico, alteración de la marcha |
raras | |
|
Exploraciones complementarias |
ALT elevada |
frecuentes |
|
AST elevada, fosfatasa alcalina en sangre aumentada |
poco frecuentes | |
|
hematíes en orina positivos, sodio disminuido en sangre, peso disminuido, recuento disminuido de neutrófilos, presencia de glucosuria, excreción urinaria aumentada |
raras |
* Náuseas y vómitos fueron parámetros de eficacia en los 5 primeros días de tratamiento post-quimioterapia y sólo después se notificaron como reacciones adversas.
Descripción de reacciones adversas seleccionadas
Los perfiles de reacciones adversas en adultos en la extensión de Ciclos Múltiples de ensayos en HEC y MEC que se prolongó durante 6 ciclos adicionales de quimioterapia fueron por lo general similares a los observados en el Ciclo 1.
En un ensayo clínico adicional con comparador activo en 1.169 pacientes adultos que estaban recibiendo el régimen de 3 días de aprepitant oral y HEC, el perfil de reacciones adversas fue generalmente similar al observado en los otros ensayos HEC con el régimen de 3 días de aprepitant oral.
Se observaron reacciones adversas adicionales en pacientes tratados con aprepitant para las náuseas y los vómitos posquirúrgicos (NVPQ), con una incidencia mayor que con ondansetrón: dolor en la zona superior del abdomen, ruidos intestinales anormales, estreñimiento*, disartria, disnea, hipoestesia, insomnio, miosis, náuseas, alteración sensitiva, molestias en el estómago, subíleo*, agudeza visual disminuida, sibilancia.
* Notificado en pacientes que tomaron una dosis más alta de aprepitant,
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
En caso de sobredosis, se debe suspender la administración de EMEND y proporcionar tratamiento de apoyo general y vigilancia. Debido a la actividad antiemética de aprepitant, es posible que la emesis inducida por un medicamento no resulte eficaz.
Aprepitant no puede eliminarse mediante hemodiálisis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antieméticos y antinauseosos, código ATC: A04AD12
Aprepitant es un antagonista selectivo de alta afinidad por los receptores de la sustancia P neurocinina 1 (NKi) humana. Fosaprepitant, un profármaco de aprepitant, cuando se administra por vía intravenosa se convierte rápidamente en aprepitant.
En base a un perfil comparativo farmacocinético/farmacodinámico, el régimen de tratamiento oral de 1 día de EMEND 165 mg se espera que tenga un perfil de eficacia similar al del régimen de tratamiento de 1 día de fosaprepitant 150 mg y al del régimen oral de 3 días de aprepitant (ver sección 5.2).
Régimen de tratamiento de 3 días de aprepitant en adultos
En 2 ensayos aleatorizados, doble ciego, en los que se incluyó un total de 1.094 pacientes adultos que recibían quimioterapia que incluía cisplatino >70 mg/m2, aprepitant en combinación con un régimen de ondansetrón/dexametasona (ver sección 4.2) se comparó con un régimen estándar (placebo más ondansetrón 32 mg administrados por vía intravenosa el día 1 más dexametasona 20 mg por vía oral el día 1 y 8 mg por vía oral dos veces al día los días 2 a 4). Aunque en ensayos clínicos se usó una dosis intravenosa de 32 mg de ondansetrón, ésta ya no es la dosis recomendada. Ver la información de producto del antagonista 5-HT3 escogido para obtener información sobre la dosis adecuada.
La eficacia se basó en la evaluación de las siguientes medidas compuestas: respuesta completa (definida como ausencia de episodios eméticos sin uso de tratamiento de rescate) principalmente
durante el Ciclo 1. Los resultados se evaluaron para cada ensayo individual y para los 2 ensayos combinados.
En la Tabla 1 se muestra un resumen de los resultados más importantes del ensayo obtenidos del análisis combinado.
Tabla 1
Porcentaje de pacientes adultos que estaban recibiendo quimioterapia altamente emetógena que respondieron por grupo de tratamiento y fase - Ciclo 1
Régimen con Tratamiento estándar Diferencias*
aprepitant (N= 524) f
MEDIDAS COMPUESTAS (N= 521) f %
%_% (IC del 95%)
|
Respuesta completa (sin emesis y |
sin tratamiento de rescate) | |||
|
Global (0-120 horas) |
67,7 |
47,8 |
19,9 |
(14,0, 25,8) |
|
0-24 horas |
86,0 |
73,2 |
12,7 |
(7,9, 17,6) |
|
25-120 horas |
71,5 |
51,2 |
20,3 |
(14,5, 26,1) |
|
MEDIDAS INDIVIDUALES | ||||
|
Sin emesis (sin episodios eméticos independientemente del |
uso de tratamiento de rescate) | |||
|
Global (0-120 horas) |
71,9 |
49,7 |
22,2 |
(16,4, 28,0) |
|
0-24 horas |
86,8 |
74,0 |
12,7 |
(8,0, 17,5) |
|
25-120 horas |
76,2 |
53,5 |
22,6 |
(17,0, 28,2) |
|
Sin náuseas significativas (EAV máxima < 25 mm en una escala de 0-100 mm) | ||||
|
Global (0-120 horas) |
72,1 |
64,9 |
7,2 |
(1,6, 12,8) |
|
25-120 horas |
74,0 |
66,9 |
7,1 |
(1,5, 12,6) |
*Los intervalos de confianza se calcularon sin ajuste por sexo ni quimioterapia concomitante, los cuales fueron incluidos en el análisis primario de cociente de posibilidades y modelos logísticos. t Sólo un paciente en el régimen de aprepitant tuvo datos en la fase aguda y se excluyó de los análisis de fase retardada y general; sólo un paciente en el régimen estándar tuvo datos en la fase retardada y se excluyó del análisis de fase aguda y general.
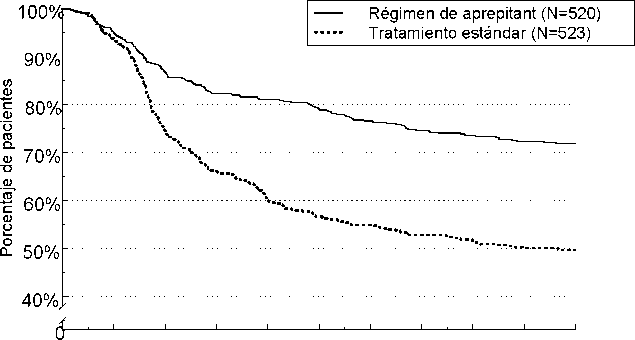
El tiempo estimado hasta la primera emesis en el análisis combinado se representa en el gráfico Kaplan-Meier de la Figura 1.
Figura 1
Porcentaje de pacientes adultos que estaban recibiendo quimioterapia altamente emetógena que siguieron sin padecer emesis con el tiempo - Ciclo 1
0 12 24 36 48 60 72 84 96 108 120
Tiempo (horas)
También se observaron diferencias estadísticamente significativas en eficacia en cada uno de los 2 ensayos individuales.
En los mismos 2 ensayos clínicos, 851 pacientes adultos continuaron en la extensión de Ciclos Múltiples durante 5 ciclos adicionales de quimioterapia. La eficacia del régimen de aprepitant se mantuvo aparentemente durante todos los ciclos.
En un ensayo aleatorizado, doble ciego con un total de 866 pacientes adultos (864 mujeres, 2 varones) que estaban recibiendo quimioterapia, que incluía ciclofosfamida 750-1.500 mg/m2; o ciclofosfamida 500-1.500 mg/m2 y doxorrubicina (<_60 mg/m2) o epirubicina (<_100 mg/m2), se comparó aprepitant en combinación con un régimen de ondansetrón/dexametasona (ver sección 4.2) con el tratamiento estándar (placebo más ondansetrón 8 mg por vía oral (dos veces en el día 1 y cada 12 horas en los días 2 y 3) más dexametasona 20 mg por vía oral en el día 1).
La eficacia se basó en la evaluación de las medidas compuestas: respuesta completa (definida como ausencia de episodios eméticos sin uso de tratamiento de rescate) principalmente durante el Ciclo 1.
En la Tabla 2 se muestra un resumen de los resultados más importantes del ensayo.
Tabla 2
Porcentaje de pacientes adultos que respondieron por grupo de tratamiento y fase - Ciclo 1 Quimioterapia moderadamente emetógena
|
Régimen |
Tratamiento | |
|
con |
estándar | |
|
MEDIDAS COMPUESTAS aprepitant |
(N= 424) | |
|
(N= 433) f |
% |
% (IC del 95%) |
|
% |
Respuesta completa (sin emesis y sin tratamiento de rescate)_
Global (0-120 horas) 50,8 42,5 8,3 (1,6, 15,0)
0-24 horas 75,7 69,0 6,7 (0,7, 12,7)
25-120 horas_55,4_49,1_6,3 (-0,4, 13,0)
MEDIDAS INDIVIDUALES_
Sin emesis (sin episodios eméticos independientemente del uso de tratamiento de rescate)
Global (0-120 horas) 75,7 58,7 17,0 (10,8, 23,2)
0-24 horas 87,5 77,3 10,2 (5,1, 15,3)
25-120 horas_80,8_69,1_11,7 (5,9, 17,5)
Sin náuseas significativas (EAV máxima < 25 mm en una escala de 0-100 mm)_
Global (0-120 horas) 60,9 55,7 5,3 (-1,3, 11,9)
0-24 horas 79,5 78,3 1,3 (-4,2, 6,8)
doxorubicina; ciclofosfamida vía intravenosa (<1.500 mg/m2); o citarabina vía intravenosa (>1 g/m2). Los pacientes que recibían el régimen de aprepitant, estaban recibiendo quimioterapia para diversos tipos de tumores incluyendo 52 % con cáncer de mama, 21 % con algún cáncer de tipo gastrointestinal incluido cáncer colorrectal, 13 % con cáncer de pulmón y 6 % con algún cáncer de tipo ginecológico. El régimen de aprepitant en combinación con un régimen de ondansetrón/dexametasona (ver sección 4.2) se comparó con el tratamiento estándar (placebo en combinación con ondansetrón 8 mg vía oral (dos veces al día el día 1, y cada 12 horas los días 2 y 3) más dexametasona 20 mg vía oral el día 1).
La eficacia se basó en la evaluación de las siguientes variables primaria y secundaria más importantes: Sin vómitos en el periodo completo (0 a 120 horas después de la quimioterapia), evaluación de la seguridad y tolerancia del régimen de aprepitant para las náuseas y los vómitos inducidos por quimioterapia (NVIQ) y respuesta completa (definida como sin vómitos y sin uso de tratamiento de rescate) en el periodo completo (0 a 120 horas después de la quimioterapia). Además, se evaluó la ausencia de náuseas significativas en el periodo completo (0 a 120 horas después de la quimioterapia) como variable exploratoria, y en las fases aguda y retardada como análisis post-hoc.
En la Tabla 3 se muestra un resumen de los resultados más importantes del ensayo.
Tabla 3
Porcentaje de pacientes adultos que respondieron por grupo de tratamiento y fase en el ensayo 2 -
Ciclo 1
Quimioterapia moderadamente emetógena
|
Régimen |
Tratamiento |
Diferencias* |
|
con |
estándar | |
|
aprepitant |
(N= 406) | |
|
(N= 425) |
% |
% (IC del 95 %) |
|
% |
Respuesta completa (sin emesis y sin tratamiento de rescate)
|
Global (0-120 horas) |
68,7 |
56,3 |
12,4 |
(5,9, 18,9) |
|
0-24 horas |
89,2 |
80,3 |
8,9 |
(4,0, 13,8) |
|
25-120 horas |
70,8 |
60,9 |
9,9 |
(3,5, 16,3) |
|
Sin emesis (sin episodios eméticos |
independientemente del uso |
de tratamiento de rescate) | ||
|
Global (0-120 horas) |
76,2 |
62,1 |
14,1 |
(7,9, 20,3) |
|
0-24 horas |
92,0 |
83,7 |
8,3 |
(3,9, 12,7) |
|
25-120 horas |
77,9 |
66,8 |
11,1 |
(5,1, 17,1) |
|
Sin nauseas significativas (EAV máxima < 25 |
mm en una escala de 0-100 mm) | |||
|
Global (0-120 horas) |
73,6 |
66,4 |
7,2 |
(1,0, 13,4) |
|
0-24 horas |
90,9 |
86,3 |
4,6 |
(0,2, 9,0) |
|
25-120 horas |
74,9 |
69,5 |
5,4 |
(-0,7, 11,5) |
*Los intervalos de confianza se calcularon sin ajuste por sexo ni región, los cuales fueron incluidos en el análisis primario utilizando modelos logísticos.
El beneficio del tratamiento en combinación de aprepitant en la población del ensayo total fue dirigido principalmente por los resultados observados en pacientes con bajo control con el régimen estándar como en mujeres, aunque los resultados fueron numéricamente mejores independientemente de la edad, tipo de tumor o sexo. La respuesta completa al régimen de aprepitant y al tratamiento estándar, respectivamente, se alcanzó en 209/324 (65 %) y 161/320 (50 %) en mujeres y 83/101 (82 %) y 68/87 (78 %) de varones.
Régimen de tratamiento de 1 día de fosaprepitant 150 mg en adultos
En un ensayo aleatorizado, paralelo, doble ciego, controlado con comparador activo, fosaprepitant 150 mg (N=1.147) se comparó con un régimen de tratamiento de 3 días con aprepitant (N=1.175) en pacientes adultos que recibían un régimen de HEC que incluía cisplatino (>70 mg/m2). El régimen de tratamiento con fosaprepitant consistió en fosaprepitant 150 mg el día 1 en combinación con ondansetrón 32 mg IV el día 1 y dexametasona 12 mg el día 1, 8 mg el día 2 y 8 mg dos veces al día los días 3 y 4. El régimen de tratamiento con aprepitant consistió en aprepitant 125 mg el día 1 y 80 mg/día los días 2 y 3 en combinación con ondansetrón 32 mg vía intravenosa el día 1 y dexametasona 12 mg el día 1 y 8 mg al día los días 2 a 4. Se usó placebo de fosaprepitant, de aprepitant y de dexametasona (por la noche los días 3 y 4) para mantener el ensayo ciego (ver sección 4.2). Aunque en ensayos clínicos se usó una dosis intravenosa de 32 mg de ondansetrón, ésta ya no es la dosis recomendada. Ver la información de producto del antagonista 5-HT3 escogido para obtener información sobre la dosis adecuada.
La eficacia se basó en la evaluación de las siguientes variables compuestas: respuesta completa tanto en la fase global como en la fase retardada y ausencia de vómitos en la fase global. Fosaprepitant 150 mg demostró no ser inferior al régimen de tratamiento de 3 días de aprepitant. En la Tabla 4 se muestra un resumen de las variables primarias y secundarias.
Tabla 4
Porcentaje de pacientes adultos que reciben quimioterapia altamente emetógena que respondieron
por grupo de tratamiento y fase — Ciclo 1
|
VARIABLES* |
Régimen con fosaprepitant (N =1.106) ** % |
Régimen con aprepitant (N =1.134)** % |
Diferenciasf % (IC del 95 % ) |
|
Respuesta completa1 | |||
|
Global§ |
71,9 |
72,3 |
-0,4 (-4,1, 3,3) |
|
Fase retardada§§ |
74,3 |
74,2 |
0,1 (-3,5, 3,7) |
|
Ausencia de vómitos | |||
|
Global§ |
72,9 |
74,6 |
-1,7 (-5,3, 2,0) |
*La variable primaria está en negrita.
**N: Número de pacientes incluidos en el análisis primario de respuesta completa. f La diferencia e intervalo de confianza (IC) se calcularon utilizando el método propuesto por Miettinen y Nurminen y ajustado por género.
{Respuesta completa = ausencia de vómitos y no uso de terapia de rescate.
§Global = 0 a 120 horas después del inicio de la quimioterapia con cisplatino.
§§Fase retardada = 25 a 120 horas después del inicio de la quimioterapia con cisplatino.
Población pediátrica
Los ensayos clínicos para evaluar el uso de aprepitant en pacientes pediátricos están en curso (ver sección 4.2 para la información sobre el uso pediátrico).
5.2 Propiedades farmacocinéticas
Aprepitant muestra una farmacocinética no lineal. Tanto el aclaramiento como la biodisponibilidad absoluta disminuyen al aumentar la dosis.
Absorción
El AUC0_<x, de aprepitant después de la administración oral de 165 mg fue equivalente al AUC0.^ de fosaprepitant 150 mg administrado por vía intravenosa, mientras que la Cmax fue 2,4 veces más baja.
Después de una dosis única oral de aprepitant 165 mg a voluntarios sanos, el AUC0.^ medio de aprepitant fue de 32,5 pg»h/ml y la concentración máxima media de aprepitant fue 1,67 pg/ml.
La concentración plasmática máxima media (Cmax) de aprepitant se alcanzó aproximadamente a las 4 horas (tmax). La administración oral de la cápsula con un desayuno estándar ligero y un desayuno rico
en grasas ocasionó un aumento de hasta un 8 % y un 47 % en el AUC0-<X) de aprepitant, respectivamente. Este aumento no se consideró clínicamente de interés.
Distribución
Aprepitant se une fuertemente a proteínas, con una media del 97 %. La media geométrica del volumen aparente de distribución en el estado equilibrio (Vdee) es aproximadamente de 66 litros en el ser humano.
Biotransformación
Aprepitant se metaboliza extensamente. En adultos jóvenes sanos, aprepitant representa aproximadamente el 19 % de la radiactividad plasmática durante 72 horas después de una dosis única intravenosa de 100 mg de [C14]-fosaprepitant, un profármaco de aprepitant, lo que indica una importante presencia de metabolitos en el plasma. En el plasma humano se han identificado doce metabolitos de aprepitant. El metabolismo de aprepitant se produce en gran medida por oxidación en el anillo de morfolina y sus cadenas laterales y los metabolitos resultantes sólo fueron débilmente activos. Estudios in vitro en los que se usaron microsomas hepáticos humanos indicaron que aprepitant se metaboliza principalmente a través de CYP3A4 y posiblemente con una contribución menor a través de CYP1A2 y CYP2C19.
Eliminación
Aprepitant no se elimina inalterado en la orina. Los metabolitos se eliminan en la orina y a través de excreción biliar en las heces. Después de una dosis única intravenosa de 100 mg de [C14]-fosaprepitant, un profármaco de aprepitant, a sujetos sanos, el 57 % de la radiactividad se recuperó en la orina y el 45 % en las heces.
El aclaramiento plasmático de aprepitant es dependiente de la dosis, disminuyendo al aumentar la dosis y oscilando aproximadamente entre 60 a 72 ml/min en el intervalo de la dosis terapéutica. La semivida terminal osciló entre aproximadamente 9 a 13 horas.
Farmacocinética en poblaciones especiales
Pacientes de edad avanzada: Tras la administración oral de una dosis única de 125 mg de aprepitant el día 1 y 80 mg una vez al día los días 2 a 5, el AUC0-24h de aprepitant fue un 21 % superior el día 1 y un 36 % superior el día 5 en pacientes de edad avanzada (>65 años) respecto de los adultos más jóvenes. La Cmax fue un 10 % superior el día 1 y un 24 % superior el día 5 en pacientes de edad avanzada respecto de los adultos más jóvenes. Estas diferencias no se consideraron clínicamente significativas. EMEND no requiere ajuste de dosis en los pacientes de edad avanzada.
Sexo: Tras la administración oral de una dosis única de 125 mg de aprepitant, la Cmax de aprepitant es un 16 % superior en las mujeres en comparación con los varones. La semivida de aprepitant es un 25 % inferior en las mujeres en comparación con los varones y su Cax se produce en aproximadamente el mismo tiempo. Estas diferencias no se consideraron clínicamente significativas. EMEND no requiere ajuste de dosis en función del sexo.
Insuficiencia hepática: La insuficiencia hepática leve (Child-Pugh clase A) no afecta a la farmacocinética de aprepitant en un grado clínicamente relevante. No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. De los datos disponibles no pueden extraerse conclusiones relativas a la influencia de la insuficiencia hepática moderada (Child-Pugh clase B) sobre la farmacocinética de aprepitant. No existen datos clínicos ni farmacocinéticos de pacientes con insuficiencia hepática grave (Child-Pugh clase C).
Insuficiencia renal: Se administró una dosis única de 240 mg de aprepitant a pacientes con insuficiencia renal grave (CrCl< 30 ml/min) y a pacientes con nefropatía terminal que requería hemodiálisis.
En los pacientes con insuficiencia renal grave, el AUC0-<X) del aprepitant total (no unido y unido a proteínas) disminuyó en un 21 % y la Cmax disminuyó en un 32 %, respecto de los sujetos sanos. En los pacientes con nefropatía terminal sometidos a hemodiálisis, el AUC0-<X) del aprepitant total disminuyó en un 42 % y la Cmax disminuyó en un 32%. Debido a los modestos descensos en la unión a proteínas de aprepitant en los pacientes con enfermedad renal, el AUC de aprepitant no unido farmacológicamente activo no se vio significativamente afectado en los pacientes con insuficiencia renal en comparación con los sujetos sanos. La hemodiálisis realizada 4 ó 48 horas después de la administración no tuvo efectos significativos sobre la farmacocinética de aprepitant; en el dializado se recuperó menos de 0,2 % de la dosis.
En pacientes con insuficiencia renal o en pacientes con nefropatía terminal sometidos a hemodiálisis no es necesario ajustar la dosis de EMEND.
Relación entre concentración y efecto
Usando un trazador altamente específico del receptor NK1, los estudios de tomografía por emisión de positrones (PET) en varones jóvenes sanos han demostrado que aprepitant penetra en el cerebro y ocupa los receptores NK1 de forma dependiente de la dosis y de la concentración plasmática. Se predice que las concentraciones plasmáticas de aprepitant alcanzadas con el régimen de 3 días de EMEND proporcionarán una ocupación superior al 95 % de los receptores NK1 cerebrales.
Un estudio de tomografía por emisión de positrones (PET) en varones jóvenes sanos a quienes se administró una dosis única oral de 165 mg de aprepitant o una dosis única intravenosa de 150 mg de fosaprepitant demostró una ocupación similar de los receptores NK1 cerebrales a la tmax (> 99 %),
24 horas (> 99 %), 48 horas (> 97 %) y 120 horas (37 a 76 %) después de la dosis. La ocupación de los receptores NK1 cerebrales por aprepitant se correlaciona bien con las concentraciones plasmáticas de aprepitant.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de toxicidad, genotoxicidad, potencial carcinogénico, toxicidad para la reproducción y el desarrollo, a dosis únicas y repetidas. Sin embargo, se deberá tener en cuenta que la exposición sistémica en roedores fue similar o incluso inferior a la exposición terapéutica en seres humanos a las dosis de 125 mg/80 mg y 165 mg. En especial, aunque no se observaron efectos adversos en los estudios sobre la reproducción a los niveles de exposición en seres humanos, las exposiciones en animales no son suficientes para hacer una valoración de riesgo adecuada en el hombre.
En un estudio de toxicidad juvenil en ratas tratadas desde el día 10 después del nacimiento hasta el día 63, aprepitant ocasionó en las hembras una apertura vaginal prematura a partir de 250 mg/kg dos veces al día y en los machos un retraso en la separación del prepucio a partir de 10 mg/kg dos veces al día. No hubo márgenes de exposición clínicamente relevantes. No se observaron efectos sobre el apareamiento, la fertilidad o la supervivencia embrionaria o fetal ni cambios patológicos en los órganos reproductores relacionados con el tratamiento. En un estudio de toxicidad juvenil en perros tratados desde el día 14 después del nacimiento hasta el día 42, se detectó una disminución del peso testicular y del tamaño de las células de Leydig en los machos con 6 mg/kg/día y un aumento del peso del útero, hipertrofia del útero y del cuello del útero y edema de los tejidos vaginales en las hembras a partir de 4 mg/kg/día. No hubo márgenes de exposición clínicamente relevantes de aprepitant. En el tratamiento a corto plazo de acuerdo con la pauta posológica recomendada, se considera poco probable que estos resultados sean clínicamente relevantes.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Contenido de la cápsula Sacarosa
Celulosa microcristalina (E-460) Hidroxipropilcelulosa (E-463)
Lauril sulfato de sodio
Cubierta de la cápsula Gelatina
Dióxido de titanio (E-171)
Carmín índigo (E-132)
Tinta para impresión Laca
Hidróxido de potasio Óxido de hierro negro (E-172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
4 años.
6.4 Precauciones especiales de conservación
Conservar en el embalaje original para protegerlo de la humedad.
6.5 Naturaleza y contenido del envase
Blister de aluminio que contiene una cápsula de 165 mg.
6 blisters de aluminio que contienen una cápsula de 165 mg cada uno.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
Ninguna especial para su eliminación.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme Ltd.
Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/262/009
EU/1/03/262/010
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 11/noviembre/2003 Fecha de la última renovación: 22/septiembre/2008
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
EMEND 125 mg cápsulas duras EMEND 80 mg cápsulas duras
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula de 125 mg contiene 125 mg de aprepitant. Cada cápsula de 80 mg contiene 80 mg de aprepitant.
Excipiente con efecto conocido
Cada cápsula contiene 125 mg de sacarosa (en la cápsula de 125 mg).
Excipiente con efecto conocido
Cada cápsula contiene 80 mg de sacarosa (en la cápsula de 80 mg).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura.
La cápsula de 125 mg es opaca con cuerpo blanco y tapa rosa con “462” y “125 mg” impreso en forma radial en tinta negra en el cuerpo. Las cápsulas de 80 mg son opacas con cuerpo y tapa blancos con “461” y “80 mg” impresos en forma radial en tinta negra en el cuerpo.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Prevención de las náuseas y los vómitos que se asocian con la quimioterapia antineoplásica moderada y altamente emetógena en adultos y adolescentes a partir de 12 años de edad.
EMEND 125 mg/80 mg se administra como parte de un tratamiento de combinación (ver sección 4.2).
4.2 Posología y forma de administración
Posología
Adultos
EMEND se administra durante 3 días como parte de un régimen que incluye un corticoesteroide y un antagonista 5-HT3. La dosis recomendada es de 125 mg por vía oral una vez al día una hora antes de empezar la quimioterapia el día 1 y 80 mg por vía oral, una vez al día por la mañana, los días 2 y 3.
Se recomiendan los siguientes regímenes de administración en adultos para la prevención de las náus eas y los vómitos asociados con la quimioterapia antineoplásica emetógena:
Régimen de quimioterapia altamente emetógena
|
Día 1 |
Día 2 |
Día 3 |
Día 4 | |
|
EMEND |
125 mg vía oral |
80 mg vía oral |
80 mg vía oral |
nada |
|
Dexametasona |
12 mg vía oral |
8 mg vía oral |
8 mg vía oral |
8 mg vía oral |
|
Antagonistas 5-HT3 |
Dosis habituales de los antagonistas 5-HT3. Ver la información de producto del antagonista 5-HT3 escogido para obtener información sobre la dosis adecuada |
nada |
nada |
nada |
|
Dexametasona se d |
ebe administrar30 minutos antes de la quimioterapia el día 1 y por la mañana los | |||
días 2 a 4. La dosis de dexametasona es responsable de las interacciones del principio activo.
Régimen de quimioterapia moderadamente emetógena
|
Día 1 |
Día 2 |
Día 3 | |
|
EMEND |
125 mg vía oral |
80 mg vía oral |
80 mg vía oral |
|
Dexametasona |
12 mg vía oral |
nada |
nada |
|
Antagonistas 5-HT3 |
Dosis habituales de los antagonistas 5-HT3. Ver la información de producto del antagonista 5-HT3 escogido para obtener información sobre la dosis adecuada |
nada |
nada |
Dexametasona se debe administrar 30 minutos antes de la quimioterapia el día 1. La dosis de dexametasona es responsable de las interacciones del principio activo.
Población pediátrica
Adolescentes (de entre 12 y 17 años de edad)
EMEND se administra durante 3 días como parte de un régimen que incluye un antagonista 5-HT3. La dosis recomendada de EMEND cápsulas es de 125 mg por vía oral el día 1 y 80 mg por vía oral los días 2 y 3. EMEND se administra por vía oral 1 hora antes de la quimioterapia los días 1,2 y 3. Si los días 2 y 3 no se administra quimioterapia, EMEND se debe administrar por la mañana. Consultar la Ficha Técnica del antagonista 5-HT3 seleccionado para obtener información sobre la dosis adecuada. Si junto con EMEND se administra un corticosteroide, como dexametasona, la dosis del corticosteroide debe ser un 50% de la dosis habitual (ver las secciones 4.5 y 5.1).
No se ha establecido la seguridad y eficacia de las cápsulas de 80 mg y 125 mg en niños menores de 12 años. No se dispone de datos. Consultar la Ficha técnica del polvo para suspensión oral para la dosis adecuada en lactantes y niños de entre 6 meses y menos de 12 años de edad.
General
Los datos de eficacia en combinación con otros corticoesteroides y antagonistas 5HT3 son limitados. Para información adicional sobre la co-administración con corticoesteroides, ver sección 4.5. Consultar la Ficha Técnica de los medicamentos antagonistas 5-HT3 coadministrados.
Poblaciones especiales
Pacientes de edad avanzada (>65 años)
No es necesario ajustar la dosis en los pacientes de edad avanzada (ver sección 5.2).
Sexo
No es necesario ajustar la dosis según el sexo (ver sección 5.2).
Insuficiencia renal
No es necesario ajustar la dosis en los pacientes con insuficiencia renal ni en los pacientes con nefropatía terminal sometidos a hemodiálisis (ver sección 5.2).
Insuficiencia hepática
No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. Existen datos limitados en pacientes con insuficiencia hepática moderada y no existen datos en pacientes con insuficiencia hepática grave. Aprepitant se debe usar con precaución en estos pacientes (ver las secciones 4.4 y 5.2).
Forma de administración
Las cápsulas duras se deben tragar enteras.
EMEND puede tomarse con o sin alimentos.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Co-administración con pimozida, terfenadina, astemizol o cisaprida (ver sección 4.5).
4.4 Advertencias y precauciones especiales de empleo Pacientes con insuficiencia hepática moderada a grave
Existen datos limitados en pacientes con insuficiencia hepática moderada y no existen datos en pacientes con insuficiencia hepática grave. EMEND se debe usar con precaución en estos pacientes (ver sección 5.2).
Interacciones con el CYP3A4
EMEND se debe usar con precaución en pacientes que estén recibiendo de forma concomitante principios activos, administrados por vía oral, metabolizados principalmente a través de CYP3A4 y con un rango terapéutico estrecho, tales como ciclosporina, tacrolimus, sirolimus, everolimus, alfentanilo, alcaloides derivados del ergot, fentanilo y quinidina (ver sección 4.5). Adicionalmente, la administración concomitante con irinotecán se debe abordar con especial prudencia ya que esta combinación puede provocar un aumento de la toxicidad.
Administración conjunta con warfarina (un sustrato de CYP2C9)
En pacientes en tratamiento crónico con warfarina, el coeficiente internacional normalizado (INR) se debe vigilar estrechamente durante el tratamiento con EMEND y durante 14 días después de cada ciclo de 3 días de EMEND (ver sección 4.5).
Administración conjunta con anticonceptivos hormonales
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de la administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo (ver sección 4.5).
Excipientes
EMEND cápsulas contiene sacarosa. Los pacientes con intolerancia hereditaria a la fructosa, problemas de absorción de glucosa o galactosa, o insuficiencia de sacarasa-isomaltasa, no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Aprepitant (125 mg/80 mg) es un sustrato, un inhibidor moderado, y un inductor de CYP3A4. Aprepitant es también un inductor de CYP2C9. Durante el tratamiento con EMEND, CYP3A4 se inhibe. Después de terminar el tratamiento, EMEND produce una inducción transitoria suave de CYP2C9, de CYP3A4 y glucuronidación. Aprepitant no parece que interaccione con el transportador de la P-glucoproteína, como sugiere la falta de interacción de aprepitant con digoxina.
Efecto de aprepitant sobre la farmacocinética de otros principios activos
Inhibición de CYP3A4
Como inhibidor moderado de CYP3A4, aprepitant (125 mg/80 mg) puede aumentar las concentraciones plasmáticas de los principios activos que se metabolizan a través de CYP3A4 cuando se administran conjuntamente. La exposición total de los sustratos de CYP3A4 que se administran por vía oral puede aumentar hasta aproximadamente tres veces durante el tratamiento de 3 días con EMEND; se estima que el efecto de aprepitant sobre las concentraciones plasmáticas de los sustratos de CYP3A4 que se administran por vía intravenosa sea menor. EMEND no se debe usar simultáneamente con pimozida, terfenadina, astemizol o cisaprida (ver sección 4.3). La inhibición de CYP3A4 por aprepitant podría dar lugar a elevaciones de las concentraciones plasmáticas de estos principios activos, lo que podría provocar reacciones graves o potencialmente mortales. Se aconseja precaución durante la administración concomitante de EMEND y principios activos, administrados por vía oral, metabolizados principalmente a través de CYP3A4 y con un rango terapéutico estrecho, tales como ciclosporina, tacrolimus, sirolimus, everolimus, alfentanilo, diergotamina, ergotamina, fentanilo y quinidina (ver sección 4.4).
Corticoesteroides
Dexametasona: La dosis habitual de dexametasona oral se debe reducir aproximadamente en un 50% cuando se administra conjuntamente con un tratamiento de 125 mg/80 mg de EMEND. La dosis de dexametasona utilizada en los ensayos clínicos de náuseas y vómitos inducidos por quimioterapia, se eligió en función de las interacciones del principio activo (ver sección 4.2). EMEND, administrado en un régimen de 125 mg conjuntamente con 20 mg de dexametasona oral el día 1, y EMEND, administrado en un régimen de 80 mg/día conjuntamente con 8 mg de dexametasona oral los días 2 a 5, aumentó el AUC de dexametasona, un sustrato de CYP3A4, 2,2 veces los días 1 y 5.
Metilprednisolona: La dosis habitual de metilprednisolona intravenosa se debe reducir aproximadamente un 25%, y la dosis habitual de metilprednisolona oral se debe reducir aproximadamente un 50% al administrarse conjuntamente con un tratamiento de 125 mg/80 mg de EMEND. EMEND, administrado en un régimen de 125 mg el día 1 y 80 mg/día los días 2 y 3, aumentó el AUC de metilprednisolona, un sustrato de CYP3A4, 1,3 veces el día 1 y 2,5 veces el día 3, al administrarse conjuntamente metilprednisolona por vía intravenosa, 125 mg el día 1, y por vía oral, 40 mg los días 2 y 3.
Durante el tratamiento continuo con metilprednisolona, el AUC de metilprednisolona puede disminuir en puntos de tiempo posteriores en el transcurso de las 2 semanas siguientes al inicio de la administración de EMEND, debido al efecto inductor de aprepitant sobre CYP3A4. Puede ser que este efecto sea más pronunciado para metilprednisolona administrada oralmente.
Antineoplásicos
En ensayos farmacocinéticos, EMEND, administrado en un régimen de 125 mg el día 1 y 80 mg/día los días 2 y 3, no influyó en la farmacocinética de docetaxel administrado por vía intravenosa el día 1 ni en la de vinorelbina administrada por vía intravenosa el día 1 o el día 8. Debido a que el efecto de EMEND sobre la farmacocinética de sustratos de CYP3A4 administrados por vía oral es mayor que el efecto de EMEND sobre la farmacocinética de sustratos de CYP3A4 administrados por vía intravenosa, no puede excluirse una interacción con medicamentos antineoplásicos administrados por vía oral que se metabolizan principal o parcialmente a través de CYP3A4 (p. ej. etopósido, vinorelbina). En pacientes que reciben medicamentos que se metabolizan principal o parcialmente a través de CYP3A4, se aconseja precaución y puede ser conveniente una vigilancia adicional (ver
sección 4.4). Se han notificado acontecimientos adversos de neurotoxicidad postcomercialización, una reacción adversa potencial de ifosfamida, tras la administración simultánea de aprepitant e ifosfamida.
Inmunosupresores
Durante el régimen de tratamiento de 3 días de las náuseas y vómitos inducidos por quimioterapia, se espera un incremento moderado transitorio seguido de una leve disminución en la exposición de los inmunosupresores metabolizados por CYP3A4 (por ej. ciclosporina, tacrolimus, everolimus y sirolimus). Dada la corta duración del régimen de tratamiento de 3 días y los cambios limitados en la exposición dependientes del tiempo, no se recomienda una reducción de dosis de los inmunosupresores durante los 3 días de la administración conjunta con EMEND.
Midazolam
Los posibles efectos de aumentos en las concentraciones plasmáticas de midazolam u otras benzodiazepinas metabolizadas a través de CYP3A4 (alprazolam, triazolam) se deben tener en cuenta al administrar estos medicamentos conjuntamente con EMEND (125 mg/80 mg).
EMEND aumentó el AUC de midazolam, un sustrato sensible de CYP3A4, 2,3 veces el día 1 y
3,3 veces el día 5, al administrarse conjuntamente una dosis oral única de 2 mg de midazolam los días 1 y 5 de un régimen de EMEND 125 mg el día 1 y 80 mg/día los días 2 a 5.
En otro ensayo con administración intravenosa de midazolam, EMEND se administró como 125 mg el día 1 y 80 mg/día los días 2 y 3, y 2 mg de midazolam se administró por vía intravenosa antes de la administración del régimen de 3 días de EMEND y en los días 4, 8 y 15. EMEND aumentó el AUC de midazolam un 25% el día 4 y disminuyó el AUC de midazolam un 19% el día 8 y un 4% el día 15. Estos efectos no se consideraron clínicamente importantes.
En un tercer ensayo con administración intravenosa y oral de midazolam, se administraron 125 mg de EMEND en el día 1 y 80 mg/día en los días 2 y 3, junto con 32 mg de ondansetrón el día 1, 12 mg de dexametasona el día 1 y 8 mg los días 2-4. Esta combinación (esto es, EMEND, ondansetrón y dexametasona) disminuyó el AUC de midazolam oral un 16 % el día 6, un 9 % el día 8, un 7 % el día 15 y un 17 % el día 22. Estos efectos no se consideraron clínicamente importantes.
Se finalizó un ensayo adicional con administración intravenosa de midazolam y EMEND. Una hora después de la administración oral de una dosis única de EMEND 125 mg, se administraron por vía intravenosa 2 mg de midazolam. El AUC plasmático de midazolam aumentó en 1,5 veces. Este efecto no se consideró clínicamente importante.
Inducción
Como inductor suave de CYP2C9, de CYP3A4 y de la glucuronidación, aprepitant puede disminuir las concentraciones plasmáticas de los sustratos eliminados por estas vías durante las dos semanas posteriores al inicio del tratamiento. Este efecto puede hacerse evidente únicamente después de finalizar el tratamiento de 3 días con EMEND. Para los sustratos de CYP2C9 y CYP3A4, la inducción es transitoria con un efecto máximo alcanzado a los 3-5 días después de finalizar el tratamiento de 3 días con EMEND. El efecto se mantiene durante unos pocos días, después desciende lentamente y es clínicamente insignificante a las 2 semanas después de finalizar el tratamiento con EMEND. La inducción suave de la glucuronidación también se observa con 80 mg de aprepitant oral administrado durante 7 días. Se carece de datos relativos a los efectos sobre CYP2C8 y CYP2C19. Se aconseja precaución al administrar durante este periodo de tiempo warfarina, acenocumarol, tolbutamida, fenitoína u otros principios activos que se sabe que son metabolizados por CYP2C9.
Warfarina
En pacientes en tratamiento crónico con warfarina, el tiempo de protrombina (INR) se debe vigilar estrechamente durante el tratamiento con EMEND y durante 2 semanas después de cada ciclo de 3 días de EMEND en el caso de náuseas y vómitos inducidos por quimioterapia (ver sección 4.4). Al administrarse una dosis única de 125 mg de EMEND el día 1 y 80 mg/día los días 2 y 3 a sujetos sanos estabilizados en un tratamiento crónico con warfarina, no se observó ningún efecto de EMEND sobre el AUC plasmático de R(+) o S(-) warfarina determinado el día 3; sin embargo, se observó un descenso del 34% en la concentración mínima de S(-) warfarina (un sustrato de CYP2C9) acompañado de un descenso del 14% en el INR 5 días después de finalizar el tratamiento con EMEND.
Tolbutamida
EMEND, administrado como 125 mg el día 1 y 80 mg/día los días 2 y 3, disminuyó el AUC de tolbutamida (un sustrato de CYP2C9) en un 23% el día 4, un 28% el día 8 y un 15% el día 15, al administrarse una dosis única de tolbutamida 500 mg por vía oral antes de la administración de un régimen de 3 días de EMEND y en los días 4, 8 y 15.
Anticonceptivos hormonales
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de la administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo.
En un ensayo clínico, dosis únicas de un anticonceptivo oral que contenía etinil estradiol y noretindrona se administraron en los días 1 hasta el 21 con EMEND, administrado con una pauta posológica de 125 mg en el día 8 y 80 mg/día en los días 9 y 10 con ondansetrón 32 mg vía intravenosa en el día 8 y dexametasona oral administrada como 12 mg en el día 8 y 8 mg/día los días 9, 10 y 11. Durante los días 9 hasta el 21 en este ensayo, hubo un descenso hasta del 64% en las concentraciones mínimas de etinil estradiol y hasta del 60% en las concentraciones mínimas de noretindrona.
Antagonistas 5-HT3
En ensayos clínicos de interacción, aprepitant no tuvo efectos clínicamente importantes sobre la farmacocinética de ondansetrón, granisetrón ni hidrodolasetrón (el metabolito activo de dolasetrón).
Efecto de otros medicamentos sobre la farmacocinética de aprepitant
La administración concomitante de EMEND con principios activos que inhiben la actividad de CYP3A4 (por ejemplo, ketoconazol, itraconazol, voriconazol, posaconazol, claritromicina, telitromicina, nefazodona e inhibidores de la proteasa) se debe abordar con precaución, ya que la combinación se espera que provoque una elevación de varias veces las concentraciones plasmáticas de aprepitant (ver sección 4.4).
La administración concomitante de EMEND con principios activos que inducen fuertemente la actividad de CYP3A4 (por ejemplo, rifampicina, fenitoína, carbamazepina, fenobarbital) se debe evitar ya que la combinación provoca reducciones en las concentraciones plasmáticas de aprepitant lo que podría provocar una disminución de la eficacia de EMEND. No se recomienda la administración concomitante de EMEND con plantas medicinales que contienen hipérico (Hypericum perforatum).
Ketoconazol
Al administrarse una dosis única de 125 mg de aprepitant el día 5 de un régimen de 10 días de 400 mg/día de ketoconazol, un potente inhibidor de CYP3A4, el AUC de aprepitant aumentó aproximadamente 5 veces y la semivida terminal media de aprepitant aumentó aproximadamente 3 veces.
Rifampicina
Al administrarse una dosis única de 375 mg de aprepitant el día 9 de un régimen de 14 días de 600 mg/día de rifampicina, un potente inductor de CYP3A4, el AUC de aprepitant disminuyó un 91% y la semivida terminal media disminuyó un 68%.
Población pediátrica
Los estudios de interacciones se han realizado sólo en adultos.
4.6 Fertilidad, embarazo y lactancia
Anticoncepción en varones y mujeres
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de su administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo (ver las secciones 4.4 y 4.5).
Embarazo
No hay datos clínicos disponibles sobre la exposición a aprepitant durante el embarazo. La capacidad de aprepitant para provocar toxicidad sobre la reproducción no se ha caracterizado completamente, ya que en los estudios animales no se pudieron alcanzar niveles de exposición por encima de la exposición terapéutica en seres humanos a la dosis de 125 mg/80 mg. Estos estudios no sugirieron efectos perjudiciales directos ni indirectos en términos del embarazo, el desarrollo embrionario/fetal, el parto o el desarrollo posnatal (ver sección 5.3). Se desconocen los posibles efectos sobre la reproducción de alteraciones en la regulación de la neurocinina. EMEND no se debe utilizar durante el embarazo excepto si fuese claramente necesario.
Lactancia
Aprepitant se excreta en la leche de ratas lactantes. Se desconoce si aprepitant se excreta en la leche materna; por consiguiente, no se recomienda la lactancia durante el tratamiento con EMEND.
Fertilidad
El potencial efecto de aprepitant sobre la fertilidad no se ha caracterizado completamente, ya que en los estudios en animales no se pudieron alcanzar niveles de exposición por encima de la exposición terapéutica en humanos. Estos estudios de fertilidad no sugirieron efectos perjudiciales directos ni indirectos en términos del estado de apareamiento, fertilidad, desarrollo embrionario/fetal, o recuento de esperma y movilidad (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de EMEND sobre la capacidad para conducir, ir en bicicleta y utilizar máquinas es pequeña. Se pueden producir mareos y fatiga después de la administración de EMEND (ver sección 4.8).
4.8 Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad de aprepitant se evaluó en aproximadamente 6.500 adultos en más de 50 ensayos y 184 niños y adolescentes en dos ensayos clínicos pediátricos pivotales.
Las reacciones adversas más frecuentes que se notificaron con una mayor incidencia en adultos tratados con el régimen de aprepitant que con el tratamiento estándar en pacientes que estaban recibiendo quimioterapia altamente emetógena (HEC) fueron: hipo (4,6 % versus 2,9 %), alanina aminotransferasa (ALT) elevada (2,8 % versus 1,1 %), dispepsia (2,6 % versus 2,0 %), estreñimiento (2,4 % versus 2,0 %), cefalea (2,0 % versus 1,8 %) y apetito disminuido (2,0 % versus 0,5 %). La reacción adversa más frecuente notificada con una mayor incidencia en pacientes tratados con el régimen de aprepitant que con el tratamiento estándar en pacientes que estaban recibiendo quimioterapia moderadamente emetógena (MEC) fue fatiga (1,4 % versus 0,9 %).
Las reacciones adversas más frecuentes que se notificaron con una mayor incidencia en pacientes pediátricos tratados con el régimen de aprepitant que con el régimen de control mientras recibían quimioterapia antineoplásica emetógena fueron: hipo (3,3 % versus 0,0 %) y sofocos (1,1 % versus 0,0 %).
Tabla de reacciones adversas
Las reacciones adversas siguientes se observaron en un análisis combinado de los ensayos en HEC y en MEC con una incidencia mayor con aprepitant que con el tratamiento estándar en adultos o en pacientes pediátricos o en el uso después de la comercialización. Las categorías de frecuencia dadas en la tabla se basan en los ensayos en adultos; las frecuencias observadas en los ensayos pediátricos fueron similares o menores, a menos que se muestre en la tabla. Algunas reacciones adversas menos frecuentes en la población adulta no se observaron en los ensayos pediátricos.
Las frecuencias se definen como: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000) y muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
|
Sistema de clasificación de órganos |
Reacción adversa |
Frecuencia |
|
Infecciones e infestaciones |
candidiasis, infección estafilocócica |
raras |
|
Trastornos de la sangre y del sistema linfático |
neutropenia febril, anemia |
poco frecuentes |
|
Trastornos del sistema inmunológico |
reacciones de hipersensibilidad incluyendo reacciones anafilácticas |
no conocida |
|
Trastornos del metabolismo y de la nutrición |
apetito disminuido |
frecuentes |
|
polidipsia |
raras | |
|
Trastornos psiquiátricos |
ansiedad |
poco frecuentes |
|
desorientación, estado de ánimo eufórico |
raras | |
|
Trastornos del sistema nervioso |
cefalea |
frecuentes |
|
mareo, somnolencia |
poco frecuentes | |
|
trastorno cognoscitivo, letargia, disgeusia |
raras | |
|
Trastornos oculares |
conjuntivitis |
raras |
|
Trastornos del oído y del laberinto |
acúfenos |
raras |
|
Trastornos cardiacos |
palpitaciones |
poco frecuentes |
|
bradicardia, trastorno cardiovascular |
raras | |
|
Trastornos vasculares |
acaloramiento/rubefacción |
poco frecuentes |
|
Trastornos respiratorios, torácicos y mediastínicos |
hipo |
frecuentes |
|
dolor orofaríngeo, estornudos, tos, goteo postnasal, irritación de garganta |
raras | |
|
Trastornos gastrointestinales |
estreñimiento, dispepsia |
frecuentes |
|
eructos, náuseasT, vómitosT, enfermedad por reflujo gastroesofágico, dolor abdominal, boca seca, flatulencia |
poco frecuentes | |
|
perforación de úlcera de duodeno, estomatitis, distensión abdominal, heces duras, colitis neutropénica |
raras | |
|
Trastornos de la piel y del tejido subcutáneo |
erupción, acné |
poco frecuentes |
|
reacción de fotosensibilidad, hiperhidrosis, seborrea, lesión de la piel, erupción prurítica, síndrome de Stevens-Johnson/necrolisis epidérmica tóxica |
raras | |
|
prurito, urticaria |
no conocida | |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
debilidad muscular, calambres musculares |
raras |
|
Trastornos renales y urinarios |
disuria |
poco frecuentes |
|
polaquiuria |
raras | |
|
Trastornos generales y alteraciones en el lugar de |
fatiga |
frecuentes |
|
astenia, malestar general |
poco frecuentes |
|
Sistema de clasificación de órganos |
Reacción adversa |
Frecuencia |
|
administración |
edema, malestar torácico, alteración de la marcha |
raras |
|
Exploraciones complementarias |
ALT elevada |
frecuentes |
|
AST elevada, fosfatasa alcalina en sangre aumentada |
poco frecuentes | |
|
hematíes en orina positivos, sodio disminuido en sangre, peso disminuido, recuento disminuido de neutrófilos, presencia de glucosuria, excreción urinaria aumentada |
raras |
T Náuseas y vómitos fueron parámetros de eficacia en los 5 primeros días de tratamiento post-quimioterapia y sólo después se notificaron como reacciones adversas.
Descripción de reacciones adversas seleccionadas
Los perfiles de reacciones adversas en adultos en la extensión de Ciclos Múltiples de ensayos en HEC y MEC que se prolongó durante 6 ciclos adicionales de quimioterapia fueron por lo general similares a los observados en el Ciclo 1.
En un ensayo clínico adicional con comparador activo en 1.169 pacientes adultos que estaban recibiendo aprepitant y HEC, el perfil de reacciones adversas fue generalmente similar al observado en los otros ensayos HEC con aprepitant.
Se observaron otras reacciones adversas en pacientes adultos tratados con aprepitant para las náuseas y los vómitos posquirúrgicos (NVPQ), con una incidencia mayor que con ondansetrón: dolor en la zona superior del abdomen, ruidos intestinales anormales, estreñimiento*, disartria, disnea, hipoestesia, insomnio, miosis, náuseas, alteración sensitiva, molestias en el estómago, subíleo*, agudeza visual disminuida, sibilancia.
*Notificado en pacientes que tomaron una dosis más alta de aprepitant.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
En caso de sobredosis, se debe suspender la administración de EMEND y proporcionar tratamiento de apoyo general y vigilancia. Debido a la actividad antiemética de aprepitant, es posible que la emesis inducida por un medicamento no resulte eficaz.
Aprepitant no puede eliminarse mediante hemodiálisis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antieméticos y antinauseosos, código ATC: A04AD12
Aprepitant es un antagonista selectivo de alta afinidad por los receptores de la sustancia P neurocinina 1 (NKi) humana.
Régimen de tratamiento de 3 días de aprepitant en adultos
En 2 ensayos aleatorizados, doble ciego, en los que se incluyó un total de 1.094 pacientes adultos que recibían quimioterapia que incluía cisplatino > 70 mg/m2, aprepitant en combinación con un régimen
de ondansetrón/dexametasona (ver sección 4.2) se comparó con un régimen estándar (placebo más ondansetrón 32 mg administrados por vía intravenosa el día 1 más dexametasona 20 mg por vía oral el día 1 y 8 mg por vía oral dos veces al día los días 2 a 4). Aunque en ensayos clínicos se usó una dosis intravenosa de 32 mg de ondansetrón, ésta ya no es la dosis recomendada. Ver la información de producto del antagonista 5-HT3 escogido para obtener información sobre la dosis adecuada.
La eficacia se basó en la evaluación de las siguientes medidas compuestas: respuesta completa (definida como ausencia de episodios eméticos sin uso de tratamiento de rescate) principalmente durante el Ciclo 1. Los resultados se evaluaron para cada ensayo individual y para los 2 ensayos combinados.
En la Tabla 1 se muestra un resumen de los resultados más importantes del ensayo obtenidos del análisis combinado.
Tabla 1
Porcentaje de pacientes adultos que estaban recibiendo quimioterapia altamente emetógena que respondieron por grupo de tratamiento y fase - Ciclo 1
Régimen con Tratamiento estándar Diferencias*
aprepitant (N= 524)f
MEDIDAS COMPUESTAS (N= 521)f % % (IC del 95%)
_%_
Respuesta completa (sin emesis y sin tratamiento de rescate)_
|
Global (0-120 horas) |
67,7 |
47,8 |
19,9 |
(14,0, 25,8) |
|
0-24 horas |
86,0 |
73,2 |
12,7 |
(7,9, 17,6) |
|
25-120 horas |
71,5 |
51,2 |
20,3 |
(14,5, 26,1) |
MEDIDAS INDIVIDUALES
|
Sin emesis (sin episodios eméticos |
independientemente del |
uso de tratamiento de |
rescate) | |
|
Global (0-120 horas) |
71,9 |
49,7 |
22,2 |
(16,4, 28,0) |
|
0-24 horas |
86,8 |
74,0 |
12,7 |
(8,0, 17,5) |
|
25-120 horas |
76,2 |
53,5 |
22,6 |
(17,0, 28,2) |
|
Sin náuseas significativas (EAV máxima < 25 mm en una escala de 0- |
100 mm) | |||
|
Global (0-120 horas) |
72,1 |
64,9 |
7,2 |
(1,6, 12,8) |
|
25-120 horas |
74,0 |
66,9 |
7,1 |
(1,5, 12,6) |
*Los intervalos de confianza se calcularon sin ajuste por sexo ni quimioterapia concomitante, los cuales fueron incluidos en el análisis primario de cociente de posibilidades y modelos logísticos. t Sólo un paciente en el régimen de aprepitant tuvo datos en la fase aguda y se excluyó de los análisis de fase retardada y general; sólo un paciente en el régimen estándar tuvo datos en la fase retardada y se excluyó del análisis de fase aguda y general.
El tiempo estimado hasta la primera emesis en el análisis combinado se representa en el gráfico Kapl an-Meier de la Figura 1.
Figura 1
Porcentaje de pacientes adultos que estaban recibiendo quimioterapia altamente emetógena que siguieron sin padecer emesis con el tiempo - Ciclo 1
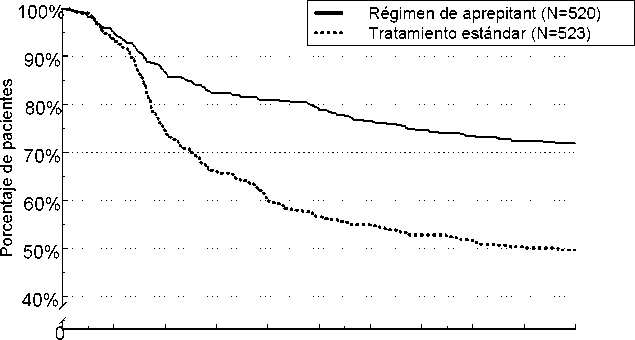
0 12 24 36 48 60 72 84 96 108 120
Tiempo (horas)
También se observaron diferencias estadísticamente significativas en eficacia en cada uno de los 2 ensayos individuales.
En los mismos 2 ensayos clínicos, 851 pacientes adultos continuaron en la extensión de Ciclos Múltiples durante 5 ciclos adicionales de quimioterapia. La eficacia del régimen de aprepitant se mantuvo aparentemente durante todos los ciclos.
En un ensayo aleatorizado, doble ciego con un total de 866 pacientes adultos (864 mujeres, 2 varones) que estaban recibiendo quimioterapia, que incluía ciclofosfamida 750-1.500 mg/m2; o ciclofosfamida 500-1.500 mg/m2 y doxorrubicina (<60 mg/m2) o epirubicina (<100 mg/m2), se comparó aprepitant en combinación con un régimen de ondansetrón/dexametasona (ver sección 4.2) con el tratamiento estándar (placebo más ondansetrón 8 mg por vía oral (dos veces en el día 1 y cada 12 horas en los días 2 y 3) más dexametasona 20 mg por vía oral en el día 1).
La eficacia se basó en la evaluación de las medidas compuestas: respuesta completa (definida como ausencia de episodios eméticos sin uso de tratamiento de rescate) principalmente durante el Ciclo 1.
Tabla 2
Porcentaje de pacientes adultos que respondieron por grupo de tratamiento y fase - Ciclo 1 Quimioterapia moderadamente emetógena
|
Régimen |
Tratamiento | |
|
con |
estándar | |
|
MEDIDAS COMPUESTAS aprepitant |
(N= 424) | |
|
(N= 433) f |
% |
% (IC del 95%) |
|
% |
Respuesta completa (sin emesis y sin tratamiento de rescate)_
Global (0-120 horas) 50,8 42,5 8,3 (1,6, 15,0)
0-24 horas 75,7 69,0 6,7 (0,7, 12,7)
25-120 horas_55,4_49,1_6,3 (-0,4, 13,0)
MEDIDAS INDIVIDUALES_
Sin emesis (sin episodios eméticos independientemente del uso de tratamiento de rescate)
Global (0-120 horas) 75,7 58,7 17,0 (10,8, 23,2)
0-24 horas 87,5 77,3 10,2 (5,1, 15,3)
25-120 horas_80,8_69,1_11,7 (5,9, 17,5)
Sin náuseas significativas (EAV máxima < 25 mm en una escala de 0-100 mm)_
Global (0-120 horas) 60,9 55,7 5,3 (-1,3, 11,9)
0-24 horas 79,5 78,3 1,3 (-4,2, 6,8)
Tabla 3
Porcentaje de pacientes adultos que respondieron por grupo de tratamiento y fase en el ensayo 2 -
Ciclo 1
Quimioterapia moderadamente emetógena
|
Régimen |
Tratamiento |
Diferencias* |
|
con |
estándar | |
|
aprepitant |
(N= 406) | |
|
(N= 425) |
% |
% (IC del 95 %) |
|
% |
Respuesta completa (sin emesis y sin tratamiento de rescate)
|
Global (0-120 horas) |
68,7 |
56,3 |
12,4 |
(5,9, 18,9) |
|
0-24 horas |
89,2 |
80,3 |
8,9 |
(4,0, 13,8) |
|
25-120 horas |
70,8 |
60,9 |
9,9 |
(3,5, 16,3) |
|
Sin emesis (sin episodios eméticos |
independientemente del uso |
de tratamiento de rescate) | ||
|
Global (0-120 horas) |
76,2 |
62,1 |
14,1 |
(7,9, 20,3) |
|
0-24 horas |
92,0 |
83,7 |
8,3 |
(3,9, 12,7) |
|
25-120 horas |
77,9 |
66,8 |
11,1 |
(5,1, 17,1) |
|
Sin nauseas significativas (EAV máxima < 25 |
mm en una escala de 0-100 mm) | |||
|
Global (0-120 horas) |
73,6 |
66,4 |
7,2 |
(1,0, 13,4) |
|
0-24 horas |
90,9 |
86,3 |
4,6 |
(0,2, 9,0) |
|
25-120 horas |
74,9 |
69,5 |
5,4 |
(-0,7, 11,5) |
*Los intervalos de confianza se calcularon sin ajuste por sexo ni región, los cuales fueron incluidos en el análisis primario utilizando modelos logísticos.
El beneficio del tratamiento en combinación de aprepitant en la población del ensayo total fue dirigido principalmente por los resultados observados en pacientes con bajo control con el régimen estándar como en mujeres, aunque los resultados fueron numéricamente mejores independientemente de la edad, tipo de tumor o sexo. La respuesta completa al régimen de aprepitant y al tratamiento estándar, respectivamente, se alcanzó en 209/324 (65 %) y 161/320 (50 %) en mujeres y 83/101 (82 %) y 68/87 (78 %) de varones.
Población pediátrica
En un ensayo clínico aleatorizado, doble ciego, controlado con comparador activo en el que se incluyó a 302 niños y adolescentes (de entre 6 meses y 17 años de edad) que recibieron quimioterapia moderada o altamente emetógena, el régimen de aprepitant se comparó con un régimen de control para la prevención de NVIQ. La eficacia del régimen de aprepitant se evaluó en un único ciclo (Ciclo 1). A los pacientes se les dio la oportunidad de recibir aprepitant en abierto en ciclos posteriores (Ciclos 2-6 opcionales); sin embargo, no se evaluó la eficacia en estos ciclos opcionales. El régimen de aprepitant para adolescentes de entre 12 y 17 años de edad (n=47) consistió en EMEND cápsulas de 125 mg por vía oral el día 1 y 80 mg/día los días 2 y 3, en combinación con ondansetrón el día 1. El régimen de aprepitant para niños de entre 6 meses y menos de 12 años de edad (n=105) consistió en EMEND polvo para suspensión oral 3,0 mg/kg (hasta 125 mg) por vía oral el día 1 y 2,0 mg/kg (hasta 80 mg) por vía oral los días 2 y 3, en combinación con ondansetrón el día 1. El régimen de control en adolescentes de entre 12 y 17 años de edad (n=48) y niños de entre 6 meses y menos de 12 años de edad (n=102) consistió en placebo de aprepitant los días 1, 2 y 3 en combinación con ondansetrón el día 1. EMEND o placebo y ondansetrón se administraron 1 hora y 30 minutos antes del inicio de la quimioterapia, respectivamente. Se permitió dexametasona intravenosa como parte del régimen antiemético en pacientes pediátricos de ambos grupos de edad, a criterio del médico. Fue necesaria una disminución de la dosis de dexametasona (50 %) en los pacientes pediátricos que recibieron aprepitant. No fue necesaria una disminución de la dosis en los pacientes pediátricos que recibieron el régimen de control. De los pacientes pediátricos, el 29 % del régimen de aprepitant y el 28 % del régimen de control utilizaron dexametasona como parte del régimen en el Ciclo 1.
La actividad antiemética de EMEND se evaluó durante un periodo de 5 días (120 horas) después del inicio de la quimioterapia el día 1. El criterio de valoración principal fue la respuesta completa en la fase retardada (de 25 a 120 horas después del comienzo de la quimioterapia) en el Ciclo 1. En la tabla 4 se muestra un resumen de los resultados más importantes del ensayo.
Tabla 4
Número (%) de pacientes pediátricos con respuesta completa y sin vómitos por grupo de tratamiento y
fase - Ciclo 1 (población por intención de tratar)
|
Régimen de aprepitant n/m (%) |
Régimen de control n/m (%) | |
|
CRITERIO DE VALORACIÓN PRINCIPAL | ||
|
Respuesta completa* - Fase retardada |
77/152 (50,7)t |
39/150 (26,0) |
|
OTROS CRITERIOS DE VALORACIÓN PRE-ESPECIFICADOS | ||
|
Respuesta completa* - Fase aguda |
101/152 (66,4); |
78/150 (52,0) |
|
Respuesta completa - Fase global |
61/152 (40,1)t |
30/150 (20,0) |
|
Sin vómitos5 - Fase global |
71/152 (46,7)t |
32/150 (21,3) |
|
*Respuesta completa = Sin vómitos, náuseas o arcadas improductivas y sin uso de medicación de | ||
|
rescate. | ||
|
Tp < 0,01 en comparación con el régimen de control *p < 0,05 en comparación con el régimen de control §Sin vómitos = Sin vómitos, náuseas o arcadas improductivas | ||
|
n/m = Número de pacientes con la respuesta deseada/número de pacientes incluidos en el punto temporal. Fase aguda: de 0 a 24 horas después del inicio de la quimioterapia. Fase retardada: de 25 a 120 horas después del inicio de la quimioterapia. Fase global: de 0 a 120 horas después del inicio de la quimioterapia. | ||
El tiempo estimado hasta el primer vómito después del comienzo del tratamiento con quimioterapia fue mayor con el régimen de aprepitant (la mediana del tiempo estimado hasta el primer vómito fue
94,5 horas) en comparación con el grupo del régimen de control (la mediana del tiempo estimado hasta el primer vómito fue 26,0 horas) tal y como se representa en las curvas de Kaplan-Meier de la Figura 2.
Figura 2
Tiempo hasta el primer episodio de vómitos desde el inicio de la administración de la quimioterapia -pacientes pediátricos en la fase global - Ciclo 1 (población por intención de tratar)
Un análisis de la eficacia en subpoblaciones en el Ciclo 1 demostró que, independientemente de la categoría de edad, sexo, uso de dexametasona para la profilaxis antiemética y emetogenicidad de la
quimioterapia, el régimen de aprepitant proporcionó un mejor control que el régimen de control con respecto a los criterios de valoración de la respuesta completa.
5.2 Propiedades farmacocinéticas
Aprepitant muestra una farmacocinética no lineal. Tanto el aclaramiento como la biodisponibilidad absoluta disminuyen al aumentar la dosis.
Absorción
La biodisponibilidad oral absoluta media de aprepitant es de 67% para la cápsula de 80 mg y de 59% para la cápsula de 125 mg. La concentración plasmática máxima media (Cmax) de aprepitant se alcanzó aproximadamente a las 4 horas (tmax). La administración oral de la cápsula con un desayuno estándar de aproximadamente 800 Kcal ocasionó un aumento de hasta el 40% en el AUC de aprepitant. Este aumento no se consideró clínicamente de interés.
La farmacocinética de aprepitant es no lineal en el intervalo de la dosis clínica. En adultos jóvenes sanos, el aumento en el AUC0-a, fue un 26% mayor que proporcional a la dosis entre las dosis únicas de 80 mg y 125 mg administradas en estado posprandial.
Tras la administración oral de una dosis única de 125 mg de EMEND el día 1 y 80 mg una vez al día los días 2 y 3, el AUC0-24h (media ± DE) fue 19,6 ± 2,5 ^g»h/ml y 21,2 ± 6,3 ^g»h/ml los días 1 y 3, respectivamente. La Cmax fue 1,6 ± 0,36 ^g/ml y 1,4 ± 0,22 ^g/ml los días 1 y 3, respectivamente.
Distribución
Aprepitant se une fuertemente a proteínas, con una media del 97%. La media geométrica del volumen aparente de distribución en el estado equilibrio (Vdee) es aproximadamente de 66 litros en el ser humano.
Biotransformación
Aprepitant se metaboliza extensamente. En adultos jóvenes sanos, aprepitant representa aproximadamente el 19% de la radiactividad plasmática durante 72 horas después de una dosis única intravenosa de 100 mg de [C14]-fosaprepitant, un profármaco de aprepitant, lo que indica una importante presencia de metabolitos en el plasma. En el plasma humano se han identificado doce metabolitos de aprepitant. El metabolismo de aprepitant se produce en gran medida por oxidación en el anillo de morfolina y sus cadenas laterales y los metabolitos resultantes sólo fueron débilmente activos. Estudios in vitro en los que se usaron microsomas hepáticos humanos indicaron que aprepitant se metaboliza principalmente a través de CYP3A4 y posiblemente con una contribución menor a través de CYP1A2 y CYP2C19.
Eliminación
Aprepitant no se elimina inalterado en la orina. Los metabolitos se eliminan en la orina y a través de excreción biliar en las heces. Después de una dosis única intravenosa de 100 mg de [C14]-fosaprepitant, un profármaco de aprepitant a sujetos sanos, el 57% de la radiactividad se recuperó en la orina y el 45% en las heces.
El aclaramiento plasmático de aprepitant es dependiente de la dosis, disminuyendo al aumentar la dosis y oscilando aproximadamente entre 60 a 72 ml/min en el intervalo de la dosis terapéutica. La semivida terminal osciló entre aproximadamente 9 a 13 horas.
Farmacocinética en poblaciones especiales
Pacientes de edad avanzada: Tras la administración oral de una dosis única de 125 mg de aprepitant el día 1 y 80 mg una vez al día los días 2 a 5, el AUC0-24h de aprepitant fue un 21% superior el día 1 y un 36% superior el día 5 en pacientes de edad avanzada (>65 años) respecto de los adultos más jóvenes. La Cmax fue un 10% superior el día 1 y un 24% superior el día 5 en pacientes de edad avanzada respecto de los adultos más jóvenes. Estas diferencias no se consideraron clínicamente significativas. EMEND no requiere ajuste de dosis en los pacientes de edad avanzada.
Sexo: Tras la administración oral de una dosis única de 125 mg de aprepitant, la Cmax de aprepitant es un 16% superior en las mujeres en comparación con los varones. La semivida de aprepitant es un 25% inferior en las mujeres en comparación con los varones y su tmax se produce en aproximadamente el mismo tiempo. Estas diferencias no se consideraron clínicamente significativas. EMEND no requiere ajuste de dosis en función del sexo.
Insuficiencia hepática: La insuficiencia hepática leve (Child-Pugh clase A) no afecta a la farmacocinética de aprepitant en un grado clínicamente relevante. No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. De los datos disponibles no pueden extraerse conclusiones relativas a la influencia de la insuficiencia hepática moderada (Child-Pugh clase B) sobre la farmacocinética de aprepitant. No existen datos clínicos ni farmacocinéticos de pacientes con insuficiencia hepática grave (Child-Pugh clase C).
Insuficiencia renal: Se administró una dosis única de 240 mg de aprepitant a pacientes con insuficiencia renal grave (CrCl<30 ml/min) y a pacientes con nefropatía terminal que requería hemodiálisis.
En los pacientes con insuficiencia renal grave, el AUC0_<» del aprepitant total (no unido y unido a proteínas) disminuyó en un 21% y la Cmax disminuyó en un 32%, respecto de los sujetos sanos. En los pacientes con nefropatía terminal sometidos a hemodiálisis, el AUC0_<» del aprepitant total disminuyó en un 42% y la Cmax disminuyó en un 32%. Debido a los modestos descensos en la unión a proteínas de aprepitant en los pacientes con enfermedad renal, el AUC de aprepitant no unido farmacológicamente activo no se vio significativamente afectado en los pacientes con insuficiencia renal en comparación con los sujetos sanos. La hemodiálisis realizada 4 ó 48 horas después de la administración no tuvo efectos significativos sobre la farmacocinética de aprepitant; en el dializado se recuperó menos de 0,2% de la dosis.
En pacientes con insuficiencia renal o en pacientes con nefropatía terminal sometidos a hemodiálisis no es necesario ajustar la dosis de EMEND.
Población pediátrica: Como parte de un régimen de 3 días de duración, la dosis de aprepitant cápsulas (125/80/80 mg) en pacientes adolescentes (de entre 12 y 17 años de edad) logró un AUC0-24h por encima de 17 ^g»h/ml el día 1 y concentraciones (Cmin) al final de los días 2 y 3 superiores a 0,4 ^g/ml en la mayoría de los pacientes. La concentración plasmática máxima media (Cmax) fue aproximadamente 1,3 ^g/ml el día 1, alcanzándose a las 4 horas aproximadamente. Como parte de un régimen de 3 días de duración, la dosis de aprepitant polvo para suspensión oral (3/2/2 mg/kg) en pacientes de entre 6 meses y menos de 12 años de edad alcanzó un AUC0-24h superior a 17 ^g»h/ml el día 1, con concentraciones (Cmin) al final de los días 2 y 3 por encima de 0,1 ^g/ml en la mayoría de los pacientes. La concentración plasmática máxima media (Cmax) fue aproximadamente 1,2 ^g/ml el día 1, alcanzándose entre 5 y 7 horas.
Un análisis farmacocinético poblacional de aprepitant en pacientes pediátricos (de entre 6 meses y 17 años de edad) sugiere que el sexo y la raza no tienen un efecto clínicamente significativo sobre la farmacocinética de aprepitant.
Relación entre concentración y efecto
Usando un trazador altamente específico del receptor NKb los estudios de tomografía por emisión de positrones (PET) en varones jóvenes sanos han demostrado que aprepitant penetra en el cerebro y ocupa los receptores NK de forma dependiente de la dosis y de la concentración plasmática. Se predice que las concentraciones plasmáticas de aprepitant alcanzadas en adultos con el régimen de 3 días de EMEND proporcionarán una ocupación superior al 95% de los receptores NK cerebrales.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de toxicidad, genotoxicidad, potencial carcinogénico, toxicidad para la reproducción y el desarrollo, a dosis únicas y repetidas. Sin embargo, se deberá tener en cuenta que la exposición sistémica en roedores fue similar o incluso inferior a la exposición terapéutica en seres humanos a la dosis de 125 mg/80 mg. En especial, aunque no se observaron efectos adversos en los estudios sobre la reproducción a los niveles de exposición en seres humanos, las exposiciones en animales no son suficientes para hacer una valoración de riesgo adecuada en el hombre.
En un estudio de toxicidad juvenil en ratas tratadas desde el día 10 después del nacimiento hasta el día 63, aprepitant ocasionó en las hembras una apertura vaginal prematura a partir de 250 mg/kg dos veces al día y en los machos un retraso en la separación del prepucio a partir de 10 mg/kg dos veces al día. No hubo márgenes de exposición clínicamente relevantes. No se observaron efectos sobre el apareamiento, la fertilidad o la supervivencia embrionaria o fetal ni cambios patológicos en los órganos reproductores relacionados con el tratamiento. En un estudio de toxicidad juvenil en perros tratados desde el día 14 después del nacimiento hasta el día 42, se detectó una disminución del peso testicular y del tamaño de las células de Leydig en los machos con 6 mg/kg/día y un aumento del peso del útero, hipertrofia del útero y del cuello del útero y edema de los tejidos vaginales en las hembras a partir de 4 mg/kg/día. No hubo márgenes de exposición clínicamente relevantes de aprepitant. En el tratamiento a corto plazo de acuerdo con la pauta posológica recomendada, se considera poco probable que estos resultados sean clínicamente relevantes.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Contenido de la cápsula Sacarosa
Celulosa microcristalina (E-460)
Hidroxipropilcelulosa (E-463)
Lauril sulfato de sodio
Cubierta de la cápsula (125 mg)
Gelatina
Dióxido de titanio (E-171)
Óxido de hierro rojo (E-172)
Óxido de hierro amarillo (E-172)
Cubierta de la cápsula (80 mg)
Gelatina
Dióxido de titanio (E-171)
Tinta para impresión Laca
Hidróxido de potasio Óxido de hierro negro (E-172)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
4 años.
6.4 Precauciones especiales de conservación
Conservar en el embalaje original para protegerlo de la humedad.
6.5 Naturaleza y contenido del envase
Están disponibles diferentes tamaños de envase con concentraciones diferentes.
Blister de aluminio que contiene una cápsula de 80 mg.
Blister de aluminio que contiene dos cápsulas de 80 mg.
5 blisters de aluminio que contienen una cápsula de 80 mg cada uno.
Blister de aluminio que contiene una cápsula de 125 mg.
5 blisters de aluminio que contienen una cápsula de 125 mg cada uno.
Blister de aluminio que contiene una cápsula de 125 mg y dos cápsulas de 80 mg.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación Ninguna especial para su eliminación.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme Ltd.
Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/262/001
EU/1/03/262/002
EU/1/03/262/003
EU/1/03/262/004
EU/1/03/262/005
EU/1/03/262/006
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 11/noviembre/2003 Fecha de la última renovación: 22/septiembre/2008
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
1. NOMBRE DEL MEDICAMENTO
EMEND 125 mg polvo para suspensión oral
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada sobre contiene 125 mg de aprepitant. Tras la reconstitución, 1 ml de suspensión oral contiene 25 mg de aprepitant.
Excipientes con efecto conocido
Cada sobre contiene aproximadamente 125 mg de sacarosa y 468,7 mg de lactosa (como lactosa anhidro).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo para suspensión oral.
Polvo de color rosa a rosa claro.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Prevención de las náuseas y los vómitos que se asocian con la quimioterapia antineoplásica moderada y altamente emetógena en lactantes y niños de entre 6 meses y menos de 12 años de edad.
EMEND polvo para suspensión oral se administra como parte de un tratamiento de combinación (ver sección 4.2).
4.2 Posología y forma de administración
Posología
Población pediátrica
Lactantes y niños (de entre 6 meses y menos de 12 años de edad y no menos de 6 kg)
EMEND se administra durante 3 días como parte de un régimen que incluye un antagonista 5-HT3. La dosis recomendada de EMEND polvo para suspensión oral se basa en el peso, como se especifica en la tabla siguiente. EMEND se administra por vía oral 1 hora antes de la quimioterapia los días 1,2 y 3. Si los días 2 y 3 no se administra quimioterapia, EMEND se debe administrar por la mañana. Consultar la Ficha Técnica del antagonista 5-HT3 seleccionado para obtener información sobre la dosis adecuada. Si junto con EMEND se administra un corticosteroide, como dexametasona, la dosis del corticosteroide debe ser un 50% de la dosis habitual (ver las secciones 4.5 y 5.1).
Dosis y volumen recomendados de EMEND suspensión oral en pacientes pediátricos de entre
6 meses y menos de 12 años de edad
|
Peso corporal |
Volumen de la dosis de suspension para administración oral | |||||
|
Día 1 |
Día 2 |
Día 3 | ||||
|
menos de 6 kg |
No recomendada | |||||
|
de 6 kg a menos de 8 kg |
1 ml |
(25 mg) |
0,6 ml |
(15 mg) |
0,6 ml |
(15 mg) |
|
de 8 kg a menos de 10 kg |
1,2 ml |
(30 mg) |
0,8 ml |
(20 mg) |
0,8 ml |
(20 mg) |
|
de 10 kg a menos de 12 kg |
1,4 ml |
(35 mg) |
1 ml |
(25 mg) |
1 ml |
(25 mg) |
|
de 12 kg a menos de 15 kg |
1,8 ml |
(45 mg) |
1,2 ml |
(30 mg) |
1,2 ml |
(30 mg) |
|
de 15 kg a menos de 20 kg |
2,4 ml |
(60 mg) |
1,6 ml |
(40 mg) |
1,6 ml |
(40 mg) |
|
de 20 kg a menos de 25 kg |
3 ml |
(75 mg) |
2 ml |
(50 mg) |
2 ml |
(50 mg) |
|
de 25 kg a menos de 30 kg |
3,6 ml |
(90 mg) |
2,4 ml |
(60 mg) |
2,4 ml |
(60 mg) |
|
30 kg y superiores |
Extraer todo el contenido del vaso de mezcla al dispensador oral (~5 ml) |
(125 mg) |
3,2 ml |
(80 mg) |
3,2 ml |
(80 mg) |
No se ha establecido la eficacia del polvo para suspensión oral de 125 mg en niños de 12 años de edad y mayores. Para adolescentes de entre 12 y 17 años, EMEND está disponible en cápsulas que contienen 80 mg o 125 mg de aprepitant.
No se ha establecido la seguridad y eficacia de EMEND polvo para suspensión oral en lactantes de menos de 6 meses de edad o de peso inferior a 6 kg. No se dispone de datos.
General
Los datos de eficacia en combinación con otros corticoesteroides y antagonistas 5HT3 son limitados. Para información adicional sobre la administración conjunta con corticoesteroides, ver sección 4.5. Consultar la Ficha Técnica de los medicamentos antagonistas 5-HT3 que se administren conjuntamente.
Poblaciones especiales Sexo
No es necesario ajustar la dosis según el sexo (ver sección 5.2).
Insuficiencia renal
No es necesario ajustar la dosis en los pacientes con insuficiencia renal ni en los pacientes con nefropatía terminal sometidos a hemodiálisis (ver sección 5.2).
Insuficiencia hepática
No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. Existen datos limitados en pacientes con insuficiencia hepática moderada y no existen datos en pacientes con insuficiencia hepática grave. Aprepitant se debe usar con precaución en estos pacientes (ver las secciones 4.4 y 5.2).
Forma de administración
La suspensión oral se puede tomar con o sin alimentos.
Para obtener más información sobre la preparación y la administración de la suspensión, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Administración conjunta con pimozida, terfenadina, astemizol o cisaprida (ver sección 4.5).
4.4 Advertencias y precauciones especiales de empleo Pacientes con insuficiencia hepática moderada a grave
Existen datos limitados en pacientes con insuficiencia hepática moderada y no existen datos en pacientes con insuficiencia hepática grave. EMEND se debe usar con precaución en estos pacientes (ver sección 5.2).
Interacciones con el CYP3A4
EMEND se debe usar con precaución en pacientes que estén recibiendo de forma concomitante principios activos, administrados por vía oral, metabolizados principalmente a través de CYP3A4 y con un rango terapéutico estrecho, tales como ciclosporina, tacrolimus, sirolimus, everolimus, alfentanilo, alcaloides derivados del ergot, fentanilo y quinidina (ver sección 4.5). Adicionalmente, la administración concomitante con irinotecán se debe abordar con especial prudencia ya que esta combinación puede provocar un aumento de la toxicidad.
Administración conjunta con warfarina (un sustrato de CYP2C9)
En pacientes en tratamiento crónico con warfarina, el coeficiente internacional normalizado (INR) se debe vigilar estrechamente durante el tratamiento con EMEND y durante 14 días después de cada ciclo de 3 días de EMEND (ver sección 4.5).
Administración conjunta con anticonceptivos hormonales
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de la administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo (ver sección 4.5).
Excipientes
EMEND polvo para suspensión oral contiene sacarosa y lactosa. Los pacientes con intolerancia hereditaria a la fructosa o a la galactosa, problemas de absorción de glucosa o galactosa, insuficiencia de lactasa de Lapp (insuficiencia observada en ciertas poblaciones de Laponia) o insuficiencia de sacarasa-isomaltasa, no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Aprepitant (125 mg/80 mg) es un sustrato, un inhibidor moderado, y un inductor de CYP3A4. Aprepitant es también un inductor de CYP2C9. Durante el tratamiento con EMEND, CYP3A4 se inhibe. Después de terminar el tratamiento, EMEND produce una inducción transitoria suave de CYP2C9, de CYP3A4 y glucuronidación. Aprepitant no parece que interaccione con el transportador de la P-glucoproteína, como sugiere la falta de interacción de aprepitant con digoxina.
Efecto de aprepitant sobre la farmacocinética de otros principios activos
Inhibición de CYP3A4
Como inhibidor moderado de CYP3A4, aprepitant (125 mg/80 mg) puede aumentar las concentraciones plasmáticas de los principios activos que se metabolizan a través de CYP3A4 cuando se administran conjuntamente. La exposición total de los sustratos de CYP3A4 que se administran por vía oral puede aumentar hasta aproximadamente tres veces durante el tratamiento de 3 días con EMEND; se estima que el efecto de aprepitant sobre las concentraciones plasmáticas de los sustratos de CYP3A4 que se administran por vía intravenosa sea menor. EMEND no se debe usar simultáneamente con pimozida, terfenadina, astemizol o cisaprida (ver sección 4.3). La inhibición de CYP3A4 por aprepitant podría dar lugar a elevaciones de las concentraciones plasmáticas de estos principios activos, lo que podría provocar reacciones graves o potencialmente mortales. Se aconseja
precaución durante la administración concomitante de EMEND y principios activos administrados por vía oral, metabolizados principalmente a través de CYP3A4 y con un rango terapéutico estrecho, tales como ciclosporina, tacrolimus, sirolimus, everolimus, alfentanilo, diergotamina, ergotamina, fentanilo y quinidina (ver sección 4.4).
Corticoesteroides
Dexametasona: La dosis habitual de dexametasona oral se debe reducir aproximadamente en un 50% cuando se administra conjuntamente con un tratamiento de 125 mg/80 mg de EMEND. La dosis de dexametasona utilizada en los ensayos clínicos de náuseas y vómitos inducidos por quimioterapia, se eligió en función de las interacciones del principio activo (ver sección 4.2). EMEND, administrado en un régimen de 125 mg conjuntamente con 20 mg de dexametasona oral el día 1 y EMEND administrado en un régimen de 80 mg/día conjuntamente con 8 mg de dexametasona oral los días 2 a 5, aumentó el AUC de dexametasona, un sustrato de CYP3A4, 2,2 veces los días 1 y 5.
Metilprednisolona: La dosis habitual de metilprednisolona intravenosa se debe reducir aproximadamente un 25%, y la dosis habitual de metilprednisolona oral se debe reducir aproximadamente un 50% al administrarse conjuntamente con un tratamiento de 125 mg/80 mg de EMEND. EMEND, administrado en un régimen de 125 mg el día 1 y 80 mg/día los días 2 y 3, aumentó el AUC de metilprednisolona, un sustrato de CYP3A4, 1,3 veces el día 1 y 2,5 veces el día 3, al administrarse conjuntamente metilprednisolona por vía intravenosa, 125 mg el día 1, y por vía oral, 40 mg los días 2 y 3.
Durante el tratamiento continuo con metilprednisolona, el AUC de metilprednisolona puede disminuir en puntos de tiempo posteriores en el transcurso de las 2 semanas siguientes al inicio de la administración de EMEND, debido al efecto inductor de aprepitant sobre CYP3A4. Puede ser que este efecto sea más pronunciado para metilprednisolona administrada oralmente.
Antineoplásicos
En ensayos farmacocinéticos, EMEND, administrado en un régimen de 125 mg el día 1 y 80 mg/día los días 2 y 3, no influyó en la farmacocinética de docetaxel administrado por vía intravenosa el día 1 ni en la de vinorelbina administrada por vía intravenosa el día 1 o el día 8. Debido a que el efecto de EMEND sobre la farmacocinética de sustratos de CYP3A4 administrados por vía oral es mayor que el efecto de EMEND sobre la farmacocinética de sustratos de CYP3A4 administrados por vía intravenosa, no se puede excluir una interacción con medicamentos antineoplásicos administrados por vía oral que se metabolizan principal o parcialmente a través de CYP3A4 (p. ej. etopósido, vinorelbina). En pacientes que reciben medicamentos que se metabolizan principal o parcialmente a través de CYP3A4, se aconseja precaución y puede ser conveniente una vigilancia adicional (ver sección 4.4). Se han notificado acontecimientos adversos de neurotoxicidad postcomercialización, una reacción adversa potencial de ifosfamida, tras la administración simultánea de aprepitant e ifosfamida.
Inmunosupresores
Durante el régimen de tratamiento de 3 días de las náuseas y vómitos inducidos por quimioterapia, se espera un incremento moderado transitorio seguido de una leve disminución en la exposición de los inmunosupresores metabolizados por CYP3A4 (por ej. ciclosporina, tacrolimus, everolimus y sirolimus). Dada la corta duración del régimen de tratamiento de 3 días y los cambios limitados en la exposición dependientes del tiempo, no se recomienda una reducción de dosis de los inmunosupresores durante los 3 días de la administración conjunta con EMEND.
Midazolam
Los posibles efectos de aumentos en las concentraciones plasmáticas de midazolam u otras benzodiazepinas metabolizadas a través de CYP3A4 (alprazolam, triazolam) se deben tener en cuenta al administrar estos medicamentos conjuntamente con EMEND (125 mg/80 mg).
EMEND aumentó el AUC de midazolam, un sustrato sensible de CYP3A4, 2,3 veces el día 1 y
3,3 veces el día 5, al administrarse conjuntamente una dosis oral única de 2 mg de midazolam los días 1 y 5 de un régimen de EMEND 125 mg el día 1 y 80 mg/día los días 2 a 5.
En otro ensayo con administración intravenosa de midazolam, EMEND se administró como 125 mg el día 1 y 80 mg/día los días 2 y 3, y 2 mg de midazolam se administró por vía intravenosa antes de la administración del régimen de 3 días de EMEND y en los días 4, 8 y 15. EMEND aumentó el AUC de midazolam un 25% el día 4 y disminuyó el AUC de midazolam un 19% el día 8 y un 4% el día 15. Estos efectos no se consideraron clínicamente importantes.
En un tercer ensayo con administración intravenosa y oral de midazolam, se administraron 125 mg de EMEND en el día 1 y 80 mg/día en los días 2 y 3, junto con 32 mg de ondansetrón el día 1, 12 mg de dexametasona el día 1 y 8 mg los días 2-4. Esta combinación (esto es, EMEND, ondansetrón y dexametasona) disminuyó el AUC de midazolam oral un 16 % el día 6, un 9 % el día 8, un 7 % el día 15 y un 17 % el día 22. Estos efectos no se consideraron clínicamente importantes.
Se finalizó un ensayo adicional con administración intravenosa de midazolam y EMEND. Una hora después de la administración oral de una dosis única de EMEND 125 mg, se administraron por vía intravenosa 2 mg de midazolam. El AUC plasmático de midazolam aumentó en 1,5 veces. Este efecto no se consideró clínicamente importante.
Inducción
Como inductor suave de CYP2C9, de CYP3A4 y de la glucuronidación, aprepitant puede disminuir las concentraciones plasmáticas de los sustratos eliminados por estas vías durante las dos semanas posteriores al inicio del tratamiento. Este efecto puede hacerse evidente únicamente después de finalizar el tratamiento de 3 días con EMEND. Para los sustratos de CYP2C9 y CYP3A4, la inducción es transitoria con un efecto máximo alcanzado a los 3-5 días después de finalizar el tratamiento de 3 días con EMEND. El efecto se mantiene durante unos pocos días, después desciende lentamente y es clínicamente insignificante a las 2 semanas después de finalizar el tratamiento con EMEND. La inducción suave de la glucuronidación también se observa con 80 mg de aprepitant oral administrado durante 7 días. Se carece de datos relativos a los efectos sobre CYP2C8 y CYP2C19. Se aconseja precaución al administrar durante este periodo de tiempo warfarina, acenocumarol, tolbutamida, fenitoína u otros principios activos que se sabe que son metabolizados por CYP2C9.
Warfarina
En pacientes en tratamiento crónico con warfarina, el tiempo de protrombina (INR) se debe vigilar estrechamente durante el tratamiento con EMEND y durante 2 semanas después de cada ciclo de 3 días de EMEND en el caso de náuseas y vómitos inducidos por quimioterapia (ver sección 4.4). Al administrarse una dosis única de 125 mg de EMEND el día 1 y 80 mg/día los días 2 y 3 a sujetos sanos estabilizados en un tratamiento crónico con warfarina, no se observó ningún efecto de EMEND sobre el AUC plasmático de R(+) o S(-) warfarina determinado el día 3; sin embargo, se observó un descenso del 34% en la concentración mínima de S(-) warfarina (un sustrato de CYP2C9) acompañado de un descenso del 14% en el INR 5 días después de finalizar el tratamiento con EMEND.
Tolbutamida
EMEND, administrado como 125 mg el día 1 y 80 mg/día los días 2 y 3, disminuyó el AUC de tolbutamida (un sustrato de CYP2C9) en un 23% el día 4, un 28% el día 8 y un 15% el día 15, al administrarse una dosis única de tolbutamida 500 mg por vía oral antes de la administración de un régimen de 3 días de EMEND y en los días 4, 8 y 15.
Anticonceptivos hormonales
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de la administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo.
En un ensayo clínico, dosis únicas de un anticonceptivo oral que contenía etinil estradiol y noretindrona se administraron en los días 1 hasta el 21 con EMEND, administrado con una pauta posológica de 125 mg en el día 8 y 80 mg/día en los días 9 y 10 con ondansetrón 32 mg vía intravenosa en el día 8 y dexametasona oral administrada como 12 mg en el día 8 y 8 mg/día los días 9, 10 y 11. Durante los días 9 hasta el 21 en este ensayo, hubo un descenso hasta del 64% en las concentraciones mínimas de etinil estradiol y hasta del 60% en las concentraciones mínimas de noretindrona.
Antagonistas 5-HT3
En ensayos clínicos de interacción, aprepitant no tuvo efectos clínicamente importantes sobre la farmacocinética de ondansetrón, granisetrón ni hidrodolasetrón (el metabolito activo de dolasetrón).
Efecto de otros medicamentos sobre la farmacocinética de aprepitant
La administración concomitante de EMEND con principios activos que inhiben la actividad de CYP3A4 (p.ej., ketoconazol, itraconazol, voriconazol, posaconazol, claritromicina, telitromicina, nefazodona e inhibidores de la proteasa) se debe abordar con precaución, ya que la combinación se espera que provoque una elevación de varias veces las concentraciones plasmáticas de aprepitant (ver sección 4.4).
La administración concomitante de EMEND con principios activos que inducen fuertemente la actividad de CYP3A4 (p.ej., rifampicina, fenitoína, carbamazepina, fenobarbital) se debe evitar ya que la combinación provoca reducciones en las concentraciones plasmáticas de aprepitant lo que podría provocar una disminución de la eficacia de EMEND. No se recomienda la administración concomitante de EMEND con plantas medicinales que contienen hipérico (Hypericum perforatum).
Ketoconazol
Al administrarse una dosis única de 125 mg de aprepitant el día 5 de un régimen de 10 días de 400 mg/día de ketoconazol, un potente inhibidor de CYP3A4, el AUC de aprepitant aumentó aproximadamente 5 veces y la semivida terminal media de aprepitant aumentó aproximadamente 3 veces.
Rifampicina
Al administrarse una dosis única de 375 mg de aprepitant el día 9 de un régimen de 14 días de 600 mg/día de rifampicina, un potente inductor de CYP3A4, el AUC de aprepitant disminuyó un 91% y la semivida terminal media disminuyó un 68%.
Población pediátrica
Los estudios de interacciones se han realizado sólo en adultos.
4.6 Fertilidad, embarazo y lactancia
Anticoncepción en varones y mujeres
La eficacia de los anticonceptivos hormonales puede disminuir durante la administración de EMEND y durante 28 días después de su administración. Durante el tratamiento con EMEND y en los 2 meses siguientes a la última dosis de EMEND, se deben usar métodos anticonceptivos alternativos no hormonales de refuerzo (ver las secciones 4.4 y 4.5).
Embarazo
No hay datos clínicos disponibles sobre la exposición a aprepitant durante el embarazo. La capacidad de aprepitant para provocar toxicidad sobre la reproducción no se ha caracterizado completamente, ya que en los estudios animales no se pudieron alcanzar niveles de exposición por encima de la exposición terapéutica en seres humanos a la dosis de 125 mg/80 mg. Estos estudios no sugirieron efectos perjudiciales directos ni indirectos en términos del embarazo, el desarrollo embrionario/fetal, el parto o el desarrollo posnatal (ver sección 5.3). Se desconocen los posibles efectos sobre la reproducción de alteraciones en la regulación de la neurocinina. EMEND no se debe utilizar durante el embarazo excepto si fuese claramente necesario.
Lactancia
Aprepitant se excreta en la leche de ratas lactantes. Se desconoce si aprepitant se excreta en la leche materna; por consiguiente, no se recomienda la lactancia durante el tratamiento con EMEND.
Fertilidad
El potencial efecto de aprepitant sobre la fertilidad no se ha caracterizado completamente, ya que en los estudios en animales no se pudieron alcanzar niveles de exposición por encima de la exposición terapéutica en humanos. Estos estudios de fertilidad no sugirieron efectos perjudiciales directos ni indirectos en términos del estado de apareamiento, fertilidad, desarrollo embrionario/fetal, o recuento y movilidad del esperma (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de EMEND sobre la capacidad para montar en bicicleta y utilizar máquinas es pequeña. Se pueden producir mareos y fatiga después de la administración de EMEND (ver sección 4.8).
4.8 Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad de aprepitant se evaluó en aproximadamente 6.500 adultos en más de 50 ensayos y 184 niños y adolescentes en 2 ensayos clínicos pediátricos pivotales.
Las reacciones adversas más frecuentes que se notificaron con una mayor incidencia en adultos tratados con el régimen de aprepitant que con el tratamiento estándar en pacientes que estaban recibiendo quimioterapia altamente emetógena (HEC) fueron: hipo (4,6 % versus 2,9 %), alanina aminotransferasa (ALT) elevada (2,8 % versus 1,1 %), dispepsia (2,6 % versus 2,0 %), estreñimiento (2,4 % versus 2,0 %), cefalea (2,0 % versus 1,8 %) y apetito disminuido (2,0 % versus 0,5 %). La reacción adversa más frecuente notificada con una mayor incidencia en pacientes tratados con el régimen de aprepitant que con el tratamiento estándar en adultos que estaban recibiendo quimioterapia moderadamente emetógena (MEC) fue fatiga (1,4 % versus 0,9 %).
Las reacciones adversas más frecuentes que se notificaron con una mayor incidencia en pacientes pediátricos tratados con el régimen de aprepitant que con el régimen de control mientras recibían quimioterapia antineoplásica emetógena fueron: hipo (3,3 % versus 0,0 %) y sofocos (1,1 % versus 0,0 %).
Tabla de reacciones adversas
Las reacciones adversas siguientes se observaron en un análisis combinado de los ensayos en HEC y en MEC con una incidencia mayor con aprepitant que con el tratamiento estándar o en el uso después de la comercialización. Las categorías de frecuencia dadas en la tabla se basan en los ensayos en adultos; las frecuencias observadas en los ensayos pediátricos fueron similares o menores, a menos que se muestre en la tabla. Algunas reacciones adversas menos frecuentes en la población adulta no se observaron en los ensayos pediátricos.
Las frecuencias se definen como: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000) y muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
|
Sistema de clasificación de órganos |
Reacción adversa |
Frecuencia |
|
Infecciones e infestaciones |
candidiasis, infección estafilocócica |
raras |
|
Trastornos de la sangre y del sistema linfático |
neutropenia febril, anemia |
poco frecuentes |
|
Trastornos del sistema inmunológico |
reacciones de hipersensibilidad incluyendo reacciones anafilácticas |
no conocida |
|
Trastornos del metabolismo y de la nutrición |
apetito disminuido |
frecuentes |
|
polidipsia |
raras | |
|
Trastornos psiquiátricos |
ansiedad |
poco frecuentes |
|
desorientación, estado de ánimo eufórico |
raras | |
|
Trastornos del sistema nervioso |
cefalea |
frecuentes |
|
mareo, somnolencia |
poco frecuentes |
|
Sistema de clasificación de órganos |
Reacción adversa |
Frecuencia |
|
trastorno cognoscitivo, letargia, disgeusia |
raras | |
|
Trastornos oculares |
conjuntivitis |
raras |
|
Trastornos del oído y del laberinto |
acúfenos |
raras |
|
Trastornos cardiacos |
palpitaciones |
poco frecuentes |
|
bradicardia, trastorno cardiovascular |
raras | |
|
Trastornos vasculares |
acaloramiento/rubefacción |
poco frecuentes |
|
Trastornos respiratorios, torácicos y mediastínicos |
hipo |
frecuentes |
|
dolor orofaríngeo, estornudos, tos, goteo postnasal, irritación de garganta |
raras | |
|
Trastornos gastrointestinales |
estreñimiento, dispepsia |
frecuentes |
|
eructos, náuseasT, vómitosT, enfermedad por reflujo gastroesofágico, dolor abdominal, boca seca, flatulencia |
poco frecuentes | |
|
perforación de úlcera de duodeno, estomatitis, distensión abdominal, heces duras, colitis neutropénica |
raras | |
|
Trastornos de la piel y del tejido subcutáneo |
erupción, acné |
poco frecuentes |
|
reacción de fotosensibilidad, hiperhidrosis, seborrea, lesión de la piel, erupción prurítica, síndrome de Stevens-Johnson/necrolisis epidérmica tóxica |
raras | |
|
prurito, urticaria |
no conocida | |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
debilidad muscular, calambres musculares |
raras |
|
Trastornos renales y urinarios |
disuria |
poco frecuentes |
|
polaquiuria |
raras | |
|
Trastornos generales y alteraciones en el lugar de administración |
fatiga |
frecuentes |
|
astenia, malestar general |
poco frecuentes | |
|
edema, malestar torácico, alteración de la marcha |
raras | |
|
Exploraciones complementarias |
ALT elevada |
frecuentes |
|
AST elevada, fosfatasa alcalina en sangre aumentada |
poco frecuentes | |
|
hematíes en orina positivos, sodio disminuido en sangre, peso disminuido, recuento disminuido de neutrófilos, presencia de glucosuria, excreción urinaria aumentada |
raras |
T Náuseas y vómitos fueron parámetros de eficacia en los 5 primeros días de tratamiento post-quimioterapia y sólo después se notificaron como reacciones adversas.
Descripción de reacciones adversas seleccionadas
Los perfiles de reacciones adversas en adultos en la extensión de Ciclos Múltiples de ensayos en HEC y MEC que se prolongó hasta un máximo de 6 ciclos adicionales de quimioterapia fueron por lo general similares a los observados en el Ciclo 1.
En un ensayo clínico adicional con comparador activo en 1.169 pacientes adultos que estaban recibiendo aprepitant y HEC, el perfil de reacciones adversas fue generalmente similar al observado en los otros ensayos HEC con aprepitant.
Se observaron otras reacciones adversas en pacientes adultos tratados con aprepitant para las náuseas y los vómitos posquirúrgicos (NVPQ), con una incidencia mayor que con ondansetrón: dolor en la zona superior del abdomen, ruidos intestinales anormales, estreñimiento*, disartria, disnea, hipoestesia, insomnio, miosis, náuseas, alteración sensitiva, molestias en el estómago, subíleo*, agudeza visual disminuida, sibilancia.
*Notificado en pacientes que tomaron una dosis más alta de aprepitant.
Notificación de sospechas de reacciones adversas
. Se invita a los sistema nacional
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento profesionales sanitarios a notificar las sospechas de reacciones adversas a través del de notificación incluido en el Apéndice V.
4.9 Sobredosis
En caso de sobredosis, se debe suspender la administración de EMEND y proporcionar tratamiento de apoyo general y vigilancia. Debido a la actividad antiemética de aprepitant, es posible que la emesis inducida por un medicamento no resulte eficaz.
Aprepitant no se puede eliminar mediante hemodiálisis.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Antieméticos y antinauseosos, código ATC: A04AD12
Aprepitant es un antagonista selectivo de alta afinidad por los receptores de la sustancia P neurocinina 1 (NKi) humana.
Régimen de tratamiento de 3 días de aprepitant en adultos
En 2 ensayos aleatorizados, doble ciego, en los que se incluyó un total de 1.094 pacientes adultos que recibían quimioterapia que incluía cisplatino > 70 mg/m2, aprepitant en combinación con un régimen de ondansetrón/dexametasona (ver sección 4.2) se comparó con un régimen estándar (placebo más ondansetrón 32 mg administrados por vía intravenosa el día 1 más dexametasona 20 mg por vía oral el día 1 y 8 mg por vía oral dos veces al día los días 2 a 4). Aunque en ensayos clínicos se usó una dosis intravenosa de 32 mg de ondansetrón, ésta ya no es la dosis recomendada. Ver la información de producto del antagonista 5-HT3 escogido para obtener información sobre la dosis adecuada.
La eficacia se basó en la evaluación de las siguientes medidas compuestas: respuesta completa (definida como ausencia de episodios eméticos sin uso de tratamiento de rescate) principalmente durante el Ciclo 1. Los resultados se evaluaron para cada ensayo individual y para los 2 ensayos combinados.
En la Tabla 1 se muestra un resumen de los resultados más importantes del ensayo obtenidos del análi sis combinado.
Tabla 1
Porcentaje de pacientes adultos que estaban recibiendo quimioterapia altamente emetógena que respondieron por grupo de tratamiento y fase - Ciclo 1
Régimen con Tratamiento estándar Diferencias*
aprepitant (N= 524) f
MEDIDAS COMPUESTAS (N= 521) f % % (IC del 95%)
%
|
Respuesta completa (sin emesis y |
sin tratamiento de rescate) | |||
|
Global (0-120 horas) |
67,7 |
47,8 |
19,9 |
(14,0, 25,8) |
|
0-24 horas |
86,0 |
73,2 |
12,7 |
(7,9, 17,6) |
|
25-120 horas |
71,5 |
51,2 |
20,3 |
(14,5, 26,1) |
|
MEDIDAS INDIVIDUALES | ||||
|
Sin emesis (sin episodios eméticos independientemente del |
uso de tratamiento de rescate) | |||
|
Global (0-120 horas) |
71,9 |
49,7 |
22,2 |
(16,4, 28,0) |
|
0-24 horas |
86,8 |
74,0 |
12,7 |
(8,0, 17,5) |
|
25-120 horas |
76,2 |
53,5 |
22,6 |
(17,0, 28,2) |
|
Sin náuseas significativas (EAV máxima < 25 mm en una escala de 0-100 mm) | ||||
|
Global (0-120 horas) |
72,1 |
64,9 |
7,2 |
(1,6, 12,8) |
|
25-120 horas |
74,0 |
66,9 |
7,1 |
(1,5, 12,6) |
*Los intervalos de confianza se calcularon sin ajuste por sexo ni quimioterapia concomitante, los cuales fueron incluidos en el análisis primario de cociente de posibilidades y modelos logísticos. t Sólo un paciente en el régimen de aprepitant tuvo datos en la fase aguda y se excluyó de los análisis de fase retardada y general; sólo un paciente en el régimen estándar tuvo datos en la fase retardada y se excluyó del análisis de fase aguda y general.
El tiempo estimado hasta la primera emesis en el análisis combinado se representa en el gráfico Kaplan-Meier de la Figura 1.
Figura 1
Porcentaje de pacientes adultos que estaban recibiendo quimioterapia altamente emetógena que siguieron sin padecer emesis con el tiempo - Ciclo 1
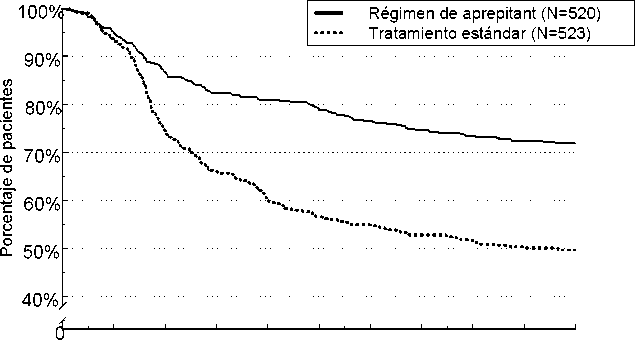
0 12 24 36 48 60 72 84 96 108 120
Tiempo (horas)
También se observaron diferencias estadísticamente significativas en eficacia en cada uno de los 2 ensayos individuales.
En los mismos 2 ensayos clínicos, 851 pacientes adultos continuaron en la extensión de Ciclos Múltiples hasta un máximo de 5 ciclos adicionales de quimioterapia. La eficacia del régimen de aprepitant se mantuvo aparentemente durante todos los ciclos.
En un ensayo aleatorizado, doble ciego con un total de 866 pacientes adultos (864 mujeres, 2 varones) que estaban recibiendo quimioterapia, que incluía ciclofosfamida 750-1.500 mg/m2; o ciclofosfamida 500-1.500 mg/m2 y doxorrubicina (<_60 mg/m2) o epirubicina (<100 mg/m2), se comparó aprepitant en combinación con un régimen de ondansetrón/dexametasona (ver sección 4.2) con el tratamiento estándar (placebo más ondansetrón 8 mg por vía oral (dos veces en el día 1 y cada 12 horas en los días 2 y 3) más dexametasona 20 mg por vía oral en el día 1).
La eficacia se basó en la evaluación de las medidas compuestas: respuesta completa (definida como ausencia de episodios eméticos sin uso de tratamiento de rescate) principalmente durante el Ciclo 1.
En la Tabla 2 se muestra un resumen de los resultados más importantes del ensayo.
Tabla 2
Porcentaje de pacientes adultos que respondieron por grupo de tratamiento y fase - Ciclo 1 Quimioterapia moderadamente emetógena
|
Régimen |
Tratamiento | |
|
con |
estándar | |
|
MEDIDAS COMPUESTAS aprepitant |
(N= 424) | |
|
(N= 433) f |
% |
% (IC del 95%) |
|
% |
Respuesta completa (sin emesis y sin tratamiento de rescate)_
Global (0-120 horas) 50,8 42,5 8,3 (1,6, 15,0)
0-24 horas 75,7 69,0 6,7 (0,7, 12,7)
25-120 horas_55,4_49,1_6,3 (-0,4, 13,0)
MEDIDAS INDIVIDUALES_
Sin emesis (sin episodios eméticos independientemente del uso de tratamiento de rescate)
Global (0-120 horas) 75,7 58,7 17,0 (10,8, 23,2)
0-24 horas 87,5 77,3 10,2 (5,1, 15,3)
25-120 horas_80,8_69,1_11,7 (5,9,17,5)
Sin náuseas significativas (EAV máxima < 25 mm en una escala de 0-100 mm)_
Global (0-120 horas) 60,9 55,7 5,3 (-1,3, 11,9)
0-24 horas 79,5 78,3 1,3 (-4,2, 6,8)
incluido cáncer colorrectal, 13 % con cáncer de pulmón y 6 % con algún cáncer de tipo ginecológico. El régimen de aprepitant en combinación con un régimen de ondansetrón/dexametasona (ver sección 4.2) se comparó con el tratamiento estándar (placebo en combinación con ondansetrón 8 mg vía oral (dos veces al día el día 1, y cada 12 horas los días 2 y 3) más dexametasona 20 mg vía oral el día 1).
La eficacia se basó en la evaluación de las siguientes variables primaria y secundaria más importantes: Sin vómitos en el periodo completo (0 a 120 horas después de la quimioterapia), evaluación de la seguridad y tolerancia del régimen de aprepitant para las náuseas y los vómitos inducidos por quimioterapia (NVIQ) y respuesta completa (definida como sin vómitos y sin uso de tratamiento de rescate) en el periodo completo (0 a 120 horas después de la quimioterapia). Además, se evaluó la ausencia de náuseas significativas en el periodo completo (0 a 120 horas después de la quimioterapia) como variable exploratoria, y en las fases aguda y retardada como análisis post-hoc.
En la Tabla 3 se muestra un resumen de los resultados más importantes del ensayo.
Tabla 3
Porcentaje de pacientes adultos que respondieron por grupo de tratamiento y fase en el ensayo 2 -
Ciclo 1
Quimioterapia moderadamente emetógena
|
Régimen con aprepitant (N= 425) % |
Tratamiento estándar (N= 406) % |
Diferencias* % (IC del 95 %) | ||
|
Respuesta completa (sin emesis y sin tratamiento de rescate) | ||||
|
Global (0-120 horas) |
68,7 |
56,3 |
12,4 |
(5,9, 18,9) |
|
0-24 horas |
89,2 |
80,3 |
8,9 |
(4,0, 13,8) |
|
25-120 horas |
70,8 |
60,9 |
9,9 |
(3,5, 16,3) |
|
Sin emesis (sin episodios eméticos independientemente del uso t |
e tratamiento | |||
|
de rescate) | ||||
|
Global (0-120 horas) |
76,2 |
62,1 |
14,1 |
(7,9, 20,3) |
|
0-24 horas |
92,0 |
83,7 |
8,3 |
(3,9, 12,7) |
|
25-120 horas |
77,9 |
66,8 |
11,1 |
(5,1, 17,1) |
|
Sin nauseas significativas (EAV máxima < 25 mm en una escala de | ||||
|
0-100 mm) | ||||
|
Global (0-120 horas) |
73,6 |
66,4 |
7,2 |
(1,0, 13,4) |
|
0-24 horas |
90,9 |
86,3 |
4,6 |
(0,2, 9,0) |
|
25-120 horas |
74,9 |
69,5 |
5,4 |
(-0,7, 11,5) |
*Los intervalos de confianza se calcularon sin ajuste por sexo ni región, los cuales fueron incluidos en el análisis primario utilizando modelos logísticos.
El beneficio del tratamiento en combinación de aprepitant en la población del ensayo total fue dirigido principalmente por los resultados observados en pacientes con bajo control con el régimen estándar como en mujeres, aunque los resultados fueron numéricamente mejores independientemente de la edad, tipo de tumor o sexo. La respuesta completa al régimen de aprepitant y al tratamiento estándar, respectivamente, se alcanzó en 209/324 (65 %) y 161/320 (50 %) en mujeres y 83/101 (82 %) y 68/87 (78 %) de varones.
Población pediátrica
En un ensayo clínico aleatorizado, doble ciego, controlado con comparador activo en el que se incluyó a 302 niños y adolescentes (de entre 6 meses y 17 años de edad) que recibieron quimioterapia moderada o altamente emetógena, el régimen de aprepitant se comparó con un régimen de control para la prevención de NVIQ. La eficacia del régimen de aprepitant se evaluó en un único ciclo (Ciclo 1). A los pacientes se les dio la oportunidad de recibir aprepitant en abierto en ciclos posteriores (Ciclos 2-6 opcionales); sin embargo, no se evaluó la eficacia en estos ciclos opcionales. El régimen de aprepitant para adolescentes de entre 12 y 17 años de edad (n=47) consistió en EMEND cápsulas de 125 mg por vía oral el día 1 y 80 mg/día los días 2 y 3, en combinación con ondansetrón el día 1. El régimen de aprepitant para niños de entre 6 meses y menos de 12 años de edad (n=105) consistió en EMEND polvo para suspensión oral 3,0 mg/kg (hasta 125 mg) por vía oral el día 1 y 2,0 mg/kg (hasta 80 mg) por vía oral los días 2 y 3, en combinación con ondansetrón el día 1. El régimen de control en adolescentes de entre 12 y 17 años de edad (n=48) y niños de entre 6 meses y menos de 12 años de edad (n=102) consistió en placebo de aprepitant los días 1,2 y 3 en combinación con ondansetrón el día 1. EMEND o placebo y ondansetrón se administraron 1 hora y 30 minutos antes del inicio de la quimioterapia, respectivamente. Se permitió dexametasona intravenosa como parte del régimen antiemético en pacientes pediátricos de ambos grupos de edad, a criterio del médico. Fue necesaria una disminución de la dosis de dexametasona (50 %) en los pacientes pediátricos que recibieron aprepitant. No fue necesaria una disminución de la dosis en los pacientes pediátricos que recibieron el régimen de control. De los pacientes pediátricos, el 29 % del régimen de aprepitant y el 28 % del régimen de control utilizaron dexametasona como parte del régimen en el Ciclo 1.
La actividad antiemética de EMEND se evaluó durante un periodo de 5 días (120 horas) después del inicio de la quimioterapia el día 1. El criterio de valoración principal fue la respuesta completa en la fase retardada (de 25 a 120 horas después del comienzo de la quimioterapia) en el Ciclo 1. En la tabla 4 se muestra un resumen de los resultados más importantes del ensayo.
Tabla 4
Número (%) de pacientes pediátricos con respuesta completa y sin vómitos por grupo de tratamiento y
fase - Ciclo 1 (población por intención de tratar)
|
Régimen de aprepitant n/m (%) |
Régimen de control n/m (%) | |
|
CRITERIO DE VALORACIÓN PRINCIPAL | ||
|
Respuesta completa* - Fase retardada |
77/152 (50,7)t |
39/150 (26,0) |
|
OTROS CRITERIOS DE VALORACIÓN PRE-ESPECIFICADOS | ||
|
Respuesta completa* - Fase aguda |
101/152 (66,4)* |
78/150 (52,0) |
|
Respuesta completa - Fase global |
61/152 (40,1)t |
30/150 (20,0) |
|
i* Sin vómitos8 - Fase global |
71/152 (46,7)t |
32/150 (21,3) |
|
*Respuesta completa = Sin vómitos, náuseas o arcadas improductivas y sin uso de medicación de | ||
|
rescate. | ||
|
Tp < 0,01 en comparación con el régimen de control *p < 0,05 en comparación con el régimen de control §Sin vómitos = Sin vómitos, náuseas o arcadas improductivas | ||
|
n/m = Número de pacientes con la respuesta deseada/número de pacientes incluidos en el punto temporal. Fase aguda: de 0 a 24 horas después del inicio de la quimioterapia. Fase retardada: de 25 a 120 horas después del inicio de la quimioterapia. Fase global: de 0 a 120 horas después del inicio de la quimioterapia. | ||
El tiempo estimado hasta el primer vómito después del comienzo del tratamiento con quimioterapia fue mayor con el régimen de aprepitant (la mediana del tiempo estimado hasta el primer vómito fue
94,5 horas) en comparación con el grupo del régimen de control (la mediana del tiempo estimado hasta el primer vómito fue 26,0 horas) tal y como se representa en las curvas de Kaplan-Meier de la Figura 2.
Figura 2
Tiempo hasta el primer episodio de vómitos desde el inicio de la administración de la quimioterapia -pacientes pediátricos en la fase global - Ciclo 1 (población por intención de tratar)

Un análisis de la eficacia en subpoblaciones en el Ciclo 1 demostró que, independientemente de la categoría de edad, sexo, uso de dexametasona para la profilaxis antiemética y emetogenicidad de la quimioterapia, el régimen de aprepitant proporcionó un mejor control que el régimen de control con respecto a los criterios de valoración de la respuesta completa.
5.2 Propiedades farmacocinéticas
Aprepitant muestra una farmacocinética no lineal. Tanto el aclaramiento como la biodisponibilidad absoluta disminuyen al aumentar la dosis.
Absorción
La biodisponibilidad oral absoluta media de aprepitant es de 67% para la cápsula de 80 mg y de 59% para la cápsula de 125 mg. La concentración plasmática máxima media (Cmax) de aprepitant se alcanzó aproximadamente a las 4 horas (tmax). La administración oral de la cápsula con un desayuno estándar de aproximadamente 800 Kcal ocasionó un aumento de hasta el 40% en el AUC de aprepitant. Este aumento no se consideró clínicamente de interés.
La farmacocinética de aprepitant es no lineal en el intervalo de la dosis clínica. En adultos jóvenes sanos, el aumento en el AUC0.^ fue un 26% mayor que proporcional a la dosis entre las dosis únicas de 80 mg y 125 mg administradas en estado posprandial.
Tras la administración oral de una dosis única de 125 mg de EMEND el día 1 y 80 mg una vez al día los días 2 y 3, el AUC0.24h (media ± DE) fue 19,6 ± 2,5 ^g»h/ml y 21,2 ± 6,3 ^g»h/ml los días 1 y 3, respectivamente. La Cmax fue 1,6 ± 0,36 ^g/ml y 1,4 ± 0,22 ^g/ml los días 1 y 3, respectivamente.
Distribución
Aprepitant se une fuertemente a proteínas, con una media del 97%. La media geométrica del volumen aparente de distribución en el estado equilibrio (Vdee) es aproximadamente de 66 litros en el ser humano.
Biotransformación
Aprepitant se metaboliza extensamente. En adultos jóvenes sanos, aprepitant representa aproximadamente el 19% de la radiactividad plasmática durante 72 horas después de una dosis única intravenosa de 100 mg de [C14]-fosaprepitant, un profármaco de aprepitant, lo que indica una importante presencia de metabolitos en el plasma. En el plasma humano se han identificado doce metabolitos de aprepitant. El metabolismo de aprepitant se produce en gran medida por oxidación en el anillo de morfolina y sus cadenas laterales y los metabolitos resultantes sólo fueron débilmente activos. Estudios in vitro en los que se usaron microsomas hepáticos humanos indicaron que aprepitant se metaboliza principalmente a través de CYP3A4 y posiblemente con una contribución menor a través de CYP1A2 y CYP2C19.
Eliminación
Aprepitant no se elimina inalterado en la orina. Los metabolitos se eliminan en la orina y a través de excreción biliar en las heces. Después de una dosis única intravenosa de 100 mg de [C14]-fosaprepitant, un profármaco de aprepitant a sujetos sanos, el 57% de la radiactividad se recuperó en la orina y el 45% en las heces.
El aclaramiento plasmático de aprepitant es dependiente de la dosis, disminuyendo al aumentar la dosis y oscilando aproximadamente entre 60 a 72 ml/min en el intervalo de la dosis terapéutica. La semivida terminal osciló entre aproximadamente 9 a 13 horas.
Farmacocinética en poblaciones especiales
Sexo: Tras la administración oral de una dosis única de 125 mg de aprepitant, la Cmax de aprepitant es un 16% superior en las mujeres en comparación con los varones. La semivida de aprepitant es un 25% inferior en las mujeres en comparación con los varones y su tmax se produce en aproximadamente el mismo tiempo. Estas diferencias no se consideraron clínicamente significativas. EMEND no requiere ajuste de dosis en función del sexo.
Insuficiencia hepática: La insuficiencia hepática leve (Child-Pugh clase A) no afecta a la farmacocinética de aprepitant en un grado clínicamente relevante. No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. De los datos disponibles no pueden extraerse conclusiones relativas a la influencia de la insuficiencia hepática moderada (Child-Pugh clase B) sobre la farmacocinética de aprepitant. No existen datos clínicos ni farmacocinéticos de pacientes con insuficiencia hepática grave (Child-Pugh clase C).
Insuficiencia renal: Se administró una dosis única de 240 mg de aprepitant a pacientes con insuficiencia renal grave (CrCl < 30 ml/min) y a pacientes con nefropatía terminal que requería hemodiálisis.
En los pacientes con insuficiencia renal grave, el AUC0.^ de aprepitant total (no unido y unido a proteínas) disminuyó en un 21% y la Cmax disminuyó en un 32%, respecto a los sujetos sanos. En los pacientes con nefropatía terminal sometidos a hemodiálisis, el AUC0.^ de aprepitant total disminuyó en un 42% y la Cmax disminuyó en un 32%. Debido a los modestos descensos en la unión a proteínas de aprepitant en los pacientes con enfermedad renal, el AUC de aprepitant no unido farmacológicamente activo no se vio significativamente afectado en los pacientes con insuficiencia renal en comparación con los sujetos sanos. La hemodiálisis realizada 4 ó 48 horas después de la administración no tuvo efectos significativos sobre la farmacocinética de aprepitant; en el dializado se recuperó menos de 0,2% de la dosis.
En pacientes con insuficiencia renal o en pacientes con nefropatía terminal sometidos a hemodiálisis no es necesario ajustar la dosis de EMEND.
Población pediátrica: Como parte de un régimen de 3 días de duración, la dosis de aprepitant cápsulas (125/80/80 mg) en pacientes adolescentes (de entre 12 y 17 años de edad) logró un AUC0-24h por encima de 17 ^g^h/ml el día 1 y concentraciones (Cmin) al final de los días 2 y 3 superiores a 0,4 ^g/ml en la mayoría de los pacientes. La concentración plasmática máxima media (Cmax) fue aproximadamente 1,3 ^g/ml el día 1, alcanzándose a las 4 horas aproximadamente. Como parte de un régimen de 3 días de duración, la dosis de aprepitant polvo para suspensión oral (3/2/2 mg/kg) en pacientes de entre 6 meses y menos de 12 años de edad alcanzó un AUC0.24h superior a 17 ^g^h/ml el día 1, con concentraciones (Cmin) al final de los días 2 y 3 por encima de 0,1 ^g/ml en la mayoría de los pacientes. La concentración plasmática máxima media (Cmax) fue aproximadamente 1,2 ^g/ml el día 1, alcanzándose entre 5 y 7 horas.
Un análisis farmacocinético poblacional de aprepitant en pacientes pediátricos (de entre 6 meses y 17 años de edad) sugiere que el sexo y la raza no tienen un efecto clínicamente significativo sobre la farmacocinética de aprepitant.
Relación entre concentración y efecto
Usando un trazador altamente específico del receptor NKi, los estudios de tomografía por emisión de positrones (PET) en varones jóvenes sanos han demostrado que aprepitant penetra en el cerebro y ocupa los receptores NK1 de forma dependiente de la dosis y de la concentración plasmática. Se predice que las concentraciones plasmáticas de aprepitant alcanzadas en adultos con el régimen de 3 días de EMEND proporcionan una ocupación superior al 95% de los receptores NK1 cerebrales.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de toxicidad, genotoxicidad, potencial carcinogénico, toxicidad para la reproducción y el desarrollo, a dosis únicas y repetidas. Sin embargo, se debe tener en cuenta que la exposición sistémica en roedores fue similar o incluso inferior a la exposición terapéutica en seres humanos a la dosis de 125 mg/80 mg. En especial, aunque no se observaron efectos adversos en los estudios sobre la reproducción a los niveles de exposición en seres humanos, las exposiciones en animales no son suficientes para hacer una valoración de riesgo adecuada en el hombre.
En un estudio de toxicidad juvenil en ratas tratadas desde el día 10 después del nacimiento hasta el día 63, aprepitant ocasionó en las hembras una apertura vaginal prematura a partir de 250 mg/kg dos veces al día y en los machos un retraso en la separación del prepucio a partir de 10 mg/kg dos veces al día. No hubo márgenes de exposición clínicamente relevantes. No se observaron efectos sobre el apareamiento, la fertilidad o la supervivencia embrionaria o fetal ni cambios patológicos en los órganos reproductores relacionados con el tratamiento. En un estudio de toxicidad juvenil en perros tratados desde el día 14 después del nacimiento hasta el día 42, se detectó una disminución del peso testicular y del tamaño de las células de Leydig en los machos con 6 mg/kg/día y un aumento del peso del útero, hipertrofia del útero y del cuello del útero y edema de los tejidos vaginales en las hembras a partir de 4 mg/kg/día. No hubo márgenes de exposición clínicamente relevantes de aprepitant. En el tratamiento a corto plazo de acuerdo con la pauta posológica recomendada, se considera poco probable que estos resultados sean clínicamente relevantes.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Hidroxipropilcelulosa (E-463)
Lauril sulfato de sodio (E-463)
Sacarosa Lactosa (anhidra)
Óxido de hierro rojo (E-172)
Estearil fumarato de sodio (E-485)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
Sobre sin abrir: 2 años.
Tras la reconstitución: La suspensión oral se puede tener a temperatura ambiente (no a más de 30°C) hasta 3 horas. También se puede conservar refrigerada (entre 2°C y 8°C) hasta 72 horas..
6.4 Precauciones especiales de conservación
Este medicamento no requiere ninguna temperatura especial de conservación. Conservar en el embalaje original para protegerlo de la humedad.
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Sobres de PET/aluminio/LLDPE.
Caja de un solo uso.
Cada caja contiene un sobre con el polvo para suspensión oral, un dispensador oral de 1 ml y otro de 5 ml (de polipropileno con un émbolo rematado con un anillo de silicona), una tapa y un vaso de mezcla (de polipropileno).
6.6 Precauciones especiales de eliminación y otras manipulaciones
El contenido de cada sobre monodosis se debe suspender en 4,6 ml de agua, dando una concentración final de 25 mg por ml.
• Para más detalles sobre la preparación y la administración de la suspensión, ver el prospecto y las instrucciones para profesionales del sector sanitario para la preparación de la suspensión oral.
• Utilizar el dispensador oral de 5 ml para medir 4,6 ml de agua, que se añaden dentro del vaso de mezcla.
• Verter el contenido completo del sobre en los 4,6 ml de agua y mezclar.
• Una vez mezclado, medir el volumen recomendado (dosis) de suspensión con el dispensador oral. Elegir el dispensador oral según la dosis. Usar el dispensador oral de 1 ml si la dosis es 1 ml o menos y usar el dispensador oral de 5 ml si la dosis es más de 1 ml. Administrar la dosis por vía oral. Si la dosis no se administra inmediatamente después de la medición, el dispensador oral lleno se puede tener refrigerado (entre 2°C y 8°C) durante un máximo de 72 horas antes de su uso.
• Antes de su administración, la suspensión oral se puede tener a temperatura ambiente (no a más de 30°C) hasta 3 horas.
• Desechar la suspensión que sobre y los materiales que hayan estado en contacto con ella.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme Ltd.
Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/262/011
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 11/noviembre/2003 Fecha de la última renovación: 22/septiembre/2008
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
ANEXO II
A. FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
Nombre y dirección del(de los) fabricante(s) responsable(s) de la liberación de los lotes
Merck Sharp & Dohme B.V. Waarderweg 39 2031 BNHaarlem Países Bajos
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica.
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y cualquier actualización posterior publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado
en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en
cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
ANEXO III
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
EMEND 40 mg cápsulas duras Aprepitant
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura contiene 40 mg de aprepitant.
3. LISTA DE EXCIPIENTES
Contiene sacarosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
1 cápsula dura 5 x 1 cápsula dura
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/262/007 1 cápsula dura EU/1/03/262/008 5 x 1 cápsula dura
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
EMEND 40 mg
INFORMACIÓN MÍNIMA A INCLUIR EN BLISTERS O TIRAS TEXTO DEL BLISTER
1. NOMBRE DEL MEDICAMENTO
EMEND 40 mg cápsulas duras Aprepitant
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
MSD
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
1. NOMBRE DEL MEDICAMENTO
EMEND 80 mg cápsulas duras Aprepitant
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura contiene 80 mg de aprepitant.
3. LISTA DE EXCIPIENTES
Contiene sacarosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
1 cápsula dura
Envase para un tratamiento de 2 días conteniendo: 2 cápsulas duras de 80 mg 5 x 1 cápsula dura
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/262/001 1 cápsula dura EU/1/03/262/002 2 x 1 cápsula dura EU/1/03/262/003 5 x 1 cápsula dura
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
EMEND 80 mg
INFORMACIÓN QUE DEBE FIGURAR EN EL ACONDICIONAMIENTO PRIMARIO (QUE INCLUYE 2 CÁPSULAS DURAS DE 80 MG)
ACONDICIONAMIENTO PRIMARIO - TRIPLE - Envase para un tratamiento de 2 días
1. NOMBRE DEL MEDICAMENTO
EMEND 80 mg cápsulas duras Aprepitant
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura contiene 80 mg de aprepitant.
3. LISTA DE EXCIPIENTES
Contiene sacarosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Envase para un tratamiento de 2 días conteniendo: 2 cápsulas duras de 80 mg
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN_
Vía oral.
Leer el prospecto adjunto antes de utilizar este medicamento.
Si desea información adicional sobre cómo tomar EMEND vea el prospecto adjunto.
CUÁNDO y CÓMO tomar EMEND
Su médico le ha recetado EMEND, un antiemético, para ayudarle a evitar las náuseas y los vómitos asociados a la quimioterapia.
CÓMO:
EMEND 80 mg cápsulas duras se toma solamente una vez al día durante 2 días consecutivos.
Las cápsulas de EMEND pueden tomarse con o sin alimentos.
No extraiga todas las cápsulas al mismo tiempo.
Para extraerlas, empuje las cápsulas desde este lado.
Comienzo del tratamiento CUÁNDO:
Tome una cápsula de EMEND 80 mg cada mañana. Empiece al día siguiente a la quimioterapia. Día 1 Día 2
EMEND 80 mg, cápsula
Dado que las náuseas y los vómitos pueden producirse en los días siguientes a la quimioterapia, es importante que tome EMEND durante 2 días consecutivos tal como le ha recetado su médico.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/262/002
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
1. NOMBRE DEL MEDICAMENTO
EMEND 80 mg cápsulas duras Aprepitant
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
MSD
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
1. NOMBRE DEL MEDICAMENTO
EMEND 125 mg cápsulas duras Aprepitant
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura contiene 125 mg de aprepitant.
3. LISTA DE EXCIPIENTES
Contiene sacarosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
1 cápsula dura 5 x 1 cápsula dura
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/262/004 1 cápsula dura EU/1/03/262/005 5 x 1 cápsula dura
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
EMEND 125 mg
1. NOMBRE DEL MEDICAMENTO
EMEND 125 mg cápsulas duras Aprepitant
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
MSD
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
1. NOMBRE DEL MEDICAMENTO
EMEND 165 mg cápsulas duras Aprepitant
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura contiene 165 mg de aprepitant.
3. LISTA DE EXCIPIENTES
Contiene sacarosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
1 cápsula dura 6x1 cápsula dura
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/262/009 1 cápsula dura EU/1/03/262/010 6x 1 cápsula dura
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
EMEND 165 mg
1. NOMBRE DEL MEDICAMENTO
EMEND 165 mg cápsulas duras Aprepitant
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
MSD
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
EMBALAJE EXTERIOR - ENVASE TRIPLE (CAJA) - Envase para un tratamiento de 3 días
1. NOMBRE DEL MEDICAMENTO
EMEND 125 mg cápsula dura EMEND 80 mg cápsulas duras Aprepitant
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura de 125 mg contiene 125 mg de aprepitant. Cada cápsula dura de 80 mg contiene 80 mg de aprepitant.
3. LISTA DE EXCIPIENTES
Contiene sacarosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Envase para un tratamiento de 3 días conteniendo:
1 cápsula dura de 125 mg y
2 cápsulas duras de 80 mg
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/262/006
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
EMEND 125 mg/80 mg
INFORMACIÓN QUE DEBE FIGURAR EN EL ACONDICIONAMIENTO PRIMARIO (QUE INCLUYE 1 CÁPSULA DURA DE 125 MG Y 2 CÁPSULAS DURAS DE 80 MG)
ACONDICIONAMIENTO PRIMARIO - TRIPLE - Envase para un tratamiento de 3 días
1. NOMBRE DEL MEDICAMENTO
EMEND 125 mg cápsula dura EMEND 80 mg cápsulas duras Aprepitant
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula dura de 125 mg contiene 125 mg de aprepitant. Cada cápsula dura de 80 mg contiene 80 mg de aprepitant.
3. LISTA DE EXCIPIENTES
Contiene sacarosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Envase para un tratamiento de 3 días conteniendo:
1 cápsula dura de 125 mg y
2 cápsulas duras de 80 mg
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía oral.
Leer el prospecto adjunto antes de utilizar este medicamento.
Si desea información adicional sobre cómo tomar EMEND vea el prospecto adjunto.
CUÁNDO y CÓMO tomar EMEND
Su médico le ha recetado EMEND, un antiemético, para ayudarle a evitar las náuseas y los vómitos asociados a la quimioterapia.
CÓMO:
EMEND se toma solamente una vez al día durante 3 días consecutivos.
Las cápsulas de EMEND pueden tomarse con o sin alimentos.
No extraiga todas las cápsulas al mismo tiempo.
Para extraerlas, empuje las cápsulas desde este lado.
Comienzo del tratamiento CUÁNDO:
Tome una cápsula de EMEND 125 mg por vía oral 1 hora ANTES de empezar la quimioterapia. Día 1
EMEND 125 mg, cápsula CUÁNDO:
Tome una cápsula de EMEND 80 mg cada mañana durante los dos días siguientes.
Día 2 Día 3
EMEND 80 mg, cápsula
Dado que las náuseas y los vómitos pueden producirse en los días siguientes a la quimioterapia, es importante que tome EMEND durante 3 días consecutivos tal como le ha recetado su médico.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/262/006
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CAJA de EMEND 125 mg polvo para suspensión oral
1. NOMBRE DEL MEDICAMENTO
EMEND 125 mg polvo para suspensión oral Aprepitant
Para niños de entre 6 meses y menos de 12 años de edad
2. PRINCIPIO(S) ACTIVO(S)
Cada sobre contiene 125 mg de aprepitant. Tras la reconstitución, 1 ml de suspensión oral contiene 25 mg de aprepitant.
3. LISTA DE EXCIPIENTES
Contiene sacarosa y lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo para suspension oral.
Un sobre, dos dispensadores orales, una tapa y un vaso de mezcla.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme Limited Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/262/011
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACION EN BRAILLE
EMEND 125 mg polvo para suspensión oral
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
Sobre de EMEND 125 mg polvo para suspensión oral
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
EMEND 125 mg polvo para suspensión oral
Aprepitant
Vía oral
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
6. OTROS
B. PROSPECTO
Prospecto: información para el usuario
EMEND 40 mg cápsulas duras
aprepitant
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero. incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4
Contenido del prospecto
1. Qué es EMEND y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar EMEND
3. Cómo tomar EMEND
4. Posibles efectos adversos
5. Conservación de EMEND
6. Contenido del envase e información adicional
1. Qué es EMEND y para qué se utiliza
EMEND contiene el principio activo aprepitant y pertenece a un grupo de medicamentos llamados “antagonistas del receptor de la neurocinina 1 (NKj)” (antieméticos y antinauseosos).
El cerebro tiene una zona específica que controla las náuseas y los vómitos. EMEND actúa bloqueando las señales a esta zona y se usa para evitar las náuseas y vómitos después de la cirugía en adultos.
2. Qué necesita saber antes de empezar a tomar EMEND No tome EMEND:
- si es alérgico a aprepitant o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
Advertencias y precauciones
Consulte a su médico, farmacéutico o enfermero antes de empezar a tomar EMEND.
Antes del tratamiento con este medicamento, informe a su médico si tiene una enfermedad del hígado, porque su hígado es importante para eliminar el medicamento de su cuerpo. Por lo tanto, su médico puede tener que controlar el estado de su hígado.
Niños y adolescentes
No dé EMEND 40 mg a niños y adolescentes menores de 18 años de edad, porque las cápsulas de 40 mg no se han estudiado en esta población.
Uso de EMEND con otros medicamentos
Este medicamento puede afectar a otros medicamentos.
Los efectos de EMEND o de otros medicamentos pueden estar influenciados si toma EMEND junto con otros medicamentos, incluyendo los indicados a continuación. Consulte a su médico o farmacéutico si está tomando alguno de los siguientes medicamentos:
- medicamentos anticonceptivos que pueden incluir píldoras anticonceptivas, parches cutáneos, implantes y ciertos dispositivos intrauterinos (DIUs) que liberan hormonas, puede que no funcionen adecuadamente cuando se toman junto con EMEND. Durante el tratamiento con EMEND y hasta 2 meses después de usar EMEND, deben utilizarse otros métodos o métodos adicionales de anticoncepción no hormonales,
- pimozida (usado para tratar enfermedades psiquiátricas),
- terfenadina; astemizol (usados para la fiebre del heno y otras enfermedades alérgicas),
- cisaprida (usado para tratar problemas digestivos),
- medicamentos que contengan alcaloides derivados del ergot (usados para tratar migrañas),
- rifampicina, claritromicina, telitromicina (antibióticos usados para tratar infecciones),
- fenitoína (un medicamento usado para tratar las convulsiones),
- carbamazepina (usado para tratar la depresión y la epilepsia),
- midazolam, fenobarbital (medicamentos usados para tranquilizar o para ayudar a dormir),
- hierba de San Juan (un preparado a base de plantas usado para tratar la depresión),
- inhibidores de la proteasa (usados para tratar infecciones por VIH),
- nefazodona (usado para tratar la depresión),
- ketoconazol, excepto champú (usado para tratar el síndrome de Cushing - cuando el cuerpo produce un exceso de cortisol),
- itraconazol, voriconazol, posaconazol (antifungicos),
- corticosteroides (tales como dexametasona)
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o pudiera tener que tomar cualquier otro medicamento.
Embarazo y lactancia
No debe utilizar este medicamento durante el embarazo a no ser que sea claramente necesario. Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento.
Para información relacionada con el control de natalidad, ver ‘Uso de EMEND con otros medicamentos’.
Se desconoce si EMEND pasa a la leche materna; por tanto, no se recomienda dar el pecho durante el tratamiento con este medicamento. Es importante informar al médico antes de tomar este medicamento si está dando el pecho a su bebé o tiene previsto hacerlo.
Conducción y uso de máquinas
Se debe tener en cuenta que algunas personas tienen mareo y sueño después de tomar EMEND. Si se siente mareado o somnoliento, evite conducir o usar máquinas después de tomar este medicamento (ver ‘Posibles efectos adversos’).
EMEND contiene sacarosa
EMEND cápsulas contiene sacarosa. Si su médico le ha indicado que padece una intolerancia a ciertos azúcares, consulte con él antes de tomar este medicamento.
3. Cómo tomar EMEND
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
La dosis oral recomendada de EMEND es una cápsula de 40 mg en las 3 horas anteriores al inicio de la anestesia.
Trague la cápsula entera con algún líquido.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Deje de tomar EMEND y acuda a su médico inmediatamente si nota alguno de los siguientes efectos adversos, que pueden ser graves, y para los que puede necesitar tratamiento médico urgente:
- Ronchas, sarpullido, picor, dificultad para respirar o tragar (frecuencia no conocida, no puede estimarse a partir de los datos disponibles): estos son signos de una reacción alérgica.
Otros efectos adversos que se han comunicado se detallan a continuación.
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas) son:
- aumento de la cantidad de enzimas del hígado en la sangre.
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas) son:
- dolor alto de estómago, ruidos intestinales anómalos, boca seca, náuseas, molestias en el estómago, estreñimiento intenso, intestino delgado que no funciona adecuadamente (subíleon),
- percepción o sensibilidad disminuida (especialmente en la piel), trastornos sensitivos, dificultad para hablar,
- disminución del tamaño de la pupila, disminución de la agudeza visual (pérdida de visión),
- incapacidad para dormir,
- ritmo cardiaco lento,
- silbidos al respirar, dificultad para respirar.
Efectos adversos de frecuencia no conocida:
- síndrome de Stevens-Johnson/necrolisis epidérmica tóxica (reacción de la piel grave rara). Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de EMEND
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en el embalaje original para protegerlo de la humedad.
No extraer la cápsula de su blister hasta el momento de tomarla.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de EMEND
- El principio activo es aprepitant. Cada cápsula contiene 40 mg de aprepitant.
- Los demás componentes son: sacarosa, celulosa microcristalina (E-460), hidroxipropilcelulosa (E-463), lauril sulfato de sodio, gelatina, dióxido de titanio (E-171), laca, hidróxido de potasio, óxido de hierro negro (E-172) y óxido de hierro amarillo (E-172).
Aspecto del producto y contenido del envase
La cápsula dura de 40 mg es opaca con cuerpo blanco y tapa amarillo mostaza, con “464” y “40 mg” impreso en forma radial en tinta negra en el cuerpo.
Las cápsulas duras de EMEND 40 mg se suministran en los siguientes tamaños de envase:
- Blister de aluminio que contiene una cápsula de 40 mg.
- 5 blisters de aluminio que contienen una cápsula de 40 mg cada uno.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización Responsable de la fabricación
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN11 9BU Reino Unido
Merck Sharp & Dohme B. V. Waarderweg 39 2031 BNHaarlem Países Bajos
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Lietuva
UAB Merck Sharp & Dohme Tel. + 370 5278 02 47 msd_lietuva@merck.com
Luxembourg/Luxemburg
MSD Belgium BVBA/SPRL Tél/Tel: +32(0)27766211 dpoc_belux@merck.com
Magyarország
MSD Pharma Hungary Kft.
Tel.: +36 1 888 5300 hungary_msd@merck.com
Malta
Merck Sharp & Dohme Cyprus Limited Tel: 8007 4433 (+356 99917558) malta_info@merck.com
Nederland
Merck Sharp & Dohme BV Tel: 0800 9999000 (+31 23 5153153) medicalinfo.nl@merck.com
Norge
MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no
Belgique/Belgie/Belgien
MSD Belgium BVBA/SPRL
Tél/Tel: 0800 38 693 (+32(0)27766211)
Etarapna
MepK fflapn h floyM Etarapna EOOfl Tea.:+359 2 819 3737 info-msdbg@merck.com
Ceská republika
Merck Sharp & Dohme s.r.o.
Tel: +420 233 010 111 dpoc_czechslovak@merck.com
Danmark
MSD Danmark ApS Tlf: + 45 4482 4000 dkmail@merck.com
Deutschland
MSD SHARP & DOHME GMBH
Tel: 0800 673 673 673 (+49 (0) 89 4561 2612)
Eesti
Merck Sharp & Dohme OÜ Tel.:+372 6144 200 msdeesti@merck.com
|
EXXáSa MSD A.OB.E.E. T^: +30 210 98 97 300 dpoc_greece@merck.com |
Osterreich Merck Sharp & Dohme Ges.m.b.H. Tel: +43 (0) 1 26 044 msd-medizin@merck.com |
|
España Merck Sharp & Dohme de España, S.A. Tel: +34 91 321 06 00 msd info@merck.com |
Polska MSD Polska Sp. z o.o. Tel:+48 22 549 51 00 msdpolska@merck.com |
|
France MSD France Tél: +33 (0) 1 80 46 40 40 |
Portugal Merck Sharp & Dohme, Lda Tel: +351 21 4465700 clic@merck.com |
|
Hrvatska Merck Sharp & Dohme d.o.o. Tel: + 385 1 6611 333 croatia info@merck.com |
Romania Merck Sharp & Dohme Romania S.R.L. Tel:+40 21 529 29 00 msdromania@merck.com |
|
Ireland Merck Sharp & Dohme Ireland (Human Health) Limited Tel: +353 (0)1 2998700 medinfo ireland@merck.com |
Slovenija Merck Sharp & Dohme, inovativna zdravila d.o.o. Tel: +386 1 5204 201 msd_slovenia@merck.com |
|
Ísland Vistor hf. Simi: +354 535 7000 |
Slovenská republika Merck Sharp & Dohme, s. r. o. Tel:+421 2 58282010 dpoc_czechslovak@merck.com |
|
Italia MSD Italia S.r.l. Tel: +39 06 361911 medicalinformation.it@merck.com |
Suomi/Finland MSD Finland Oy Puh/Tel: +358 (0)9 804 650 info@msd.fi |
|
Kúrcpog Merck Sharp & Dohme Cyprus Limited TpV: 800 00 673 (+357 22866700) cyprus info@merck.com |
Sverige Merck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488 medicinskinfo@merck.com |
|
Latvija SIA Merck Sharp & Dohme Latvija Tel: +371 67364224 msd lv@merck.com |
United Kingdom Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com |
|
Fecha de la última revisión de este prospecto: |
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Prospecto: información para el usuario
EMEND 80 mg cápsulas duras
aprepitant
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted. Si es usted el progenitor de un niño que está tomando EMEND, lea esta información detenidamente.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted o al niño, y no debe dárselo a otras personas aunque tengan los mismos síntomas, ya que puede perjudicarles.
- Si usted o el niño experimentan efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4
Contenido del prospecto
1. Qué es EMEND y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar o dar EMEND
3. Cómo tomar EMEND
4. Posibles efectos adversos
5. Conservación de EMEND
6. Contenido del envase e información adicional
1. Qué es EMEND y para qué se utiliza
EMEND contiene el principio activo aprepitant y pertenece a un grupo de medicamentos llamados "antagonistas del receptor de la neurocinina 1 (NKj)". El cerebro tiene una zona específica que controla las náuseas y los vómitos. EMEND actúa bloqueando las señales a esta zona, y por tanto, reduciendo las náuseas y los vómitos. EMEND cápsulas se usa en adultos y adolescentes a partir de 12 años de edad, en combinación con otros medicamentos para evitar las náuseas y los vómitos que provoca un tipo de quimioterapia (tratamiento del cáncer) que desencadena de forma fuerte y moderada náuseas y vómitos (como cisplatino, ciclofosfamida, doxorubicina o epirubicina).
2. Qué necesita saber antes de empezar a tomar o dar EMEND No tome EMEND:
- si usted o el niño son alérgicos a aprepitant o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
- con medicamentos que contengan pimozida (utilizado para tratar enfermedades psiquiátricas), terfenadina y astemizol (utilizados para la rinitis alérgica y otros trastornos alérgicos), cisaprida (utilizado para tratar problemas digestivos). Informe al médico si usted o el niño están tomando estos medicamentos ya que el tratamiento debe ser modificado antes de que usted o el niño empiecen a tomar EMEND.
Advertencias y precauciones
Consulte a su médico, farmacéutico o enfermero antes de empezar a tomar EMEND o de dar al niño este medicamento.
Antes del tratamiento con este medicamento, informe al médico si usted o el niño tienen una enfermedad del hígado, porque el hígado es importante para eliminar el medicamento del cuerpo. Por lo tanto, el médico puede tener que controlar el estado de su hígado o el del niño.
Niños y adolescentes
No dé EMEND 80 mg cápsulas a niños menores de 12 años de edad, porque las cápsulas de 80 mg no se han estudiado en esta población.
Uso de EMEND con otros medicamentos
EMEND puede afectar a otros medicamentos durante y después del tratamiento con EMEND. Hay algunos medicamentos que no deben tomarse con EMEND (tales como pimozida, terfenadina, astemizol y cisaprida) o que requieren un ajuste en la dosis (véase también ‘No tome EMEND’).
Los efectos de EMEND o de otros medicamentos pueden estar influenciados si usted o el niño toman EMEND junto con otros medicamentos, incluyendo los indicados a continuación. Consulte a su médico o farmacéutico si usted o el niño están tomando alguno de los siguientes medicamentos:
- medicamentos anticonceptivos que pueden incluir píldoras anticonceptivas, parches cutáneos, implantes y ciertos dispositivos intrauterinos (DIUs) que liberan hormonas, puede que no funcionen adecuadamente cuando se toman junto con EMEND. Durante el tratamiento con EMEND y hasta 2 meses después de usar EMEND, deben utilizarse otros métodos o métodos adicionales de anticoncepción no hormonales,
- ciclosporina, tacrolimus, sirolimus, everolimus (inmunosupresores),
- alfentanilo, fentanilo (usados para tratar el dolor),
- quinidina (usado para tratar los latidos irregulares),
- irinotecán, etopósido, vinorelbina, ifosfamida (medicamentos usados para tratar el cáncer),
- medicamentos que contengan alcaloides derivados del ergot, tales como ergotamina y diergotamina (usados para tratar migrañas),
- warfarina, acenocumarol (diluyentes de la sangre; se pueden necesitar análisis de sangre),
- rifampicina, claritromicina, telitromicina (antibióticos usados para tratar infecciones),
- fenitoína (un medicamento usado para tratar las convulsiones),
- carbamazepina (usado para tratar la depresión y la epilepsia),
- midazolam, triazolam, fenobarbital (medicamentos usados para tranquilizar o para ayudar a dormir),
- hierba de San Juan (un preparado a base de plantas usado para tratar la depresión),
- inhibidores de la proteasa (usados para tratar infecciones por VIH),
- ketoconazol,excepto champú (usado para tratar el síndrome de Cushing - cuando el cuerpo produce un exceso de cortisol),
- itraconazol, voriconazol, posaconazol (antifúngicos),
- nefazodona (usado para tratar la depresión),
- corticosteroides (tales como dexametasona y metilprednisolona),
- medicamentos para la ansiedad (tales como alprazolam),
- tolbutamida (un medicamento usado para tratar la diabetes)
Informe a su médico o farmacéutico si usted o el niño están tomando, han tomado recientemente o pudieran tener que tomar cualquier otro medicamento.
Embarazo y lactancia
No debe utilizar este medicamento durante el embarazo a no ser que sea claramente necesario. Si usted o la niña están embarazadas o en periodo de lactancia, podrían estar embarazadas o tienen intención de quedarse embarazadas, consulte a su médico antes de utilizar este medicamento.
Para información relacionada con el control de natalidad, ver ‘Uso de EMEND con otros medicamentos’.
Se desconoce si EMEND pasa a la leche materna; por tanto, no se recomienda dar el pecho durante el tratamiento con este medicamento. Es importante informar al médico antes de tomar este medicamento si usted o la niña están dando el pecho a su bebé o tienen previsto hacerlo.
Conducción y uso de máquinas
Se debe tener en cuenta que algunas personas tienen mareo y sueño después de tomar EMEND. Si usted o el niño se sienten mareados o somnolientos, eviten conducir, montar en bicicleta o usar máquinas o herramientas después de tomar este medicamento (ver ‘Posibles efectos adversos’).
EMEND contiene sacarosa
EMEND cápsulas contiene sacarosa. Si su médico le ha indicado que usted o el niño padecen una intolerancia a ciertos azúcares, consulte con él antes de tomar este medicamento.
3. Cómo tomar EMEND
Siga exactamente las instrucciones de administración de este medicamento indicadas para usted o para el niño por su médico, farmacéutico o enfermero. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero. Tome siempre EMEND junto con otros medicamentos para prevenir las náuseas y vómitos. Después del tratamiento con EMEND, el médico puede pedir que usted o el niño continúen tomando otros medicamentos para prevenir las náuseas y los vómitos, incluyendo corticosteroides (como dexametasona) y un ‘antagonista 5-HT3’ (como ondansetrón). En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero.
La dosis oral recomendada de EMEND es Día 1:
- una cápsula de 125 mg 1 hora antes de empezar la sesión de quimioterapia y
Días 2 y 3:
- una cápsula de 80 mg cada día.
- si no le están administrando quimioterapia, tome EMEND por la mañana.
- si le están administrando quimioterapia, tome EMEND 1 hora antes de comenzar su sesión de quimioterapia.
EMEND se puede tomar con o sin alimentos.
Trague la cápsula entera con algún líquido.
Si toma más EMEND del que debe
No tome más cápsulas de las que el médico recomienda. Si usted o el niño han tomado demasiadas cápsulas, contacte con su médico inmediatamente.
Si olvidó tomar EMEND
Si usted o el niño han olvidado una dosis, pida consejo a su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Deje de tomar EMEND y acuda a su médico inmediatamente si usted o el niño notan alguno de los siguientes efectos adversos, que pueden ser graves, y para los que usted o el niño pueden necesitar tratamiento médico urgente:
- Ronchas, sarpullido, picor, dificultad para respirar o tragar (frecuencia no conocida, no puede estimarse a partir de los datos disponibles): estos son signos de una reacción alérgica.
Otros efectos adversos que se han comunicado se detallan a continuación.
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas) son:
- estreñimiento, indigestión,
- cefaleas,
- cansancio,
- pérdida de apetito,
- hipo,
- aumento de la cantidad de enzimas del hígado en la sangre.
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas) son:
- mareo, somnolencia,
- acné, sarpullido,
- ansiedad,
- eructos, náuseas, vómitos, ardor de estómago, dolor de estómago, boca seca, flatulencias,
- aumento de la micción dolorosa o que escuece,
- debilidad, malestar general,
- sofocos/enrojecimiento de la cara o de la piel,
- latidos rápidos o irregulares,
- fiebre con riesgo elevado de infección, disminución de los glóbulos rojos.
Efectos adversos raros (pueden afectar hasta 1 de cada 1.000 personas) son:
- dificultad para pensar, falta de energía, alteración del gusto,
- sensibilidad de la piel al sol, sudor excesivo, piel grasa, llagas en la piel, erupción con picor, síndrome de Stevens-Johnson/necrolisis epidérmica tóxica (reacción cutánea grave rara),
- euforia (sensación de felicidad extrema), desorientación,
- infección bacteriana, infección por hongos,
- estreñimiento intenso, úlcera de estómago, inflamación del intestino delgado y colon, llagas en la boca, hinchazón del vientre,
- frecuentes ganas de orinar, orinar más de lo normal, presencia de azúcar o sangre en la orina,
- molestias en el pecho, hinchazón, cambios en la manera de andar,
- tos, mucosidad en la parte de atrás de la garganta, irritación de la garganta, estornudos, dolor de garganta,
- secreción y picor oculares,
- zumbido de oídos,
- espasmos musculares, debilidad muscular,
- sed excesiva,
- latidos lentos, problemas de los vasos del corazón y de las venas,
- disminución de los glóbulos blancos, niveles bajos de sodio en la sangre, pérdida de peso, Comunicación de efectos adversos
Si usted o el niño experimentan cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de EMEND
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en el embalaje original para protegerlo de la humedad.
No extraer la cápsula de su blister hasta el momento de tomarla.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de EMEND
- El principio activo es aprepitant. Cada cápsula contiene 80 mg de aprepitant.
- Los demás componentes son: sacarosa, celulosa microcristalina (E-460), hidroxipropilcelulosa (E-463), lauril sulfato de sodio, gelatina, dióxido de titanio (E-171), laca, hidróxido de potasio y óxido de hierro negro (E-172).
Aspecto del producto y contenido del envase
La cápsula dura de 80 mg es opaca con cuerpo y tapa blancos con “461” y “80 mg” impreso en forma radial en tinta negra en el cuerpo.
Las cápsulas duras de EMEND 80 mg se suministran en los siguientes tamaños de envase:
- Blister de aluminio que contiene una cápsula de 80 mg.
- Envase para un tratamiento de 2 días que contiene dos cápsulas de 80 mg.
- 5 blisters de aluminio que contienen una cápsula de 80 mg cada uno.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización Responsable de la fabricación
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN11 9BU Reino Unido
Merck Sharp & Dohme B. V. Waarderweg 39 2031 BNHaarlem Países Bajos
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgique/Belgie/Belgien
MSD Belgium BVBA/SPRL
Tél/Tel: 0800 38 693 (+32(0)27766211)
Etarapna
MepK fflapn u floyM Etarapua EOOfl Tea.:+359 2 819 3737 info-msdbg@merck.com
Ceská republika
Merck Sharp & Dohme s.r.o.
Tel: +420 233 010 111 dpoc_czechslovak@merck.com
Danmark
MSD Danmark ApS Tlf: + 45 4482 4000 dkmail@merck.com
Lietuva
UAB Merck Sharp & Dohme Tel. + 370 5278 02 47 msd_lietuva@merck.com
Luxembourg/Luxemburg
MSD Belgium BVBA/SPRL Tél/Tel: +32(0)27766211 dpoc_belux@merck.com
Magyarország
MSD Pharma Hungary Kft.
Tel.: +36 1 888 5300 hungary_msd@merck.com
Malta
Merck Sharp & Dohme Cyprus Limited Tel: 8007 4433 (+356 99917558) malta_info@merck.com
Deutschland
MSD SHARP & DOHME GMBH
Tel: 0800 673 673 673 (+49 (0) 89 4561 2612)
Eesti
Merck Sharp & Dohme OÜ Tel.:+372 6144 200 msdeesti@merck.com
EXXáSa
MSD A.OB.E.E.
T^: +30 210 98 97 300 dpoc_greece@merck.com
España
Merck Sharp & Dohme de España, S.A. Tel: +34 91 321 06 00 msd_info@merck.com
France
MSD France
Tél: +33 (0) 1 80 46 40 40
Hrvatska
Merck Sharp & Dohme d.o.o.
Tel: + 385 1 6611 333 croatia_info@merck.com
Ireland
Merck Sharp & Dohme Ireland (Human Health) Limited
Tel: +353 (0)1 2998700 medinfo_ireland@merck.com
Ísland
Vistor hf.
Simi: +354 535 7000
Italia
MSD Italia S.r.l.
Tel: +39 06 361911 medicalinformation.it@merck.com
Kúrcpog
Merck Sharp & Dohme Cyprus Limited TpV: 800 00 673 (+357 22866700) cyprus_info@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: +371 67364224 msd_lv@merck.com
Nederland
Merck Sharp & Dohme BV Tel: 0800 9999000 (+31 23 5153153) medicalinfo.nl@merck.com
Norge
MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no
Osterreich
Merck Sharp & Dohme Ges.m.b.H.
Tel: +43 (0) 1 26 044 msd-medizin@merck.com
Polska
MSD Polska Sp. z o.o.
Tel:+48 22 549 51 00 msdpolska@merck.com
Portugal
Merck Sharp & Dohme, Lda Tel: +351 21 4465700 clic@merck.com
Romania
Merck Sharp & Dohme Romania S.R.L.
Tel:+40 21 529 29 00 msdromania@merck.com
Slovenija
Merck Sharp & Dohme, inovativna zdravila d.o.o.
Tel: +386 1 5204 201 msd_slovenia@merck.com
Slovenská republika
Merck Sharp & Dohme, s. r. o.
Tel:+421 2 58282010 dpoc_czechslovak@merck.com
Suomi/Finland
MSD Finland Oy Puh/Tel: +358 (0)9 804 650 info@msd.fi
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488 medicinskinfo@merck.com
United Kingdom
Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Prospecto: información para el usuario
EMEND 125 mg cápsulas duras
aprepitant
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted. Si es usted el progenitor de un niño que está tomando EMEND, lea esta información detenidamente.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted o al niño, y no debe dárselo a otras personas aunque tengan los mismos síntomas, ya que puede perjudicarles.
- Si usted o el niño experimentan efectos adversos, consulte a su médico, farmacéutico o enfermero. incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4
Contenido del prospecto
1. Qué es EMEND y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar o dar EMEND
3. Cómo tomar EMEND
4. Posibles efectos adversos
5. Conservación de EMEND
6. Contenido del envase e información adicional
1. Qué es EMEND y para qué se utiliza
EMEND contiene el principio activo aprepitant y pertenece a un grupo de medicamentos llamados "antagonistas del receptor de la neurocinina 1 (NKj)". El cerebro tiene una zona específica que controla las náuseas y los vómitos. EMEND actúa bloqueando las señales a esta zona, y por tanto, reduciendo las náuseas y los vómitos. EMEND cápsulas se usa en adultos y adolescentes a partir de 12 años de edad, en combinación con otros medicamentos para evitar las náuseas y los vómitos que provoca un tipo de quimioterapia (tratamiento del cáncer) que desencadena de forma fuerte y moderada náuseas y vómitos (como cisplatino, ciclofosfamida, doxorubicina o epirubicina).
2. Qué necesita saber antes de empezar a tomar o dar EMEND No tome EMEND:
- si usted o el niño son alérgicos a aprepitant o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
- con medicamentos que contengan pimozida (utilizado para tratar enfermedades psiquiátricas), terfenadina y astemizol (utilizados para la rinitis alérgica y otros trastornos alérgicos), cisaprida (utilizado para tratar problemas digestivos). Informe al médico si usted o el niño están tomando estos medicamentos ya que el tratamiento debe ser modificado antes de que usted o el niño empiecen a tomar EMEND.
Advertencias y precauciones
Consulte a su médico, farmacéutico o enfermero antes de empezar a tomar EMEND o de dar al niño este medicamento.
Antes del tratamiento con EMEND, informe al médico si usted o el niño tienen una enfermedad del hígado, porque el hígado es importante para eliminar el medicamento del cuerpo. Por lo tanto, su médico puede tener que controlar el estado de su hígado o el del niño.
Niños y adolescentes
No dé EMEND 125 mg cápsulas a niños menores de 12 años de edad, porque las cápsulas de 125 mg no se han estudiado en esta población.
Uso de EMEND con otros medicamentos
EMEND puede afectar a otros medicamentos durante y después del tratamiento con EMEND. Hay algunos medicamentos que no deben tomarse con EMEND (tales como pimozida, terfenadina, astemizol y cisaprida) o que requieren un ajuste en la dosis (véase también No tome EMEND).
Los efectos de EMEND o de otros medicamentos pueden estar influenciados si usted o el niño toman EMEND junto con otros medicamentos, incluyendo los indicados a continuación. Consulte a su médico o farmacéutico si usted o el niño están tomando alguno de los siguientes medicamentos:
- medicamentos anticonceptivos que pueden incluir píldoras anticonceptivas, parches cutáneos, implantes y ciertos dispositivos intrauterinos (DIUs) que liberan hormonas, puede que no funcionen adecuadamente cuando se toman junto con EMEND. Durante el tratamiento con EMEND y hasta 2 meses después de usar EMEND, deben utilizarse otros métodos o métodos adicionales de anticoncepción no hormonales,
- ciclosporina, tacrolimus, sirolimus, everolimus (inmunosupresores),
- alfentanilo, fentanilo (usados para tratar el dolor),
- quinidina (usado para tratar los latidos irregulares),
- irinotecán, etopósido, vinorelbina, ifosfamida (medicamentos usados para tratar el cáncer),
- medicamentos que contengan alcaloides derivados del ergot, tales como ergotamina y diergotamina (usados para tratar migrañas),
- warfarina, acenocumarol (diluyentes de la sangre; se pueden necesitar análisis de sangre),
- rifampicina, claritromicina, telitromicina (antibióticos usados para tratar infecciones),
- fenitoína (un medicamento usado para tratar las convulsiones),
- carbamazepina (usado para tratar la depresión y la epilepsia),
- midazolam, triazolam, fenobarbital (medicamentos usados para tranquilizar o para ayudar a dormir),
- hierba de San Juan (un preparado a base de plantas usado para tratar la depresión),
- inhibidores de la proteasa (usados para tratar infecciones por VIH),
- ketoconazol, excepto champú (usado para tratar el síndrome de Cushing - cuando el cuerpo produce un exceso de cortisol),
- itraconazol, voriconazol, posaconazol (antifungicos),
- nefazodona (usado para tratar la depresión),
- corticosteroides (tales como dexametasona y metilprednisolona),
- medicamentos para la ansiedad (tales como alprazolam),
- tolbutamida (un medicamento usado para tratar la diabetes)
Informe a su médico o farmacéutico si usted o el niño están tomando, han tomado recientemente o pudieran tener que tomar cualquier otro medicamento.
Embarazo y lactancia
No debe utilizar este medicamento durante el embarazo a no ser que sea claramente necesario. Si usted o la niña están embarazadas o en periodo de lactancia, podrían estar embarazadas o tienen intención de quedarse embarazadas, consulte a su médico antes de utilizar este medicamento.
Para información relacionada con el control de natalidad, ver ‘Uso de EMEND con otros medicamentos’.
Se desconoce si EMEND pasa a la leche materna; por tanto, no se recomienda dar el pecho durante el tratamiento con este medicamento. Es importante informar al médico antes de tomar este medicamento si usted o la niña están dando el pecho a su bebé o tienen previsto hacerlo.
Conducción y uso de máquinas
Se debe tener en cuenta que algunas personas tienen mareo y sueño después de tomar EMEND. Si usted o el niño se sienten mareados o somnolientos, eviten conducir, montar en bicicleta o usar máquinas o herramientas después de tomar este medicamento (ver ‘Posibles efectos adversos’).
EMEND contiene sacarosa
EMEND cápsulas contiene sacarosa. Si su médico le ha indicado que usted o el niño padecen una intolerancia a ciertos azúcares, consulte con él antes de tomar este medicamento.
3. Cómo tomar EMEND
Siga exactamente las instrucciones de administración de este medicamento indicadas para usted o para el niño por su médico, farmacéutico o enfermero. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero. Tome siempre EMEND junto con otros medicamentos para prevenir las náuseas y vómitos. Después del tratamiento con EMEND, su médico puede pedir que usted o el niño continúen tomando otros medicamentos para prevenir las náuseas y los vómitos, incluyendo corticosteroides (como dexametasona) y un ‘antagonista 5-HT3’ (como ondansetrón). En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero.
La dosis oral recomendada de EMEND es Día 1:
- una cápsula de 125 mg 1 hora antes de empezar la sesión de quimioterapia y
Días 2 y 3:
- una cápsula de 80 mg cada día.
- si no le están administrando quimioterapia, tome EMEND por la mañana.
- si le están administrando quimioterapia, tome EMEND 1 hora antes de comenzar su sesión de quimioterapia.
EMEND se puede tomar con o sin alimentos.
Trague la cápsula entera con algún líquido.
Si toma más EMEND del que debe
No tome más cápsulas de las que el médico recomienda. Si usted o el niño han tomado demasiadas cápsulas, contacte con su médico inmediatamente.
Si olvidó tomar EMEND
Si usted o el niño han olvidado una dosis, pida consejo a su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Deje de tomar EMEND y acuda a su médico inmediatamente si usted o el niño notan alguno de los siguientes efectos adversos, que pueden ser graves, y para los que usted o el niño pueden necesitar tratamiento médico urgente:
- Ronchas, sarpullido, picor, dificultad para respirar o tragar (frecuencia no conocida, no puede estimarse a partir de los datos disponibles): estos son signos de una reacción alérgica.
Otros efectos adversos que se han comunicado se detallan a continuación.
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas) son:
- estreñimiento, indigestión,
- cefaleas,
- cansancio,
- pérdida de apetito,
- hipo,
- aumento de la cantidad de enzimas del hígado en la sangre.
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas) son:
- mareo, somnolencia,
- acné, sarpullido,
- ansiedad,
- eructos, náuseas, vómitos, ardor de estómago, dolor de estómago, boca seca, flatulencias,
- aumento de la micción dolorosa o que escuece,
- debilidad, malestar general,
- sofocos/enrojecimiento de la cara o de la piel,
- latidos rápidos o irregulares,
- fiebre con riesgo elevado de infección, disminución de los glóbulos rojos.
Efectos adversos raros (pueden afectar hasta 1 de cada 1.000 personas) son:
- dificultad para pensar, falta de energía, alteración del gusto,
- sensibilidad de la piel al sol, sudor excesivo, piel grasa, llagas en la piel, erupción con picor, síndrome de Stevens-Johnson/necrolisis epidérmica tóxica (reacción cutánea grave rara),
- euforia (sensación de felicidad extrema), desorientación,
- infección bacteriana, infección por hongos,
- estreñimiento intenso, úlcera de estómago, inflamación del intestino delgado y colon, llagas en la boca, hinchazón del vientre,
- frecuentes ganas de orinar, orinar más de lo normal, presencia de azúcar o sangre en la orina,
- molestias en el pecho, hinchazón, cambios en la manera de andar,
- tos, mucosidad en la parte de atrás de la garganta, irritación de la garganta, estornudos, dolor de garganta,
- secreción y picor oculares,
- zumbido de oídos,
- espasmos musculares, debilidad muscular,
- sed excesiva,
- latidos lentos, problemas de los vasos del corazón y de las venas,
- disminución de los glóbulos blancos, niveles bajos de sodio en la sangre, pérdida de peso, Comunicación de efectos adversos
Si usted o el niño experimentan cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de EMEND
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en el embalaje original para protegerlo de la humedad.
No extraer la cápsula de su blister hasta el momento de tomarla.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de EMEND
- El principio activo es aprepitant. Cada cápsula dura de 125 mg contiene 125 mg de aprepitant.
- Los demás componentes son: sacarosa, celulosa microcristalina (E-460), hidroxipropilcelulosa (E-463), lauril sulfato de sodio, gelatina, dióxido de titanio (E-171), laca, hidróxido de potasio, óxido de hierro negro (E-172), óxido de hierro rojo (E-172) y óxido de hierro amarillo (E-172).
Aspecto del producto y contenido del envase
La cápsula dura de 125 mg es opaca con cuerpo blanco y tapa rosa con “462” y “125 mg” impreso en forma radial en tinta negra en el cuerpo.
Las cápsulas duras de EMEND 125 mg se suministran en los siguientes tamaños de envase:
- Blister de aluminio que contiene una cápsula de 125 mg.
- 5 blisters de aluminio que contienen una cápsula de 125 mg cada uno.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización Responsable de la fabricación
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN11 9BU Reino Unido
Merck Sharp & Dohme B. V. Waarderweg 39 2031 BNHaarlem Países Bajos
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgique/Belgie/Belgien
MSD Belgium BVBA/SPRL
Tél/Tel: 0800 38 693 (+32(0)27766211)
EfcnrapHH
MepK fflapn h floyM Etnrapna EOOfl Ten.:+359 2 819 3737 info-msdbg@merck.com
Ceská republika
Merck Sharp & Dohme s.r.o.
Tel: +420 233 010 111 dpoc_czechslovak@merck.com
Danmark
MSD Danmark ApS Tlf: + 45 4482 4000 dkmail@merck.com
Lietuva
UAB Merck Sharp & Dohme Tel. + 370 5278 02 47 msd_lietuva@merck.com
Luxembourg/Luxemburg
MSD Belgium BVBA/SPRL Tél/Tel: +32(0)27766211 dpoc_belux@merck.com
Magyarország
MSD Pharma Hungary Kft.
Tel.: +36 1 888 5300 hungary_msd@merck.com
Malta
Merck Sharp & Dohme Cyprus Limited Tel: 8007 4433 (+356 99917558) malta_info@merck.com
Deutschland
MSD SHARP & DOHME GMBH
Tel: 0800 673 673 673 (+49 (0) 89 4561 2612)
Eesti
Merck Sharp & Dohme OÜ Tel.:+372 6144 200 msdeesti@merck.com
EXXáSa
MSD A.O.B.EE.
T^: +30 210 98 97 300 dpoc_greece@merck.com
España
Merck Sharp & Dohme de España, S.A. Tel: +34 91 321 06 00 msd_info@merck.com
France
MSD France
Tél: +33 (0) 1 80 46 40 40
Hrvatska
Merck Sharp & Dohme d.o.o.
Tel: + 385 1 6611 333 croatia_info@merck.com
Ireland
Merck Sharp & Dohme Ireland (Human Health) Limited
Tel: +353 (0)1 2998700 medinfo_ireland@merck.com
Ísland
Vistor hf.
Simi: +354 535 7000
Italia
MSD Italia S.r.l.
Tel: +39 06 361911 medicalinformation.it@merck.com
Kúrcpog
Merck Sharp & Dohme Cyprus Limited TpV: 800 00 673 (+357 22866700) cyprus_info@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: +371 67364224 msd_lv@merck.com
Nederland
Merck Sharp & Dohme BV Tel: 0800 9999000 (+31 23 5153153) medicalinfo.nl@merck.com
Norge
MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no
Osterreich
Merck Sharp & Dohme Ges.m.b.H.
Tel: +43 (0) 1 26 044 msd-medizin@merck.com
Polska
MSD Polska Sp. z o.o.
Tel:+48 22 549 51 00 msdpolska@merck.com
Portugal
Merck Sharp & Dohme, Lda Tel: +351 21 4465700 clic@merck.com
Romania
Merck Sharp & Dohme Romania S.R.L.
Tel:+40 21 529 29 00 msdromania@merck.com
Slovenija
Merck Sharp & Dohme, inovativna zdravila d.o.o.
Tel: +386 1 5204 201 msd_slovenia@merck.com
Slovenská republika
Merck Sharp & Dohme, s. r. o.
Tel:+421 2 58282010 dpoc_czechslovak@merck.com
Suomi/Finland
MSD Finland Oy Puh/Tel: +358 (0)9 804 650 info@msd.fi
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488 medicinskinfo@merck.com
United Kingdom
Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Prospecto: información para el usuario
EMEND 165 mg cápsulas duras
aprepitant
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4
Contenido del prospecto
1. Qué es EMEND y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar EMEND
3. Cómo tomar EMEND
4. Posibles efectos adversos
5. Conservación de EMEND
6. Contenido del envase e información adicional
1. Qué es EMEND y para qué se utiliza
EMEND contiene el principio activo aprepitant y pertenece a un grupo de medicamentos llamados "antagonistas del receptor de la neurocinina 1 (NKj)". El cerebro tiene una zona específica que controla las náuseas y los vómitos. EMEND actúa bloqueando las señales a esta zona, y por tanto, reduciendo las náuseas y los vómitos. EMEND se usa en adultos, en combinación con otros medicamentos para evitar las náuseas y los vómitos que provoca un tipo de quimioterapia (tratamiento del cáncer) que contiene cisplatino (fuerte desencadenante de náuseas y vómitos) u otra quimioterapia que desencadena de forma moderada náuseas y vómitos (como ciclofosfamida, doxorubicina o epirubicina).
2. Qué necesita saber antes de empezar a tomar EMEND
No tome EMEND:
- si es alérgico a aprepitant o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
- con medicamentos que contengan pimozida (utilizado para tratar enfermedades psiquiátricas), terfenadina y astemizol (utilizados para la rinitis alérgica y otros trastornos alérgicos), cisaprida (utilizado para tratar problemas digestivos). Informe a su médico si está tomando estos medicamentos ya que su tratamiento debe ser modificado antes de empezar a tomar EMEND.
Advertencias y precauciones
Consulte a su médico, farmacéutico o enfermero antes de empezar a tomar este medicamento.
Antes del tratamiento con EMEND, informe a su médico si tiene una enfermedad del hígado, porque
su hígado es importante para eliminar el medicamento de su cuerpo. Por lo tanto, su médico puede
tener que controlar el estado de su hígado.
Niños y adolescentes
No dé EMEND 165 mg a niños y adolescentes menores de 18 años de edad, porque las cápsulas de
165 mg no se han estudiado en esta población.
Uso de EMEND con otros medicamentos
EMEND puede afectar a otros medicamentos durante y después del tratamiento con EMEND. Hay algunos medicamentos que no deben tomarse con EMEND (tales como pimozida, terfenadina, astemizol y cisaprida) o que requieren un ajuste en la dosis (véase también No tome EMEND).
Los efectos de EMEND o de otros medicamentos pueden estar influenciados si toma EMEND junto con otros medicamentos, incluyendo los indicados a continuación. Consulte a su médico o farmacéutico si está tomando alguno de los siguientes medicamentos:
- medicamentos anticonceptivos que pueden incluir píldoras anticonceptivas, parches cutáneos, implantes y ciertos dispositivos intrauterinos (DIUs) que liberan hormonas, puede que no funcionen adecuadamente cuando se toman junto con EMEND. Durante el tratamiento con EMEND y hasta 2 meses después de usar EMEND, deben utilizarse otros métodos o métodos adicionales de anticoncepción no hormonales,
- ciclosporina, tacrolimus, sirolimus, everolimus (inmunosupresores),
- alfentanilo, fentanilo (usados para tratar el dolor),
- quinidina (usado para tratar los latidos irregulares),
- irinotecán, etopósido, vinorelbina, ifosfamida (medicamentos usados para tratar el cáncer),
- medicamentos que contengan alcaloides derivados del ergot, tales como ergotamina y diergotamina (usados para tratar migrañas),
- warfarina, acenocumarol (diluyentes de la sangre; se pueden necesitar análisis de sangre),
- rifampicina, claritromicina, telitromicina (antibióticos usados para tratar infecciones),
- fenitoína (un medicamento usado para tratar las convulsiones),
- carbamazepina (usado para tratar la depresión y la epilepsia),
- midazolam, triazolam, fenobarbital (medicamentos usados para tranquilizar o para ayudar a dormir),
- hierba de San Juan (un preparado a base de plantas usado para tratar la depresión),
- inhibidores de la proteasa (usados para tratar infecciones por VIH),
- ketoconazol, excepto champú (usado para tratar el síndrome de Cushing - cuando el cuerpo produce un exceso de cortisol),
- itraconazol, voriconazol, posaconazol (antifungicos),
- nefazodona (usado para tratar la depresión),
- corticosteroides (tales como dexametasona y metilprednisolona),
- medicamentos para la ansiedad (tales como alprazolam),
- tolbutamida (un medicamento usado para tratar la diabetes)
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o pudiera tener que tomar cualquier otro medicamento.
Embarazo y lactancia
No debe utilizar este medicamento durante el embarazo a no ser que sea claramente necesario. Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento.
Para información relacionada con el control de natalidad, ver ‘Uso de EMEND con otros medicamentos’.
Se desconoce si EMEND pasa a la leche materna; por tanto, no se recomienda dar el pecho durante el tratamiento con este medicamento. Es importante informar al médico antes de tomar este medicamento si está dando el pecho a su bebé o tiene previsto hacerlo.
Conducción y uso de máquinas
Se debe tener en cuenta que algunas personas tienen mareo y sueño después de tomar EMEND. Si se siente mareado o somnoliento, evite conducir o usar máquinas después de tomar este medicamento (ver ‘Posibles efectos adversos’).
EMEND contiene sacarosa
EMEND cápsulas contiene sacarosa. Si su médico le ha indicado que padece una intolerancia a ciertos azúcares, consulte con él antes de tomar este medicamento.
3. Cómo tomar EMEND
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico o farmacéutico. Tome la cápsula de EMEND el primer día de su ciclo de quimioterapia, junto con otros medicamentos, para prevenir las náuseas y los vómitos. Su médico le pedirá que continúe tomando otros medicamentos para prevenir las náuseas y los vómitos, incluyendo corticosteroides (como dexametasona) y un ‘antagonista 5-HT3’ (como ondansetrón) los siguientes tres días.
La dosis oral recomendada de EMEND es
una cápsula de 165 mg el primer día de su ciclo de quimioterapia. Tome la cápsula 1 hora antes de empezar la sesión de quimioterapia.
EMEND se puede tomar con o sin alimentos.
Trague la cápsula entera con algún líquido.
Si toma más EMEND del que debe
No tome más cápsulas de las que el médico recomienda. Si ha tomado demasiadas cápsulas, contacte con su médico inmediatamente.
Si olvidó tomar EMEND
No tome una dosis doble para compensar las dosis olvidadas.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Deje de tomar EMEND y acuda a su médico inmediatamente si nota alguno de los siguientes efectos adversos, que pueden ser graves, y para los que puede necesitar tratamiento médico urgente:
- Ronchas, sarpullido, picor, dificultad para respirar o tragar (frecuencia no conocida, no puede estimarse a partir de los datos disponibles): estos son signos de una reacción alérgica.
Otros efectos adversos que se han comunicado se detallan a continuación.
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas) son:
- estreñimiento, indigestión,
- cefaleas,
- cansancio,
- pérdida de apetito,
- hipo,
- aumento de la cantidad de enzimas del hígado en la sangre.
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas) son:
- mareo, somnolencia,
- acné, sarpullido,
- ansiedad,
- eructos, náuseas, vómitos, ardor de estómago, dolor de estómago, boca seca, flatulencias,
- aumento de la micción dolorosa o que escuece,
- debilidad, malestar general,
- sofocos,
- latidos rápidos o irregulares,
- fiebre con riesgo elevado de infección, disminución de los glóbulos rojos.
Efectos adversos raros (pueden afectar hasta 1 de cada 1.000 personas) son:
- dificultad para pensar, falta de energía, alteración del gusto,
- sensibilidad de la piel al sol, sudor excesivo, piel grasa, llagas en la piel, erupción con picor, síndrome de Stevens-Johnson/necrolisis epidérmica tóxica (reacción cutánea grave rara),
- euforia (sensación de felicidad extrema), desorientación,
- infección bacteriana, infección por hongos,
- estreñimiento intenso, úlcera de estómago, inflamación del intestino delgado y colon, llagas en la boca, hinchazón del vientre,
- frecuentes ganas de orinar, orinar más de lo normal, presencia de azúcar o sangre en la orina,
- molestias en el pecho, hinchazón, cambios en la manera de andar,
- tos, mucosidad en la parte de atrás de la garganta, irritación de la garganta, estornudos, dolor de garganta,
- secreción y picor oculares,
- zumbido de oídos,
- espasmos musculares, debilidad muscular,
- sed excesiva,
- latidos lentos, problemas de los vasos del corazón y de las venas,
- disminución de los glóbulos blancos, niveles bajos de sodio en la sangre, pérdida de peso, Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de EMEND
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el blister y caja después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en el embalaje original para protegerlo de la humedad.
No extraer la cápsula de su blister hasta el momento de tomarla.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de EMEND
- El principio activo es aprepitant. Cada cápsula dura contiene 165 mg de aprepitant.
- Los demás componentes son: sacarosa, celulosa microcristalina (E-460), hidroxipropilcelulosa (E-463), lauril sulfato de sodio, gelatina, dióxido de titanio (E-171), carmín índigo (E-132), laca, hidróxido de potasio y óxido de hierro negro (E-172).
Aspecto del producto y contenido del envase
La cápsula dura de 165 mg es opaca con una tapa azul claro y un cuerpo blanco con “466” y “165 mg” impreso en forma radial en tinta negra en una parte del cuerpo y el logo de Merck en la otra parte.
Las cápsulas duras de EMEND 165 mg se suministran en cajas en los siguientes tamaños de envase:
- Blister de aluminio que contiene una cápsula de 165 mg.
- 6 blisters de aluminio que contienen una cápsula de 165 mg cada uno.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización Merck Sharp & Dohme Ltd.
Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Reino Unido
Responsable de la fabricación Merck Sharp & Dohme B. V. Waarderweg 39 2031 BN Haarlem Países Bajos
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgique/Belgie/Belgien
MSD Belgium BVBA/SPRL
Tél/Tel: 0800 38 693 (+32(0)27766211)
Etarapna
MepK fflapn h floyM Etarapna EOOfl Tea.:+359 2 819 3737 info-msdbg@merck.com
Ceská republika
Merck Sharp & Dohme s.r.o.
Tel: +420 233 010 111 dpoc_czechslovak@merck.com
Danmark
MSD Danmark ApS Tlf: + 45 4482 4000 dkmail@merck.com
Deutschland
MSD SHARP & DOHME GMBH
Tel: 0800 673 673 673 (+49 (0) 89 4561 2612)
Eesti
Merck Sharp & Dohme OÜ Tel.:+372 6144 200 msdeesti@merck.com
EXXáSa
MSD A.O.B.EE.
Tp^: +30 210 98 97 300 dpoc_greece@merck.com
Lietuva
UAB Merck Sharp & Dohme Tel. + 370 5278 02 47 msd_lietuva@merck.com
Luxembourg/Luxemburg
MSD Belgium BVBA/SPRL Tél/Tel: +32(0)27766211 dpoc_belux@merck.com
Magyarország
MSD Pharma Hungary Kft.
Tel.: +36 1 888 5300 hungary_msd@merck.com
Malta
Merck Sharp & Dohme Cyprus Limited Tel: 8007 4433 (+356 99917558) malta_info@merck.com
Nederland
Merck Sharp & Dohme BV Tel: 0800 9999000 (+31 23 5153153) medicalinfo.nl@merck.com
Norge
MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no
Osterreich
Merck Sharp & Dohme Ges.m.b.H.
Tel: +43 (0) 1 26 044 msd-medizin@merck.com
España
Merck Sharp & Dohme de España, S.A. Tel: +34 91 321 06 00 msd_info@merck.com
France
MSD France
Tél: +33 (0) 1 80 46 40 40
Hrvatska
Merck Sharp & Dohme d.o.o.
Tel: + 385 1 6611 333 croatia_info@merck.com
Ireland
Merck Sharp & Dohme Ireland (Human Health) Limited
Tel: +353 (0)1 2998700 medinfo_ireland@merck.com
Ísland
Vistor hf.
Simi: +354 535 7000
Italia
MSD Italia S.r.l.
Tel: +39 06 361911 medicalinformation.it@merck.com
Kúrcpog
Merck Sharp & Dohme Cyprus Limited TpV: 800 00 673 (+357 22866700) cyprus_info@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: +371 67364224 msd_lv@merck.com
Polska
MSD Polska Sp. z o.o.
Tel:+48 22 549 51 00 msdpolska@merck.com
Portugal
Merck Sharp & Dohme, Lda Tel: +351 21 4465700 clic@merck.com
Romanía
Merck Sharp & Dohme Romania S.R.L.
Tel:+40 21 529 29 00 msdromania@merck.com
Slovenija
Merck Sharp & Dohme, inovativna zdravila d.o.o.
Tel: +386 1 5204 201 msd_slovenia@merck.com
Slovenská republika
Merck Sharp & Dohme, s. r. o.
Tel:+421 2 58282010 dpoc_czechslovak@merck.com
Suomi/Finland
MSD Finland Oy Puh/Tel: +358 (0)9 804 650 info@msd.fi
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488 medicinskinfo@merck.com
United Kingdom
Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Prospecto: información para el usuario
EMEND 125 mg cápsulas duras EMEND 80 mg cápsulas duras
aprepitant
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted. Si es usted el progenitor de un niño que está tomando EMEND, lea esta información detenidamente.
• Conserve este prospecto, ya que puede tener que volver a leerlo.
• Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
• Este medicamento se le ha recetado solamente a usted o al niño, y no debe dárselo a otras personas aunque tengan los mismos síntomas, ya que puede perjudicarles.
• Si usted o el niño experimentan efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4
Contenido del prospecto
1. Qué es EMEND y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar o dar EMEND
3. Cómo tomar EMEND
4. Posibles efectos adversos
5. Conservación de EMEND
6. Contenido del envase e información adicional
1. Qué es EMEND y para qué se utiliza
EMEND contiene el principio activo aprepitant y pertenece a un grupo de medicamentos llamados "antagonistas del receptor de la neurocinina 1 (NKj)". El cerebro tiene una zona específica que controla las náuseas y los vómitos. EMEND actúa bloqueando las señales a esta zona, y por tanto, reduciendo las náuseas y los vómitos. EMEND cápsulas se usa en adultos y adolescentes a partir de 12 años de edad, en combinación con otros medicamentos para evitar las náuseas y los vómitos que provoca un tipo de quimioterapia (tratamiento del cáncer) que desencadena de forma fuerte y moderada náuseas y vómitos (como cisplatino, ciclofosfamida, doxorubicina o epirubicina).
2. Qué necesita saber antes de empezar a tomar o dar EMEND No tome EMEND:
- si usted o el niño son alérgicos a aprepitant o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
- con medicamentos que contengan pimozida (utilizado para tratar enfermedades psiquiátricas), terfenadina y astemizol (utilizados para la rinitis alérgica y otros trastornos alérgicos), cisaprida (utilizado para tratar problemas digestivos). Informe al médico si usted o el niño están tomando estos medicamentos ya que el tratamiento debe ser modificado antes de que usted o el niño empiecen a tomar EMEND.
Advertencias y precauciones
Consulte a su médico, farmacéutico o enfermero antes de empezar a tomar EMEND o de dar al niño este medicamento.
Antes del tratamiento con EMEND, informe al médico si usted o el niño tienen una enfermedad del hígado, porque el hígado es importante para eliminar el medicamento del cuerpo. Por lo tanto, su médico puede tener que controlar el estado de su hígado o el del niño.
Niños y adolescentes
No dé EMEND 80 mg y 125 mg cápsulas a niños menores de 12 años de edad, porque las cápsulas de 80 mg y 125 mg no se han estudiado en esta población.
Uso de EMEND con otros medicamentos
EMEND puede afectar a otros medicamentos durante y después del tratamiento con EMEND. Hay algunos medicamentos que no deben tomarse con EMEND (tales como pimozida, terfenadina, astemizol y cisaprida) o que requieren un ajuste en la dosis (véase también ‘No tome EMEND’).
Los efectos de EMEND o de otros medicamentos pueden estar influenciados si usted o el niño toman EMEND junto con otros medicamentos, incluyendo los indicados a continuación. Consulte a su médico o farmacéutico si usted o el niño están tomando alguno de los siguientes medicamentos:
- medicamentos anticonceptivos que pueden incluir píldoras anticonceptivas, parches cutáneos, implantes y ciertos dispositivos intrauterinos (DIUs) que liberan hormonas, puede que no funcionen adecuadamente cuando se toman junto con EMEND. Durante el tratamiento con EMEND y hasta 2 meses después de usar EMEND, deben utilizarse otros métodos o métodos adicionales de anticoncepción no hormonales,
- ciclosporina, tacrolimus, sirolimus, everolimus (inmunosupresores),
- alfentanilo, fentanilo (usados para tratar el dolor),
- quinidina (usado para tratar los latidos irregulares),
- irinotecán, etopósido, vinorelbina, ifosfamida (medicamentos usados para tratar el cáncer),
- medicamentos que contengan alcaloides derivados del ergot, tales como ergotamina o diergotamina (usados para tratar migrañas),
- warfarina, acenocumarol (diluyentes de la sangre; se pueden necesitar análisis de sangre),
- rifampicina, claritromicina, telitromicina (antibióticos usados para tratar infecciones),
- fenitoína (un medicamento usado para tratar las convulsiones),
- carbamazepina (usado para tratar la depresión y la epilepsia),
- midazolam, triazolam, fenobarbital (medicamentos usados para tranquilizar o para ayudar a dormir),
- hierba de San Juan (un preparado a base de plantas usado para tratar la depresión),
- inhibidores de la proteasa (usados para tratar infecciones por VIH),
- ketoconazol, excepto champú (usado para tratar el síndrome de Cushing - cuando el cuerpo produce un exceso de cortisol),
- itraconazol, voriconazol, posaconazol (antifúngicos),
- nefazodona (usado para tratar la depresión),
- corticosteroides (tales como dexametasona y metilprednisolona),
- medicamentos para la ansiedad (tales como alprazolam),
- tolbutamida (un medicamento usado para tratar la diabetes)
Informe a su médico o farmacéutico si usted o el niño están tomando, han tomado recientemente o pudieran tener que tomar cualquier otro medicamento.
Embarazo y lactancia
No debe utilizar este medicamento durante el embarazo a no ser que sea claramente necesario. Si usted o la niña están embarazadas o en periodo de lactancia, podrían estar embarazadas o tienen intención de quedarse embarazadas, consulte a su médico antes de utilizar este medicamento.
Para información relacionada con el control de natalidad, ver ‘Uso de EMEND con otros medicamentos’.
Se desconoce si EMEND pasa a la leche materna; por tanto, no se recomienda dar el pecho durante el tratamiento con este medicamento. Es importante informar al médico antes de tomar este medicamento si usted o la niña están dando el pecho a su bebé o tienen previsto hacerlo.
Conducción y uso de máquinas
Se debe tener en cuenta que algunas personas tienen mareo y sueño después de tomar EMEND. Si usted o el niño se sienten mareados o somnolientos, eviten conducir, montar en bicicleta o usar máquinas o herramientas después de tomar este medicamento (ver ‘Posibles efectos adversos’).
EMEND contiene sacarosa
EMEND cápsulas contiene sacarosa. Si su médico le ha indicado que usted o el niño padecen una intolerancia a ciertos azúcares, consulte con él antes de tomar este medicamento.
3. Cómo tomar EMEND
Siga exactamente las instrucciones de administración de este medicamento indicadas para usted o para el niño por su médico, farmacéutico o enfermero. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero. Tome siempre EMEND junto con otros medicamentos para prevenir las náuseas y vómitos. Después del tratamiento con EMEND, el médico puede pedir que usted o el niño continúen tomando otros medicamentos para prevenir las náuseas y los vómitos, incluyendo corticosteroides (como dexametasona) y un ‘antagonista 5-HT3’ (como ondansetrón). En caso de dudaconsulte de nuevo a su médico, farmacéutico o enfermero.
La dosis oral recomendada de EMEND es Día 1:
- una cápsula de 125 mg 1 hora antes de empezar la sesión de quimioterapia y
Días 2 y 3:
- una cápsula de 80 mg cada día.
- si no le están administrando quimioterapia, tome EMEND por la mañana.
- si le están administrando quimioterapia, tome EMEND 1 hora antes de comenzar su sesión de quimioterapia.
EMEND se puede tomar con o sin alimentos.
Trague la cápsula entera con algún líquido.
Si toma más EMEND del que debe
No tome más cápsulas de las que el médico recomienda. Si usted o el niño han tomado demasiadas cápsulas, contacte con su médico inmediatamente.
Si olvidó tomar EMEND
Si usted o el niño han olvidado una dosis, pida consejo a su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Deje de tomar EMEND y acuda a su médico inmediatamente si usted o el niño notan alguno de los siguientes efectos adversos, que pueden ser graves, y para los que usted o el niño pueden necesitar tratamiento médico urgente:
- Ronchas, sarpullido, picor, dificultad para respirar o tragar (frecuencia no conocida, no puede estimarse a partir de los datos disponibles): estos son signos de una reacción alérgica.
Otros efectos adversos que se han comunicado se detallan a continuación.
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas) son:
- estreñimiento, indigestión,
- cefaleas,
- cansancio,
- pérdida de apetito,
- hipo,
- aumento de la cantidad de enzimas del hígado en la sangre.
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas) son:
- mareo, somnolencia,
- acné, sarpullido,
- ansiedad,
- eructos, náuseas, vómitos, ardor de estómago, dolor de estómago, boca seca, flatulencias,
- aumento de la micción dolorosa o que escuece,
- debilidad, malestar general,
- sofocos/enrojecimiento de la cara o de la piel,
- latidos rápidos o irregulares,
- fiebre con riesgo elevado de infección, disminución de los glóbulos rojos.
Efectos adversos raros (pueden afectar hasta 1 de cada 1.000 personas) son:
- dificultad para pensar, falta de energía, alteración del gusto,
- sensibilidad de la piel al sol, sudor excesivo, piel grasa, llagas en la piel, erupción con picor, síndrome de Stevens-Johnson/necrolisis epidérmica tóxica (reacción cutánea grave rara),
- euforia (sensación de felicidad extrema), desorientación,
- infección bacteriana, infección por hongos,
- estreñimiento intenso, úlcera de estómago, inflamación del intestino delgado y colon, llagas en la boca, hinchazón del vientre,
- frecuentes ganas de orinar, orinar más de lo normal, presencia de azúcar o sangre en la orina,
- molestias en el pecho, hinchazón, cambios en la manera de andar,
- tos, mucosidad en la parte de atrás de la garganta, irritación de la garganta, estornudos, dolor de garganta,
- secreción y picor oculares,
- zumbido de oídos,
- espasmos musculares, debilidad muscular,
- sed excesiva,
- latidos lentos, problemas de los vasos del corazón y de las venas,
- disminución de los glóbulos blancos, niveles bajos de sodio en la sangre, pérdida de peso, Comunicación de efectos adversos
Si usted o el niño experimentan cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de EMEND
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en el embalaje original para protegerlo de la humedad.
No extraer la cápsula de su blister hasta el momento de tomarla.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de EMEND
- El principio activo es aprepitant. Cada cápsula dura de 125 mg contiene 125 mg de aprepitant. Cada cápsula dura de 80 mg contiene 80 mg de aprepitant.
- Los demás componentes son: sacarosa, celulosa microcristalina (E-460), hidroxipropilcelulosa (E-463), lauril sulfato de sodio, gelatina, dióxido de titanio (E-171), laca, hidróxido de potasio y óxido de hierro negro (E-172); la cápsula dura de 125 mg también contiene óxido de hierro rojo (E-172) y óxido de hierro amarillo (E-172).
Aspecto del producto y contenido del envase
La cápsula dura de 125 mg es opaca con cuerpo blanco y tapa rosa con “462” y “125 mg” impreso en forma radial en tinta negra en el cuerpo.
La cápsula dura de 80 mg es opaca con cuerpo y tapa blancos con “461” y “80 mg” impreso en forma radial en tinta negra en el cuerpo.
Las cápsulas duras de EMEND 125 mg y 80 mg se suministran en el siguiente tamaño de envase:
- Envase para un tratamiento de 3 días que contiene una cápsula de 125 mg y dos cápsulas de 80 mg
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización Merck Sharp & Dohme Ltd.
Hertford Road, Hoddesdon Hertfordshire EN11 9BU Reino Unido
Responsable de la fabricación Merck Sharp & Dohme B. V. Waarderweg 39 2031 BNHaarlem Países Bajos
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgique/Belgie/Belgien
MSD Belgium BVBA/SPRL
Tél/Tel: 0800 38 693 (+32(0)27766211)
Etarapna
MepK fflapn h floyM Etarapna EOOfl Tea.:+359 2 819 3737 info-msdbg@merck.com
Lietuva
UAB Merck Sharp & Dohme Tel. + 370 5278 02 47 msd_lietuva@merck.com
Luxembourg/Luxemburg
MSD Belgium BVBA/SPRL Tél/Tel: +32(0)27766211 dpoc_belux@merck.com
Ceská republika
Merck Sharp & Dohme s.r.o. Tel: +420 233 010 111 dpoc_czechslovak@merck.com
Magyarország MSD Pharma Hungary Kft. Tel.: +36 1 888 5300 hungary_msd@merck.com
Danmark
MSD Danmark ApS Tlf: + 45 4482 4000 dkmail@merck.com
Deutschland
MSD SHARP & DOHME GMBH
Tel: 0800 673 673 673 (+49 (0) 89 4561 2612)
Eesti
Merck Sharp & Dohme OÜ Tel.:+372 6144 200 msdeesti@merck.com
EXXáSa
MSD A.O.B.EE.
T^: +30 210 98 97 300 dpoc_greece@merck.com
España
Merck Sharp & Dohme de España, S.A. Tel: +34 91 321 06 00 msd_info@merck.com
France
MSD France
Tél: +33 (0) 1 80 46 40 40
Hrvatska
Merck Sharp & Dohme d.o.o.
Tel: + 385 1 6611 333 croatia_info@merck.com
Ireland
Merck Sharp & Dohme Ireland (Human Health) Limited
Tel: +353 (0)1 2998700 medinfo_ireland@merck.com
Ísland
Vistor hf.
Simi: +354 535 7000
Italia
MSD Italia S.r.l.
Tel: +39 06 361911 medicalinformation.it@merck.com
Kúrcpog
Merck Sharp & Dohme Cyprus Limited TpV: 800 00 673 (+357 22866700) cyprus_info@merck.com
Malta
Merck Sharp & Dohme Cyprus Limited Tel: 8007 4433 (+356 99917558) malta_info@merck.com
Nederland
Merck Sharp & Dohme BV Tel: 0800 9999000 (+31 23 5153153) medicalinfo.nl@merck.com
Norge
MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no
Osterreich
Merck Sharp & Dohme Ges.m.b.H.
Tel: +43 (0) 1 26 044 msd-medizin@merck.com
Polska
MSD Polska Sp. z o.o.
Tel:+48 22 549 51 00 msdpolska@merck.com
Portugal
Merck Sharp & Dohme, Lda Tel: +351 21 4465700 clic@merck.com
Romanía
Merck Sharp & Dohme Romania S.R.L.
Tel:+40 21 529 29 00 msdromania@merck.com
Slovenija
Merck Sharp & Dohme, inovativna zdravila d.o.o.
Tel: +386 1 5204 201 msd_slovenia@merck.com
Slovenská republika
Merck Sharp & Dohme, s. r. o.
Tel:+421 2 58282010 dpoc_czechslovak@merck.com
Suomi/Finland
MSD Finland Oy Puh/Tel: +358 (0)9 804 650 info@msd.fi
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488 medicinskinfo@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: +371 67364224 msd_lv@merck.com
United Kingdom
Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Prospecto: información para el usuario
EMEND 125 mg polvo para suspensión oral
aprepitant
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted. Este prospecto se ha redactado para el progenitor o cuidador que va a administrar este medicamento al niño, lea esta información detenidamente.
• Conserve este prospecto, ya que puede tener que volver a leerlo.
• Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
• Este medicamento se le ha recetado solamente al niño, y no debe dárselo a otras personas aunque tengan los mismos síntomas, ya que puede perjudicarles.
• Si el niño experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4
Contenido del prospecto
1. Qué es EMEND y para qué se utiliza
2. Qué necesita saber antes de empezar a dar EMEND
3. Cómo dar EMEND
4. Posibles efectos adversos
5. Conservación de EMEND
6. Contenido del envase e información adicional
1. Qué es EMEND y para qué se utiliza
EMEND contiene el principio activo aprepitant. Pertenece a un grupo de medicamentos llamados ‘antagonistas del receptor de la neurocinina 1 (NKi)’.
El cerebro tiene una zona específica que controla las náuseas y los vómitos. EMEND actúa bloqueando las señales a esta zona, y por tanto, reduciendo las náuseas y los vómitos.
El polvo para suspensión oral se usa en niños de entre 6 meses y menos de 12 años de edad, en combinación con otros medicamentos para evitar las náuseas y los vómitos que provoca un tipo de quimioterapia (tratamiento del cáncer) que desencadena de forma fuerte y moderada náuseas y vómitos (como cisplatino, ciclofosfamida, doxorubicina o epirubicina).
2. Qué necesita saber antes de empezar a dar EMEND No dé EMEND:
- si el niño es alérgico a aprepitant o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
- si el niño está usando medicamentos que contienen ‘pimozida’ (para problemas de salud mental).
- si el niño está usando ‘terfenadina’ o ‘astemizol’ (para la rinitis alérgica y otras alergias).
- si el niño está usando ‘cisaprida’ (para problemas con la digestión).
No dé este medicamento si el niño se encuentra en alguna de las situaciones anteriores e informe al médico del niño si está utilizando alguno de los medicamentos citados arriba. Esto es debido a que su tratamiento se tendrá que modificar antes de empezar el tratamiento con este medicamento. Si no está seguro, consulte a su médico, farmacéutico o enfermero antes de dar este medicamento.
Advertencias y precauciones
Consulte a su médico, farmacéutico o enfermero antes de dar este medicamento al niño.
Problemas del hígado
Si el niño tiene problemas del hígado, consulte con el médico antes de empezar el tratamiento con EMEND. Esto es porque el hígado es importante para eliminar el medicamento del cuerpo. El médico puede que tenga que controlar el estado del hígado del niño durante el tratamiento.
Niños y adolescentes
No dé EMEND polvo para suspensión oral a niños menores de 6 meses de edad o que pesen menos de 6 kg o a adolescentes de entre 12 y 18 años, ya que el polvo para suspensión oral no se ha estudiado en esta población.
Uso de EMEND con otros medicamentos
Informe a su médico, farmacéutico o enfermero si el niño está utilizando, ha utilizado recientemente o pudiera tener que utilizar cualquier otro medicamento. Esto es porque EMEND puede afectar la actividad de otros medicamentos, durante y después del tratamiento con EMEND. Además, algunos medicamentos pueden afectar la forma en que actúa este medicamento.
No dé EMEND e informe al médico o farmacéutico si el niño está utilizando alguno de los siguientes medicamentos (ver además ‘No dé EMEND’). Esto es porque será necesario modificar su tratamiento antes de empezar con EMEND:
- pimozida - para problemas de salud mental
- terfenadina y astemizol - para la rinitis alérgica y otras alergias
- cisaprida - para problemas con la digestión
No dé este medicamento y consulte al médico o farmacéutico si algo de lo citado anteriormente es aplicable al niño.
Informe al médico, farmacéutico o enfermero si el niño está tomando alguno de los siguientes medicamentos:
- medicamentos que afectan al sistema inmunitario - como ciclosporina, tacrolimus, sirolimus, everolimus
- alfentanilo, fentanilo - para el dolor
- quinidina - para los latidos irregulares
- medicamentos para el cáncer - como irinotecán, etopósido, vinorelbina, ifosfamida
- medicamentos que contengan ‘alcaloides derivados del ergot’ - como ergotamina o diergotamina - para las migrañas
- medicamentos que diluyen la sangre - como warfarina, acenocumarol. El niño puede necesitar análisis de sangre durante el tratamiento con EMEND
- antibióticos para tratar infecciones - como rifampicina, claritromicina, telitromicina
- fenitoína - para las convulsiones
- carbamazepina - para la depresión y la epilepsia
- midazolam, triazolam, fenobarbital - para tranquilizar o para ayudar a dormir
- hierba de San Juan - medicamento a base de plantas para la depresión
- inhibidores de la proteasa - para infecciones por VIH
- ketoconazol, excepto champú (usado para tratar el síndrome de Cushing - cuando el cuerpo produce un exceso de cortisol)
- medicamentos antifungicos como itraconazol, voriconazol, posaconazol
- nefazodona - para la depresión
- corticosteroides - como dexametasona y metilprednisolona
- medicamentos para la ansiedad como alprazolam
- tolbutamida - para la diabetes
- medicamentos anticonceptivos que incluyen píldoras, parches, implantes y algunos dispositivos intrauterinos (DIUs) que liberan hormonas. Estos puede que no funcionen adecuadamente cuando se toman junto con este medicamento. Es posible que necesite utilizar otro método diferente o un método adicional de anticoncepción no hormonal durante el tratamiento con este medicamento y hasta 2 meses después de que el tratamiento haya finalizado.
Informe al médico, farmacéutico o enfermero antes de dar este medicamento si algo de lo citado anteriormente es aplicable al niño (o en caso de duda).
Embarazo y lactancia
No debe utilizar este medicamento durante el embarazo y la lactancia a no ser que sea claramente necesario.
Para información relacionada con el embarazo, la lactancia y la anticoncepción, pida consejo a su médico.
Conducción y uso de máquinas
Se debe tener en cuenta que algunas personas pueden sentir mareo y sueño después de tomar EMEND. Si el niño se siente mareado o somnoliento, no debe montar en bicicleta ni usar herramientas o máquinas.
EMEND contiene sacarosa y lactosa
El polvo para suspensión oral contiene sacarosa y lactosa. Si su médico le ha indicado que el niño padece una intolerancia a ciertos azúcares, consulte con él antes de dar al niño este medicamento.
3. Cómo dar EMEND
Profesionales del sector sanitario: Ver al final de este prospecto las instrucciones para profesionales del sector sanitario para la preparación de la suspensión oral. En ellas se le indica cómo preparar la dosis de EMEND en forma de suspensión oral.
Progenitores y cuidadores: Siga exactamente las instrucciones de administración de este medicamento indicadas para el niño por su médico, farmacéutico o enfermero. En caso de duda, consulte de nuevo al médico, farmacéutico o enfermero del niño.
Es muy importante que este medicamento se dé exactamente como se indica a continuación.
Para cada dosis de EMEND, tendrá un dispensador oral precargado que contiene la dosis prescrita al niño.
Mantener el dispensador oral en la nevera (entre 2°C y 8°C) hasta que dé el medicamento al niño.

Use este medicamento durante los 2 días posteriores a recibir el medicamento del profesional del sector sanitario.
Antes de su administración, el medicamento se puede tener a temperatura ambiente (no a más de 30°C) hasta 3 horas.

El color del medicamento en el dispensador oral puede ser de diferentes tonos de rosa (de rosa claro a rosa oscuro). Esto es normal y el medicamento se puede usar.
- Quite la tapa del dispensador oral.
- Coloque la punta del dispensador oral en la boca del niño en la cara interna de la mejilla, en el lado derecho o en el izquierdo.
- Empuje el émbolo lentamente hasta abajo del todo del dispensador oral para dar al niño todo el medicamento del dispensador oral.
Si el niño no puede tomar la dosis completa, llame al profesional sanitario del niño.
Los medicamentos no se deben tirar por los desagües ni a la basura, cuando haya terminado. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
Cuánto tomar
- El médico calculará la dosis correcta de polvo para suspensión oral en función del peso del niño.
- No modifique la dosis ni interrumpa el tratamiento sin consultar primero con el médico, farmacéutico o enfermero.
Cuándo tomar
Día 1:
- Dé este medicamento una hora antes del comienzo de la sesión de quimioterapia.
Día 2 y Día 3:
- Si el niño no va a tener quimioterapia - dé este medicamento por la mañana.
- Si el niño va a tener quimioterapia - dé este medicamento una hora antes de comenzar la sesión de quimioterapia.
EMEND se puede tomar con o sin alimentos.
Dé siempre este medicamento junto con otros medicamentos, para prevenir las náuseas y vómitos. Después del tratamiento con EMEND, el médico puede indicar que el niño continúe tomando otros medicamentos para prevenir las náuseas y los vómitos, tales como:
- un corticosteroide - como dexametasona y
- un ‘antagonista 5-HT3’ - como ondansetrón
En caso de duda pregunte al médico, farmacéutico o enfermero.
Si da más EMEND del que debe
No dé al niño más medicamento del recomendado por el médico. Si le da al niño más del que debe, contacte con el médico inmediatamente.
Si olvidó dar EMEND
Si el niño no toma una dosis de este medicamento, consulte al médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Efectos adversos graves
Deje de dar este medicamento y acuda a un médico inmediatamente si usted o el niño notan alguno de los siguientes efectos adversos graves - el niño puede necesitar tratamiento médico de urgencia:
- reacción alérgica - los signos pueden incluir ronchas, sarpullido, picor, dificultad para respirar o tragar (se desconoce la frecuencia con que suceden).
Deje de dar este medicamento y acuda a un médico inmediatamente si nota alguno de los efectos adversos graves citados anteriormente.
Otros efectos adversos
Informe al médico, farmacéutico o enfermero si usted o el niño notan alguno de los siguientes efectos adversos:
Frecuentes: pueden afectar hasta 1 de cada 10 personas
- estreñimiento o indigestión
- dolor de cabeza
- sensación de cansancio
- pérdida de apetito
- hipo
- aumento de la cantidad de enzimas del hígado en la sangre (se observa en los análisis).
Poco frecuentes: pueden afectar hasta 1 de cada 100 personas
- sensación de mareo o somnolencia
- acné, sarpullido
- sensación de ansiedad
- eructos, náuseas, vómitos, ardor de estómago, dolor de estómago, boca seca, flatulencias
- dolor o escozor al orinar
- sensación de debilidad, malestar general
- sofocos/enrojecimiento de la cara o de la piel
- latidos rápidos o irregulares
- fiebre con riesgo elevado de infección, número bajo de glóbulos rojos (se observa en los análisis).
Raros: pueden afectar hasta 1 de cada 1.000 personas
- dificultad para pensar, falta de energía, cambios en el gusto
- sensibilidad de la piel al sol, sudor excesivo, piel grasa, llagas en la piel, erupción con picor, síndrome de Stevens-Johnson o necrolisis epidérmica tóxica (reacciones cutáneas graves raras)
- euforia (sensación de felicidad extrema), sensación de confusión
- infección bacteriana, infección por hongos
- estreñimiento intenso, úlcera de estómago, intestino delgado y colon inflamados, llagas en la boca, hinchazón del vientre
- orinar más frecuentemente o más de lo normal, azúcar o sangre en la orina
- molestias en el pecho, hinchazón, cambios en la manera de andar
- tos, mucosidad en la parte de atrás de la garganta, irritación de la garganta, estornudos, dolor de garganta
- secreción y picor oculares
- zumbido de oídos
- espasmos musculares, debilidad muscular
- sensación de mucha sed
- latidos lentos, problemas de los vasos del corazón y de las venas
- número bajo de glóbulos blancos, niveles bajos de sodio en la sangre, pérdida de peso. Comunicación de efectos adversos
Si el niño experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de EMEND
Mantener este medicamento fuera de la vista y del alcance de los niños.
Antes de la reconstitución:
Emend será conservado generalmente por profesionales del sector sanitario. Los detalles de conservación, si los necesita, son los siguientes:
No dé este medicamento al niño después de la fecha de caducidad que aparece en la caja y el sobre después de CAD. La fecha de caducidad es el último día del mes que se indica.
Este medicamento no requiere ninguna temperatura especial de conservación.
Conservar en el embalaje original para protegerlo de la humedad.
Después de la reconstitución:
Antes de su administración, la suspensión oral se puede tener a temperatura ambiente (no a más de 30°C) hasta 3 horas. También se puede conservar refrigerada (entre 2°C y 8°C) hasta 72 horas.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
Composición de EMEND
- El principio activo es aprepitant. Cada sobre contiene 125 mg de aprepitant. Tras la reconstitución, 1 ml de suspensión oral contiene 25 mg de aprepitant.
- Los demás componentes son: hidroxipropilcelulosa (E-463), lauril sulfato de sodio (E-487), sacarosa y lactosa (ver en la sección 2 “EMEND contiene sacarosa y lactosa”), óxido de hierro rojo (E-172) y estearil fumarato de sodio (E-485).
Aspecto del producto y contenido del envase
El polvo para suspensión oral es un polvo de color rosa a rosa claro en un sobre de un solo uso.
Caja de un solo uso
La caja contiene un sobre, un dispensador oral de 1 ml y otro de 5 ml (de polipropileno con un émbolo
rematado con un anillo de silicona), una tapa y un vaso de mezcla (de polipropileno).
Titular de la autorización de comercialización y responsable de la fabricación
|
Titular de la autorización de comercialización Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN11 9BU Reino Unido |
Responsable de la fabricación Merck Sharp & Dohme B. V. Waarderweg 39 2031 BN Haarlem Países Bajos |
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgique/Belgie/Belgien MSD Belgium BVBA/SPRL Tél/Tel: 0800 38 693 (+32(0)27766211) |
Lietuva UAB Merck Sharp & Dohme Tel.:+370 5 278 02 47 msd_lietuva@merck.com |
|
Etarapna MepK fflapn h floyM Etarapna EOOfl Tea.: +359 2 819 3737 info-msdbg@merck.com |
Luxembourg/Luxemburg MSD Belgium BVBA/SPRL Tél/Tel: +32(0)27766211 dpoc_belux@merck.com |
|
Ceská republika Merck Sharp & Dohme s.r.o. Tel.: +420 233 010 111 dpoc_czechslovak@merck.com |
Magyarország MSD Pharma Hungary Kft. Tel.: +36 1 888 53 00 hungary_msd@merck.com |
|
Danmark MSD Danmark ApS Tlf: +45 44824000 dkmail@merck.com |
Malta Merck Sharp & Dohme Cyprus Limited Tel: 8007 4433 (+356 99917558) malta info@merck.com |
|
Deutschland MSD SHARP & DOHME GMBH Tel: 0800 673 673 673 (+49 (0) 89 4561 2612) |
Nederland Merck Sharp & Dohme BV Tel: 0800 9999000 (+31 23 5153153) |
|
Eesti Merck Sharp & Dohme OÜ Tel.: +372 6144 200 msdeesti@merck.com |
Norge MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no |
|
EXXáSa MSD A.O.B.EE. Tp^: + 30 210 98 97 300 dpoc_greece@merck.com |
Osterreich Merck Sharp & Dohme Ges.m.b.H. Tel: +43 (0) 1 26 044 msd-medizin@merck.com |
|
España Merck Sharp & Dohme de España, S.A. Tel: +34 91 321 06 00 msd_info@merck.com |
Polska MSD Polska Sp.z o.o. Tel:+48 22 549 51 00 msdpolska@merck.com |
|
France MSD France Tél: +33 (0) 1 80 46 40 40 |
Portugal Merck Sharp & Dohme, Lda Tel: +351 21 4465700 clic@merck.com |
Hrvatska
Merck Sharp & Dohme d.o.o.
Tel: + 385 1 6611 333 croatia_info@merck.com
Ireland
Merck Sharp & Dohme Ireland (Human Health) Limited
Tel: +353 (0)1 2998700 medinfo_ireland@merck.com
Ísland
Vistor hf.
Simi: +354 535 7000
Italia
MSD Italia S.r.l.
Tel: +39 06 361911 medicalinformation.it@merck.com
Kúrcpog
Merck Sharp & Dohme Cyprus Limited T^: 80000 673 (+357 22866700) cyprus_info@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: +371 67364 224 msd_lv@merck.com
Romanía
Merck Sharp & Dohme Romania S.R.L.
Tel: + 4021 529 29 00 msdromania@merck.com
Slovenija
Merck Sharp & Dohme, inovativna zdravila d.o.o.
Tel:+ 386 1 5204201 msd_slovenia@merck.com
Slovenská republika
Merck Sharp & Dohme, s. r. o.
Tel.: +421 2 58282010 dpoc_czechslovak@merck.com
Suomi/Finland
MSD Finland Oy Puh/Tel: +358 (0)9 804650 info@msd.fi
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 (0) 77 5700488 medicinskinfo@merck.com
United Kingdom
Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com
Fecha de la última revisión de este prospecto:
Otras fuentes de información
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Esta información está destinada únicamente a profesionales del sector sanitario:
Instrucciones para profesionales del sector sanitario sobre la preparación de la suspensión oral
Cada caja de EMEND contiene un sobre con el polvo para suspensión oral, un dispensador oral de 1 ml y otro de 5 ml, una tapa y un vaso de mezcla.

1.
Llene el vaso de mezcla con agua potable a temperatura ambiente.
2.
Llene el dispensador oral de 5 ml con 4,6 ml de agua del vaso de mezcla.
Compruebe que no hay aire en el dispensador oral (si hay aire, elimínelo).
3.
Tire toda el agua no utilizada que quede en el vaso de mezcla.
4.
Añada de nuevo los 4,6 ml de agua del dispensador oral al vaso de mezcla.

5.
Cada sobre de EMEND para suspensión oral contiene 125 mg de aprepitant, que se usa para hacer una suspensión en 4,6 ml de agua, obteniéndose una concentración final de 25 mg/ml.
Sujete recto el sobre de EMEND polvo para suspensión oral y agite el contenido hacia la parte inferior antes de abrir el sobre.
6. Vacíe todo el contenido del sobre en los 4,6 ml de agua del vaso de mezcla y cierre la tapa.
7.
Mezcle la suspensión de EMEND suavemente, moviendo el vaso de forma lateral 20 veces; luego gire el vaso de mezcla suavemente 5 veces.
Para evitar espuma, no agite el vaso de mezcla. La mezcla será de color entre rosa turbio y rosa claro.
Línea de apertura

|
8. Compruebe la mezcla de EMEND por si tiene grumos o espuma: - Si hay grumos, repita el paso 7 hasta que no haya grumos. - Si hay espuma, espere a que ésta desaparezca antes de pasar al paso 9. | ||||||
|
Peso corporal |
Volumen de la dosis de suspensión para administración por vía oral | |||||
|
Día 1 |
Día 2 |
Día 3 | ||||
|
menos de 6 kg |
No recomendada | |||||
|
de 6 kg a menos de 8 kg |
1 ml (25 mg) |
0,6 ml (15 mg) |
0,6 ml (15 mg) | |||
|
de 8 kg a menos de 10 kg |
1,2 ml (30 mg) |
0,8 ml (20 mg) |
0,8 ml (20 mg) | |||
|
de 10 kg a menos de 12 kg |
1,4 ml (35 mg) |
1 ml (25 mg) |
1 ml (25 mg) | |||
|
de 12 kg a menos de 15 kg |
1,8 ml (45 mg) |
1,2 ml (30 mg) |
1,2 ml (30 mg) | |||
|
de 15 kg a menos de 20 kg |
2,4 ml (60 mg) |
1,6 ml (40 mg) |
1,6 ml (40 mg) | |||
|
de 20 kg a menos de 25 kg |
3 ml (75 mg) |
2 ml (50 mg) |
2 ml (50 mg) | |||
|
de 25 kg a menos de 30 kg |
3,6 ml (90 mg) |
2,4 ml (60 mg) |
2,4 ml (60 mg) | |||
|
30 kg y superiores |
Extraer todo (125 mg) el contenido del vaso de mezcla al dispensador oral (~ 5 ml) |
3,2 ml (80 mg) |
3,2 ml (80 mg) | |||
|
9. Llene el dispensador oral con suspensión del vaso de mezcla, con la cantidad de dosis prescrita de acuerdo con la tabla de arriba. - Elija el dispensador oral según la dosis: - Use del dispensador oral de 1 ml si la dosis es 1 ml o menos. - Use el dispensador oral de 5 ml si la dosis es más de 1 ml. - Es frecuente que quede medicamento sobrante en el vaso. Compruebe que no haya aire en el dispensador oral (si hay aire, elimínelo). Compruebe que el dispensador oral contiene la dosis prescrita. |
1 i | Dispensador_ 3 O i""1_Dispensador; v* _ de 1 ml de 5 ml : | |||||
|
10. Ajuste la tapa en el dispensador oral hasta que encaje. 11. Si la dosis no se administra inmediatamente después de medirla, conserve el(los) dispensador(es) oral(es) lleno(s) en la nevera entre 2°C y 8°C durante un máximo de 72 horas antes de su uso. Cuando se dispense(n) la(s) dosis a los cuidadores, indíqueles que conserven el(los) dispensador(es) oral(es) refrigerado(s) hasta que estén listos para administrar la dosis. 12. La suspensión oral se puede tener a temperatura ambiente (no a más de 30oC) hasta 3 horas, antes de su administración. |
[■'..............................i...... |
-\ | ||||
Deseche la suspensión que sobre y los materiales que hayan estado en contacto con ella. La eliminación del medicamento no utilizado y todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
135
25-120 horas_653_61,5_3,9 (-2,6, 10,3)
Los intervalos de confianza se calcularon sin ajuste por categoría de edad (< 55 años, > 55 años) ni por grupo del investigador, los cuales fueron incluidos en el análisis primario de cociente de posibilidades y modelos logísticos.
tSólo un paciente en el régimen de aprepitant tuvo datos en la fase aguda y se excluyó de los análisis de fase retardada y general.
En el mismo ensayo clínico, 744 pacientes adultos continuaron en la extensión de Ciclos Múltiples durante 3 ciclos adicionales de quimioterapia. La eficacia del régimen de aprepitant se mantuvo aparentemente durante todos los ciclos.
En un segundo ensayo clínico multicéntrico, aleatorizado, doble ciego, con grupo paralelo, el régimen de aprepitant se comparó con el tratamiento estándar en 848 pacientes adultos (652 mujeres,
196 varones) que recibían un régimen de quimioterapia que incluía cualquier dosis vía intravenosa de oxaliplatino, carboplatino, epirubicina, idarubicina, ifosfamida, irinotecán, daunorubicina,
25-120 horas_653_61,5_3,9 (-2,6, 10,3)
Los intervalos de confianza se calcularon sin ajuste por categoría de edad (< 55 años, > 55 años) ni por grupo del investigador, los cuales fueron incluidos en el análisis primario de cociente de posibilidades y modelos logísticos.
tSólo un paciente en el régimen de aprepitant tuvo datos en la fase aguda y se excluyó de los análisis de fase retardada y general.
En el mismo ensayo clínico, 744 pacientes adultos continuaron en la extensión de Ciclos Múltiples durante 3 ciclos adicionales de quimioterapia. La eficacia del régimen de aprepitant se mantuvo aparentemente durante todos los ciclos.
En un segundo ensayo clínico multicéntrico, aleatorizado, doble ciego, con grupo paralelo, el régimen de aprepitant se comparó con el tratamiento estándar en 848 pacientes adultos (652 mujeres,
196 varones) que recibían un régimen de quimioterapia que incluía cualquier dosis vía intravenosa de oxaliplatino, carboplatino, epirubicina, idarubicina, ifosfamida, irinotecán, daunorubicina, doxorubicina; ciclofosfamida vía intravenosa (< 1.500 mg/m2); o citarabina vía intravenosa (> 1 g/m2). Los pacientes que recibían el régimen de aprepitant, estaban recibiendo quimioterapia para diversos tipos de tumores incluyendo 52 % con cáncer de mama, 21 % con algún cáncer de tipo gastrointestinal incluido cáncer colorrectal, 13 % con cáncer de pulmón y 6 % con algún cáncer de tipo ginecológico. El régimen de aprepitant en combinación con un régimen de ondansetrón/dexametasona (ver sección 4.2) se comparó con el tratamiento estándar (placebo en combinación con ondansetrón 8 mg vía oral (dos veces al día el día 1, y cada 12 horas los días 2 y 3) más dexametasona 20 mg vía oral el día 1).
La eficacia se basó en la evaluación de las siguientes variables primaria y secundaria más importantes: Sin vómitos en el periodo completo (0 a 120 horas después de la quimioterapia), evaluación de la seguridad y tolerancia del régimen de aprepitant para las náuseas y los vómitos inducidos por quimioterapia (NVIQ) y respuesta completa (definida como sin vómitos y sin uso de tratamiento de rescate) en el periodo completo (0 a 120 horas después de la quimioterapia). Además, se evaluó la ausencia de náuseas significativas en el periodo completo (0 a 120 horas después de la quimioterapia) como variable exploratoria, y en las fases aguda y retardada como análisis post-hoc.
25-120 horas_653_61,5_3,9 (-2,6, 10,3)
Los intervalos de confianza se calcularon sin ajuste por categoría de edad (< 55 años, > 55 años) ni por grupo del investigador, los cuales fueron incluidos en el análisis primario de cociente de posibilidades y modelos logísticos.
tSólo un paciente en el régimen de aprepitant tuvo datos en la fase aguda y se excluyó de los análisis de fase retardada y general.
En el mismo ensayo clínico, 744 pacientes adultos continuaron en la extensión de Ciclos Múltiples hasta un máximo de 3 ciclos adicionales de quimioterapia. La eficacia del régimen de aprepitant se mantuvo aparentemente durante todos los ciclos.
En un segundo ensayo clínico multicéntrico, aleatorizado, doble ciego, con grupo paralelo, el régimen de aprepitant se comparó con el tratamiento estándar en 848 pacientes adultos (652 mujeres,
196 varones) que recibían un régimen de quimioterapia que incluía cualquier dosis vía intravenosa de oxaliplatino, carboplatino, epirubicina, idarubicina, ifosfamida, irinotecán, daunorubicina, doxorubicina; ciclofosfamida vía intravenosa (< 1.500 mg/m2); o citarabina vía intravenosa (> 1 g/m2). Los pacientes que recibían el régimen de aprepitant, estaban recibiendo quimioterapia para diversos tipos de tumores incluyendo 52 % con cáncer de mama, 21 % con algún cáncer de tipo gastrointestinal