Eligard Semestral 45 Mg Polvo Y Disolvente Para Solucion Inyectable
.Uí1.
"I
an
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
ELIGARD SEMESTRAL 45 mg polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Una jeringa precargada con polvo para solución inyectable contiene 45 mg de leuprorelina acetato, equivalente a 41,7 mg de leuprorelina.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo (Jeringa B):
Jeringa precargada con un polvo blanco o blanquecino.
Disolvente (Jeringa A):
Jeringa precargada con una solución transparente, de incolora a amarillo pálido.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
ELIGARD SEMESTRAL 45 mg está indicado en el tratamiento del carcinoma de próstata avanzado hormonodependiente y en el tratamiento del carcinoma de próstata localizado de alto riesgo y localmente avanzado hormonodependiente en combinación con radioterapia.
4.2 Posología y forma de administración
Posología
Varones adultos
ELIGARD SEMESTRAL 45 mg debe administrarse bajo la dirección de un profesional sanitario que cuente con la experiencia adecuada para monitorizar la respuesta al tratamiento.
ELIGARD SEMESTRAL 45 mg se administra como una sola inyección subcutánea cada seis meses. La solución inyectada forma un depósito sólido de administración del medicamento que permite la liberación continua de leuprorelina acetato durante un periodo de seis meses.
Como norma, el tratamiento del carcinoma de próstata avanzado con ELIGARD SEMESTRAL 45 mg implica un tratamiento prolongado por lo que no debe interrumpirse cuando se produce remisión o mejoría.
ELIGARD SEMESTRAL 45 mg se puede utilizar como tratamiento neoadyuvante o adyuvante en combinación con radioterapia en el carcinoma de próstata localizado de alto riesgo y localmente avanzado.
La respuesta a ELIGARD SEMESTRAL 45 mg debe controlarse mediante parámetros clínicos y mediante la determinación de las concentraciones séricas del antígeno prostático específico (PSA). Los estudios clínicos demostraron que las concentraciones de testosterona aumentaban durante los 3 primeros días de
*2
Jll^a
sm
tratamiento en la mayoría de los pacientes no orquiectomizados y que después descendían hasta concentraciones por debajo de la castración médica en el plazo de 3 a 4 semanas. Una vez alcanzados, los niveles de castración se mantuvieron mientras continuó el tratamiento farmacológico (escapes de testosterona < 1%). En el caso de que la respuesta de un paciente no fuera óptima, deberá confirmarse que las concentraciones de testosterona sérica han alcanzado o se mantienen en los niveles de castración. Como se puede producir falta de eficacia debido a una incorrecta preparación, reconstitución o administración, se deben evaluar los niveles de testosterona en los casos en los que se sospeche o se conozca que ha tenido lugar una incorrecta manipulación (ver sección 4.4).
Población pediátrica
No se ha establecido la seguridad y eficacia en niños de 0 a 18 años (ver también sección 4.3).
Poblaciones de pacientes específicas
No se han realizado estudios clínicos en pacientes con alteración de la función hepática o renal.
Forma de administración
ELIGARD SEMESTRAL 45 mg sólo se debe preparar, reconstituir y administrar por profesionales sanitarios que estén familiarizados con estos procedimientos. Ver sección 6.6: Precauciones especiales de eliminación y otras manipulaciones. Si el producto no se prepara adecuadamente, no se debe administrar.
El contenido de las dos jeringas estériles precargadas debe mezclarse inmediatamente antes de la administración de ELIGARD SEMESTRAL 45 mg mediante inyección subcutánea.
Basándonos en datos obtenidos de estudios en animales debe evitarse totalmente la administración intrarterial o intravenosa.
Como ocurre con otros medicamentos que se administran por inyección subcutánea, el lugar de la inyección deberá cambiarse periódicamente.
4.3 Contraindicaciones
ELIGARD SEMESTRAL 45 mg está contraindicado en mujeres y pacientes pediátricos.
Hipersensibilidad a la leuprorelina acetato, a otros análogos LHRH o a alguno de los excipientes incluidos en la sección 6.1.
En pacientes que hayan sido sometidos previamente a orquiectomía (como ocurre con otros análogos LHRH, en casos de castración quirúrgica, ELIGARD SEMESTRAL 45 mg no da lugar a un descenso adicional de la testosterona sérica).
Como tratamiento único en pacientes con cáncer de próstata con compresión de la médula espinal o evidencia de metástasis espinal (ver también apartado 4.4).
4.4 Advertencias y precauciones especiales de empleo
Se puede producir falta de eficacia clínica debido a una incorrecta reconstitución del producto. Para las instrucciones de preparación y administración del producto, y para la evaluación de los niveles de testosterona en los casos en los que se sospeche o se conozca que ha tenido lugar una incorrecta manipulación, ver sección 4.2 y sección 6.6.
El tratamiento de deprivación androgénica puede producir una prolongación del intervalo QT.
.<s&>
JIM a
•m
En pacientes con antecedentes o factores de riesgo de prolongación del intervalo QT, y en pacientes tratados con medicación concomitante que pueda producir una prolongación del intervalo QT (ver sección 4.5), el médico debe evaluar la relación beneficio riesgo, incluyendo el riesgo potencial de Torsade de pointes, antes de iniciar el tratamiento con ELIGARD SEMESTRAL 45 mg.
Leuprorelina acetato, al igual que otros análogos LHRH, produce un aumento transitorio de las concentraciones séricas de testosterona, dihidrotestosterona y fosfatasa ácida durante la primera semana de tratamiento. Los pacientes pueden experimentar un empeoramiento de los síntomas existentes o la aparición de síntomas nuevos, que pueden incluir dolor óseo, neuropatía, hematuria u obstrucción ureteral u obstrucción de la salida vesical (ver apartado 4.8). Estos síntomas generalmente remiten al continuar el tratamiento.
Se debe considerar la administración adicional de un antiandrógeno apropiado comenzando 3 días antes del tratamiento con leuprorelina y continuando durante las dos o tres primeras semanas de tratamiento. Se ha descrito que previene las secuelas de un aumento inicial de la testosterona sérica.
Tras la castración quirúrgica, la administración de ELIGARD SEMESTRAL 45 mg no conlleva un descenso adicional en las concentraciones de testosterona sérica en los pacientes varones.
Con el uso de los análogos LHRH se han comunicado casos de obstrucción ureteral y compresión de la médula espinal, que pueden contribuir a la aparición de parálisis con o sin complicaciones mortales. Si aparece compresión de la médula espinal o alteración de la función renal, deberá establecerse el tratamiento específico estandarizado para estas complicaciones.
Los pacientes con metástasis vertebrales y/o cerebrales, así como los pacientes con obstrucción del tracto urinario deberán ser monitorizados estrechamente durante las primeras semanas de tratamiento.
Una proporción de pacientes presentará tumores no sensibles a la manipulación hormonal. La ausencia de mejoría clínica pese a una supresión adecuada de testosterona, es una señal de esta condición, para la que un tratamiento adicional con ELIGARD no supondría ningún beneficio.
En la literatura médica se ha descrito la disminución de la densidad ósea en varones que habían sido orquiectomizados o tratados con análogos LHRH (ver el apartado 4.8).
La terapia antiandrogénica aumenta significativamente el riesgo de fracturas debidas a osteoporosis. Sobre este aspecto sólo se dispone de datos limitados. Se observaron fracturas debidas a osteoporosis en un 5% de los pacientes después de 22 meses de deprivación farmacológica de andrógenos y en un 4% de los pacientes después de 5 a 10 años de tratamiento. El riesgo de fracturas debidas a osteoporosis es generalmente mayor que el riesgo de fracturas patológicas. Aparte del déficit prolongado de testosterona, tanto el aumento de la edad como el tabaquismo y el consumo de bebidas alcohólicas, la obesidad y la falta de ejercicio pueden influir en la aparición de osteoporosis.
Durante la vigilancia post-autorización, raramente se han comunicado casos de apoplejía hipofisaria (un síndrome clínico derivado del infarto de la hipófisis) tras la administración de análogos LHRH, ocurriendo la mayoría dentro de las 2 semanas siguientes a la primera dosis, y algunos dentro de la primera hora. En estos casos, la apoplejía hipofisaria se ha presentado como cefalea súbita, vómitos, cambios visuales, oftalmoplejía, alteración del estado mental, y algunas veces colapso cardiovascular. Se requiere actuación médica inmediata.
Hiperglucemia y diabetes: Se han notificado casos de hiperglucemia y un aumento del riesgo de desarrollar diabetes en hombres tratados con análogos de la hormona liberadora de gonadotropina (GnRH). La hiperglucemia puede dar lugar al desarrollo de diabetes mellitus o empeoramiento del control glucémico en pacientes diabéticos. En estos pacientes, se debe monitorizar periódicamente la glucosa en sangre y/o la hemoglobina glicosilada (HbA1c) y tratar la hiperglucemia o la diabetes de acuerdo con la práctica habitual.
ÍTTI
Enfermedades cardiovasculares: Se ha notificado un aumento del riesgo de sufrir infarto de miocardio, muerte súbita de origen cardíaco y accidente cerebrovascular, asociado al uso de análogos de la GnRH en hombres. El riesgo parece ser bajo, de acuerdo a la razón de probabilidad notificada. Cuando se determina un tratamiento para los pacientes con cáncer de próstata, este riesgo debe evaluarse cuidadosamente junto con los factores de riesgo cardiovasculares. Se deben monitorizar los síntomas y signos indicativos de la aparición de una enfermedad cardiovascular en pacientes que reciben análogos GnRH y tratar de acuerdo con la práctica clínica habitual.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios farmacocinéticos de interacciones medicamentosas con ELIGARD SEMESTRAL 45 mg. No se dispone de informes relativos a interacciones de leuprorelina acetato con otros medicamentos.
Debido a que el tratamiento de deprivación androgénica puede producir una prolongación del intervalo QT, el uso concomitante de ELIGARD SEMESTRAL 45 mg con medicamentos que producen una prolongación del intervalo QT o de medicamentos capaces de inducir Torsades de pointes, tales como antiarrítmicos de clase IA (ej. quinidina, disopiramida) o de clase III (ej. amiodarona, sotalol, dofetilida, ibutilida), metadona, moxifloxacino, antipsicóticos, etc, deben ser cuidadosamente evaluados (ver sección 4.4).
4.6 Fertilidad, embarazo y lactancia
No procede puesto que ELIGARD SEMESTRAL 45 mg está contraindicado en mujeres.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios de los efectos de ELIGARD SEMESTRAL 45 mg sobre la capacidad para conducir y utilizar máquinas.
La capacidad para conducir y utilizar máquinas puede verse alterada debido a cansancio, mareos y trastornos de la visión, que pueden ser posibles reacciones adversas del tratamiento o consecuencia de la enfermedad subyacente.
4.8 Reacciones adversas
Las reacciones adversas que se observan con ELIGARD se deben principalmente a la acción farmacológica específica de la leuprorelina, es decir aumentos y descensos en los niveles de ciertas hormonas. Las reacciones adversas comunicadas con mayor frecuencia son sofocos, nauseas, malestar general y cansancio, así como irritación local pasajera en el lugar de la inyección. Los sofocos leves o moderados se producen aproximadamente en un 58% de los pacientes.
Lista tabulada de reacciones adversas
Durante los ensayos clínicos con ELIGARD realizados en pacientes con carcinoma de próstata avanzado se comunicaron los siguientes acontecimientos adversos. En función de su frecuencia, los acontecimientos adversos se clasifican como muy frecuentes (>1/10), frecuentes (> 1/100, <1/10), poco frecuentes (>1/1.000, <1/100), raros (>1/10.000, <1/1.000) y muy raros (<1/10.000), frecuencia no conocida (no se puede estimar a partir de los datos disponibles).
|
Tabla 1: Reacciones adversas en estudios clínicos con Eligard | |
|
Infecciones e infestaciones | |
|
frecuentes |
nasofaringitis |
|
poco frecuentes |
infección del tracto urinario, infección local de la piel |
|
Tabla 1: Reacciones adversas en estudios clínicos con Eligard | |
|
Trastornos del metabolismo y de la nutrición poco frecuentes |
empeoramiento de la diabetes mellitus |
|
Trastornos psiquiátricos poco frecuentes |
sueños anormales, depresión, disminución de la libido |
|
Trastornos del sistema nervioso poco frecuentes raras |
mareos, cefalea, insomnio, trastornos del gusto, trastornos del olfato, hipoestesia movimientos involuntarios anormales |
|
Trastornos cardiacos no conocida |
prolongación del intervalo QT (ver secciones 4.4 y 4.5) |
|
Trastornos vasculares muy frecuentes poco frecuentes raras |
sofocos hipertensión, hipotensión síncope , colapso |
|
Trastornos respiratorios, torácicos y mediastínicos poco frecuentes |
rinorrea, disnea |
|
Trastornos gastrointestinales frecuentes poco frecuentes raras |
náuseas, diarrea estreñimiento, sequedad de boca, vómitos, dispepsia flatulencia, eructos |
|
Trastornos de la piel y del tejido subcutáneo muy frecuentes frecuentes poco frecuentes raras |
equimosis, eritema prurito, sudores nocturnos sensación de bochorno, aumento de la sudoración alopecia, erupción cutánea |
|
Trastornos musculoesqueléticos y del tejido conjuntivo frecuentes poco frecuentes |
artralgia, dolor de las extremidades, mialgia dolor lumbar, calambres musculares |
|
Trastornos renales y urinarios frecuentes poco frecuentes |
frecuencia urinaria disminuida, dificultad de micción, disuria, nocturia, oliguria espasmos vesicales, hematuria, aumento de la frecuencia miccional, retención de orina |
|
Trastornos del aparato reproductor y de la mama frecuentes poco frecuentes raras |
sensibilidad mamaria, atrofia testicular, dolor testicular, infertilidad, hipertrofia mamaria ginecomastia, impotencia, trastorno testicular dolor mamario |
|
Trastornos generales y alteraciones en el lugar de administración muy frecuentes frecuentes |
cansancio, quemazón en el lugar de la inyección, parestesia en el lugar de la inyección Malestar general, dolor en el lugar de la inyección, hematoma en el lugar de inyección, escozor en el lugar de la inyección, rigidez, debilidad |
|
Tabla 1: Reacciones adversas en estudios poco frecuentes raras muy raras |
clínicos con Eligard prurito en el lugar de la inyección, letargo, dolor, pirexia ulceración en el lugar de la inyección necrosis en el lugar de la inyección |
|
Trastornos de la sangre y del sistema linfático frecuentes |
cambios hematológicos |
|
Exploraciones complementarias frecuentes poco frecuentes |
aumento de la creatinina fosfoquinasa sanguínea, prolongación del tiempo de coagulación aumento de la alanina aminotransferasa, aumento de los triglicéridos sanguíneos, prolongación del tiempo de protrombina, aumento de peso |
Se han comunicado otros acontecimientos adversos en general con el tratamiento de leuprorelina acetato, entre ellos edema periférico, embolia pulmonar, palpitaciones, mialgia, trastornos de la sensibilidad en la piel, debilidad muscular, escalofríos, vértigo periférico, erupción cutánea, amnesia, y trastornos visuales. Raramente se han comunicado casos de infarto de apoplejía hipofisaria preexistente tras la administración de análogos LHRH de larga y corta acción. Se han comunicado casos raros de trombocitopenia y leucopenia. Se han comunicado cambios en la tolerancia a la glucosa.
Los acontecimientos adversos locales comunicados después de la inyección de ELIGARD son similares a los acontecimientos adversos locales asociados a productos similares de administración mediante inyección subcutánea.
En general, estos acontecimientos adversos localizados después de la inyección subcutánea son leves y se han descrito como de corta duración.
Cambios en la densidad ósea
En la literatura médica se ha descrito disminución de la densidad ósea en varones que habían sido orquiectomizados o que habían sido tratados con análogos LHRH. Puede esperarse que el tratamiento prolongado con leuprorelina revele signos crecientes de osteoporosis. En relación con el mayor riesgo de fracturas debidas a osteoporosis (ver el apartado 4.4).
Exacerbación de los signos y síntomas de la enfermedad
El tratamiento con leuprorelina puede dar lugar a una exacerbación de los signos y síntomas de la enfermedad durante las primeras semanas. En el caso de que afecciones como las metástasis vertebrales y/o la obstrucción urinaria o la hematuria empeorasen, podrían surgir problemas neurológicos como debilidad y/o parestesia de las extremidades inferiores o empeoramiento de los síntomas urinarios.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: www.notificaRAM.es.
4.9 Sobredosis
ELIGARD SEMESTRAL 45 mg no tiene potencial de abuso y la sobredosis deliberada es improbable. No existen informes de que en la práctica clínica se haya producido abuso o sobredosis con leuprorelina, pero en el caso de que se produjese una exposición excesiva se recomienda proceder a una observación y tratamiento sintomático de soporte.
ÍTTI
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Análogos de la hormona liberadora de Gonadotropina Código ATC: L02A E02
Leuprorelina acetato es un nonapéptido sintético agonista de la hormona liberadora de gonadotropina natural (GnRH o LHRH) que cuando se administra de forma continua, inhibe la secreción de gonadotropina hipofisaria y suprime la esteroidogénesis testicular en los varones. Este efecto es reversible cuando se suspende el tratamiento con este medicamento. Sin embargo, el agonista posee mayor potencia que la hormona natural y el tiempo hasta la recuperación de las concentraciones de testosterona puede variar dependiendo del paciente.
La administración de leuprorelina acetato produce un aumento inicial de las concentraciones circulantes de la hormona luteinizante (LH) y la hormona foliculoestimulante (FSH), lo que da lugar a un aumento transitorio de los niveles de esteroides gonadales, testosterona y dihidrotestosterona en los varones. La administración continuada de leuprorelina acetato da lugar a una disminución de las cifras de LH y FSH.
En los varones, la testosterona se reduce por debajo del nivel de castración (< 50 ng/dl). Estas disminuciones se producen entre la tercera y la cuarta semana después de iniciarse el tratamiento. Los niveles medios de testosterona a los seis meses, son de 10,4 (± 0,53) ng/dl, comparables a los niveles tras una orquiectomía bilateral. Todos los pacientes menos uno que recibieron la dosis completa de 45 mg de leuprorelina en el ensayo clínico pivotal, alcanzaron niveles de castración a las cuatro semanas. En la gran mayoría de los pacientes, se observaron niveles de testosterona por debajo de 20 ng/dl, aunque todavía no se ha establecido el completo beneficio de estos niveles bajos. Los niveles de PSA disminuyeron en un 97% pasados seis meses.
Los estudios a largo plazo han demostrado que la continuación del tratamiento mantiene la testosterona por debajo de los niveles de castración durante periodos de hasta siete años, y lo más probable que de forma indefinida.
No se hicieron mediciones directas del tamaño del tumor durante el programa del ensayo clínico, pero hubo una respuesta indirecta beneficiosa del tumor, como se ve en la reducción del PSA medio en un 97% para ELIGARD SEMESTRAL 45 mg.
En un ensayo clínico en fase III aleatorizado con 970 pacientes incluidos con cáncer de próstata localmente avanzado (principalmente pacientes T2c-T4 con algún T1c a T2b con afectación linfática regional) de los cuales 483 fueron asignados a una supresión androgénica a corto plazo (6 meses) en combinación con radioterapia, y 487 a terapia a largo plazo (3 años), un análisis de no inferioridad comparó el tratamiento hormonal concomitante y adyuvante a corto y largo plazo con agonistas de la GnRH (triptorelina o goserelina). La mortalidad global a 5 años fue del 19,0% y 15,2% en los grupos a corto y largo plazo respectivamente. El Hazard Ratio observado de 1,42 (IC 95,71% unilateral superior de 1,79; o IC 95,71% bilateral: 1,09 - 1,85; p = 0,65 para no inferioridad), demuestra que la combinación de radioterapia con 6 meses de terapia de deprivación androgénica proporciona una supervivencia inferior que radioterapia con 3 años de terapia de deprivación androgénica. La supervivencia global a 5 años del tratamiento a largo y corto plazo muestra una supervivencia del 84,8% y 81,0% respectivamente. La calidad de vida global, usando QLQ-C30, no difirió significativamente entre los dos grupos (p= 0,37). Los resultados están dominados por la población de pacientes con tumores localmente avanzados.
La evidencia de la indicación en carcinoma de próstata localizado de alto riesgo está basada en estudios publicados sobre radioterapia en combinación con los análogos de la GnRH, incluyendo leuprorelina acetato. Se analizaron los datos clínicos de cinco estudios publicados (EORTC 22863, RTOG 85-31, RTOG 92-02, RTOG 8610, y D’Amico et al., JAMA, 2004), y todos demuestran un beneficio de la
ÍTTI
combinación de los análogos de la GnRH con radioterapia. En los estudios publicados, no fue posible una clara diferenciación entre las respectivas poblaciones del estudio para las indicaciones de carcinoma de próstata localmente avanzado y carcinoma de próstata localizado de alto riesgo.
Los datos clínicos han demostrado que la radioterapia seguida de 3 años de tratamiento con deprivación androgénica es preferible a radioterapia seguida de 6 meses de tratamiento con deprivación androgénica.
La duración recomendada de la terapia de deprivación androgénica en las guías médicas para pacientes T3-T4 que están recibiendo radioterapia es de 2-3 años.
5.2 Propiedades farmacocinéticas
Absorción: En pacientes con carcinoma de próstata avanzado, las concentraciones medias de leuprorelina sérica después de la inyección inicial se elevan hasta 82 ng/ml a las 4,4 h (Cmáx) después de la inyección. Tras el aumento inicial después de cada inyección (fase de equilibrio estacionario del día 3 al 168 después de cada dosis), las concentraciones séricas se mantienen relativamente constantes (0,2 - 2 ng/ml). No existen pruebas de acumulación durante la administración continuada.
Distribución: El volumen medio de distribución en equilibrio de leuprorelina tras la administración de un bolo intravenoso a voluntarios varones sanos fue de 27 litros. La unión a proteínas plasmáticas humanas in vitro osciló entre 43% y 49%.
Eliminación: En voluntarios varones sanos, un bolo de 1 mg de leuprorelina acetato administrado por vía intravenosa reveló que el aclaramiento sistémico medio era de 8,34 l/h, con una semivida de eliminación terminal de aproximadamente 3 horas basado en un modelo bicompartimental.
No se han llevado a cabo estudios de excreción con ELIGARD.
No se ha realizado ningún estudio de metabolismo con ELIGARD
5.3 Datos preclínicos sobre seguridad
Estudios preclínicos con leuprorelina acetato revelaron en ambos sexos efectos sobre el sistema reproductor, lo cual era de esperar debido a las conocidas propiedades farmacológicas. Se demostró que estos efectos eran reversibles después de suspender el tratamiento y de un periodo de regeneración apropiado. Leuprorelina acetato no mostró teratogenicidad. Se observó embriotoxicidad/mortalidad en conejos, en línea con los efectos farmacológicos de leuprorelina acetato sobre el sistema reproductor.
Se realizaron estudios de carcinogenia en ratas y ratones durante 24 meses. En ratas, se observó un aumento relacionado con la dosis de apoplejía hipofisaria después de la administración subcutánea, a dosis de 0,6 a 4 mg/kg/día. Este tipo de efecto no se observó en ratones.
Leuprorelina acetato y su producto de administración mensual ELIGARD MENSUAL 7,5 mg no resultaron mutagénicos en un conjunto de valoraciones in vivo e in vitro.
6 . DATOS FARMACÉUTICOS 6.1 Lista de excipientes
Disolvente (jeringa A): Ácido poli (DL-láctico-co-glicólico) (85:15)
N-metil-pirrolidona
Polvo (jeringa B): Ninguno
6.2 Incompatibilidades
La leuprorelina presente en la jeringa B sólo debe mezclarse con el disolvente en la jeringa A y no debe mezclarse con otros medicamentos.
6.3 Periodo de validez
2 años
Una vez que el producto se ha sacado de la nevera, se puede conservar en el embalaje original a temperatura ambiente (por debajo de 25°C) hasta 4 semanas.
Después de abrir la bandeja, el polvo y el disolvente para la solución inyectable se deben reconstituir inmediatamente y administrar al paciente.
Una vez reconstituido: usar inmediatamente, ya que la viscosidad de la solución aumenta con el tiempo.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C); en el embalaje original para protegerlo de la humedad. Este producto debe estar a temperatura ambiente antes de la inyección. Sacar de la nevera aproximadamente 30 minutos antes de usar. Una vez fuera de la nevera, este producto se puede conservar en su embalaje original a temperatura ambiente (por debajo de 25°C) hasta 4 semanas.
6.5 Naturaleza y contenido del envase
Dos jeringas de copolímero de olefina cíclica/polipropileno precargadas, una de ellas contiene el polvo (jeringa B) y la otra contiene el disolvente (jeringa A). Las dos jeringas juntas conforman un sistema de mezclado.
La jeringa A consta de un émbolo con un extremo de caucho termoplástico, cubierto por un capuchón de polietileno o polipropileno Luer-Lok. El extremo del capuchón y los dos extremos del émbolo de la jeringa B están compuestos de caucho de clorobutilo.
Están disponibles los siguientes tamaños de envase:
• Un estuche que consiste en dos bandejas termoformadas insertadas en un soporte de cartón. Una bandeja contiene una jeringa de polipropileno precargada (jeringa A), un émbolo largo para la jeringa B y una bolsa con desecante. La otra bandeja contiene una jeringa de copolímero de olefina cíclica precargada (jeringa B), una aguja estéril de calibre 18 y una bolsa con desecante.
• Un envase múltiple que contiene 2 estuches de 2 jeringas de polipropileno/copolímero de olefina cíclica precargadas (1 jeringa A; 1 jeringa B)
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Espere a que el producto alcance la temperatura ambiente sacándolo de la nevera aproximadamente 30 minutos antes de usar.
Por favor primero prepare al paciente para la inyección, siguiendo a continuación con la preparación del producto, usando las siguientes instrucciones. Si el producto no se prepara utilizando la técnica adecuada, no se debe administrar, ya que se puede producir falta de eficacia clínica debido a una incorrecta reconstitución del producto.
an
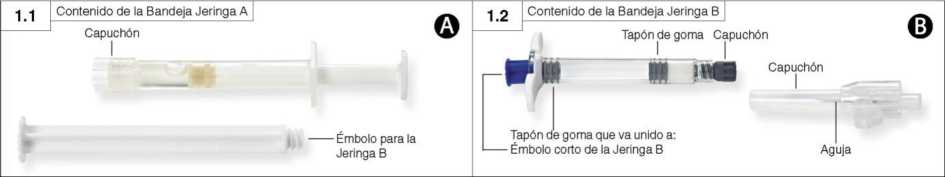
Paso 1: Abra ambas bandejas (retire el aluminio por la esquina identificada con una pequeña burbuja) y vacíe el contenido en una superficie limpia (dos bandejas que contienen la Jeringa A (Figura 1.1) y la Jeringa B (Figura 1.2)). Deseche las bolsas de desecante.
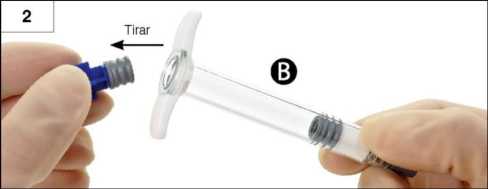
Paso 2: Tire, sin desenroscar, del émbolo corto de color azul de la jeringa B junto con el tapón gris al que va unido y deséchelos (Figura 2). No intente mezclar el producto si quedan dos tapones en la jeringa
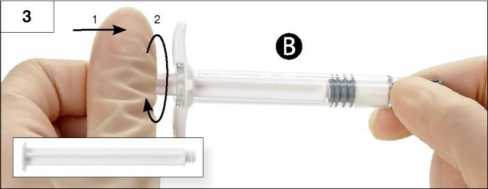
Paso 3: Enrosque suavemente el émbolo blanco de la Jeringa B al único tapón gris que ha quedado en la Jeringa B (Figura 3).
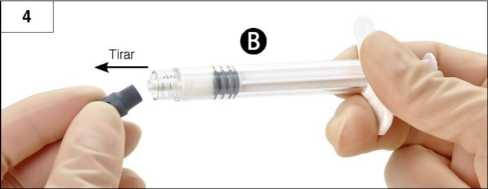
Paso 4: Retire el capuchón de goma gris de la Jeringa B y déjela sobre una superficie (Figura 4).
Paso 5: Sujete la Jeringa A en posición vertical para asegurarse que no se derrama líquido y desenrosque su capuchón transparente (Figura 5).

Desenroscar
O

Paso 6: Acople ambas jeringas apretando y girando la Jeringa B sobre la Jeringa A hasta que estén bien sujetas (Figura 6a y 6b). No apriete en exceso.
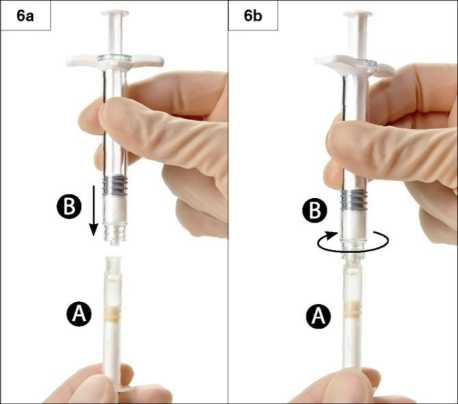
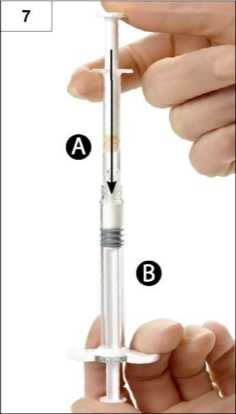
Paso 7: Invierta el sistema acoplado de jeringas, manténgalo verticalmente con la Jeringa B abajo, y transfiera el líquido de la Jeringa A a la B que contiene el polvo ( leuprorelina acetato) (Figura 7).
11 de 16 MINISTERIO DE
SANIDAD, POLITICA SOCIAL E IGUALDAD Agencia es paneta de medicamentos y proouctos san-ianos
an
Paso 8: En posición horizontal, mezcle minuciosamente el producto transfiriendo suavemente el contenido de una jeringa a otra (60 veces en total, aproximadamente 60 segundos) hasta obtener una solución viscosa homogénea (Figura 8). No doble el sistema de jeringas (por favor tenga en cuenta que podría salirse producto si se desenroscan parcialmente las jeringas).
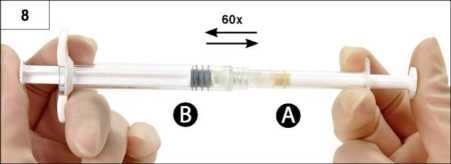
Cuando esté minuciosamente mezclada, la solución viscosa tendrá un color dentro de la gama del incoloro al blanco - amarillo pálido (que podría incluir tonos desde el blanco al amarillo pálido).
Importante: después de mezclar, continúe con el siguiente paso inmediatamente ya que la viscosidad del producto aumenta con el tiempo. No refrigerar el producto reconstituido.
Por favor tenga en cuenta: El producto debe mezclarse como se describe; agitar NO proporcionará una mezcla adecuada del producto.
Paso 9: Sujete las jeringas verticalmente con la Jeringa B en la parte inferior. Las jeringas deben permanecer bien acopladas. Transfiera todo el producto a la Jeringa B (jeringa ancha) apretando el émbolo de la Jeringa A y retrayendo ligeramente el émbolo de la Jeringa B (Figura 9).

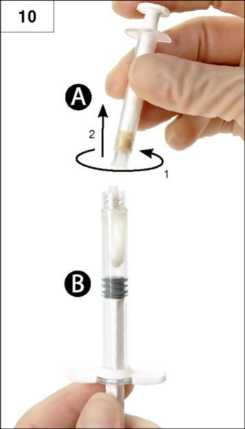
Paso 10: Desenrosque la Jeringa A sin dejar de apretar su émbolo (Figura 10). Asegúrese que no se sale producto ya que entonces la aguja no se ajustará adecuadamente cuando se acople.
Por favor tenga en cuenta: En la formulación puede quedar una burbuja de aire grande o varias pequeñas -esto es aceptable. ¡Por favor no purgue las burbujas de aire de la jeringa B en este momento, ya que se perdería producto!
Paso 11: Mantenga la Jeringa B en posición vertical. Abra el envase de la aguja de seguridad retirando la etiqueta de papel y sáquela. Fíjela sujetando la Jeringa B y girando la aguja en el sentido de las agujas del reloj hasta ajustarla completamente (Figura 11). No apriete en exceso.
13 de 16 MINISTERIO DE
SANIDAD, POLITICA SOCIAL E IGUALDAD Agencia es paño® de medicamentos y proouctos san-ianos

Paso 12: Antes de la administración quite el capuchón protector de la aguja (Figura 12). Importante: No manipule el mecanismo de la aguja de seguridad antes de la administración.
12
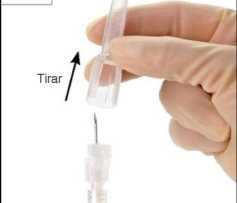
O s
1/ M
Paso 13: Antes de la administración, purgue cualquier burbuja grande de aire de la Jeringa B. Administre el producto por vía subcutánea. Por favor asegúrese de inyectar todo el producto de la Jeringa B.
Paso 14: Después de la inyección bloquee el mecanismo de seguridad usando cualquiera de los métodos de activación mencionados a continuación.
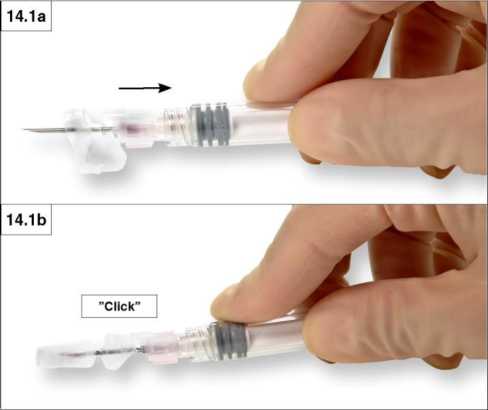
1. Cierre sobre una superficie plana
Presione la cubierta de seguridad con el deslizador hacia abajo, contra una superficie plana (Figura 14.1a y b) para cubrir la aguja y bloquear la cubierta.
Verifique la posición de bloqueo mediante un “click” audible y táctil. La posición de bloqueo cubrirá completamente la punta de la aguja (figura 14.1b).
14 de 16 MINISTERIO DE
SANIDAD, POLITICA SOCIAL E IGUALDAD Agencia es paño® de medicamentos y proouctos san-lanos
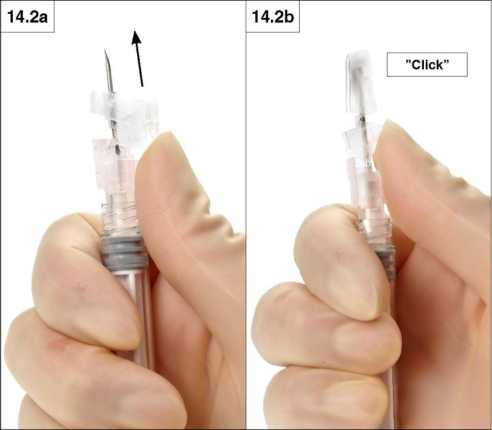
2. Cierre con su pulgar
Poniendo su pulgar sobre el deslizador, arrastre suavemente la cubierta de seguridad hacia la punta de la aguja (Figura 14.2a y b) para cubrir la aguja y bloquear la cubierta.
Verifique la posición de bloqueo mediante un “click” audible y táctil. La posición de bloqueo cubrirá completamente la punta de la aguja (figura 14.2b).
Paso 15: Una vez bloqueada la cubierta de seguridad, deseche inmediatamente la aguja y la jeringa en un contenedor autorizado para objetos punzantes.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Astellas Pharma S.A.
Paseo del Club Deportivo, n° 1, Bloque 14,
28223 Pozuelo de Alarcón Madrid
an
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
69.357
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización 23/Octubre/ 2007 Fecha de la última renovación 20/Diciembre/2009
10. FECHA DE LA REVISIÓN DEL TEXTO
Abril 2015
16 de 16