Cotellic 20 Mg Comprimidos Recubiertos Con Pelicula
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Cotellic 20 mg comprimidos recubiertos con película
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene cobimetinib hemifumarato equivalente a 20 mg de cobimetinib.
Excipiente con efecto conocido:
Cada comprimido recubierto con película contiene 36 mg de lactosa monohidrato.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película.
Comprimidos recubiertos con película, blancos, redondos, con un diámetro aproximado de 6,6 mm y con la inscripción grabada "COB" en una de las caras.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Cotellic está indicado en combinación con vemurafenib para el tratamiento de pacientes adultos con melanoma no resecable o metastásico con una mutación BRAF V600 (ver secciones 4.4 y 5.1).
4.2 Posología y forma de administración
El tratamiento con Cotellic en combinación con vemurafenib se debe iniciar y ser supervisado por un médico especializado en el uso de medicamentos anticancerígenos.
Antes de comenzar este tratamiento, los pacientes deben tener un diagnóstico de mutación BRAF V600 positiva en el tumor, confirmado por un test validado (ver secciones 4.4 y 5.1).
Posología
La dosis recomendada de Cotellic es de 60 mg (3 comprimidos de 20 mg) una vez al día.
Cotellic se toma en un ciclo de 28 días. Cada dosis consiste en tres comprimidos de 20 mg (60 mg) y se deben tomar una vez al día durante 21 días seguidos (días 1 a 21-período de tratamiento); seguidos de un descanso de 7 días (días 22 a 28-pausa del tratamiento). Cada ciclo siguiente del tratamiento con Cotellic se debería iniciar después de que haya terminado el descanso de 7 días sin tratamiento.
Para información sobre la posología de vemurafenib, consulte su Ficha Técnica.
El tratamiento con Cotellic se debe continuar hasta que el paciente deje de obtener beneficios del mismo o hasta que aparezca una toxicidad no aceptable (ver la Tabla 1 a continuación).
Dosis olvidadas
Si se olvida tomar una dosis, puede tomarla hasta 12 horas antes de la siguiente dosis para mantener el régimen posológico de una administración al día.
Vómitos
En caso de vómitos tras la administración de Cotellic, el paciente no debe tomar una dosis adicional ese día y se debe continuar el tratamiento tal y como se ha prescrito al día siguiente.
Modificaciones generales de la dosis
La decisión de reducir o no la dosis de uno o ambos tratamientos se debe basar en la evaluación por parte del médico de la seguridad o tolerabilidad individual del paciente. La modificación de la dosis de Cotellic es independiente de la modificación de la dosis de vemurafenib.
Si se omiten dosis por toxicidad, estas dosis no se deben sustituir. Una vez se ha reducido la dosis, no se debe aumentar posteriormente.
A continuación la Tabla 1 proporciona una recomendación general para la modificación de la dosis de Cotellic.
Tabla 1 Modificaciones de dosis recomendadas de Cotellic
|
Grado (CTC-Reacción Adversa)* |
Dosis recomendada de Cotellic |
|
Grado 1 o Grado 2 (tolerable) |
Sin reducción de la dosis. Mantener Cotellic a una dosis de 60 mg una vez al día (3 comprimidos) |
|
Grado 2 (intolerable) o Grado 3/4 | |
|
1a aparición |
Interrumpir el tratamiento hasta el Grado < 1, reiniciar el tratamiento con 40 mg una vez al día (2 comprimidos) |
|
2a aparición |
Interrumpir el tratamiento hasta el Grado < 1, reiniciar el tratamiento con 20 mg una vez al día (1 comprimido) |
|
3a aparición |
Considerar la interrupción permanente |
* La intensidad de las reacciones adversas clínicas se clasifican según los Criterios Terminológicos Comunes para Reacciones Adversas v4.0 (CTC-AE)
Recomendación sobre la modificación de la dosis para disfunción ventricular izquierda
Se debe considerar interrumpir permanentemente el tratamiento con Cotellic si se atribuyen síntomas cardíacos a Cotellic y estos no mejoran tras una interrupción temporal.
Tabla 2 Modificaciones de las dosis recomendadas de Cotellic en pacientes con una disminución de la fracción de eyección del ventrículo izquierdo (FEVI) con respecto al inicio
|
Paciente |
Valor FEVI |
Modificación de dosis recomendada de Cotellic |
Valor FEVI tras pausa en el tratamiento |
Dosis diaria recomendada de Cotellic |
|
Asintomático |
> 50% (o 40%-49% y disminución total < 10% con respecto al inicio) |
Continuar con la dosis actual |
N/A |
N/A |
|
< 40% (o 40%-49% y disminución total > 10% con respecto al inicio) |
Interrumpir el tratamiento durante 2 semanas |
Disminución total < 10% con respecto al inicio |
1a aparición: 40 mg | |
|
2a aparición: 20 mg | ||||
|
3 a aparición: Interrupción permanente. | ||||
|
< 40% (o disminución total > 10% con respecto al inicio) |
Interrupción permanente | |||
|
Sintomático |
N/A |
Interrumpir el tratamiento durante 4 semanas |
Asintomático y disminución total < 10% con respecto al inicio |
1a aparición: 40 mg |
|
2a aparición: 20 mg | ||||
|
3 a aparición: Interrupción permanente | ||||
|
Asintomático y < 40% (o disminución total > 10% con respecto al inicio) |
Interrupción permanente | |||
|
Sintomático independientemente de FEVI |
Interrupción permanente |
N/A = No aplica
Se puede continuar el tratamiento con vemurafenib cuando se modifique el tratamiento con Cotellic, si está clínicamente indicado.
Recomendación sobre la modificación de la dosis de Cotellic cuando se utiliza con vemurafenib Alteraciones analíticas hepáticas
En el caso de alteraciones analíticas hepáticas de Grado 1 y 2, se debe continuar el tratamiento con Cotellic y vemurafenib según la dosis prescrita.
Grado 3: Se debe continuar el tratamiento con Cotellic según la dosis prescrita. Se puede reducir la dosis de vemurafenib cuando resulte clínicamente adecuado. Consultar la Ficha Técnica de vemurafenib.
Grado 4:
Se debe interrumpir el tratamiento con Cotellic y el tratamiento con vemurafenib. Si las alteraciones analíticas hepáticas mejoran hasta un Grado < 1 en 4 semanas, se debe reiniciar el tratamiento con Cotellic con una dosis reducida de 20 mg y con vemurafenib a una dosis clínicamente adecuada, según su Ficha Técnica.
Se debe suspender el tratamiento con Cotellic y el tratamiento con vemurafenib si las alteraciones analíticas hepáticas no se resuelven hasta un Grado < 1 en 4 semanas o si reaparecen las alteraciones analíticas hepáticas de Grado 4 después de la mejoría inicial.
Elevaciones de creatina fosfoquinasa (CPK)
No se debe modificar ni suspender la dosis de Cotellic para controlar elevaciones de CPK asintomáticas.
Fotosensibilidad
Se debe controlar la fotosensibilidad de Grado < 2 (tolerable) con cuidados complementarios.
Fotosensibilidad de Grado 2 (intolerable) o Grado > 3: Se debe interrumpir el tratamiento con Cotellic y vemurafenib hasta que se resuelva a Grado < 1. Se puede reiniciar el tratamiento con Cotellic sin ningún cambio en la dosis. La dosis de vemurafenib se debe reducir según resulte clínicamente adecuado, para más información, consultar su ficha técnica.
Rash
Se pueden producir casos de erupciones debidas tanto al tratamiento con Cotellic como a vemurafenib. Se puede interrumpir y/o reducir temporalmente la dosis de Cotellic y/o vemurafenib cuando esté clínicamente indicado. Además, en el caso de:
Una erupción de Grado < 2 (tolerable) se debe controlar con cuidados complementarios. Se puede continuar la administración de la dosis de Cotellic sin modificaciones.
Una erupción acneiforme de Grado 2 (intolerable) o Grado > 3: Se deben seguir las recomendaciones generales de las modificaciones de dosis de Cotellic que figuran en la Tabla 1 . Se puede continuar la administración de la dosis de vemurafenib cuando se modifique el tratamiento con Cotellic (si está clínicamente indicado).
Erupción no-acneiforme o maculopapular de Grado 2 (intolerable) o Grado > 3: Se puede continuar sin modificaciones la administración de la dosis de Cotellic si está clínicamente indicado. La administración de la dosis de vemurafenib se puede interrumpir y/o reducir temporalmente, para más información, consultar su ficha técnica.
Prolongación del intervalo QT
Si durante el tratamiento el intervalo QTc es superior a 500 ms, por favor consulte la Ficha Técnica de vemurafenib (sección 4.2) para modificar la dosis de vemurafenib. No se requiere modificar la dosis de Cotellic cuando se toma en combinación con vemurafenib.
Poblaciones especiales
Pacientes de edad avanzada
No se requiere ningún ajuste de la dosis para pacientes con edad >65 años.
Insuficiencia renal
No se recomienda ningún ajuste de la dosis en pacientes con insuficiencia renal leve o moderada basado en un análisis farmacocinético poblacional (ver sección 5.2). Se dispone de pocos datos sobre Cotellic en pacientes con insuficiencia renal grave, por lo que no se puede descartar algún efecto. Cotellic se debe utilizar con precaución en pacientes con insuficiencia renal grave.
Insuficiencia hepática
No se recomienda ningún ajuste de dosis en pacientes con insuficiencia hepática. Los pacientes con insuficiencia hepática grave podrían presentar concentraciones plasmaticas elevadas de cobimetinib no ligado en comparación con las de los pacientes con una función hepática normal (ver sección 5.2). Se pueden producir alteraciones analíticas hepáticas cuando se usa Cotellic por lo que se debe tener precaución en pacientes con insuficiencia hepática de cualquier grado (ver sección 4.4).
Pacientes no caucásicos
No se ha establecido la seguridad y eficacia de Cotellic en pacientes no-caucásicos.
Población pediátrica
No se ha establecido la seguridad y eficacia de Cotellic en niños y adolescentes menores de 18 años. No se dispone de datos.
Forma de administración
Cotellic es para uso por vía oral. Los comprimidos se deben ingerir enteros con agua. Se pueden tomar con o sin alimentos.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Antes de tomar Cotellic en combinación con vemurafenib, se debe haber confirmado por un test validado que los pacientes tienen un tumor con una mutación BRAF V600 positiva.
Cotellic en combinación con vemurafenib en pacientes que han progresado con un inhibidor BRAF
Existen pocos datos de pacientes en combinación de Cotellic con vemurafenib que han progresado a un tratamiento previo con un inhibidor de BRAF. Estos datos muestran que la eficacia de la combinación será menor en estos pacientes (ver sección 5.1). Por tanto, se deben considerar otras opciones terapéuticas antes de tratar con la combinación a esta población tratada previamente con un inhibidor de BRAF. No se ha establecido la secuencia de tratamientos tras progresión con un tratamiento inhibidor de BRAF.
Cotellic en combinación con vemurafenib en pacientes con metástasis cerebrales.
La seguridad y eficacia de la combinación de Cotellic y vemurafenib no se ha evaluado en pacientes con melanoma BRAF V600 positivo con metástasis en el cerebro. De hecho, se desconoce la actividad intracraneal de cobimetinib (ver secciones 5.1 y 5.2).
Retinopatía serosa
Se ha observado retinopatía serosa (acumulación de fluidos en las capas de la retina) en pacientes tratados con inhibidores de MEK, incluido Cotellic (ver sección 4.8). La mayoría de los casos fueron notificados como coriorretinopatía o desprendimiento de retina.
La mediana de tiempo para la aparición inicial de casos de retinopatía serosa fue de 1 mes (rango de 09 meses). La mayor parte de los casos observados en ensayos clínicos se resolvieron, o mejoraron hasta Grado 1 asintomático, tras la interrupción o la reducción de la dosis.
En cada visita los pacientes deben ser evaluados de nuevos síntomas de alteraciones visuales o empeoramiento de los mismos. Se recomienda un examen oftalmológico si se identifican nuevos síntomas de alteraciones visuales o empeoramiento de los mismos. Si se diagnostica retinopatía serosa, se debería retirar el tratamiento con Cotellic hasta que los síntomas visuales mejoren hasta Grado < 1. La retinopatía serosa se puede controlar mediante la interrupción del tratamiento, la reducción de la dosis o la suspensión del tratamiento (ver la Tabla 1 en la sección 4.2).
Disfunción ventrícular izquierda
Se ha notificado una disminución de la FEVI con respecto al inicio en pacientes que reciben Cotellic (ver sección 4.8). La mediana de tiempo para la aparición inicial de los casos fue de 4 meses (113 meses).
Se debe evaluar la FEVI antes de iniciar el tratamiento para establecer los valores de referencia, y, posteriormente, tras el primer mes de tratamiento y, como mínimo, cada 3 meses o cuando esté clínicamente indicado hasta la suspensión del tratamiento. La disminución de la FEVI con respecto al inicio se puede controlar mediante la interrupción del tratamiento, la reducción de la dosis o la suspensión del tratamiento (ver sección 4.2).
Se deben realizar mediciones de FEVI a todos los pacientes que reinicien el tratamiento con reducción de la dosis de Cotellic después de aproximadamente 2 semanas, 4 semanas, 10 semanas y 16 semanas y, posteriormente, cuando esté clinicamente indicado.
No se han estudiado pacientes con una FEVI inicial por debajo del límite inferior de la normalidad (LLN) ni por debajo del 50%.
Alteraciones analíticas hepáticas
Se pueden producir alteraciones analíticas hepáticas cuando se utiliza Cotellic en combinación con vemurafenib y con el uso de vemurafenib en monoterapia (consultar su ficha técnica).
Se han observado alteraciones analíticas hepáticas, especialmente aumentos de la alanina aminotransferasa (ALT), aspartato aminotransferasa (AST) y fosfatasa alcalina (ALP) en pacientes tratados con Cotellic más vemurafenib (ver sección 4.8).
Las alteraciones de la función hepática se deben monitorizar mediante pruebas analíticas hepáticas antes de iniciar el tratamiento en combinación y cada mes durante el tratamiento, o con mayor frecuencia si está clínicamente indicado (ver sección 4.2).
Las alteraciones analíticas hepáticas de Grado 3 se deben controlar mediante la interrupción del tratamiento con vemurafenib o la reducción de la dosis. Controlar las alteraciones analíticas hepáticas de Grado 4 con interrupción del tratamiento, reducción de la dosis o con suspensión del tratamiento tanto de Cotellic como de vemurafenib (ver sección 4.2).
Diarrea
Se han notificado casos de diarrea de Grado >3 y diarrea grave en pacientes tratados con Cotellic. La diarrea se debe controlar con agentes antidiarreicos y cuidados complementarios. Para la diarrea de Grado >3 producida a pesar de los cuidados complementarios, se debe suspender el tratamiento con Cotellic y vemurafenib hasta que la diarrea mejore a Grado <1. Si reaparece la diarrea de Grado >3, se debe reducir la dosis de Cotellic y vemurafenib (ver sección 4.2).
Intolerancia a la lactosa
Este medicamento contiene lactosa. Los pacientes con raros problemas hereditarios de intolerancia a la galactosa, deficiencia congénita de lactasa o malabsorción de glucosa-galactosa deben consultar con su médico y determinar si los beneficios superan a los riesgos en cada caso particular.
Se debe evitar el uso concomitante de inhibidores potentes de CYP3A durante el tratamiento con Cotellic. Se debe tener precaución si se coadministra un inhibidor moderado de CYP3A con Cotellic. Si es inevitable el uso concomitante de inhibidores potentes o moderados de CYP3A, los pacientes deben ser cuidadosamente monitorizados por su seguridad y aplicadas modificaciones de dosis si está clínicamente indicado (ver tabla 1 en la sección 4.2).
Prolongación del intervalo QT
Si durante el tratamiento el intervalo QTc es superior a 500 ms, por favor consulte las secciones 4.2 y
4.4 de la Ficha Técnica de vemurafenib.
4.5 Interacción con otros medicamentos y otras formas de interacción
Efectos de otros medicamentos sobre cobimetinib
Inhibidores de CYP3A
Cobimetinib es metabolizado por CYP3A y el AUC de cobimetinib aumentó, aproximadamente 7 veces en presencia de un inhibidor potente de CYP3A (itraconazol) en pacientes sanos. La magnitud de interacción en los pacientes podría ser potencialmente menor.
Inhibidores_potentes de CYP3A (ver sección 4.4): evitar el uso concomitante con inhibidores potentes de CYP3A durante el tratamiento con cobimetinib. Entre los inhibidores potentes de CYP3A se incluyen, pero no se limitan sólo a estos, ritonavir, cobicistat, telaprevir, lopinavir, itraconazol, voriconazol, claritromicina, telitromicina, posaconzaol, nefazodona y zumo de pomelo. Si es inevitable el uso concomitante de inhibidores potentes de CYP3A, los pacientes deben ser cuidadosamente monitorizados por su seguridad. Para el uso de inhibidores potentes de CYP3A durante un corto período de tiempo (7 días o menos), hay que considerar la interrupción del tratamiento de cobimetinib durante el tiempo del uso del inhibidor.
Inhibidores moderados CYP3A (ver sección 4.4): Se debe tener precaución si cobimetinib se coadministra con un inhibidor moderado de CYP3A. Entre los inhibidores moderados de CYP3A se incluyen, pero no se limitan sólo a estos, amiodarona, eritromicina, fluconazol, miconazol, diltiazem, verapamilo, delavirdina, amprenavir, fosamprenavir, imatinib. Cuando cobimetinib se coadministra con un inhibidor moderado de CYP3A, los pacientes deben ser cuidadosamente monitorizados por su seguridad.
Inhibidores leves de CYP3A: Cobimetinib puede ser coadministrado con inhibidores leves de CYP3A sin ningún ajuste de dosis.
Inductores de CYP3A
La coadministración de cobimetinib con un inductor potente de CYP3A no se ha evaluado en ningún estudio clínico. Sin embargo, es probable que se produzca una reducción en la exposición a cobimetinib. Por lo tanto, se debe evitar el uso concomitante de inductores moderados y potentes de CYP3A (por ejemplo, carbamazepina, rifampicina, fenitoína y hierba de San Juan). Se deben considerar agentes alternativos sin inducción de CYP3A o con una inducción mínima. Dado que es probable que las concentraciones de cobimetinib se reduzcan significativamente cuando se coadministre con inductores de la CYP3A de moderados a potentes, se puede ver comprometida la eficacia en el paciente.
Cobimetinib es un sustrato de la glicoproteína-P (P-gp). La administración concomitante de inhibidores de la P-gp, como la ciclosporina y el verapamilo, puede aumentar las concentraciones plasmáticas de cobimetinib.
Efectos de cobimetinib sobre otros medicamentos
Sustratos de CYP3A y CYP2D6
Un estudio de interacción entre medicamentos (DDI) en pacientes con cáncer reveló que las concentraciones plasmáticas de midazolam (un sustrato sensible de CYP3A) y dextrometorfano (un sustrato sensible de CYP2D6) no se vieron alteradas en presencia de cobimetinib.
Sustratos de CYP1A2
In vitro, cobimetinib es un inductor potente de CYP1A2 y, por lo tanto, podría reducir la exposición de sustratos de este enzima e.j. teofilina. No se han llevado a cabo estudios clínicos de interacción entre medicamentos para evaluar la relevancia clínica de este hallazgo.
Sustratos de la BCRP
In vitro, cobimetinib es un inhibidor moderado de la BCRP (proteína de resistencia al cáncer de mama). No se han llevado a cabo estudios clínicos de interacción entre medicamentos para evaluar este hallazgo, y no se puede descartar una inhibición clínicamente relevante de la BCRP a nivel intestinal.
Otros agentes anticancerígenos
Vemurafenib
No existe evidencia de ninguna interacción clínicamente significativa entre cobimetinib y vemurafenib en pacientes con melanoma no resecable o metastásico y, por lo tanto, no se recomienda ajustar la dosis.
Efectos de cobimetinib en los sistemas transportadores de fármacos
Estudios in vitro demuestran que cobimetinib no es un sustrato de los transportadores hepáticos OATP1B1,OATP1B3 y OCT1. No obstante, cobimetinib inhibe débilmente estos transportadores. No se ha investigado la importancia clínica de estos hallazgos.
Población pediátrica
Los estudios de interacciones se han realizado sólo en adultos.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil
Se debe indicar a las mujeres en edad fértil que utilicen dos métodos anticonceptivos eficaces, como el preservativo u otro método de barrera (con espermicida, si es posible) durante el tratamiento con Cotellic y durante al menos tres meses después de la suspensión del tratamiento.
Embarazo
No hay datos relativos al uso de Cotellic en mujeres embarazadas. Los estudios realizados con animales han mostrado casos de embrioletalidad y malformaciones fetales de los grandes vasos y el cráneo (ver sección 5.3). No se debe usar Cotellic durante el embarazo a menos que sea claramente necesario y tras una cuidadosa evaluación de las necesidades de la madre y del riesgo para el feto.
Lactancia
Se desconoce si cobimetinib se excreta en la leche materna. No se puede excluir el riesgo en los recién nacidos/lactantes. Se debe decidir si es necesario suspender la lactancia o el tratamiento con Cotellic teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre.
Fertilidad
No hay datos en humanos para cobimetinib. No se han llevado a cabo estudios de fertilidad en animales, pero se han observado efectos adversos en los órganos reproductores (ver sección 5.3). Se desconoce su relevancia clínica.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Cotellic sobre la capacidad para conducir y utilizar máquinas es pequeña. Se han notificado casos de trastornos visuales en algunos pacientes tratados con cobimetinib durante los ensayos clínicos (ver secciones 4.4 y 4.8). Se debe aconsejar a los pacientes que no conduzcan ni utilicen máquinas si experimentan trastornos visuales o algún otro efecto adverso que pueda afectar a su capacidad para conducir y utilizar máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Se ha evaluado la seguridad de Cotellic en combinación con vemurafenib en 247 pacientes con melanoma avanzado con mutación BRAF V600 en el Estudio GO28141. La mediana de tiempo para la aparición de los primeros acontecimientos adversos de Grado > 3 fue de 0,6 meses en el grupo tratado con Cotellic más vemurafenib frente a los 0,8 meses del grupo tratado con placebo más vemurafenib.
También se ha evaluado la seguridad de Cotellic en combinación con vemurafenib en 129 pacientes con melanoma avanzado con mutación BRAF V600 en el Estudio NO25395. El perfil de seguridad del estudio NO25395 fue coherente con el observado en el Estudio GO28141.
En el estudio GO28141, las reacciones adversas más comunes ( 20% ) que se observaron con una frecuencia mayor en el grupo de Cotellic más vemurafenib fueron diarrea, rash, nauseas, pirexia, reacción de fotosensibilidad, aumento de alanina aminotransferasa, aumento de aspartato aminotransferasa, aumento en sangre de creatina fosfoquinasa, y vómitos. Las reacciones adversas más comunes ( 20%) que se observaron con una frecuencia mayor en el grupo de placebo más vemurafenib fueron artralgia, alopecia, e hiperqueratosis. La fatiga se observó en ambos grupos de manera similar.
Por favor, consultar la Ficha Técnica de vemurafenib para una descripción completa de todas las reacciones adversas asociadas al tratamiento con vemurafenib.
Tabla de reacciones adversas
Las reacciones adversas (RAM) se basan en los resultados de un Estudio Fase III (GO28141), multicéntrico, aleatorizado, doble ciego, controlado con placebo que evaluó la seguridad y la eficacia de Cotellic en combinación con vemurafenib en comparación con vemurafenib en monoterapia en pacientes con mutación BRAF V600 positiva que no habían sido previamente tratados y que padecían melanoma no resecable localmente avanzado (estadío IIIc) o melanoma metastásico (estadío IV).
Las frecuencias de las reacciones adversas (RAM) se basan en los análisis de seguridad de los pacientes tratados con cobimetinib mas vemurafenib con una mediana de seguimiento de 11,2 meses (fecha corte de datos 19 Septiembre de 2014).
Las reacciones adversas (RAM) que fueron notificadas en los pacientes con melanoma se han enumerado bajo la clasificación de órganos del sistema MedDRA , frecuencia y grado de gravedad. La siguiente clasificación se ha utilizado para ordenarlas por frecuencia:
Muy frecuentes > 1/10 Frecuentes > 1/100 a < 1/10 Poco frecuentes > 1/1.000 a < 1/100 Raras > 1/10.000 a < 1/1.000 Muy raras < 1/10.000
La Tabla 3 recoge las reacciones adversas relacionadas con el uso de Cotellic. Dentro de cada grupo de frecuencias, se presentan las reacciones adversas en orden decreciente de gravedad y se notificaron de acuerdo con NCI-CTCAE v 4.0 (criterios comunes de toxicidad) para evaluar la toxicidad en el Estudio GO28141.
Tabla 3 Reacciones adversas en pacientes tratados con Cotellic en combinación con vemurafenib en el Estudio GO28141A
|
Sistema de Clasificación de Órganos |
Muy frecuentes |
Frecuentes |
|
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos) |
Carcinoma de células basales, carcinoma de células escamosas cutáneo **, queratoacantoma** | |
|
Trastornos de la sangre y el sistema linfático |
Anemia | |
|
Trastornos del metabolismo y de la nutrición |
Deshidratación, hipofosfatemia, hiponatremia, hiperglucemia. | |
|
Trastornos visuales |
Retinopatía serosa a, visión borrosa |
Alteración visual. |
|
Trastornos vasculares |
Hipertensión, hemorragia* | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Neumonitis | |
|
Trastornos gastrointestinales |
Diarrea, náuseas, vómitos | |
|
Trastornos de la piel y del tejido subcutáneo |
Fotosensibilidadb, erupción, erupción maculopapular, dermatitis acneiforme, hiperqueratosis** | |
|
Trastornos generales y alteraciones en el lugar de administración |
Pirexia, escalofríos | |
|
Exploraciones complementarias |
Aumento de CPK en sangre, aumento de la ALT, aumento de la AST, aumento de la Gamma Glutamiltransferasa (GGT), aumento de ALP en sangre |
Disminución de la fracción de eyección, aumento de la bilirrubina en sangre |
A Fecha corte de datos 19 Septiembre de 2014
* Consultar el párrafo Hemorragia en la sección “Descripción de las reacciones adversas seleccionadas”.
** Consultar el párrafo Carcinoma cutáneo de células escamosas, queratoacantoma e hiperqueratosis en la sección “Descripción de las reacciones adversas seleccionadas”.
a Incluye casos de coriorretinopatia y desprendimiento de retina indicativos de retinopatía serosa (ver sección 4.4)
b Cifra combinada incluye informes de reacción de fotosensibilidad, quemaduras solares, dermatitis solar, elastosis actínica
Descripción de las reacciones adversas seleccionadas
Hemorragia
Se han notificado casos de hemorragia con mayor frecuencia en el grupo tratado con Cotellic más vemurafenib que en el grupo tratado con placebo más vemurafenib (todos los tipos y grados: 13% frente al 7%). En el grupo tratado con Cotellic más vemurafenib se observaron mayores frecuencias de hemorragia cerebral (1% frente al 0%), hemorragia del tracto gastrointestinal (4% frente al 1%), hemorragia del sistema reproductor (2% frente <1%) y hematuria (3% frente al 1%).
La mayor parte de los casos fueron de Grado 1 ó 2 y no graves (12% de los pacientes en el grupo tratado con Cotellic más vemurafenib frente al 7% de los pacientes en el grupo tratado con placebo más vemurafenib). Un 1% de los pacientes de cada brazo sufrió casos de Grado 3-4.
La mediana de tiempo hasta el primer acontecimiento fue de 4,4 meses (rango de 0,0 a 12,7 meses) en el grupo tratado con Cotellic más vemurafenib.
Fotosensibilidad
Se ha observado fotosensibilidad con mayor frecuencia en el grupo tratado con Cotellic más vemurafenib respecto al grupo tratado con placebo más vemurafenib (47% frente al 35%). La mayor parte de los casos fueron de Grado 1 ó 2, mientras que los casos de Grado > 3 ocurrieron en el 4% de los pacientes del grupo tratado con Cotellic más vemurafenib frente al 0% de los pacientes del grupo tratado con placebo más vemurafenib.
No se apreciaron tendencias en el tiempo de aparición de los casos de Grado > 3. Los casos de fotosensibilidad de Grado > 3 en el grupo tratado con Cotellic más vemurafenib se trataron con medicamentos tópicos primarios junto con interrupciones de la dosis tanto de cobimetinib como de vemurafenib (ver sección 4.2).
No se observó evidencia de fototoxicidad con Cotellic en monoterapia.
Carcinoma de células escamosas cutáneo, queratoacantoma e hiperqueratosis
Se han notificado casos de carcinoma de células escamosas cutáneo con menor frecuencia en el grupo tratado con Cotellic más vemurafenib respecto al grupo tratado con placebo más vemurafenib (todos los grados: 3% frente al 13%). Se han notificado casos de queratoacantoma con menor frecuencia en el grupo tratado con Cotellic más vemurafenib respecto al grupo tratado con placebo más vemurafenib (todos los grados: 2% frente al 9%). Se han notificado casos de hiperqueratosis con menor frecuencia en el grupo tratado con Cotellic más vemurafenib respecto al grupo tratado con placebo más vemurafenib (todos los grados: 11% frente al 30%).
Retinopatía serosa
Se han notificado casos de retinopatía serosa en pacientes tratados con Cotellic (ver sección 4.4). Para pacientes que notifican trastornos visuales nuevos o empeoramiento de los mismos, se recomienda un examen oftalmológico. La retinopatía serosa se puede controlar mediante la interrupción del tratamiento, la reducción de la dosis o la suspensión del tratamiento (ver la Tabla 1 en la sección 4.2).
Disfunción ventrícular izquierda
Se han notificado casos de disminución de la FEVI con respecto al inicio en pacientes tratados con Cotellic (ver sección 4.4). Se debe evaluar la FEVI antes de iniciar el tratamiento para establecer los valores de referencia, y posteriormente, tras el primer mes de tratamiento y, como mínimo, cada 3 meses o cuando esté clínicamente indicado hasta la suspensión del tratamiento. La disminución de la FEVI con respecto al inicio se puede controlar mediante la interrupción del tratamiento, la reducción de la dosis o la suspensión del tratamiento (ver sección 4.2).
Alteraciones en pruebas de laboratorio Alteraciones analíticas hepáticas
Se han observado alteraciones analíticas hepáticas, específicamente ALT, AST y ALP, en pacientes tratados con Cotellic en combinación con vemurafenib (ver sección 4.4).
Se deben monitorizar las pruebas analíticas hepáticas antes de iniciar el tratamiento en combinación y cada mes durante el tratamiento, o con mayor frecuencia si está clínicamente indicado (ver sección 4.2).
Aumento de la creatina fosfoquinasa en sangre
Se observaron aumentos asintomáticos de los niveles de CPK en sangre con mayor frecuencia en el grupo tratado con Cotellic más vemurafenib respecto al grupo tratado con placebo más vemurafenib en el Estudio GO28141 (ver sección 4.2). Se observó un caso de rabdomiolisis en cada grupo de tratamiento de este estudio con los consecuentes aumentos de CPK en sangre.
La Tabla 4 muestra la frecuencia de las alteraciones analíticas hepáticas medidas y el aumento de creatina fosfoquinasa para todos los grados y los Grados 3-4.
Tabla 4 Función hepática y otras pruebas analíticas observadas en el Estudio Fase III GO28141
|
Cambios en los datos de laboratorio notificados |
Cobimetinib más Vemurafenib (n = 247) (%) |
Plac Vemi (n |
ebo más urafenib = 246) %) | |
|
Todos los grados |
Grados 3-4 |
Todos los grados |
Grados 3-4 | |
|
Pruebas de Función Hepática | ||||
|
Aumento de la ALP |
69 |
7 |
55 |
3 |
|
Aumento de la ALT |
67 |
11 |
54 |
5 |
|
Aumento de la AST |
71 |
7 |
43 |
2 |
|
Aumento de la GGT |
62 |
20 |
59 |
17 |
|
Aumento de la bilirrubina en sangre |
33 |
2 |
43 |
1 |
|
Otras alteraciones Analíticas | ||||
|
Aumento de CPK en sangre |
70 |
12 |
14 |
<1 |
Poblaciones especiales
Pacientes de edad avanzada
En el estudio Fase III con Cotellic en combinación con vemurafenib en pacientes con melanoma no resecable o metastásico (n=247), 183 pacientes (74%) eran 65 años, 44 pacientes (18%) tenía entre 65-74 años, 16 (6%) tenían entre 75-84 años, y 4 pacientes (2%) eran >85 años. La proporción de pacientes que sufrieron reacciones adversas (RAM) fue similar en pacientes 65 años que en pacientes >65 años. Los pacientes >65 años fueron más propensos que los pacientes 65 años a sufrir reacciones adversas graves (RAG) y reacciones adversas que produjeron la suspensión del tratamiento con cobimetinib.
Insuficiencia Renal
No se ha llevado a cabo ningún ensayo farmacocinético en sujetos con insuficiencia renal. No se recomienda ajustar la dosis para la insuficiencia renal deleve a moderada en base a los resultados del análisis farmacocinético poblacional. Se dispone de pocos datos sobre Cotellic en pacientes con insuficiencia renal grave. Cotellic se debe usar con precaución en pacientes con insuficiencia renal grave.
Insuficiencia Hepática
No se recomienda ningún ajuste de dosis en pacientes con insuficiencia hepática (ver sección 5.2). Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
No existen experiencias de sobredosis en ensayos clínicos con humanos. En el caso de sospecha de sobredosis, se debe suspender el tratamiento con cobimetinib e iniciarse cuidados complementarios. No existe ningún antídoto específico para tratar la sobredosis de cobimetinib.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agentes antineoplásticos código ATC: L01XE38 Mecanismo de acción
Cobimetinib es un inhibidor reversible, selectivo, alostérico y oral que bloquea la ruta de las proteínquinasas activadas por mitógenos (MAPK) dirigiéndose a la quinasa activada por mitógenos reguladora de la señal extracelular (MEK) 1 y MEK 2, lo que provoca una inhibición de la fosforilación de la quinasa reguladora de la señal extracelular (ERK) 1 y ERK 2. Por lo tanto, cobimetinib bloquea la proliferación de células inducida por la ruta de la MAPK mediante la inhibición de la señalización a nivel de MEK1/2.
En los modelos preclínicos, la combinación de cobimetinib y vemurafenib mostró que la acción simúltanea sobre las proteínas BRAFV600 mutadas y las proteínas MEK en las células del melanoma, la combinación de los dos productos que inhiben la reactivación de la ruta MAPK a través de MEK1/2, provoca una mayor inhibición de la señalización intracelular y una disminución de la proliferación de las células tumorales.
Eficacia clínica y seguridad
No se disponen de datos sobre la seguridad o eficacia de Cotellic en combinación con vemurafenib en pacientes con metastasis de sistema nervioso central o en pacientes con melanoma maligno no-cutáneo.
Estudio GO28141 (coBRIM)
El estudio GO28141 es un estudio Fase III, multicéntrico, aleatorizado, doble ciego, controlado con placebo que evalua la seguridad y la eficacia de Cotellic en combinación con vemurafenib en comparación con vemurafenib más placebo en pacientes con mutación BRAF V600 positiva que no habían sido previamente tratados y que padecían melanoma no resecable localmente avanzado (estadío IIIc) o melanoma metastásico (estadío IV).
En el Estudio GO28141 solamente se incluyeron pacientes con un estado de desarrollo ECOG de 0 y 1. Se excluyeron del estudio pacientes con un estado de desarrollo ECOG de 2 o superior.
Tras la confirmación de la mutación BRAF V600, mediante el test de Cobas® 4800 para la mutación BRAF V600, se aleatorizaron a 495 pacientes no tratados previamente con melanoma no resecable localmente avanzado o melanoma metastásico para recibir:
• Placebo una vez al día en los días 1-21 de cada ciclo de tratamiento de 28 días y 960 mg de vemurafenib dos veces al día en los días 1-28, o bien
• Cotellic 60 mg una vez al día en los días 1-21 de cada ciclo de tratamiento de 28 días y 960 mg de vemurafenib dos veces al día en los días 1-28.
La supervivencia libre de progresión (SLP) evaluada por el investigador (INV) fue la variable principal. Las variables secundarias de eficacia incluyeron la supervivencia global (SG), la tasa de respuesta objetiva, la duración de la respuesta (DdR) evaluado por el INV y la SLP evaluada por un Comité de Revisión Independiente (CRI).
Las principales características basales incluyeron: el 58% de los pacientes eran varones, la mediana de edad era de 55 años (rango de 23 a 88 años), el 60% tenía melanoma metastásico en estadío M1c y la proporción de pacientes con LDH elevado era del 46,3% en el grupo tratado con cobimetinib más vemurafenib y del 43,0% en el grupo tratado con placebo más vemurafenib.
En el Estudio GO28141 había 89 pacientes (18,1%) de 65-74 años, 38 pacientes (7,7%) de 75-84años y 5 pacientes (1,0%) de 85 años o mayores.
Tabla 5 Resultados de eficacia del Estudio GO28141 (coBRIM)
|
Cotellic + vemurafenib |
Placebo + vemurafenib | |
|
N=247 |
N=248 | |
|
Variable principal3^ | ||
|
Supervivencia Libre de Progresión (SLP) | ||
|
Mediana (meses) |
12,3 |
7,2 |
|
(IC del 95%) |
(9,5; 13,4) |
(5,6; 7,5) |
|
Hazard ratio (IC del 95%)b |
0,58 (0,46; 0,72) | |
|
Variables secundarias^ | ||
|
Supervivencia Global (SG)g | ||
|
Mediana (meses) |
22,3 |
17,4 |
|
(IC del 95%) |
(20,3; NE) |
(15,0; 19,8) |
|
0,70 (95% CI: 0,55; 0,90) | ||
|
Hazard ratio (IC del 95%)b |
(valor-p= |
0,0050e) |
|
Tasa de respuesta objetiva (TRO) |
172 (69,6%) |
124 (50,0%) |
|
(IC del 95%) para la TROc |
(63,5%; 75,3%) |
(43,6%,; 56,4%) |
|
Diferencia en TRO % (IC del 95%)d |
19,6% (11,0; 28,3) | |
|
Mejor respuesta global | ||
|
Respuesta completa |
39 (15,8%) |
26 (10,5%) |
|
Respuesta parcial |
133 (53,8%) |
98 (39,5%) |
|
Enfermedad estable |
44 (17,8%) |
92 (37,1%) |
|
Duración de Respuesta (DdR) | ||
|
Mediana DdR (meses) |
13 |
9,2 |
|
(IC del 95%) para la mediana |
(11,1; 16,6) |
(7,5; 12,8) |
NE= No evaluable
a Evaluada y confirmada por el investigador (INV) utilizando RECIST v1.1 b Análisis estratificado por región geográfica y clasificación metastásica (estadío de la enfermedad) c Utilizando el método Clopper-Pearson d Utilizando el método Hauck-Anderson
e El valor-p de SG (0,0050) superó el límite pre-especificado (valor-p <0,0499)
f La fecha de corte de datos para el análisis actualizado de SLP y para las variables secundarias de TRO, MRG y DdR es el 16 de Enero de 2015. La mediana del seguimiento fue de 14,2 meses.
g La fecha de corte de datos para el análisis final de SG es el 28 de agosto de 2015 y la mediana del seguimiento fue de 18,5 meses
Los análisis iniciales para el Estudio GO28141 se realizaron con una fecha de corte de datos 9 de mayo de 2014. Se observó una mejoría significativa en la variable principal, SLP valorada por investigador, en los pacientes asignados al grupo de Cotellic más vemurafenib en comparación con el grupo de placebo más vemurafenib (HR 0,51 (0,39; 0,68); valor p 0,0001).
La mediana estimada para la SLP valorada por el investigador fue 9,9 meses para el grupo de Cotellic más vemurafenib vs. 6,2 meses para el grupo de placebo más vemurafenib. La mediana estimada para la SLP por el revisor independiente fue 11,3 meses para el grupo de Cotellic más vemurafenib vs. 6,0 meses para el grupo de placebo más vemurafenib (HR 0,60 (0,45; 0,79); valor p = 0,0003). La tasa de respuesta objetiva (TRO) para el grupo de Cotellic más vemurafenib fue de 67,6% vs 44,8% para el grupo de placebo más vemurafenib. La diferencia en TRO fue 22,9% (valor p 0,0001).
El análisis de SG final para el Estudio GO28141 se realizaró con una fecha de corte de datos 28 de agosto de 2015. Se observó una mejoría significativa en SG en los pacientes asignados al grupo Cotellic más vemurafenib en comparación con el grupo de placebo más vemurafenib (Figura 1). Las SG estimadas a 1 año (75%) y a 2 años (48%) para el grupo Cotellic más vemurafenib fueron mayores que las de para el grupo de placebo más vemurafenib (64% y 38% respectivamente).
Figura 1 Curvas de Kaplan-Meier de supervivencia global final - Población intención de tratar
(fecha de corte de datos: 28 agosto 2015)
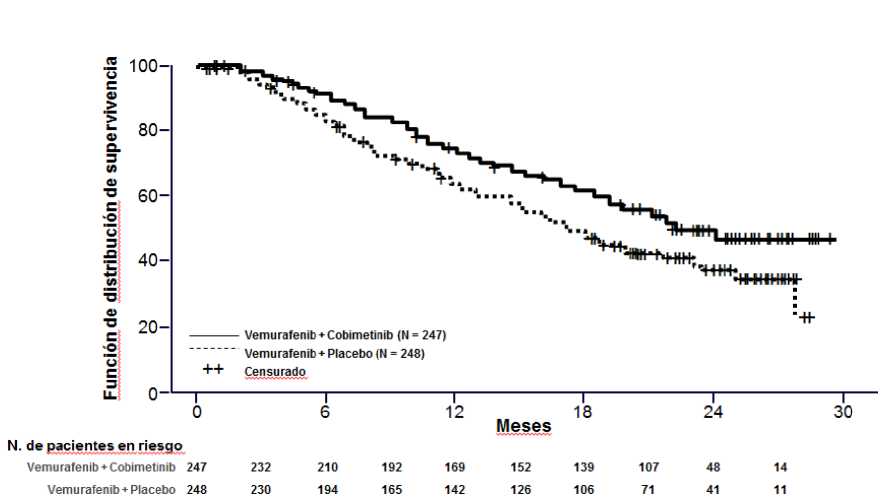
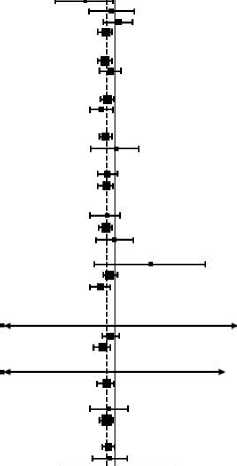
Figura 2: Gráficos de Hazard Ratios de supervivencia global final en los análisis de subgrupo-Población intención de tratar (Fecha de corte de datos: 28 agosto de 2015)
Placebo + vemurafenib Cobimetinlb+vemurafenib
n =248 n=247
Mediana Mediana Hazard
|
Factores de rtesao básales |
N |
n |
Eventos |
(meses) |
n |
Eventos |
(meses) |
Ratio |
Cl del 95% |
|
Toctos los pacientes * |
495 |
248 |
141 |
17-4 |
247 |
114 |
22-3 |
0-70 |
(0-54-0-89) |
|
Fase de la enfermedad | |||||||||
|
tile """"""....."" |
34 |
13 |
6 |
19-1 |
21 |
4 |
NE |
0-29 |
(0-08-1-03) |
|
M1A |
80 |
40 |
9 |
NE |
40 |
8 |
NE |
0-85 |
(0-33-2-19) |
|
MtB |
82 |
42 |
20 |
23-3 |
40 |
22 |
19-4 |
1-13 |
(0-62-2-08) |
|
MIC |
299 |
153 |
106 |
14-8 |
146 |
30 |
18-9 |
0-65 |
(0-48-0-87) |
|
Fase de fa enfermedad HIcj'MIafMIb.or M1c) | |||||||||
|
MtC |
299 |
153 |
106 |
14-8 |
146 |
80 |
18-9 |
0-65 |
(0-48-0-87) |
|
No resecable HIC/M1A/M1B |
196 |
95 |
35 |
NE |
101 |
34 |
NE |
0-63 |
(0-52-1-33) |
|
Grupo de edad (años] | |||||||||
|
<65 |
362 |
179 |
99 |
18-3 |
183 |
85 |
22-1 |
0-75 |
(0-56-1-01) |
|
>65 |
133 |
69 |
42 |
14-7 |
64 |
29 |
24-1 |
0-56 |
(0-35-0-91) |
|
Raza | |||||||||
|
Blanco |
462 |
235 |
135 |
17-4 |
227 |
104 |
22-8 |
0-68 |
(0-53-0-88) |
|
No blanco |
33 |
13 |
6 |
NE |
20 |
10 |
22-3 |
1-00 |
(0-36-2-76) |
|
Sexo | |||||||||
|
Mujer |
209 |
108 |
54 |
22-7 |
101 |
40 |
NE |
0-72 |
(0-48-1-08) |
|
Hombre |
286 |
140 |
87 |
15-0 |
146 |
74 |
21-1 |
0-66 |
(0-48-0-90) |
|
Reoión Geoaráfíca | |||||||||
|
AustralraíNueva Zelanda/Otros |
78 |
38 |
16 |
23-0 |
40 |
13 |
NE |
0-71 |
(0-34-1-48) |
|
Europa |
366 |
184 |
111 |
16-1 |
182 |
87 |
22-8 |
0-67 |
(0-51-0-89) |
|
N. America |
51 |
26 |
14 |
22-7 |
25 |
14 |
19-2 |
0-95 |
(0-45-2-00) |
|
ECQG estado funcional Descono cirro |
8 |
4 |
2 |
NE |
4 |
3 |
15-7 |
4-34 |
(0-42, 44.42) |
|
Ó |
348 |
164 |
83 |
19.8 |
184 |
83 |
23-8 |
0-30 |
(0-59-1-09) |
|
1 |
138 |
SO |
56 |
11.7 |
58 |
28 |
21-8 |
0-53 |
(0-34-0-84) |
|
2 |
1 |
1 |
0 |
NE |
NE |
(NE-NE) | |||
|
Detección en suero LDH | |||||||||
|
desconocido ' |
11 |
6 |
5 |
9-4 |
5 |
0 |
NE |
<0-01 |
(0-00-NE) |
|
Elevado |
216 |
104 |
70 |
11-2 |
112 |
73 |
14-8 |
0-77 |
(0-56-1-07) |
|
Normal |
268 |
138 |
66 |
23-3 |
130 |
41 |
NE |
0-59 |
(0-40-0-87) |
|
Me test. cerebrales previamente tratad os |
3 |
2 |
1 |
NE |
1 |
0 |
NE |
<0-01 |
(0,00-NE) |
|
No |
492 |
246 |
140 |
17-4 |
246 |
114 |
22-3 |
0-70 |
(0-55-0-89) |
|
Terap(a adyuyante previa |
48 |
24 |
13 |
19-1 |
24 |
10 |
NE |
0-76 |
(0-33-1-75) |
|
No |
447 |
224 |
128 |
17-4 |
223 |
104 |
22-3 |
0-69 |
(0-53-0-89) |
|
Estado mutación BRA Fvecc | |||||||||
|
V600E |
344 |
174 |
101 |
17-5 |
170 |
82 |
21-9 |
0-73 |
(0-55-0-98) |
|
V600K |
56 |
32 |
17 |
16-7 |
24 |
11 |
24-1 |
0-79 |
(0-37-1-69) |
Mejor Mejor
Cobjmetirib+ Placebo + vem urafen jb yemu rafe nib
+
i
o-i ....... ......Yo
El estado de salud global/la calidad de vida relacionada con la salud notificada por el paciente se midió mediante el Cuestionario de Calidad de Vida EORTC - Core 30 (QLQ-C30). Los resultados para todos los ámbitos funcionales y la mayor parte de los síntomas (pérdida de apetito, estreñimiento, náuseas y vómitos, disnea, dolor, fatiga) mostraron que el cambio medio desde el inicio fue similar entre los dos grupos de tratamiento y no mostraron un cambio clínicamente significativo (todos los resultados fueron < 10 puntos de cambio con respecto al inicio).
Estudio NO25395 (BRIM7)
Se evaluó la eficacia de Cotellic en el Estudio Fase Ib NO25395, que se diseñó para evaluar la seguridad, tolerabilidad, farmacocinética y eficacia de Cotellic cuando se combina con vemurafenib para el tratamiento de pacientes con melanoma no resecable o metastásico con mutación BRAFV600 positiva (detectado por el test de Cobas® 4800 para la mutación BRAF V600) no resecable o metastásico.
En este estudio se trataron 129 pacientes con Cotellic y vemurafenib: 63 eran pacientes sin tratamiento previo con un inhibidor BRAF (BRAFi) y 66 eran pacientes que habían progresado previamente con un tratamiento anterior de vemurafenib. De los 63 pacientes sin tratamiento previo con BRAFi, 20 habían recibido un tratamiento sistémico anterior para melanoma avanzado, siendo la mayoría (el 80%) inmunoterapia.
Los resultados de la población sin tratamineto previo con BRAFi del Estudio NO25395, por lo general, eran consistentes con los del Estudio GO28141. Los pacientes sin tratamiento previo con BRAFi (n=63) alcanzaron una tasa de respuesta objetiva del 87%, incluyendo una respuesta completa en el 16% de los pacientes. La mediana de duración de la respuesta fue de 14,3 meses. La mediana de la SLP para los pacientes sin tratamiento previo con BRAFi fue de 13,8 meses, con una mediana de tiempo de seguimiento de 20,6 meses.
Entre los pacientes que habían progresado con vemurafenib (n=66), la tasa de respuesta objetiva fue del 15%. La mediana de duración de la respuesta fue de 6,8 meses. La mediana de la SLP para los pacientes que habían progresado con vemurafenib fue de 2,8 meses, con una mediana de tiempo de seguimiento de 8,1 meses.
En los pacientes sin tratamiento previo con inhibidor BRAF, la mediana de supervivencia global fue 28,5 meses (IC del 95% 23,3-34,6). En los pacientes que habían progresado con la terapia del inhibidor BRAF, la mediana de supervivencia global fue 8,4 meses (IC del 95% 6,7-11,1).
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Cotellic en uno o más grupos de población pediátrica en tumores sólidos malignos (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Tras la administración oral de 60 mg en pacientes con cáncer, cobimetinib mostró una tasa moderada de absorción con una mediana de Tmáx de 2,4 horas. La Cmáx media en el estado estacionario y AUC0_24 fue de 273 ng/ml y de 4340 ng.h/ml, respectivamente. El ratio de acumulación medio en estado estacionario fue de aproximadamente 2,4 veces.
Cobimetinib tiene una farmacocinética lineal en el rango de dosis de ~3,5 mg a 100 mg.
La biodisponibilidad absoluta de cobimetinib fue del 45,9% (IC del 90%: 39,7%, 53,1%) en voluntarios sanos. Se realizó un estudio de balance de masas en voluntatios sanos, el cual demostró que cobimetinib se metabolizaba ampliamente y se eliminaba en forma de heces. La fracción absorbida fue del ~88%, lo que indica una alta absorción y metabolismo de primer paso.
La farmacocinética de cobimetinib no se altera cuando se administra en el estado de plenitud (comida rica en grasas) comparado con el estado de ayunas en sujetos sanos. Puesto que los alimentos no alteran la farmacocinética de cobimetinib, este se puede administrar con o sin ella.
Distribución
In vitro cobimetinib se une en un 94,8% a las proteínas plasmáticas humanas. No se observó ninguna unión preferente a los glóbulos rojos humanos (relación sangre - plasma: 0,93).
El volumen de distribución fue de 1.050 l en sujetos sanos a los que se administró una dosis intravenosa de 2 mg. El volumen de distribución aparente fue de 806 l en pacientes con cáncer en base al análisis farmacocinético poblacional.
Cobimetinib es un sustrato de P-gp in vitro. Se desconoce el transporte a través de la barrera hemato encefálica.
Biotransformación
La oxidación mediante CYP3A y la glucuronidación mediante UGT2B7 parecen ser las rutas metabólicas principales de cobimetinib. Cobimetinib es la fracción predominante en el plasma. No se observaron metabolitos oxidativos superiores al 10% de la radioactividad circulante total, ni metabolitos humanos específicos en el plasma. El fármaco inalterado en las heces y la orina representa el 6,6% y el 1,6% de la dosis administrada, respectivamente, lo que indica que cobimetinib es principalmente metabolizado y tiene una eliminación renal mínima. In vitro los datos muestran que cobimetinib no es un inhibidor de OAT1, OAT3 u OCT2.
Eliminación
Cobimetinib y sus metabolitos se caracterizaron en un estudio de balance de masas en sujetos sanos. Como promedio, se recuperó el 94% de la dosis en un plazo de 17 días. Cobimetinib en gran parte se metabolizó y se eliminó en forma de heces.
Tras la administración intravenosa de una dosis de 2 mg de cobimetinib, el aclaramiento plasmático medio (Cl) fue de 10,7 l/h. El Cl aparente medio tras la administración de una dosis oral de 60 mg en pacientes con cáncer fue de 13,8 l/h.
La semivida de eliminación media tras la administración oral de cobimetinib fue de 43,6 horas (rango:
23,1 a 69,6 horas). En consecuencia, cobimetinib puede tardar hasta 2 semanas tras interrumpir el tratamiento en eliminarse completamente de la circulación sistémica.
Poblaciones especiales
Basándose en un análisis farmacocinético poblacional, el sexo, la raza, la etnia, el ECOG basal, la insuficiencia renal leve y moderada no afectaron a la farmacocinética de cobimetinib. La edad y el peso corporal basal se identificaron como covariables estadísticamente significativas respecto al aclaramiento y el volumen de distribución de cobimetinib, respectivamente. Sin embargo, el análisis de sensibilidad sugiere que ninguna de estas covariables tuvo un impacto clínicamente significativo en la exposición en el estado estacionario.
Sexo
El sexo no tienen ningún efecto en la exposición de cobimetinib, basándose en un análisis farmacocinético poblacional que incluye a 210 mujeres y 277 hombres.
Edad avanzada
La edad no tiene ningún efecto en la exposición de cobimetinib, basándose en un análisis farmacocinético poblacional que incluye a 133 pacientes > 65 años.
Basándose en datos preclínicos y en el estudio de balance de masas en humanos, cobimetinib principalmente es metabolizado, con una eliminación renal mínima. No se ha realizado ningún estudio farmacocinético formal en pacientes con insuficiencia renal.
Un análisis farmacocinético poblacional con datos de 151 pacientes con insuficiencia renal leve (aclaramiento de creatinina (ClCr) de 60 a menos de 90 ml/min), 48 pacientes con insuficiencia renal moderada (ClCr de 30 a menos de 60 ml/min), y 286 pacientes con función renal normal (ClCr superior o igual a 90 ml/min) mostró que el ClCr no tuvo ninguna influencia significativa en la exposición de cobimetinib.
La insuficiencia renal de leve a moderada no influye en la exposición de cobimetinib basándose en el análisis farmacocinético poblacional. Se dispone de pocos datos sobre Cotellic en pacientes con insuficiencia renal grave.
Insuficiencia hepática
Se evaluó la farmacocinética de cobimetinib en 6 sujetos con insuficiencia hepática leve (Child Pugh A), 6 sujetos con insuficiencia hepática moderada (Child Pugh B), 6 sujetos con insuficiencia hepática grave (Child Pugh C) y en 10 sujetos sanos. Las exposiciones sistémicas totales de cobimetinib después de una única dosis fueron similares en pacientes con insuficiencia hepática leve o moderada en comparación con los sujetos sanos, mientras que los sujetos con insuficiencia hepática grave tuvieron exposiciones totales a cobimetinib inferiores (proporción de media geométrica AUC0-<X) 0,69 en comparación con sujetos sanos) que no se consideran clínicamente significativas. Las exposiciones de cobimetinib no ligado fueron similares entre sujetos con insuficiencia hepática moderada y leve en comparación con la de los sujetos con función hepática normal mientras que sujetos con insuficiencia hepática grave tuvieron aproximadamente exposiciones dos veces más altas (ver sección 4.2).
Población pediátrica
No se ha realizado ningún estudio para investigar la farmacocinética de cobimetinib en pacientes pediátricos.
5.3 Datos preclínicos sobre seguridad
No se han realizado estudios de carcinogenicidad con cobimetinib. Los estudios de genotoxicidad estándar con cobimetinib dieron resultado negativo.
No se ha realizado ningún estudio de fertilidad específico en animales con cobimetinib. En los estudios toxicológicos, se observaron cambios degenerativos en los tejidos reproductivos, incluyendo un aumento de la apoptosis/necrosis del cuerpo lúteo y vesícula seminal, células epiteliales vaginales y epididimales en ratas y células epiteliales epididimales en perros. Se desconoce su importancia clínica.
Cuando se administró a ratas embarazadas, cobimetinib causó embrioletalidad y malformaciones fetales de los grandes vasos y el cráneo en exposiciones sistémicas similares a la exposición humana con la dosis recomendada.
No se ha evaluado la seguridad cardiovascular de cobimetinib en combinación con vemurafenib in vivo. In vitro, cobimetinib produjo una inhibición moderada del canal del ión hERG (IC50 0,5 pM
[266 ng/ml]), lo cual es aproximadamente 18 veces superior a las concentraciones plasmáticas máximas (Cmáx) a los 60 mg que es la dosis que se va a comercializar (Cmáxno ligada=14 ng/ml [0,03 pM]).
Los estudios de toxicidad en ratas y perros identificaron cambios degenerativos generalmente reversibles en la médula ósea, el tracto gastrointestinal, la piel, el timo, la glándula adrenal, el hígado, el bazo, los ganglios linfáticos, los riñones, el corazón, los ovarios y la vagina en exposiciones plasmáticas inferiores a los niveles clínicos eficaces. Las efectos tóxicos que limitan la dosis incluyen
ulceraciones cutáneas, exudados superficiales y acantosis en ratas e inflamación activa crónica y degeneración del esófago asociada con grados variables de gastroenteropatía en perros.
En un estudio de toxicidad de dosis repetidas en ratas jóvenes, las exposiciones sistémicas a cobimetinib fueron de 2 a 11 veces superiores a los 10 días después de nacer que a los 38 días, cuando las exposiciones fueron similares a las de las ratas adultas. En las ratas jóvenes, la administración de cobimetinib dio lugar a cambios similares a los vistos en los estudios de toxicidad pivotales en ratas adultas, incluyendo cambios degenerativos reversibles en el timo y el hígado, descenso del peso de la tiroides/paratiroides y del bazo, aumento del fósforo, la bilirrubina y la masa sanguínea de glóbulos rojos, y descenso de los triglicéridos. En animales jóvenes se produjo una mortalidad a una dosis (3 mg/kg) que no dio lugar a mortalidad en animales adultos.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo del comprimido Lactosa monohidrato Celulosa microcristalina (E460)
Croscarmelosa sódica (E468)
Estearato de magnesio (E470b)
Recubrimiento con película Alcohol de polivinilo Dióxido de titanio (E171)
Macrogol 3350 Talco (E553b)
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
4 años.
6.4. Precauciones especiales de conservación
Este medicamento no requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Blísters transparentes de PVC/PVDC que contienen 21 comprimidos. Cada envase contiene 63 comprimidos.
6.6. Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/15/1048/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
20 de Noviembre 2015
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
Nombre y dirección del fabricante responsable de la liberación de los lotes
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (Ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2. de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo,o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CARTONAJE
1. NOMBRE DEL MEDICAMENTO
Cotellic 20 mg comprimidos recubiertos con película cobimetinib
2. PRINCIPIOACTIVO
Cada comprimido recubierto con película contiene cobimetinib hemifumarato equivalente a 20 mg de cobimetinib.
3. LISTA DE EXCIPIENTES
Los comprimidos también contienen lactosa. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
63 comprimidos recubiertos con película
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral
Se debe seguir el tratamiento durante 21 días consecutivos, seguidos de una pausa de 7 días
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO
UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE
COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/15/1048/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
cotellic
17. IDENTIFICADOR ÚNICO - CÓDIGO DE BARRAS 2D
Incluido el código de barras 2D que lleva el identificador único
18. IDENTIFICADOR ÚNICO - INFORMACIÓN EN CARACTERES VISUALES
PC:
SN:
NN:
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS BLÍSTER
1. NOMBRE DEL MEDICAMENTO
Cotellic 20 mg comprimidos recubiertos con película cobimetinib
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Ltd
3. FECHA DE CADUCIDAD
EXP
4. NÚMERO DE LOTE
Lot
5. OTROS
B. PROSPECTO
Prospecto: Información para el paciente
Cotellic 20 mg comprimidos recubiertos con película cobimetinib
▼ Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
• Conserve este prospecto, ya que puede tener que volver a leerlo.
• Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
• Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
• Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es Cotellic y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Cotellic
3. Cómo tomar Cotellic
4. Posibles efectos adversos
5. Conservación de Cotellic
6. Contenidos del envase e información adicional
1. Qué es Cotellic y para qué se utiliza Qué es Cotellic
Cotellic es un medicamento contra el cáncer que contiene el principio activo cobimetinib.
Para qué se utiliza Cotellic
Cotellic se utiliza para tratar a pacientes adultos con un tipo de cáncer de piel llamado melanoma, que se ha extendido a otras partes del cuerpo o no puede ser extraído por cirugía.
• Se usa en combinación con otro medicamento contra el cáncer llamado vemurafenib.
• Solo se puede utilizar en pacientes cuyo cáncer tenga un cambio (mutación) en una proteína llamada "BRAF". Antes de empezar el tratamiento, su médico le hará una prueba para esta mutación. Este cambio puede hacer que el melanoma se desarrolle.
Cómo funciona Cotellic
Cotellic actúa sobre una proteína llamada "MEK", que es importante para controlar el crecimiento de las células cancerosas. Cuando se utiliza Cotellic en combinación con vemurafenib (que actúa en la proteína "BRAF" cambiada), ralentiza todavía más o detiene el crecimiento de su cáncer.
2. Qué necesita saber antes de empezar a tomar Cotellic No tome Cotellic:
• Si es alérgico al cobimetinib o a alguno de los demás componentes de este medicamento (incluidos en la sección 6).
Si tiene dudas, consulte a su médico, farmacéutico o enfermero antes de tomar Cotellic.
Advertencias y precauciones
Consulte a su médico, farmacéutico o enfermero antes de empezar a tomar Cotellic si tiene:
• Un problema visual
• Problemas cardíacos
• Problemas hepáticos.
Si le ha sucedido algo de lo anterior (o no está seguro), consulte a su médico, farmacéutico o enfermero antes de tomar Cotellic.
• Problemas visuales
Cotellic puede causar problemas visuales (ver también “Problemas visuales” en la sección 4).
Informe a su médico inmediatamente si tiene los siguientes síntomas: visión borrosa, distorsionada, parcialmente deficitaria o cualquier otro cambio en la vista durante el tratamiento. Su médico debe examinar sus ojos si experimenta algún problema visual nuevo o un empeoramiento del mismo mientras toma Cotellic.
• Problemas cardíacos
Cotellic puede disminuir la cantidad de sangre que bombea su corazón (ver también “Problemas cardíacos” en la sección 4). Su médico debe realizarle pruebas antes y durante el tratamiento con Cotellic para comprobar que su corazón puede bombear bien la sangre. Informe inmediatamente a su médico si siente su corazón acelerado, vibrante o late de forma desigual o si experimenta mareos, vahídos, dificultad para respirar, cansancio o hinchazón en las piernas.
• Problemas hepáticos
Cotellic puede aumentar la cantidad de algunas enzimas hepáticas en la sangre durante el tratamiento. Su médico le realizará análisis de sangre para controlar estas cantidades y si su hígado funciona bien.
• Diarrea
Informe inmediatamente a su médico si tiene diarrea. La diarrea grave puede provocar la pérdida de líquidos corporales (deshidratación). Siga las instrucciones de su médico para saber que hacer para ayudar a prevenir o tratar la diarrea.
Niños y adolescentes
No se recomienda el uso de Cotellic en niños y adolescentes. Se desconocen los efectos de Cotellic en niños menores de 18 años.
Uso de Cotellic con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o pudiera tener que tomar cualquier otro medicamento. Esto se debe a que Cotellic puede afectar al funcionamiento de otros medicamentos. También otros medicamentos pueden afectar al funcionamiento de Cotellic.
Consulte a su médico antes de tomar Cotellic si está tomando:
|
Medicamento o principio activo |
Indicación del medicamento |
|
Itraconazol, claritromicina, eritromicina, telitromicina, voriconazol, rifampicina, posaconazol, fluconazol, miconazol |
para algunas infecciones fúngicas o bacterianas |
|
ritonavir,cobicistat, lopinavir, delavirdina, amprenavir, fosamprenavir |
para infecciones VIH |
|
telaprevir |
para hepatitis C |
|
nefazodona |
para depresión |
|
amiodarona |
para ritmo cardíaco irregular |
|
diltiazem, verapamilo |
para tensión arterial elevada |
|
imatinib |
para cáncer |
|
carbamazepina, fenitoína |
para convulsiones (ataques) |
|
Hierba de San Juan |
medicamento a base de plantas utilizado para el tratamiento de la depresión. Está disponible sin receta médica. |
Uso de cotellic con alimentos y bebidas
Evitar tomar Cotellic con zumo de pomelo. La razón es porque puede aumentar la cantidad de Cotellic en su sangre.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento.
• No se recomienda tomar Cotellic durante el embarazo. Aunque no se han estudiado los efectos de Cotellic en mujeres embarazadas, puede causar daño permanente o defectos congénitos en el feto.
• Si se queda embarazada durante el tratamiento con Cotellic o en los 3 meses posteriores a su última dosis, informe inmediatamente a su médico.
• Se desconoce si Cotellic pasa a la leche materna. Si está dando el pecho, su médico le informará de los beneficios y los riesgos de tomar Cotellic.
Métodos anticonceptivos
Las mujeres en edad fértil deben utilizar dos métodos anticonceptivos eficaces, como el preservativo u otro método de barrera (con espermicida, si es posible) durante el tratamiento y durante al menos 3 meses tras finalizar el tratamiento. Consulte a su médico sobre el mejor método anticonceptivo para usted.
Conducción y uso de máquinas
Cotellic puede afectar a su capacidad para conducir o usar máquinas. Evite conducir o usar máquinas si tiene algún problema en la visión u otros problemas que podrían afectar su capacidad e.j. si siente mareo o cansancio . Consulte a su médico si no está seguro.
Cotellic contienen lactosa
Los comprimidos de Cotellic contienen lactosa (un tipo de azúcar). Si su médico le ha indicado que padece una intolerancia a ciertos azúcares, informe a su médico antes de tomar este medicamento.
3. Cómo tomar Cotellic
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Cuánto se tiene que tomar
La dosis recomendada es de 3 comprimidos (un total de 60 mg) una vez al día.
• Tome los comprimidos cada día durante 21 días (lo que se llama un "período de tratamiento").
• Después de 21 días, no tome ningún comprimido de Cotellic durante 7 días. Durante esta pausa de 7 días en el tratamiento de Cotellic, debe continuar tomando vemurafenib, tal y como le indicó su médico.
• Empiece su período de tratamiento de 21 días con Cotellic después de la pausa de 7 días.
• Si experimenta algún efecto adverso, su médico puede considerar disminuir la dosis, interrumpir el tratamiento temporalmente o permanentemente. Siga exactamente las instrucciones de administración de Cotellic indicadas por su médico.
Toma del medicamento
• Trague los comprimidos enteros con agua.
• Cotellic se puede tomar con o sin alimentos.
Si siente náuseas
Si siente náuseas (vomita) después de tomar Cotellic, no tome una dosis adicional de Cotellic ese día. Continúe tomando Cotellic de forma normal al día siguiente.
Si toma más Cotellic del que debe
Si toma más Cotellic del que debe, informe inmediatamente a su médico. Lleve el envase del medicamento y este prospecto con usted.
Si olvidó tomar Cotellic
• Si faltan más de 12 horas hasta la siguiente dosis, tome la dosis olvidada tan pronto como lo acuerde.
• Si faltan menos de 12 horas hasta la siguiente dosis, sáltese la dosis olvidada y siga tomando Cotellic según la pauta habitual.
• No tome una dosis doble para compensar la dosis olvidada.
Si interrumpe el tratamiento con Cotellic
Es importante tomar Cotellic durante el tiempo de tratamiento recetado por su médico .
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Si experimenta algún efecto adverso, su médico puede considerar disminuir la dosis, interrumpir el tratamiento temporalmente o permanentemente.
Consulte también el prospecto de vemurafenib, que se utiliza en combinación con Cotellic.
Efectos adversos graves
Informe inmediatamente a su médico si presenta alguno de los efectos adversos que aparecen a continuación o si alguno de estos empeora durante el tratamiento.
Problemas visuales (muy frecuentes: pueden afectar a más de 1 de cada 10 personas)
Cotellic puede causar problemas visuales. Alguno de estos problemas visuales puede dar lugar a “retinopatía serosa” (acumulación de fluido debajo de la retina del ojo). Los síntomas de retinopatía serosa incluyen:
• visión borrosa
• visión distorsionada
• visión parcialmente deficiente
• cualquier otro cambio en la vista.
Problemas cardíacos (frecuentes: pueden afectar hasta a 1 de cada 10 personas)
Cotellic puede disminuir la cantidad de sangre bombeada por el corazón. Los síntomas pueden incluir:
• sensación de mareo
• sensación de desmayo
• sensación de falta de aire
• sensación de cansancio
• sensación de ritmo cardíaco acelerado, vibrante o si su corazón late de forma desigual
• hinchazón en las piernas.
Diarrea (muy frecuentes: pueden afectar a más de 1 de cada 10 personas)
Informe inmediatamente a su médico si tiene diarrea y siga las instrucciones de su médico para saber qué hacer para ayudar a prevenir o tratar la diarrea.
Otros efectos adversos
Informe a su médico, farmacéutico o enfermero si experimenta alguno de los efectos adversos siguientes:
Muy frecuentes (pueden afectar a más de 1 de cada 10 personas)
• aumento de la sensibilidad de la piel a la luz del sol
• erupción cutánea
• sensación de mareo (náusea)
• fiebre
• escalofríos
• aumento de las enzimas hepáticas (se muestra en los análisis de sangre)
• alteraciones en los resultados de análisis de sangre relacionadas concreatina fosfoquiinasa, una enzima que se encuentra principalmente en el corazón, cerebro y músculo esquelético.
• vómitos
• erupción cutánea con una zona descolorida plana o bultos como acné
• tensión arterial alta
• anemia (nivel bajo de glóbulos rojos en sangre)
• hemorragia
• espesor cutáneo anormal.
Frecuentes: pueden afectar hasta 1 de cada 10 personas
• algunos tipos de cáncer de piel como carcinoma de células basales, carcinoma cutáneo de células escamosas y queratoacantoma
• deshidratación, una afección en la que su cuerpo no tiene tanto fluido como debería
• disminución de los niveles de fosfato y sodio (se muestran en los análisis sangre)
• aumento del nivel de azúcar ( se muestran en los análisis de sangre)
• aumento de un pigmento hepático llamado bilirrubina en la sangre. Los síntomas incluyen coloración amarillenta de la piel y ojos
• inflamación de los pulmones que puede provocar dificultad para respirar y puede resultar mortal llamada “neumonitis”.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
Conservación de Cotellic
5.
• Mantener este medicamento fuera de la vista y del alcance de los niños.
• No tome este medicamento después de la fecha de caducidad que aparece en el blíster y caja después de CAD. La fecha de caducidad es el último día del mes que se indica.
• Este medicamento no requiere condiciones especiales de conservación.
• Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Cotellic
• El principio activo es cobimetinib. Cada comprimido recubierto con película contiene cobimetinib hemifumarate equivalente a 20 mg de cobimetinib.
• Los demás componentes son:
• lactosa monohidrato, celulosa microcristalina, croscarmelosa sódica y estearato de magnesio en el núcleo del comprimido; y
• alcohol de polivinilo, dióxido de titanio, macrogol y talco en el recubrimiento con película. Aspecto de Cotellic y contenido del envase
Los comprimidos recubiertos con película de Cotellic son blancos, redondos y tienen la inscripción grabada "COB" en una de las caras. Hay disponible un tamaño de envase: 63 comprimidos (3 blísters de 21 comprimidos).
Titular de la Autorización de Comercialización
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
Responsable de la fabricación
Roche Pharma AG Emil-Barell-Strasse 1 D-79639
Grenzach-Wyhlen
Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien N.V. Roche S.A. Tél/Tel: +32 (0) 2 525 82 11 |
Lietuva UAB “ Roche Lietuva” Tel.: +370 5 2546799 |
|
Etarapun Pom Etarapna EOOfl Tea: +359 2 818 44 44 |
Luxembourg/Luxemburg (Voir/siehe Belgique/Belgien) |
|
Ceská republika Roche s. r. o. Tel.: +420 - 2 20382111 |
Magyarország Roche (Magyarország) Kft. Tel.: +36 - 23 446 800 |
|
Danmark Roche a/s Tel.: +45 - 36 39 99 99 |
Malta (ver United Kingdom) |
|
Deutschland Roche Pharma AG Tel.: +49 (0) 7624 140 |
Nederland Roche Nederland B.V. Tel.: +31 (0) 348 438050 |
|
Eesti Roche Eesti OÜ Tel.: + 372 - 6 177 380 |
Norge Roche Norge AS Tel.: +47 - 22 78 90 00 |
|
EXlába Roche (Hellas) A.E. Tel.: +30 210 61 66 100 |
Osterreich Roche Austria GmbH Tel: +43 (0) 1 27739 |
|
España Roche Farma S.A. Tel: +34 - 91 324 81 00 |
Polska Roche Polska Sp.z o.o. Tel.: +48 - 22 345 18 88 |
|
France Roche Tél: +33 (0) 1 47 61 40 00 |
Portugal Roche Farmacéutica Química, Lda Tel: +351 - 21 425 70 00 |
|
Hrvatska Roche d.o.o. Tel: +385 1 4722 333 |
Romania Roche Romania S.R.L. Tel: +40 21 206 47 01 |
|
Ireland Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700 |
Slovenija Roche farmacevtska druzba d.o.o. Tel: +386 - 1 360 26 00 |
|
Ísland Roche a/s c/o Icepharma hf Sími: +354 540 8000 Italia Roche S.p.A. Tel: +39 - 039 2471 |
Slovenská republika Roche Slovensko, s.r.o. Tel: +421 - 2 52638201 Suomi/Finland Roche Oy Puh/Tel: +358 (0) 10 554 500 |
|
Kúnpoq r.A.Exapáxqg & Era AxS. Ttfk: +357 - 22 76 62 76 |
Sverige Roche AB Tel: +46 (0) 8 726 1200 |
|
Latvija Roche Latvija SIA Tel: +371 - 6 7039831 |
United Kingdom Roche Products Ltd. Tel: +44 (0) 1707 366000 |
Fecha de la última revisión de este prospecto: {MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
39