Elocta 250Ui Polvo Y Disolvente Para Solucion Inyectable
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
ELOCTA 250 UI polvo y disolvente para solución inyectable ELOCTA 500 UI polvo y disolvente para solución inyectable ELOCTA 750 UI polvo y disolvente para solución inyectable ELOCTA 1000 UI polvo y disolvente para solución inyectable ELOCTA 1500 UI polvo y disolvente para solución inyectable ELOCTA 2000 UI polvo y disolvente para solución inyectable ELOCTA 3000 UI polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
ELOCTA 250 UI polvo y disolvente para solución inyectable
Cada vial contiene nominalmente 250 UI de efmoroctocog alfa. Después de la reconstitución, cada ml de solución inyectable contiene aproximadamente 83 UI de efmoroctocog alfa.
ELOCTA 500 UI polvo y disolvente para solución inyectable
Cada vial contiene nominalmente 500 UI de efmoroctocog alfa. Después de la reconstitución, cada ml de solución inyectable contiene aproximadamente 167 UI de efmoroctocog alfa.
ELOCTA 750 UI polvo y disolvente para solución inyectable
Cada vial contiene nominalmente 750 UI de efmoroctocog alfa. Después de la reconstitución, cada ml de solución inyectable contiene aproximadamente 250 UI de efmoroctocog alfa.
ELOCTA 1000 UI polvo y disolvente para solución inyectable
Cada vial contiene nominalmente 1000 UI de efmoroctocog alfa. Después de la reconstitución, cada ml de solución inyectable contiene aproximadamente 333 UI de efmoroctocog alfa.
ELOCTA 1500 UI polvo y disolvente para solución inyectable
Cada vial contiene nominalmente 1500 UI de efmoroctocog alfa. Después de la reconstitución, cada ml de solución inyectable contiene aproximadamente 500 UI de efmoroctocog alfa.
ELOCTA 2000 UI polvo y disolvente para solución inyectable
Cada vial contiene nominalmente 2000 UI de efmoroctocog alfa. Después de la reconstitución, cada ml de solución inyectable contiene aproximadamente 667 UI de efmoroctocog alfa.
ELOCTA 3000 UI polvo y disolvente para solución inyectable
Cada vial contiene nominalmente 3000 UI de efmoroctocog alfa. Después de la reconstitución, cada ml de solución inyectable contiene aproximadamente 1000 UI de efmoroctocog alfa.
La potencia (Unidades Internacionales) se determina utilizando el ensayo de sustrato cromogénico de la Farmacopea Europea frente a un estándar interno referenciado al estándar del factor VIII de la OMS. La actividad específica de ELOCTA es de 4.000 - 10.200 UI/mg de proteína.
Efmoroctocog alfa (factor VIII de coagulación recombinante humano, proteína de fusión Fc [rFVIIIFc]) consta de 1.890 aminoácidos. Se produce mediante tecnología de DNA recombinante en una línea celular embrionaria de riñón humano (HEK) sin la adición de ninguna proteína exógena de origen humano o animal en el proceso de cultivo celular, la purificación o la formulación final.
Excipiente con efecto conocido
0,6 mmol (o 14 mg) de sodio por vial.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: liofilizado, suelto o sólido de color blanco a blanquecino.
Disolvente: agua para preparaciones inyectables, una disolución transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII).
ELOCTA se puede usar en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento se debe iniciar bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia.
Pacientes sin tratamiento previo
No se ha establecido todavía la seguridad y eficacia de ELOCTA en pacientes no tratados previamente. No se dispone de datos.
Posología
La dosis y la duración del tratamiento de sustitución dependen de la gravedad de la deficiencia de factor VIII, de la localización y la magnitud de la hemorragia y del estado clínico del paciente.
El número de unidades de Fc de factor VIII recombinante administradas se expresa en Unidades Internacionales (UI), en relación con el estándar actual de la OMS para los productos de factor VIII. La actividad plasmática del factor VIII se expresa como un porcentaje (en relación con el plasma humano normal) o en Unidades Internacionales (en relación con un estándar internacional para el factor VIII plasmático).
La actividad de 1 UI de Fc de factor VIII recombinante equivale a la cantidad de factor VIII presente en 1 ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis necesaria de Fc de factor VIII recombinante se basa en el hallazgo empírico de que 1 Unidad Internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad plasmática del factor VIII en 2 UI/dl. La dosis necesaria se determina mediante la siguiente fórmula:
Unidades necesarias = peso corporal (kg) x aumento deseado del factor VIII (%) (UI/dl) x 0,5 (UI/kg por UI/dl)
La dosis y la frecuencia de administración se establecerán siempre en función de la eficacia clínica observada en cada caso (ver sección 5.2). No se prevé un retraso en el tiempo transcurrido hasta la actividad máxima.
En el caso de los siguientes episodios hemorrágicos, la actividad del factor VIII no debe caer por debajo del nivel de actividad plasmática establecido (en % del nivel normal o UI/dl) durante el periodo correspondiente. La Tabla 1 se puede usar como guía posológica en los episodios hemorrágicos y durante la cirugía:
Tabla 1: Guía posológica de ELOCTA para el tratamiento de los episodios hemorrágicos y durante la cirugía
|
Grado de hemorragia / Tipo de procedimiento quirúrgico |
Nivel requerido de factor VIII (%) (UI/dl) |
Frecuencia de dosificación (horas) / Duración del tratamiento (días) |
|
Hemorragia Hemartrosis precoz, hemorragia muscular o hemorragia oral |
20 - 40 |
Repetir la inyección cada 12 a 24 horas durante al menos 1 día, hasta que, en función del dolor, el episodio hemorrágico se haya resuelto o hasta que se produzca la cicatrización. 1 |
|
Hemartrosis, hemorragia muscular o hematoma más extensos |
30 - 60 |
Repetir la inyección cada 12 a 24 horas durante 3-4 días o más hasta que el dolor y la discapacidad aguda se hayan resuelto. 1 |
|
Hemorragias potencialmente mortales |
60 - 100 |
Repetir la inyección cada 8 a 24 horas hasta que desaparezca el riesgo. |
|
Cirugía Cirugía menor, incluidas las extracciones dentales |
30 - 60 |
Repetir la inyección cada 24 horas, durante al menos 1 día hasta que se produzca la cicatrización. |
|
Cirugía mayor |
80 - 100 (antes y después de la cirugía) |
Repetir la inyección cada 8 a 24 horas según sea necesario hasta la cicatrización adecuada de la herida y después continuar el tratamiento al menos otros 7 días para mantener una actividad del factor VIII del 30 % al 60 % (UI/dl). |
1 En ciertos pacientes y circunstancias, el intervalo de dosificación se puede prolongar hasta 36 horas. Ver la sección 5.2 para consultar los datos farmacocinéticos.
Profilaxis
Para la profilaxis a largo plazo, la dosis recomendada es de 50 UI/kg cada 3 a 5 días. La dosis se puede ajustar en función de la respuesta del paciente dentro de un intervalo comprendido entre 25 y 65 UI/kg (ver secciones 5.1 y 5.2). En algunos casos, especialmente en pacientes jóvenes, es posible que sea necesario acortar los intervalos de administración o usar dosis más elevadas.
Control del tratamiento
Durante el transcurso del tratamiento se recomienda controlar adecuadamente los niveles de factor VIII (mediante el ensayo de coagulación en una fase o el ensayo cromogénico) para estimar la dosis que se debe administrar y la frecuencia de repetición de las inyecciones. La respuesta individual de los pacientes al factor VIII puede variar y presentar distintas semividas y niveles de recuperación. La dosis basada en el peso corporal puede precisar ajustes en los pacientes con bajo peso o sobrepeso. En el caso de las intervenciones de cirugía mayor en particular, es indispensable controlar con precisión el tratamiento de sustitución mediante pruebas de coagulación (actividad plasmática del factor VIII).
Cuando se utilice una prueba de coagulación de una etapa basada en el tiempo de tromboplastina (TTPa) in vitro para determinar la actividad del factor VIII en las muestras de sangre de los pacientes, los resultados de la actividad plasmática del factor VIII se pueden ver afectados considerablemente tanto por el tipo de reactivo TTPa como por el patrón de referencia utilizado en el análisis. Esto es importante, sobre todo cuando se cambia de laboratorio o de reactivo para la prueba.
Personas de edad avanzada
Se dispone de experiencia limitada en los pacientes >65 años.
Población _ pediátrica
Los niños menores de 12 años pueden precisar dosis más altas o más frecuentes (ver sección 5.1). En los adolescentes de edad igual o superior a 12 años, las recomendaciones posológicas son las mismas que en los adultos.
Forma de administración Vía intravenosa.
ELOCTA se debe inyectar por vía intravenosa durante varios minutos. La velocidad de administración se debe determinar en función del grado de comodidad del paciente y no debe superar los 10 ml/min.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo (factor VIII de coagulación recombinante humano, o dominio Fc) o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Hipersensibilidad
Es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico con ELOCTA. Si aparecen síntomas de hipersensibilidad, se debe aconsejar a los pacientes que interrumpan inmediatamente el uso del medicamento y se pongan en contacto con su médico.
Se debe informar a los pacientes sobre los signos de las reacciones de hipersensibilidad, como habón urticarial, urticaria generalizada, tirantez de pecho, sibilancia, hipotensión y anafilaxia.
En caso de choque anafiláctico, se debe instaurar el tratamiento médico estándar para el choque.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) contra el factor VIII es una complicación conocida del tratamiento de los pacientes con hemofilia A. Estos inhibidores son generalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, que se cuantifica en Unidades Bethesda (UB) por ml de plasma, utilizando el ensayo modificado. El riesgo de desarrollar inhibidores se correlaciona con la exposición al factor VIII y es mayor en los primeros 20 días de exposición. Raramente se pueden desarrollar inhibidores tras los primeros 100 días de exposición.
Se han observado casos de inhibidor recurrente (titulación baja) después de cambiar desde un producto de factor VIII a otro, en pacientes tratados anteriormente con más de 100 días de exposición que tenían antecedentes de desarrollo de inhibidores. Por lo tanto, se recomienda vigilar cuidadosamente a todos los pacientes en cuanto a la aparición de inhibidores después de cualquier cambio de producto.
En general, todos los pacientes tratados con productos del factor VIII de coagulación deben ser controlados cuidadosamente por si desarrollan inhibidores, mediante las observaciones clínicas y pruebas analíticas adecuadas. Si no se alcanzan los niveles esperados de actividad plasmática del factor VIII o si la hemorragia no se controla con la dosis adecuada, se deben realizar pruebas para descartar la presencia de inhibidores del factor VIII. En los pacientes con niveles altos de inhibidor, el tratamiento con factor VIII puede no ser eficaz y se deben considerar otras opciones terapéuticas. El tratamiento de estos pacientes deben dirigirlo médicos con experiencia en pacientes con hemofilia y en inhibidores del factor VIII.
Acontecimientos cardiovasculares
En pacientes con factores de riesgo cardiovascular ya existentes, el tratamiento de sustitución con FVIII puede aumentar el riesgo cardiovascular.
Complicaciones asociadas al catéter
Si para la administración es necesario un dispositivo de acceso venoso central (DAVC), se debe tener en cuenta el riesgo de complicaciones relacionadas con el DAVC, incluidas las infecciones locales, la bacteriemia y la trombosis en el lugar de colocación del catéter.
Registro del número de lote
Se recomienda encarecidamente que cada vez que se administre ELOCTA a un paciente, se registren el nombre y el número de lote del producto con el fin de tener una relación entre el paciente y el lote del medicamento.
Población pediátrica
Las advertencias y precauciones descritas son válidas tanto para adultos como para niños.
Consideraciones relativas a los excipientes
Este medicamento contiene 0,6 mmol (o 14 mg) de sodio por vial, lo que debe ser tenido en cuenta en pacientes con dietas pobres en sodio.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han notificado interacciones del factor VIII de coagulación humano (rDNA) con otros medicamentos. No se han realizado ensayos de interacciones con ELOCTA.
4.6 Fertilidad, embarazo y lactancia
Embarazo y lactancia
No se han realizado estudios de reproducción animal con ELOCTA. Se realizó un estudio de transferencia placentaria en ratones (ver sección 5.3). Dado que la hemofilia A aparece raramente en las mujeres, no se dispone de experiencia sobre el uso del factor VIII durante el embarazo y la lactancia. Por lo tanto, solo se debe usar el factor VIII durante el embarazo y la lactancia si está claramente indicado.
Fertilidad
No se dispone de datos sobre la fertilidad. No se han realizado estudios de fertilidad en animales con ELOCTA.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de ELOCTA sobre la capacidad para conducir y utilizar máquinas es nula.
Resumen del perfil de seguridad
Se han observado raramente reacciones alérgicas o de hipersensibilidad (entre las que se pueden encontrar hinchazón de la cara, erupción, habón urticarial, tirantez de pecho y dificultad para respirar, escozor y punzadas en el lugar de la perfusión, escalofríos, rubefacción, urticaria generalizada, cefalea, hipotensión, letargia, náuseas, inquietud y taquicardia), que en algunos casos pueden evolucionar a anafilaxia grave (incluido choque).
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) contra el factor VIII. Si aparecen dichos inhibidores, la situación se manifestará en forma de respuesta clínica insuficiente. En estos casos, se recomienda ponerse en contacto con un centro especializado en hemofilia.
Tabla de reacciones adversas
Las frecuencias mostradas en la Tabla 2 siguiente se observaron en un total de 233 pacientes con hemofilia A grave en ensayos clínicos de fase 3 y en un estudio de extensión. El número total de días de exposición fue de 34.746 días, con una mediana de 129 (intervalo 1 - 326) días de exposición por sujeto.
La Tabla 2, que figura a continuación, está ordenada conforme a la clasificación de órganos del sistema MedDRA (COS y nivel terminológico preferido).
Las frecuencias se han evaluado según la siguiente convención: muy frecuente (>1/10); frecuente (>1/100 a <1/10); poco frecuente (>1/1.000 a <1/100); rara (>1/10.000 a <1/1.000); muy rara (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Clasificación de órganos del sistema MedDRA |
Reacciones adversas |
Categoría de frecuencia |
|
Trastornos del sistema nervioso |
Cefalea Mareo Disgeusia |
Poco frecuente Poco frecuente Poco frecuente |
|
Trastornos cardiacos |
Bradicardia |
Poco frecuente |
|
Trastornos vasculares |
Hipertensión Sofoco Angiopatía1 |
Poco frecuente Poco frecuente Poco frecuente |
|
Trastornos respiratorios, torácicos y mediastínicos |
Tos |
Poco frecuente |
|
Trastornos gastrointestinales |
Dolor abdominal inferior |
Poco frecuente |
|
Trastornos de la piel y del tejido subcutáneo |
Erupción |
Poco frecuente |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgias Mialgias Dolor de espalda Tumefacción articular |
Poco frecuente Poco frecuente Poco frecuente Poco frecuente |
|
Trastornos generales y alteraciones en el lugar de administración |
Malestar general Dolor torácico Sensación de frío Sensación de calor |
Poco frecuente Poco frecuente Poco frecuente Poco frecuente |
|
Exploraciones complementarias |
Positivo a anticuerpos antifactor VIII2 |
Poco frecuente |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Hipotensión por procedimiento terapéutico |
Poco frecuente |
1 Término del investigador: dolor vascular tras la inyección de ELOCTA
2 Un sujeto adulto tuvo un resultado positivo en la prueba de anticuerpos antifactor VIII que coincidió con una única determinación de una titulación de anticuerpo neutralizante de 0,73 Unidades Bethesda/ml en la semana 14. El anticuerpo neutralizante no se confirmó al repetir la prueba 18 días después y fue negativo en las siguientes visitas. Hubo un aumento del aclaramiento (CL) en la semana 14 que desapareció continuando el tratamiento con rFVIIIFc.
Experiencia post-comercialización
En la experiencia post-comercialización se ha observado el desarrollo de inhibidores del factor VIII. Población pediátrica
No se han observado diferencias específicas debidas a la edad en los sujetos pediátricos y los adultos con respecto a las reacciones adversas.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
No se han notificado síntomas de sobredosis.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos, factor VIII de coagulación sanguínea, código ATC: B02BD02
Mecanismo de acción
El complejo factor VIII/factor de von Willebrand consta de 2 moléculas (factor VIII y factor de von Willebrand) con diferentes funciones fisiológicas. Tras la activación de la cascada de la coagulación, el factor VIII se convierte en factor VIII activado y se libera del factor de von Willebrand. El factor VIII activado actúa como cofactor del factor IX activado, lo que acelera la conversión del factor X a factor X activado en las superficies fosfolipídicas. El factor X activado convierte la protrombina en trombina. La trombina convierte a su vez el fibrinógeno en fibrina, lo que permite la formación del coágulo sanguíneo.
La hemofilia A es un trastorno hereditario de la coagulación sanguínea ligado al cromosoma X que se debe a una disminución de los niveles de factor VIII funcional, lo que da lugar a hemorragias en las articulaciones, los músculos o los órganos internos, ya sea de forma espontánea o como consecuencia de un traumatismo accidental o quirúrgico. El tratamiento de sustitución aumenta los niveles plasmáticos del factor VIII, lo que permite corregir temporalmente la deficiencia de este factor y la tendencia a las hemorragias.
ELOCTA (efmoroctocog alfa) es una proteína de fusión totalmente recombinante con una semivida extendida. ELOCTA está constituida por factor VIII de coagulación humano recombinante con deleción del dominio B, unido covalentemente al dominio Fc de la inmunoglobulina humana G1. La región Fc de la inmunoglobulina humana G1 se une al receptor Fc neonatal. Este receptor se expresa durante toda la vida y forma parte de una vía natural que protege a las inmunoglobulinas de la degradación lisosómica al reciclar estas proteínas de vuelta a la circulación, lo que causa su prolongada semivida plasmática. Efmoroctocog alfa se une al receptor Fc neonatal, de modo que utiliza esta misma vía natural para retrasar su degradación lisosómica y alcanzar una semivida plasmática más prolongada que la del factor VIII endógeno.
Eficacia clínica y seguridad
La seguridad, eficacia y farmacocinética de ELOCTA se evaluaron en 2 estudios pivotales multinacionales abiertos: un estudio de fase 3, al que se hará referencia como estudio I, y un estudio pediátrico de fase 3, al que se hará referencia como estudio II (ver Población pediátrica).
En el estudio I se comparó la eficacia de 2 pautas de tratamiento profilácticas (individualizada y semanal) con el tratamiento a demanda. En el estudio se inscribió a un total de 165 pacientes varones previamente tratados (de 12 a 65 años) con hemofilia A grave. Los sujetos que estaban recibiendo pautas profilácticas antes de su entrada en el estudio se asignaron al grupo de profilaxis individualizada. Los sujetos que estaban recibiendo tratamiento a demanda antes de su entrada se incorporaron al grupo de profilaxis individualizada o se aleatorizaron a los grupos de profilaxis semanal o tratamiento a demanda. En el grupo de profilaxis individualizada, los sujetos comenzaron con una pauta de dos dosis por semana que consistía en la administración de 25 UI/kg en el primer día seguidos de 50 UI/kg en el cuarto día. Las dosis y los intervalos de la profilaxis individualizada se ajustaron dentro de un intervalo de entre 25 y 65 UI/kg cada 3 a 5 días. La dosis de la profilaxis semanal fue de 65 UI/kg. Además, en el estudio I se evaluó la eficacia hemostática en el tratamiento de los episodios hemorrágicos y se determinó la eficacia hemostática durante el tratamiento perioperatorio de los sujetos sometidos a intervenciones de cirugía mayor.
Profilaxis individualizada: En los 117 sujetos evaluables inscritos en el grupo de profilaxis individualizada del estudio I, la mediana del intervalo de dosificación fue 3,51 (intervalo intercuartílico: 3,17 - 4,43) días y la mediana de la dosis semanal total fue 77,90 (intervalo intercuartílico: 72,35 - 91,20) UI/kg.
Las medianas de las tasas de hemorragia anualizadas en los sujetos evaluables para la eficacia fueron de 1,60 (intervalo intercuartílico: 0,0 - 4,69) para los del grupo de profilaxis individualizada, 3,59 (1,86 - 8,36) para los del grupo de profilaxis semanal y 33,57 (21,14 - 48,69) para los del grupo de tratamiento a demanda. No se observaron episodios hemorrágicos en el 45,3% de los sujetos durante el régimen de profilaxis individualizada ni en el 17,4% de los sujetos durante el régimen de profilaxis semanal.
Tratamiento de las hemorragias: De los 757 episodios hemorrágicos observados durante el estudio I, el 87,3% se controlaron con 1 inyección y el 97,8% con 2 inyecciones o menos. La mediana de la dosis por inyección para tratar un episodio hemorrágico fue 27,35 (intervalo intercuartílico: 22,73 - 32,71) UI/kg. La mediana de la dosis global para tratar un episodio hemorrágico fue 31,32 UI/kg (23,53 - 52,53) en los grupos de profilaxis individualizada y de profilaxis semanal y 27,35 IU/kg (22,59 - 32,71) en el grupo de tratamiento a demanda.
Tratamiento _perioperatorio (profilaxis quirúrgica): Se realizaron y evaluaron un total de 23 intervenciones de cirugía mayor en 22 sujetos del estudio I y de un estudio de extensión. La mayoría de los sujetos (95,7%) recibieron una única dosis preoperatoria para mantener la hemostasia durante la intervención. La mediana de la dosis por inyección para mantener la hemostasia durante la intervención fue 58,3 (intervalo: 45 - 102) UI/kg. La mayoría de los sujetos recibieron una segunda inyección el día de la intervención. La dosis total administrada el día de la intervención osciló entre 50,8 y 126,6 UI/kg.
Población pediátrica <12 años
En el estudio II se inscribió a un total de 71 pacientes pediátricos varones previamente tratados que padecían hemofilia A grave. De los 71 pacientes inscritos, 69 recibieron al menos 1 dosis de ELOCTA y fueron evaluables para la eficacia. Los sujetos eran menores de 12 años (35 eran <6 años y 34 tenían entre 6 y <12 años). La pauta profiláctica de inicio consistió en la administración de 25 UI/kg en el primer día seguidos de 50 UI/kg en el cuarto día. Se permitió una dosificación de hasta 80 UI/kg e intervalos de dosificación tan cortos como de dos días, y se utilizó en un número limitado de pacientes del estudio.
Profilaxis individualizada: En los sujetos pediátricos tratados con la pauta de profilaxis individualizada, la mediana del intervalo de dosificación fue 3,49 (intervalo intercuartílico: 3,46 - 3,51) días y la mediana de la dosis semanal total fue 91,63 (intervalo intercuartílico: 84,72 - 104,56) UI/kg en los sujetos <6 años y 86,88 (intervalo intercuartílico: 79,12 - 103,08) UI/kg en los sujetos de 6 a <12 años. La mayoría de los pacientes (78,3%) permanecieron en una pauta de administración con dosis alternas (mediana de 31,73 UI/kg de la dosis baja y de 55,87 UI/kg de la dosis alta). La mediana de la tasa de hemorragia anualizada global fue 1,96 (intervalo intercuartílico: 0,00 - 3,96). El 46,4% de los pacientes pediátricos no presentaron episodios hemorrágicos.
Tratamiento de las hemorragias: De los 86 episodios hemorrágicos observados durante el estudio II, el 81,4% se controlaron con 1 inyección y el 93,0% con 2 inyecciones o menos. La mediana de la dosis por inyección para tratar un episodio hemorrágico fue 49,69 (intervalo intercuartílico: 29,41 - 56,82) UI/kg. La mediana de la dosis global para tratar un episodio hemorrágico fue 54,90 UI/kg (29,41 - 71,09).
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con ELOCTA en uno o más grupos de la población pediátrica en el tratamiento de la deficiencia hereditaria del factor VIII (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
Todos los estudios farmacocinéticos con ELOCTA se realizaron en pacientes previamente tratados que padecían hemofilia A grave. Los datos que se presentan en esta sección se obtuvieron mediante los ensayos cromogénico y de coagulación en una fase. Los parámetros farmacocinéticos derivados de los resultados del ensayo cromogénico fueron similares a los obtenidos con el ensayo en una fase.
Las propiedades farmacocinéticas se evaluaron en 28 sujetos (>15 años) tratados con ELOCTA (rFVIIIFc). Tras un periodo de lavado de al menos 96 horas (4 días), los sujetos recibieron una dosis única de 50 UI/kg de ELOCTA. Se recogieron muestras farmacocinéticas antes de la dosis y con posterioridad a la misma en 7 puntos temporales que se extendieron hasta 120 horas (5 días) después de la dosis. Los parámetros farmacocinéticos después de una dosis de 50 UI/kg de ELOCTA se presentan en las Tablas 3 y 4.
Tabla 3: Parámetros farmacocinéticos de ELOCTA utilizando el ensayo de coagulación en una fase
|
Parámetros farmacocinéticos1 |
ELOCTA (IC del 95 %) |
|
N = 28 | |
|
Recuperación incremental (UI/dl por UI/kg) |
2,24 (2,11 - 2,38) |
|
AUC/Dosis |
51,2 |
|
(UI*h/dl por UI/kg) |
(45,0 - 58,4) |
|
Cmax (UI/dl) |
108 (101 - 115) |
|
CL (ml/h/kg) |
1,95 (1,71 - 2,22) |
|
t/ (h) |
19,0 (17,0 - 21,1) |
|
TRM (h) |
25,2 (22,7 - 27,9) |
|
Vee (ml/kg) |
49,1 (46,6 - 51,7) |
|
Parámetros farmacocinéticos1 |
ELOCTA (IC del 95 %) |
|
N = 27 | |
|
Recuperación incremental (UI/dl por UI/kg) |
2,49 (2,28 - 2,73) |
|
AUC/Dosis |
47,5 |
|
(UI*h/dl por UI/kg) |
(41,6 - 54,2) |
|
Cmax (UI/dl) |
131 (104 - 165) |
|
CL (ml/h/kg) |
2,11 (1,85 - 2,41) |
|
t/ (h) |
20,9 (18,2 - 23,9) |
|
TRM (h) |
25,0 (22,4 - 27,8) |
|
Vee (ml/kg) |
52,6 (47,4 - 58,3) |
1 Los parámetros farmacocinéticos se presentan en medias geométricas (IC del 95%)
Abreviaturas: IC = intervalo de confianza; Cmax= actividad máxima; AUC = área bajo la curva de actividad del FVffl - tiempo; t/= semivida terminal; CL = aclaramiento; Vee = volumen de distribución en el estado estacionario; TRM = tiempo de residencia medio.
Los datos farmacocinéticos demuestran que ELOCTA tiene una semivida de circulación prolongada. Población pediátrica
Los parámetros farmacocinéticos de ELOCTA se determinaron en los adolescentes del estudio I (el muestreo farmacocinético se realizó antes de la dosis, seguido de una evaluación en múltiples puntos temporales que se extendieron hasta 120 horas [5 días] después de la dosis) y en los niños del estudio II (el muestreo farmacocinético se realizó antes de la dosis, seguido de una evaluación en múltiples puntos temporales que se extendieron hasta 72 horas [3 días] después de la dosis). En las Tablas 5 y 6 se presentan los parámetros farmacocinéticos calculados a partir de los datos pediátricos de los sujetos menores de 18 años.
Tabla 5: Parámetros farmacocinéticos de ELOCTA en los pacientes pediátricos utilizando el ensayo de coagulación en una fase _^_
|
Estudio II |
Estudio I* | |||
|
Parámetros farmacocinéticos1 |
<6 años |
De 6 a <12 años |
De 12 a <18 años | |
|
N = 23 |
N = 31 |
N = 11 | ||
|
Recuperación incremental (UI/dl por UI/kg) |
1,90 (1,79 - 2,02) |
2,30 (2,04 - 2,59) |
1,81 (1,56 - 2,09) | |
|
AUC/Dosis (UI*h/dl por UI/kg) |
28,9 (25,6 - 32,7) |
38,4 (33,2 - 44,4) |
38,2 (34,0 - 42,9) | |
|
t/ (h) |
12,3 (11,0 - 13,7) |
13,5 (11,4 - 15,8) |
16,0 (13,9 - 18,5) | |
|
TRM (h) |
16,8 (15,1 - 18,6) |
19,0 (16,2 - 22,3) |
22,7 (19,7 - 26,1) | |
|
CL (ml/h/kg) |
3,46 (3,06 - 3,91) |
2,61 (2,26 - 3,01) |
2,62 (2,33 - 2,95) | |
|
Vee (ml/kg) |
57,9 (54,1 - 62,0) |
49,5 (44,1 - 55,6) |
59,4 (52,7 - 67,0) | |
|
1 Los parámetros farmacocinéticos se presentan en medias geométricas (IC del |
95%) | |||
Abreviaturas: IC = intervalo de confianza; AUC = área bajo la curva de actividad del FVIII - tiempo; t/ = semivida terminal; CL = aclaramiento; MRT = tiempo de residencia medio; Vee = volumen de distribución en el estado estacionario
*Los parámetros farmacocinéticos de los pacientes de 12 a <18 años incluían a sujetos de todos los grupos del estudio I con diferentes programas de muestreo
Tabla 6: Parámetros farmacocinéticos de ELOCTA en los pacientes pediátricos utilizando el ensayo cromogénico__^_
|
Estudio II |
Estudio I* | |||
|
Parámetros farmacocinéticos1 |
<6 años |
De 6 a <12 años |
De 12 a <18 años | |
|
N = 24 |
N = 27 |
N = 11 | ||
|
Recuperación incremental (UI/dl por UI/kg) |
1,88 (1,73 - 2,05) |
2,08 (1,91 - 2,25) |
1,91 (1,61 - 2,27) | |
|
AUC/Dosis (UI*h/dl por UI/kg) |
25,9 (23,4 - 28,7) |
32,8 (28,2 - 38,2) |
40,8 (29,3 - 56,7) | |
|
t/ (h) |
14,3 (12,6 - 16,2) |
15,9 (13,8 - 18,2) |
17,5 (12,7 - 24,0) | |
|
TRM (h) |
17,2 (15,4 - 19,3) |
20,7 (18,0 - 23,8) |
23,5 (17,0 - 32,4) | |
|
CL (ml/h/kg) |
3,86 (3,48 - 4,28) |
3,05 (2,62 - 3,55) |
2,45 (1,76 - 3,41) | |
|
Vee (ml/kg) |
66,5 (59,8 - 73,9) |
63,1 (56,3 - 70,9) |
57,6 (50,2 - 65,9) | |
|
1 Los parámetros farmacocinéticos se presentan en medias geométricas (IC del |
95%) | |||
Abreviaturas: IC = intervalo de confianza; AUC = área bajo la curva de actividad del FVIII - tiempo; t/ = semivida terminal; CL = aclaramiento; MRT = tiempo de residencia medio; Vee = volumen de distribución en el estado estacionario
*Los parámetros farmacocinéticos de los pacientes de 12 a <18 años incluían a sujetos de todos los grupos del estudio I con diferentes programas de muestreo
En comparación con los adolescentes y adultos, los niños menores de 12 años pueden presentar un mayor aclaramiento y una semivida más corta, lo que concuerda con los datos observados para otros factores de la coagulación. Estas diferencias se deben tener en cuenta para la pauta posológica.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios de toxicidad aguda y a dosis repetidas (que incluyeron evaluaciones de la toxicidad local y de la farmacología de seguridad). No se han realizado estudios para investigar la genotoxicidad, la carcinogenicidad, la toxicidad para la reproducción o el desarrollo fetoembrionario. En los estudios de transferencia placentaria, se ha constatado que ELOCTA atraviesa la placenta en pequeñas cantidades en los ratones.
6. DATOS FARMACÉUTICOS 6.1 Lista de excipientes
Polvo
Sacarosa Cloruro de sodio
L-histidina
Cloruro de calcio dihidrato Polisorbato 20
Hidróxido de sodio (para ajuste del pH)
Ácido clorhídrico (para ajuste del pH)
Disolvente
Agua para preparaciones inyectables
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
Solo se debe usar el equipo de perfusión suministrado, ya que el factor VIII de coagulación se puede adsorber a las superficies internas de otros equipos de inyección con el consiguiente fracaso terapéutico.
6.3 Periodo de validez
Vial sin abrir 3 años
Durante el periodo de validez, el medicamento se puede conservar a temperatura ambiente (hasta 30 °C) durante un periodo único que no supere los 6 meses. Se debe anotar en la caja la fecha de extracción del medicamento de la nevera. Tras la conservación a temperatura ambiente, el medicamento no se puede reintroducir en la nevera. No utilizar después de la fecha de caducidad impresa en el vial o seis meses después de retirar la caja de la nevera, según cuál de estas circunstancias se produzca primero.
Tras la reconstitución
Tras la reconstitución, la estabilidad química y física se ha demostrado durante 6 horas cuando se conservaba a temperatura ambiente (hasta 30 °C). Proteger el medicamento de la luz solar directa. Tras la reconstitución, si el medicamento no se utiliza en un plazo de 6 horas, se debe desechar. Desde el punto de vista microbiológico, el medicamento se debe utilizar inmediatamente después de la reconstitución. De lo contrario, los tiempos de conservación durante el uso y las condiciones previas al uso serán responsabilidad del usuario.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C). No congelar. Conservar el vial en el embalaje exterior para protegerlo de la luz.
Para las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase y de los equipos especiales para su utilización, administración o implantación
Cada envase contiene:
- polvo en un vial de vidrio tipo 1 con un tapón de goma de clorobutilo sin látex
- 3 ml de disolvente en una jeringa precargada de vidrio tipo 1 con un tapón de émbolo de goma de bromobutilo sin látex
- un vástago del émbolo
- un adaptador del vial estéril para la reconstitución
- un equipo de perfusión estéril
- dos toallitas con alcohol
- dos tiritas
- una gasa
Tamaño de envase: 1.
6.6 Precauciones especiales de eliminación y otras manipulaciones
El polvo liofilizado inyectable del vial se debe reconstituir con el disolvente suministrado (agua para preparaciones inyectables) de la jeringa precargada utilizando el adaptador del vial estéril para la reconstitución.
El vial se debe mover suavemente en círculos hasta que todo el polvo se haya disuelto.
Consultar el prospecto para información adicional sobre la reconstitución y la administración.
La solución reconstituida debe ser transparente a ligeramente opalescente e incolora. Las soluciones que presenten un aspecto turbio o contengan depósitos no se deben utilizar. El medicamento reconstituido se debe inspeccionar visualmente en busca de material particulado y signos de cambio de color antes de la administración.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm Suecia
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/15/1046/001 EU/1/15/1046/002 EU/1/15/1046/003 EU/1/15/1046/004 EU/1/15/1046/005 EU/1/15/1046/006 EU/1/15/1046/007
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 19 de noviembre de 2015
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTE(S) DEL (DE LOS) PRINCIPIO(S) ACTIVO(S) BIOLÓGICO(S) Y FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE(S) DEL (DE LOS) PRINCIPIO(S) ACTIVO(S) BIOLÓGICO(S) Y FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante del principio activo biológico.
Biogen Inc 250 Binney Street Cambridge, MA 02142 EEUU.
Nombre y dirección del fabricante responsable de la liberación de los lotes
Swedish Orphan Biovitrum AB (publ)
Strandbergsgatan 49 SE-112 76 Stockholm Suecia
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
El Titular de la Autorización de Comercialización (TAC) presentará el primer informe periódico de seguridad para este medicamento en un plazo de 6 meses después de la autorización.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CAJA
1. NOMBRE DEL MEDICAMENTO_
ELOCTA 250 UI polvo y disolvente para solución inyectable ELOCTA 500 UI polvo y disolvente para solución inyectable ELOCTA 750 UI polvo y disolvente para solución inyectable ELOCTA 1000 UI polvo y disolvente para solución inyectable ELOCTA 1500 UI polvo y disolvente para solución inyectable ELOCTA 2000 UI polvo y disolvente para solución inyectable ELOCTA 3000 UI polvo y disolvente para solución inyectable efmoroctocog alfa
factor VIII de coagulación recombinante, proteína de fusión Fc
2. PRINCIPIO(S) ACTIVO(S)
Polvo: 250 UI de efmoroctocog alfa (aprox. 83 UI/ml tras reconstitución) Polvo: 500 UI de efmoroctocog alfa (aprox. 167 UI/ml tras reconstitución) Polvo: 750 UI de efmoroctocog alfa (aprox. 250 UI/ml tras reconstitución) Polvo: 1000 UI de efmoroctocog alfa (aprox. 333 UI/ml tras reconstitución)
Polvo: 1500 UI de efmoroctocog alfa (aprox. 500 UI/ml tras reconstitución) Polvo: 2000 UI de efmoroctocog alfa (aprox. 667 UI/ml tras reconstitución) Polvo: 3000 UI de efmoroctocog alfa (aprox. 1000 UI/ml tras reconstitución)
3. LISTA DE EXCIPIENTES_
Polvo: sacarosa, cloruro de sodio, L-histidina, cloruro de calcio dihidrato, polisorbato 20, hidróxido de sodio y ácido clorhídrico.
Disolvente: agua para preparaciones inyectables
Polvo y disolvente para solución inyectable
Contenido: 1 vial de polvo, 3 ml de disolvente en una jeringa precargada, 1 vástago del émbolo, 1 adaptador del vial, 1 equipo de perfusión, 2 toallitas con alcohol, 2 tiritas, 1 gasa.
Vía intravenosa, tras la reconstitución.
Leer el prospecto antes de utilizar este medicamento.
Un vídeo con instrucciones sobre cómo preparar y administrar ELOCTA está disponible al escanear el código QR con un teléfono inteligente o a través de la página web.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD
Usar en un plazo máximo de 6 horas tras la reconstitución.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Conservar en nevera.
No congelar.
Se puede conservar a temperatura ambiente (hasta 30 °C) durante un periodo único de hasta 6 meses. No se debe reintroducir en la nevera tras la conservación a temperatura ambiente. Fecha de extracción de la nevera:
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm
Suecia
EU/1/15/1046/001 EU/1/15/1046/002 EU/1/15/1046/003 EU/1/15/1046/004 EU/1/15/1046/005 EU/1/15/1046/006 EU/1/15/1046/007
Lote
16. INFORMACIÓN EN BRAILLE
ELOCTA 250 ELOCTA 500 ELOCTA 750 ELOCTA 1000 ELOCTA 1500 ELOCTA 2000 ELOCTA 3000
ELOCTA 250 UI polvo para solución inyectable
ELOCTA 500 UI polvo para solución inyectable ELOCTA 750 UI polvo para solución inyectable
ELOCTA 1000 UI polvo para solución inyectable
ELOCTA 1500 UI polvo para solución inyectable ELOCTA 2000 UI polvo para solución inyectable
ELOCTA 3000 UI polvo para solución inyectable efmoroctocog alfa
factor VIII de coagulación recombinante IV
EXP
Lot
1000 UI 1500 UI 2000 UI 3000 UI 6. OTROS
ETIQUETA DE LA JERINGA PRECARGADA
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Disolvente para ELOCTA
agua para preparaciones inyectables
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
3 ml
6. OTROS
B. PROSPECTO
Prospecto: información para el usuario
ELOCTA 250 UI polvo y disolvente para solución inyectable ELOCTA 500 UI polvo y disolvente para solución inyectable ELOCTA 750 UI polvo y disolvente para solución inyectable ELOCTA 1000 UI polvo y disolvente para solución inyectable ELOCTA 1500 UI polvo y disolvente para solución inyectable ELOCTA 2000 UI polvo y disolvente para solución inyectable ELOCTA 3000 UI polvo y disolvente para solución inyectable
efmoroctocog alfa (factor VIII de coagulación recombinante)
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
• Conserve este prospecto, ya que puede tener que volver a leerlo.
• Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
• Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
• Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es ELOCTA y para qué se utiliza
2. Qué necesita saber antes de empezar a usar ELOCTA
3. Cómo usar ELOCTA
4. Posibles efectos adversos
5. Conservación de ELOCTA
6. Contenido del envase e información adicional
7. Instrucciones de preparación y administración
1. Qué es ELOCTA y para qué se utiliza
ELOCTA contiene el principio activo efmoroctocog alfa, un factor VIII de coagulación recombinante, proteína de fusión Fc. El factor VIII es una proteína producida de forma natural por el cuerpo y es necesaria para que la sangre forme coágulos y detener las hemorragias.
ELOCTA es un medicamento utilizado para el tratamiento y la prevención de las hemorragias en los pacientes de todos los grupos de edad con hemofilia A (un trastorno hemorrágico hereditario causado por una deficiencia del factor VIII).
ELOCTA se prepara mediante tecnología recombinante sin la adición de ningún componente de origen humano o animal en el proceso de fabricación.
Como actúa ELOCTA
En los pacientes con hemofilia A, el factor VIII está ausente o no funciona adecuadamente. ELOCTA se utiliza para sustituir el factor VIII ausente o deficiente. ELOCTA aumenta las concentraciones de factor VIII en la sangre y corrige temporalmente la tendencia a sufrir hemorragias.
2. Qué necesita saber antes de empezar a usar ELOCTA No use ELOCTA:
• si es alérgico al efmoroctocog alfa o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
Advertencias y precauciones
Consulte a su médico, farmacéutico o enfermero antes de empezar a usar ELOCTA.
• Existe una pequeña posibilidad de que sufra una reacción anafiláctica (una reacción alérgica grave y repentina) a ELOCTA. Entre los signos de las reacciones alérgicas se encuentran picor generalizado, ronchas, sensación de opresión en el pecho, dificultad para respirar y presión arterial baja. Si aparece cualquiera de estos síntomas, interrumpa inmediatamente la inyección y póngase en contacto con su médico.
• Consulte a su médico si cree que no está consiguiendo controlar las hemorragias con la dosis que recibe, ya que pueden existir varios motivos para ello. Por ejemplo, la formación de anticuerpos (también conocidos como inhibidores) contra el factor VIII es una complicación conocida que puede ocurrir durante el tratamiento de la hemofilia A. Los anticuerpos impiden que ELOCTA actúe adecuadamente. Su médico comprobará si esto es así. No aumente la dosis total de ELOCTA para controlar las hemorragias sin hablar antes con su médico.
Complicaciones relacionadas con el catéter
Si necesita un dispositivo de acceso venoso central (DAVC), se debe tener en cuenta el riesgo de complicaciones relacionadas con el DAVC, incluidas las infecciones locales, la presencia de bacterias en la sangre y la trombosis en el lugar de inserción del catéter.
Documentación
Le recomendamos encarecidamente que cada vez que se administre ELOCTA, se anoten el nombre y el número de lote del producto.
Uso de ELOCTA con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o pudiera tener que utilizar cualquier otro medicamento.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
Conducción y uso de máquinas
No se han observado efectos sobre la capacidad para conducir o utilizar máquinas.
ELOCTA contiene sodio
Este medicamento contiene 14 mg de sodio por vial tras la preparación. Consulte a su médico si sigue una dieta pobre en sodio.
3. Cómo usar ELOCTA
El tratamiento con ELOCTA lo iniciará un médico con experiencia en el cuidado de pacientes con hemofilia. Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico (ver sección 7). En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero.
ELOCTA se administra mediante inyección en una vena. Su médico calculará su dosis de ELOCTA (en Unidades Internacionales o “UI”), dependiendo de sus necesidades individuales de tratamiento de sustitución del factor VIII y de si se utiliza para la prevención o el tratamiento de las hemorragias.
Consulte a su médico si cree que no está consiguiendo controlar las hemorragias con la dosis que recibe.
Con qué frecuencia necesitará una inyección, dependerá del grado de eficacia que ELOCTA esté mostrando con usted. Su médico le realizará las pruebas de laboratorio pertinentes para asegurarse de que tiene concentraciones adecuadas de factor VIII en la sangre.
Tratamiento de las hemorragias
La dosis de ELOCTA se calcula en función de su peso corporal y de las concentraciones de factor VIII que se desean conseguir. Las concentraciones objetivo de factor VIII dependen de la gravedad y la localización de la hemorragia.
Prevención de las hemorragias
• La dosis habitual de ELOCTA es de 50 UI por kg de peso corporal, administradas cada 3 a 5 días.
Su médico puede ajustar la dosis en un intervalo comprendido entre 25 y 65 UI por kg de peso corporal. En algunos casos, especialmente en los pacientes más jóvenes, puede ser necesario usar intervalos de dosificación más cortos o dosis mayores.
Uso en niños y adolescentes
ELOCTA se puede utilizar en niños y adolescentes de todas las edades. En los niños menores de 12 años, pueden ser necesarias dosis más altas o inyecciones más frecuentes.
Si usa más ELOCTA del que debe
Informe a su médico lo antes posible. Siga siempre exactamente las instrucciones de administración de ELOCTA indicadas por su médico. En caso de duda, consulte de nuevo a su médico, farmacéutico o enfermero.
Si olvidó usar ELOCTA
No tome una dosis doble para compensar las dosis olvidadas. Tome su dosis tan pronto se acuerde y después reanude su pauta normal de dosificación. Si no está seguro de lo que debe hacer, consulte a su médico o farmacéutico.
Si interrumpe el tratamiento con ELOCTA
No interrumpa el tratamiento con ELOCTA sin consultar a su médico. Si interrumpe el tratamiento con ELOCTA, es posible que ya no esté protegido contra las hemorragias o que una hemorragia ya existente no se detenga.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Si se producen reacciones alérgicas graves y repentinas (reacción anafiláctica), la inyección se debe interrumpir inmediatamente. Se debe poner en contacto con su médico de inmediato si presenta alguno de los siguientes síntomas de las reacciones alérgicas: hinchazón de la cara, erupción, picor generalizado, ronchas, sensación de tirantez en el pecho, dificultad para respirar, escozor y pinchazos en el lugar de la inyección, escalofríos, sofocos, dolor de cabeza, presión arterial baja, sensación general de malestar, náuseas, agitación y latido cardiaco rápido, sensación de mareo o pérdida del conocimiento.
Con este medicamento pueden aparecer los siguientes efectos adversos.
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas): dolor de cabeza, mareo, alteraciones del gusto, latido cardiaco lento, presión arterial alta, sofocos, dolor vascular después de la inyección, tos, dolor abdominal, erupción cutánea, hinchazón articular, dolor muscular, dolor de espalda, dolor articular, malestar general, dolor en el pecho, sensación de frío, sensación de calor y presión arterial baja.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de ELOCTA
Mantener este medicamento fuera de la vista y del alcance de los niños.
Conservar en nevera (entre 2 °C - 8 °C). No congelar. Conservar en el embalaje original para protegerlo de la luz.
Alternativamente, ELOCTA se puede conservar a temperatura ambiente (hasta 30 °C) durante un periodo único que no supere los 6 meses. Anote en la caja la fecha en la que se extrajo ELOCTA de la nevera y se dejó a temperatura ambiente. Tras la conservación a temperatura ambiente, el medicamento no se debe volver a introducir en la nevera.
No utilice este medicamento después de la fecha de caducidad que aparece en la caja y en la etiqueta del vial después de “CAD/EXP”. La fecha de caducidad es el último día del mes que se indica. No utilice este medicamento si se ha conservado a temperatura ambiente durante más de 6 meses.
Una vez haya preparado ELOCTA, debe utilizarlo inmediatamente. Si no puede usar la solución preparada de ELOCTA de inmediato, debe utilizarla en un plazo máximo de 6 horas. No refrigere la solución preparada. Proteja la solución preparada de la luz solar directa.
La solución preparada debe ser transparente a ligeramente opalescente e incolora. No utilice este medicamento si observa que está turbio o contiene partículas visibles.
Elimine adecuadamente cualquier resto de solución no utilizada. Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de ELOCTA
Polvo:
• El principio activo es efmoroctocog alfa (factor VIII de coagulación recombinante, proteína de fusión Fc). Cada vial de ELOCTA contiene nominalmente 250, 500, 750, 1.000, 1.500, 2.000 o 3.000 UI de efmoroctocog alfa.
• Los demás componentes son sacarosa, cloruro de sodio, L-histidina, cloruro de calcio dihidrato, polisorbato 20, hidróxido de sodio y ácido clorhídrico. Ver sección 2 si sigue una dieta pobre en sodio.
Disolvente:
3 ml de agua para preparaciones inyectables
Aspecto del producto y contenido del envase
ELOCTA se presenta en forma de polvo y disolvente para solución inyectable. El polvo es un polvo o torta de color blanco a blanquecino. El disolvente suministrado para la preparación de la solución inyectable es una disolución transparente e incolora. Tras la preparación, la solución para inyectar es de transparente a ligeramente opalescente e incolora.
Cada envase de ELOCTA contiene 1 vial de polvo, 3 ml de disolvente en una jeringa precargada, 1 vástago del émbolo, 1 adaptador del vial, 1 equipo de perfusión, 2 toallitas con alcohol, 2 tiritas y 1 gasa.
Titular de la autorización de comercialización y responsable de la fabricación
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm,
Suecia
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Swedish Orphan Biovitrum BVBA
Tél/Tel: + 32 2880 6119 e-mail: benelux@sobi.com
Lietuva
Oy Swedish Orphan Biovitrum Ab
c/o UAB CentralPharma Communications
Tel: +370 5 2430444
e-mail: centralpharma@centralpharma.lt
|
Etnrapna Cyugum Op^aH EuoBHTpyM Ktoh Etnrapua OOfl |
Luxembourg/Luxemburg Swedish Orphan Biovitrum BVBA |
|
Ten.: +420 257 222 034 e-mail: mail.bg@sobi.com |
Tél/Tel: + 32 2880 6119 e-mail: benelux@sobi.com |
|
Ceská republika Swedish Orphan Biovitrum s.r.o. Tel: +420 257 222 034 e-mail: mail.cz@sobi.com |
Magyarország Swedish Orphan Biovitrum s.r.o. Magyarországi Fióktelepe Tel: +420 257 222 034 e-mail: mail.hu@sobi.com |
|
Danmark Swedish Orphan Biovitrum A/S Tlf: + 45 32 96 68 69 e-mail: mail.dk@sobi.com |
Malta Swedish Orphan Biovitrum S.r.l. Tel: +39 0521 19 111 e-mail: mail.it@sobi.com |
|
Deutschland Swedish Orphan Biovitrum GmbH Tel: +49 89 55066760 e-mail: mail.de@sobi.com |
Nederland Swedish Orphan Biovitrum BVBA Tel: + 32 288 06119 e-mail: benelux@sobi.com |
|
Eesti Oy Swedish Orphan Biovitrum Ab c/o CentralPharma Communications OÜ |
Norge Swedish Orphan Biovitrum AS |
|
Tel. +372 6 015 540 e-mail: centralpharma@centralpharma.ee |
Tlf: +47 66 82 34 00 e-mail: mail.no@sobi.com |
|
EXXába Swedish Orphan Biovitrum S.r.l. T@: +39 0521 19 111 e-mail: mail.it@sobi.com |
Osterreich Swedish Orphan Biovitrum GmbH Tel: +49 89 55066760 e-mail: mail.de@sobi.com |
|
España Swedish Orphan Biovitrum S.L Tel: + 34 913 91 35 80 e-mail: mail.es@sobi.com |
Polska Swedish Orphan Biovitrum Sp. z o.o. Oddzial w Polsce Tel: +420 257 222 034 e-mail: mail.pl@sobi.com |
|
France Swedish Orphan Biovitrum SARL Tél: +33 1 85 78 03 40 e-mail: mail.fr@sobi.com |
Portugal Swedish Orphan Biovitrum S.L Tel: + 34 913 91 35 80 e-mail: mail.es@sobi.com |
|
Hrvatska SWEDISH ORPHAN BIOVITRUM, Glavna Podruznica Zagreb Tel: +420 257 222 034 e-mail: mail.hr@sobi.com |
Romania Swedish Orphan Biovitrum s.r.o. Praga - Sucursala Bucuresti Tel: +420 257 222 034 e-mail: mail.ro@sobi.com |
|
Ireland Swedish Orphan Biovitrum Ltd Tel: + 44 1223 891854 e-mail: mail.uk@sobi.com |
Slovenija Swedish Orphan Biovitrum s.r.o. - Podruznica v Sloveniji Tel: +420 257 222 034 e-mail : mail.si@sobi.com |
|
Ísland Swedish Orphan Biovitrum A/S Tlf: + 45 32 96 68 69 e-mail: mail.dk@sobi.com |
Slovenská republika Swedish Orphan Biovitrum o.z. Tel: +420 257 222 034 e-mail: mail.sk@sobi.com |
|
Italia Swedish Orphan Biovitrum S.r.l. Tel: +39 0521 19 111 e-mail: mail.it@sobi.com |
Suomi/Finland Oy Swedish Orphan Biovitrum Ab Puh/Tel: +358 201 558 840 e-mail: mail.fi@sobi.com |
|
Kúnpoq Swedish Orphan Biovitrum S.r.l. T@: +39 0521 19 111 e-mail: mail.it@sobi.com |
Sverige Swedish Orphan Biovitrum AB (publ) Tel: +46 8 697 20 00 e-mail: mail.se@sobi.com |
|
Latvija Oy Swedish Orphan Biovitrum Ab c/o CentralPharma Communications SIA |
United Kingdom Swedish Orphan Biovitrum Ltd |
|
Tel. +371 67 450 497 e-mail: centralpharma@centralpharma.lv |
Tel: +44 1223 891854 e-mail: mail.uk@sobi.com |
|
Fecha de la última revisión de este prospecto: |
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Dé la vuelta al prospecto para consultar la sección 7. Instrucciones de preparación y administración
Instrucciones de preparación y administración
7.
ELOCTA se administra mediante inyección intravenosa (IV) después de disolver el polvo inyectable con el disolvente suministrado en la jeringa precargada. El envase de ELOCTA contiene:
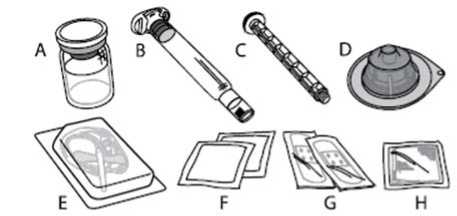
A) 1 vial de polvo
B) 3 ml de disolvente en una jeringa precargada
C) 1 vástago del émbolo
D) 1 adaptador del vial
E) 1 equipo de perfusión
F) 2 toallitas con alcohol
G) 2 tiritas
H) 1 gasa
ELOCTA no debe mezclarse con otras soluciones inyectables o para perfusión.
Lávese las manos antes de abrir el envase.
Preparación:
|
1. Compruebe el nombre y la dosis del medicamento en el envase, para asegurarse de que contiene el medicamento adecuado. Compruebe la fecha de caducidad en la caja de ELOCTA. No utilice el medicamento si está caducado. | ||
|
2. Si ELOCTA se ha conservado en la nevera, deje que el vial de ELOCTA (A) y la jeringa de disolvente (B) alcancen la temperatura ambiente antes del uso. No utilice calor externo. | ||
|
3. Coloque el vial sobre una superficie limpia y plana. Retire la cápsula de cierre desprendible de plástico del vial de ELOCTA. | ||
|
4. Limpie la parte superior del vial con una de las toallitas con alcohol (F) suministradas en el envase y deje que se seque al aire. No toque la parte superior del vial ni permita que entre en contacto con nada una vez la haya limpiado. |
¥ | |
5. Desprenda la tapa protectora de papel del adaptador del vial de plástico transparente (D). No extraiga el adaptador de su cápsula de cierre protectora. No toque el interior del envase del adaptador del vial.
6.
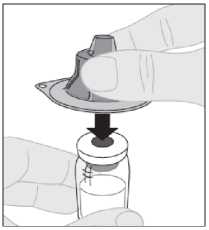
Sostenga el adaptador del vial en su cápsula de cierre protectora y colóquelo directamente sobre la parte superior del vial. Presione firmemente hacia abajo hasta que el adaptador encaje en la parte superior del vial, con el perforador del adaptador atravesando el tapón del vial.
7.
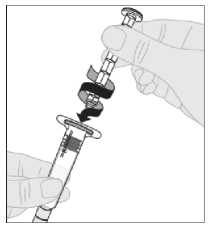
Acople el vástago del émbolo (C) a la jeringa de disolvente insertando la punta del vástago en la apertura del émbolo de la jeringa. Gire el vástago del émbolo firmemente en el sentido de las agujas del reloj hasta que quede bien asentado en el émbolo de la jeringa.
8.
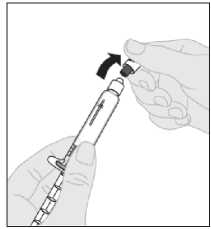
Desprenda la cápsula de cierre de seguridad inviolable de plástico blanco de la jeringa de disolvente doblándola por la cápsula de cierre de perforación hasta que se rompa. Deje la cápsula de cierre aparte colocándola con la parte de arriba mirando hacia abajo sobre una superficie plana. No toque el interior de la cápsula de cierre ni la punta de la jeringa.
9.
Retire la cápsula de cierre protectora del adaptador levantándola y deséchela.
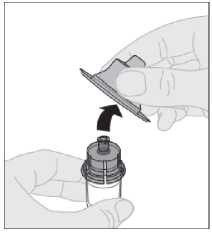
Conecte la jeringa de disolvente al adaptador del vial insertando la punta de la jeringa en la apertura del adaptador. Empuje firmemente y gire la jeringa en el sentido de las agujas del reloj hasta que quede bien conectada.
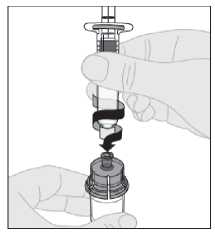
11. Presione lentamente hacia abajo el vástago del émbolo para inyectar todo el disolvente en el vial de ELOCTA.
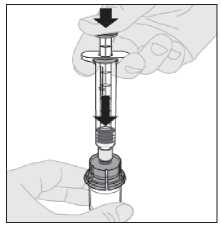
12. Con la jeringa todavía conectada al adaptador y el vástago del
émbolo presionado hacia abajo, mueva suavemente en círculos el vial hasta que el polvo se haya disuelto.
No lo agite.
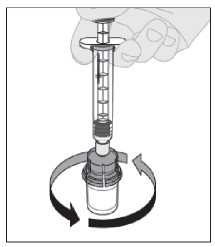
13. La solución final se debe inspeccionar visualmente antes de la administración. La solución debe ser de transparente a ligeramente opalescente e incolora. No utilice la solución si está turbia o contiene partículas visibles.
14. Asegurándose de que el vástago del émbolo de la jeringa siga completamente presionado hacia abajo, invierta el vial. Tire lentamente del vástago del émbolo para trasladar toda la solución al interior de la jeringa a través del adaptador del vial.
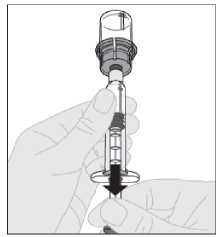
15. Desacople la jeringa del adaptador del vial tirando suavemente del vial al tiempo que lo gira en el sentido contrario a las agujas del reloj.
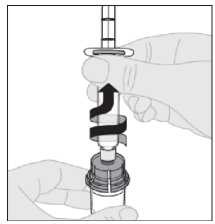
Nota: si usa más de un vial de ELOCTA por inyección, cada vial se debe preparar por separado conforme a las instrucciones previas (pasos 1 a 13) y la jeringa de disolvente se debe retirar, dejando el adaptador del vial colocado en su posición. Se puede utilizar una única jeringa luer lock más grande para extraer el contenido preparado de cada uno de los viales.
16. Deseche el vial y el adaptador.
Nota: si la solución no se va a utilizar inmediatamente, la cápsula de cierre de la jeringa se debe volver a colocar cuidadosamente sobre la punta de la jeringa. No toque la punta de la jeringa ni el interior de la cápsula de cierre.
Tras la preparación, ELOCTA se puede conservar a temperatura ambiente durante un máximo de 6 horas antes de la administración. Una vez transcurrido este tiempo, la solución preparada de ELOCTA se debe desechar. Protéjala de la luz solar directa.
Administración (inyección intravenosa):
ELOCTA se debe administrar utilizando el equipo de perfusión (E) suministrado en el envase.
1.
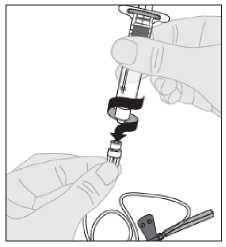
Abra el envase del equipo de perfusión y retire la cápsula de cierre del extremo del tubo. Acople la jeringa con la solución preparada de ELOCTA al extremo del tubo del equipo de perfusión girando en el sentido de las agujas del reloj.
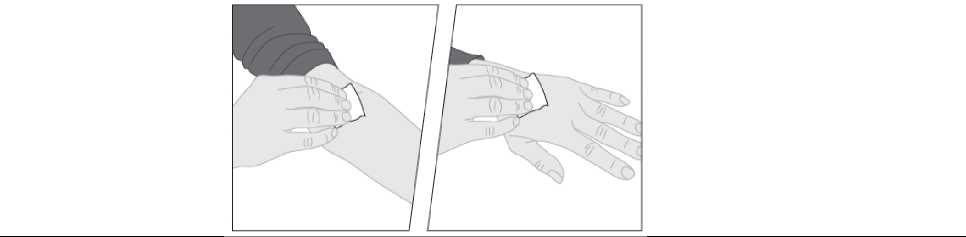
2. Si es necesario, aplique un torniquete y prepare el lugar de la inyección limpiando bien la piel con la otra toallita con alcohol suministrada en el envase.
3. Extraiga todo el aire del tubo del equipo de perfusión presionando lentamente el vástago del émbolo hacia abajo hasta que el líquido haya alcanzado la aguja del equipo de perfusión. No empuje la solución a través de la aguja. Retire la cubierta protectora de plástico transparente de la aguja.
4. Inserte la aguja del equipo de perfusión en una vena, tal como le ha indicado su médico o
enfermero, y retire el torniquete. Si lo prefiere, puede usar una de las tiritas (G) suministradas en el envase para sujetar las alas de plástico de la aguja en posición en el lugar de la inyección. El producto preparado se debe inyectar por vía intravenosa durante varios minutos. Su médico puede modificar la velocidad de inyección recomendada para que le resulte más cómoda.
5.
Una vez terminada la inyección y retirada la aguja, debe replegar el protector de la aguja y encajarlo sobre esta.
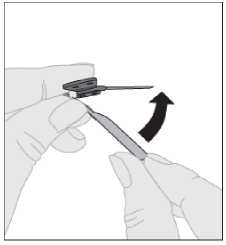
6. Deseche de forma segura la aguja usada, todo resto de solución no utilizado, la jeringa y el vial vacío en un contenedor de residuos médicos apropiado, ya que estos materiales pueden causar daños a otras personas si no se eliminan adecuadamente. No reutilice el instrumental.
42
Los parámetros farmacocinéticos se presentan en medias geométricas (IC del 95%)
Abreviaturas: IC = intervalo de confianza; Cmax= actividad máxima; AUC = área bajo la curva de actividad del FVIII - tiempo; t/2= semivida terminal; CL = aclaramiento; Vee = volumen de distribución en el estado estacionario; TRM = tiempo de residencia medio.