Dacogen 50Mg Polvo Para Concentrado Para Solucion Para Perfusion
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Dacogen 50 mg polvo para concentrado para solución para perfusión.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene 50 mg de decitabina en polvo.
Tras la reconstitución con 10 ml de agua para preparaciones inyectables, cada ml del concentrado contiene 5 mg de decitabina.
Excipientes con efecto conocido: Cada vial contiene 0,5 mmol de potasio (E340) y 0,29 mmol de sodio (E524).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo para concentrado para solución para perfusión (polvo para perfusión). Polvo liofilizado de color blanco o casi blanco.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Dacogen está indicado para el tratamiento de pacientes adultos con diagnóstico reciente de leucemia mieloide aguda (LMA) de novo o secundaria, según la clasificación de la Organización Mundial de la Salud (OMS), que no son candidatos a quimioterapia de inducción convencional.
4.2 Posología y forma de administración
La administración de Dacogen debe iniciarse bajo la supervisión de un médico con experiencia en el uso de fármacos quimioterápicos.
Posología
En un ciclo de tratamiento, Dacogen se administra en una dosis de 20 mg/m2 de superficie corporal mediante perfusión intravenosa durante 1 hora al día durante 5 días consecutivos (es decir, un total de 5 dosis por ciclo de tratamiento). La dosis diaria total no debe superar los 20 mg/m2 y la dosis total por ciclo de tratamiento no podrá ser mayor de 100 mg/m2. Si se omite una dosis, el tratamiento debe reanudarse lo antes posible. El ciclo se repetirá cada 4 semanas, en función de la respuesta clínica del paciente y de la toxicidad observada. Se recomienda tratar a los pacientes durante un mínimo de 4 ciclos; sin embargo, se puede tardar más de 4 ciclos en conseguir una remisión parcial o completa.
El tratamiento puede ser continuado mientras el paciente muestre respuesta, siga beneficiándose o presente enfermedad estable, es decir, en ausencia de progresión aparente.
Si después de 4 ciclos, los valores hematológicos del paciente (por ejemplo, recuento de plaquetas o recuento absoluto de neutrófilos) no han regresado a los valores previos al primer tratamiento o si aparece progresión de la enfermedad (el recuento de blastos periféricos está aumentado o el recuento de blastos en la médula ósea está empeorando), se considerará que el paciente no ha respondido y se deben considerar opciones terapéuticas alternativas a Dacogen.
No se recomienda sistemáticamente el tratamiento previo para la prevención de las náuseas y los vómitos, pero puede administrarse en caso necesario.
Tratamiento de la mielosupresióny otras complicaciones relacionadas
La mielosupresión y los acontecimientos adversos relacionados con ella (trombocitopenia, anemia, neutropenia y neutropenia febril) son frecuentes en los pacientes con LMA tratados y sin tratar. Las complicaciones de la mielosupresión comprenden infecciones y hemorragias. El tratamiento puede retrasarse a criterio del médico responsable si el paciente presenta complicaciones asociadas a mielosupresión, como las que se describen a continuación:
• Neutropenia febril (temperatura > 38,5 °C y recuento absoluto de neutrófilos < 1.000/^l)
• Infección vírica, bacteriana o micótica activa (es decir, con necesidad de antiinfecciosos intravenosos o de tratamiento de soporte exhaustivo)
• Hemorragia (digestiva, genitourinaria, pulmonar con plaquetas < 25.000/^l o cualquier hemorragia que afecte al sistema nervioso central)
El tratamiento con Dacogen podrá reanudarse una vez que estas afecciones hayan mejorado o se hayan estabilizado con el tratamiento adecuado (antiinfecciosos, transfusiones o factores de crecimiento).
En estudios clínicos, aproximadamente un tercio de los pacientes que recibieron Dacogen, necesitaron un retraso de la dosis. La reducción de la dosis no está recomendada.
Población pediátrica
No se han establecido todavía la seguridad y eficacia de Dacogen en niños menores de 18 años. No se dispone de datos.
Insuficiencia hepática
No se han realizado estudios en pacientes con insuficiencia hepática. No se ha evaluado la necesidad de ajustar la dosis en pacientes con insuficiencia hepática. Si se produce un empeoramiento de la función hepática, los pacientes deben ser estrechamente vigilados (ver secciones 4.4 y 5.2).
Insuficiencia renal
No se han realizado estudios en pacientes con insuficiencia renal. No se ha evaluado la necesidad de ajustar la dosis en pacientes con insuficiencia renal (ver secciones 4.4 y 5.2).
Forma de administración
Dacogen se administra mediante perfusión intravenosa. No se precisa un catéter venoso central.
Para consultar las instrucciones de reconstitución y dilución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Lactancia (ver sección 4.6).
4.4 Advertencias y precauciones especiales de empleo
Mielosupresión
La mielosupresión y sus complicaciones, incluidas las infecciones y las hemorragias que se producen en los pacientes con LMA, pueden empeorar con el tratamiento con Dacogen. Por consiguiente, los pacientes tienen un elevado riesgo de infecciones graves (debidas a cualquier patógeno tales como bacteriano, micótico y vírico), con un desenlace potencialmente mortal (ver sección 4.8). Se debe vigilar a los pacientes en los signos y síntomas de infección y se deben tratar rápidamente.
En estudios clínicos, la mayoría de los pacientes tenían mielosupresión de Grado 3/4 en el momento basal. Se observó empeoramiento de la mielosupresión en la mayoría de los pacientes con alteraciones de Grado 2 en el momento basal, y más frecuentemente que en pacientes con alteraciones de Grado 1 ó 0 en el momento basal. La mielosupresión causada por Dacogen es reversible. Se realizarán hemogramas y recuentos de plaquetas periódicamente, cuando esté clínicamente indicado y antes de cada ciclo de tratamiento. En presencia de mielosupresión o sus complicaciones, se puede interrumpir el tratamiento con Dacogen y/o instaurar medidas de apoyo (ver secciones 4.2 y 4.8).
Insuficiencia hepática
No se ha establecido el uso de Dacogen en pacientes con insuficiencia hepática. Se recomienda precaución durante la administración de Dacogen a pacientes con insuficiencia hepática así como realizar controles estrictos a estos pacientes (ver secciones 4.2 y 5.2).
Insuficiencia renal
No se ha estudiado el uso de Dacogen en pacientes con insuficiencia renal grave. Se recomienda precaución durante la administración de Dacogen a pacientes con insuficiencia renal grave (Aclaramiento de creatinina [CrCl] <30 ml/min) así como realizar controles estrictos a estos pacientes (ver sección 4.2).
Enfermedad cardiaca
Los pacientes con antecedentes de insuficiencia cardíaca congestiva grave o de cardiopatía clínicamente inestable fueron excluidos de los estudios clínicos y, por tanto, no se han establecido la seguridad ni la eficacia de Dacogen en estos pacientes.
Excipientes
Este medicamento contiene 0,5 mmol de potasio por vial. Tras la dilución de la solución reconstituida para perfusión intravenosa, este medicamento contiene entre 1-10 mmol de potasio por dosis en función del volumen para perfusión de la dilución. Los pacientes que presenten un deterioro de la función renal o los que sigan una dieta con control del potasio deben tenerlo en cuenta.
Este medicamento contiene 0,29 mmol de sodio por vial. Tras la dilución de la solución reconstituida para perfusión intravenosa, este medicamento contiene entre 0,6-6 mmol de sodio por dosis en función del volumen para perfusión de la dilución. Los pacientes que sigan una dieta con control del sodio deben tenerlo en cuenta.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios clínicos formales de interacciones farmacológicas con decitabina.
Existe la posibilidad de una interacción fármaco-fármaco con otros agentes que también se activan por fosforilación secuencial (a través de las actividades intracelulares de la fosfoquinasa) y/o que se metabolizan por enzimas implicadas en la inactivación de la decitabina (por ejemplo, citidina deaminasa). Por lo tanto, se debe tener precaución si estos medicamentos se combinan con Dacogen.
Efecto de los medicamentos administrados de forma concomitante sobre decitabina No se espera que se produzcan interacciones metabólicas mediadas por el citocromo P450 (CYP) dado que el metabolismo de la decitabina no está mediado por este sistema, sino por desaminación oxidativa.
Efecto de la decitabina sobre los medicamentos administrados de forma concomitante Teniendo en cuenta su escasa unión a las proteínas plasmáticas in vitro (< 1 %), es improbable que la decitabina impida la unión a las proteínas plasmáticas de los medicamentos administrados de forma concomitante. Se ha demostrado que la decitabina es un inhibidor débil del transporte mediado por la
P-gp in vitro, por lo que no se espera que afecte a los medicamentos administrados concomitantemente cuyo transporte está mediado por la P-gp (ver sección 5.2).
4.6 Fertilidad, embarazo y lactancia
Anticoncepción en hombres y mujeres
Las mujeres en edad fértil deben emplear métodos anticonceptivos eficaces y evitar quedarse embarazadas mientras reciban tratamiento con Dacogen. Se desconoce el periodo que debe transcurrir después del tratamiento con Dacogen antes de que sea seguro concebir. Los hombres deben utilizar métodos anticonceptivos eficaces y se les aconsejará que no engendren un hijo mientras estén recibiendo Dacogen y durante 3 meses después de la finalización del tratamiento (ver sección 5.3).
No se ha estudiado el uso de Dacogen con anticonceptivos hormonales.
Embarazo
No existen datos suficientes sobre la utilización de Dacogen en mujeres embarazadas. Los estudios han revelado que la decitabina es teratógena en ratas y ratones (ver sección 5.3). Se desconoce el posible riesgo para el ser humano. Basándose en los resultados de los estudios en animales y en su mecanismo de acción, Dacogen no debe utilizarse durante el embarazo ni en mujeres en edad fértil que no empleen métodos anticonceptivos eficaces. Si se usa Dacogen durante el embarazo, o si una paciente se queda embarazada mientras recibe este medicamento, se debe informar a la paciente sobre el potencial riesgo para el feto.
Lactancia
Se desconoce si la decitabina o sus metabolitos se excretan en la leche materna. Dacogen está contraindicado durante la lactancia; por tanto, si se necesita tratamiento con Dacogen, se debe interrumpir la lactancia (ver sección 4.3).
Fertilidad
No existen datos en seres humanos acerca del efecto de la decitabina sobre la fertilidad. En estudios preclínicos realizados en animales, la decitabina afectó a la fertilidad masculina y fue mutágeno. Debido a la posibilidad de infertilidad como consecuencia del tratamiento con Dacogen, los hombres deben recibir asesoramiento sobre la conservación de espermatozoides y las mujeres en edad fértil deben recibir asesoramiento sobre la crioconservación de ovocitos antes de iniciar el tratamiento con Dacogen.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Dacogen sobre la capacidad para conducir y utilizar máquinas puede ser moderada.
Se debe advertir a los pacientes que pueden experimentar efectos adversos como anemia durante el tratamiento. Por consiguiente, se debe recomendar precaución al conducir o utilizar máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Las reacciones adversas más frecuentes (> 35 %) notificadas durante el tratamiento con Dacogen son pirexia, anemia y trombocitopenia.
Las reacciones adversas de Grado 3/4 más frecuentes (> 20 %) incluyeron neumonía, trombocitopenia, neutropenia, neutropenia febril y anemia.
En los estudios clínicos, el 30% de los pacientes tratados con Dacogen y el 25% de los pacientes tratados en el grupo comparador sufrieron efectos adversos con un resultado de muerte durante el tratamiento o dentro de los 30 días posteriores a la última dosis del medicamento del estudio.
En el grupo de tratamiento con Dacogen, hubo una incidencia mayor de la interrupción del tratamiento en mujeres que en hombres debido a acontecimientos adversos (43% versus 32%).
Lista tabulada de reacciones adversas
Las reacciones adversas notificadas en 293 pacientes con LMA tratados con Dacogen se resumen en la Tabla 1. En la siguiente tabla se recogen los datos de los estudios clínicos en pacientes con LMA y de la experiencia postcomercialización. Las reacciones adversas se enumeran por orden de frecuencia.
Las categorías de frecuencia se definen de la manera siguiente: Muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100), raras (> 1/10.000 a < 1/1.000), muy raras (< 1/10.000), no conocida (la frecuencia no puede estimarse a partir de los datos disponibles).
Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden de gravedad decreciente.
Tabla 1: Reacciones adversas identificadas con Dacogen
|
Sistema de Clasificación de Órganos |
Frecuencia (todos los grados) |
Reacción adversa al fármaco |
Frecuencia | |
|
Todos los grados3 (%) |
Grados 3-4a (%) | |||
|
Infecciones e infestaciones |
Muy frecuentes |
neumonía* |
24 |
20 |
|
infección urinaria* |
15 |
7 | ||
|
Todas las infecciones restantes (vírica, bacteriana, micótica)*,b,c,d |
63 |
39 | ||
|
Frecuentes |
shock séptico* |
6 |
4 | |
|
sepsis* |
9 |
8 | ||
|
sinusitis |
3 |
1 | ||
|
Trastornos de la sangre y del sistema linfático |
Muy frecuentes |
neutropenia febril* |
34 |
32 |
|
neutropenia* |
32 |
30 | ||
|
trombocitopenia*,e |
41 |
38 | ||
|
anemia |
38 |
31 | ||
|
leucopenia |
20 |
18 | ||
|
Poco frecuentes |
pancitopenia* |
<1 |
<1 | |
|
Trastornos del sistema inmunológico |
Frecuentes |
hipersensibilidad, incluida reacción anafilácticaf |
1 |
<1 |
|
Trastornos del sistema nervioso |
Muy frecuentes |
cefalea |
16 |
1 |
|
Trastornos respiratorios, torácicos y mediastínicos |
Muy frecuentes |
epistaxis |
14 |
2 |
|
Trastornos gastrointestinales |
Muy frecuentes |
diarrea |
31 |
2 |
|
vómitos |
18 |
1 | ||
|
náuseas |
33 |
<1 | ||
|
Frecuentes |
estomatitis |
7 |
1 | |
|
No conocida |
enterocolitis, incluyendo colitis neutropénica, tiflitis* |
No conocida |
No conocida | |
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuentes |
dermatosis neutrofílica febril aguda (síndrome de Sweet) |
<1 |
NP |
|
Trastornos generales y alteraciones en el lugar de administración |
Muy frecuentes |
fiebre |
48 |
9 |
Se incluyen los términos preferentes hipersensibilidad, hipersensibilidad al fármaco, reacción anafiláctica, shock anafiláctico, reacción anafilactoide, shock anafilactoide.
* Incluye los acontecimientos con desenlace mortal NP = No procede
Descripción de reacciones adversas seleccionadas
Reacciones adversas hematológicas
Las reacciones adversas hematológicas más frecuentes asociadas al tratamiento con Dacogen fueron neutropenia febril, trombocitopenia, neutropenia, anemia y leucopenia.
Se han notificado reacciones adversas graves relacionadas con hemorragias, algunas de las cuales tuvieron un desenlace mortal, como hemorragia en el sistema nervioso central (SNC; 2 %) y hemorragia digestiva (2 %), en el contexto de una trombocitopenia grave, en pacientes tratados con Dacogen.
Las reacciones adversas hematológicas se deben tratar mediante controles periódicos con hemogramas completos y la administración precoz de tratamientos de apoyo según sea necesario. En caso de neutropenia y transfusiones en caso de anemia o trombocitopenia, los tratamientos de apoyo comprenden la administración de antibióticos profilácticos y/o apoyo con factores de crecimiento (por ejemplo, G-CSF), de acuerdo con las directrices del centro. Para consultar las situaciones en las que debe retrasarse la administración de decitabina, ver sección 4.2.
Reacciones adversas infecciones e infestaciones
Se han notificado reacciones adversas graves, con desenlace potencialmente mortal, como shock séptico, sepsis, neumonía y otras infecciones (vírica, bacteriana y micótica), en pacientes tratados con Dacogen.
Trastornos gastrointestinales
Se ha notificado la aparición de enterocolitis, incluida colitis neutropénica, tiflitis durante el tratamiento con decitabina. La enterocolitis puede conducir a complicaciones por septicemia y se puede asociar con desenlace mortal.
Notificación de sospechas de reacciones adversas
. Se invita a los sistema nacional
a
b
Peor Grado según los Criterios Terminológicos Comunes para Acontecimientos Adversos del National Cáncer Institute.
Se excluye neumonía, infección urinaria, sepsis, shock séptico y sinusitis.
c
d
e
Las “infecciones restantes” notificadas con más frecuencia en el ensayo DACO-016 fueron: herpes oral, candidiasis oral, faringitis, infección de vías respiratorias altas, celulitis, bronquitis, nasofaringitis.
Se incluye enterocolitis infecciosa
Se incluye hemorragia asociada a trombocitopenia, incluidos casos mortales
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento profesionales sanitarios a notificar las sospechas de reacciones adversas a través del de notificación incluido en el Anexo V.
4.9 Sobredosis
No existe experiencia directa de sobredosis en el ser humano y no se dispone de ningún antídoto específico. Sin embargo, los datos de la literatura publicada, procedentes de los primeros estudios clínicos con dosis más de 20 veces mayores que la dosis terapéutica actual, notificaron una mayor incidencia de mielosupresión, incluyendo neutropenia y trombocitopenia prolongadas. Es probable que la toxicidad se manifieste en forma de exacerbaciones de las reacciones adversas al fármaco, principalmente la mielosupresión. El tratamiento de la sobredosis debe ser de apoyo.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Fármacos antineoplásicos, antimetabolitos, análogos de las pirimidinas; Código ATC: L01BC08
Mecanismo de acción
La decitabina (5-aza-2'-desoxicitidina) es un análogo de la citidina desoxinucleósida que inhibe de forma selectiva las metiltransferasas del ADN en dosis bajas, lo que produce la hipometilación de promotores génicos, que puede causar una reactivación de genes supresores tumorales, la inducción de diferenciación celular o la senescencia celular, seguida de la muerte celular programada.
Experiencia, clínica.
El uso de Dacogen se investigó en un ensayo de Fase III abierto, aleatorizado y multicéntrico (DACO-016) en sujetos con diagnóstico reciente de LMA de novo o secundaria según la clasificación de la OMS. Dacogen (n = 242) se comparó con un tratamiento de elección (TE, n = 243) que consistió, dependiendo de la elección del paciente con asesoramiento del médico, en tratamiento de apoyo solo (n = 28, 11,5%) o 20 mg/m2 de citarabina por vía subcutánea una vez al día durante 10 días consecutivos, repetido cada 4 semanas (n = 215; 88,5 %). Dacogen se administró en una perfusión intravenosa de 20 mg/m2 durante 1 hora, una vez al día, durante 5 días consecutivos, repetida cada 4 semanas.
No se incluyeron en el estudio sujetos que se consideraban candidatos a la quimioterapia de inducción convencional, como demuestran las siguientes características basales. La mediana de edad de la población por intención de tratar (IT) era de 73 años (intervalo de 64 a 91 años). El 36 % de los sujetos presentaban características citogenéticas de alto riesgo en el momento basal. El resto de los sujetos tenían características citogenéticas de riesgo intermedio. No se incluyó en el estudio a pacientes con citogenética favorable. El 25 % de los sujetos presentaban un estado funcional del ECOG >2. El 81 % de los sujetos padecían enfermedades concomitantes importantes (por ejemplo, infección, insuficiencia cardíaca, insuficiencia pulmonar). El número de pacientes tratados con Dacogen por grupo étnico fue de 209 blancos (86,4 %) y 33 asiáticos (13,6 %).
El criterio de valoración principal del estudio fue la supervivencia global. El criterio de valoración secundario fue la tasa de remisiones completas, que se evaluó mediante una revisión de expertos independientes. La supervivencia libre de progresión y la supervivencia sin acontecimientos fueron criterios de valoración terciarios.
La mediana de la supervivencia global en la población por intención de tratar fue de 7,7 meses en los sujetos tratados con Dacogen en comparación con 5,0 meses en los del grupo de TE (Hazard ratio, 0,85; IC del 95 %: 0,69; 1,04; p = 0,1079). La diferencia no alcanzó significación estadística; sin embargo, se observó una tendencia a la mejoría de la supervivencia, con una reducción del riesgo de muerte del 15 %, en los sujetos del grupo de Dacogen (Figura 1). Cuando se censuró con respecto al tratamiento posterior con modificadores de la enfermedad (es decir, quimioterapia de inducción o hipometilantes), el análisis de la supervivencia global reveló una reducción del riesgo de muerte del 20 % en los sujetos del grupo de Dacogen [HR = 0,80; (IC del 95 %: 0,64; 0,99), valor p = 0,0437)].
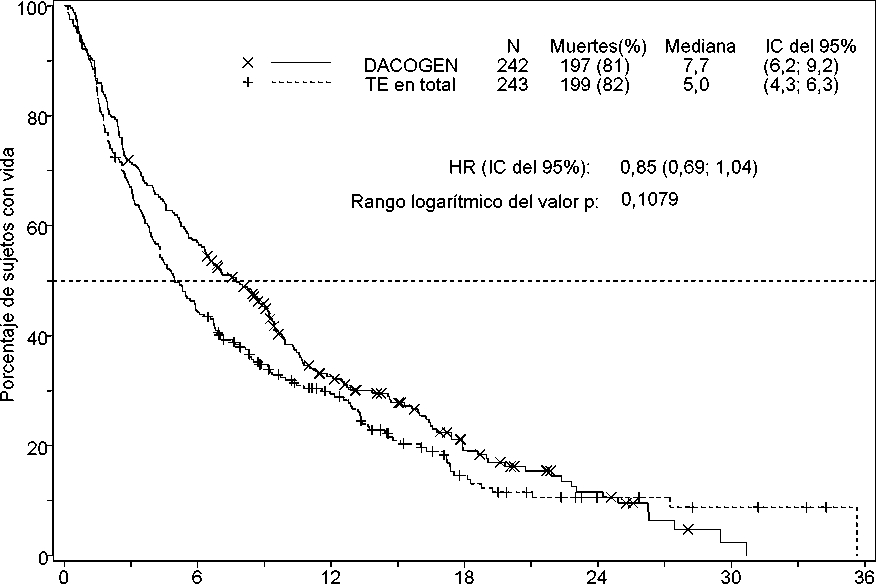
Tiempo (meses)
N.° de sujetos con riesgo DACOGEN 242 137
TE en total 243 107
65
55
28
19
12
7
En un análisis que incluyó los datos de supervivencia maduros de 1 año adicional, el efecto de Dacogen en la supervivencia global demostró una mejoría clínica en comparación con el grupo de TE (7,7 meses frente a 5,0 meses, respectivamente; hazard ratio= 0,82; IC del 95 %: 0,68; 0,99, valor nominal de p = 0,0373, Figura 2).
____L___,
N.° de sujetos con riesgo DACOGEN 242 TE en total 243
|
6 |
12 |
18 |
24 Tiempo (meses) |
30 |
36 |
42 |
48 |
|
137 |
78 |
50 |
28 |
11 |
2 |
0 |
0 |
|
107 |
68 |
35 |
20 |
10 |
4 |
2 |
0 |
100-f
HR(IC del 95%): Rango logarítmico del valor p:
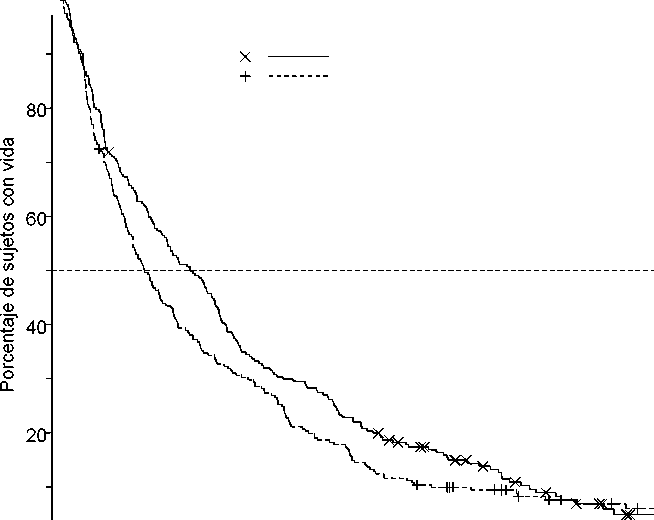
0,82 (0,68; 0,99) 0,0373
DACOGEN TE en total
|
N |
Muertes (%) |
Mediana |
IC del 95% |
|
242 |
219 (90) |
7,7 |
(6,2; 9,2) |
|
243 |
227 (93) |
5,0 |
(4,3; 6,3) |
0
Basándose en el análisis inicial realizado en la población por intención de tratar, se alcanzó una diferencia estadísticamente significativa en la tasa de remisión completa (RC + RCp) a favor de los sujetos del grupo de Dacogen, 17,8 % (43/242), en comparación con el grupo de TE, 7,8 % (19/243); diferencia entre los tratamientos del 9,9 % (IC del 95 %: 4,07; 15,83), p = 0,0011. La mediana del tiempo hasta la mejor respuesta y la mediana de la duración de la mejor respuesta en los pacientes que alcanzaron una RC o una RCp fueron de 4,3 y 8,3 meses, respectivamente. La supervivencia libre de progresión (SLP) fue significativamente más prolongada en los sujetos del grupo de Dacogen,
3,7 meses (IC del 95 %: 2,7; 4,6) en comparación con los sujetos del grupo de TE, 2,1 meses (IC del 95 %: 1,9; 3,1); hazard ratio 0,75 (IC del 95 %: 0,62; 0,91), p = 0,0031. Estos resultados, así como otros criterios de valoración, se recogen en la Tabla 2.
Tabla 2: Otros criterios de valoración de la eficacia del estudio DACO-016 (población IT)
|
Resultados |
Dacogen n = 242 |
TE (grupo combinado) n = 243 |
Valor p |
|
RC + RCp |
43 (17,8 %) |
19 (7,8 %) |
0,0011 |
|
RP (1,4 |
P = 2,5 0; 4,78)b | ||
|
RC |
38(15,7%) |
18 (7,4%) |
- |
|
SLEa |
3,5 (2,5; 4,1)b |
2,1 (1,9; 2,8)b |
0,0025 |
|
HR = 0,75 (0,62; 0,90)b | |||
|
SLPa |
3,7 |
2,1 |
0,0031 |
|
(2,7; 4,6)b |
(1,9; 3,1)b | ||
|
HR = 0,75 | |||
|
(0,62; 0,91)b | |||
RC = remisión completa; RCp = remisión completa con recuperación plaquetaria incompleta, SLE = supervivencia libre de enfermedad, SLP = supervivencia libre de progresión, RP = razón de probabilidades, HR = hazard ratio - = No evaluable
Indicado como mediana en meses b Intervalos de confianza del 95 %
Las tasas de supervivencia global y de remisión completa en los subgrupos preespecificados relacionados con la enfermedad (es decir, riesgo citogenético, puntuación del Eastern Cooperative Oncology Group [ECOG], edad, tipo de LMA y recuento basal de blastocitos en médula ósea) coincidieron con los resultados obtenidos en la población total del estudio.
Los sujetos tratados con Dacogen (11%, 24/223) experimentaron un empeoramiento de la hiperglucemia en comparación con los sujetos del grupo de TE (6%, 13/212).
También se evaluó el uso de Dacogen como tratamiento inicial en un estudio de Fase II abierto, con un solo grupo (DACO-017) en 55 sujetos de más de 60 años con LMA según la clasificación de la OMS. El criterio de valoración principal fue la tasa de remisiones completas (RC), que se evaluó mediante una revisión de expertos independientes. El criterio de valoración secundario del estudio fue la supervivencia global. Dacogen se administró en una perfusión intravenosa de 20 mg/m2 durante 1 hora, una vez al día, durante 5 días consecutivos, repetida cada 4 semanas. En el análisis por intención de tratar, se observó una tasa de RC del 23,6 % (IC del 95 %: 13,2; 37) en 13/55 pacientes tratados con Dacogen. La mediana de tiempo hasta la RC fue de 4,1 meses y la mediana de la duración de la RC, de 18,2 meses. La mediana de supervivencia global en la población por intención de tratar fue de 7,6 meses (IC del 95 %: 5,7; 11,5).
No se han evaluado la eficacia y la seguridad de Dacogen en pacientes con leucemia promielocítica aguda o leucemia del SNC.
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Dacogen en uno o más grupos de la población pediátrica para el tratamiento de la leucemia mieloide aguda. Ver sección 4.2 para consultar la información sobre el uso en población pediátrica.
5.2 Propiedades farmacocinéticas
Se agruparon los parámetros de farmacocinética (FC) poblacional de decitabina obtenidos en 3 estudios clínicos realizados en 45 pacientes con LMA o síndrome mielodisplásico (SMD) en los que se utilizó la pauta de 5 días. En cada estudio, la FC de la decitabina se evaluó el quinto día del primer ciclo de tratamiento.
Distribución
La farmacocinética de decitabina tras la administración intravenosa en una perfusión de 1 hora se describió mediante un modelo lineal bicompartimental, caracterizado por una rápida eliminación del fármaco del compartimento central y por una distribución relativamente lenta desde el compartimento periférico. En la Tabla 3, presentada a continuación, se muestran los parámetros farmacocinéticos de decitabina para un paciente típico (peso de 70 kg y superficie corporal de 1,73 m2).
Tabla 3: Resumen del análisis de FC poblacional para un paciente típico tratado con infusiones diarias de 1 hora de Dacogen 20 mg/m2 durante 5 días cada 4 semanas
|
Parámetro3 |
Valor teórico |
IC del 95 % |
|
Cmax (ng/ml) |
107 |
88,5 - 129 |
|
AUCcum (ng.h/ml) |
580 |
480 - 695 |
|
t1/2 (min) |
68,2 |
54,2 - 79,6 |
|
Vdss (l) |
116 |
84,1 - 153 |
|
CL (l/h) |
298 |
249 - 359 |
La dosis total por ciclo fue de 100 mg/m2
Decitabina presenta una farmacocinética lineal y, tras la perfusión intravenosa, las concentraciones en estado estacionario se alcanzan en el plazo de 0,5 horas. En base a una simulación de modelos, los parámetros FC fueron independientes del tiempo (es decir, no se modificaron entre un ciclo y otro) y no se observó acumulación con esta pauta posológica. La unión de decitabina a las proteínas plasmáticas es insignificante (<1 %). El Vdss de decitabina es amplio en los pacientes con cáncer, lo que indica la distribución del medicamento en los tejidos periféricos. No hubo evidencia de dependencia de la edad, del aclaramiento de creatinina, de la bilirrubina total ni de la enfermedad.
Biotransformación
En el compartimento intracelular, decitabina es activada mediante fosforilación secuencial a través de las actividades de fosfoquinasa dando lugar al trifosfato correspondiente, que posteriormente es incorporado por la polimerasa del ADN. Los datos de metabolismo in vitro y los resultados del estudio de equilibrio de masas en el ser humano indicaron que el sistema del citocromo P450 no interviene en el metabolismo de decitabina. Es probable que la vía principal de metabolismo sea la desaminación por la citidina desaminasa en el hígado, el riñón, el epitelio intestinal y la sangre. Los resultados del estudio de equilibrio de masas en el ser humano demostraron que la decitabina inalterada en plasma representaba aproximadamente el 2,4 % de la radiactividad total en el plasma. Se cree que los principales metabolitos circulantes no son farmacológicamente activos. La presencia de estos metabolitos en la orina, junto con el elevado aclaramiento corporal total y la baja excreción urinaria del fármaco intacto (~4 % de la dosis), indican que decitabina se metaboliza de forma apreciable in vivo. Los estudios in vitro revelan que la decitabina no inhibe ni induce las enzimas del CYP 450 en más de 20 veces la concentración plasmática máxima terapéutica observada (Cmax). Por tanto, no se espera que se produzcan interacciones farmacológicas metabólicas mediadas por el CYP y es poco probable que decitabina interaccione con fármacos metabolizados por estas vías. Además, los datos in vitro indican que decitabina es un mal sustrato de la P-gp.
Eliminación
La eliminación plasmática media tras la administración intravenosa en sujetos con cáncer fue > 200 l/h, observándose una moderada variabilidad interindividual (el Coeficiente de variación [CV] es aproximadamente del 50 %). La excreción del fármaco intacto parece tener solo un pequeño papel en la eliminación de decitabina.
Los resultados de un estudio de equilibrio de masas con 14C-decitabina radiactiva en pacientes con cáncer mostraron que el 90 % de la dosis administrada de decitabina (fármaco intacto en un 4 %) se excreta en la orina.
Información adicional sobre poblaciones especiales
No se han estudiado formalmente los efectos de la insuficiencia renal o hepática, el sexo, la edad ni la raza sobre la farmacocinética de decitabina. La información relativa a las poblaciones especiales se determinó a partir de los datos farmacocinéticos de los 3 estudios citados anteriormente y de un estudio en Fase I realizado en sujetos con SMD (N = 14; 15 mg/m2x 3 horas cada 8 horas x 3 días).
Pacientes de edad avanzada
El análisis de farmacocinética poblacional demostró que la farmacocinética de la decitabina no depende de la edad (intervalo de edad estudiado de 40 a 87 años; mediana de 70 años).
Sexo
El análisis de la farmacocinética poblacional de decitabina no mostró diferencias clínicamente importantes entre los varones y las mujeres.
Raza
La mayoría de los pacientes estudiados eran de raza blanca. Sin embargo, el análisis de farmacocinética poblacional de decitabina indicó que la raza no tenía efectos aparentes sobre la exposición a decitabina.
Insuficiencia hepática
La FC de la decitabina no se ha estudiado formalmente en pacientes con insuficiencia hepática. Los resultados de un estudio de equilibrio de masas en el ser humano y los experimentos in vitro mencionados anteriormente indican que es improbable que las enzimas del CYP intervengan en el metabolismo de decitabina. Además, los pocos datos obtenidos en el análisis de FC poblacional indicaron que no existe dependencia significativa de ningún parámetro FC de la concentración de bilirrubina total a pesar de una amplia variedad de niveles de bilirrubina total. Por tanto, no es probable que la exposición a decitabina resulte afectada en los pacientes con deterioro de la función hepática.
Insuficiencia renal
La FC de decitabina no se ha estudiado formalmente en pacientes con insuficiencia renal. El análisis de FC poblacional realizado con los pocos datos de decitabina disponibles indicó que no existe dependencia significativa de los parámetros FC del aclaramiento de creatinina normalizado, un indicador de la función renal. Por tanto, no es probable que la exposición a decitabina resulte afectada en los pacientes con deterioro de la función renal.
5.3 Datos preclínicos sobre seguridad
No se han realizado estudios de carcinogenicidad formales con decitabina. Los datos de la bibliografía indican que decitabina tiene potencial carcinogénico. Los datos disponibles procedentes de estudios in vitro e in vivo aportan pruebas suficientes de que decitabina tiene potencial genotóxico. Los datos obtenidos de la bibliografía indican también que decitabina ejerce efectos adversos en todos los aspectos del ciclo reproductor, incluidas la fertilidad, el desarrollo embrionario y fetal y el desarrollo posnatal. Los estudios de toxicidad de dosis repetidas y ciclos múltiples en ratas y conejos mostraron que la toxicidad principal fue la mielosupresión, incluyendo efectos sobre la médula ósea, que fue reversible al suspender el tratamiento. Se observó asimismo toxicidad digestiva y, en los machos, atrofia testicular que no se corrigió en los períodos de recuperación previstos. La administración de decitabina a ratas neonatales/jóvenes mostró un perfil de toxicidad general similar al observado en ratas de más edad. El desarrollo neuroconductual y la capacidad reproductora no se vieron afectadas cuando las ratas neonatales/jóvenes fueron tratadas con niveles de dosis que inducen la mielosupresión. Ver Sección 4.2 para información sobre uso en población pediátrica.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Dihidrógeno fosfato de potasio (E340)
Hidróxido de sodio (E524)
Ácido clorhídrico (para ajustar el pH)
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección 6.6.
6.3 Periodo de validez
Vial sin abrir 3 años.
Solución reconstituida y diluida
En el plazo de 15 minutos desde la reconstitución, el concentrado (en 10 ml de agua estéril para preparaciones inyectables) se debe diluir posteriormente utilizando líquidos para perfusión refrigerados (2°C - 8°C). Esta solución diluida preparada para perfusión intravenosa puede ser conservada a 2°C - 8°C durante un máximo de 3 horas, seguidas de hasta 1 hora a temperatura ambiente (20°C - 25°C) antes de la administración.
Desde el punto de vista microbiológico, el producto debe ser utilizado dentro del plazo recomendado anteriormente. Es responsabilidad del usuario seguir los tiempos y las condiciones de conservación recomendados y garantizar que la reconstitución se haya realizado en condiciones asépticas.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 25°C.
Para las condiciones de conservación tras la reconstitución y dilución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Vial de vidrio de tipo I incoloro de 20 ml, cerrado con un tapón de goma de bromobutilo y un precinto de aluminio con tapa de plástico desprendible que contiene 50 mg de decitabina.
Tamaño del envase: 1 vial.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Recomendaciones para una manipulación segura
Debe evitarse el contacto de la piel con la solución y deben utilizarse guantes protectores. Se adoptarán los procedimientos habituales para el manejo de fármacos antineoplásicos.
Procedimiento de reconstitución
El polvo debe reconstituirse en condiciones asépticas con 10 ml de agua para preparaciones inyectables. Tras la reconstitución, cada mililitro contiene aproximadamente 5 mg de decitabina a un pH de 6,7 a 7.3. En el plazo de 15 minutos desde la reconstitución, la solución debe ser diluida posteriormente con líquidos para perfusión refrigerados [solución inyectable de cloruro sódico 9 mg/ml (0,9 %) o solución inyectable de glucosa al 5 %] hasta lograr una concentración final de 0,1a 1,0 mg/ml. Para el periodo de validez y las precauciones de conservación tras la reconstitución, ver sección 6.3.
Dacogen no se debe perfundir a través del mismo acceso/vía intravenosa con otros medicamentos. Eliminación
Este medicamento es únicamente para un solo uso. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse Bélgica
8. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/792/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 20/septiembre/2012
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/
A. FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del (de los) fabricante(s) responsable(s) de la liberación de los lotes
Janssen Pharmaceutica N.V. Turnhoutseweg 30 B-2340 Beerse Bélgica
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (Ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107ter, (párrafo 7), de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias
según lo acordado en la versión del PGR incluido en el Módulo 1.8.2. de la Autorización de
Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden
presentar conjuntamente.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CAJA DE CARTÓN
1. NOMBRE DEL MEDICAMENTO
Dacogen 50 mg polvo para concentrado para solución para perfusión decitabina
2. PRINCIPIO(S) ACTIVO(S)
Cada vial contiene 50 mg de decitabina.
Tras la reconstitución, 1 ml de concentrado contiene 5 mg de decitabina.
3. LISTA DE EXCIPIENTES
Excipientes: dihidrógeno fosfato de potasio (E340), hidróxido de sodio (E524), y ácido clorhídrico.
Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo para concentrado para solución para perfusión. 1 vial
5. FORMA Y VÍA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Exclusivamente para un solo uso.
Vía intravenosa.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Citotóxico
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Vial sin abrir: No conservar a temperatura superior a 25°C.
Consultar en el prospecto el periodo de validez del medicamento reconstituido y diluido.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Janssen Cilag International NV Turnhoutseweg 30 B-2340 Beerse Bélgica
12. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/792/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille.
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETA DEL VIAL
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Dacogen 50 mg polvo para perfusión
decitabina
IV
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
50 mg
6. OTROS
Citotóxico
B. PROSPECTO
Prospecto: Información para el usuario
Dacogen 50 mg polvo para concentrado para solución para perfusión
decitabina
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Dacogen y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Dacogen
3. Cómo usar Dacogen
4. Posibles efectos adversos
5. Conservación de Dacogen
6. Contenido del envase e información adicional
1. Qué es Dacogen y para qué se utiliza Qué es Dacogen
Dacogen contiene el principio activo “decitabina”. Es un medicamento contra el cáncer.
Para qué se utiliza Dacogen
Dacogen se utiliza para tratar un tipo de cáncer llamado “leucemia mieloide aguda” o “LMA”. Es un tipo de cáncer que afecta a las células sanguíneas. Se le administrará Dacogen cuando se le diagnostique por primera vez LMA. Este medicamento se utiliza únicamente en adultos.
Cómo actúa Dacogen
Dacogen actúa impidiendo el crecimiento de las células cancerosas. También destruye las células del cáncer.
Si tiene alguna pregunta sobre cómo actúa Dacogen o por qué le han recetado este medicamento, consulte a su médico o enfermero.
2. Qué necesita saber antes de empezar a usar Dacogen No use Dacogen:
• si es alérgico a decitabina o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
• si está dando el pecho.
Si no está seguro de si las condiciones anteriores le aplican a usted, consulte a su médico, farmacéutico o enfermero antes de utilizar Dacogen.
Advertencias y precauciones
Consulte a su médico, farmacéutico o enfermero antes de empezar a usar Dacogen si tiene
• número bajo de plaquetas, glóbulos rojos o glóbulos blancos,
• una infección,
• una enfermedad hepática,
• un trastorno renal grave,
• un trastorno cardíaco.
Si no está seguro de si las condiciones anteriores le aplican a usted, consulte a su médico, farmacéutico o enfermero antes de utilizar Dacogen.
Niños y adolescentes
Dacogen no debe utilizarse en niños ni adolescentes menores de 18 años.
Uso de Dacogen con otros medicamentos
Informe a su médico, enfermero o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento, incluso los adquiridos sin receta y las plantas medicinales. Dacogen puede afectar a la manera en que otros medicamentos actúan. Además, algunos otros medicamentos pueden afectar a la manera en que Dacogen actúa.
Pruebas o controles
Se le harán análisis de sangre antes de empezar el tratamiento con Dacogen y al comienzo de cada ciclo de tratamiento. Estas pruebas sirven para comprobar que:
• tiene suficientes células sanguíneas, y
• el hígado y los riñones funcionan correctamente.
Embarazo y lactancia
• Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento.
• No debe utilizar Dacogen si está embarazada porque puede dañar a su hijo. Informe a su médico de inmediato si se queda embarazada durante el tratamiento con Dacogen.
• No puede dar el pecho a su hijo si está utilizando Dacogen, ya que se desconoce si el medicamento pasa a la leche materna.
Fertilidad masculina y femenina y anticoncepción
• Los hombres no deben engendrar un hijo mientras estén utilizando Dacogen.
• Los hombres deben usar métodos anticonceptivos eficaces durante el tratamiento y durante 3 meses después de haber suspendido el tratamiento.
• Consulte a su médico si desea conservar su semen antes de comenzar el tratamiento.
• Las mujeres deben utilizar métodos anticonceptivos eficaces durante el tratamiento. Se desconoce cuándo es seguro para las mujeres quedarse embarazadas después de que se haya suspendido el tratamiento.
• Consulte con su médico si desea congelar sus óvulos antes de comenzar el tratamiento. Conducción y uso de máquinas
Es posible que se sienta cansado o débil después del uso de Dacogen. Si es así, no conduzca ni utilice herramientas o máquinas.
Dacogen contiene potasio y sodio
• Este medicamento contiene 0,5 mmol de potasio por vial. Tras la dilución de la solución reconstituida para perfusión intravenosa, este medicamento contiene entre 1-10 mmol de potasio por dosis en función del volumen para perfusión de la dilución. Los pacientes que presenten un deterioro de la función renal o los que sigan una dieta con control del potasio deben tenerlo en cuenta.
• Este medicamento contiene 0,29 mmol de sodio por vial. Tras la dilución de la solución reconstituida para perfusión intravenosa, este medicamento contiene entre 0,6-6 mmol de sodio por dosis en función del volumen para perfusión de la dilución. Los pacientes que sigan una dieta con control del sodio deben tenerlo en cuenta.
3. Cómo usar Dacogen
Dacogen le será administrado por un médico o enfermero preparado para administrar este tipo de medicamento.
Dosis recomendada
• Su médico calculará su dosis de Dacogen, que dependerá de su estatura y su peso (superficie corporal).
• La dosis es de 20 mg/m2 de superficie corporal.
• Recibirá Dacogen a diario durante 5 días, seguidos de 3 semanas sin fármaco. Esto se denomina un “ciclo de tratamiento” y se repetirá cada 4 semanas.
• Normalmente recibirá como mínimo 4 ciclos de tratamiento.
Su médico podrá retrasar la dosis y modificar el número total de ciclos, dependiendo de cómo responda al tratamiento.
Cómo se administra Dacogen
La solución se administra por vía intravenosa (como perfusión) durante una hora.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no
todas las personas los sufran. Con este medicamento pueden aparecer los siguientes efectos adversos.
Avise inmediatamente a su médico o enfermero si experimenta alguno de los siguientes efectos
adversos graves:
• Fiebre: puede ser un signo de una infección causada por niveles bajos de glóbulos blancos (muy frecuente).
• dolor en el pecho o dificultad para respirar (con o sin fiebre o tos): pueden ser signos de una infección de los pulmones denominada “neumonía” (muy frecuente).
• Hemorragia: incluyendo sangre en las heces. Puede ser un signo de hemorragia en el estómago o el intestino (frecuente).
• hemorragia dentro de la cabeza: sus síntomas pueden ser dificultad para moverse, para hablar o entender o para ver; dolor de cabeza intenso y repentino, convulsiones, entumecimiento o debilidad en cualquier parte del cuerpo (frecuente).
• dificultad para respirar, hinchazón de los labios, picor o erupción cutánea. Se pueden deber a una reacción alérgica (hipersensibilidad) (frecuente).
Consulte inmediatamente a su médico o enfermero si experimenta alguno de los efectos adversos
graves citados.
Otros efectos adversos de Dacogen son
Muy frecuentes (pueden afectar a más de 1 de cada 10 pacientes)
• infección de orina
• otra infección en cualquier parte del cuerpo, causada por bacterias, virus u hongos
• hemorragia o formación de hematomas con mayor facilidad - pueden ser signos de una disminución del número de plaquetas de la sangre (trombocitopenia)
• sensación de cansancio o palidez - pueden ser signos de una disminución del número de glóbulos rojos (anemia)
• dolor de cabeza
• hemorragia nasal
• diarrea
• vómitos
• náuseas
• fiebre
Frecuentes (pueden afectar hasta a 1 de cada 10 pacientes)
• infección de la sangre provocada por bacterias - puede ser un signo de un nivel bajo de glóbulos blancos
• dolor de nariz o moqueo, dolor de los senos nasales
• llagas en la boca o la lengua
Poco frecuentes (pueden afectar hasta a 1 de cada 100 pacientes)
• disminución del número de glóbulos rojos, glóbulos blancos y plaquetas (pancitopenia)
• placas rojas, elevadas y dolorosas en la piel, fiebre, aumento de los glóbulos blancos - pueden ser signos de “Dermatosis Neutrofílica Febril Aguda” o “Síndrome de Sweet”.
No conocida (la frecuencia no puede estimarse a partir de los datos disponibles)
• intestino inflamado (enterocolitis, colitis y tiflitis), con síntomas de dolor abdominal, meteorismo (gases intestinales), o diarrea. La enterocolitis puede conducir a complicaciones por septicemia (respuesta del organismo ante una infección) y se puede asociar con desenlace mortal.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero,
incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede
comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V.
Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información
sobre la seguridad de este medicamento.
5. Conservación de Dacogen
• Su médico, enfermero o farmacéutico son los responsables de la conservación de Dacogen.
• Mantenga este medicamento fuera de la vista y del alcance de los niños.
• No utilice este medicamento después de la fecha de caducidad que aparece en la caja y en la etiqueta del vial después de CAD. La fecha de caducidad es el último día del mes que se indica.
• No conservar a temperatura superior a 25°C.
• Tras la reconstitución, el concentrado se debe diluir posteriormente en el plazo de 15 minutos utilizando líquidos para perfusión refrigerados. Esta solución diluida preparada para perfusión intravenosa puede ser conservada refrigerada a 2°C - 8°C durante un máximo de 3 horas, seguidas de hasta 1 hora a temperatura ambiente (20°C - 25°C) antes de la administración.
• Su médico, enfermero o farmacéutico es responsable de la correcta eliminación de Dacogen no utilizado.
6. Contenido del envase e información adicional
Composición de Dacogen
• El principio activo es decitabina. Cada vial de polvo contiene 50 mg de decitabina. Tras la reconstitución con 10 ml de agua para preparaciones inyectables, cada ml del concentrado contiene 5 mg de decitabina.
• Los demás componentes son dihidrógeno fosfato de potasio (E340), hidróxido de sodio (E524), y ácido clorhídrico (para ajustar el pH).
Aspecto del producto y contenido del envase
Dacogen es un polvo para concentrado para solución para perfusión de color blanco o casi blanco. Se
presenta en un vial de vidrio de 20 ml que contiene 50 mg de decitabina. Cada envase contiene 1 vial.
Titular de la autorización de comercialización
Janssen-Cilag International NV Turnhoutseweg 30
B-2340 Beerse Bélgica
Responsable de la fabricación
Janssen Pharmaceutica NV Turnhoutseweg 30 B-2340 Beerse Bélgica
Pueden solicitar más información respecto a este titular de la autorización de comercialización:
Belgie/Belgique/Belgien
Janssen-Cilag NV Tel/Tél: +32 14 64 94 11
Etnrapna
„fl^OHCtH & fl^OHCtH E'bnrapHa” EOOfl Ten.: +359 2 489 94 00
Ceská republika JANSSEN-CILAG s.r.o.
Tel: +420 227 012 227
Danmark
JANSSEN-CILAG A/S Tlf: +45 45 94 82 82
Deutschland
JANSSEN-CILAG GmbH Tel: +49 2137 955-955
Eesti
UAB "JOHNSON & JOHNSON" Eesti filiaal Tel: +372 617 7410
EXláSa
JANSSEN-CILAG Oap^aKswix^ A.E.B.E. T^: +30 210 80 90 000
España
JANSSEN-CILAG, S.A.
Tel: +34 91 722 81 00
France
JANSSEN-CILAG
Tél: 0 800 25 50 75 / +33 1 55 00 40 03 Hrvatska
Johnson & Johnson S.E. d.o.o.
Tel: +385 1 6610 700
Ireland
JANSSEN-CILAG Ltd.
Tel: +44 1 494 567 444
medicamento dirigiéndose al representante local del
Lietuva
UAB "JOHNSON & JOHNSON"
Tel: +370 5 278 68 88
Luxembourg/Luxemburg
Janssen-Cilag NV Tél/Tel: +32 14 64 94 11
Magyarország JANSSEN-CILAG Kft.
Tel: +36 1 884 2858
Malta
AM MANGION LTD.
Tel: +356 2397 6000
Nederland
JANSSEN-CILAG B.V.
Tel: +31 13 583 73 73
Norge
JANSSEN-CILAG AS Tlf: +47 24 12 65 00
Osterreich
JANSSEN-CILAG Pharma GmbH Tel: +43 1 610 300
Polska
JANSSEN-CILAG Polska Sp. z o.o.
Tel: +48 22 237 60 00
Portugal
JANSSEN-CILAG FARMACÉUTICA, LDA. Tel: +351 21 43 68 835
Romania
Johnson & Johnson Romania SRL Tel: +40 21 207 1800
Slovenija
Johnson & Johnson d.o.o.
Tel: +386 1 401 18 30
Ísland
JANSSEN-CILAG AB c/o Vistor hf.
Sími: +354 535 7000
Italia
JANSSEN-CILAG SpA Tel: +39 02 2510 1
Kúrcpog
BapváPag Xar^nnavay^g AxS,
T^: +357 22 207 700
Latvija
UAB "JOHNSON & JOHNSON" filia Tel: +371 678 93561
Slovenská republika
Johnson & Johnson s.r.o. Tel: +421 232 408 400
Suomi/Finland
JANSSEN-CILAG OY Puh/Tel: +358 207 531 300
Sverige
JANSSEN-CILAG AB Tel: +46 8 626 50 00
United Kingdom
Latvija JANSSEN-CILAG Ltd.
Tel: +44 1 494 567 444
Fecha de la última revisión de este prospecto: MM/YYYY
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu/.
Esta información está destinada únicamente para profesionales del sector sanitario:
1. RECONSTITUCIÓN
Se debe evitar el contacto de la piel con la solución y se deben utilizar guantes protectores. Se deben adoptar los procedimientos habituales para el manejo de fármacos antineoplásicos.
El polvo debe ser reconstituido en condiciones asépticas con 10 ml de agua para preparaciones inyectables. Tras la reconstitución, cada mililitro contiene aproximadamente 5 mg de decitabina a un pH de 6,7 a 7,3. En el plazo de 15 minutos desde la reconstitución, la solución se debe diluir posteriormente con líquidos para perfusión refrigerados (2°C - 8°C) (solución inyectable de cloruro sódico 9 mg/ml (0,9 %) o solución inyectable de glucosa al 5 %) hasta lograr una concentración final de 0,1 a 1,0 mg/ml.
Para el periodo de validez y las precauciones de conservación tras la reconstitución, ver sección 5 del prospecto.
2. ADMINISTRACIÓN
Perfundir la solución reconstituida por vía intravenosa durante 1 hora.
3. ELIMINACIÓN
Los viales son para un solo uso y se debe desechar cualquier solución sobrante.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
29