Constella 290 Microgramos Capsulas Duras
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
Constella 290 microgramos cápsulas duras.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula contiene 290 microgramos de linaclotida.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Cápsula dura.
Cápsula opaca de color blanco a blanquecino y naranja (18 mm x 6,35 mm) con la inscripción «290» en tinta gris.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Constella está indicado para el tratamiento sintomático del síndrome del intestino irritable con estreñimiento (SII-E) entre moderado y grave en adultos.
4.2 Posología y forma de administración
Posología
La dosis recomendada es una cápsula (290 microgramos), una vez al día.
El médico deberá evaluar periódicamente la necesidad de continuar con el tratamiento. La eficacia de linaclotida quedó demostrada en estudios de hasta seis meses de duración realizados a doble ciego y controlados con placebo. Si los pacientes no han experimentado una mejora de los síntomas tras cuatro semanas de tratamiento, se recomienda que se los someta a una nueva exploración y que se reconsideren los beneficios y riesgos de proseguir con la administración del medicamento.
Poblaciones especiales
Pacientes con insuficiencia renal o hepática
No es necesario un ajuste de la dosis en los pacientes con insuficiencia hepática o renal (ver sección 5.2).
Pacientes de edad avanzada
En los pacientes de edad avanzada, aunque no es necesario un ajuste de la dosis, el tratamiento debe vigilarse con atención y controlarse periódicamente (ver sección 4.4).
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de linaclotida en niños de 0 a 18 años. No se dispone de datos.
Constella no se debe utilizar en niños ni en adolescentes (ver las secciones 4.4 y 5.1).
Forma de administración
Vía oral. La cápsula debe tomarse al menos treinta minutos antes de una comida (ver sección 4.5).
4.3 Contraindicaciones
Hipersensibilidad a linaclotida o a alguno de los excipientes incluidos en la sección 6.1.
Pacientes en los que exista certeza o sospecha de la existencia de obstrucción gastrointestinal mecánica.
4.4 Advertencias y precauciones especiales de empleo
Constella debe utilizarse después de haber descartado enfermedades orgánicas y confirmado el diagnóstico de SII-E de moderada a grave (ver sección 5.1).
Los pacientes deben ser conscientes de la posible aparición de diarrea y hemorragia digestiva baja durante el tratamiento y es necesario indicarles que, en casos de diarrea grave o prolongada o hemorragia digestiva baja, deben informar a su médico (ver sección 4.8).
En caso que el paciente sufra diarrea prolongada (por ejemplo, de más de una semana) o grave, deberá considerarse la necesidad de suspender temporalmente el tratamiento con linaclotida hasta que el episodio de diarrea haya remitido y pedir consejo médico. Asimismo, se recomienda tomar precauciones adicionales en pacientes que tengan tendencia a sufrir alteraciones del equilibrio hídrico o electrolítico (como pueden ser ancianos, o los pacientes con enfermedades cardiovasculares, diabetes o hipertensión) y evaluar la posibilidad de realizar un control seguimiento de electrolitos.
No se han llevado a cabo estudios de linaclotida en pacientes con trastornos inflamatorios crónicos del tracto gastrointestinal como la enfermedad de Crohn y la colitis ulcerosa, por lo que no se aconseja emplear el medicamento en estos pacientes.
Pacientes de edad avanzada
Los datos en pacientes de edad avanzada son escasos (ver sección 5.1). Dado el alto riesgo de padecer diarrea observado en los ensayos clínicos (ver sección 4.8), debe prestarse especial atención a estos pacientes y evaluar cuidadosa y periódicamente la relación beneficio-riesgo.
Población pediátrica
No se ha estudiado la administración de Constella en niños y adolescentes, por tanto, no se debe utilizar en esta población. Dado que se tiene constancia de la sobreexpresión del receptor de la guanilato ciclasa C (GC-C) en edades tempranas, los niños menores de dos años pueden ser especialmente sensibles a los efectos de linaclotida.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han realizado estudios de interacciones. Linaclotida rara vez resulta detectable en el plasma tras su administración a las dosis clínicas recomendadas, y en estudios realizados in vitro se ha demostrado que no es ni sustrato ni inhibidor/inductor del sistema enzimático del citocromo P450 y no interacciona con los transportadores más relevantes que participan en la captación y eflujo de medicamentos (ver sección 5.2).
Por otro lado, en un estudio clínico sobre interacciones alimentarias llevado a cabo con individuos sanos se puso de manifiesto que, a las dosis terapéuticas, linaclotida no resultaba detectable en plasma tras haber comido ni en ayunas. La toma de Constella produjo deposiciones más frecuentes y sueltas, así como más acontecimientos adversos gastrointestinales, tras haber comido que cuando se administró en ayunas (ver sección 5.1). La cápsula debe tomarse 30 minutos antes de una comida (ver sección 4.2).
El tratamiento concomitante con inhibidores de la bomba de protones, laxantes o AINE puede aumentar el riesgo de diarrea.
En los casos de diarrea grave o prolongada es posible que se vea afectada la absorción de otros medicamentos administrados por vía oral. La eficacia de los anticonceptivos orales puede disminuir por lo que se recomienda el uso de un método anticonceptivo adicional que evite el fracaso de la anticoncepción oral (ver la ficha técnica del anticonceptivo oral). Deben tomarse precauciones especiales cuando se prescriban medicamentos que se absorban en el tracto intestinal y con un estrecho índice terapéutico como la levotiroxina ya que su eficacia puede verse reducida.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Los datos sobre el uso de linaclotida en mujeres embarazadas son escasos. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para la reproducción (ver sección 5.3). Como medida de precaución, es preferible evitar el uso durante el embarazo.
Lactancia
Puesto que la exposición sistémica a linaclotida es mínima, no resulta probable su excreción en la leche materna, si bien no se ha evaluado este factor. Aunque a las dosis terapéuticas no se prevé efecto alguno en recién nacidos o lactantes alimentados con leche materna, en ausencia de información referida a seres humanos no se aconseja el uso durante la lactancia.
Fertilidad
Los estudios realizados en animales indican que el medicamento no afecta a la fertilidad masculina ni a la femenina.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de Constella sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
En estudios clínicos controlados se suministró linaclotida por vía oral a 1166 pacientes afectados de SII-E, de los cuales 892 recibieron linaclotida a la dosis recomendada de 290 microgramos diarios. La exposición total comprendida en el programa de desarrollo clínico fue superior a 1,5 pacientes-año. La reacción adversa notificada con mayor frecuencia asociada al tratamiento con Constella fue diarrea, principalmente de intensidad leve a moderada, que se observó en menos del 20% de los pacientes. Raramente en los casos más graves, ésta puede, como consecuencia, conducir a deshidratación, hipopotasemia, disminución de la concentración de bicarbonato en sangre, mareo e hipotensión ortostática.
Otras reacciones adversas frecuentes (> 1%) fueron dolor abdominal, distensión abdominal y flatulencia.
Tabla resumen de las reacciones adversas
En estudios clínicos controlados en los que se administró la dosis recomendada de 290 microgramos al día, se comunicaron las reacciones adversas enumeradas a continuación, cuya frecuencia se define utilizando el siguiente convenio: muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000), muy raras (<1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
|
Clasificación MedDRA por órganos y sistemas |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
Frecuencia no conocida |
|
Infecciones e infestaciones |
Gastroenteritis vírica | ||||
|
Trastornos gastrointestinales |
Diarrea |
Dolor abdominal Flatulencia Distensión abdominal |
Incontinencia fecal Urgencia defecatoria Hemorragia digestiva baja, incluida hemorragia hemorroidal y hemorragia rectal Náuseas Vómitos | ||
|
Trastornos de la piel y del tejido subcutáneo |
Erupción cutánea | ||||
|
Trastornos del metabolismo y de la nutrición |
Hipopotasemia Deshidratación Disminución del apetito | ||||
|
Trastornos del sistema nervioso |
Mareo | ||||
|
Trastornos vasculares |
Hipotensión ortostática | ||||
|
Exploraciones complementarias |
Reducción del bicarbonato en sangre |
Descripción de determinadas reacciones adversas
La diarrea constituye la reacción adversa más frecuente y responde a la acción farmacológica del principio activo. El 2% de los pacientes tratados experimentaron diarrea grave y el 5% de los pacientes discontinuaron el tratamiento debido a diarrea en los ensayos clínicos.
La mayoría de los casos de diarrea referidos fueron de leves (43%) a moderados (47%); el 2% de los pacientes tratados experimentaron una diarrea grave. Aproximadamente la mitad de los episodios de diarrea comenzaron durante la primera semana de tratamiento.
La diarrea desapareció dentro de los siete días siguientes en aproximadamente un tercio de los pacientes, sin embargo 80 pacientes (el 50%) experimentaron diarrea con una duración de más de 28 días (lo que representa el 9,9% de todos los pacientes tratados con linaclotida).
El 5% de los pacientes discontinuaron el tratamiento debido a diarrea en los ensayos clínicos. En los pacientes cuya diarrea motivó la retirada del tratamiento, el trastorno se resolvió al cabo de unos días de la retirada.
En comparación con la población global de individuos afectados de SII-E que se incluyeron en los ensayos clínicos, los pacientes ancianos (> 65 años), hipertensos y diabéticos reportaron diarrea con mayor frecuencia.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
La sobredosis puede provocar síntomas resultantes de una exageración de los efectos farmacodinámicos conocidos del medicamento, principalmente diarrea. En un estudio llevado a cabo con voluntarios sanos que recibieron una dosis única de 2897 microgramos (hasta diez veces la dosis terapéutica recomendada), el perfil de seguridad de estos sujetos fue similar al de la población global tratada, siendo la diarrea el acontecimiento adverso comunicado con mayor frecuencia.
En caso de sobredosis, el paciente deberá ser tratado con la terapia y medidas sintomáticas que resulten necesarias.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Otros agentes contra el estreñimiento, código ATC: A06AX04 Mecanismo de acción
Linaclotida es un agonista del receptor de la guanilato ciclasa C (GC-C) que presenta actividad analgésica visceral y secretora.
Linaclotida es un péptido sintético de 14 aminoácidos que está relacionado estructuralmente con la familia de péptidos endógenos de la guanilina. Tanto linaclotida como su metabolito activo se unen al receptor de la GC-C en la superficie luminal del epitelio intestinal. Se ha demostrado que linaclotida, mediante su acción en la GC-C, reduce el dolor visceral y acelera el tránsito gastrointestinal en modelos animales y el colónico en humanos. Con la activación de la GC-C se produce un incremento de las concentraciones de monofosfato de guanosina cíclico (GMP cíclico) en el ámbito extracelular e intracelular; a escala extracelular, el GMP cíclico disminuye la actividad de las fibras nociceptivas, con lo que en modelos animales se verificó menos dolor visceral. En el plano intracelular, el GMP cíclico causa la secreción de cloruro y bicarbonato en la luz intestinal mediante la activación del regulador de la conductancia transmembranaria de la fibrosis quística (RTFQ), lo que aumenta la cantidad de líquido intestinal y acelera el tránsito.
Efectos farmacodinámicos
En un estudio de diseño cruzado sobre interacciones alimentarias se administró Constella de 290 microgramos a 18 voluntarios sanos durante siete días tanto tras haber comido como en ayunas. Cuando Constella se tomó inmediatamente después de un desayuno rico en grasas, se observaron heces más frecuentes y sueltas, así como más acontecimientos adversos gastrointestinales, que en el caso de la toma en ayunas.
Eficacia clínica y seguridad
La eficacia de linaclotida se demostró en dos estudios clínicos de fase III, aleatorizados, con doble enmascaramiento y controlados con placebo realizados en pacientes afectados de SII-E. En uno de estos estudios (estudio 1) se trató a 802 pacientes con Constella de 290 microgramos o placebo, tomados una vez al día durante 26 semanas; en el segundo estudio clínico (estudio 2) participaron 800 pacientes que recibieron tratamiento durante 12 semanas y fueron sometidos después a otro proceso de aleatorización para pasar a un período de tratamiento adicional de 4 semanas. En las dos semanas iniciales previas al tratamiento, los pacientes experimentaron un dolor abdominal cuya puntuación media fue de 5,6 (en una escala de 0 a 10), con un 2,2% de días sin dolor, una hinchazón de 6,6 de media (en una escala de 0 a 10) y un promedio de 1,8 movimientos intestinales espontáneos (MIE) por semana.
Las características de los pacientes que tomaron parte en los ensayos clínicos de fase III fueron las siguientes: media de edad de 43,9 años [intervalo de 18-87 años, con un 5,3% > 65 años], siendo mujeres el 90,1% del total. Todos los pacientes cumplían los criterios de Roma II para el SII-E y debieron referir una media de dolor abdominal > 3 en una escala de puntuación numérica de 0 a 10 (criterios que corresponden a una población con SII de moderado a grave), < 3 movimientos intestinales espontáneos completos y < 5 MIE por semana a lo largo de un período inicial de dos semanas.
En ambos estudios clínicos, los criterios de valoración principales (múltiples) fueron la existencia de una tasa de pacientes que presentasen alivio del SII durante 12 semanas y una tasa de pacientes que refiriesen dolor abdominal o incomodidad a lo largo del mismo período. Por paciente que presentase un grado de alivio del SII se entendió aquel cuyos síntomas se aliviasen considerable o completamente durante al menos el 50% del período de tratamiento, mientras que los pacientes que refiriesen dolor abdominal o incomodidad serían los que experimentasen una mejora del 30% o más en por lo menos el 50% de dicho período.
En los datos de 12 semanas, el estudio 1 revela que el 39% de los pacientes tratados con linaclotida , frente al 17% de los tratados con placebo, experimentaron alivio del SII (p < 0,0001), y el 54% de los pacientes tratados con linaclotida , frente al 39% de los que recibieron el placebo, mostraron una respuesta al dolor abdominal y a la incomodidad (p < 0,0001). El estudio 2 pone de manifiesto que el 37% de los pacientes que habían recibido el tratamiento con linaclotida, en comparación con el 19% de los tratados con placebo, manifestaron alivio del SII (p < 0,0001) y el 55% de los tratados con linaclotida , en comparación con el 42% de los tratados con placebo, mostraron respuesta al dolor abdominal y a la incomodidad (p < 0,0002).
En los datos de 26 semanas, el estudio 1 revela que el 37% y el 54% de los pacientes tratados con linaclotida, frente al 17% y el 36% de los tratados con placebo, mostraron alivio del SII (p < 0,0001) y respuesta al dolor abdominal y a la incomodidad (p < 0,0001) respectivamente.
En ambos estudios estas mejoras se observaron ya antes de finalizar la semana 1 y se mantuvieron durante todo el período de tratamiento (figuras 1 y 2). Se ha observado que linaclotida no causa efecto rebote cuando el tratamiento se interrumpió al cabo de 3 meses de tratamiento continuo.
Fig. 1: Respuesta del paciente al alivio del SII
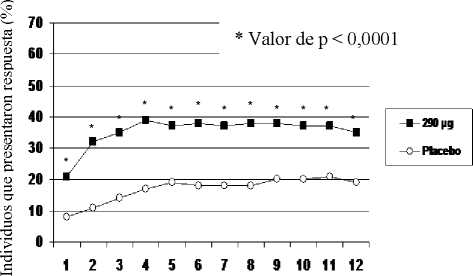
Semanadel ensayo
Fig. 2: Respuesta del paciente al dolor andommel y a la sensación de incomodidad
|
* Valor de p < 0,0001 | |
|
: | |
|
^290 |ig | |
|
0/~ |
-C^ Placebo |
0
1 0-1-,-,-,-,-,-,-,-,-,-,-,-
| 1 2 3 4 5 6 7 8 9 10 11 12
£
Semana del ensayo
Estudios clínicos combinados de fase III relativos a 1a eficacia (estudios 1 y 2) Estudios clínicos combinados di! fase III relativos a la eficacia (estudios 1 y 2)
Método: observación de casos (población de intención de tratar) Método: obsemación dp casos (ooblación de intención de tratar)
En comparación con los pacientes tratados con placebo, aquellos a los que se les administró linaclotida experimentaron una mejora (p < 0,0001) en otros signos y síntomas del SII-E (hinchazón, frecuencia de movimientos intestinales espontáneos completos [MIEC], esfuerzo y consistencia de las heces), según se indica en la tabla que figura a continuación. Estos efectos se alcanzaron al cabo de una semana y se mantuvieron a lo largo de la totalidad de los períodos de tratamiento.
Efectos de Constella en los síntomas de SII-E durante las 12 primeras semanas de tratamiento en los estudios clínicos combinados de fase III relativos a la eficacia (estudios 1 y 2)
|
Principales parámetros secundarios de la eficacia |
Placebo (N = 797) |
Linaclotida (N = 805) | |||||
|
Perío-do inicial Media |
12 semana s Media |
Cambio con respecto al período inicial Media |
Perío-do inicial Media |
12 semanas Media |
Cambio con respect o al período inicial Media |
Diferen -cia media de MC | |
|
Hinchazón (EPN de 11 puntos) |
6,5 |
5,4 |
-1,0 |
6,7 |
4,6 |
-1,9 |
-0,9* |
|
MIEC/semana |
0,2 |
1,0 |
0,7 |
0,2 |
2,5 |
2,2 |
1,6* |
|
Consistencia de las heces (puntuación de la escala de heces de Bristol; BSFS) |
2,3 |
3,0 |
0,6 |
2,3 |
4,4 |
2,0 |
1,4* |
|
Esfuerzo (escala ordinal de 5 puntos) |
3,5 |
2,8 |
-0,6 |
3,6 |
2,2 |
-1,3 |
-0,6* |
* p < 0,0001, linaclotida frente a placebo. MC: mínimos cuadrados MIEC: movimiento intestinal espontáneo completo
El tratamiento con linaclotida también conllevó una mejora significativa en la medida de la calidad de vida validada y específica para este trastorno (SII-QoL, calidad de vida en el síndrome del intestino irritable; p < 0.0001) y en el EuroQoL (p = 0,001); así, en el 54% de los pacientes tratados con linaclotida se alcanzó una respuesta clínicamente significativa en el SII-QoL (> 14 puntos de diferencia), frente a un 39% en el caso de los pacientes a los que se les administró placebo.
Población pediátrica
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos clínicos realizados con Constella en uno o más grupos de la población pediátrica con estreñimiento funcional; ver sección 4.2 para consultar la información sobre el uso en población pediátrica.
5.2 Propiedades farmacocinéticas
Absorción
En general, linaclotida es apenas detectable en el plasma tras la administración oral de las dosis terapéuticas y, por lo tanto, no pueden calcularse los parámetros farmacocinéticos típicos.
Después de la toma de dosis únicas de hasta 966 microgramos y dosis múltiples de un máximo de 290 microgramos de linaclotida, no se observaron niveles detectables en plasma del compuesto inalterado ni del metabolito activo (pérdida de la tirosina terminal, destirosina). Al administrarse 2897 microgramos en el día 8 tras un ciclo de 290 microgramos al día durante 7 días, linaclotida pudo detectarse en solo 2 de 18 sujetos en concentraciones que apenas superaron el límite inferior de cuantificación de 0,2 ng/ml (y que oscilaron entre 0,212 y 0,735 ng/ml). En los dos estudios fundamentales de fase III en los que se suministró a los pacientes una dosis de 290 microgramos de linaclotida, una vez al día, linaclotida se detectó únicamente en 2 de 162 pacientes aproximadamente 2 horas después de la administración de la dosis inicial de linaclotida (las concentraciones fueron de 0,241 a 0,239 ng/ml), y en ninguno de los 162 pacientes tras 4 semanas de tratamiento. En ningún momento se detectó el metabolito activo en ninguno de los 162 pacientes.
Distribución
Dado que linaclotida rara vez resulta detectable en el plasma tras su administración a las dosis terapéuticas, no se han llevado a cabo estudios sobre su distribución típica; con todo, se prevé que la distribución sistémica de linaclotida sea insignificante o nula.
Biotransformación
Linaclotida se metaboliza localmente en el tubo digestivo y da lugar a su principal metabolito activo, destirosina. Tanto linaclotida como el metabolito activo destirosina son reducidos y proteolizados en el tubo digestivo por enzimas que los transforman en péptidos más pequeños y aminoácidos que pueden estar presentes de manera natural.
Se ha investigado in vitro la actividad inhibidora que puedan ejercer linaclotida y su metabolito activo principal, el MM-419447, sobre los transportadores humanos de eflujo BCRP, MRP2, MRP3 y MRP4, así como sobre los transportadores humanos de captación OATP1B1, OATP1B3, OATP2B1, PEPT1 y OCTN1; los resultados de este estudio han demostrado que ninguno de los péptidos es inhibidor, en concentraciones clínicamente relevantes, de los transportadores más relevantes de captación y eflujo de medicamentos.
También se han realizado estudios in vitro del efecto de linaclotida y su metabolito en la inhibición de enzimas intestinales (CYP2C9 y CYP3A4) y hepáticas (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 y 3A4) más comunes, o en la inducción de enzimas hepáticas (CYP1A2, 2B6 y 3A4/5). Los resultados de estos estudios han puesto de manifiesto que linaclotida y el metabolito destirosina no son inhibidores ni inductores del sistema enzimático del citocromo P450.
Eliminación
Tras la administración por vía oral de una dosis única de 2897 microgramos de linaclotida en el día 8, después de un ciclo de 7 días de tratamiento con 290 microgramos diarios a 18 voluntarios sanos, en las heces se recuperó entre el 3 y el 5% de la dosis, en su práctica totalidad bajo la forma del metabolito activo.
Edad y sexo
No se han efectuado estudios clínicos para determinar el impacto de la edad y el sexo en la farmacocinética clínica de linaclotida, puesto que rara vez resulta detectable en el plasma; en cualquier caso, no se prevé que el sexo vaya a influir en la posología. Para información relacionada con la edad, ver las secciones 4.2., 4.4. y 4.8.
Insuficiencia renal
No se ha estudiado Constella en pacientes con insuficiencia renal. Linaclotida solo se detecta en plasma en raras ocasiones, por lo que no se prevé que esta insuficiencia vaya a influir en la eliminación del compuesto inalterado ni en la de su metabolito.
Insuficiencia hepática
No se ha estudiado Constella en pacientes que sufran de insuficiencia hepática. Linaclotida solo se detecta en plasma en raras ocasiones y no es metabolizada por las enzimas hepáticas del citocromo P450, por lo tanto, no se prevé que esta insuficiencia vaya a influir en la eliminación del compuesto inalterado ni en la de su metabolito.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas, genotoxicidad, potencial carcinogénico, toxicidad para la reproducción y el desarrollo.
DATOS FARMACÉUTICOS
6.
6.1 Lista de excipientes
Contenido de las cápsulas Celulosa microcristalina Hipromelosa
Cloruro de calcio dihidrato Leucina
Recubrimiento de las cápsulas Dióxido de titanio (E171)
Gelatina
Óxido de hierro rojo (E172)
Óxido de hierro amarillo (E172)
Tinta de las cápsulas Goma laca Propilenglicol
Disolución de amoníaco concentrado Hidróxido de potasio Dióxido de titanio (E171)
Óxido de hierro negro (E172)
6.2 Incompatibilidades
No procede.
6.3 Período de validez
Envase sin abrir: 3 años.
Una vez abierto el envase, las cápsulas deben tomarse en un plazo de 18 semanas.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30 °C. Mantener el frasco perfectamente cerrado para protegerlo de la humedad.
El frasco contiene uno o más botes sellados de gel de sílice destinados a preservar la sequedad de las cápsulas; estos botes no deben sacarse del recipiente.
6.5 Naturaleza y contenido del envase
Frasco de polietileno blanco de alta densidad (HDPE) con precinto de seguridad y tapón de rosca resistente a su apertura por niños junto con uno o más botes desecantes de gel de sílice.
Tamaños del envase: 10, 28, 60, 90 cápsulas y envases múltiples que contienen 112 (4x28) cápsulas. Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Allergan Pharmaceuticals International Limited
Clonshaugh Industrial Estate
Coolock
Dublin 17
Irlanda
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/801/001
EU/1/12/801/002
EU/1/12/801/003
EU/1/12/801/004
EU/1/12/801/005
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 26/noviembre/2012
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
Nombre y dirección del fabricante responsable de la liberación de los lotes
Industrias Farmacéuticas Almirall, S.A.
Ctra. Nacional II, Km. 593 08740 Sant Andreu de la Barca Barcelona España
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica.
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva
información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR CAJA CON UN ÚNICO FRASCO
1. NOMBRE DEL MEDICAMENTO
Constella 290 microgramos cápsulas duras Linaclotida
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula contiene 290 microgramos de linaclotida
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsula dura. 10 cápsulas 28 cápsulas 60 cápsulas 90 cápsulas
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Una vez abierto el frasco, las cápsulas deben utilizarse en el plazo de 18 semanas
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Allergan Pharmaceuticals International Limited
Clonshaugh Industrial Estate
Coolock
Dublin 17
Irlanda
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/801/001 10 cápsulas EU/1/12/801/002 28 cápsulas EU/1/12/801/003 60 cápsulas EU/1/12/801/004 90 cápsulas
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
constella 290 ^g
CAJA EXTERIOR QUE CONTIENE 4 FRASCOS DE 28 CÁPSULAS (ENVASE MÚLTIPLE)
CON BLUE BOX
|
1. |
NOMBRE DEL MEDICAMENTO |
Constella 290 microgramos cápsulas duras Linaclotida
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula contiene 290 microgramos de linaclotida
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsula dura.
112 (4x28) cápsulas (envase múltiple).
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Una vez abierto el frasco, las cápsulas deben utilizarse en el plazo de 18 semanas
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Allergan Pharmaceuticals International Limited
Clonshaugh Industrial Estate
Coolock
Dublin 17
Irlanda
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/801/005 112 (4x28) cápsulas (envase múltiple)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
constella 290 mcg
CAJA INTERIOR QUE CONTIENE FRASCOS DE 28 CÁPSULAS (ENVASE MÚLTIPLE)
SIN BLUE BOX
|
1. |
NOMBRE DEL MEDICAMENTO |
Constella 290 microgramos cápsulas duras Linaclotida
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula contiene 290 microgramos de linaclotida
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsula dura.
28 cápsulas. Subunidad de un envase múltiple, no puede venderse por separado.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE
FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
Una vez abierto el frasco, las cápsulas deben utilizarse en el plazo de 18 semanas
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Allergan Pharmaceuticals International Limited
Clonshaugh Industrial Estate
Coolock
Dublin 17
Irlanda
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/801/005 112 (4x28) cápsulas (envase múltiple)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
constella 290 mcg
INFORMACIÓN QUE DEBE FIGURAR EN EL ACONDICIONAMIENTO PRIMARIO FRASCO DE HDPE
1. NOMBRE DEL MEDICAMENTO
Constella 290 microgramos cápsulas duras Linaclotida
2. PRINCIPIO(S) ACTIVO(S)
Cada cápsula contiene 290 microgramos de linaclotida
3. LISTA DE EXCIPIENTES
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Cápsula dura. 10 cápsulas 28 cápsulas 60 cápsulas 90 cápsulas
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento Vía oral
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Allergan Pharmaceuticals International Limited
Clonshaugh Industrial Estate
Coolock
Dublin 17
Irlanda
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/801/001 10 cápsulas EU/1/12/801/002 28 cápsulas EU/1/12/801/003 60 cápsulas EU/1/12/801/004 90 cápsulas
EU/1/12/801/005 112 (4x28) cápsulas (envase múltiple)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
B. PROSPECTO
Prospecto: información para el paciente
Constella 290 microgramos cápsulas duras
Linaclotida
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a tomar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Constella y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Constella
3. Cómo tomar Constella
4. Posibles efectos adversos
5. Conservación de Constella
6. Contenido del envase e información adicional
1. Qué es Constella y para qué se utiliza Para qué se utiliza Constella
Constella contiene el principio activo linaclotida. Se emplea para tratar los síntomas del síndrome del intestino irritable (a menudo denominado simplemente “SII”) de moderado a grave con estreñimiento en pacientes adultos.
El SII constituye un trastorno intestinal frecuente. Los síntomas principales del SII con estreñimiento son los siguientes:
• dolor estomacal o abdominal,
• sensación de hinchazón,
• deposiciones (heces) infrecuentes, duras, pequeñas o en forma de bolitas.
Estos síntomas pueden variar de una persona a otra.
Cómo actúa Constella
Constella actúa localmente en el intestino, aliviando el dolor y la hinchazón y restaurando el normal funcionamiento intestinal. No es absorbido por el organismo, sino que se une a un receptor de la superficie intestinal, llamado guanilato ciclasa C. Al unirse a este receptor, bloquea la sensación de dolor y permite que pase líquido al intestino, con lo cual reblandece las heces y aumenta sus movimientos intestinales.
2. Qué necesita saber antes de empezar a tomar Constella No tome Constella
- si es alérgico a linaclotida o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- si usted o su médico saben que padece obstrucción estomacal o intestinal.
Advertencias y precauciones
Su médico le ha dado este medicamento a usted tras descartar otras enfermedades, especialmente para sus intestinos y concluyendo que padece de SII con estreñimiento. Dado que estas otras enfermedades pueden tener los mismos síntomas que el SII, es importante que informe inmediatamente a su médico de cualquier cambio o irregularidad en los síntomas.
Si sufre diarrea grave o prolongada (expulsión frecuente de heces de consistencia líquida durante 7 días o más), deje de tomar Constella y consulte a su médico (ver sección 4); no olvide beber líquidos en abundancia para reponer el agua y electrolitos, como el potasio, perdidos con la diarrea.
Consulte a su médico si sufre hemorragia intestinal o del recto.
Tome precauciones especiales si es mayor de 65 años, ya que tiene un mayor riesgo de sufrir diarrea.
Tome también precauciones especiales si padece diarrea grave o prolongada y una enfermedad adicional, como presión arterial elevada, una enfermedad previa del corazón y de los vasos sanguíneos (por ejemplo, infartos anteriores) o diabetes.
Comunique a su médico si padece enfermedades intestinales como la enfermedad de Crohn o la colitis ulcerosa, dado que no se recomienda la toma de Constella en estos pacientes.
Niños y adolescentes
No dé este medicamento a niños ni adolescentes menores de 18 años, ya que no se ha determinado si Constella es seguro y eficaz en este grupo de edad.
Toma de Constella con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento:
■ Algunos medicamentos pueden no funcionar correctamente si padece diarrea grave o de forma prolongada, como:
- Anticonceptivos orales. Si tiene una diarrea muy grave, la píldora anticonceptiva puede no funcionar correctamente y se recomienda el uso de un método anticonceptivo adicional. Consulte las instrucciones del prospecto de la píldora anticonceptiva que esté tomando.
- Medicamentos que necesitan una administración cuidadosa y exacta, como la levotiroxina (una hormona para tratar la función reducida de la glándula tiroidea).
■ Algunos medicamentos pueden aumentar el riesgo de diarrea cuando se toman junto Constella, como:
- Medicamentos para el tratamiento de las úlceras de estómago o de la excesiva producción de ácido en el estómago, que se denominan inhibidores de la bomba de protones.
- Medicamentos para el tratamiento del dolor y de la inflamación, denominados AINE.
- Laxantes.
Constella aumenta la frecuencia de los movimientos intestinales y causa diarrea (heces más sueltas) en mayor medida cuando se toma con alimentos que cuando se ingiere con el estómago vacío (ver sección 3).
Embarazo y lactancia
Se dispone de escasa información relativa a los efectos de Constella en mujeres embarazadas y que se encuentren en período de lactancia.
No tome este medicamento si está embarazada, cree que podría estar embarazada o tiene intención de quedarse embarazada, a menos que su médico le aconseje hacerlo.
Si se encuentra en periodo de lactancia, no tome Constella, a menos que se lo indique su médico. Conducción y uso de máquinas
Constella no influirá en la capacidad de conducción ni de utilización de maquinaria.
3. Cómo tomar Constella
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
La dosis recomendada es de una cápsula una vez al día. La cápsula debe tomarse al menos 30 minutos antes de una comida.
Si toma más Constella del que debe
El efecto más probable de una toma excesiva de Constella es la diarrea. Póngase en contacto con su médico o farmacéutico si ha ingerido demasiado de este medicamento.
Si olvidó tomar Constella
No tome una dosis doble para compensar las dosis olvidadas. Simplemente ingiera la siguiente en el momento que corresponda y continúe con el tratamiento como de costumbre.
Si interrumpe el tratamiento con Constella
Antes de interrumpir el tratamiento es preferible que consulte con su médico; no obstante, puede abandonar la toma de Constella en cualquier momento con total seguridad.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico o farmacéutico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, Constella puede producir efectos adversos, aunque no todas las personas los sufran.
Efectos adversos muy frecuentes (que pueden afectar a más de 1 de cada 10 personas):
• Diarrea
La diarrea suele ser de poca duración; sin embargo, si sufre un episodio grave o prolongado (deposiciones frecuentes o de consistencia líquida durante 7 días o más) y se siente aturdido o mareado o se desmaya, deje de tomar Constella y póngase en contacto con su médico.
Efectos adversos frecuentes (que pueden afectar hasta 1 de cada 10 personas):
• Dolor estomacal o abdominal
• Sensación de hinchazón
• Flatulencia
• Gastroenteritis vírica (gripe estomacal)
• Sensación de mareo
Efectos adversos poco frecuentes (que pueden afectar hasta 1 de cada 100 personas):
• Falta de control de las deposiciones (incontinencia fecal)
• Urgencia defecatoria
• Sensación de mareo al levantarse rápidamente
• Deshidratación
• Baja concentración de potasio en sangre
• Disminución del apetito
• Hemorragia rectal
• Hemorragia intestinal o del recto, incluidas hemorragias de hemorroides
• Náuseas
• Vómitos
Efectos adversos raros (que pueden afectar hasta 1 de cada 1.000 personas):
• Disminución de la concentración de bicarbonato en sangre Efectos adversos de frecuencia no conocida:
• Erupción cutánea Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Constella
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la caja y en el envase después de “CAD”. La fecha de caducidad es el último día del mes que se indica.
Una vez abierto el envase, las cápsulas deben tomarse en un plazo de 18 semanas.
No conservar a temperatura superior a 30 °C. Mantener el frasco perfectamente cerrado para protegerlo de la humedad.
Advertencia: El frasco contiene uno o más botes de gel de sílice destinados a preservar la sequedad de las cápsulas; estos botes no deben sacarse del recipiente ni ser ingeridos._
No utilice este medicamento si observa que el frasco está dañado o aprecia algún cambio en el aspecto de las cápsulas.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Constella
- El principio activo es linaclotida. Cada cápsula contiene 290 microgramos de linaclotida.
- Los demás componentes son:
Contenido de las cápsulas: celulosa microcristalina, hipromelosa, cloruro de calcio dihidrato y leucina.
Recubrimiento de las cápsulas: óxido de hierro rojo (E172), dióxido de titanio (E171), óxido de hierro amarillo (E172) y gelatina.
Tinta de las cápsulas: goma laca, propilenglicol, disolución de amoníaco concentrado, hidróxido de potasio, dióxido de titanio (E171) y óxido de hierro negro (E172).
Aspecto del producto y contenido del envase
Constella se presenta en cápsulas opacas de color blanco a blanquecino y naranja con la inscripción “290” en tinta gris.
El producto se presenta en un frasco de polietileno blanco de alta densidad (HDPE) con precinto de seguridad y tapón de rosca resistente a su apertura por niños, junto con uno o más botes desecantes de gel de sílice.
Constella se encuentra disponible en envases que contienen 10, 28, 60 ó 90 cápsulas y en envases múltiples de 112 cápsulas, los cuales incluyen 4 cajas de 28 cápsulas cada una. Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización
Allergan Pharmaceuticals International Limited
Clonshaugh Industrial Estate
Coolock
Dublin 17
Irlanda
Fabricante
Industrias Farmacéuticas Almirall, S.A.
Ctra. Nacional II, Km. 593,
E-08740 Sant Andreu de la Barca, Barcelona España
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien/Luxembourg/Luxemburg/ Nederland
Allergan n.v
Tél/Tel: +32 (0)2 351 2424
Ireland/Malta/ United Kingdom
Allergan Ltd
Tel: + 44 (0) 1628 494026
Bt^rapna
AnepraH Etnrapua EOOfl Tea.: +359 (0) 800 20 280
Ísland
Actavis ehf.
Sími: +354 550 3300
Ceská republika
Allergan CZ s.r.o.
Tel: +420 800 188 818
Italia
Allergan S.p.A Tel: + 39 06 509 561
Danmark/ Norge/ Suomi/Finland/Sverige
Allergan Norden AB Tlf/Puh/Tel: + 46 (0)8 594 100 00
Magyarország
Allergan Hungary Kft.
Tel.: +36 80 100 101
Deutschland Pharm-Allergan GmbH Tel: + 49 69 92038-10
Eesti
Allergan Baltics UAB Tel: + 372 56955144
Latvija
Allergan Baltics UAB Tel: +371 27331152
Lietuva
Allergan Baltics UAB Tel: +370 62660247
Osterreich
Pharm-Allergan GmbH Tel: +43 1 99460 6355
Polska
Allergan Sp. z o.o.
Tel: +48 22 256 37 00
EXXáSa/Kúnpoq
Allergan Hellas Pharmaceuticals S.A.
Tqk +30 210 74 73 300
Portugal
Profarin Lda.
Tel: + 351 21 425 3242
España
Allergan S.A.
Tel: + 34 91 807 6130
Romanía
Allergan S.R.L.
Tel: +40 21 301 53 02
France
Allergan France SAS Tél: +33 (0)1 49 07 83 00
Sloveníja
Ewopharma d.o.o.
Tel: + 386 (0) 590 848 40
Hrvatska
Ewopharma d.o.o.
Tel: +385 1 6646 563
Slovenská republíka Allergan SK s.r.o.
Tel: +421 800 221 223
Fecha de la última revisión de este prospecto:
Otras fuentes de ínformacíón
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
31