Cyramza 10Mg/Ml Concentrado Para Solucion Para Perfusion
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
^ Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Cyramza 10 mg/ml concentrado para solución para perfusión.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un ml de concentrado para solución para perfusión contiene 10 mg de ramucirumab
Cada vial de 10 ml contiene 100 mg de ramucirumab.
Cada vial de 50 ml contiene 500 mg de ramucirumab.
Ramucirumab es un anticuerpo IgG1monoclonal recombinante humano producido por células murinas (NS0) mediante tecnología de ADN recombinante.
Excipiente(s) con efecto conocido:
Cada vial de 10 ml contiene aproximadamente 17 mg de sodio.
Cada vial de 50 ml contiene aproximadamente 85 mg de sodio.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Concentrado para solución para perfusión (concentrado estéril).
El concentrado es una solución de aspecto transparente a ligeramente opalescente y de tinte incoloro a ligeramente amarillo con pH 6.0.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Cyramza en combinación con paclitaxel está indicado para el tratamiento de pacientes adultos con cáncer gástrico avanzado o adenocarcinoma de la unión gastroesofágica con progresión de la enfermedad tras quimioterapia previa con platino y fluoropirimidina (ver sección 5.1).
Cyramza en monoterapia está indicado para el tratamiento de pacientes adultos con cáncer gástrico avanzado o adenocarcinoma de la unión gastroesofágica con progresión de la enfermedad tras quimioterapia previa con platino o fluoropirimidina, para quienes el tratamiento en combinación con paclitaxel no es apropiado (ver sección 5.1).
Cyramza en combinación con FOLFIRI (irinotecán, ácido folínico y 5-fluorouracilo), está indicado para el tratamiento de pacientes adultos con cáncer colorrectal metastásico (metastatic colorectal cáncer, mCRC, por sus siglas en inglés) con progresión de la enfermedad durante o tras terapia previa con bevacizumab, oxaliplatino y una fluoropirimidina.
Cyramza en combinación con docetaxel está indicado para el tratamiento de pacientes adultos con cáncer de pulmón no microcítico localmente avanzado o metastásico con progresión de la enfermedad tras quimioterapia basada en platino.
4.2 Posología y forma de administración
El tratamiento con ramucirumab se debe iniciar y estar bajo la supervisión de médicos con experiencia en oncología.
Posología
Cáncer gástrico y adenocarcinoma de la unión gastroesofágica (gastro-oesophageal _junction, GEJ _por sus siglas en inglés)
Cyramza en combinación con paclitaxel
La dosis recomendada de ramucirumab es de 8 mg/kg los días 1 y 15 de un ciclo de 28 días, antes de la perfusión de paclitaxel. La dosis recomendada de paclitaxel es de 80 mg/m2 administrado por perfusión intravenosa durante aproximadamente 60 minutos los días 1, 8 y 15 de un ciclo de 28 días. Antes de cada perfusión de paclitaxel se debe realizar un hemograma completo y bioquímica sanguínea al paciente para evaluar la función hepática. En la Tabla 1 se muestran los criterios que se deben cumplir antes de cada perfusión con paclitaxel.
Tabla 1: Criterios que se deben cumplir antes de cada administración de paclitaxel
|
Criterios | |
|
Neutrófilos |
Día 1: >1,5 x 109/l Días 8 y 15: >1,0 x 109/l |
|
Plaquetas |
Día 1: >100 x 109/l Días 8 y 15: >75 x 109/l |
|
Bilirrubina |
<1,5 veces el valor del límite superior de la normalidad (upper limit of normal value, ULN) |
|
Aspartato aminotransferasa (AST) /Alanina aminotransferasa (ALT) |
Sin metástasis hepática: ALT/AST <3 x ULN Con metástasis hepática: ALT/AST <5 x ULN |
Cyramza como agente único
La dosis recomendada de ramucirumab como agente único es de 8 mg/kg cada 2 semanas.
Cáncer colorrectal
La dosis recomendada de ramucirumab es 8 mg/kg cada 2 semanas administrada por perfusión intravenosa antes de la administración de FOLFIRI. Antes de la quimioterapia, se debe realizar un hemograma completo al paciente. En la Tabla 2 se muestran los criterios que se deben cumplir antes de la administración de FOLFIRI.
Tabla 2: Criterios que se deben cumplir antes de la administración de FOLFIRI
|
Criterios | |
|
Neutrófilos |
>1,5 x 109/l |
|
Plaquetas |
>100 x 109/l |
|
Toxicidad gastrointestinal relacionada con la quimioterapia |
< Grado 1 (criterios de terminología frecuente para reacciones adversas en cáncer del Instituto Nacional del Cáncer, National Cancer Institute Common Terminology Criteria for Adverse Events [NCI CTCAE]por sus siglas en inglés) |
La dosis recomendada de ramucirumab es 10 mg/kg el día 1 de un ciclo de 21 días, antes de la perfusión de docetaxel. La dosis recomendada de docetaxel es 75 mg/m2 administrada por perfusión intravenosa durante aproximadamente 60 minutos el día 1 de un ciclo de 21 días. Para pacientes de Asia Oriental, se debeconsiderar empezar con una dosis reducida de docetaxel de 60 mg/m2 el día 1 de un ciclo de 21 días. Consultar la información de producto (ficha técnica) de docetaxel para consejos de dosificación específicos.
Duración del tratamiento
Se recomienda que el tratamiento continúe hasta progresión de la enfermedad o hasta toxicidad inaceptable.
Medicación previa
Antes de la perfusión de ramucirumab, se recomienda administrar al paciente un antagonista de histamina H1 (p.ej. difenhidramina) como medicación previa. Si un paciente presenta reacciones relacionadas con la perfusión de Grado 1 o 2, debe recibir medicación previa en todas las perfusiones posteriores. Se debe administrar dexametasona (o equivalente) si un paciente presenta una segunda reacción relacionada con la perfusión (infusion relates reactions, IRR) de Grado 1 o 2; e luego, para las siguientes perfusiones se deben utilizar como medicación previa los siguientes medicamentos o equivalentes: un antagonista de histamina H1 intravenoso (por ejemplo difenhidramina hidrocloruro), paracetamol y dexametasona.
Ver la información de producto (ficha técnica) de paclitaxel, de los componentes de FOLFIRI y de docetaxel según proceda, para sus requerimientos sobre medicación previa e información adicional.
Ajuste de dosis _para ramucirumab Reacciones relacionadas con la perfusión
Si el paciente experimenta una IRR de Grado 1 o 2, la velocidad de perfusión de ramucirumab se debe reducir en un 50% así como en todas las perfusiones posteriores. En caso de IRR de Grado 3 o 4, se debe interrumpir el tratamiento con ramucirumab de forma inmediata y permanente (ver sección 4.4).
Hipertensión
Antes de cada administración de ramucirumab, se debe controlar y tratar la tensión arterial según indicación clínica correspondiente. En caso de hipertensión grave, el tratamiento con ramucirumab se debe interrumpir temporalmente hasta que se controle con medicación. En caso de hipertensión médicamente significativa que no pueda ser controlada de forma segura con antihipertensivos, el tratamiento con ramucirumab se debe interrumpir de forma permanente (ver sección 4.4).
Proteinuria
Durante el tratamiento con ramucirumab se debe controlar el desarrollo o empeoramiento de la proteinuria en el paciente. Si los niveles de proteína en orina son >2+ en una tira reactiva de orina, se debe recoger una muestra de orina de 24 horas. El tratamiento con ramucirumab debe interrumpirse temporalmente en caso de niveles de proteína en orina >2 g/24 horas. Una vez que los niveles de proteínas en orina se hayan restablecido a valores < 2 g/24 horas, se debe reiniciar el tratamiento con una dosis reducida (ver Tabla 3). Si reaparecen niveles de proteína en orina >2 g/24 horas, se recomienda una segunda reducción de la dosis (ver Tabla 3).
Si el nivel de proteinuria es > 3 g/24 horas o existe síndrome nefrótico, el tratamiento con ramucirumab se debe interrumpir de forma permanente.
Tabla 3: Reducciones de dosis de Ramucirumab debido a proteinuria
|
Dosis de ramucirumab inicial: |
Primera reducción de la dosis a: |
Segunda reducción de la dosis a: |
|
8 mg/kg |
6 mg/kg |
5 mg/kg |
|
10 mg/kg |
8 mg/kg |
6 mg/kg |
Cirugía programada o dificultad en la curación de heridas
Se debe interrumpir el tratamiento con ramucirumab temporalmente durante al menos 4 semanas previas a una cirugía programada. Si existen complicaciones en la curación de las heridas, el tratamiento con ramucirumab se debe interrumpir temporalmente hasta que la herida esté completamente curada (ver sección 4.4).
El tratamiento con ramucirumab se debe interrumpir permanentemente en los casos de:
Enfermedad tromboembólica arterial grave (ver sección 4.4).
Perforaciones gastrointestinales (ver sección 4.4).
Hemorragias graves: hemorragias Grado 3 o 4 según NCI CTCAE (ver sección 4.4).
Desarrollo espontáneo de fístulas (ver sección 4.4).
Ajuste de dosis de paclitaxel
Se deben aplicar reducciones de dosis de paclitaxel en función del grado de toxicidad del paciente. Para toxicidad hematológica Grado 4 según NCI CTCAE o toxicidad no hematológica relacionada con paclitaxel de Grado 3, se recomienda reducir la dosis de paclitaxel en 10 mg/m2 para todos los ciclos posteriores. Si la toxicidad persiste o es recurrente, se recomienda realizar una segunda reducción adicional de la dosis de 10 mg/m2.
Ajuste de dosis de FOLFIRI
En el caso de toxicidades específicas, pueden aplicarse reducciones de dosis de los componentes individuales de FOLFIRI. Las modificaciones de las dosis de cada componente de FOLFIRI se deben aplicar de forma independiente y se muestran en la Tabla 4. La Tabla 5 muestra más información sobre retrasos de la administración o reducciones de dosis de los componentes de FOLFIRI para el siguiente ciclo basándose en el grado máximo de las reacciones adversas específicas.
Tabla 4: Reducciones de dosis de FOLFIRI
|
Componente3 de FOLFIRI |
Nivel de dosis | |||
|
Dosis inicial |
-1 |
-2 |
-3 | |
|
Irinotecán |
180 mg/m2 |
150 mg/m2 |
120 mg/m2 |
100 mg/m2 |
|
5-FU en bolo |
400 mg/m2 |
200 mg/m2 |
0 mg/m2 |
0 mg/m2 |
|
5-FU en perfusión |
2.400 mg/m2 durante 46-48 horas |
2.000 mg/m2 durante 46-48 horas |
1.600 mg/m2 durante 46-48 horas |
1.200 mg/m2 durante 46-48 horas |
a 5-FU = 5-fluorouracilo.
Tabla 5: Modificaciones de la dosis de los componentes de FOLFIRI debido a RAMs (reacciones adversas a medicamentos) específicas
|
RAM |
Grado según NCI CTCAE |
Modificación de la dosis el día 1 del ciclo siguiente tras la RAM | |
|
Diarrea |
2 |
Si la diarrea se ha recuperado a Grado <1, reducir 5-FU 1 nivel de dosis. En el caso de diarreas recurrentes de Grado 2, reducir 5-FU e irinotecán.1 nivel de dosis. | |
|
3 |
Si la diarrea se ha recuperado a Grado <1, reducir 5-FU e irinotecán 1 nivel de dosis. | ||
|
4 |
Si la diarrea se ha recuperado a Grado <1, reducir 5-FU e irinotecán 2 niveles de dosis. Si la diarrea no se ha resuelto de Grado 4 a Grado <1, posponer la administración de 5-FU e irinotecán durante un máximo de 28* días hasta que se resuelva a Grado <1 | ||
|
Neutropenia o T rombocitopenia |
Se cumplen los criterios hematológicos de la Tabla 2 |
No se cumplen los criterios hematológicos de la Tabla 2 | |
|
2 |
No hay modificación de la dosis. |
Reducción 5-FU e irinotecán 1 nivel de dosis. | |
|
3 |
Reducción de 5-FU e irinotecán 1 nivel de dosis. |
Posponer la administración de 5-FU e irinotecán durante un máximo de 28 días hasta que se resuelva a Grado <1, después reducir 5-FU e irinotecán 1 nivel de dosis. | |
|
4 |
Reducción 5-FU e irinotecán 2 niveles de dosis. |
Retrasar la administración de 5-FU e irinotecán durante un máximo de 28* días hasta que se resuelva a Grado <1, después reducir 5-FU e irinotecán 2 niveles de dosis. | |
|
Estomatitis/Mucositis |
2 |
Si la estomatitis/mucositis se ha recuperado a Grado <1, reducir 5-FU 1 nivel de dosis. En el caso de estomatitis recurrente de Grado 2, reducir 5-FU 2 niveles de dosis. | |
|
3 |
Si la estomatitis/mucositis se ha recuperado a Grado <1, reducir 5-FU 1 nivel de dosis. Si la mucositis/estomatitis de Grado 3 no se resuelve a Grado <1, retrasar la administración de 5-FU durante un máximo de 28* días hasta resolución a Grado <1, después reducir 5-FU 2 niveles de dosis. | ||
|
4 |
Posponer la administración de 5-FU durante un máximo de 28* días hasta resolución a Grado <1, después reducir 5-FU 2 niveles de dosis. | ||
|
Neutropenia febril |
Se cumplen los criterios hematológicos de la Tabla 2 y la fiebre desaparece |
No se cumplen los criterios hematológicos de la Tabla 2 y la fiebre desaparece | |
|
Reducir 5-FU e irinotecán 2 niveles de dosis |
Retrasar la administración de 5-FU e irinotecán durante un máximo de 28* días hasta que se resuelva a Grado <1, después | ||
|
reducir 5-FU e irinotecán 2 niveles de dosis. Valorar el uso de factor estimulante de colonias antes del siguiente ciclo. |
*El periodo de 28 días comienza el día 1 del ciclo siguiente a la RAM
Ajuste de dosis de docetaxel
Las reducciones de dosis de docetaxel se pueden aplicar basándose en el grado de toxicidad que ha presentado el paciente. El tratamiento con docetaxel se debe posponer hasta que la toxicidad se haya resuelto, en aquellos pacientes que durante el tratamiento presenten neutropenia febril, neutrófilos <500 células/mm3 durante más de 1 semana, reacciones cutáneas graves o acumulativas u otras toxicidades Grado 3 o 4 no hematológicas. Se recomienda reducir la dosis de docetaxel en 10 mg/m2 para el resto de ciclos. Se recomienda una segunda reducción en 15 mg/m2 si las toxicidades persisten o son recurrentes. En este caso, los pacientes de Asia Oriental con una dosis de inicio de 60 mg/m2 deben interrumpir el tratamiento con docetaxel (ver Posología).
Poblaciones especiales
Pacientes de edad avanzada
Los ensayos pivotales no han mostrado suficiente evidencia de que los pacientes de 65 años o mayores tengan un mayor riesgo de sufrir reacciones adversas comparando con los pacientes menores de 65 años. No se recomienda reducción de la dosis (ver las secciones 4.4 y 5.1).
Pacientes con insuficiencia renal
No se han llevado a cabo estudios específicos con Cyramza en pacientes con insuficiencia renal. Los datos clínicos sugieren que no se necesita un ajuste de dosis en pacientes con insuficiencia renal leve, moderada o insuficiencia renal grave (ver las secciones 4.4 y 5.2). No se recomienda reducir la dosis.
Pacientes con insuficiencia hepática
No se han llevado a cabo estudios específicos con Cyramza en pacientes con insuficiencia hepática. Los datos clínicos indican que no son necesarios ajustes de dosis en pacientes con insuficiencia hepática leve o moderada. No existen datos sobre la administración de ramucirumab en pacientes con insuficiencia hepática grave (ver las secciones 4.4 y 5.2). No se recomienda reducir la dosis.
Población pediátrica
No se ha establecido la seguridad y eficacia de Cyramza en niños y adolescentes (<18 años). No se dispone de datos.
No existe una recomendación de uso específica para ramucirumab en la población pediátrica para la indicación de cáncer gástrico avanzado o adenocarcinoma gastroesofágico, adenocarcinoma de colon y recto y carcinoma de pulmón.
Forma de administración
Tras dilución, Cyramza se administra en perfusión intravenosa durante aproximadamente 60 minutos. No se debe administrar en bolo intravenoso o inyección rápida. Para alcanzar la duración de perfusión requerida de 60 minutos aproximadamente, la velocidad de perfusión máxima no debe exceder los 25 mg/minuto; en caso contrario la duración de la perfusión debe ser incrementada. Durante la perfusión, los pacientes deben ser monitorizados ante la posible aparición de signos de reacciones relacionadas con la perfusión (ver sección 4.4), así como se debe asegurar la disponibilidad de un equipo de reanimación adecuado.
Para consultar las instrucciones de dilución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
Para pacientes con CPNM, ramucirumab está contraindicado donde haya cavitación del tumor o afectación tumoral de los grandes vasos sanguíneos (ver sección 4.4).
4.4 Advertencias y precauciones especiales de empleo
Enfermedad tromboembólica arterial
En los ensayos clínicos se han notificado casos graves, ocasionalmente mortales, de Enfermedad Tromboembólica Arterial (Arterial thromboembolic events, ATEs) incluyendo infarto de miocardio, parada cardiaca, accidente cerebrovascular e isquemia cerebral. Ramucirumab se debe interrumpir de forma permanente en aquellos pacientes que presenten ATE grave (ver sección 4.2).
Perforaciones gastrointestinales
Ramucirumab es un tratamiento antiangiogénico y puede aumentar el riesgo de perforaciones gastrointestinales. Se han notificado casos de perforación gastrointestinal en pacientes tratados con ramucirumab. El tratamiento con ramucirumab se debe interrumpir de forma permanente en pacientes que presenten perforaciones gastrointestinales (ver sección 4.2).
Hemorragia grave
Ramucirumab es un tratamiento antiangiogénico y puede aumentar el riesgo de hemorragia grave. El tratamiento con ramucirumab se debe interrumpir de forma permanente en aquellos pacientes que presenten hemorragia de Grado 3 o 4 (ver sección 4.2). En aquellos pacientes con enfermedades que predispongan al sangrado y en aquellos tratados con anticoagulantes u otros medicamentos que incrementan el riesgo hemorrágico se deben monitorizar los parámetros de hemograma y coagulación.
En pacientes con cáncer gástrico tratados con ramucirumab en combinación con paclitaxel y en pacientes con mCRC tratados con ramucirumab en combinación con FOLFIRI, se han notificado hemorragias gastrointestinales graves incluyendo casos mortales.
Hemorragia pulmonar en CPNM
Los pacientes con histología escamosa tienen un riesgo mayor de desarrollar hemorragia pulmonar grave, sin embargo, en el estudio REVE L en pacientes tratados con ramucirumab con histología escamosa, no se observó un exceso de hemorragia pulmonar Grado 5. En los ensayos clínicos se excluyeron aquellos pacientes con CPNM con hemorragia pulmonar reciente (>2,5 ml o sangre roja brillante) así como pacientes con evidencia basal de cavitación tumoral, independientemente de la histología, o aquellos con evidencia de invasión tumoral o confinamiento de grandes vasos sanguíneos (ver sección 4.3). Los pacientes que recibieron cualquier tipo de tratamiento anticoagulante y/o tratamiento crónico con fármacos antiinflamatorios no esteroideos o agentes antiplaquetarios fueron excluidos del ensayo clínico REVEL en CPNM. Se permitió el uso de Aspirina a dosis de hasta 325 mg/día (ver sección 5.1).
Reacciones relacionadas con la perfusión
Se han notificado reacciones relacionadas con la perfusión en ensayos clínicos con ramucirumab. La mayoría de los casos ocurrieron durante o tras una primera o segunda perfusión de ramucirumab. Durante la perfusión, los pacientes deben ser monitorizados ante la posible aparición de signos o síntomas de hipersensibilidad. Estos incluyeron temblores, espasmos/dolor de espalda, dolor y/u opresión torácica, escalofríos, rubor, disnea, sibilancias, hipoxia y parestesias. En casos graves, estos incluyeron broncoespasmo, taquicardia supraventricular e hipotensión. El tratamiento con ramucirumab se debe interrumpir de forma inmediata y permanente en pacientes que presenten IRR de Grado 3 o 4 (ver sección 4.2).
Hipertensión
Se notificó un aumento en la incidencia de casos de hipertensión grave en pacientes que recibieron ramucirumab comparado con placebo. En la mayoría de los casos, la hipertensión se manejó utilizando un tratamiento antihipertensivo estándar. Los pacientes con hipertensión no controlada fueron excluidos de los ensayos: no se debe comenzar el tratamiento con ramucirumab en estos pacientes hasta que la hipertensión preexistente se controle. La tensión arterial se debe monitorizar en los pacientes tratados con ramucirumab. En caso de hipertensión grave, el tratamiento con ramucirumab se debe interrumpir temporalmente hasta que se controle con medicación. En caso de hipertensión médicamente significativa que no pueda ser controlada con antihipertensivos, el tratamiento con ramucirumab se debe interrumpir de forma permanente (ver sección 4.2).
Dificultad en la curación de heridas
El impacto de ramucirumab no se ha evaluado en pacientes con heridas no curadas o graves. En un ensayo llevado a cabo en animales, ramucirumab no dificultó la curación de heridas. Sin embargo, dado que ramucirumab es un tratamiento antiangiogénico y puede potencialmente afectar de forma negativa a la curación de heridas, el tratamiento con ramucirumab se debe interrumpir durante al menos las 4 semanas previas a una cirugía programada. La decisión de reanudar el tratamiento con ramucirumab tras la intervención quirúrgica debe basarse en el juicio clínico para lograr una adecuada curación de la herida.
Si durante el tratamiento un paciente desarrolla complicaciones en la curación de heridas, ramucirumab se debe interrumpir hasta que la herida esté completamente curada (ver sección 4.2).
Insuficiencia hepática
Ramucirumab se debe utilizar con precaución en pacientes con cirrosis hepática grave (Child-Pugh B o C), cirrosis con encefalopatía hepática, ascitis debida a la cirrosis clínicamente significativa o síndrome hepatorrenal. En estos pacientes, ramucirumab se debe utilizar únicamente si se considera que el beneficio potencial del tratamiento, supera el riesgo potencial de progresión a fallo hepático.
Fístulas
Los pacientes tratados con Cyramza pueden presentar un riesgo mayor de desarrollar fístulas. Se debe interrumpir el tratamiento con ramucirumab en pacientes que desarrollan una fístula (ver sección 4.2).
Proteinuria
En pacientes que recibieron ramucirumab se notificó un aumento en la incidencia de proteinuria comparado con los que recibieron placebo. Durante el tratamiento con ramucirumab, se debe controlar el desarrollo o empeoramiento de la proteinuria en el paciente. Si los niveles de proteína en orina son >2+ en una tira reactiva de orina, se debe recoger una muestra de orina de 24 horas. El tratamiento con ramucirumab se debe interrumpir temporalmente en caso de niveles de proteína en orina >2 g/24 horas. Una vez que los niveles de proteínas en orina se hayan restablecido a valores < 2 g/24 horas, se debe reiniciar el tratamiento con una dosis reducida. Si reaparecen niveles de proteína en orina >2 g/24 horas, se recomienda una segunda reducción de la dosis. Si el nivel de proteinuria es > 3 g/24 horas o existe síndrome nefrótico, el tratamiento con ramucirumab se debe interrumpir de forma permanente (ver sección 4.2).
Estomatitis
Se notificó un aumento de la incidencia de estomatitis en pacientes que recibieron ramucirumab en combinación con quimioterapia comparado con pacientes tratados con placebo más quimioterapia. Si la estomatitis aparece, el tratamiento sintomático se debe instaurar lo antes posible.
Insuficiencia renal
Los datos de seguridad disponibles para pacientes con insuficiencia renal grave (aclaramiento de creatinina 15 a 29 ml/min) tratados con ramucirumab son limitados (ver las secciones 4.2 y 5.2).
Dietas restrictivas en sodio
Cada vial de 10 ml contiene aproximadamente 17 mg de sodio y cada vial de 50 ml contiene aproximadamente 85 mg de sodio. Esto debe tenerse en cuenta en pacientes con dietas restrictivas en sodio.
Pacientes de edad avanzada con CPNM
Se ha obsevado una tendencia a menor eficacia conforme aumenta la edad del paciente que recibe ramucirumab más docetaxel para el tratamiento de CPNM avanzado con progresión de la enfermedad tras quimioterapia basada en platino (ver sección 5.1).
Antes del comienzo de tratamiento en pacientes de edad avanzada, se debe evaluar cuidadosamente las comorbilidades, el estado funcional (performance status, PSpor sus siglas en inglés) y la potencial tolerabilidad a la quimioterapia (ver las secciones 4.2 y 5.1).
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han observado interacciones farmacológicas entre ramucirumab y paclitaxel. La farmacocinética de paclitaxel no se vio afectada cuando se administró de forma conjunta con ramucirumab y la farmacocinética de ramucirumab no se vio afectada cuando se administró de forma conjunta con paclitaxel. La farmacocinética de irinotecán y la de su metabolito activo, SN-38, no se vieron afectadas cuando se administró junto con ramucirumab. La farmacocinética de docetaxel no se vio afectada cuando se administró junto con ramucirumab.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil/Anticoncepción en mujeres
Se debe aconsejar a las mujeres en edad fértil que eviten el embarazo durante el tratamiento con Cyramza y deben ser informadas del daño potencial al embarazo y al feto. Las mujeres en edad fértil deben usar métodos anticonceptivos eficaces durante y hasta los 3 meses posteriores a la última dosis del tratamiento con ramucirumab.
Embarazo
No existen datos del uso de ramucirumab en mujeres embarazadas. Los estudios en animales son insuficientes con respecto a la toxicidad reproductiva (ver sección 5.3). La angiogénesis es crítica durante el mantenimiento del embarazo y el desarrollo fetal, por lo que la inhibición de la angiogénesis tras la administración de ramucirumab puede provocar efectos adversos sobre el embarazo, incluyendo al feto. Solo se debe utilizar Cyramza si el beneficio potencial a la madre justifica el riesgo potencial durante el embarazo. Si la paciente llega a quedarse embarazada durante el tratamiento con ramucirumab, se le debe informar del riesgo potencial de mantener el embarazo y del riesgo para el feto. No se recomienda Cyramza durante el embarazo ni en mujeres en edad fértil que no usan anticonceptivos.
Lactancia
Se desconoce si ramucirumab se excreta en la leche materna. Se espera que la excreción en la leche y la absorción oral sean bajas. Dado que no se puede excluir el riesgo en lactantes, la lactancia se debe interrumpir durante el tratamiento con Cyramza y durante al menos 3 meses tras la última dosis.
Fertilidad
No existen datos del efecto de ramucirumab sobre la fertilidad humana. Según estudios en animales, se considera probable que la fertilidad en mujeres se pueda ver comprometida durante el tratamiento con ramucirumab (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Se desconoce la influencia de Cyramza sobre la capacidad para conducir y utilizar máquinas. Si el paciente sufre síntomas que afectan su capacidad de reacción o su habilidad para concentrarse, se recomienda que no conduzca o utilice máquinas hasta que los efectos disminuyan.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Las reacciones adversas más graves asociadas al tratamiento con ramucirumab (como agente único o en combinación con quimioterapia citotóxica) fueron:
Perforación gastrointestinal (ver sección 4.4)
Hemorragia gastrointestinal grave (ver sección 4.4)
Enfermedad tromboembólica arterial (ver sección 4.4)
Las reacciones adversas más frecuentes observadas en los pacientes tratados con ramucirumab son: neutropenia, fatiga/astenia, leucopenia, epistaxis, diarrea y estomatitis.
Tabla de reacciones adversas
Las reacciones adversas a medicamentos (RAMs) notificadas en pacientes con cáncer gástrico avanzado, mCRC o CPNM se incluyen a continuación según el sistema de clasificación de órganos del sistema MedDRA según su frecuencia y grado de intensidad. La siguiente convención es la utilizada para clasificar su frecuencia:
Muy frecuentes (>1/10)
Frecuentes (>1/100 a <1/10)
Poco frecuentes (>1/1.000 a <1/100)
Raras (>1/10.000 a <1/1.000)
Muy raras (<1/10.000)
Dentro de cada grupo de frecuencia, las RAMs se presentan en orden decreciente de gravedad.
Cáncer gástrico
Ramucirumab en combinación con _paclitaxel
La siguiente tabla muestra la frecuencia y gravedad de las RAMs basándose en los resultados obtenidos del estudio RAINBOW, ensayo clínico en fase 3 en pacientes adultos con cáncer gástrico avanzado aleatorizados al tratamiento con ramucirumab en combinación con paclitaxel o placebo más paclitaxel.
Tabla 6: RAMs notificadas en > 5 % de los pacientes tratados con ramucirumab en el estudio RAINBOW
|
Sistema de clasificación de órganos |
Frecuencia |
RAM |
Cyramza más paclitaxel (N=327) |
Placebo más paclitaxel (N=329) | ||
|
Todos los grados de toxicidad (%) |
Toxicidad Grado >3 (%) |
Todos los grados de toxicidad (%) |
Toxicidad Grado >3 (%) | |||
|
Trastornos de la sangre |
Muy frecuentes |
Neutropenia |
54,4 |
40,7 |
31,0 |
18,8 |
|
y del sistema linfático |
Muy frecuentes |
Leucopenia |
33,9 |
17,4 |
21,0 |
6,7 |
|
Muy frecuentes |
T rombocitopenia |
13,1 |
1,5 |
6,1 |
1,8 | |
|
Trastornos del metabolismo y de la nutrición |
Muy frecuentes |
Hipoalbuminemia |
11,0 |
1,2 |
4,9 |
0,9 |
|
Trastornos vasculares |
Muy frecuentes |
Hipertensión3 |
25,1 |
14,7 |
5,8 |
2,7 |
|
Trastornos respiratorios, torácicos y mediastínicos |
Muy frecuentes |
Epistaxis |
30,6 |
0,0 |
7,0 |
0,0 |
|
Trastornos gastrointestinales |
Muy frecuentes |
Casos de hemorragia gastrointestinal13 |
10,1 |
3,7 |
6,1 |
1,5 |
|
Muy frecuentes |
Estomatitis |
19,6 |
0,6 |
7,3 |
0,6 | |
|
Muy frecuentes |
Diarrea |
32,4 |
3,7 |
23,1 |
1,5 | |
|
Trastornos renales y urinarios |
Muy frecuentes |
Proteinuria |
16,8 |
1,2 |
6,1 |
0,0 |
|
Trastornos generales y alteraciones en el lugar de la administración |
Muy frecuentes |
Fatiga/Astenia |
56,9 |
11,9 |
43,8 |
5,5 |
|
Muy frecuentes |
Edema periférico |
25,1 |
1,5 |
13,7 |
0,6 |
a Incluye miocardiopatía hipertensiva.
b Los términos según MedDRA son hemorragia anal, diarrea hemorrágica, hemorragia gástrica, hemorragia gastrointestinal, hematemesis, rectorragia, hemorragia hemorroidal, síndrome de Mallory-Weiss, melenas, hemorragia esofágica, hemorragia rectal y hemorragia digestiva alta.
Las RAMs clínicamente relevantes notificadas en > 1% y < 5% de los pacientes tratados con ramucirumab más paclitaxel en el estudio RAINBOW fueron perforación gastrointestinal (1,2% ramucirumab más paclitaxel frente al 0,3 % placebo más paclitaxel) y sepsis (3,1% ramucirumab más paclitaxel frente a 1,8% de placebo más paclitaxel).
Ramucirumab como asente único
La siguiente tabla muestra la frecuencia y gravedad de las RAMs basándose en los resultados obtenidos en el estudio REGARD, un ensayo clínico en fase 3 en pacientes adultos con cáncer gástrico avanzado aleatorizados al tratamiento con ramucirumab en monoterapia más el mejor tratamiento de soporte (Best Supportive Care, BSC) o placebo más BSC.
Tabla7: RAMs notificadas en > 5 % de los pacientes tratados con ramucirumab en el ensayo REGARD
|
Sistema de clasificación de órganos |
Frecuencia |
RAMa,b |
Cyramza (N=236) |
Placebo (N=115) | ||
|
Todos los gradosc de toxicidad (%) |
Toxicidad Grado 3-4 (%) |
Todos los grados de toxicidad (%) |
Toxicidad Grado 34 (%) | |||
|
Trastornos del metabolismo y de la nutrición |
Frecuentes |
Hipopotasemiad |
5,9 |
2,1 |
5,2 |
0,9 |
|
Frecuentes |
Hiponatremia |
5,5 |
3,4 |
1,7 |
0,9 | |
|
Trastornos del sistema nervioso |
Frecuentes |
Cefalea |
9,3 |
0 |
3,5 |
0 |
|
Trastornos vasculares |
Muy frecuentes |
Hipertensióne |
16,1 |
7,6 |
7,8 |
2,6 |
|
Trastornos gastrointestinales |
Muy frecuentes |
Dolor abdominalf |
28,8 |
5,9 |
27,8 |
2,6 |
|
Muy frecuentes |
Diarrea |
14,4 |
0,8 |
8,7 |
1,7 | |
|
a Término según MedD |
RA (Versión 15.0) | |||||
b No se produjeron RAMs de Grado 5 con Cyramza. Se produjo una RA Grado 4 de hipopotasemia y otra de hiponatremia.
c Según los Criterios NCI CTCAE (Versión 4.0) para cada Grado de toxicidad.
d Los términos según MedDRA son: descenso de los niveles de potasio en sangre e
hipopotasemia.
e Los términos según MedDRA son: aumento de la tensión arterial e hipertensión.
f Los términos según MedDRA son: dolor abdominal, dolor en abdomen inferior, dolor en
abdomen superior y dolor hepático.
Las RAMs clínicamente relevantes notificadas en > 1% y < 5% de los pacientes tratados con ramucirumab en el estudio REGARD fueron: neutropenia, enfermedad tromboembólica arterial (ver secciones 4.2 y 4.4), obstrucción intestinal, epistaxis y exantema.
Las reacciones clínicamente relevantes (incluyendo las de Grado > 3) asociadas a terapia antiangiogénica y observadas en pacientes tratados con ramucirumab durante los ensayos clínicos fueron: perforación gastrointestinal, reacciones relacionadas con la perfusión y proteinuria (ver secciones 4.2 y 4.4).
Cáncer colorrectal
Ramucirumab en combinación con FOLFIRI
La siguiente tabla muestra la frecuencia y gravedad de las RAMs basadas en los resultados de RAISE, un estudio en fase 3 en pacientes adultos con mCRC aleatorizados al tratamiento con ramucirumab más FOLFIRI o placebo más FOLFIRI.
Tabla 8: RAMs notificadas en >5% de pacientes tratados con ramucirumab en el ensayo RAISE
|
Sistema de clasificación de órganos |
Frecuencia |
RAM |
Cyr n FOLFIF |
amza iás J (N=529) |
Placebo más FOLFIRI (N=528) | |
|
Todos los grados de toxicidad (%) |
Toxicidad Grado >3 (%) |
Todos los grados de toxicidad (%) |
Toxicidad Grado >3 (%) | |||
|
Trastornos de la sangre y del sistema linfático |
Muy frecuentes |
Neutropenia |
58,8 |
38,4 |
45,6 |
23,3 |
|
Muy frecuentes |
T rombocitopenia |
28,4 |
3,0 |
13,6 |
0,8 | |
|
Trastornos del metabolismo y de la nutrición |
Frecuentes |
Hipoalbuminemia |
5,9 |
1,1 |
1,9 |
0,0 |
|
Trastornos vasculares |
Muy frecuentes |
Hipertensión |
26,1 |
11,2 |
8,5 |
2,8 |
|
Trastornos respiratorios, torácicos y mediastínicos |
Muy frecuentes |
Epistaxis |
33,5 |
0,0 |
15,0 |
0,0 |
|
Trastornos gastrointestinales |
Muy frecuentes |
Casos de hemorragia gastrointestinal |
12,3 |
1,9 |
6,8 |
1,1 |
|
Muy frecuentes |
Estomatitis |
30,8 |
3,8 |
20,8 |
2,3 | |
|
Trastornos renales y urinarios |
Muy frecuentes |
Proteinuriaa |
17,0 |
3,0 |
4,5 |
0,2 |
|
Trastornos de la piel y del tejido subcutáneo |
Muy frecuentes |
Síndrome de eritrodisestesia palmo-plantar |
12,9 |
1,1 |
5,5 |
0,4 |
|
Trastornos generales y alteraciones en el lugar de la administración |
Muy frecuentes |
Edema periférico |
20,4 |
0,2 |
9,1 |
0,0 |
Incluye casos de síndrome nefrótico.
Las RAMs clínicamente relevantes notificadas en >1% y <5% de los pacientes tratados con ramucirumab más FOLFIRI en el estudio RAISE fueron: perforación gastrointestinal (1,7 % ramucirumab más FOLFIRI frente a 0,6 % para placebo más FOLFIRI).
En el estudio RAISE, en pacientes con mCRC tratados con ramucirumab más FOLFIRI, la RAM más frecuente (>1%) que llevó a la interrupción de ramucirumab fue proteinuria (1,5%). Las RAMs más frecuentes (>1%) que llevaron a la interrupción de uno o más componentes de FOLFIRI fueron: neutropenia (12,5%), trombocitopenia (4,2%), diarrea (2,3%) y estomatitis (2,3%). El componente de FOLFIRI interrumpido de forma más frecuente fue 5-FU en bolo.
CPNM
Ramucirumab en combinación con docetaxel
La siguiente tabla muestra la frecuencia y gravedad de las RAMs basándose en los resultados de REVEL, un estudio fase 3 en pacientes adultos con CPNM aleatorizados a tratamiento con ramucirumab en combinación con docetaxel o placebo más docetaxel.
Tabla 9: RAMs notificadas en >5% de pacientes tratados con ramucirumab en el ensayo REVEL
|
Sistema de clasificación de órganos |
Frecuencia |
RAM |
Cyramza más docetaxel (N=627) |
Placebo más docetaxel (N=618) | ||
|
Todos los grados de toxicidad (%) |
Toxicidad Grado 3-4 (%) |
Todos los grados de toxicidad (%) |
Toxicidad Grado 3-4 (%) | |||
|
Trastornos de la sangre |
Muy frecuentes |
Neutropenia febril |
15,9 |
15,9 |
10,0 |
10,0 |
|
y del sistema linfático |
Muy frecuentes |
Neutropenia |
55,0 |
48,8 |
46,0 |
39,8 |
|
Muy frecuentes |
T rombocitopenia |
13,4 |
2,9 |
5,2 |
0,6 | |
|
Trastornos vasculares |
Muy frecuentes |
Hipertensión |
10,8 |
5,6 |
4,9 |
2,1 |
|
Trastornos respiratorios, torácicos y mediastínicos |
Muy frecuentes |
Epistaxis |
18,5 |
0,3 |
6,5 |
0,2 |
|
Trastornos gastrointestinales |
Muy frecuentes |
Estomatitis |
23,3 |
4,3 |
12,9 |
1,6 |
|
Trastornos generales y |
Muy frecuentes |
Fatiga/Astenia |
54,7 |
14,0 |
50,0 |
10,5 |
|
alteraciones en el lugar |
Muy frecuentes |
Inflamación de la mucosa |
16,1 |
2,9 |
7,0 |
0,5 |
|
de la administración |
Muy frecuentes |
Edema periférico |
16,3 |
0 |
8,6 |
0,3 |
Las RAMs clínicamente relevantes notificadas en >1% y <5% de los pacientes tratados con ramucirumab más docetaxel en el estudio REVEL, fueron hiponatremia (4,8 % ramucirumab más docetaxel frente a 2,4% para placebo más docetaxel), proteinuria (3,3 % ramucirumab más docetaxel frente a 0,8 % placebo más docetaxel) y perforación gastrointestinal (1 % ramucirumab más docetaxel frente a 0,3 % placebo más docetaxel).
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
No existen datos de sobredosis en humanos. En un estudio fase 1, se administró Cyramza hasta 10 mg/kg cada dos semanas sin alcanzar la dosis máxima tolerada. En caso de sobredosis, se debe utilizar un tratamiento sintomático.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agentes antineoplásicos, anticuerpos monoclonales. Código ATC: L01XC21. Mecanismo de acción
El Receptor tipo 2 del factor de crecimiento del endotelio vascular (Vascular Endothelial Growth Factor, VEGF) es el mediador clave de la angiogénesis inducida por el VEGF. Ramucirumab es un anticuerpo humano dirigido a receptores que se une específicamente al Receptor 2 del VEGF bloqueando la unión de los ligandos VEGF-A, VEGF-C y VEGF-D. Como resultado, ramucirumab inhibe la activación ligando-dependiente del Receptor 2 de VEGF y sus componentes posteriores de la cascada de señalización, incluyendo las proteínas kinasas activadas por mitógeno p44/p42, la proliferación ligando inducida y la migración de las células endoteliales humanas.
Eficacia clínica y seguridad
Cáncer gástrico:
RAINBOW
RAINBOW es un ensayo clínico global, aleatorizado, doble ciego de Cyramza más paclitaxel frente a placebo más paclitaxel, llevado a cabo en 665 pacientes con cáncer localmente recurrente e irresecable o cáncer gástrico metastásico (incluyendo adenocarcinoma de GEJ) tras quimioterapia con platino y fluoropirimidina, con o sin antraciclinas. La variable primaria fue supervivencia global (overall survival,
OS) y las variables secundarias incluyeron supervivencia libre de progresión (progression-free survival,
PFS) y la tasa de respuesta global (overall response rate, ORR). Se requería que los pacientes hubiesen presentado progresión de la enfermedad durante el tratamiento en primera línea o dentro de los 4 meses tras la última dosis del mismoy tuvieran ECOG PS 0-1. Los pacientes fueron asignados de forma aleatoria en un ratio 1:1 para recibir Cyramza más paclitaxel (n= 330) o placebo más paclitaxel (n= 335). La aleatorización se estratificó por región geográfica, tiempo a la progresión desde el comienzo del tratamiento en primera línea (< 6 meses frente >6 meses) y cuantificación de la enfermedad. Tanto Cyramza 8mg/kg como placebo, fueron administrados por perfusión intravenosa cada 2 semanas (los días 1 y 15) de un ciclo de 28 días. Paclitaxel 80 mg/ m2 se administró por perfusión intravenosa los días 1, 8 y 15 de cada ciclo de 28 días.
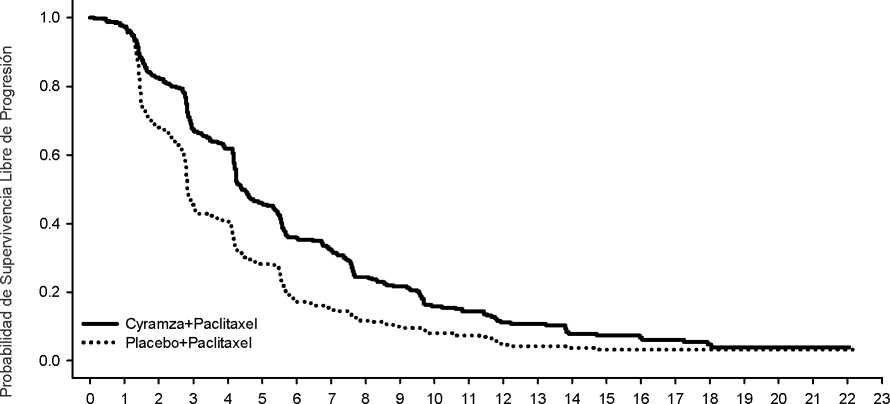
La mayoría (75%) de los pacientes aleatorizados en el estudio, recibieron previamente una terapia combinada de platino y fluoropirimidina sin antraciclina. El resto (25%), recibieron previamente una terapia combinada de platino y fluoropirimidina con antraciclina. Dos tercios de los pacientes mostraron progresión de la enfermedad, cuando aún se encontraban en tratamiento de primera línea (66,8%). Las características basales demográficas y de la enfermedad estaban globalmente balanceadas entre los brazos del estudio: la mediana de edad fue 61 años; 71% de pacientes eran hombres; 61% eran caucásicos, 35% asiáticos; ECOG PS fue 0 para el 39% de los pacientes, 1 para el 61%; 81% de los pacientes tenía enfermedad cuantificable y el 79% presentaba cáncer gástrico, 21% presentaba adenocarcinoma de GEJ. La mayoría de los pacientes (76%) había presentado progresión de la enfermedad durante los 6 meses desde el comienzo del tratamiento en primera línea. Para los pacientes tratados con Cyramza más paclitaxel, la mediana de la duración del tratamiento fue de 19 semanas y para pacientes tratados con placebo más paclitaxel, la mediana de duración del tratamiento fue de 12 semanas. La mediana de la intensidad de dosis relativa de Cyramza fue 98,6% y de placebo fue 99,6%. La mediana de la intensidad de dosis relativa de paclitaxel fue 87,7% en el brazo de Cyramza más paclitaxel y 93,2% en el brazo de placebo más paclitaxel. Un porcentaje parecido de pacientes interrumpió el tratamiento debido a reacciones adversas: 12% de los pacientes tratados con Cyramza más paclitaxel comparado con el 11% de pacientes tratados con placebo más paclitaxel. En el 47,9% de los pacientes que recibieron Cyramza más paclitaxel se administró un tratamiento anticanceroso sistémico tras la interrupción del tratamiento, frente al 46,0% de los pacientes que recibieron placebo más paclitaxel.
La supervivencia global fue mejorada de forma estadísticamente significativa en pacientes que recibieron Cyramza más paclitaxel comparado con aquellos que recibieron placebo más paclitaxel (Hazard Ratio, HR 0,807; IC 95%: 0,678 a 0,962; p=0,0169). Hubo un aumento de la mediana de supervivencia de 2,3 meses a favor del brazo de tratamiento de Cyramza más paclitaxel: 9,63 meses en el brazo de Cyramza más paclitaxel y 7,36 meses en el brazo de placebo más paclitaxel. La supervivencia libre de progresión mejoró de forma estadísticamente significativa para los pacientes que recibieron Cyramza más paclitaxel comparada con aquellos que recibieron placebo más paclitaxel (HR 0,635; IC 95% 0,536 a 0,752; p<0,0001). Hubo un aumento en la mediana de PFS de 1,5 meses a favor del brazo de tratamiento de Cyramza: 4,4 meses en el brazo de Cyramza más paclitaxel y 2,9 meses en el brazo de placebo más paclitaxel. La tasa de respuesta objetiva [ORR (respuesta completa (RC) + respuesta parcial (RP))] mejoró de forma significativa en pacientes que recibieron Cyramza más paclitaxel comparado con aquellos que recibieron placebo más paclitaxel (Odds ratio 2,140; IC 95%: 1,499 a 3,160; p=0,0001). El ORR en el brazo de Cyramza más paclitaxel fue de 27,9% y 16,1% en el brazo de placebo más paclitaxel. En subgrupos predefinidos según edad, sexo, raza se observaron mejoras de forma consistente en OS y PFS así como en la mayoría de otros subgrupos predefinidos. Los datos de eficacia se muestran en la Tabla 10.
Tabla 10: Resumen de los datos de eficacia- población por Intención de Tratar (ITT)
|
Cyramza más paclitaxel N=330 |
Placebo más paclitaxel N=335 | |
|
Supervivencia global, meses | ||
|
Mediana (IC 95%) |
9,6 (8,5, 10,8) |
7,4 (6,3, 8,4) |
|
Hazard ratio (IC 95%) |
0,807 (0,678, 0,962) | |
|
Valor de p estratificado (Log-rank) |
0,0169 | |
|
Supervivencia Libre de Progresión, meses | ||
|
Mediana (IC 95%) |
4,4 (4,2, 5,3) |
2,9 (2,8, 3,0) |
|
Hazard ratio (IC 95%) |
0,635 (0,536, 0,752) | |
|
Valor de p estratificado (Log-rank) |
<0,0001 | |
|
Tasa de respuesta objetiva (RC +RP) | ||
|
Valor de la tasa en porcentaje (IC 95%) |
27,9 (23,3, 33,0) |
16,1 (12,6, 20,4) |
|
Odd ratio |
2,140 (1,449, 3,160) | |
|
Valor de p estratificado (CMH) |
0,0001 | |
Abreviaturas: IC= intervalo de confianza, RC= respuesta completa, RP= respuesta parcial, CMH= Cochran-Mantel-Haenszel
Figura 1: Curvas de Kaplan-Meier para supervivencia global de Cyramza más paclitaxel frente a placebo más paclitaxel en RAINBOW
1.0 -
0.8 -
0.6 -
0.4 -
0.2 -

9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28
Tiempo Desde Aleatorización (Meses)
o
(O
O
Q_
0.0
|
0 1 |
2 : |
! 4 : |
5 6 7 |
8 | |
|
Número en Riesgo | |||||
|
Cyramza+Paclitaxel |
330 |
308 |
267 |
228 |
185 |
|
Placebo+Paclitaxel |
335 |
294 |
241 |
180 |
143 |
148
109
116
81
78
64
60
47
41
30
24
22
13
13
Tiempo Desde Aleatorización (Meses)
Número en Riesgo
Cyramza+Pacl itaxel 330 259 188 104 70 43 28 15 11 7 3 1
Placebo+Paclitaxel 335 214 124 50 34 21 12 8 5 3 3 3
REGARD
REGARD es un ensayo clínico multinacional, aleatorizado, doble ciego de Cyramza más BSC frente a placebo más BSC, llevado a cabo en 355 pacientes con cáncer localmente recurrente e irresecable o cáncer gástrico metastásico (incluyendo adenocarcinoma de GEJ) tras quimioterapia con platino o fluoropirimidina. La variable primaria fue OS y las variables secundarias incluyeron PFS. Se requería que los pacientes hubiesen presentado progresión de la enfermedad durante o dentro de los 4 meses tras la última dosis del tratamiento, en primera línea para la enfermedad metastásica, o durante o dentro de los 6 meses tras la última dosis de la terapia adyuvante y tuvieran ECOG PS 0-1. Para ser incluidos en el estudio, se requería que los pacientes presentaran niveles de bilirrubina total < 1,5mg/dl y AST y ALT < 3 veces ULN, o < 5 veces ULN si existía metástasis hepática.
Los pacientes fueron asignados de forma aleatoria en un ratio 2:1 para recibir una perfusión intravenosa de Cyramza 8 mg/kg (n= 238) o placebo (n= 117) cada 2 semanas. La aleatorización se estratificó por pérdida de peso durante los 3 meses anteriores (> 10% frente a <10%), región geográfica y localización del tumor primario (gástrico frente GEJ).
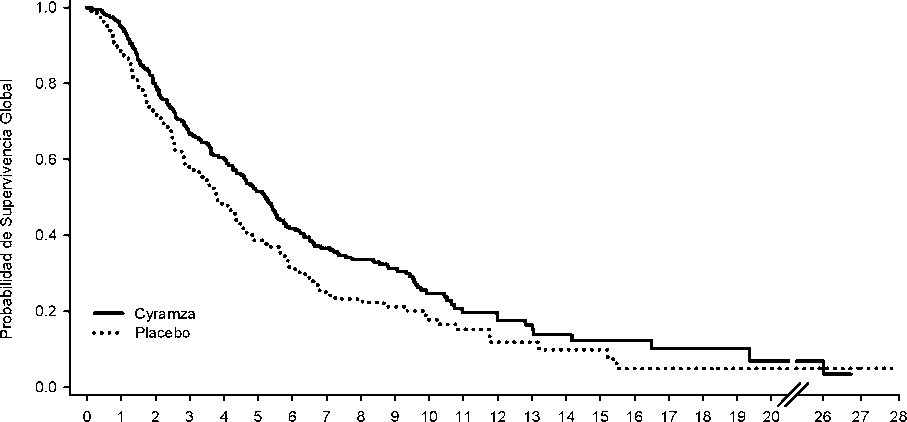
Las características basales demográficas y de la enfermedad estaban balanceadas. ECOG PS fue 1 para el 72% de los pacientes. No se reclutaron en el estudio REGARD pacientes con cirrosis hepática Child-Pugh B o C. El 11% de los pacientes tratados con Cyramza y el 6% de los pacientes tratados con placebo interrumpieron el tratamiento debido a las reacciones adversas. La supervivencia global mejoró de forma estadísticamente significativa en los pacientes que recibieron Cyramza en comparación con aquéllos que recibieron placebo (HR 0,776; IC 95%: 0,603 a 0,998; p= 0,0473), correspondiendo a una reducción del riesgo de muerte del 22% y a un aumento de la mediana de la supervivencia de 5,2 meses para Cyramza y 3,8 meses para placebo. La supervivencia libre de progresión mejoró de forma estadísticamente significativa en pacientes que recibieron Cyramza comparado con aquéllos que recibieron placebo (HR 0,483; IC: 95%: 0,376 a 0,620; p < 0,0001), correspondiendo a una reducción del riesgo de progresión o muerte del 52% y a un aumento en la mediana de PFS de 2,1 meses para Cyramza y de 1,3 meses para placebo. En la Tabla 11 se muestran los resultados de eficacia.
Tabla 11: Resumen de los datos de eficacia - población ITT
|
Cyramza N=238 |
Placebo N=117 | |
|
Supervivencia Global, meses | ||
|
Mediana (IC 95%) |
5,2 (4,4, 5,7) |
3,8 (2,8, 4,7) |
|
Hazard ratio (IC 95%) |
0,776 (0,603, 0,998) | |
|
Valor de p estratificado (Log-rank) |
0,0473 | |
|
Supervivencia Libre de Progresión, | ||
|
meses | ||
|
Mediana (IC 95%) |
2,1 (1,5, 2,7) |
1,3 (1,3, 1,4) |
|
Hazard ratio (IC 95%) |
0,483 (0,376, 0,620) | |
|
Valor de p estratificado (Log-rank) |
<0,0001 | |
|
Rango de % PFS a las 12 semanas |
40,1 (33,6, 46,4) |
15,8 (9,7, 23,3) |
Abreviaturas: IC= intervalo de confianza
Figura 3: Curvas de Kaplan-Meier para supervivencia global de Cyramza frente a placebo en REGARD
Número en Riesgo
Cyramza 238 154
Placebo 117 66
Tiempo Desde Aleatorización (Meses)
92
34
49
20
17
7
Partiendo de los limitados datos disponibles del estudio REGARD sobre pacientes con cáncer gástrico HER2 positivo o adenocarcinoma de GEJ y sobre pacientes previamente tratados con trastuzumab (en RAINBOW), se considera poco probable que Cyramza tenga un efecto perjudicial o que no tenga efecto en pacientes con cáncer gástrico HER2 positivo. Los análisis de subgrupos no estratificados realizados a posteriori de los pacientes del estudio RAINBOW previamente tratados con trastuzumab (n=39) sugirieron un beneficio en la supervivencia de pacientes (HR 0,679, IC 95% 0,327, 1,419) y demostraron un beneficio en la supervivencia libre de progresión (PFS) (HR 0,399, IC 95% 0,194, 0,822).
Cáncer colorrectal
RAISE
RAISE fue un ensayo global, aleatorizado, doble ciego de Cyramza más FOLFIRI frente a placebo más FOLFIRI, en pacientes con mCRC, que tuvieron progresión de la enfermedad durante o tras la primera línea de tratamiento con bevacizumab, oxaliplatino y una fluoropirimidina. Los pacientes debían presentar ECOG
PS 0 o 1 y progresión de la enfermedad en los 6 meses tras la última dosis del tratamiento de primera línea. Los pacientes debían presentar una función hepática, renal y una coagulación adecuadas. Se excluyeron a los pacientes con antecedentes hereditarios o adquiridos de sangrado no controlado o de trastornos trombóticos, antecedentes recientes de sangrado grave (Grado >3) o a aquellos que presentaban enfermedad tromboembólica arterial (ATE) en los 12 meses anteriores a la aleatorización. También se excluyeron a aquellos pacientes que presentaron: un ATE, hipertensión Grado 4, proteinuria Grado 3, acontecimiento hemorrágico grado 3-4 o perforación intestinal durante el tratamiento en primera línea con bevacizumab.
Un total de 1.072 pacientes fueron aleatorizados (1:1) a recibir Cyramza (n=536) a 8 mg/kg o placebo (n=536), en combinación con FOLFIRI. Todos los medicamentos se administraron de forma intravenosa. El régimen con FOLFIRI fue: irinotecán 180 mg/m2 administrado durante 90 minutos y ácido folínico 400 mg/ m2 administrado de forma simultánea durante 120 minutos, seguido de un bolo de 5-fluorouracilo (5-FU)
400 mg/m2 durante 2 a 4 minutos, seguido de 5-FU 2.400 mg/ m2 administrados por perfusión continua durante 46 a 48 horas. Los ciclos de tratamiento en ambos brazos se repitieron cada 2 semanas. A los pacientes que interrumpieron uno o más componentes del tratamiento debido a una reacción adversa, se les permitió continuar el tratamiento con el resto de componente(s) hasta progresión de la enfermedad o toxicidad inaceptable. La variable primaria fue OS y la variable secundaria incluyó PFS, tasa de respuesta objetiva (ORR) y calidad de vida (QoL) usando el criterio de la Organización Europea para la Investigación del Tratamiento del Cáncer (European Organisation for Research and Treatment of Cáncer, EORTC, por sus siglas en inglés) QLQ-C30. La aleatorización se estratificó según región geográfica, mutación KRAS (presencia de mutación o no) y tiempo hasta progresión de la enfermedad (time to disease progression, TTP) tras comienzo de la primera línea de tratamiento (<6 meses frente a >6 meses).
Las características demográficas y basales de la población ITT fueron similares en ambos brazos del tratamiento. La mediana de edad fue 62 años y el 40 % de pacientes tenían >65 años; 57 % de los pacientes eran hombres; 76 % eran blancos y el 20 % asiáticos; 49 % tuvieron ECOG PS 0; 49 % de los pacientes presentaron mutación KRAS en el tumor y 24 % de los pacientes tuvieron TTP < 6 meses tras el comienzo del tratamiento en primera línea. Se administró tratamiento anticanceroso sistémico posinterrupción al 54% de los pacientes que recibieron Cyramza más FOLFIRI y al 56 % de los pacientes que recibieron placebo más FOLFIRI.
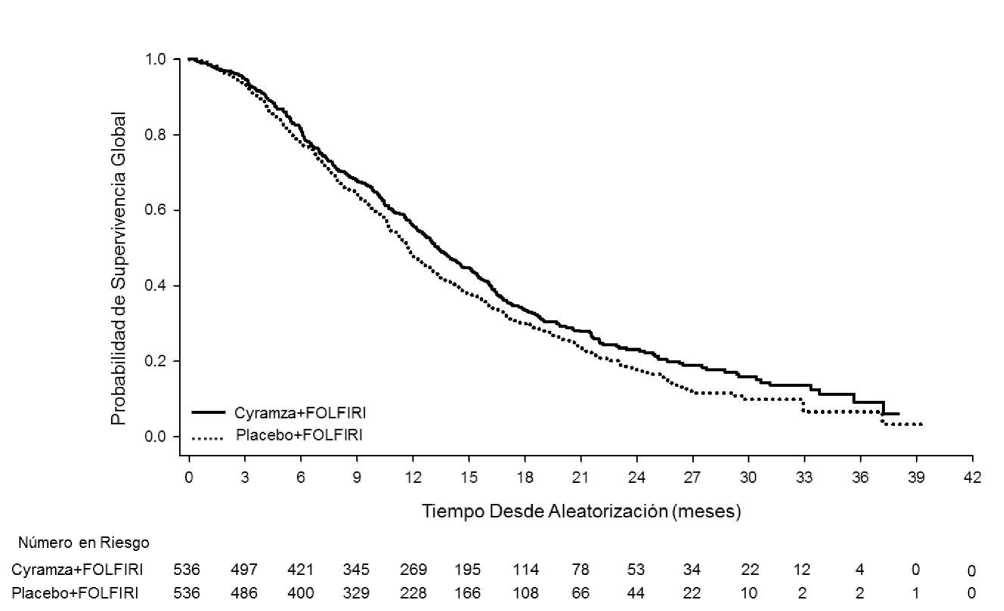
La supervivencia global mejoró de forma estadísticamente significativa en pacientes que recibieron Cyramza más FOLFIRI comparada con aquellos que recibieron placebo más FOLFIRI (HR 0,844; IC 95%: 0,730 a 0,976; p=0,0219). Hubo un aumento en la mediana de supervivencia de 1,6 meses a favor del brazo de Cyramza más FOLFIRI: 13,3 meses en el brazo de Cyramza más FOLFIRI y 11,7 meses en el brazo de placebo más FOLFIRI. La supervivencia libre de progresión mejoró de forma estadísticamente significativa en pacientes que recibieron Cyramza más FOLFIRI comparado con aquellos que recibieron placebo más FOLFIRI (HR 0,793; IC 95%: 0,697 a 0,903; p=0,0005). Hubo un aumento en la mediana de PFS de 1,2 meses a favor del brazo de Cyramza más FOLFIRI: 5,7 meses en el brazo de Cyramza más FOLFIRI y 4,5 meses en el brazo de placebo más FOLFIRI. Los resultados de eficacia se muestran en la Tabla 12 y en las Figuras 4 y 5.
Se llevaron a cabo análisis preespecificados para OS y PFS según factores de estratificación. El HR de OS fue 0,82 (IC 95%: 0,67 a 1,0) en pacientes con tumor KRAS no mutado y 0,89 (IC 95%: 0,73 a 1,09) en pacientes con un tumor con la presencia de la mutación KRAS. Para pacientes con TTP >6 meses tras comenzar el tratamiento de primera línea, el HR de OS fue 0,86 (IC 95%: 0,73 a 1,01) y en pacientes con TTP < 6 meses tras comenzar la primera línea de tratamiento, el HR fue 0,86 (IC 95%: 0,64 a 1,13). Los análisis por subgrupos preespecificados tanto para PFS como OS según la edad (< 65 años y >65 años), sexo, raza, ECOG PS (0 o >1), número de órganos afectados, únicamente metástasis hepática, localización del tumor primario (colon o recto), niveles de antígenos carcinoembrionarios (<200 pg/l, >200 pg/l), mostraron un efecto favorable del tratamiento con Cyramza más FOLFIRI sobre placebo más FOLFIRI. En 32 de los 33 análisis por subgrupos preespecificados para OS, el HR fue < 1,0. El único subgrupo con HR > 1 fue el de pacientes con progresión de la enfermedad desde el comienzo del tratamiento en primera línea con bevacizumab < 3 meses (HR 1,02 [IC 95%: 0,68 a 1,55]). Este único subgrupo puede considerarse que presenta una enfermedad agresiva relativamente refractaria al tratamiento en primera línea. En ambos brazos de tratamiento, los pacientes que presentaron neutropenia, tuvieron una mediana de OS más larga que la de aquellos que no presentaron neutropenia. La mediana de OS fue mayor en los pacientes con neutropenia de cualquier grado en el brazo de ramucirumab (16,1 meses) que en el brazo de placebo (12,6 meses). La mediana de OS en pacientes que no presentaron neutropenia fue 10,7 meses en ambos brazos.
Tabla 12: Resumen de los datos de eficacia- población ITT
|
Cyramza más FOLFIRI N=536 |
Placebo más FOLFIRI N=536 | |
|
Supervivencia Global, meses | ||
|
Mediana (IC 95%) |
13,3 (12,4, 14,5) |
11,7 (10,8, 12,7) |
|
Hazard ratio (IC 95%) |
0,84 (0,73, 0,98) | |
|
Valor de p estratificado (Log-rank) |
0,022 | |
|
Supervivencia libre de progresión, meses | ||
|
Mediana (IC 95%) |
5,7 (5,5, 6,2) |
4,5 (4,2, 5,4) |
|
Hazard ratio (IC 95%) |
0,79 (0,70, 0,90) | |
|
Valor de p estratificado (Log-rank) |
<0,001 | |
Abreviaturas: IC= intervalo de confianza
Figura 4: Curvas de Kaplan-Meier para supervivencia global de Cyramza más FOLFIRI frente a placebo más FOLFIRI en RAISE
0.8 -
Cyramza+FOLFIRI
0.0 -
P acebo+FOLF R
Tiempo Desde Aleatorizacion (meses
Numero en Riesgo
Cyramza+FOLFIRI
536
38
234
142
P acebo+FOLF R
536
345
182
El ORR fue similar en ambos brazos de tratamiento (13,4 % frente a 12,5 %, ramucirumab más FOLFIRI frente a placebo más FOLFIRI, respectivamente). La tasa de control de la enfermedad (respuesta completa más respuesta parcial más enfermedad estable) fue numéricamente más alta en pacientes en el brazo de ramucirumab más FOLFIRI comparado con el brazo de placebo más FOLFIRI (74,1% frente a 68,8%, respectivamente). Para la EORTC QLQ-C30, los pacientes en el brazo de tratamiento de ramucirumab más FOLFIRI notificaron un descenso transitorio en QoL en la mayoría de las escalas comparado con los pacientes en el brazo de tratamiento de placebo más FOLFIRI. Las diferencias entre los brazos se notificaron tras el primer mes de tratamiento.
CPNM
REVEL
REVEL, es un ensayo aleatorizado, doble ciego de Cyramza más docetaxel frente a placebo más docetaxel, llevado a cabo en 1.253 pacientes con CPNM localmente avanzado o metastásico escamoso o no escamoso con progresión de la enfermedad durante o tras tratamiento basado en platino. La variable primaria fue OS. Los pacientes fueron aleatorizados en un ratio 1:1 para recibir Cyramza más docetaxel (n=628) o placebo más docetaxel (n=625). La aleatorización fue estratificada por región geográfica, sexo, mantenimiento previo y ECOG PS. Cyramza a 10 mg/kg o placebo y docetaxel a 75 mg/m2 fueron cada uno administrados por perfusión intravenosa el día 1 de un ciclo de 21 días. Los hospitales en Asia Oriental administraron una dosis reducida de docetaxel a 60 mg/m2 cada 21 días. Se excluyeron pacientes con hemorragia pulmonar o gastrointestinal grave y reciente, sangrado posquirúrgico, evidencia de hemorragias del SNC, compromiso tumoral de las vías respiratorias principales o vasos sanguíneos, cavitación intratumoral y antecedentes de sangrados significativos o trastornos trombóticos incontrolados. También se excluyeron pacientes que habían recibido cualquier tipo de terapia anticoagulante y/o terapia crónica con fármacos antiinflamatorios no esteroideos u otros agentes antiplaquetarios o aquellos con metástasis del SNC/cerebral clínicamente inestable no tratada. Se permitió el uso de Aspirina a dosis de hasta 325 mg/día (ver sección 4.4). Se incluyeron un número limitado de no caucásicos, especialmente pacientes negros (2,6%). Por lo tanto, existe
experiencia limitada con la combinación de ramucirumab y docetaxel en esos pacientes con CPNM avanzado así como en pacientes con insuficiencia renal, enfermedad cardiovascular y obesidad.
Las características demográficas basales de los pacientes y de la enfermedad estuvieron generalmente equilibradas entre los brazos: la mediana de edad fue 62 años; 67 % de los pacientes fueron hombres; 82 % fueron caucásicos, 13 % asiáticos; el ECOG PS fue 0 para el 32 % de los pacientes, 1 para el 67 % de los pacientes; el 73 % de los pacientes tuvieron histología no escamosa y el 26 % tuvieron histología escamosa. La terapia previa más frecuente fue pemetrexed (38%), gemcitabina (25%), taxano (24%) y bevacizumab (14%); 22 % de los pacientes recibieron terapia de mantenimiento previa. La mediana de la duración de la terapia de docetaxel fue 14,1 semanas para el brazo de ramucirumab más docetaxel (con una mediana de 4,0 perfusiones recibidas) y 12,0 semanas para el brazo de placebo más docetaxel (con una mediana de perfusiones recibidas de 4,0).
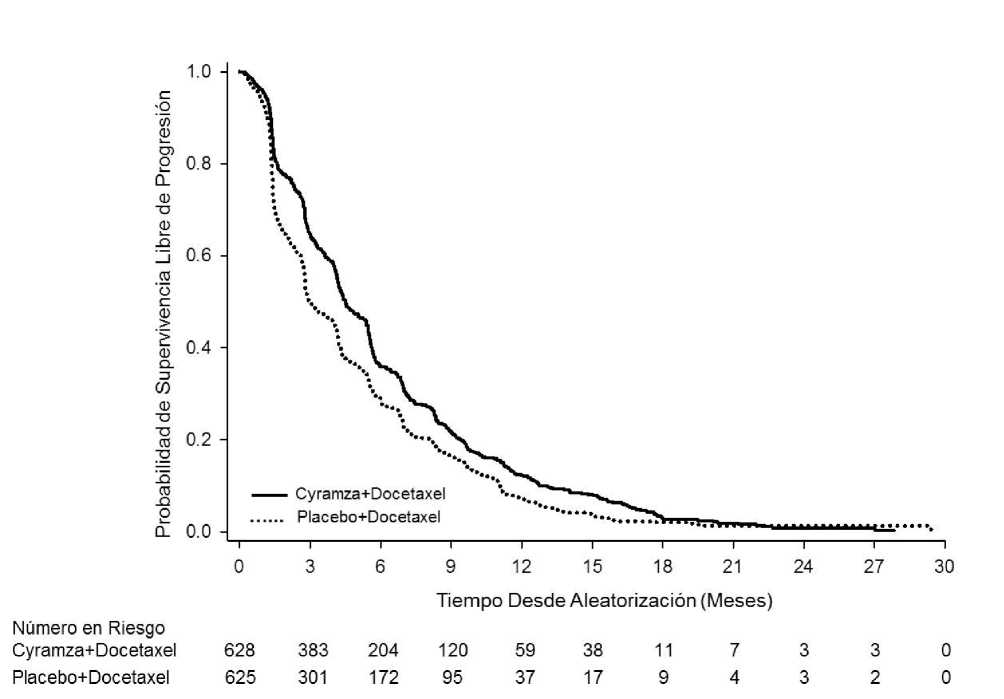
La OS aumentó de forma estadísticamente significativa en pacientes que recibieron Cyramza más docetaxel comparado con aquellos que recibieron placebo más docetaxel (HR 0,857; IC 95%: 0,751 a 0,979; p=0,024). Hubo un aumento en la mediana de supervivencia de 1,4 meses a favor del brazo de Cyramza más docetaxel: 10,5 meses en el brazo de Cyramza más docetaxel y 9,1 meses en el brazo de placebo más docetaxel. PFS aumentó de forma estadísticamente significativa en pacientes que recibieron Cyramza más docetaxel comparado con aquellos que recibieron placebo más docetaxel (HR 0,762; IC 95%: 0,677 a 0,859; p<0,001). Hubo un aumento en la mediana de PFS de 1,5 meses a favor del brazo de Cyramza más docetaxel: 4,5 meses en el brazo de Cyramza más docetaxel y 3 meses en el brazo de placebo más docetaxel. ORR mejoró de forma significativa en pacientes que recibieron Cyramza más docetaxel comparada con aquellos que recibieron placebo más docetaxel (22,9% vs. 13,6%, p<0,001). El análisis primario de QoL mostró un tiempo hasta el deterioro similar entre los brazos de tratamiento según todas las puntuaciones de las Escalas de Síntomas de Cáncer de Pulmón (Lung Cáncer Symptom Scale, LCSS).
Se observó una mejora invariable (ramucirumab más docetaxel vs. placebo más docetaxel) en subgrupos importantes para PFS y OS. Los resultados de los subgrupos para OS mostraron: histología no escamosa (HR 0,83;IC 95%: 0,71 a 0,97; mediana de OS [mOS]: 11,1 vs. 9,7 meses) e histología escamosa (HR 0,88; IC 95%: 0,69 a 1,13; mOS: 9,5 vs. 8,2 meses); pacientes con mantenimiento previo (HR 0,69; IC 95%: 0,51 a 0,93; mOS: 14,4 vs. 10,4 meses); tiempo desde el comienzo de la terapia previa <9 meses (HR 0,75; IC 95%: 0,64 a 0,88; mOS: 9,3 vs. 7,0 meses); pacientes < 65 años (HR 0,74, IC 95%: 0,62, 0,87; mOS: 11,3 vs.
8,9 meses). Se ha observado una tendencia a menor eficacia conforme aumenta la edad del paciente que recibe ramucirumab más docetaxel para el tratamiento de CPNM avanzado con progresión de la enfermedad tras quimioterapia basada en platino (ver sección 5.1).
No se observaron diferencias de eficacia entre los brazos de tratamiento en los subgrupos de pacientes >65 años (OS HR 1,10, IC 95%: 0,89, 1,36; mediana de OS [mOS]: 9,2 vs. 9,3 meses, ver sección 4.4), pacientes tratados previamente con taxanos (HR 0,81; IC 95%:0,62 a 1,07; mOS 10,8 vs. 10,4 meses) y aquellos con tiempo desde comienzo de la terapia previa >9 meses (HR 0,95; IC 95%: 0,75 a 1,2; mOS: 13,7 vs. 13,3 meses). Los resultados de eficacia se muestran en la Tabla 13.
Tabla 13: Resumen de los datos de eficacia- población ITT
|
Cyramza más docetaxel N=628 |
Placebo más docetaxel N=625 | |
|
Supervivencia Global, meses | ||
|
Mediana - meses (IC 95%) |
10,5 (9,5, 11,2) |
9,1 (8,4, 10,0) |
|
Hazard ratio (IC 95%) |
0,857 (0,751, 0,979) | |
|
Valor de p estratificado (Log-rank) |
0,024 | |
|
Supervivencia Libre de Progresión, meses | ||
|
Mediana (IC 95%) |
4,5 (4,2, 5,4) |
3,0 (2,8, 3,9) |
|
Hazard Ratio (IC 95%) |
0,762 (0,677, 0,859) | |
|
Valor de p estratificado (Log-rank) |
<0,001 | |
|
Ratio de respuesta objetiva (CR + PR) | ||
|
Ratio - porcentaje (IC 95%) |
22,9 (19,7, 26,4) |
13,6 (11,0, 16,5) |
|
Valor de p estratificado (CMH) |
<0,001 | |
Abreviaturas: IC= intervalo de confianza, CR= respuesta completa, PR= respuesta parcial, CMH = Cochran-Mantel-Haenszel
Figura 6: Curvas de Kaplan-Meier para supervivencia global de Cyramza más docetaxel frente a placebo más docetaxel en REVEL
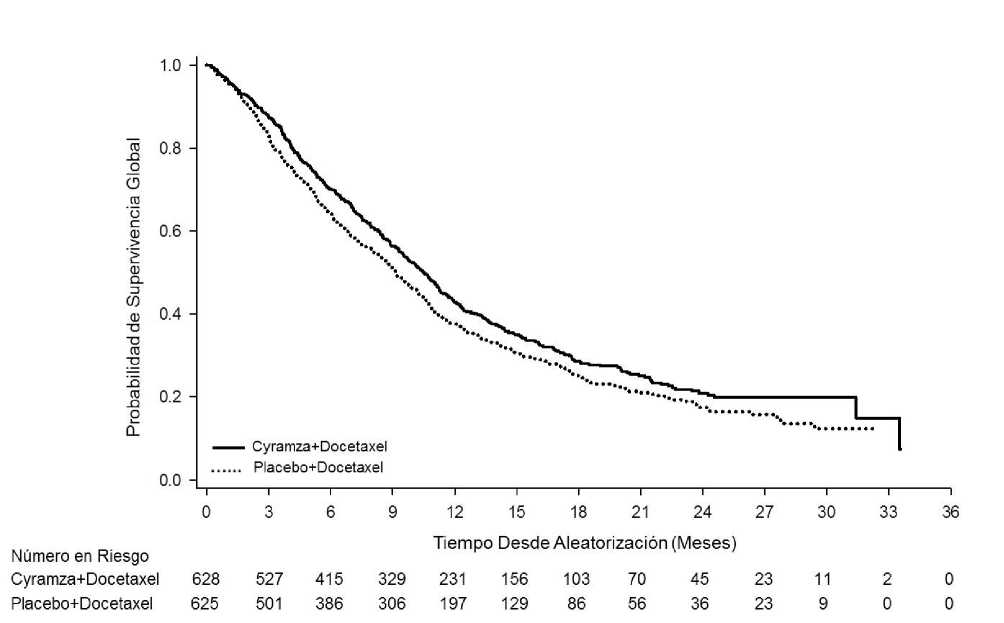
Figura 7: Curvas de Kaplan-Meier para supervivencia libre de progresión de Cyramza más docetaxel
frente a placebo más docetaxel en REVEL
Pacientes con estado funcional (PS) >2 según la escala Eastern Cooperative Oncology Group (ECOG)
Los pacientes con un ECOG PS >2 fueron excluidos de los ensayos pivotales en todas las indicaciones, por lo que se desconoce la seguridad y eficacia de Cyramza en este grupo de pacientes.
Inmunogenicidad
Los pacientes en dos estudios fase 3, RAINBOW y REGARD fueron examinados en numerosos momentos para comprobar los anticuerpos antifármaco (anti-drug antibodies, ADAs). Se evaluaron muestras de 956 pacientes: 527 pacientes tratados con ramucirumab y 429 pacientes del grupo control. Once (2,2%) de los pacientes tratados con ramucirumab y dos (0,5%) de los pacientes control desarrollaron ADAs. Ninguno de los pacientes con ADAs presentó un IRR. Ningún paciente desarrolló anticuerpos neutralizadores de ramucirumab. No hay datos suficientes para evaluar los efectos de ADAs sobre la eficacia o seguridad de ramucirumab.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Cyramza en todos los grupos de la población pediátrica en adenocarcinoma gástrico, adenocarcinoma de colon y recto y en carcinoma de pulmón (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Tras la pauta posológica de 8 mg/kg cada 2 semanas, la media geométrica de la concentración mínima de ramucirumab (Cmin) fue 49,5 pg/ml (rango de 6,3-228 pg/ml) y 74,4 pg/ml (rango 13,8-234 pg/ml) en suero en pacientes con cáncer gástrico avanzado, antes de la administración de la cuarta y séptima dosis respectivamente de ramucirumab como agente único.
Tras la pauta posológica de 8 mg/kg cada 2 semanas de ramucirumab en combinación con FOLFIRI, las medias geométricas de la Cmin en el suero de pacientes con mCRC de ramucirumab fueron 46,3 pg/ml (rango de 7,7-119 pg/ml) y 65,1 pg/ml (rango de 14,5-205 pg/ml) antes de la administración de la tercera y quinta dosis respectivamente.
Tras la pauta posológica de 10 mg/kg cada 3 semanas de ramucirumab, las medias geométricas de la Cmin de ramucirumab en el suero de pacientes con CPNM fueron 28,3 pg/ml (rango de 2,5-108 pg/ml) y 38,4 pg/ml (rango de 3,1-128 pg/ml) antes de la administración de la tercera y quinta dosis, respectivamente de ramucirumab administrado en combinación con docetaxel.
Absorción
Cyramza se administra por perfusión intravenosa. No se han llevado a cabo estudios con otras vías de administración.
Distribución
El volumen de distribución medio en el estado estacionario fue de 5,4 l (15%) (% coeficiente de variación [CV%]) basado en la aproximación por un modelo farmacocinético poblacional (population pharmacokinetic approach, PopPK).
Biotransformación
No se ha estudiado el metabolismo de ramucirumab. El aclaramiento de los anticuerpos es principalmente por catabolismo.
Eliminación
El aclaramiento medio (CV%) de ramucirumab fue 0,015 l/hora (30%) y el valor medio de vida media fue 14 días (20%), según PopPK.
Dependencia de tiempo y dosis
No hubo una desviación clara de la proporcionalidad de dosis en la farmacocinética de ramucirumab desde 6 mg/kg a 20 mg/kg. Se observó un ratio acumulado de 1,5 para ramucirumab cuando se administró una dosis cada 2 semanas. Según simulaciones usando el modelo PopPK, el estado estacionario debería alcanzarse en la sexta dosis.
Pacientes de edad avazada
No hubo diferencias en la exposición a ramucirumab en pacientes > 65 años comparado con pacientes < 65 años, según el modelo PopPK.
Insuficiencia renal
No se han llevado a cabo estudios específicos para evaluar el efecto de la insuficiencia renal sobre la farmacocinética de ramucirumab. Según modelos PopPK, la exposición a ramucirumab fue parecida en pacientes con insuficiencia renal leve (aclaramiento de creatina (creatinine clearance, CrCl) > 60 a < 90 ml/min), moderada (CrCl > 30 a < 60 ml/min) o grave (CrCl 15 a 29 ml/min) comparado con pacientes con función renal normal (CrCl > 90 ml/min).
Insuficiencia hepática
No se han llevado a cabo estudios específicos para evaluar los efectos de la insuficiencia hepática sobre la farmacocinética de ramucirumab. Según modelos PopPK, la exposición a ramucirumab en pacientes con insuficiencia hepática leve (niveles de bilirrubina total > 1,0-1,5 veces el valor de ULN y cualquier valor de AST o bilirrubina total de <1,0 veces el valor de ULN y AST > ULN) o con insuficiencia hepática moderada (niveles de bilirrubina total > 1,5-3,0 veces el valor ULN y cualquier nivel de AST) fue similar a la de pacientes con función hepática normal (niveles de bilirrubina total y AST < a ULN). Ramucirumab no se ha estudiado en pacientes con insuficiencia hepática grave (niveles de bilirrubina total >3,0 veces ULN y cualquier valor de AST).
Otras poblaciones especiales
Según modelos PopPK, se encontró que las siguientes covariables no presentaron impacto sobre el comportamiento farmacocinético de ramucirumab: edad, sexo , raza, peso corporal y niveles de albúmina. Relación exposición-respuesta
Eficacia
Los análisis de la exposición-respuesta a través de los estudios pivotales, mostraron que la eficacia estaba relacionada con la exposición a ramucirumab. La eficacia, medida por incrementos en OS y PFS, se asoció con el aumento del rango de exposición a ramucirumab al administrarse a una dosis de 8 mg/kg cada 2 semanas y de 10 mg/kg de ramucirumab cada 3 semanas.
Seguridad
En el estudio RAINBOW, la incidencia de hipertensión, neutropenia y leucopenia de Grado >3, aumentó con una mayor exposición a ramucirumab.
En el estudio RAISE, la incidencia de neutropenia Grado >3 aumentó con una mayor exposición a ramucirumab.
En el estudio REVEL, la incidencia de neutropenia febril e hipertensión Grado >3 aumentó con una mayor exposición a ramucirumab.
5.3 Datos preclínicos sobre seguridad
No se han llevado a cabo estudios en animales para evaluar el potencial carcinogénico o genotoxicidad de ramucirumab.
Los órganos identificados como diana en estudios de toxicidad a dosis repetidas en monos cynomolgus fueron riñón (glomerulonefritis), hueso (engrosamiento y osificación endocondral anormal de la placa de crecimiento epifisaria) y órganos reproductivos femeninos (descenso del peso de ovarios y útero). Se observó un grado mínimo de inflamación y/o infiltración de células mononucleares en varios órganos.
No se llevaron a cabo estudios de toxicidad reproductiva con ramucirumab, sin embargo existen modelos animales que asocian la angiogénesis, el VEGF y el Receptor 2 de VEGF a aspectos críticos de la reproducción en mujeres, el desarrollo embriofetal y el desarrollo postnatal. Basándose en el mecanismo de acción de ramucirumab, es probable que en animales, ramucirumab pueda inhibir la angiogénesis y derivar en efectos adversos sobre la fertilidad (ovulación), desarrollo de la placenta, desarrollo del feto y desarrollo postnatal.
Un dosis única de ramucirumab no perjudicó la curación de heridas en monos usando un modelo incisional de espesor completo.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Histidina
Monohidrocloruro de histidina Cloruro de sodio Glicina (E640)
Polisorbato 80 (E433)
Agua para preparaciones inyectables
6.2 Incompatibilidades
Cyramza no debe administrarse o mezclarse con soluciones de dextrosa.
Este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección 6.6.
6.3 Periodo de validez
Vial sin abrir 3 años
Tras dilución
La solución para perfusión de Cyramza no contiene conservantes antimicrobianos, cuando la solución se prepara según las recomendaciones.
Se ha demostrado la estabilidad química y física rutinaria de Cyramza una vez diluido con una solución inyectable de cloruro de sodio 9 mg/ml (0,9%) entre 2 °C y 8 °C durante 24 horas o durante 4 horas a 25 °C. Desde un punto de vista microbiológico, el medicamento se debe utilizar inmediatamente. Si no se utiliza de forma inmediata, el tiempo y las condiciones de almacenamiento previas al uso son responsabilidad del usuario y normalmente no deberían ser superiores a 24 horas entre 2 °C y 8 °C, a no ser que la dilución haya tenido lugar en condiciones asépticas controladas y validadas.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Para las condiciones de conservación del medicamento tras la dilución, ver sección 6.3.
6.5 Naturaleza y contenido del envase
10 ml de solución en un vial (vidrio Tipo I) con tapa de goma de clorobutilo, un sello de aluminio y un tapón de polipropileno.
50 ml de solución en un vial (vidrio Tipo I) con tapa de goma de clorobutilo, un sello de aluminio y un tapón de polipropileno.
Envase de 1 vial de 10 ml.
Envase de 2 viales de 10 ml.
Envase de 1 vial de 50 ml.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
No agitar el vial.
Preparar la solución para perfusión usando técnicas asépticas para garantizar la esterilidad de la disolución preparada.
Cada vial es para un solo uso. Antes de la dilución se debe comprobar el contenido de los viales para detectar la posible existencia de partículas o cambios de color (el concentrado para solución para perfusión debe ser de transparente a ligeramente opalescente y de tinte incoloro a ligeramente amarillo sin partículas visibles).
51 se identifican partículas o alteraciones del color, el vial se debe descartar.
Calcular la dosis y el volumen de ramucirumab necesarios para preparar la solución para perfusión. Los viales contienen 100 mg o 500 mg en solución a 10 mg/ml de ramucirumab. Utilizar únicamente como diluyente cloruro de sodio 9 mg/ml (0,9 %) solución inyectable.
En caso de uso de un envase precargado para perfusión intravenosa
Según el volumen de ramucirumab calculado, retirar el volumen correspondiente de solución inyectable de cloruro de sodio 9 mg/ml (0,9 %) del envase precargado de 250 ml para perfusión intravenosa.
El paso del volumen de ramucirumab calculado al envase para perfusión intravenosa se debe realizar asépticamente. El volumen total final del envase debe ser 250 ml. El envase se debe invertir cuidadosamente para garantizar una mezcla adecuada. NO CONGELAR NI AGITAR la solución para perfusión. NO diluir con otras soluciones o coperfundir con otros medicamentos o electrolitos.
En caso de uso de un envase vacío para perfusión intravenosa
El paso de volumen de ramucirumab calculado al envase para perfusión intravenosa vacío se debe realizar asépticamente. Añadir una cantidad suficiente de solución inyectable de cloruro de sodio 9 mg/ml (0,9 %) al envase para alcanzar un volumen total de 250 ml. El envase se debe invertir cuidadosamente para garantizar un mezcla adecuada. NO CONGELAR NI AGITAR. NO diluir con otras soluciones o coperfundir con otros medicamentos o electrolitos.
Los medicamentos de administración parenteral, se deben examinar visualmente antes de la administración para descartar la presencia de partículas. Si se identifican partículas, el vial se debe descartar.
Desechar cualquier porción de ramucirumab remanente en el vial dado que el medicamento no contiene conservantes antimicrobianos.
Administrar a través de una bomba de perfusión. Se debe utilizar una vía de perfusión separada con un filtro de entrada de baja unión a proteínas de 0,22 micrones para la perfusión y al finalizar la perfusión se debe lavar la vía con solución inyectable de cloruro de sodio 9 mg/ml (0,9%).
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Eli Lilly Nederland B.V.
Papendorpseweg 83 3528 BJ Utrecht Países Bajos
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/957/001-003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 19 Diciembre 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección de los fabricantes del principio activo biológico
ImClone Systems LLC 33 ImClone Drive,
Branchburg New Jersey NJ 08876
ESTADOS UNIDOS
Eli Lilly S.A.
Dunderrow Kinsale County Cork Irlanda
Nombre y dirección del fabricante responsable de la liberación de los lotes Lilly, S.A.
Avda de la Industria, 30 Alcobendas 28108 Madrid España
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva
información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
• Obligación de llevar a cabo medidas post-autorización
El TAC deberá llevar a cabo, dentro del plazo establecido, las siguientes medidas:
|
Descripción |
Fecha límite |
|
El TAC debe presentar los resultados de un estudio aleatorizado farmacocinético de rango de dosis y seguridad de ramucirumab en monoterapia (14T-MC-JVDB). Este estudio en fase 2 evaluará la farmacocinética y seguridad de varios regímenes de ramucirumab, incluyendo dosis más altas que la dosis aprobada de 8 mg/kg cada 2 semanas en segunda línea de tratamiento para adenocarcinoma gástrico. |
01/04/2017 (Resultados farmacocinéticos) 01/04/2018 (Informe final del ensayo y resultados de seguridad) |
|
Estudio posautorización de eficacia (PAES): Para investigar la posible relación entre las mediciones de biomarcadores (VEGF-C, VEGF-D, sVEGFR1, sVEGFR2 y sVEGFR3 en plasma, inmunohistoquímica (IHC) de VEGFR2, además de mutación de KRAS, NRAS y BRAF) y los resultados de eficacia (PFS, OS). El TAC debería presentar los resultados de un estudio de biomarcadores procedente del estudio traslacional en la población del RAISE. - La relación con VEGF-C, VEGF-D, sVEGFR1, sVEGFR2 y sVEGFR3 en plasma, inmunohistoquímica (IHC) de VEGFR2 se presentará el - La relación de forma adicional con KRAS, NRAS y mutaciones BRAF se presentarán el |
30 de junio de 2016 30 de septiembre de 2016 |
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
Cyramza 10 mg/ml concentrado para solución para perfusión ramucirumab
2. PRINCIPIO ACTIVO
Un ml de concentrado contiene 10 mg de ramucirumab.
3. LISTA DE EXCIPIENTES
Excipientes: histidina, monohidrocloruro de histidina, cloruro de sodio, glicina, polisorbato 80, agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Concentrado para solución para perfusión
100 mg/10 ml
1 vial
2 viales
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía intravenosa tras dilución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
No agitar.
8. FECHA DE CADUCIDAD
CAD
Conservar en nevera.
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Eli Lilly Nederland B.V. Papendorpseweg 83 3528 BJ Utrecht Países Bajos
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/957/001 - 1 vial de 10 ml.
EU/1/14/957/002 - 2 viales de 10 ml.
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille
1. NOMBRE DEL MEDICAMENTO
Cyramza 10 mg/ml concentrado para solución para perfusión ramucirumab
2. PRINCIPIO ACTIVO
Un ml de concentrado contiene 10 mg de ramucirumab.
3. LISTA DE EXCIPIENTES
Excipientes: histidina, monohidrocloruro de histidina, cloruro de sodio, glicina, polisorbato 80, agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Concentrado para solución para perfusión
500 mg/50 ml 1 vial
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía intravenosa tras dilución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
No agitar.
8. FECHA DE CADUCIDAD
CAD
Conservar en nevera.
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Eli Lilly Nederland B.V. Papendorpseweg 83 3528 BJ Utrecht Países Bajos
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/957/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille
ETIQUETA DEL VIAL- Vial de 10 ml
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Cyramza 10 mg/ml concentrado estéril
ramucirumab
Vía IV tras dilución.
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
100 mg/10 ml
6. OTROS
ETIQUETA DEL VIAL- Vial de 50 ml
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Cyramza 10 mg/ml concentrado estéril
ramucirumab
Vía IV tras dilución.
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
500 mg/50 ml
6. OTROS
B. PROSPECTO
Prospecto: información para el usuario
Cyramza 10 mg/ml concentrado para solución para perfusión
ramucirumab
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Si experimenta efectos adversos, consulte a su médico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Cyramza y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Cyramza
3. Cómo usar Cyramza
4. Posibles efectos adversos
5. Conservación de Cyramza
6. Contenido del envase e información adicional
1. Qué es Cyramza y para qué se utiliza
Cyramza es un medicamento para el cáncer que contiene ramucirumab como principio activo, el cual es un anticuerpo monoclonal. Es una proteína especializada que puede reconocer y unirse a otra proteína que se encuentra en los vasos sanguíneos llamada “Receptor de VEGF tipo 2”. Este receptor es necesario en el desarrollo de nuevos vasos sanguíneos. Para crecer, el cáncer necesita desarrollar nuevos vasos sanguíneos. Al unirse y bloquear al “Receptor de VEGF tipo 2”, el medicamento interrumpe el aporte sanguíneo a las células cancerígenas.
Cyramza también se utiliza en combinación con paclitaxel, otro medicamento anticanceroso, para el tratamiento del cáncer de estómago avanzado (o cáncer de la unión entre el esófago y el estómago) en adultos cuya enfermedad ha empeorado tras el tratamiento con medicamentos para tratar el cáncer.
Cyramza se utiliza para el tratamiento del cáncer de estómago avanzado (o cáncer de la unión entre el esófago y el estómago) en adultos cuya enfermedad ha empeorado después de tratamiento con medicamentos para tratar el cáncer y para quienes el tratamiento de Cyramza en combinación con paclitaxel no sea adecuado.
Cyramza se utiliza para el tratamiento de cáncer de colon o recto (partes del intestino grueso) en adultos. Se administra con otro medicamento llamado “FOLFIRI”, que incluye “5-fluorouracilo”, “ácido folínico” e “irinotecán”.
Cyramza se administra en combinación con docetaxel, otro medicamento anticanceroso, para el tratamiento de pacientes adultos con cáncer de pulmón en estado avanzado cuya enfermedad ha empeorado tras el tratamiento con medicamentos para tratar el cáncer.
2. Qué necesita saber antes de empezar a usar Cyramza
No use Cyramza:
- si es alérgico a ramucirumab o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- si los rayos-X muestran signos de que el cáncer de pulmón tiene una cavidad o agujero o si el cáncer de pulmón se encuentra pegado a vasos sanguíneos mayores.
Advertencias y precauciones
Consulte a su médico o enfermero antes de empezar a usar Cyramza si:
- tiene una enfermedad que aumenta el riesgo de hemorragia. También consulte a su médico si está tomando otros medicamentos que aumentan el riesgo de hemorragia o que afectan a la capacidad de coagulación de la sangre. En estos casos, su médico llevará a cabo análisis de sangre de forma regular para controlar el riesgo de hemorragia.
tiene cáncer de pulmón y ha tenido un sangrado en el pulmón recientemente (tos con sangre roja brillante) o está tomando de forma continua medicamentos antiinflamatorios no esteroideos o medicamentos que afectan a la capacidad de coagulación de la sangre.
- tiene la tensión sanguínea alta. Cyramza puede aumentar la aparición de tensión sanguínea alta. Su médico se asegurará de que si ya presenta una tensión sanguínea alta esta tiene que estar bajo control antes de empezar el tratamiento con Cyramza. Durante el tratamiento con Cyramza, su médico controlará su tensión sanguínea y ajustará la medicación antihipertensiva si fuese necesario. Se puede interrumpir temporalmente el tratamiento con Cyramza hasta que la tensión sanguínea alta esté controlada con medicamentos o se puede interrumpir de forma permanente si no se puede controlar adecuadamente.
- va a ser operado, si usted ha sido operado recientemente o si tras la operación su herida no se está curando bien. Cyramza puede aumentar el riesgo de problemas con la curación de heridas. No debe recibir Cyramza durante al menos 4 semanas antes de que se lleve a cabo la operación programada y su médico decidirá cuando reanudar el tratamiento. Si durante el tratamiento tiene una herida que cura mal, la dosis de Cyramza se interrumpirá hasta que la herida esté completamente curada.
- tiene una enfermedad del hígado grave (“cirrosis”) y enfermedades asociadas tales como acumulación excesiva de fluidos en su abdomen (“ascitis”). Su médico le comentará si los beneficios potenciales del tratamiento justifican el aumento de posibles riesgos para usted.
- tiene problemas de riñón graves. Los datos disponibles sobre el uso de Cyramza en pacientes con trastorno grave de la función renal son limitados.
Consulte con su médico o enfermero inmediatamente si alguna de las siguientes afirmaciones le afectan (o
no está seguro) durante el tratamiento con Cyramza o en cualquier otro momento después:
- Bloqueo de las arterias por un trombo (“enfermedad tromboembólica arterial”):
Cyramza puede provocar un trombo en sus arterias. Los trombos en las arterias pueden causar enfermedades graves, incluyendo infarto cardiaco o embolia cerebral. Los síntomas del infarto cardiaco pueden incluir dolor u opresión en el pecho. Los síntomas de una embolia cerebral pueden incluir entumecimiento repentino o debilidad del brazo, piernas o cara, confusión, dificultad para hablar o entender a los demás, dificultad repentina al caminar o pérdida de equilibrio o descoordinación o mareo repentino. Si desarrolla un trombo en sus arterias, el tratamiento con Cyramza se interrumpirá de forma permanente.
- Peforación en la pared del intestino (“perforación gastrointestinal”): Cyramza puede aumentar el riesgo de que se desarrollen perforaciones en la pared de su intestino. Los síntomas incluyen dolor abdominal grave, vómitos, fiebre o escalofríos. Si se desarrolla una perforación en la pared del intestino, el tratamiento con Cyramza se interrumpirá de forma permanente.
- Hemorragia grave: Cyramza puede aumentar el riesgo de sangrado grave. Los síntomas pueden incluir: cansancio extremo, debilidad, mareo o cambios en el color de sus heces. Si sufre hemorragia grave, el tratamiento con Cyramza se interrumpirá de forma permanente.
- Reacciones relacionadas con la perfusión: pueden ocurrir reacciones relacionadas con la perfusión durante el tratamiento, debido a que Cyramza se administra mediante perfusión intravenosa por goteo (ver sección 3). Su médico o enfermero controlorá la aparición de efectos adversos durante la perfusión. Los síntomas pueden incluir: aumento de la tensión muscular, dolor de espalda, dolor y/u opresión en el pecho, escalofríos, enrojecimiento, dificultad para respirar, silbidos al respirar y sensación de hormigueo o entumecimiento en las manos o los pies. En casos graves, los síntomas pueden incluir dificultad respiratoria producida por un estrechamiento de las vías respiratorias, pulso acelerado y sensación de desmayo. Si sufre una reacción grave relacionada con la perfusión, el tratamiento con Cyramza se interrumpirá de forma permanente.
- Conductos anómalos en el interior del cuerpo (“fístula”): Cyramza puede aumentar el riesgo de aparición de conductos anómalos en el interior del cuerpo entre órganos internos y la piel u otros tejidos. Si desarrolla una fístula, el tratamiento con Cyramza se interrumpirá de forma permanente.
- Prueba de orina anormal (“proteinuria”): Cyramza puede aumentar el riesgo de desarrollar o empeorar los niveles anormales de proteína en orina. El tratamiento con Cyramza puede necesitar ser interrumpido temporalmente hasta que los niveles de proteína en orina desciendan y entonces ser reanudado a dosis más bajas, o interrumpirse de forma permanente si los niveles de proteínas en orina no se reducen lo suficiente.
- Inflamación de la boca (“estomatitis”): Cyramza, cuando se administra en combinación con quimioterapia puede aumentar el riesgo de desarrollar inflamación de la boca. Los síntomas pueden incluir sensación de quemazón en la boca, úlceras, ampollas o inflamación. Su médico puede recetarle un tratamiento para ayudarle con los síntomas.
- Fiebre o infección: Durante el tratamiento puede tener una temperatura de 38 °C o más (dado que puede tener una cantidad menor de lo normal de células blancas de la sangre lo cual es muy común). Los síntomas pueden incluir sudoración u otros signos de infección, tales como dolor de cabeza, dolor en las extremidades o disminución del apetito. La infección (sepsis) podría ser grave y llevar a la muerte.
Paciente de edad avanzada con cáncer de pulmón: Su médico considerará cuidadosamente el tratamiento más apropiado para usted.
Niños y adolescentes
Cyramza no se debe administrar a pacientes menores de 18 años debido a que no hay información sobre cómo funciona en este grupo de edad.
Uso de Cyramza con otros medicamentos
Informe a su médico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento. Esto incluye medicamentos obtenidos sin receta y a base de plantas.
Embarazo, lactancia y fertilidad
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento. Debe evitar quedarse embarazada mientras recibe este medicamento y durante al menos 3 meses tras recibir la última dosis de Cyramza. Consulte a su médico sobre el mejor método anticonceptivo para usted.
Como Cyramza impide el desarrollo de nuevos vasos sanguíneos, puede disminuir la probabilidad de quedarse embarazada o mantener un embarazo. Puede también dañar al feto. No debe usar este medicamento durante el embarazo. Si llega a quedarse embarazada durante el tratamiento con Cyramza, su médico comentará con usted si los beneficios del tratamiento son mayores que cualquier posible riesgo para usted o su bebé.
No se conoce si el medicamento pasa a la leche materna y podría afectar al lactante. Por lo tanto, no debe dar el pecho a su bebé durante el tratamiento con Cyramza y durante al menos 3 meses tras recibir la última dosis.
Conducción y uso de máquinas
Se desconoce si Cyramza puede afectar a su habilidad para conducir o utilizar máquinas. Si usted presenta cualquier síntoma que afecte a su habilidad para concentrarse y reaccionar, no conduzca ni use máquinas hasta que los síntomas desaparezcan.
Cyramza contiene sodio
Este medicamento contiene cloruro de sodio.
Cada vial de 10 ml contiene aproximadamente 17 mg de sodio (menos de 1 mmol).
Cada vial de 50 ml contiene aproximadamente 85 mg de sodio (3,7 mmol). Esto debe tenerse en cuenta en pacientes con dietas bajas en sodio.
3. Cómo usar Cyramza
Este tratamiento contra el cáncer le será administrado por su médico o enfermero.
Dosis y frecuencia de administración
La cantidad adecuada de Cyramza necesaria para tratar su enfermedad será calculada por su médico o farmacéutico hospitalario dependiendo de su peso corporal.
La dosis recomendada de Cyramza para el tratamiento del cáncer gástrico y para el tratamiento de cáncer avanzado de colon o recto es 8 mg por kilo de peso una vez cada 2 semanas.
La dosis recomendada de Cyramza para el tratamiento de cáncer de pulmón es de 10 mg por kilo de peso una vez cada 3 semanas.
El número de perfusiones que recibirá dependerá de cómo esté respondiendo al tratamiento. Su médico lo comentará con usted.
Vía y forma de administración
Cyramza es un concentrado para solución para perfusión (también llamado “concentración estéril”). Un farmacéutico hospitalario, enfermero o médico diluirán el contenido del vial en una solución de cloruro de sodio 9 mg/ml (0,9%) antes de usarlo. Este medicamento se administra mediante perfusión por goteo durante aproximadamente 60 minutos.
Medicación previa
Puede recibir otro medicamento para reducir el riesgo de reacciones relacionadas con la perfusión antes de recibir Cyramza. Si presenta una reacción relacionada con la perfusión durante el tratamiento con Cyramza, debe recibir medicación previa en todas las perfusiones posteriores.
Ajuste de dosis
Durante cada perfusión, su médico o enfermero comprobarán si presenta efectos adversos.
Si presenta una reacción relacionada con la perfusión durante el tratamiento, la duración final de la perfusión aumentará para el resto de esa perfusión y para las siguientes.
La cantidad de proteínas en la orina será comprobada regularmente durante el tratamiento. Cyramza se puede interrumpir de forma temporal según la medida del nivel de proteínas. Una vez que los niveles de proteína en orina han disminuido hasta ciertos niveles, el tratamiento se puede reanudar a una dosis menor.
El tratamiento con Cyramza se interrumpirá de forma temporal si:
- presenta tensión sanguínea alta, hasta que se controle con antihipertensivos.
- presenta problemas para la curación de heridas, hasta que la herida se cure o antes de una cirugía
programada.
- tiene una cirugía programada, 4 semanas antes de la misma.
El tratamiento con Cyramza se interrumpirá de forma permanente si:
- presenta un trombo en sus arterias
- presenta una perforación en la pared del intestino
- experimenta hemorragia grave
- experimenta reacciones relacionadas con la perfusión
- presenta tensión sanguínea alta que no se puede controlar con medicamentos
- está pasando a su orina más cantidad de proteínas de lo normal o presenta una enfermedad grave en los riñones (síndrome nefrótico)
- presenta un conducto anómalo en el interior de su cuerpo entre órganos internos y la piel u otros tejidos (fístula).
Cuando reciba Cyramza en combinación con paclitaxel o docetaxel
Paclitaxel y también docetaxel se administran por goteo en vena (perfusión intravenosa) durante un periodo de aproximadamente 60 minutos. Si recibe Cyramza en combinación con paclitaxel o docetaxel el mismo día,Cyramza se le administrará primero.
La cantidad de paclitaxel o docetaxel necesaria depende de su superficie corporal. Su médico o farmacéutico hospitalario calculará su superficie corporal midiendo su talla y peso y calculará la dosis adecuada para usted. La dosis recomendada de paclitaxel es de 80 mg por metro cuadrado (m2) de su superficie corporal una vez a la semana durante 3 semanas seguido de 1 semana sin tratamiento.
La dosis recomendada de docetaxel es 75 mg por metro cuadrado (m2) de su superficie corporal una vez cada 3 semanas. Si es de Asia Oriental, puede recibir una dosis de inicio de docetaxel reducida de 60 mg por m2 de su superficie corporal una vez cada 3 semanas.
Antes de comenzar con cualquier perfusión con paclitaxel, se realizará una analítica para comprobar que el recuento de sus células sanguíneas es suficientemente alto y que su hígado funciona correctamente.
Lea el prospecto de paclitaxel o docetaxel para más información.
Cuando reciba Cyramza en combinación con FOLFIRI
La quimioterapia con FOLFIRI se administra por perfusión intravenosa una vez finalizada la perfusión de Cyramza. Por favor, lea el prospecto del resto de medicamentos que son parte de su tratamiento para comprobar si son apropiados para usted. Si no está seguro, consulte a su médico, farmacéutico o enfermero si hay alguna razón por la que usted no puede utilizar estos medicamentos.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Consulte con su médico inmediatamente si sufre cualquiera de los siguientes efectos adversos graves que han sido observados durante el tratamiento con Cyramza (ver también Qué necesita saber antes de empezar a usar Cyramza):
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas):
- perforaciones en la pared del intestino: Perforación que se desarrolla en el estómago o intestino. Los síntomas incluyen dolor abdominal grave, vómitos, fiebre o escalofrios.
- hemorragia grave en el intestino: los síntomas pueden incluir cansancio extremo, debilidad, mareo o cambios en el color de sus heces.
- trombos en arterias: los trombos en las arterias puede causar un infarto cardiaco o embolia cerebral. Los síntomas de un infarto cardiaco pueden incluir dolor u opresión en el pecho. Los síntomas de una embolia cerebral pueden incluir entumecimiento repentino o debilidad del brazo, piernas o cara, confusión, dificultad para hablar o entender a los demás, dificultad repentina al caminar o pérdida de equilibrio o descoordinación o mareo repentino.
Consulte a su médico si experimenta alguno de los siguientes efectos adversos:
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
- recuento bajo de células blancas en la sangre (puede aumentar el riesgo de infección)
- cansancio o debilidad
- hemorragia nasal
- diarrea
- inflamación de la boca
- dolor abdominal
- recuento bajo de plaquetas (células sanguíneas que ayudan a que la sangre coagule)
- tensión sanguínea alta
- hinchazón de manos, pies y piernas debido a retención de líquidos
- proteínas en la orina (prueba de orina anómala)
- inflamación de las membranas mucosas tales como tractos digestivos y respiratorios
- fiebre acompañada de recuento bajo de células blancas sanguíneas
- eritema, inflamación, entumecimiento/hormigueo o dolor y/o descamación de la piel de las manos y/o los pies (llamado síndrome mano-pie)
- niveles bajos en sangre de una proteína llamada albúmina
Efectos adversos frecuentes (pueden afectar hasta 1 de cada 10 personas):
- dolor de cabeza
- niveles bajos de potasio en sangre (hipopotasemia) que puede causar debilidad muscular, espasmos o ritmo cardiaco anormal
- niveles bajos de sodio en sangre (hiponatremia) que puede causar cansancio y confusión o espasmos musculares
- sarpullido
- infecciones graves (sepsis)
- obstrucción intestinal; los síntomas pueden incluir estreñimiento y dolor abdominal
Cyramza se ha asociado con reacciones relacionadas con el lugar de la perfusión.
Cyramza puede producir cambios en las pruebas de laboratorio. Estos cambios pueden ser, de entre los efectos adversos mencionados anteriormente: recuento bajo de células blancas en sangre, recuento bajo de plaquetas en sangre, niveles bajos de albúmina, potasio o sodio en sangre, presencia de proteínas en la orina.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Cyramza
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el cartonaje y la etiqueta del vial después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
No congelar ni agitar la solución para perfusión. No administrar la solución si nota cualquier partícula o un color extraño.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Cyramza
- El principio activo es ramucirumab. Un ml de concentrado para solución para perfusión contiene 10 mg de ramucirumab.
- Cada vial de 10 ml contiene 100 mg de ramucirumab.
- Cada vial de 50 ml contiene 500 mg de ramucirumab.
- Los demás componentes son histidina, monihidrocloruro de histidina, cloruro de sodio, glicina (E640), polisorbato 80 (E433) y agua para preparaciones inyectables (ver en la sección 2 “Cyramza contiene sodio”).
Aspecto del producto y contenido del envase
El concentrado para solución para perfusión (o concentrado estéril) es una solución de aspecto transparente a ligeramente opalescente y de tinte incoloro a ligeramente amarillo que se presenta en un vial de vidrio con un tapón de goma.
Cyramza está disponible en envases de:
- 1 vial de 10 ml
- 2 viales de 10 ml
- 1 vial de 50 ml
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Eli Lilly Nederland B.V.
Papendorpseweg 83 3528 BJ Utrecht Países Bajos
Responsable de la fabricación
Lilly, S.A.
Avda de la Industria, 30 Alcobendas 28108 Madrid España
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Lietuva
Eli Lilly Holdings Limited atstovybé Tel. +370 (5) 2649600
Luxembourg/Luxemburg
Eli Lilly Benelux S.A./N.V.
Tél/Tel: + 32-(0)2 548 84 84
Magyarország
Lilly Hungária Kft.
Tel: + 36 1 328 5100
Belgique/Belgie/Belgien
Eli Lilly Benelux S.A./N.V.
Tél/Tel: + 32-(0)2 548 84 84
Etnrapun
Tn "Enn Runu HegepnaHg" E.B. - Etnrapna Ten. + 359 2 491 41 40
Ceská republika ELI LILLY CR, s.r.o.
Tel: + 420 234 664 111
|
Danmark Eli Lilly Danmark A/S Tlf: +45 45 26 60 00 |
Malta Charles de Giorgio Ltd. Tel: + 356 25600 500 |
|
Deutschland Lilly Deutschland GmbH Tel. + 49-(0) 6172 273 2222 |
Nederland Eli Lilly Nederland B.V. Tel: + 31 -(0) 30 60 25 800 |
|
Eesti Eli Lilly Holdings Limited Eesti filiaal Tel: +372 6 817 280 |
Norge Eli Lilly Norge A.S. Tlf: + 47 22 88 18 00 |
|
ELXába OAPMAIEPB-AIAAY A.E.B.E. T r(k: +30 210 629 4600 |
Osterreich Eli Lilly Ges.m.b.H. Tel: + 43-(0) 1 711 780 |
|
España Lilly S.A. Tel: + 34-91 663 50 00 |
Polska Eli Lilly Polska Sp. z o.o. Tel: +48 22 440 33 00 |
|
France Lilly France SAS Tél: +33-(0) 1 55 49 34 34 |
Portugal Lilly Portugal Produtos Farmacéuticos, Lda Tel: + 351-21-4126600 |
|
Hrvatska Eli Lilly Hrvatska d.o.o. Tel: +385 1 2350 999 |
Romania Eli Lilly Romania S.R.L. Tel: + 40 21 4023000 |
|
Ireland Eli Lilly and Company (Ireland) Limited Tel: + 353-(0) 1 661 4377 |
Slovenija Eli Lilly farmacevtska druzba, d.o.o. Tel: +386 (0)1 580 00 10 |
|
Ísland Icepharma hf. Sími + 354 540 8000 |
Slovenská republika Eli Lilly Slovakia, s.r.o. Tel: + 421 220 663 111 |
|
Italia Eli Lilly Italia S.p.A. Tel: + 39- 055 42571 |
Suomi/Finland Oy Eli Lilly Finland Ab Puh/Tel: + 358-(0) 9 85 45 250 |
|
Kúrcpog Phadisco Ltd Tr|7,: +357 22 715000 |
Sverige Eli Lilly Sweden AB Tel: + 46-(0) 8 7378800 |
|
Latvija Eli Lilly Holdings Limited párstávniedba Latvija Tel: +371 67364000 |
United Kingdom Eli Lilly and Company Limited Tel: + 44-(0) 1256 315000 |
|
Fecha de la última revisión de este prospecto: Otras fuentes de información |
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Esta información está destinada únicamente a profesionales del sector sanitario:
No agitar el vial.
Preparar la solución para perfusión usando técnicas asépticas para garantizar la esterilidad de la disolución preparada.
Cada vial es de un solo uso. Antes de la dilución se debe comprobar el contenido de los viales para detectar la posible existencia de partículas o decoloración (el concentrado para solución para perfusión debe ser de transparente a ligeramente opalescente y de tinte incoloro a ligeramente amarillo sin partículas visible). Si se identifican partículas o alteraciones del color, el vial se debe descartar.
Calcular la dosis y el volumen de ramucirumab necesarios para preparar la solución para perfusión. Los viales contienen 100 mg o 500 mg en solución a 10 mg/ml de ramucirumab. Utilizar únicamente como diluyente cloruro de sodio 9 mg/ml (0,9 %) solución inyectable.
En caso de uso de un envase precargado para perfusión intravenosa
Según el volumen de ramucirumab calculado, retirar el volumen correspondiente de solución inyectable de cloruro de sodio 9 mg/ml (0,9 %) del envase precargado de 250 ml para perfusión intravenosa.
El paso del volumen de ramucirumab calculado al envase para perfusión intravenosa se debe realizar asépticamente. El volumen total final del envase debe ser 250 ml. El envase se debe invertir cuidadosamente para garantizar una mezcla adecuada. NO CONGELAR NI AGITAR la solución para perfusión. NO diluir con otras soluciones o coperfundir con otros medicamentos o electrolitos.
En caso de uso de un envase vacío para perfusión intravenosa
El paso de volumen de ramucirumab calculado al envase para perfusión intravenosa vacío se debe realizar asépticamente. Añadir una cantidad suficiente de solución inyectable de cloruro de sodio 9 mg/ml (0,9 %) al envase para alcanzar un volumen total de 250 ml. El envase se debe invertir cuidadosamente para garantizar un mezcla adecuada. NO CONGELAR NI AGITAR. NO diluir con otras soluciones o coperfundir con otros medicamentos o electrolitos.
Tras su dilución y preparación, el medicamento se debe utilizar inmediatamente. Si no lo utiliza inmediatamente, el tiempo y las condiciones de almacenamiento previas al uso son responsabilidad del usuario y normalmente no deberían ser superiores a 24 horas entre 2°C y 8°C.
Los medicamentos de administración parenteral, se deben examinar visualmente antes de la administración para descartar la presencia de partículas. Si se identifican partículas, el vial se debe descartar.
Desechar cualquier porción de ramucirumab remanente en el vial dado que el medicamento no contiene conservantes antimicrobianos.
Administrar a través de una bomba de perfusión. Se debe utilizar una vía de perfusión separada con un filtro de entrada de baja unión a proteínas de 0,22 micrones para la perfusión y al finalizar la perfusión se debe lavar la vía con solución inyectable de cloruro de sodio 9 mg/ml (0,9%).
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
50