Clexane 120 Mg (12.000 Ui) Solucion Inyectable En Jeringa Precargada
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Clexane 120 mg (12.000 UI) solución inyectable en jeringa precargada Clexane 150 mg (15.000 UI) solución inyectable en jeringa precargada
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Composición cualitativa y cuantitativa
Clexane 120 mg (12.000 UI) solución inyectable en jeringa precargada
Cada jeringa precargada contiene:
- Enoxaparina sódica: 120 mg (equivalente a 12.000 UI)1
- Agua para preparaciones inyectables c.s.p. 0,8 ml
Clexane 150 mg (15.000 UI) solución inyectable en jeringa precargada
Cada jeringa precargada contiene:
- Enoxaparina sódica: 150 mg (equivalente a 15.000 UI)1
- Agua para preparaciones inyectables c.s.p. 1 ml
* Valorada frente al primer estándar internacional OMS, para heparina de bajo peso molecular (HBPM), con el método anti-Xa amidolítico con sustratos específicos y utilizando el patrón internacional LMWHs (NIBSC)
La concentración de enoxaparina en estas presentaciones es de 150 mg/ml.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Solución inyectable contenida en jeringas precargadas
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
- Tratamiento de la trombosis venosa profunda establecida (con o sin embolia pulmonar).
- Prevención de la coagulación en el circuito de circulación extracorpórea en la hemodiálisis.
4.2 Posología y forma de administración
Las diferentes heparinas de bajo peso molecular no son necesariamente equivalentes. En consecuencia, se debe respetar la dosificación y el modo de empleo específico de cada una de estas especialidades farmacéuticas (ver sección 4.4).
Un miligramo de enoxaparina tiene una actividad anti-Xa de 100 UI, aproximadamente.
Tratamiento de la trombosis venosa profunda establecida (con o sin embolia pulmonar):
Clexane está indicado para la pauta posológica de 1,5 mg (150 UI) por kg de peso al día. En pacientes con trastornos tromboembólicos complicados se recomienda la dosis de 1 mg (100 UI) por kg de peso de enoxaparina dos veces al día. La duración del tratamiento es, generalmente, de 10 días. Salvo contraindicación expresa, debe iniciarse tratamiento anticoagulante por vía oral lo antes posible y continuar el tratamiento con enoxaparina hasta que se haya alcanzado el efecto anticoagulante terapéutico (2 a 3 de INR).
Prevención de la coagulación en el circuito de circulación extracorpórea en la hemodiálisis:
- En los pacientes sometidos a sesiones de hemodiálisis repetidas, la prevención de la coagulación en el circuito de circulación extracorpórea se obtiene inyectando una dosis de 0,6 a 1 mg/kg (60 - 100 UI/kg) en la línea arterial del circuito de diálisis, al comienzo de la sesión [0,8 a 1 mg (80 - 100 UI/kg) para los casos de flujos bajos, unipunción, o diálisis superior a 4 horas]. En general, para un paciente tipo de unos 60 kg de peso, una dosis de 40 mg (4000 UI) es eficaz y bien tolerada. En caso de aparición de anillos de fibrina, se practicará una nueva inyección de 0,5 a 1 mg/kg (50-100 UI/kg), en función del tiempo que reste hasta el final de la diálisis.
- En pacientes de alto riesgo hemorrágico (en particular diálisis pre o post-operatorias), o que presenten un síndrome hemorrágico en evolución, las sesiones de diálisis se podrán efectuar utilizando una dosis de 0,4-0,5 mg/kg (40-50 UI/kg) (bipunción) o de 0,5-0,75 mg/kg (50 - 75 UI/kg) (unipunción).
Observaciones generales:
Las jeringas precargadas están listas para su empleo y no deben ser purgadas antes de la inyección.
Pacientes de edad avanzada: en los pacientes de edad avanzada no se necesita ninguna reducción de la dosis, a menos que la función renal esté alterada (ver sección 4.4: Hemorragia en pacientes de edad avanzada; sección 5.2: Insuficiencia renal; y en esta sección 4.2: Insuficiencia renal).
Niños: la seguridad y la eficacia de la enoxaparina en niños no han sido establecidas.
Insuficiencia hepática: en estos pacientes no se necesita ningún ajuste posológico a las dosis usadas en la profilaxis (ver sección 5.2).
Insuficiencia renal:
(ver sección 4.4: Insuficiencia renal y sección 5.2: Insuficiencia renal). 1
En pacientes con insuficiencia renal grave (aclaramiento de creatinina <30ml/min) se requiere un ajuste posológico, según se indica a continuación, ya que la exposición a la enoxaparina se incrementa significativamente en estos pacientes.
Se recomiendan los siguientes ajustes posológicos en el tratamiento de la trombosis venosa profunda:
|
Posología estándar |
Insuficiencia renal grave |
|
1 mg/kg SC dos veces al día |
1 mg/kg SC una vez al día |
|
1,5 mg/kg SC una vez al día |
1 mg/kg SC una vez al día |
Estos ajustes posológicos recomendados, no afectan a la indicación de hemodiálisis.
• Insuficiencia renal leve o moderada
Aunque no se necesita ningún ajuste posológico en pacientes con insuficiencia renal moderada (aclaramiento de creatinina 30-50 ml/min) o leve (aclaramiento de creatinina 50-80 ml/min), se recomienda una monitorización clínica cuidadosa.
Anestesia espinal/epidural
Para pacientes que estén recibiendo anestesia espinal/epidural, ver sección 4.4: Anestesia espinal/epidural. Forma de administración:
El paciente deberá estar en decúbito supino y la administración de enoxaparina debe ser realizada mediante inyección subcutánea profunda, normalmente en la pared abdominal antero-lateral o postero-lateral, alternativamente del lado derecho y del lado izquierdo. La aguja deberá introducirse verticalmente en toda su longitud, en el espesor de un pliegue cutáneo tomado entre el pulgar y el índice del operador. Este pliegue cutáneo se debe mantener mientras se administra la inyección. Tras la administración, no frotar el punto de inyección.
4.3 Contraindicaciones
- Hipersensibilidad a la enoxaparina sódica, a la heparina o sus derivadas incluyendo otras heparinas de bajo peso molecular o a cualquiera de los demás componentes de este medicamento, incluidos en la sección 6.
- Hemorragias intensas activas o condiciones de alto riesgo de hemorragia incontrolada, incluyendo ictus hemorrágico reciente.
- Historia de trombocitopenia o trombosis secundaria a la enoxaparina.
- Endocarditis séptica.
4.4 Advertencias y precauciones especiales de empleo
- No administrar por vía intramuscular.
- Hemorragias
Como con cualquier otro anticoagulante, puede producirse sangrado en cualquier parte del cuerpo (ver sección 4.8).
En caso de sangrado, debe investigarse el origen de la hemorragia e instaurarse el tratamiento adecuado.
- No intercambiar Clexane con otras heparinas de bajo peso molecular dado que difieren en su proceso de fabricación, pesos moleculares, actividades antiXa específicas, unidades y dosis, y consecuentemente, en su farmacocinética y actividades biológicas asociadas (por ej. actividad antiIIa, e interacciones plaquetarias). Se requiere, por lo tanto, especial atención y cumplimiento de las instrucciones específicas de uso proporcionadas por el laboratorio.
- Al igual que otros anticoagulantes, la inyección de enoxaparina debe usarse con extrema precaución en las situaciones con aumento de riesgo de hemorragia, tales como alteraciones de la coagulación, insuficiencia hepática, historia de úlcera péptica, hipertensión arterial grave no controlada, retinopatía hipertensiva o diabética, anestesia espinal o epidural, permanencia de catéteres intratecales o postoperatorio inmediato oftalmológico o neurológico, uso concomitante de medicación que tenga efecto sobre la homeostasis (ver sección 4.5).
- Anestesia espinal/epidural: En pacientes sometidos a anestesia espinal/epidural o a punción lumbar, la
administración de enoxaparina sódica con fines profilácticos se ha asociado raramente a la aparición de hematomas neuroaxiales, con el resultado final de parálisis prolongada o permanente. Este riesgo se incrementa por el uso de enoxaparina sódica a dosis elevadas, por el uso de catéteres epidurales o espinales postoperatorios, la administración concomitante de medicamentos con efecto sobre la coagulación como antiinflamatorios no esteroídicos (AINES) (ver sección 4.5), antiagregantes plaquetarios o anticoagulantes, y por las punciones neuroaxiales traumáticas o repetidas o en pacientes con un historial de cirugía espinal o deformidad espinal.
o Para reducir el riesgo potencial de sangrado asociado al uso concomitante de enoxaparina sódica y la anestesia/analgesia epidural o espinal, se deberá considerar el perfil farmacocinético del fármaco (ver sección 5.2). La inserción y retirada del catéter se realizará mejor cuando el efecto anticoagulante de la enoxaparina sea bajo; sin embargo, no se conoce el tiempo exacto para alcanzar un efecto anticoagulante lo suficientemente bajo.
o A la hora de decidir el intervalo de tiempo que debe transcurrir entre la administración de enoxaparina y la inserción o retirada de un catéter espinal/epidural, deben tenerse en cuenta las características del paciente y del producto, debiendo de transcurrir al menos de 12 horas después de la administración de enoxaparina a dosis más bajas (20 mg una vez al día, 30 mg una vez o dos veces al día o 40 mg una vez al día), y al menos 24 horas después de la administración a dosis superiores (0,75 mg/kg dos veces al día, 1 mg/kg dos veces al día, o 1,5 mg/kg una vez al día). Los niveles Anti-Xa aún son detectables en estos puntos de tiempo, y este retraso no es una garantía de que el hematoma neuroaxial será evitado. Los pacientes que reciban la dosis de 0,75 mg/kg dos veces al día o la dosis de 1 mg/kg dos veces al día no deben recibir la segunda dosis de enoxaparina en el régimen “dos veces al día” para permitir un retraso mayor antes de la inserción o retirada del catéter. Igualmente, aunque no se puede realizar una recomendación específica para programar una dosis posterior de enoxaparina después de la retirada del catéter, se debe considerar el retraso de la siguiente dosis durante al menos 4 horas, basándose en una evaluación del riesgo-beneficio considerando tanto el riesgo para trombosis como el riesgo de sangrado, en el contexto del procedimiento y los factores de riesgo del paciente. Para pacientes con aclaramiento de creatinina <30ml/min, son necesarias consideraciones adicionales debido a que la eliminación de enoxaparina es más prolongada; se debe considerar duplicar el tiempo de retirada de un catéter, al menos 24 horas para la dosis más baja de enoxaparina prescrita (20 mg una vez al día) y al menos 48 horas para la dosis más alta (1 mg/kg/día).
o Si bajo criterio médico se decide administrar tratamiento anticoagulante durante un procedimiento anestésico espinal/epidural o punción lumbar, se debe controlar de forma frecuente al paciente para detectar precozmente cualquier signo o síntoma de déficit neurológico, como dolor lumbar, déficit sensorial y motor (entumecimiento y debilidad de extremidades inferiores) y trastornos funcionales del intestino o vejiga. El personal de enfermería debe ser entrenado para detectar tales signos y síntomas. Asimismo, se advertirá a los pacientes que informen inmediatamente al médico o personal de enfermería si experimentan cualquiera de los síntomas antes descritos.
o Si se sospecha la aparición de algún signo o síntoma sugestivo de hematoma espinal o epidural deben realizarse las pruebas diagnósticas con carácter de urgencia e instaurar el tratamiento adecuado, incluyendo la descompresión medular.
- Hemorragia en pacientes de edad avanzada
En pacientes de edad avanzada, no se observó aumento de la tendencia a la hemorragia, a las dosis usadas en la profilaxis. En pacientes de edad avanzada (especialmente los pacientes con edad igual o mayor de 80 años) puede aumentar el riesgo de complicaciones hemorrágicas a la dosis terapéutica.
Se recomienda una cuidadosa monitorización clínica (ver sección 4.2: Pacientes de edad avanzada y sección 5.2: Pacientes de edad avanzada).
- Insuficiencia renal
En pacientes con insuficiencia renal, existe un aumento de la exposición a la enoxaparina sódica, con la consecuente elevación del riesgo de hemorragia. En pacientes con insuficiencia renal grave (aclaramiento de creatinina <30 ml/min) se recomienda ajustar la posología terapéutica, ya que, la exposición a enoxaparina sódica está significativamente aumentada en estos pacientes.
Aunque no se requiere ningún ajuste posológico en pacientes con insuficiencia renal moderada (aclaramiento de creatinina 30-50 ml/min) y leve (aclaramiento de creatinina 50-80 ml/min), se recomienda una cuidadosa monitorización clínica (ver sección 4.2: Insuficiencia renal y sección 5.2: Insuficiencia renal).
- Pacientes de bajo peso
En mujeres de bajo peso corporal (menos de 45 kg) y en hombres de bajo peso corporal (menos de 57 kg) se ha observado un aumento de la exposición a enoxaparina sódica a las dosis usadas en la profilaxis (no ajustadas según el peso), lo cual puede aumentar el riesgo de hemorragia. Por lo tanto se recomienda una cuidadosa monitorización clínica en estos pacientes (ver sección 5.2: Peso).
- Pacientes obesos
Los pacientes obesos tienen un mayor riesgo de sufrir tromboembolismo. No se ha determinado completamente la seguridad y eficacia de las dosis profilácticas en pacientes obesos (IMC > 30 kg/m2) y no existe ningún consenso para el ajuste de la dosis. Estos pacientes deben ser observados cuidosamente para detectar signos y síntomas de tromboembolismo.
- Procedimientos de revascularización coronaria percutánea
Para minimizar el riesgo de hemorragia después de la instrumentación vascular durante el tratamiento de la angina inestable e infarto de miocardio sin onda Q, la guía de acceso vascular debe mantenerse en su lugar durante 6 a 8 horas después de la dosis vía subcutánea (SC) de enoxaparina sódica. La siguiente dosis programada no debe administrarse antes de 6 a 8 horas después de la retirada de la guía. La zona donde se ha realizado el proceso debe observarse para detectar signos de hemorragia o formación de hematomas.
- Válvulas protésicas cardíacas
El uso de Clexane como tromboprofilaxis en pacientes con prótesis valvulares cardíacas no ha sido suficientemente estudiado. Se han notificado casos aislados de trombosis en prótesis valvulares cardíacas en pacientes en los que se administró enoxaparina como profilaxis. Ciertos factores confusos, incluyendo patologías de base y falta de datos clínicos limitan la evaluación de estos casos. Algunos de estos casos se dieron en embarazadas en las que las trombosis condujeron a un desenlace fatal tanto para la madre como para el feto Las mujeres embarazadas con prótesis valvulares cardíacas pueden tener un mayor riesgo de tromboembolismo (ver en esta sección Mujeres embarazadas con válvulas protésicas cardiacas).
- Mujeres embarazadas con válvulas protésicas cardiacas
El uso de Clexane como tromboprofilaxis en mujeres embarazadas con prótesis valvulares cardíacas no ha sido suficientemente estudiado. En un ensayo clínico en el que se administró enoxaparina sódica (1 mg/kg dos veces al día) a 8 mujeres embarazadas con válvulas protésicas cardiacas para reducir el riesgo de tromboembolismo, 2 de ellas desarrollaron coágulos que bloquearon la válvula que condujeron al desenlace fatal tanto para la madre como para el feto. Se han notificado casos aislados en postmarketing de trombosis en mujeres embarazadas con prótesis valvulares cardíacas en los que se administró enoxaparina como tromboprofilaxis. Las mujeres embarazadas con prótesis valvulares cardiacas pueden tener un mayor riesgo de tromboembolismo (ver en esta sección Válvulas protésicas cardiacas).
- Pruebas de laboratorio
En las dosis empleadas para la profilaxis del tromboembolismo venoso, la enoxaparina sódica no modifica de forma significativa las pruebas de tiempo de sangrado y coagulación sanguínea global, ni afecta a la agregación plaquetaria o la unión de fibrinógeno a plaquetas.
A dosis más elevadas, pueden aparecer incrementos en el tiempo de tromboplastina parcial activada (PTT) y en el tiempo de coagulación activado (ACT). Los aumentos de PTT y ACT no se correlacionan de forma lineal con el incremento de actividad antitrombótica de enoxaparina sódica y por tanto no son adecuados ni fiables para la monitorización de la actividad de enoxaparina sódica.
- Monitorización del recuento de plaquetas
Con las heparinas de bajo peso molecular también existe el riesgo de trombocitopenia inducida por la heparina y mediada por anticuerpos. Estas trombopenias aparecen habitualmente entre el día 5 y 21 después del comienzo del tratamiento con enoxaparina.
Por lo tanto, es recomendable efectuar un recuento de plaquetas antes del comienzo de la terapia y después regularmente, a lo largo del tratamiento con enoxaparina. Si, pasado este tiempo, es necesario prolongar el tratamiento, el recuento de plaquetas tiene que realizarse una vez a la semana, hasta el final del tratamiento.
En la práctica, ante cualquier descenso significativo (30 a 50 % del valor inicial) del recuento de plaquetas tratamiento con enoxaparina debe interrumpirse inmediatamente e instaurarse otra terapia de sustitución.
En pacientes con historia de trombocitopenia tras un tratamiento con heparina, con o sin trombosis, la enoxaparina debe ser utilizada con extrema precaución. El riesgo de trombocitopenia inducida por heparina puede durar varios años. Si se sospecha de trombocitopenia inducida por heparina, un test in vitro de agregación plaquetaria tiene un valor predictivo limitado. La decisión de utilizar enoxaparina en tales casos debe realizarse consultando con un experto en el campo.
4.5 Interacción con otros medicamentos y otras formas de interacción
Se recomienda, antes del tratamiento con enoxaparina sódica, interrumpir la utilización de aquellos fármacos que afecten a la hemostasia a menos que estén estrictamente indicados.
Sustancias que interfieren los mecanismos de la coagulación:
- Ácido acetilsalicílico, otros salicilatos y antiinflamatorios no esteroides (vía sistémica), incluido ketorolaco
- Anticoagulantes orales y trombolíticos
- Glucocorticoides (vía sistémica): la administración de enoxaparina aumenta el riesgo hemorrágico propio de la corticoterapia a altas dosis o en tratamientos prolongados.
Inhibidores de la agregación plaquetaria:
- Ticlopidina, dipiridamol, sulfinpirazona
- Dextrano 40 (vía parenteral), clopidogrel
- Otros agentes antiplaquetarios como son los antagonistas IIb/IIIa.
Si la asociación de estos medicamentos y enoxaparina sódica es necesaria se recomienda proceder a una estrecha monitorización clínica y de laboratorio.
4.6 Fertilidad, embarazo y lactancia
Embarazo
Los estudios realizados en animales no han mostrado ninguna evidencia de acción teratógena o fetotóxica. En la rata preñada, el paso de 35S-enoxaparina a través de la placenta hasta el feto es mínimo.
En humanos, no hay ninguna evidencia de que la enoxaparina sódica atraviese la barrera placentaria durante el segundo trimestre de embarazo. No se dispone de información sobre el primer y el tercer trimestre. Al no poder realizarse estudios controlados adecuados en mujeres embarazadas y dado que los estudios realizados en animales no siempre son predictivos de la respuesta en humanos, este medicamento sólo debe utilizarse durante el embarazo si el médico ha establecido una clara necesidad. Si se realiza una anestesia epidural, el tratamiento con enoxaparina debe ser interrumpido.
Ver también sección 4.4: Mujeres embarazada con válvulas protésicas cardiacas.
Lactancia
En la rata, durante el periodo de lactancia, la concentración de 35S-enoxaparina en la leche es muy baja. No se sabe si la enoxaparina se excreta en la leche humana, en las madres durante el periodo de lactancia. Sin embargo, como precaución en este periodo, las madres lactantes que reciban enoxaparina deberían ser aconsejadas a evitar la lactancia materna.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La enoxaparina no tiene efectos sobre la capacidad para conducir y utilizar máquinas.
4.8 Reacciones adversas
Se ha evaluado la enoxaparina en más de 15.000 pacientes que recibieron enoxaparina en ensayos clínicos. Estos incluyeron 1.776 para profilaxis de trombosis venosa profunda sometidos a cirugía ortopédica o abdominal en pacientes con riesgo de complicaciones tromboembólicas, 1.169 para la profilaxis de trombosis venosa profunda en pacientes no quirúrgicos con patología aguda y movilidad gravemente restringida, 559 para el tratamiento de la trombosis venosa profunda con o sin embolismo pulmonar, 1.578 para el tratamiento de angina inestable e infarto de miocardio sin onda Q y 10.176 para el tratamiento de de infarto agudo de miocardio con elevación del segmento ST.
El régimen posológico de enoxaparina sódica administrado en estos ensayos clínicos varía dependiendo de las indicaciones. La dosis de enoxaparina sódica fue de 40 mg SC (vía subcutánea) una vez al día para la profilaxis de la trombosis venosa profunda después de cirugía o pacientes no quirúrgicos con patología aguda y movilidad gravemente restringida. En el tratamiento de la trombosis venosa profunda (TVP) con o sin embolismo pulmonar (EP), los pacientes que recibían enoxaparina fueron tratados con una dosis de 1 mg/kg SC cada 12 horas o con una dosis de 1,5 mg/kg SC una vez al día. En los estudios clínicos para el tratamiento de angina inestable e infarto de miocardio sin onda Q, la dosis fue de 1 mg/kg SC cada 12 horas y en el estudio clínico para el tratamiento del infarto agudo de miocardio con elevación del segmento ST, el régimen posológico de enoxaparina sódica fue de 30 mg IV en bolo, seguido de 1 mg/kg SC cada 12 horas.
La reacciones adversas observadas en estos estudios clínicos y notificadas en la experiencia postcomercialización se detallan a continuación.
Las frecuencias se definen de la siguiente forma: muy frecuentes (> 1/10); frecuentes (> 1/100 a <1/10); poco frecuentes (> 1/1.000 a <1/100); raras (> 1/10.000 a <1/1.000); muy raras (<1/10.000) o frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas postcomercialización están designadas con una frecuencia “no conocida”.
Hemorragias
En estudios clínicos, las hemorragias fueron la reacción más frecuentemente notificada. Éstas incluyeron hemorragias mayores, notificadas como máximo en el 4,2 % de los pacientes (pacientes quirúrgicos1). Algunos de estos casos tuvieron un desenlace fatal.
Al igual que otros anticoagulantes, la hemorragia puede suceder en presencia de factores de riesgo asociados tales como: lesiones orgánicas susceptibles de sangrar, procesos invasivos o algunas asociaciones medicamentosas que afectan a la hemostasia (ver sección 4.4 y sección 4.5).
|
Clasificación de órganos del sistema MedDRA |
Tratamiento en pacientes TVP con o sin EP |
|
Trastornos vasculares |
Muy frecuentes: Hemorragia* Poco frecuentes: Hemorragia intracraneal, hemorragia retroperitoneal |
*: tales como hematoma, equimosis en sitio diferente al lugar de administración, hematoma con herida, hematuria, epistaxis y hemorragia gastrointestinal.
'En pacientes quirúrgicos, las complicaciones hemorrágicas se consideraron mayores: (1) si la hemorragia causó un evento clínico significativo, o (2) si estaban acompañadas por una disminución de la hemoglobina > 2 g/dL o transfusión de 2 o más unidades de productos sanguíneos. Las hemorragias retroperitoneales e intracraneales siempre se consideraron como mayores.
Trombocitopenia y trombocitosis
|
Clasificación de órganos del sistema MedDRA |
Tratamiento en pacientes TVP con o sin EP |
|
Trastornos |
Muy frecuentes: |
|
de la sangre |
Trombocitosis* |
|
y del sistema | |
|
linfático |
Frecuentes: |
|
Trombocitopenia |
* Incremento de plaquetas > 400 G/L
Otras reacciones adversas clínicamente relevantes
Estas reacciones se describen a continuación, independientemente de las indicaciones, por clasificación de órganos del sistema, y enumeradas en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Clasificación de órganos del sistema MedDRA |
Todas las indicaciones |
|
Trastornos del sistema inmunológico |
Frecuentes: Reacción alérgica Raras: Reacción anafilactoide / anafiláctica (ver también Experiencia postcomercialización) |
|
Trastornos hepatobiliares |
Muy frecuentes: Aumento de enzimas hepáticas (principalmente transaminasas**) |
|
Trastornos de la piel y del tejido subcutáneo |
Frecuentes: Urticaria, prurito, eritema Poco frecuentes: Dermatitis bullosa |
|
Trastornos generales y alteraciones en el lugar de administración |
Frecuentes: Hematoma en el punto de inyección, dolor en el punto de inyección, otras reacciones en el punto de inyección* Poco frecuentes: Irritación local; necrosis cutánea en el punto de inyección |
|
Exploraciones complementarias |
Raras: Hipercaliemia |
*; tales como edema en el punto de inyección, hemorragia, hipersensibilidad, inflamación, masa, dolor o reacción (no especificado de otra forma)
**: niveles de transaminasas > 3 veces del límite superior de normalidad Experiencia post-comercialización
Se han identificado las siguientes reacciones adversas durante el uso de Clexane después de su autorización. Las reacciones adversas derivan de notificaciones espontáneas y por tanto, la frecuencia es “no conocida” (no puede estimarse a partir de los datos disponibles).
• Trastornos del sistema inmunológico
Reacción anafilactoide/anafiláctica incluyendo shock.
• Trastornos del sistema nervioso Dolor de cabeza.
• Trastornos vasculares
Se han notificado casos de hematoma intradural (o hematoma neuroaxial) con el uso concomitante de enoxaparina sódica y anestesia intradural/epidural o punción intradural. Estas reacciones resultaron en diversos grados de daños neurológicos a largo plazo o en parálisis permanente (ver sección 4.4: Anestesia espinal/epidural).
• Trastornos de la sangre y del tejido linfático
- Anemia hemorrágica
- Casos de trombocitopenia inmunoalérgica con trombosis; en algunos de ellos la trombosis se complicó con infartos de órganos o isquemia de las extremidades (ver sección 4.4, Monitorización del recuento de plaquetas)
- Eosinofilia.
• Trastornos de la piel y del tejido subcutáneo
- Vasculitis cutánea, necrosis cutánea normalmente sobrevenida en el punto de inyección (estos fenónemos habitualmente son precedidos por la aparición de púrpura o de placas eritomatosas, infiltradas y dolorosas). Se debe suspender el tratamiento con enoxaparina sódica.
- Nódulos en el lugar de inyección (nódulos inflamados, que no consisten en un enquistamiento de enoxaparina)
Estos problemas desaparecen en unos días y no debe interrumpirse el tratamiento por ellos.
- Alopecia.
• Trastornos hepatobiliares
- Lesión hepática hepatocelular
- Lesión hepática colestásica.
• Trastornos músculo esqueléticos y del tejido conjuntivo
- Osteoporosis después del tratamiento a largo plazo (mayor de 3 meses).
Notificación de sospechas de reacciones adversas:
Es importante notificar las sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano: https://www.notificaram.es.
4.9 Sobredosis
La sobredosis accidental, tras la administración intravenosa, subcutánea, o extracorpórea, puede conducir a complicaciones hemorrágicas. Los efectos pueden ser ampliamente neutralizados por la inyección intravenosa lenta de protamina (sulfato o clorhidrato). La dosis de protamina depende de la dosis de enoxaparina sódica inyectada, 1 mg de protamina neutraliza el efecto anticoagulante de 1 mg de enoxaparina sódica, dentro de las 8 horas siguientes a la administración de la enoxaparina sódica. En caso de superar las 8 horas tras la administración de la enoxaparina, o si es necesaria una segunda dosis de protamina, se podrá proceder a la infusión de 0,5 mg protamina por 1 mg de enoxaparina. Después de 12 horas de la administración de la enoxaparina sódica, ya no será necesario administrar protamina. No obstante, incluso con dosis elevadas de protamina, la actividad anti-Xa nunca es totalmente neutralizada (máximo 60 %).
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agentes antitrombóticos: heparinas, Código ATC: B01AB05
La enoxaparina es una heparina de bajo peso molecular. Se caracteriza por un peso molecular medio de 4.500 daltons (3.500 - 5.500). Es una sal sódica. La distribución del peso molecular es la siguiente: fracciones <2.000 daltons: <20 %, fracciones de 2.000 a 8.000 daltons: >68 % y fracciones >8.000 daltons: <18 %. En sistema purificado in vitro, enoxaparina posee una actividad anti-Xa elevada (alrededor de 100 UI/mg) y una débil actividad anti-IIa o antitrombínica (aproximadamente 28 UI/mg). La relación entre estas dos actividades es de 3,6, aproximadamente.
Estas actividades anticoagulantes están mediadas a través de la antitrombina III (ATIII) teniendo en humanos actividad antitrombótica.
Más allá de su actividad anti-Xa/IIa, se han identificado más propiedades antitrombóticas y antiinflamatorias de enoxaparina en individuos sanos y pacientes, así como en modelos no clínicos.
Esto incluye la inhibición ATIII dependiente de otros factores de coagulación como el factor VIIa, la inducción de la liberación del inhibidor de las vías del factor tisular (TFPI) así como una liberación reducida del factor de von Willebrand (vWF) del endotelio vascular a la circulación sanguínea. Se sabe que estos factores contribuyen al efecto antitrombótico global de la enoxaparina.
Enoxaparina sódica se obtiene por despolimerización alcalina del éster bencílico de heparina obtenido de la mucosa intestinal porcina. Su estructura se caracteriza por un grupo ácido 2-O-sulfo-4-enepiranosurónico en el extremo no reductor y un grupo 2-N, 6-O-disulfo-D-glucosamina en el extremo reductor de la cadena. Aproximadamente el 20% (entre el 15% y el 25%) de la estructura de la enoxaparina contiene un derivado 1,6 anhidro en el extremo reductor de la cadena de polisacáridos.
A las posologías utilizadas para la profilaxis del tromboembolismo, la enoxaparina no influye negativamente de forma significativa sobre los tests globales de coagulación. No modifica la agregación plaquetaria ni la fijación del fibrinógeno sobre las plaquetas.
5.2 Propiedades farmacocinéticas
Los parámetros farmacocinéticos de la enoxaparina han sido estudiados a partir de la evolución de las actividades plasmáticas anti-Xa y anti-IIa, a los intervalos de dosis recomendados tras la administración subcutánea única y repetida y tras la administración intravenosa única.
La determinación de la actividad farmacocinética de anti Xa y anti IIa se efectúa por método amidolítico validado, con substratos específicos y un estándar de enoxaparina calibrado frente al patrón internacional LMWH1 (NIBSC).
Biodisponibilidad y Absorción
La biodisponibilidad, de enoxaparina sódica tras la inyección por vía subcutánea, basada en la actividad anti-Xa, está próxima al 100 %.El volumen de inyección y la concentración de dosis en el rango de 100200 mg/ml no afecta a los parámetros farmacocinéticos en voluntarios sanos.
La actividad anti-Xa plasmática máxima se observa 3 a 5 horas después de la inyección subcutánea y alcanza 0,2, 0,4, 1,0 y 1,3 UI anti-Xa/ml, tras la administración subcutánea única de dosis de 20 mg (2000 UI), 40 mg (4000 UI) 1 mg/kg (100 Ul/kg) y 1,5 mg/kg (150 UI), respectivamente.
La farmacocinética de enoxaparina, parece ser lineal en los intervalos de dosis recomendados. La variabilidad intra-paciente e inter-paciente es baja. Tras administraciones repetidas subcutáneas de 40 mg una vez al día y pautas posológicas de 1,5 mg/kg una vez al día en voluntarios sanos, la situación de equilibrio se alcanzó a los 2 días con un promedio de niveles de exposición un 15% más elevados que los alcanzados tras la dosis única. Después de administraciones subcutáneas repetidas con la pauta posológica de 1 mg/kg dos veces al día, el equilibrio se alcanza del día 3 al 4, con una exposición alrededor del 65% más elevada que tras una única dosis y con niveles medios de pico y valle de 1,2 y 0,52 UI/ml, respectivamente. Basándonos en la farmacocinética de la enoxaparina, esta diferencia del estado estacionario es la esperada y está dentro del rango terapéutico.
La actividad plasmática anti-IIa tras la administración subcutánea es aproximadamente 10 veces menor que la actividad anti-Xa. El máximo de la actividad anti-IIa se observa aproximadamente de 3 a 4 horas después de la administración subcutánea y alcanza 0,13 UI/ml y 0,19 UI/ml tras la administración repetida de 1 mg/kg dos veces al día y 1,5 mg/kg una vez al día, respectivamente.
Distribución
El volumen de distribución de la actividad anti-Xa de enoxaparina sódica es de aproximadamente 5 litros y está cercano al volumen de sangre.
Metabolismo
La enoxaparina sódica se metaboliza en un primer paso en el hígado por desulfatación y/o despolimerización hasta tipos con peso molecular más bajo, consecuentemente con potencia biológica muy reducida.
Eliminación
La enoxaparina sódica es un principio activo con un aclaramiento bajo; el aclaramiento plasmático medio anti-Xa es de 0,74 L/h después de una perfusión intravenosa durante 6 h de 1,5 mg/kg.
La eliminación parece ser monofásica con una semivida de aproximadamente 4 horas tras una única dosis hasta aproximadamente 7 h, tras una dosificación repetida.
El aclaramiento renal de fragmentos activos representa aproximadamente el 10% de la dosis administrada, y el total de la excreción renal de los fragmentos activos y no activos el 40% de la dosis.
Excreción
En voluntarios varones sanos, con dosis única de enoxaparina de 20 mg (2000 UI) ó 40 mg (2000 UI), por vía subcutánea, la excreción urinaria basada en actividad anti-Xa es del 7,4 % y 9,3 %, respectivamente, de la dosis administrada. La eliminación de la enoxaparina y sus metabolitos sucede por las vías renal y biliar.
Poblaciones de riesgo
Pacientes de edad avanzada
Según los resultados de un análisis farmacocinético de la población, el perfil cinético de la enoxaparina sódica no es diferente en pacientes de edad avanzada respecto a sujetos jóvenes cuando la función renal es normal. Sin embargo, como se sabe que la función renal disminuye con la edad, los pacientes de edad avanzada podrían mostrar una reducción en la eliminación de enoxaparina sódica (ver secciones 4.2 Pacientes de edad avanzada y 4.4: Hemorragia en pacientes de edad avanzada; ver Insuficiencia renal en esta misma sección).
Insuficiencia renal
En estado de equilibrio, se ha observado una relación lineal entre el aclaramiento del plasma con anti-Xa y el aclaramiento de creatinina, lo que indica una disminución en el aclaramiento de enoxaparina sódica en pacientes con la función renal disminuida.
En estado de equilibrio, la exposición a anti-Xa representada por AUC está aumentada de modo marginal en pacientes con insuficiencia renal leve (aclaramiento de creatinina 50-80 ml/min) y moderada (aclaramiento de creatinina 30-50 ml/min) tras administraciones subcutáneas repetidas de la dosis de 40 mg una vez al día. En pacientes con insuficiencia renal grave (aclaramiento de creatinina <30 ml/min), la AUC en estado de equilibrio está significativamente aumentada en una media de un 65% mayor en el día 4 (IC90%, 137-199%,p<0,001) tras administraciones subcutáneas repetidas de la dosis de 40 mg una vez al día (ver secciones 4.4 y 4.2: Insuficiencia renal).
Peso
Tras repetidas administraciones subcutáneas de 1,5 mg/kg una vez al día, la AUC media de la actividad anti-Xa es marginalmente más elevado en el estado de equilibrio en voluntarios sanos obesos (BMI 30-48 Kg/ m2) comparado con sujetos control no obesos (un 16 % en el día 4 de tratamiento), mientras que Amax no estaba aumentada. Hay un menor aclaramiento ajustado al peso en personas obesas con administración subcutánea.
Cuando se administró una dosis no ajustada al peso, se encontró que, la exposición anti-Xa es un 52% superior en las mujeres con bajo peso (<45 Kg) y un 27% superior en hombres con bajo peso (<57 kg) tras una dosis única de 40 mg, cuando se comparó con sujetos control con peso normal (ver sección 4.4: Pacientes de bajo peso).
Hemodiálisis
En un estudio único, la velocidad de eliminación fue similar pero la AUC fue dos veces mayor respecto a la población control, después de una dosis intravenosa única de 0,25 ó 0,50 mg/kg.
Interacciones farmacocinéticas
No se observaron interacciones farmacocinéticas entre enoxaparina y los fármacos trombolíticos cuando se administraron concomitantemente.
5.3 Datos preclínicos sobre seguridad
No se han realizado estudios a largo plazo en animales para evaluar el potencial carcinogénico de la enoxaparina.
Enoxaparina no fue mutagénica en los ensayos in vitro que incluyen, test de Ames, test de mutación en células de linfoma de ratón, test de aberración cromosómica en linfocitos humanos, y test de aberración cromosómica en médula ósea de rata.
Enoxaparina resultó no tener efecto sobre la fertilidad o capacidad reproductora de ratas hembras o machos cuando se administraron dosis SC de hasta 20 mg/kg/día. Se han realizado estudios teratológicos en ratas y conejos gestantes a dosis SC de enoxaparina de hasta 30 mg/kg/día. No hubo evidencia de efectos teratogénicos o de fetotoxicidad debido a la enoxaparina.
Además de los efectos anticoagulantes de la enoxaparina, no existe evidencia de efectos adversos en estudios de toxicidad subcutánea en ratas y perros a dosis de 15 mg/kg/día durante 13 semanas y en estudios de toxicidad subcutánea e intravenosa en ratas y monos a dosis de 10 mg/kg/día durante 26 semanas.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Agua estéril para preparaciones inyectables.
6.2 Incompatibilidades
No mezclar con otros productos.
6.3 Periodo de validez 24 meses.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 25° C. No congelar.
6.5 Naturaleza y contenido del envase Clexane 120 mg (12000 UI)/0,8 ml jeringas
Caja con 10 jeringas precargadas de 0,8 ml, conteniendo cada una 120 mg (12000 UI) de enoxaparina sódica.
Caja con 30 jeringas precargadas de 0,8 ml, conteniendo cada una 120 mg (12000 UI) de enoxaparina sódica.
Clexane 150 mg (15000 UI)/1 ml jeringas
Caja con 10 jeringas precargadas de 1ml, conteniendo cada una 150 mg (15000 UI) de enoxaparina sódica. Caja con 30 jeringas precargadas de 1ml, conteniendo cada una 150 mg (15000 UI) de enoxaparina sódica.
6.6 Precauciones especiales de eliminación y otras manipulaciones Instrucciones de uso para la técnica de inyección subcutánea
Ver sección 4.2.
En caso de que el paciente vaya a administrarse él mismo la inyección (autoinyección), el profesional sanitario le mostrará cómo hacerlo, antes de abandonar el hospital. Es esencial que el paciente siga exactamente estas instrucciones. En caso de dudas el paciente debe preguntar al profesional sanitario para que se las aclare.
Es necesario realizar una inyección subcutánea (bajo la piel) de forma correcta con el fin de reducir el dolor y hematoma en el punto de inyección.
Para evitar cualquier pinchazo accidental después de la inyección, las jeringas precargadas van equipadas con un dispositivo de seguridad automático.
Preparación del lugar de inyección
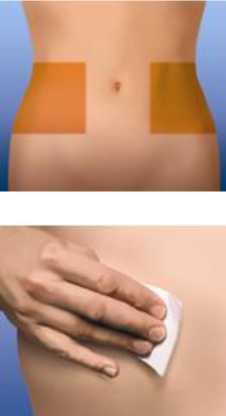
El lugar recomendado para la inyección es la zona del tejido graso del abdomen bajo, al menos a 5 cm del ombligo y hacia cualquiera de ambos costados.
Antes de la inyección lávese las manos. Limpie (no frote), la zona elegida para realizar la inyección, con un trozo de algodón con alcohol. Elija una zona diferente del abdomen bajo para cada inyección.
Preparación de la jeringa antes de la inyección
Compruebe la caducidad en el envase o en la jeringa. Si ha caducado no se debe utilizar. Verifique que la jeringa no está dañada y que el producto es una solución clara sin partículas. Si la jeringa estuviera dañada o el producto no fuera claro utilice otra jeringa.
- Quite el capuchón tirando del mismo.
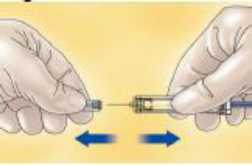
- Ajuste la dosis que tiene que ser inyectada (si fuera necesario).
La cantidad de solución que hay que inyectar debe ajustarse dependiendo del peso del paciente; por lo tanto se debe eliminar cualquier exceso de medicamento antes de administrar la inyección. Mantenga la jeringa apuntando hacia abajo (para mantener la burbuja de aire en la jeringa) y expulse el exceso de medicamento en un contenedor adecuado.
NOTA: Si el exceso de medicamento no se expulsa antes de la inyección, no se podrá activar el dispositivo de seguridad al finalizar la inyección.
Cuando no es necesario ajustar la dosis, la jeringa precargada está lista para ser utilizada. No elimine el aire de la jeringa antes de administrar la inyección.
Podría aparecer una gota en el extremo de la aguja. Si esto sucede, hay que eliminar la gota antes de administrar la inyección. Para ello se debe dar golpecitos suaves con el dedo a la jeringa, siempre con la aguja apuntando hacia abajo, hasta que se desprenda la gota.
Administración de la inyección
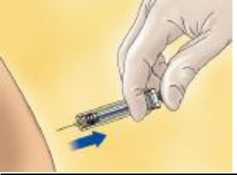
Mientras que se está tumbado o sentado en una posición cómoda, tomar un pliegue cutáneo entre el dedo pulgar y el índice.
Mantener la aguja en un ángulo adecuado respecto al pliegue cutáneo y pinchar en dicho pliegue. Este pliegue cutáneo debe mantenerse mientras se administra la inyección. Completar la administración de la inyección utilizando todo el medicamento de la jeringa.
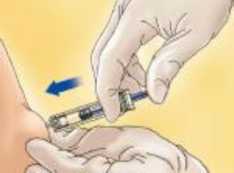
Una vez administrada toda la inyección, extraer la jeringa del lugar de inyección manteniendo el dedo en el émbolo.
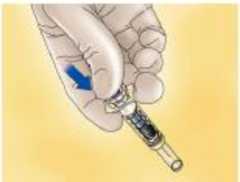
Para jeringas con dispositivo de seguridad:
Orientar la jeringa hacia abajo, alejada de sí mismo y de otras personas, y presionar firmemente el émbolo par activar el sistema de seguridad. La funda protectora cubrirá la aguja automáticamente a la vez que se escuchará un CLIC que confirma la activación del sistema de seguridad.
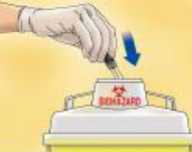
Depositar la jeringa inmediatamente en el contenedor más cercano para eliminación de agujas.
"I
an
Para mayor información, contacte con su médico.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
sanofi-aventis, S.A.
Josep Pla, 2 08019 Barcelona
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
Clexane 120 mg (12.000 UI) solución inyectable en jeringa precargada: N° Reg 63002 Clexane 150 mg (15.000 UI) solución inyectable en jeringas precargada: N° Reg 63000
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Clexane 120 mg (12.000 UI) solución inyectable en jeringa precargada: 24 abril 2000/ octubre 2009 Clexane 150 mg (15.000 UI) solución inyectable en jeringa precargada: 22 mayo 2000/ noviembre 2009
10. FECHA DE LA REVISIÓN DEL TEXTO
Enero 2015
La información detallada y actualizada de este medicamento está disponible en la página web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) http://www.aemps.gob.es/
19 de 19
Insuficiencia renal grave: