Bondenza 150 Mg, Comprimidos Recubiertos Con Pelicula
6^
ANEXO I
RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
/
■o^

1. NOMBRE DEL MEDICAMENTO
Bondenza 150 mg comprimidos recubiertos con película.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 150 mg de ácido ibandrónico (como monohidrato sódico).
Excipientes con efecto conocido:
Contiene 162,75 mg de lactosa monohidrato (equivalente a154,6 mg de lactosa anhidra).
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto con película
Comprimidos recubiertos con película, de color blanco o blanquecino, con forma oblonga, marcados con la inscripción “BNVA” en una cara y “150” en la otra.
4. DATOS CLÍNICOS 4.1 Indicaciones terapéuticas
Tratamiento de la osteoporosis en mujeres posmenopáusicas con riesgo elevado de fractura (ver sección 5.1).
Se ha demostrado una reducción en el riesgo de fracturas vertebrales, la eficacia en fracturas de cuello femoral no ha sido establecida.
rP
4.2 Posología y forma de administración
Posología
La dosis recomendada es de un comprimido con cubierta pelicular de 150 mg una vez al mes. El comprimido debe tomarse preferiblemente el mismo día de cada mes.
Bondenza debe tomarse después del ayuno nocturno (como mínimo, de 6 horas) y 1 hora antes del desayuno o de la primera bebida (distinta del agua) del día (ver sección 4.5) o de cualquier otro medicamento o suplemento por vía oral (incluido el calcio).
Se deberá indicar a las pacientes que, en caso de olvido de una dosis, tomen un comprimido de Bondenza 150 mg, la mañana siguiente al día que recuerden que olvidaron la dosis, a menos que les queden 7 días o menos para la administración de la siguiente dosis. Después las pacientes deberán volver a tomar su dosis el mismo día del mes que iniciaron originalmente el tratamiento.
Si les quedaran de 1 a 7 días para la administración de la siguiente dosis, las pacientes deberán esperar hasta la próxima dosis y entonces continuarán tomando la dosis mensual en la fecha originalmente elegida.
Las pacientes no deberán tomar dos comprimidos en una misma semana.
Si el aporte dietético es insuficiente (ver secciones 4.4 y 4.5), las pacientes deberían recibir suplementos de calcio y/o vitamina D.
No se ha establecido la duración óptima del tratamiento con bifosfonatos para la osteoporosis. La necesidad de continuar con el tratamiento debe ser reevaluada periódicamente considerando los beneficios y riesgos potenciales de Bondenza para cada paciente de forma individualizada, sobre todo tras 5 o más años de uso.
Poblaciones especiales Pacientes con insuficiencia renal
No se recomienda el uso de Bondenza en pacientes con un aclaramiento de creatinina inferior a 30 ml/min, debido a que la experiencia clínica es limitada (ver sección 4.4 y sección 5.2).
No es preciso un ajuste de dosis en pacientes con insuficiencia renal leve o moderada si el aclaramiento de creatinina es mayor o igual de 30 ml/min.
Pacientes con alteraciones de la función hepática No se precisa ningún ajuste de dosis (ver sección 5.2).
Pacientes de edad avanzada (> 65 años)
No se requiere ningún ajuste de dosis (ver sección 5.2).
Población pediátrica

No hay un uso relevante de Bondenza en niños menores de 18 años. Bondenza no ha sido estudiado en esta población (ver sección 5.1 y sección 5.2).
Forma de administración Por vía oral.
Los comprimidos se deben tragar enteros con un vaso de agua (de 180 a 240 ml) mientras la paciente está sentada o de pie. No se debe utiliza agua con una alta concentración de calcio. Si hay duda en cuanto a los niveles de calcio potencialmente altos en el agua del grifo (aguas duras), se recomienda usar agua embotellada con bajo contenido mineral.
Las pacientes no se podrán tumbar hasta 1 hora después de tomar Bondenza.El agua es la única bebida que se puede administrar con Bondenza.
Los comprimidos no se deben masticar ni chupar debido al peligro potencial de úlceras bucofaríngeas.
4.3 Contraindicaciones
Hipersensibilidad al ácido ibandrónico o a alguno de los excipientes incluidos en la sección 6.1. Hipocalcemia
Anormalidades esofágicas que retrasen el vaciamiento esofágico, como la estenosis o la acalasia Imposibilidad de permanecer erguido, tanto de pie como sentado, durante al menos 60 minutos
4.4 Advertencias y precauciones especiales de empleo
Hipocalcemia
Antes de iniciar el tratamiento con Bondenza, hay que corregir la hipocalcemia; así como deben tratarse de manera adecuada otros trastornos del metabolismo óseo y mineral. El aporte suficiente de calcio y vitamina D es esencial para todas las pacientes.
Irritación gastrointestinal
La administración oral de bifosfonatos puede causar irritación local de la mucosa gastrointestinal superior. Debido a estos posibles efectos irritantes y al potencial de empeoramiento de las enfermedades subyacentes, Bondenza debe administrarse con precaución a los pacientes con trastornos activos de la parte superior del aparato digestivo (ej. Esófago de Barrett diagnosticado, disfagia, otras enfermedades esofágicas, gastritis, duodenitis o úlceras).
En pacientes que reciben tratamiento oral de bifosfonatos, se han notificado reacciones adversas tales como esofagitis, úlceras esofágicas y erosiones esofágicas. En algunos casos fueron graves y requirieron hospitalización, raramente con sangrado o seguidas de estenosis esofágica o perforación.
El riesgo de experiencias adversas esofágicas graves parece ser mayor en pacientes que no cumplen
con las instrucciones posológicas y/o siguen tomando bifosfonatos por vía oral después de desarrollar síntomas indicativos de irritación esofágica. Los pacientes deben prestar especial atención y cumplir las instrucciones posológicas (ver sección 4.2).
Los médicos han de estar atentos a cualquier signo o síntoma que indique una posible reacción esofágica y los pacientes deben recibir instrucciones precisas para suspender el tratamiento con Bondenza y acudir al médico si desarrollan disfagia, odinofagia, dolor retroesternal o pirosis reciente o progresiva.
Aunque en los ensayos clínicos controlados no se ha observado incremento del riesgo, tras la comercialización, se han notificado casos de úlceras gástricas y duodenales con el uso de los bifosfonatos por vía oral, algunos graves y con complicaciones.
Como tanto los Antiinflamatorios No Esteroideos como los bifosfonatos se asocian, ambos, con irritación gastrointestinal, se recomienda tener precaución cuando se administren concomitantemente.
Osteonecrosis mandibular
Se ha observado casos de osteonecrosis mandibular generalmente asociados con extracciones dentales y/o infecciones locales (incluyendo osteomielitis) en pacientes oncológicos tratados con regímenes que incluían principalmente bifosfonatos de administración intravenosa. La mayoría de estos pacientes también recibieron quimioterapia y corticosteroides. También se ha observado osteonecrosis mandibular en pacientes con osteoporosis tratados con bifosfonatos de administración oral.
En aquellos pacientes con factores de riesgo concomitantes (p.ej. cáncer, quimioterapia, radioterapia, corticosteroides, higiene bucal pobre) deberá considerarse un examen dental con una apropiada odontología preventiva, antes de iniciar el tratamiento con bifosfonatos.
Durante el tratamiento, si es posible, estos pacientes deb en evitar procesos dentales invasivos. La cirugía dental puede agravar la situación en pacientes que desarrollen osteonecrosis mandibular durante la terapia con bifosfonatos. No hay datos disponibles que indiquen que la interrupción del tratamiento con bifosfonatos reduce el riesgo de osteonecrosis mandibular en pacientes que precisen procesos dentales. La valoración clínica del facultativo, debe orientar sobre cómo proceder con cada paciente según la valoración individual de la relación beneficio-riesgo.
ípicas subtrocantéricas y diafisarias del fémur asociadas al palmente en pacientes en tratamiento prolongado para la sversales u oblicuas cortas pueden ocurrir en cualquier parte a lo ebajo del trocánter menor hasta justo por encima de la cresta ras se producen después de un traumatismo mínimo o en ausencia de él y
Fracturas atípicas de fémur Se han notificado casos de fract tratamiento con bifosfonatos osteoporosis. Estas fractur largo del fémur, desde j supracondílea. Estas
algunos pacientes tienen dolor en el muslo o en la ingle, a menudo asociado con imágenes características de fracturas por sobrecarga, semanas a meses antes de que se presente la fractura femoral completa. Las fracturas son generalmente bilaterales; por lo tanto, el fémur del lado opuesto debe ser examinado en los pacientes tratados con bifosfonatos que hayan tenido una fractura de la diálisis femoral. También se ha notificado un bajo índice de consolidación de estas fracturas. Debe considerarse la interrupción del tratamiento con bifosfonatos, valorando de forma individualizada el balance beneficio/riesgo, en aquellos pacientes en los que exista sospecha de fractura atípica de fémur pendiente de evaluación.
Durante el tratamiento con bifosfonatos debe advertirse a los pacientes que notifiquen cualquier dolor en el muslo, cadera o ingle. En cualquier paciente que presente dichos síntomas deberá valorarse si existe una fractura de fémur incompleta.
Insuficiencia renal
Debido a la limitada experiencia clínica, no se recomienda el uso de Bondenza en pacientes con un aclaramiento de creatinina inferior a 30 ml/min (ver sección 5.2.).
Intolerancia a la galactosa
Los pacientes con problemas hereditarios raros de intolerancia a la galactosa, deficiencia de Lapp lactosa o malabsorción de glucosa-galactosa no deben tomar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Interacción entre el Medicamento-y los AlimentosLa biodisponibilidad oral del ácido ibandrónico disminuye en general con la ingesta de alimentos. En concreto, los productos que contienen calcio, incluida la leche, y otros cationes multivalentes (como aluminio, magnesio y hierro), pueden interferir en la absorción de Bondenza, como se ha demostrado en los estudios con animales. Así pues, se recomienda ayuno nocturno (como mínimo, de 6 horas) antes de tomar Bondenza y su mantenimiento durante una hora después (ver sección 4.2).
Interacciones con otros medicamentos
Las interacciones metabólicas son consideradas poco probables, puesto que el ácido ibandrónico no inhibe las principales isoenzimas del citocromo P450 hepático humano y tampoco induce el sistema hepático del citocromo P450 de las ratas (ver sección 5.2). El ácido ibandrónico se excreta sólo por vía renal y no se biotransforma.
Suplementos de calcio, antiácidos y otros medicamentos para administración oral que contienen cationes multivalentes
Es muy probable que los suplementos de calcio, los antiácidos y otros medicamentos para administración oral que contienen cationes multivalentes (como aluminio, magnesio y hierro) dificulten la absorción de Bondenza. Por eso, las pacientes no podrán tomar ningún otro medicamento por vía oral desde, por lo menos, 6 horas antes hasta 1 hora después de administrar Bondenza.
íP
Ácido acetilsalicílico y AINES En un ensayo a dos años llevado a cabo en mujeres posmenopáusicas con osteoporosis (BM 16549), la incidencia de acontecimientos en el tracto gastrointestinal superior tras la administración conjunta con ácido acetilsalicílico o AINES, fue similar en tratamientos con una dosis diaria de 2,5 mg de ácido ibandrónico y Bondenza 150 mg dosis mensual, tras uno y dos años.
Bloqueadores H2 o inhibidores de la bomba de protones
De las 1500 pacientes incluidas en el ensayo BM 16549 en el que se comparaban las pautas posológicas de la administración mensual con la administración diaria de ácido ibandrónico, un 14 % y un 18 % de estas pacientes tomaban antihistamínicos (H2) o inhibidores de la bomba de protones tras uno y dos años, respectivamente. Dentro de este grupo de pacientes, la incidencia de acontecimientos en el tracto gastrointestinal superior fue similar, independientemente de si habían recibido Bondenza 150 mg una vez al mes o diariamente 2,5 mg de ácido ibandrónico.
La ranitidina administrada por vía intravenosa aumentó la biodisponibilidad del ácido ibandrónico en varones voluntarios sanos y de mujeres posmenopáusicas en un 20 %, probablemente por el descenso de la acidez gástrica. Sin embargo, dado que este aumento se encuentra dentro del intervalo normal de variación en la biodisponibilidad del ácido ibandrónico, no es necesario un ajuste de la dosis de Bondenza cuando se administre con antagonistas H2 o con otros principios activos que aumente el pH del estómago.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No existen datos suficientes acerca del uso del ácido ibandrónico por las mujeres embarazadas. Los estudios con ratas han revelado cierta toxicidad sobre la función reproductora (ver sección 5.3). Se desconoce el posible riesgo para la especie humana.
No se debe utilizar Bondenza durante el embarazo.
Lactancia
Se desconoce si el ácido ibandrónico se excreta con la leche humana. Los estudios con ratas lactantes han demostrado la presencia de valores reducidos del ácido ibandrónico en la leche después de su administración intravenosa.
Se desaconseja el uso de Bondenza durante la lactancia.
Fertilidad
No hay datos de los efectos del ácido ibandrónico en humanos. En estudios sobre la función reproductora en ratas por vía oral, el ácido ibandrónico disminuyó la fertilidad. En estudios en ratas por vía intravenosa, el ácido ibandrónico disminuyó la fertilidad a dosis diarias altas (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Tomando como base el perfil farmacocinético y farmacodinámico y las reacciones adversas notificadas, se espera que Bondenza no tenga efecto o éste sea despreciable sobre la capacidad de conducir y utilizar máquinas.
&
4.8 Reacciones adversas

os y de la experiencia das fueron artralgia y
Resumen del perfil de seguridad
El perfil de seguridad de Bondenza procede de los ensayos clínicos contro post-comercialización. Las reacciones adversas más frecuentemente notific
síntomas seudogripales. Estos síntomas están típicamente asociados a la primera dosis, generalmente son de corta duración, de intensidad leve o moderada, y que se suelen resolver con la continuación del tratamiento sin requerir medidas adicionales (por favor ver párrafo “enfermedad seudogripal”).
Tabla de reacciones adversas
En la tabla 1 se muestra una visión general de reacciones adversas. La seguridad del tratamiento oral con 2,5 mg de ácido ibandrónico administrados diariamente, se evaluó en 1.251 pacientes tratados en 4 ensayos clínicos controlados con placebo; procediendo la gran mayoría de los pacientes del ensayo pivotal sobre fracturas a lo largo de tres años (MF4411).
En un ensayo a dos años en mujeres posmenopáusicas con osteoporosis (BM 16549), la seguridad general de Bondenza 150 mg dosis mensual y 2,5 mg de ácido ibandrónico administrados diariamente, fue similar. El porcentaje total de pacientes que experimentaron una reacción adversa, representó un 22,7 % y un 25,0 % para Bondenza 150 mg dosis mensual, tras uno y dos años respectivamente. En la mayor parte de los casos no fue necesaria la suspensión del tratamiento.
Tabla 1:Reacciones adversas ocurridas en los ensayos fase III BM16549, MF4411 y en la experiencia post-comercialización en mujeres postmenopáusicas que recibieron Bondenza 150 mg una _vez al mes o 2,5 mg de ácido ibandrónico diariamente._
|
Las reacciones adversas se enumeran de acuerdo al sistema de clasificación de órganos y categoría de frecuencia MedDRA Se definen las categorías de frecuencia usando la siguiente convención: Muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000), muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Dentro de cada grupo de frecuencia se presentan las reacciones adversas en orden decreciente de gravedad. | ||||
|
Sistema de Clasificación Órganos |
Frecuentes |
Poco frecuentes |
Raras |
Muy raras |
|
Trastornos del sistema inmunológico |
Reacciones de hipersensibilidad |
Reacción/ schock anafiláctico *f | ||
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Mareos |
!> | |
|
Trastornos oculares |
Inflamación ocular*f | |||
|
Trastornos gastrointestinales* |
Esofagitis, Gastritis, Reflujo gastroesofágico, Dispepsia, Diarrea, Dolor abdominal, Náuseas |
Esofagitis incluyendo ulceraciones o estenosis esofágicas y disfagia, Vómitos, Flatulencia |
Duodenitis | |
|
Trastornos de la piel y del tejido subcutáneo |
Erupción cutánea cf |
<Ír K |
Angioedema, Edema facial, Urticaria | |
|
Trastornos musculoesqueléticos, y del tejido conjuntivo |
Artralgia, Mialgia, Dolor, xQ,' musculoesquelético, Calambres musculares, Rigidez musculoesquelética |
Dolor de espalda |
Fracturas atípicas subtrocantéri-cas y diafisarias del fémurf ) |
Osteonecrosis mandibular*f |
|
Trastornos generales y alteraciones en el lugar de administración |
Enfermedad seudogripal* |
Fatiga | ||
*Ver abajo más información
fIdentificado en la experiencia post-comercialización
Descripción de las reacciones adversas seleccionadas
Reacciones adversas gastrointestinales
En el ensayo de tratamiento con la dosis mensual se incluyeron las pacientes con antecedentes de enfermedad gastrointestinal incluyendo las pacientes con úlcera péptica sin sangrado reciente u hospitalización y las pacientes con dispepsia o reflujo controlado con medicación. No se observó diferencia en la incidencia de los acontecimientos adversos en el tracto gastrointestinal superior entre las pacientes tratadas con Bondenza 150 mg dosis mensual y las tratadas con dosis diarias de 2,5 mg de ácido ibandrónico.
Enfermedad seudogripal
. El síndrome seudogripal incluye los acontecimientos ocurridos durante la fase aguda de la reacción y síntomas como mialgia, artralgia, fiebre, escalofríos, fatiga, nauseas, pérdida del apetito o dolor óseo.
Se ha observado osteonecrosis mandibular en pacientes tratados con bifosfonatos. La mayoría de los informes se refieren a pacientes oncológicos, pero también algunos casos han sido observados en pacientes tratados de osteoporosis. La osteonecrosis mandibular se asocia generalmente con extracciones dentales y/o infecciones locales (incluyendo osteomielitis). También se consideran factores de riesgo el diagnóstico de cáncer, quimioterapia, radioterapia, corticosteroides, y una higiene bucal pobre (ver sección 4.4).
Inflamación ocular
Se han notificado casos de inflamación ocular como uveítis, episcleritis y escleritis con el tratamiento con ácido ibandrónico. En algunos casos estos acontecimientos no se resolvieron hasta que se interrumpió el tratamiento con ácido ibandrónico.
&
Reacción/shock anafiláctico Se han notificado casos de reacción/schock anafiláctico, incluyendo eventos mortales, en pacientes tratados con ac. ibandrónico intravenoso.
4.9 Sobredosis
No se dispone de información concreta sobre el tratamiento de la sobredosis de Bondenza.
No obstante, según los datos conocidos sobre este grupo terapéutico, la sobredosis por vía oral puede ocasionar complicaciones de la parte alta del tubo digestivo (dolor de estómago, dispepsia, esofagitis, gastritis o úlceras) o hipocalcemia. Se debe administrar leche o antiácidos que se fijen a Bondenza y aplicar un tratamiento sintomático de las posibles reacciones adversas. Dado el riesgo de irritación esofágica, no conviene inducir el vómito y la paciente deberá permanecer totalmente erguida.
íP
5. PROPIEDADES FARMACOLÓGICAS

5.1 Propiedades farmacodinámicas
ara el tratamiento de las enfermedades óseas, bifosfonatos,
Grupo farmacoterapéutico: Medicament código ATC: M05BA06
Mecanismo de acción El ácido ibandrónico es nitrogenados; actúa de f

sfonato muy potente que pertenece al grupo de bifosfonatos selectiva sobre el tejido óseo y, en concreto, inhibe la actividad osteoclástica sin influir directamente en la formación de hueso. No impide el reclutamiento de los osteoclastos. El ácido ibandrónico conduce a un aumento neto progresivo de la masa ósea y reduce la incidencia de fracturas de las mujeres posmenopáusicas hasta los valores previos a la menopausia, gracias a la disminución del recambio óseo elevado.
Efectos farmacodinámicos
La acción farmacodinámica del ácido ibandrónico se basa en una inhibición de la resorción ósea. En condiciones in vivo, el ácido ibandrónico evita la destrucción ósea experimental causada por el cese de la función gonadal, los retinoides, los tumores o los extractos tumorales. Asimismo, inhibe la resorción de hueso endógeno en las crías de rata (en fase de crecimiento rápido), con lo que aumenta la masa ósea normal en comparación con la de los animales no tratados.
En los modelos con animales se ha confirmado que el ácido ibandrónico es un inhibidor muy potente de la actividad osteoclástica. No se han detectado indicios de mineralización anómala en las crías de rata, ni siquiera después de administrar dosis más de 5000 veces superiores a las utilizadas en la osteoporosis.
La administración diaria y la administración duradera e intermitente (con intervalos largos sin tratamiento) a ratas, perros y monos se asoció con la formación de hueso nuevo de calidad normal, que conservaba o aumentaba su fuerza mecánica, incluso en dosis superiores a las dosis farmacológicas previstas (es decir, dosis que se hallaban dentro del intervalo tóxico). En humanos, la eficacia de la
administración diaria y de la intermitente con un intervalo libre de dosis de ácido ibandrónico entre 910 semanas, ha sido confirmada en un ensayo clínico (MF 4411), en el cual el ácido ibandrónico demostró tener eficacia antifractura.
En modelos animales, el ácido ibandrónico ocasionó alteraciones bioquímicas indicativas de una inhibición de la resorción ósea proporcional a la dosis, incluida la supresión de los marcadores bioquímicos urinarios de la degradación del colágeno óseo (como la desoxipiridinolina y los N-telopéptidos entrecruzados del colágeno de tipo I (NTX)).
En un estudio de bioequivalencia de fase 1 realizado en 72 mujeres posmenopáusicas que recibían una dosis oral de 150 mg cada 28 días hasta un total de 4 dosis, se observó una inhibición de los telopéptidos carboxiterminales entrecruzados del colágeno de tipo I (CTX) a las 24 horas de la administración (inhibición media del 28 %), con una inhibición media máxima (69 %) observada a los 6 días después de la administración. En la tercera y cuarta dosis, la inhibición media máxima a los 6 días de la administración de la dosis fue del 74 % con una reducción de la inhibición media de un 56 % observado a los 28 días siguientes de la administración de la cuarta dosis. Dejando de administrar dosis posteriores, se produce una perdida de la supresión de los marcadores bioquímicos de la resorción ósea.
Eficacia clínica
Para identificar a las mujeres con un riesgo elevado de sufrir fracturas deben considerarse factores de riesgo independientes, tales como, baja densidad de masa ósea (DMO), edad, existencia de fracturas previas, antecedentes familiares de fracturas, alto recambio óseo y bajo índice de masa corporal.
Cr
Bondenza 150 mg, dosis mensual
;<y
Densidad mineral ósea (DMO)
En un ensayo a dos años multicéntrico, doble ciego realizado en mujeres posmenopáusicas con osteoporosis (BM 16549), Bondenza 150 mg administrado una vez al mes ha demostrado ser igual de eficaz aumentando la DMO que 2,5 mg de ácido ibandrónico administrados diariamente (valor lumbar basal, T-score por debajo de -2,5 DE).
Esto se demostró tanto en el análisis fundamental a un año como en el análisis confirmatorio a dos
años (Tabla 2).
Tabla 2: Cambios relativos medios en el valor de referencia de la DMO de columna lumbar, cadera completa, cuello femoral y trocánter tras un año (análisis fundamental) y dos años de tratamiento (Por Protocolo Poblacional) en el ensayo BM 16549.
|
Datos a un año del ensayo BM 16549 |
Datos a dos años del ensayo BM 16549 | |||
|
Cambios relativos medios en el valor de referencia % [95% CI] |
2,5 mg de ácido ibandrónico dosis diaria (N=318) |
Bondenza 150 mg dosis mensual (N=320) |
2,5 mg de ácido ibandrónico dosis diaria (N=294) |
Bondenza 150 mg dosis mensual (N=291) |
|
Columna lumbar L2-L4 DMO |
3,9 [3,4, 4,3] |
4,9 [4,4, 5,3] |
5,0 [4,4, 5,5] |
6,6 [6,0, 7,1] |
|
Cadera Completa DMO |
2,0 [1,7, 2,3] |
3,1 [2,8, 3,4] |
2,5 [2,1, 2,9] |
4,2 [3,8, 4,5] |
|
Cuello femoral DMO |
1,7 [1,3, 2,1] |
2,2 [1,9, 2,6] |
1,9 [1,4, 2,4] |
3,1 [2,7, 3,6] |
|
Trocánter BMO |
3,2 [2,8, 3,7] |
4,6 [4,2, 5,1] |
4,0 [3,5, 4,5] |
6,2 [5,7, 6,7] |
Además, según el análisis prospectivo a un año (p=0,002) y a dos años p<0,001, Bondenza 150 mg, dosis mensual demostró ser superior a 2,5 mg de ácido ibandrónico administrados diariamente en incrementos de la DMO lumbar.
A un año (análisis fundamental), el 91,3 % de las pacientes que recibieron la dosis mensual de Bondenza 150 mg fueron respondedores (aquellos que mantuvieron o aumentaron el valor de DMO lumbar sobre los valores de referencia), frente al 84 % de pacientes respondedores que recibieron una dosis diaria de 2,5 mg de ácido ibandrónico (p=0,005). A dos años, el 93,5 % y 86,4 % de las pacientes que recibieron la dosis mensual de Bondenza 150 mg o la dosis diaria de 2,5 mg de ácido ibandrónico respectivamente, fueron respondedores.
A un año, en cadera completa, el 90 % (p<0,001) de las pacientes que recibieron la dosis mensual de Bondenza 150 mg y el 76,7 % de las pacientes que recibieron la dosis diaria de 2,5 mg de ácido ibandrónico presentaron una DMO por encima o igual a los valores de referencia. A dos años el
93.4 % (p<0,001) de las pacientes que recibieron la dosis mensual de Bondenza 150 mg y el 78,4 % de las pacientes que recibieron la dosis diaria de 2,5 mg de ácido ibandrónico presentaban una DMO por encima o igual a los valores de referencia.
Si tenemos en cuenta un criterio más estricto que combina ambos valores de DMO (columna lumbar y cadera completa) encontramos que resultan respondedores, a un año, un 83,9 % (p<0,001) y un 65,7 % de las pacientes que reciben la dosis mensual de Bondenza 150 mg y de las pacientes que reciben la dosis diaria de 2,5 mg de ácido ibandrónico, respectivamente. A dos años, un 87,1 % (p<0,001) y un
70.5 % de los pacientes cumplen con este criterio en los brazos de dosis mensual de 150 mg y dosis diaria de 2,5 mg respectivamente.
Marcadores bioquímicos del recambio óseo
En todos los tiempos de medida se han observado reducciones clínicamente significativas de los niveles de CTX en suero, es decir, a los 3, 6,12 y 24 meses. Tras un año (análisis fundamental) la mediana de los cambios relativos con respecto a los valores de referencia fue de -76 % para la dosis mensual de Bondenza 150 mg y de -67 % para la dosis diaria de 2,5 mg de ácido ibandrónico. A dos años la mediana de los cambios relativos fue de -68 % y -62 % en los brazos de dosis mensual de 150 mg y dosis diaria de 2,5 mg respectivamente.
A un año el 83,5 % (p=0,006) de las pacientes que recibieron la dosis mensual de Bondenza 150 mg y el 73,9 % de las pacientes que recibieron diariamente 2,5 mg de ácido ibandrónico, resultaron respondedores (definido como un descenso >50 % del valor de referencia). A dos años 78,7 % (p=0,002) y 65,6 % de las pacientes resultaron respondedores para la dosis mensual de 150 mg y la dosis diaria de 2,5 mg respectivamente.
Según los resultados del ensayo BM 16549 se espera que la dosis mensual de Bondenza 150 mg sea igual de efectiva en la prevención de fracturas que la dosis diaria de2,5 mg de ácido ibandrónico.
Dosis diaria de 2,5 mg de ácido ibandrónico
En el primer ensayo a 3 años, aleatorizado, doble ciego, controlado con placebo, sobre fracturas (MF 4411) se observó un descenso estadística y clínicamente significativo de la incidencia de nuevas fracturas vertebrales radiológicas (morfométricas) y clínicas (tabla 3). En este ensayo se evaluó el ácido ibandrónico en dosis orales de 2,5 mg al día y dosis intermitentes de 20 mg como búsqueda de dosis. El ácido ibandrónico se administró 60 minutos antes del desayuno o de la primera bebida del día (período de ayuno posterior a la dosis). En este ensayo se reclutó a mujeres de 55 a 80 años, que llevaban, por lo menos, 5 años desde la menopausia y mostraban una DMO de 2 a 5 DE por debajo de la media premenopáusica (T-score) de, al menos, una vértebra lumbar [L1-L4] y que habían sufrido de una a cuatro fracturas vertebrales prevalentes. Todas las pacientes recibieron 500 mg de calcio y 400 UI de vitamina D al día. Se evaluó la eficacia entre 2.928 pacientes.
La incidencia de nuevas fracturas vertebrales se redujo de forma estadística y clínicamente significativa con la pauta de 2,5 mg de ácido ibandrónico administrados diariamente. Esta pauta redujo la aparición de nuevas fracturas vertebrales radiológicas en un 62 % (p=0,0001) durante los tres años del ensayo. La reducción del riesgo relativo alcanzó el 61 % al cabo de 2 años (p=0,0006) de tratamiento. No se obtuvieron diferencias estadísticamente significativas después de 1 año de tratamiento (p=0,056). El efecto profiláctico de las fracturas se mantuvo durante todo el ensayo. No se hallaron indicios de que el efecto se disipara con el tiempo.
La incidencia de fracturas vertebrales clínicas también se redujo en un 49 % (p=0,011). El fuerte efecto sobre las fracturas vertebrales quedó reflejado, asimismo, en una reducción estadísticamente significativa de la pérdida de talla, en comparación con el placebo (p<0,0001).
Tabla 3: resultados del ensayo MF 4411 de fracturas a los 3 años (%, IC del 95 %)
|
Placebo (N=974) |
2,5 mg de ácido ibandrónico dosis diaria (N=977) | |
|
Reducción del riesgo relativo Nuevas fracturas vertebrales morfométricas |
62 % (40,9,75,1) | |
|
Incidencia de nuevas fracturas vertebrales morfométricas |
9,56 % (7,5 , 11,7) |
4,68 % (3,2,6,2) |
|
Reducción del riesgo relativo de las fracturas vertebrales clínicas |
49 % (14,03,69,49) | |
|
Incidencia de fracturas vertebrales clínicas |
5,33 % (3,73,6,92) |
(1,61,3,89) |
|
DMO: diferencia media a los 3 años con respecto al valor lumbar basal |
1,26 % (0,8 , 1,7) |
6,54 % (6,1,7,0) |
|
DMO: diferencia media a los 3 años con respecto al valor basal de toda la cadera |
-0,69 % (-1,0 , -0,4) __ |
3,36 % (3,0,3,7) |
El efecto del tratamiento con ácido ibandrónico fue evaluado en un análisis de subpoblación de pacientes que tenían el valor lumbar basal DMO T-score por debajo de - 2,5 la reducción del riesgo de fracturas vertebrales fue considerado consistente con lo visto para la población global.
cr
Tabla 4: Resultado del ensayo MF 4411 de fractura a los 3 años (% IC 95 % ) para pacientes con valor lumbar basal DMO T-score por debajo de -2,5
|
Placebo (N=587) |
2,5 mg de ácido ibandrónico dosis diaria (N=575) | |
|
Reducción del riesgo relativo Nuevas fracturas vertebrales morfométricas |
59 % (34,5, 74,3) | |
|
Incidencia de nuevas fracturas vertebrales morfométricas |
12,54 % (9,53 , 15,55) |
5,36 % (3,31,7,41) |
|
Reducción del riesgo relativo de las fracturas vertebrales clínicas |
50 % (9,49,71,91) | |
|
Incidencia de fracturas vertebrales clínicas |
6,97 % (4,67,9,27) |
3,57 % (1,89,5,24) |
|
DMO: diferencia media a los 3 años con respecto al valor lumbar basal |
1,13 % (0,6 , 1,7) |
7,01 % (6,5,7,6) |
|
DMO: diferencia media a los 3 años con respecto al valor basal de toda la cadera |
-0,70 % (-1,1 , -0,2) |
3,59 % (3,1,4,1) |
En el total de la población de pacientes incluidos en el ensayo MF4411, no se observó ningún descenso en el número de fracturas no vertebrales, sin embargo, la toma diaria de ácido ibandrónico pareció ser efectiva en una subpoblación de alto riesgo (DMO en cuello femoral T-score < -3,0), en la que se observó una reducción del 69% en el riesgo de sufrir fracturas no vertebrales.
El tratamiento diario con 2,5 mg aumentó de forma progresiva la DMO vertebral y no vertebral.
El incremento de la DMO lumbar a los 3 años, en relación con el placebo, representó 5,3 % y 6,5 % con respecto al valor basal. El aumento de la DMO de la cadera, en relación con el valor basal, resultó del 2,8 % en el cuello femoral, del 3,4 % en toda la cadera y del 5,5 % en el trocánter.
Los marcadores bioquímicos del recambio óseo (como la CTX urinaria y la osteocalcina sérica) manifestaron el patrón previsible de supresión hasta las cifras premenopáusicas y alcanzaron la supresión máxima a lo largo de 3 a 6 meses.
Los marcadores bioquímicos de la resorción ósea experimentaron un descenso clínicamente relevante del 50 % ya durante el primer mes de tratamiento con 2,5 mg de ácido ibandrónico administrados diariamente.
Después de suspender el tratamiento, la tasa de elevación de la resorción ósea, asociada con la osteoporosis posmenopáusica, revirtió hasta los valores patológicos previos al tratamiento.
El análisis histológico de las muestras de biopsia ósea de las mujeres posmenopáusicas, efectuado a los dos y tres años del tratamiento, mostró un hueso de calidad normal y ningún defecto de la mineralización.
Población pediátrica (ver sección 4.2 y sección 5.2)Bondenza no ha sido estudiado en población pediátrica por lo tanto, no hay datos de eficacia o seguridad disponibles para esta población.
5.2 Propiedades farmacocinéticas

Los efectos farmacológicos fundamentales del ácido iband relación directa con las concentraciones plasmáticas re estudios con animales y seres humanos.
Absorción
El ácido ibandrónico se absorbe en seguida administración y las concentraciones plasmá
sobre el hueso no guardan una omo se ha demostrado en diversos
parte alta del tubo digestivo después de su as aumentan de forma proporcional hasta la dosis de 50 mg, con incrementos mayores a la proporcionalidad de la dosis una vez alcanzada esta dosis. Las concentraciones plasmáticas máximas se observaron al cabo de 0,5 a 2 horas (mediana de 1 hora) en ayunas y la biodisponibilidad absoluta llegó al 0,6 %. El grado de absorción se altera cuando se toma junto con alimentos o bebidas (que no sean agua). La biodisponibilidad disminuye casi en un 90 % si el ácido ibandrónico se administra con un desayuno habitual y no en ayunas. La biodisponibilidad apenas disminuye si el ácido ibandrónico se toma 60 minutos antes del desayuno. Tanto la biodisponibilidad como el incremento de la DMO disminuyen si el desayuno o las bebidas se ingieren menos de 60 minutos después de tomar ácido ibandrónico.
Distribución
Después de la exposición sistémica inicial, el ácido ibandrónico se une rápidamente al hueso o se excreta en la orina. El volumen terminal aparente de distribución en la especie humana resulta, como mínimo, de 90 l y la cantidad de la dosis que llega al hueso se estima como el 40-50 % de la dosis circulante. La unión a las proteínas del plasma humano es aproximadamente un 85 % - 87 % (determinada en condiciones in vitro, con concentraciones terapéuticas), por lo que la posibilidad de interacción con otros medicamentos por desplazamiento es mínima.
Biotransformación
No hay pruebas de que el ácido ibandrónico se metabolice en los animales o en la especie humana.
Eliminación
La fracción absorbida del ácido ibandrónico desaparece de la circulación a través de la absorción ósea (40-50 % en mujeres posmenopáusicas) y el resto se elimina inalterado por los riñones. La fracción no absorbida del ácido ibandrónico se excreta de forma intacta con las heces.
El intervalo de las semividas aparentes observadas es amplio pero, por regla general, la semivida terminal aparente se sitúa en el intervalo de 10 a 72 horas. Como los valores calculados están principalmente en función de la duración del estudio de la dosis administrada y de la sensibilidad analítica, la semivida terminal real es probable que sea sustancialmente más larga al igual que ocurre con otros bifosfonatos. Los valores plasmáticos iniciales descienden en seguida para alcanzar el 10 % de los valores máximos a las 3 y a las 8 horas de su administración intravenosa u oral, respectivamente.
El aclaramiento total del ácido ibandrónico es reducida: los valores medios se sitúan dentro del margen de 84-160 ml/min. La depuración renal (aprox. 60 ml/min entre mujeres posmenopáusicas sanas) explica del 50 al 60 % de la depuración total y se relaciona con el aclaramiento de creatinina. La diferencia entre el aclaramiento total y la depuración renal refleja, con toda seguridad, la captación por el hueso.
La vía secretora no incluye, en principio, ningún sistema de transporte ácido o alcalino que intervenga en la eliminación de otros principios activos. Además, el ácido ibandronico no inhibe las principales isoenzimas del citocromo P450 hepático humano y tampoco induce el sistema hepático del citocromo P450 de las ratas.
Farmacocinética en situaciones clínicas especiales Sexo
La biodisponibilidad y la farmacocinética del ácido ibandrónico se asi
i
Raza
xO
No hay pruebas de que existan diferencias étnicas de interés clínico en la disposición del ácido ibandrónico por los asiáticos y los blancos. Hay muy pocos datos sobre pacientes de origen africano.
Pacientes con insuficiencia renal &........ . .
El aclaramiento renal del ácido ibandrónico entre pacientes con distintos grados de insuficiencia renal se relaciona linealmente con el aclaramiento de creatinina.
Según se demostró en el ensayo BM 16549 donde la mayoría de las pacientes tenían insuficiencia renal leve a moderada, no es necesario un ajuste de dosis en pacientes con insuficiencia renal leve o moderada (CLCr igual o mayor de 30 ml/min).
Los sujetos con insuficiencia renal grave (CLCr menor de 30 ml/min) que reciban 10 mg de ácido ibandrónico al día por vía oral durante 21 días tienen concentraciones plasmáticas de 2 a 3 veces mayores que aquellos con una función renal normal; la depuración total del ácido ibandrónico llegó a 44 ml/min. Tras la administración intravenosa de 0,5 mg, la depuración total, renal y extrarrenal se redujo en un 67 %, 77 % y 50 %, respectivamente, entre los sujetos con insuficiencia renal grave pero la tolerabilidad relacionada con esta mayor exposición no disminuyó. Dada la limitada experiencia clínica, no se recomienda el uso de Bondenza en pacientes con insuficiencia renal grave (véase la sección 4.2 y sección 4.4). No se ha evaluado la farmacocinética del ácido ibandrónico entre pacientes con enfermedad renal terminal tratada por medios distintos a la hemodiálisis. La farmacocinética del ácido ibandrónico en estos casos se ignora; bajo ningún concepto, debe administrarse este preparado a estas pacientes.
Pacientes con alteraciones de la función hepática (ver sección 4.2)
No hay datos farmacocinéticos sobre el ácido ibandrónico en casos de alteración hepática. El hígado no desempeña ningún papel importante para la depuración del ácido ibandrónico, que no se metaboliza sino que se elimina mediante excreción renal y captación ósea. Por consiguiente, no es necesario ajustar la dosis de las pacientes con alteraciones hepáticas.
Pacientes de edad avanzada (ver sección 4.2)
En un estudio multivariable, la edad no resultó un factor independiente para ninguno de los parámetros farmacocinéticos examinados. Como la función renal disminuye con la edad, aquél es el único factor que merece consideración (ver sección sobre insuficiencia renal).
Población pediátrica (ver sección 4.2 y sección 5.1)
No se dispone de datos sobre el uso de Bondenza en estos grupos de edad.
5.3 Datos preclínicos sobre seguridad
Los efectos tóxicos, por ejemplo, signos de daños renales, se manifestaron en perros sólo con exposiciones que excedían suficientemente la máxima exposición humana, lo que indica una relevancia clínica mínima.
Mutagenesis/carcinogenesis:
No se hallaron indicios de poder cancerígeno. Los ensayos de genotoxicidad tampoco revelaron pruebas de la actividad genética del ácido ibandrónico.
Toxicidad sobre la función reproductora:
Durante el tratamiento oral de ratas y de conejos no se encontraron pruebas de ningún efecto fetotóxico o teratógeno directo del ácido ibandrónico y tampoco se advirtieron efectos secundarios para el desarrollo de la generación F1 de las ratas que recibieron una exposición, como mínimo, 35 veces mayor que la humana según los datos extrapolados. Los efectos sobre la función reproductora de la rata en estudios por vía oral, consistieron en un aumento de pérdidas preimplantación a dosis de 1 mg/kg/día y superiores. En estudios sobre la función reproductora de las ratas por vía intravenosa, el ácido ibandrónico disminuyó el recuento de esperma a dosis de 0,3 y 1 mg/kg/día y disminuyó la fertilidad en los machos a 1 mg / kg / día y en las hembras a 1,2 mg / kg / día. Los efectos adversos del ácido ibandrónico en los estudios sobre la toxicidad de la función reproductora de la rata son los mismos que los de los bifosfonatos como grupo. Se caracterizan por un descenso del número de lugares de implantación, dificultades para el parto natural (distocia) y aumento de las variaciones viscerales (síndrome de la pelvis renal y de los uréteres).
ÍN
6
Núcleo del comprimido Lactosa monohidrato Povidona
Celulosa microcristalina Crospovidona Ácido esteárico
hO
c/
Sílice coloidal anhid ra
Cubierta del c

ido
Hipromelosa Dióxido de titanio (E 171) Talco
Macrogol 6000
5 años.
6.4 Precauciones especiales de conservación
Este medicamento no requiere condiciones especiales de conservación.
6.5 Naturaleza y contenido del envase
Bondenza 150 mg comprimidos con cubierta pelicular se presentan en blísteres (PVC/PVDC, sellado con una lámina de aluminio) con 1 ó 3 unidades.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local. La eliminación de productos farmacéuticos en el medio ambiente se debe reducir al mínimo.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN

<C
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City
AL7 1TW Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COM

EU/1/03/266/003
EU/1/03/266/004

2. 2004
9. FECHA DE LA PRIMERA rCIÓN/RENOVACIÓN DE LA
AUTORIZACIÓN

10. FECHA DE
VISIÓN DEL TEXTO
DE
ón detallada d edicamentos
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu
1. NOMBRE DEL MEDICAMENTO
Bondenza 3 mg solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Una jeringa precargada de 3 ml de solución contiene 3 mg de ácido ibandrónico (como monohidrato sódico).
La concentración de ácido ibandrónico en la solución inyectable es de 1 mg por ml.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
&
Solución inyectable.
La solución es clara e incolora.
4. DATOS CLÍNICOS 4.1 Indicaciones terapéuticas
n rie
Tratamiento de la osteoporosis en mujeres posmenopáusicas con riesgo elevado de fractura (ver sección 5.1).
Se ha demostrado una reducción en el riesgo de fractur femoral no ha sido establecida.
:<U
verteb
ebrales, la eficacia en fracturas de cuello
4.2 Posología y forma de administración
Posología
La dosis recomendada es de 3 mg de ácido ibandrónico, administrado como inyección intravenosa durante 15 - 30 segundos, cada tres meses.
Las pacientes deben recibir suplementos de calcio y vitamina D (ver sección 4.4 y sección 4.5).
Si usted olvidó una dosis, se debería administrar la inyección tan pronto como sea posible. Después las inyecciones se deberían programar cada tres meses a partir de la fecha de la última inyección.
No se ha establecido la duración óptima del tratamiento con bifosfonatos para la osteoporosis. La necesidad de continuar con el tratamiento debe ser reevaluada periódicamente considerando los beneficios y riesgos potenciales de Bondenza para cada paciente de forma individualizada, sobre todo tras 5 o más años de uso.
Poblaciones especiales Pacientes con insuficiencia renal
No se recomienda la administración de Bondenza inyectable a pacientes con creatinina sérica superior a 200 pmol/l (2.3 mg/dl) o con aclaramiento de creatinina (medida o estimada) inferior a 30ml/min debido a que los datos clínicos disponibles de estudios incluyendo este tipo de pacientes son limitados (ver sección 4.4 y sección 5.2).
No es preciso un ajuste de dosis en pacientes con insuficiencia renal leve o moderada si la creatinina sérica es menor o igual de 200 pmol/l (2,3 mg/dl) o si el aclaramiento de creatinina (medido o estimado) es mayor o igual de 30 ml/min.
Pacientes con alteraciones de la función hepática No se precisa ningún ajuste de dosis (ver sección 5.2).
Pacientes de edad avanzada ( >65 años)No se requiere ningún ajuste de dosis (ver sección 5.2). Población pediátrica
No hay un uso relevante de Bondenza en niños menores de 18 años y Bondenza no ha sido estudiado en esta población (ver sección 5.1y sección 5.2).
Forma de administración Por vía intravenosa.
Es necesario respetar rigurosamente la vía de administración intravenosa (ver sección 4.4).
4.3 Contraindicaciones
- Hipersensibilidad al ácido ibandrónico o a alguno de los excipientes incluidos en la sección 6.1.
- Hipocalcemia
4.4 Advertencias y precauciones especiales de empleo
rS>
Fallos en la administración
Se debe tener cuidado para no administrar las inyecciones de Bondenza vía intrarterial o paravenosa ya que esto podría causar daño en los tejidos.
intraveno
Hipocalcemia
osa, pueden provocar un descenso
Bondenza como otros bifosfonatos de administración i transitorio de los valores de calcio sérico. N:
Antes de iniciar el tratamiento con Bondenza inyectable, hay que tratar la hipocalcemia, así como otros trastornos del metabolismo óseo y mi
Todas las pacientes deben recibir un suplemento adecuado de calcio y Vitamina D.
Reacción/shock anafiláctico
Se han notificado casos de reacción/schock anafiláctico, incluyendo eventos mortales, en pacientes tratados con ac. ibandrónico Intravenoso.
Cuando se administre la inyección intravenosa de Bondenza se debe disponer fácilmente de apoyo médico apropiado y medidas de monitorización. Si se producen reacciones anafilácticas u otras reacciones graves alérgicas/hipersensibilidad se debe interrumpir la inyección e iniciar tratamiento apropiado.
Insuficiencia renal
Las pacientes que presenten enfermedades concomitantes o que utilicen medicamentos con potenciales reacciones adversas sobre el riñón deben ser estrechamente vigiladas durante el tratamiento de acuerdo a la práctica clínica habitual.
Debido a la limitada experiencia clínica, no se recomienda el uso de Bondenza inyectable en pacientes con creatinina sérica por encima de 200 pmol (2,3 mg/dl) o con un aclaramiento de creatinina inferior a 30 ml/min (ver sección 4.2 y sección 5.2.).
Pacientes con alteración cardíaca
En pacientes con riesgo de fallo cardíaco se debe evitar la hiperhidratación
Osteonecrosis mandibular
Se ha observado casos de osteonecrosis mandibular generalmente asociados con extracciones dentales y/o infecciones locales (incluyendo osteomielitis) en pacientes oncológicos tratados con regímenes que incluían principalmente bifosfonatos de administración intravenosa. La mayoría de estos pacientes también recibieron quimioterapia y corticosteroides. También se ha observado osteonecrosis mandibular en pacientes con osteoporosis tratados con bifosfonatos de administración oral.
En aquellos pacientes con factores de riesgo concomitantes (p.ej. cáncer, quimioterapia, radioterapia, corticosteroides, higiene bucal pobre) deberá considerarse un examen dental con una apropiada odontología preventiva, antes de iniciar el tratamiento con bifosfonatos.
Durante el tratamiento, si es posible, estos pacientes deben evitar procesos dentales invasivos. La cirugía dental puede agravar la situación en pacientes que desarrollen osteonecrosis mandibular durante la terapia con bifosfonatos. No hay datos disponibles que indiquen que la interrupción del tratamiento con bifosfonatos reduce el riesgo de osteonecrosis mandibular en pacientes que precisen procesos dentales. La valoración clínica del facultativo, debe orientar sobre cómo proceder con cada paciente según la valoración individual de la relación beneficio-riesgo.
Fracturas atípicas de fémur
Se han notificado casos de fracturas atípicas subtrocantéricas y diafisarias del fémur asociadas al tratamiento con bifosfonatos, principalmente en pacientes en tratamiento prolongado para la osteoporosis. Estas fracturas transversales u oblicuas cortas pueden ocurrir en cualquier parte a lo largo del fémur, desde justo debajo del trocánter menor hasta justo por encima de la cresta supracondílea. Estas fracturas se producen después de un traumatismo mínimo o en ausencia de él y algunos pacientes tienen dolor en el muslo o en la ingle, a menudo asociado con imágenes características de fracturas por sobrecarga, semanas a meses antes de que se presente la fractura femoral completa. Las fracturas son generalmente bilaterales; por lo tanto, el fémur del lado opuesto debe ser examinado en los pacientes tratados con bifosfonatos que hayan tenido una fractura de la diáfisis femoral. También se ha notificado un bajo índice de consolidación de estas fracturas. Debe considerarse la interrupción del tratamiento con bifosfonatos, valorando de forma individualizada el balance beneficio/riesgo, en aquellos pacientes en los que exista sospecha de fractura atípica de fémur pendiente de evaluación..
paciente que presente dichos síntomas deberá valorarse si
Durante el tratamiento con bifosfonatos debe advertirse a los pacientes que notifiquen cualquier dolor en el muslo, cadera o ingle. En cualquier existe una fractura de fémur incompleta.
cento de sodio.
Bondenza está esencialmen
4.5 Interacción c
os medicamentos y otras formas de interacción
Las interacciones metabólicas son consideradas poco probables, puesto que el ácido ibandrónico no inhibe las principales isoenzimas del citocromo P450 hepático humano; tampoco induce el sistema hepático del citocromo P450 de las ratas (ver sección 5.2).El ácido ibandrónico se excreta sólo por vía renal y no se biotransforma.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No existen datos suficientes acerca del uso del ácido ibandrónico por las mujeres embarazadas. Los estudios con ratas han revelado cierta toxicidad sobre la función reproductora (véase la sección 5.3). Se desconoce el posible riesgo para la especie humana.
No se debe utilizar Bondenza durante el embarazo.
Lactancia
Se desconoce si el ácido ibandrónico se excreta con la leche humana. Los estudios con ratas lactantes han demostrado la presencia de valores reducidos del ácido ibandrónico en la leche después de su administración intravenosa. Se desaconseja el uso de Bondenza durante la lactancia.
Fertilidad
No hay datos de los efectos del ácido ibandrónico en humanos. En estudios sobre la función reproductora de las ratas por vía oral, el ácido ibandrónico disminuyó la fertilidad. En estudios en ratas por vía intravenosa, el ácido ibandrónico disminuyó la fertilidad a dosis diarias altas (ver sección 5.3).
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Tomando como base el perfil farmacocinético y farmacodinámico y las reacciones adversas notificadas, se espera que Bondenza no tenga efecto o éste sea despreciable sobre la capacidad de conducir y utilizar máquinas
4.8 Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad de Bondenza procede de los ensayos clínicos controlados y de la experiencia post-comercialización. Las reacciones adversas más frecuentemente notificadas fueron artralgia y síntomas de seudogripales. Estos síntomas están típicamente asociados a la primera dosis, generalmente son de corta duración, de intensidad leve o moderada, y que se suelen resolver con la continuación del tratamiento sin requerir medidas adicionales (por favor véase párrafo “enfermedad seudogripal”).
Tabla de reacciones adversas
En la tabla 1 se muestra una visión general de reacciones adve
La seguridad del tratamiento oral con 2,5 mg de ácido ibandrónico administrados diariamente, se evaluó en 1.251 pacientes tratados en 4 ensayos clínicos controlados con placebo; procediendo la gran mayoría de los pacientes del ensayo pivotal sobre fracturas a lo largo de tres años (MF4411)
En un ensayo pivotal a dos años en mujeres posmenopáusicas con osteoporosis (BM16550), la seguridad general de Bondenza 3 mg inyección intravenosa cada 3 meses y 2,5 mg de ácido ibandrónico administrados diariamente por vía oral fue similar. El porcentaje total de pacientes que experimentaron una reacción adversa, fue 26,0 % y 28,6 % para Bondenza 3 mg/3 ml inyectable cada tres meses después de uno y dos años, respectivamente. En la mayor parte de los casos no fue necesaria la suspensión del tratamiento.
\cr
e.
Tabla 1: Reacciones adversas ocurridas en los ensayos fase III BM16550, MF4411 y en la experiencia post-comercialización en mujeres postmenopáusicas que recibieron Bondenza 3 mg inyectable cada 3 meses o 2,5 mg de ácido ibandrónico diariamente.
|
Las reacciones adversas se enumeran de acuerdo al sistema de clasificación de órgano y categoría de frecuencia MedDRA Se definen las categorías de frecuencia usando la siguiente convención: Muy frecuentes (>1/10), frecuentes (>1/100 a <1/10), poco frecuentes (>1/1.000 a <1/100), raras (>1/10.000 a <1/1.000), muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Dentro de cada grupo de frecuencia se presentan las reacciones adversas en orden decreciente de gravedad. | ||||
|
Sistema de Clasificación de Órganos |
Frecuentes |
Poco frecuentes |
Raras |
Muy raras |
|
Trastornos del sistema inmunológico |
Reacción de hipersensibilidad |
Reacción /schock anafiláctico*f | ||
|
Trastornos del sistema nervioso |
Dolor de cabeza |
& | ||
|
Trastornos oculares |
/ |
Inflamación ocular*f | ||
|
Trastornos vasculares |
Flebitis/tromboflebitis | |||
|
Trastornos gastrointestinales |
Gastritis, Dispepsia, Diarrea, Dolor abdominal, Náuseas, Estreñimiento | |||
|
Trastornos de la piel y del tejido subcutáneo |
Erupción cutánea |
/ |
Angioedema, Hinchazón facial/edema, Urticaria | |
|
Trastornos musculoesqueléticos, del tejido conjuntivo y de los huesos |
Artralgia, Mialgia, Dolor musculoesquelético, Dolor de espalda |
Dolor de huesos |
Fracturas atípicas subtrocantéricas y diafisarias del fémurf ) |
Osteonecrosis mandibular*f |
|
Trastornos generales y alteraciones en el lugar de administración |
seudogripal*, Fatiga c£_ |
Reacciones en el lugar de la inyección, Astenia | ||
|
*Ver abajo más informa |
ción | |||
fIdentificado en la experiencia post-comercialización Descripción de reacciones adversas seleccionadas
Enfermedad seudogripal
El síndrome seudogripal incluye los acontecimientos notificados similares a la reacción de la fase aguda o a los síntomas, incluyendo mialgias, artralgia, fiebre, escalofríos, fatiga, nauseas, pérdida de apetito y dolor de huesos.
Osteonecrosis mandibular
Se ha observado osteonecrosis mandibular en pacientes tratados con bifosfonatos. La mayoría de los informes se refieren a pacientes oncológicos, pero también algunos casos han sido observados en pacientes tratados de osteoporosis. La osteonecrosis mandibular se asocia generalmente con extracciones dentales y/o infecciones locales (incluyendo osteomielitis). También se consideran factores de riesgo el diagnóstico de cáncer, quimioterapia, radioterapia, corticosteroides, y una higiene bucal pobre (ver sección 4.4).
Inflamación ocular
Se han notificado casos de inflamación ocular como uveítis, episcleritis y escleritis con el tratamiento con ácido ibandrónico. En algunos casos estos acontecimientos no se resolvieron hasta que se interrumpió el tratamiento con ácido ibandrónico.
Reacción/shock anafiláctico
Se han notificado casos de reacción/schock anafiláctico, incluyendo eventos mortales, en pacientes tratados con ac. ibandrónico intravenoso.
4.9 Sobredosis
No se dispone de información concreta sobre el tratamiento de la sobredosis de Bondenza.
Según los datos conocidos sobre este grupo terapéutico, la sobredosis por vía intravenosa puede ocasionar complicaciones como hipocalcemia, hipopotasemia y hipomagnesemia. La reducción clínicamente relevante de los niveles séricos de calcio, fosfatos y magnesio debería ser tratados con la administración intravenosa de gluconato cálcico, fosfato potásico o fosfato sódico y sulfato magnésico respectivamente.
.
5. PROPIEDADES FARMACOLOGICAS
Grupo farmacoterapéutico: Medicamentos para el tratamiento de las enfermedades óseas, bifosfonatos,
5.1 Propiedades farmacodinámicas
código ATC: M05BA06
Mecanismo de acción ......
s eo elevado.
El ácido ibandrónico es un bisfosfonato muy potente que pertenece al grupo de bifosfonatos nitrogenados; actúa de forma selectiva sobre el tejido óseo y, en concreto, inhibe la actividad osteoclástica sin influir directamente en la formación de hueso. No impide el reclutamiento de los osteoclastos. El ácido ibandrónico conduce a un aumento neto progresivo de la masa ósea y reduce la incidencia de fracturas de las mujeres posmenopáusicas hasta los valores previos a la menopausia, gracias a la disminución del recambio óse
Efectos farmacodinámicos La acción farmacodinámic
del á
ca del ácido ibandrónico se basa en una inhibición de la resorción ósea. En condiciones in vivo, el ácido ibandrónico previene la destrucción ósea experimental causada por el cese de la función gonadal, los retinoides, los tumores o los extractos tumorales. Asimismo, inhibe la resorción de hueso endógeno de las crías de rata (en fase de crecimiento rápido), con lo que aumenta la masa ósea normal en comparación con la de los animales no tratados.
En los modelos con animales se ha confirmado que el ácido ibandrónico es un inhibidor muy potente de la actividad osteoclástica. No se han detectado indicios de mineralización anómala de las crías de rata, ni siquiera después de administrar dosis más de 5000 veces superiores a las utilizadas en la osteoporosis.
La administración a largo plazo tanto de dosis diarias como intermitentes (con intervalos prolongados libres de dosis) a ratas, perros y monos se asoció con la formación de hueso nuevo de calidad normal, que conservaba o aumentaba su fuerza mecánica, incluso en dosis superiores a las dosis farmacológicas previstas (es decir, dosis que se hallaban dentro del intervalo tóxico). En humanos, la eficacia de la administración tanto diaria como intermitente con intervalos libres de dosis de ácido ibandrónico entre 9 - 10 semanas, se han confirmado en un ensayo clínico (MF 4411), en el cual el ácido ibandrónico demostró eficacia anti-fractura.
En modelos animales, el ácido ibandrónico ocasionó alteraciones bioquímicas indicativas de una inhibición de la resorción ósea proporcional a la dosis, incluida la supresión de los marcadores bioquímicos urinarios de la degradación del colágeno óseo (como la desoxipiridinolina y los telopéptidos aminoterminales entrecruzados del colágeno de tipo I (NTX)).
Tanto la administración oral diaria como la intermitente (con intervalos libres de dosis de entre 9 - 10 semanas por trimestre), así como la administración de ácido ibandrónico intravenoso en mujeres posmenopáusicas produjeron cambios bioquímicos indicativos de una inhibición de la resorción ósea dosis dependiente.
Bondenza inyección intravenosa disminuye los niveles séricos de telopéptidos carboxiterminales de la cadena alfa del colágeno tipo I (CTX) en los 3 - 7 primeros días de tratamiento y disminuye los niveles de osteocalcina en los 3 primeros meses.
Tras la interrupción del tratamiento existe una reversión a los valores patológicos elevados de la tasa de resorción ósea asociadas a osteoporosis posmenopáusica.
El análisis histológico de biopsias óseas tras 2 y 3 años de tratamiento de mujeres posmenopáusicas con 2,5 mg de ácido ibandrónico administrados diariamente por vía oral y dosis intravenosas intermitentes de hasta 1 mg cada 3 meses, muestra calidad ósea normal y ningún indicio de defecto en
la mineralización. Tras 2 años de tratamiento con Bondenza 3 mg inyectable, disminución esperada del recambio óseo, calidad ósea normal y ausencia de mineralización.
én se observó una en la
fracturas deben considerarse factores de O), edad, existencia de fracturas y bajo índice de masa corporal.
Eficacia clínica
Para identificar a las mujeres con un riesgo elevado de sufrir riesgo independientes, tales como, baja densidad de masa óse previas, antecedentes familiares de fracturas, alto recambio ó
Bondenza 3 mg inyectable cada 3 meses
Densidad mineral ósea (DMO)

En el ensayo (BM16550) a dos años, randomizado, doble ciego, multicéntrico de no inferioridad, realizado en mujeres posmenopáusicas (1.386 mujeres entre 55-80 años) con osteoporosis (valor lumbar basal, T- score por debajo de -2,5 DE), Bondenza 3 mg intravenoso administrado cada 3 meses demostró ser al menos igual de eficaz que 2,5 mg de ácido ibandrónico diarios administrados por vía oral. Esto se demostró tanto en el análisis fundamental a un año como en el análisis confirmatorio a dos años (Tabla 2).
El análisis fundamental de los datos del ensayo BM16550 a un año y el análisis confirmatorio a 2 años demostraron la no inferioridad del régimen de dosificación de 3 mg inyectable cada 3 meses en comparación con la dosis oral diaria de 2,5 mg, respecto al aumento en la media de la DMO de la columna lumbar, cadera completa, cuello femoral y trocánter (Tabla 2).
Tabla 2: Cambios relativos medios en el valor de referencia de la BMO de la columna lumbar, cadera completa, cuello femoral y trocánter tras un año (análisis fundamental) y dos años de tratamiento (por protocolo poblacional) en el ensayo BM 16550.
|
Datos a un año del ensayo BM 16550 |
Datos a dos años del ensayo BM 16550 | |||
|
Cambios relativos medios en el valor de referencia % [95% CI] |
2,5 mg de ácido ibandrónico dosis diaria (N=377) |
Bondenza 3 mg inyectable cada 3 meses (N=365) |
2,5 mg de ácido ibandrónico dosis diaria (N=334) |
Bondenza 3 mg inyectable cada 3 meses (N=334) |
|
Columna Lumbar BMO |
3,8 [3,4, 4,2] |
4,8 [4,5, 5,2] |
4,8 [4,3, 5,4] |
6,3 [5,7, 6,8] |
|
Cadera completa DMO |
1,8 [1,5, 2,1] |
2,4 [2,0, 2,7] |
2,2 [1,8, 2,6] |
3,1 [2,6, 3,6] A |
|
Cuello femoral DMO |
1,6 [1,2, 2,0] |
2,3 [1,9, 2,7] |
2,2 [1,8, 2,7] |
2,8 [2,3, 3,3] |
|
Trocánter DMO |
3,0 [2,6, 3,4] |
3,8 [3,2, 4,4] |
3,5 [3,0, 4,0] |
4,9 [4,1, 5,7] |
Además, según el análisis prospectivo a un año, p<0,001 y a dos años p<0,001 Bondenza 3 mg inyectable demostró ser superior a la dosis diaria de 2,5 mg de ácido ibandrónico administrada por vía oral.
j$r
En la DMO lumbar, el 92,1 % de las pacientes que recibieron la dosis de 3 mg inyectable cada tres meses aumentaron o mantuvieron su DMO después de 1 año de tratamiento (es decir fueron respondedores) comparado con 84,9 % de las pacientes que recibieron la dosis oral de 2,5 mg diarios (p=0,002). Tras dos años de tratamiento, el 92,8 % de las pacientes que recibieron la dosis de 3 mg inyectables y el 84,7 % de las pacientes que recibieron la dosis oral de 2,5 mg diarios aumentaron o mantuvieron la DMO lumbar (p=0,001).
cr
En la DMO en cadera completa, el 82,3 % de las pacientes que recibieron la dosis de 3 mg inyectable cada 3 meses fueron respondedores , frente al 77,0 % de las pacientes que recibieron la dosis
oral de 2,5 mg diarios (p=0,02). Tras dos años de tratamiento, el 85,6 % de las pacientes que recibieron la dosis de 3 mg cada 3 meses y el 77,0 % de las pacientes que recibieron la dosis oral de
2,5 mg diarios tuvieron un aumento o mantenimiento de la DMO en cadera (p=0,004).
La proporción de pacientes que aumentaron o mantuvieron su DMO lumbar y cadera total a un año de tratamiento fue 76,2 % en el brazo de 3 mg inyectable cada 3 meses y 67,2 % en el brazo de dosis oral de 2,5 mg diarios (p= 0,007). A los dos años, un 80,1 % y 68,8 % de las pacientes cumplen con este criterio en la administración de 3 mg inyectables cada 3 meses.
Marcadores bioquímicos del recambio óseo
En todos los tiempos de medidas se han observado reducciones clínicamente significativas de los niveles de CTX en suero. A los 12 meses la mediana de los cambios relativos con respecto a los valores de referencia fue de -58,6 % para la dosis de 3 mg intravenoso cada 3 meses y de 62,6 % para la dosis oral de 2,5 mg diarios. Además el 64,8% de las pacientes que recibieron 3 mg inyectable cada 3 meses resultaron respondedores (definido como un descenso >50 % del valor de referencia), frente al
64,9 % de las pacientes que recibieron dosis oral de 2,5 mg diarios. La reducción de CTX en suero se mantuvo durante los 2 años, con más de la mitad de los pacientes identificados como respondedores en ambos grupos de tratamiento.
Según los resultados del ensayo BM 16550 se espera que Bondenza 3 mg inyección intravenosa, administrado cada tres meses sea igual de efectivo en la prevención de fracturas que la administración oral diaria de 2,5 mg de ácido ibandrónico.
Comprimidos de 2,5 mg de ácido ibandrónico administrados diariamente
En un ensayo inicial de 3 años, aleatorizado, doble ciego, controlado con placebo, sobre fracturas (MF 4411,) se observó un descenso estadística y clínicamente significativo de la incidencia de nuevas fracturas vertebrales radiológicas (morfométricas) y clínicas (tabla 3). En este ensayo se evaluó el ácido ibandrónico en dosis orales de 2,5 mg al día y dosis intermitentes de 20 mg como régimen exploratorio. El ácido ibandrónico se administró 60 minutos antes del desayuno o de la primera bebida del día (período de ayuno posterior a la dosis). En este ensayo se reclutó a mujeres de 55 a 80 años, que llevaban, por lo menos, 5 años desde la menopausia y mostraban una DMO de 2 a 5 DE por debajo de la media premenopáusica (T-score) de, al menos, una vértebra lumbar [L1-L4] y que habían sufrido de una a cuatro fracturas vertebrales prevalentes. Todas las pacientes recibieron 500 mg de calcio y 400 UI de vitamina D al día. Se evaluó la eficacia entre 2.928 pacientes. La incidencia de nuevas fracturas vertebrales se redujo de forma estadística y clínicamente significativa con la pauta de
2,5 mg de ácido ibandrónico administrados diariamente. Esta pauta redujo la aparición de nuevas fracturas vertebrales radiológicas en un 62 % (p=0,0001) durante los tres años del ensayo. La reducción del riesgo relativo alcanzó el 61 % al cabo de 2 años (p=0,0006) de tratamiento. No se obtuvieron diferencias estadísticamente significativas después de 1 año de tratamiento (p=0,056). El efecto profiláctico de las fracturas se mantuvo durante todo el ensayo. No se hallaron indicios de que el efecto se disipara con el tiempo.
La incidencia de fracturas vertebrales clínicas también se redujo en un 49 % después de 3 años (p=0,011). El fuerte efecto sobre las fracturas vertebrales quedó reflejado, asimismo, en una reducción estadísticamente significativa de la pérdida de talla, en comparación con el placebo (p<0,0001).
Tabla 3 resultados del ensayo MF 4411 de fracturas a los 3 años (%, IC del 95 %)
|
Placebo (N=974) |
2,5 mg de ácido ibandrónico dosis diaria (N=977) | |
|
Reducción del riesgo relativo Nuevas fracturas vertebrales morfométricas |
62 % (40,9,75,1) | |
|
Incidencia de nuevas fracturas vertebrales morfométricas |
9 9,56 % (7,5 , 11,7) |
4,68 % (3,2,6,2) |
|
Reducción del riesgo relativo de las fracturas vertebrales clínicas |
49 % (14,03,69,49) | |
|
Incidencia de fracturas vertebrales clínicas |
5,33 % (3,73,6,92) |
2,75 % (1,61 , 3,89) |
|
DMO: diferencia media a los 3 años con respecto al valor lumbar basal |
1,26 % (0,8 , 1,7) |
6,54 % (6,1,7,0) |
|
DMO: diferencia media a los 3 años con respecto al valor basal de toda la cadera |
-0,69 % (-1,0 , -0,4) |
3,36 % (3,0,3,7) |
El efecto del tratamiento con ácido ibandrónico fue evaluado en un análisis de subpoblación de pacientes que tenían el valor lumbar basal DMO T-score por debajo de - 2,5 (tabla 4). La reducción del riesgo de fracturas vertebrales fue considerada consistente con lo visto para la población global.
Tabla 4: Resultado del ensayo MF 4411 de fractura a los 3 años (% IC 95 % ) para pacientes con valor lumbar basal DMO T-score por debajo de -2,5
|
Placebo (N=587) |
2,5 mg de ácido ibandrónico dosis diaria (N=575) | |
|
Reducción del riesgo relativo Nuevas fracturas vertebrales morfométricas |
59 % (34,5, 74,3) | |
|
Incidencia de nuevas fracturas vertebrales morfométricas |
12,54 % (9,53 , 15,55) |
5,36 % (3,31,7,41) |
|
Reducción del riesgo relativo de las fracturas vertebrales clínicas |
50 % (9,49,71,91) | |
|
Incidencia de fracturas vertebrales clínicas |
6,97 % (4,67,9,27) |
3,57 % (1,89,5,24) |
|
DMO: diferencia media a los 3 años con respecto al valor lumbar basal |
1,13 % (0,6 , 1,7) |
7,01 % (6,5,7,6) >$> |
|
DMO: diferencia media a los 3 años con respecto al valor basal de toda la cadera |
-0,70 % (-1,1 , -0,2) |
3,59 % (3,1,4,1) |
En el total de la población de pacientes incluidos en el ensayo MF4411, no se observó ningún descenso en el número de fracturas no vertebrales, sin embargo, la toma diaria de ácido ibandrónico pareció ser efectiva en una subpoblación de alto riesgo (DMO en cuello femoral T-score < -3,0), en la que se observó una reducción del 69% en el riesgo de sufrir fracturas no vertebrales.
El tratamiento oral diario con comprimidos de 2,5 mg de ácido ibandrónico aumentó de forma progresiva la DMO vertebral y no vertebral.
¿y
El incremento de la DMO lumbar a los 3 años, en relación con el placebo, representó 5,3 % y 6,5 % con respecto al valor basal. El aumento de la DMO de la cadera, en relación con el valor basal, resultó del 2,8 % en el cuello femoral, del 3,4 % en toda la cadera y del 5,5 % en el trocánter.
Los marcadores bioquímicos del recambio óseo (como la CTX urinaria y la osteocalcina sérica) manifestaron el patrón previsible de supresión hasta las cifras premenopáusicas y alcanzaron la supresión máxima a lo largo de 3 a 6 meses administrando diariamente 2,5 mg de ácido ibandrónico.
diátricc
Los marcadores bioquímicos de la resorción ósea experimentaron un descenso clínicamente relevante del 50 % ya durante el primer mes de tratamiento con 2,5 mg de ácido ibandrónico administrados diariamente. n<2>
Población pediátrica (ver sección 4.2 y sección 5.1)Bondenza no ha sido estudiado en población pediátrica por lo tanto, no hay datos de eficacia o seguridad disponibles para esta población.
5.2 Propiedades farmacocinéticas
Los efectos farmacológicos fundamentales del ácido ibandrónico sobre el hueso no guardan una relación directa con las concentraciones plasmáticas reales, como se ha demostrado en diversos estudios con animales y seres humanos.
Las concentraciones en plasma de ácido ibandrónico aumentan de manera proporcional después de la administración intravenosa de 0,5 mg a 6 mg.
Absorción No procede
Distribución
Después de la exposición sistémica inicial, el ácido ibandrónico se une rápidamente al hueso o se excreta en la orina. El volumen terminal aparente de distribución en la especie humana resulta, como mínimo, de 90 L y la cantidad de la dosis que llega al hueso se estima como el 40-50 % de la dosis circulante. La unión a las proteínas del plasma humano es aproximadamente un 85 % - 87 % (determinada en condiciones in vitro, a concentraciones terapéuticas de ácido ibandrónico), por lo que la posibilidad de interacción con otros medicamentos por desplazamiento es mínima.
Biotransformación
No hay pruebas de que el ácido ibandrónico se metabolice en los animales o en la especie humana.
Eliminación
El ácido ibandrónico se elimina de la circulación vía absorción ósea (se estima un 40 - 50 % en mujeres posmenopáusicas) y el resto se elimina de forma inalterado por el riñón.
El intervalo de las semividas aparentes observadas es amplio, por regla general, la semivida terminal aparente se sitúa en el intervalo de 10 a 72 horas. Como los valores calculados están en función de la duración del estudio, de la dosis utilizada y de la sensibilidad del ensayo, la semivida terminal real es probable que sea substancialmente mayor como ocurre con otros bifosfonatos. Los valores plasmáticos iniciales descienden en seguida para alcanzar el 10 % de los valores máximos a las 3 y a las 8 horas de su administración intravenosa u oral, respectivamente.
El aclaramiento total del ácido ibandrónico es reducido: los valores medios se sitúan dentro del margen de 84 - 160 ml/min. La depuración renal (aprox. 60 ml/min entre mujeres posmenopáusicas sanas) explica del 50 al 60 % de la depuración total y se relaciona con el aclaramiento de creatinina. La diferencia entre el aclaramiento total y la depuración renal refleja, con toda seguridad, la captación por el hueso.
La vía secretora no incluye, en principio, ningún sistema de transporte ácido o alcalino que intervenga en la eliminación de otros principios activos. Además, el ácido ibandronico no inhibe las principales isoenzimas principales del citocromo P450 hepático humano y tampoco induce el sistema hepático del
citocromo P450 de las ratas
Farmacocinética en situaciones clínicas especiales
&
Sexo
La farmacocinética del áci
ido iban
ndrónico se asemeja en ambos sexos.
Raza No hay pruebas ibandrónico p
xistan diferencias étnicas de interés clínico en la disposición del ácido siáticos y los blancos. Los datos sobre pacientes de origen africano son limitados.
Pacientes con insuficiencia renal
El aclaramiento renal del ácido ibandrónico entre pacientes con distintos grados de insuficiencia renal se relaciona linealmente con el aclaramiento de creatinina.
No es necesario un ajuste de dosis en pacientes con insuficiencia renal leve o moderada (CLCr igual o mayor de 30 ml/min).
Los sujetos con insuficiencia renal grave (CLCr menor de 30 ml/min) que reciban 10 mg de ácido ibandrónico al día por vía oral durante 21 días tienen concentraciones plasmáticas de 2 a 3 veces mayores que aquellos con una función renal normal; la depuración total del ácido ibandrónico llegó a 44 ml/min. Tras la administración intravenosa de 0,5 mg de ácido ibandrónico, la depuración total, renal y extrarrenal se redujo en un 67 %, 77 % y 50 %, respectivamente, entre los sujetos con insuficiencia renal grave pero la tolerabilidad relacionada con esta mayor exposición no disminuyó. Dada la limitada experiencia clínica, no se recomienda el uso de Bondenza en pacientes con insuficiencia renal grave (ver sección 4.2 y sección 4.4). Sólo se ha evaluado la farmacocinética del ácido ibandrónico en un número limitado de pacientes con enfermedad renal terminal tratada por hemodiálisis, por lo tanto se desconoce la farmacocinética del ácido ibandrónico en los pacientes no tratados por hemodiálisis. El ácido ibandrónico no debería utilizarse en todos los pacientes con una enfermedad renal terminal debido a que los datos disponibles son limitados.
Pacientes con alteraciones de la función hepática (ver sección 4.2)
No hay datos farmacocinéticos sobre el ácido ibandrónico en casos de alteración hepática. El hígado no desempeña ningún papel importante para la depuración del ácido ibandrónico, que no se metaboliza sino que se elimina mediante excreción renal y captación ósea. Por consiguiente, no es necesario ajustar la dosis de las pacientes con alteraciones hepáticas.
Pacientes de edad avanzada (ver sección 4.2)
En un estudio multivariable, la edad no resultó un factor independiente para ninguno de los parámetros farmacocinéticos examinados. Como la función renal disminuye con la edad, la función renal es el único factor que merece consideración (ver sección sobre insuficiencia renal).
&
Población pediátrica (ver sección 4.2 y sección 5.1)
No se dispone de datos sobre el uso de Bondenza en estos grupos de edad.
5.3 Datos preclínicos sobre seguridad
on en
Los efectos tóxicos, por ejemplo, signos de daños renales, se manifest aron en perros sólo con exposiciones que excedían suficientemente la máxima exposición humana, lo que indica una relevancia clínica mínima.
s de g
Mutagenesis/carcinogenesis:
genotoxicidad tampoco revelaron
No se hallaron indicios de poder cancerígeno. Los ensayos ( pruebas de la actividad genética del ácido ibandrónico.
Toxicidad sobre la función reproductora:
No se han realizado estudios específicos para el régimen posológico de 3 meses. En estudios para dosis diarias IV no se encontraron pruebas de ningún efecto fetotóxico o teratógeno directo del ácido ibandrónico en ratas y conejos. El aumento de peso corporal disminuyó en la generación F1 de las ratas. Los efectos sobre la función uctora de la rata en estudios por vía oral, consistieron en un aumento de pérdidas preimplantación a dosis de 1 mg/kg/día y superiores. En estudios sobre la función reproductora de las ratas por vía intravenosa, el ácido ibandrónico disminuyó el recuento de esperma a dosis de 0,3 y 1 mg/kg/día y disminuyó la fertilidad en los machos a 1 mg / kg / día y en las hembras a 1,2 mg / kg / día. Otras reacciones adversas del ácido ibandrónico en los estudios sobre la toxicidad de la función reproductora de la rata son los mismos que los de los bifosfonatos como grupo. Se caracterizan por un descenso del número de lugares de implantación, dificultades para el parto natural (distocia) y aumento de las variaciones viscerales (síndrome de la pelvis renal y de los uréteres).
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Cloruro de sodio
Ácido acético glacial
Acetato de sodio trihidrato
Agua para preparaciones inyectables
6.2 Incompatibilidades
Bondenza solución inyectable no se debe mezclar con soluciones que contengan calcio o con otros medicamentos de administración intravenosa.
6.3 Período de validez 2 años.
6.4 Precauciones especiales de conservación
Este medicamento no requiere ninguna condición especial de conservación.
6.5 Naturaleza y contenido del envase
Jeringa precargada (5 ml) de vidrio incoloro tipo I, con 3 ml de solución inyectable, dotada de un tapón gris tipo émbolo y capuchón de caucho butílico laminado de fluororresina.
Disponible en envase de 1 jeringa precargada y aguja para inyección ó 4 jeringas precargadas y 4 agujas para inyección.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Cuando el medicamento se administre a través de una vía intravenosa ya restringir tanto a soluciones salinas isotónicas como a soluciones de 50 m
50 m ,yl<
tente, la infusión se debe de glucosa al 5%. Esto

es aplicable también a las soluciones utilizadas para enjuagar la aguja y los otros dispositivos.
nga y la aguja para inyección deberían ión de pro
La solución que no se use en la inyección, así como la jeringa
desecharse de acuerdo con la normativa local. La eliminación de productos farmacéuticos en el medio
Se deben seguir estrictamente los siguientes puntos para el uso y eliminación de jeringas y otros dispositivos de medicamentos que sean punzantes:
• Nunca se deben reutilizar las agujas y jeringas.
• Desechar las agujas y jeringas en contenedores de objetos punzantes (contenedor de residuos a prueba de pinchazos).
• Mantener el contenedor fuera del alcance de los niños.
• Se debe evitar tirar los contenedores de objetos punzantes a la basura.
• Desechar el contenedor entero de acuerdo con la normativa local o según le haya indicado su profesional sanitario.
ambiente se debe reducir al mínimo.
TITUL
AR D
E LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/266/005
EU/1/03/266/006
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 23.02. 2004 Fecha de la última renovación: 20 .02.2009
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu

ANEXO II
&
A.
B.
RESPONSABLEDE LA LIBERACIÓN DE LOS LOTES
CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
ST
C.
OTRAS CONDICIONE AUTORIZACIÓN DE <
Y REQUISITOS DE LA MERCIALIZACIÓN

A. FABRICANTE (S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del (de los) fabricantc(s) responsables de la liberación de los lotes
Comprimidos recubiertos con película:
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Alemania
Solución inyectable en jeringa precargada:
&
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Alemania
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica.
COMERCIALIZACIÓN
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE
Sistema de Farmacovigilancia
El Titular de la Autorización de Comercialización (TAC) debe asegurar que el Sistema de Farmacovigilancia, incluida en el Módulo 1.8.1. de la Autorización de Comercialización, esté instaurado y en funcionamiento antes de que el medicamento se comercialice y durante el tiempo que
permanezca en el mercado.
Plan de Gestión de Riesgos (PG R

El TAC realizará los estudios y las actividades adicionales de farmacovigilancia detalladas en el Plan de Farmacovigilancia, de acuerdo con el PGR incluido en el Módulo 1.8.2. de la Autorización de Comercialización y cualquier actualización posterior del PGR acordada con el Comité de Medicamentos de Uso Humano (CHMP).
De acuerdo con la directriz del CHMP sobre Sistemas de Gestión de Riesgos para medicamentos de uso humano, el PGR actualizado se debe presentar junto con el siguiente Informe Periódico de Seguridad (IPS).
Además se debe presentar un PGR actualizado:
• Cuando se reciba nueva información que pueda afectar a las especificaciones de seguridad vigentes, al Plan de Farmacovigilancia o a las actividades de minimización de riesgos.
• Dentro de los 60 días posteriores a la consecución de un hito importante (farmacovigilancia o minimización de riesgos)
• A petición de la Agencia Europea de Medicamentos
IPSs
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
• CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
No procede


ANEXO III
ETIQUETADO Y PR OSPEC TO

/


1. NOMBRE DEL MEDICAMENTO
Bondenza 150 mg comprimidos recubiertos con película Ácido ibandrónico
2. PRINCIPIO(S) ACTIVO(S)

No chupar, masticar o machacar los comprimidos Leer el prospecto antes de utilizar este medicamento Comprimido mensual Vía oral
Mes 1 / / 3 comprimidos recubiertos
Mes 2 / / 3 comprimidos recubiertos
Mes 3 / / 3 comprimidos recubiertos
Escriba la fecha de administración del comprimido
con película con película con película
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park

Bondenza 150 mg
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN LOS BLÍSTERS O TIRAS Blíster
1. NOMBRE DEL MEDICAMENTO
Bondenza 150 mg comprimidos recubiertos con película Ácido ibandrónico
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Ltd.
|
3. |
FECHA DE CADUCIDAD | |
|
EXP |
/ | |
|
4. |
NÚMERO DE LOTE | |
|
Lot |
i & | |
|
5. |
OTROS |

1. NOMBRE DEL MEDICAMENTO
Bondenza 3 mg solución inyectable Ácido ibandrónico
2. PRINCIPIO(S) ACTIVO(S)
Una jeringa precargada de 3 ml de solución contiene 3 mg de ácido ibandrónico (como monohidrato sódico).
_
3. LISTA DE EXCIPIENTES _
También contiene cloruro de sodio, ácido acético glacial, acetato de sodio trihidrato, agua para preparaciones inyectables. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DE
L E
ENV
ASE
Solución inyectable 1 jeringa precargada + 1 aguja para inyección 4 jeringas precargadas + 4 agujas para inyección
5
FORMA Y VÍA(S) DE ADMINI
STRA
CIÓN
Leer el prospecto antes de utilizar este medicamento Sólo para administración intravenosa
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido

INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
JERINGAS PRECARGADAS
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Bondenza 3 mg solución para inyección
Ácido ibandrónico
Sólo para administración I.V.
|
2. |
FORMA DE ADMINISTRACIÓN | |
|
Leer el prospecto antes de utilizar este medicamento. | ||
|
3. |
FECHA DE CADUCIDAD |
_ |
|
EXP |
& | |
|
4. |
NÚMERO DE LOTE „ 0)^ | |
|
Lot |
/ | |
|
5. |
CONTENIDO EN PESO, VOLUMEN O EN UNIDADES | |
|
3 mg/3 ml | ||
|
6 |
OTROS | |

Prospecto: información para el usuario Bondenza
150 mg comprimidos recubiertos con película
Ácido ibandrónico
Lea todo el prospecto detenidamente antes de empezar a tomar el medicamento porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte con su médico o farmacéutico.
- Este medicamento se le ha recetado solamente a usted y no debe dárselo a otras personas, aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto.
Contenido del prospecto:
Planificación de la toma de Bondenza
con pegatinas autoadhesivas a usar en su calendario personal
1. Qué es Bondenza y para qué se utiliza
2. Qué necesita saber antes de empezar a tomar Bondenza
3. Cómo tomar Bondenza
4. Posibles efectos adversos
5. Conservación de Bondenza
6. Contenido del envase e información adicional
&
1. QUÉ ES BONDENZA Y PARA QUÉ SE UTILIZA
Bondenza pertenece a un grupo de medicamentos denominados bifosfonatos. Contiene el principio activo ácido ibandrónico..
Bondenza puede invertir la pérdida de hueso ya que impide que se pierda más hueso y aumenta la masa de hueso en la mayoría de las mujeres que lo toman, aún incluso cuando éstas no son capaces de ver o apreciar la diferencia. Bondenza puede ayudar a reducir los casos de rotura de huesos (fracturas). Esta reducción ha sido demostrada en fracturas vertebrales pero no en las de cadera.
Se le ha recetado Bondenza para tratar su osteoporosis posmenopáusica porque tiene un riesgo elevado de sufrir fracturas. La osteoporosis consiste en un adelgazamiento y debilitamiento de los huesos, hecho frecuente entre las mujeres después de la menopausia. En la menopausia, los ovarios dejan de producir la hormona femenina —los estrógenos— que ayuda a conservar la salud del esqueleto.
Cuanto antes llegue una mujer a la menopausia, mayor es el riesgo de que sufra fracturas por osteoporosis. Otros factores que aumentan el riesgo de sufrir fracturas son:
- aporte insuficiente de calcio y de vitamina D en la dieta
- tabaquismo o consumo excesivo de alcohol
- pocos paseos u otros ejercicios con carga de peso
- antecedentes familiares de osteoporosis
Los hábitos saludables de vida también facilitan los efectos favorables del tratamiento. Entre éstas se encuentran:
- una alimentación equilibrada, rica en calcio y en vitamina D
- los paseos o cualquier otro ejercicio con carga
- no fumar y un consumo no excesivo de alcohol.
2. Qué necesita saber antes de empezar a tomar Bondenza No tome Bondenza:
- Si es alérgico al ácido ibandrónico o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- Si tiene ciertos problemas con su garganta/tubo por donde pasan los alimentos (esófago), tales como estrechamiento o dificultad al tragar.
- Si no puede permanecer erguido, tanto de pie como sentado, durante al menos una hora seguida (60 minutos).
- Si tiene o ha tenido valores bajos del calcio en la sangre. Por favor consulte con su médico.
Advertencias y precauciones
Algunas personas precisan cuidados especiales durante el tratamiento con Bondenza. Consulte a su médico antes de empezar a tomar Bondenza:
- Si padece algún trastorno del metabolismo mineral (por ejemplo, carencia de vitamina D).
l, comu
- Si tiene algún problema en los riñones.
- Si tiene cualquier problema al tragar o problemas digestivos.
- Si se encuentra bajo tratamiento dental o tiene prevista alguna cirugía dental, comunique a su dentista que está en tratamiento con Bondenza.
Puede producirse irritación, inflamación o ulceración de la garganta/tubo por donde pasan los alimentos (esófago) a menudo con síntomas de dolor intenso en el pecho, dolor intenso después de tragar comida y/o bebida, náuseas intensas o vómitos, especialmente si los pacientes no beben un vaso de agua y/o si se tumban antes de que transcurra una hora tras la toma de Bondenza. Si desarrolla estos síntomas, deje de tomar Bondenza e informe a su médico inmediatamente(ver sección 3).
Niños y adolescentes
No administre Bondenza a niños o adolescentes me
enores de 18 años.
Otros medicamentos y Bondenza 'Zr
Informe a su médico o farmacéutico si está tomando o ha tomado recientemente o podría tener que tomar cualquier otro medicamento. Especialmente:
- Suplementos que contengan calcio, magnesio, hierro o aluminio ya que posiblemente podrían influir en los efectos de Bondenza.
- Ácido acetilsalicílico y otros antiinflamatorios no esteroideos (AINEs) (como el ibuprofeno, el diclofenaco sódico y el naproxeno) que pueden irritar el estómago y el intestino; al igual que los bifosfonatos (como Bondenza). Por lo tanto, tenga mucho cuidado cuando tome analgésicos o antiinflamatorios al mismo tiempo que Bondenza.
Después de ingerir el comprimido mensual de Bondenza, espere 1 hora para tomar cualquier otro medicamento, incluidos los comprimidos contra la indigestión, los suplementos de calcio y las vitaminas.
Bondenza con los alimentos y bebidas:
No tome Bondenza con los alimentos. Bondenza pierde eficacia si se toma con los alimentos. Puede beber agua pero no otros líquidos (ver el punto 3 Cómo tomar Bondenza).
Embarazo y lactancia
No tome Bondenza si está embarazada o se encuentra en período de lactancia. Si está en periodo de lactancia, posiblemente tenga que suspenderla para tomar Bondenza.
Consulte a su médico o farmacéutico antes de utilizar este medicamento.
Conducción y uso de máquinas
Puede conducir y utilizar máquinas ya que se espera que Bondenza no tenga efecto o éste sea despreciable sobre su capacidad para conducir y utilizar máquinas.
Bondenza contiene lactosa.
Si su médico le ha indicado que no puede tolerar o digerir algunos azúcares (p. ej. si tiene una intolerancia a la galactosa, deficiencia de Lap lactasa o problemas de absorción de glucosa-galactosa) consulte con su médico antes de tomar este medicamento.
3. Cómo tomar Bondenza
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico o farmacéutico..
La dosis habitual de Bondenza es de un comprimido al mes.
Cómo tomar el comprimido mensual
&
Es importante que siga con atención estas instrucciones. Están diseñadas para que Bondenza llegue en seguida al estómago y ocasione menos irritación.
- Tome un comprimido de Bondenza 150 mg una vez al mes.
- Elija el día del mes que le resulte más fácil de recordar. Puede elegir la misma fecha (ya sea el primer día de cada mes) o siempre el mismo día (como el primer domingo de cada mes). Elija lo que mejor se ajuste a su rutina.
- Tome el comprimido de Bondenza después de un mínimo de 6 horas tras la última comida o bebida (exceptuando el agua)
- Tome el comprimido de Bondenza
- nada más levantarse, y
- antes de desayunar o de ingerir líquidos (con el estómago vacío)
- Trague el comprimido con un vaso de agua (como mínimo, 180 ml).
dr
No tome el comprimido con agua con una alta concentración de calcio, zumo de fruta u otras bebidas. Si tuviera duda sobre los niveles de calcio potencialmente altos del agua del grifo (aguas duras), se recomienda usar agua embotellada con un bajo contenido mineral
- Trague el comprimido entero-no lo mastique, triture ni lo disuelva dentro de la boca.
Durante la hora siguiente (60 minutos) después de haber ingerido el comprimido

tumb
no se tumbe; si no permanece erguida (de pie o sentada), parte del medicamento podría regresar al esófago
X
no coma nada
no beba nada (salvo agua, si la necesita) no tome ningún otro medicamento
- Después de esperar 1 hora, usted podrá tomar el desayuno y la primera bebida del día. Una vez que haya comido, puede, si lo desea, tumbarse y tomar los demás medicamentos que necesite.
No tome este medicamento a la hora de acostarse ni antes de levantarse.
Continuación del tratamiento con Bondenza
Es importante que tome Bondenza todos los meses, hasta que se lo indique el médico.
Bondenza sirve para tratar la osteoporosis sólo mientras usted toma este medicamento.
Si toma más Bondenza del que debe
Si ha tomado, por error, algún comprimido de más, beba un vaso entero de leche y comuníqueselo de inmediato a su médico.
No se induzca el vómito ni se tumbe porque podría irritarse el esófago.
Si olvidó tomar Bondenza
Si olvida tomar el comprimido en la mañana del día que ha elegido, no ingiera el comprimido más tarde. En su lugar, consulte su calendario para ver cuando le corresponde tomar su próxima dosis:
Si su próxima dosis es dentro de 1 a 7 días...
ndario.
Jo
Espere hasta que le corresponda tomar la siguiente dosis y tómela de manera habitual, después vuelva a tomar un comprimido al mes según los días marcados en su calendario C'
Si su próxima dosis es dentro de más de 7 días...
Tome un comprimido la mañana siguiente al día que recuerde que olvidó la dosis, después vuelva a tomar un comprimido al mes según los días marcados en su calendario.
Nunca tome dos comprimidos de Bondenza dentro de la misma semana.
te m
4. Posibles efectos adversos
edicamento puede producir efectos adversos, aunque no
Al igual que todos los medicamentos,
todas las personas los sufran. ¿i"..........
Informe a su médico o enfermera inmediatamente si nota cualquiera de los siguientes efectos “ gruves ya que^neces¡t,r,r^¡ent0 murgente:
Frecuentes (pueden afectar hasta 1 de cada 10 personas):
• síntomas seudo-gripales, incluyendo fiebre, escalofríos y tiritona, sensación de malestar, dolor de huesos y de músculos y articulaciones. Consulte a su enfermera o médico si cualquier efecto llega a ser molesto o dura más de un par de días.
• erupción cutánea. Puede que esté teniendo una reacción alérgica al medicamento.
Poco frecuentes (pueden afectar hasta 1 de cada 100 personas)
• dolor intenso en el pecho, dolor intenso al tragar comida o bebida, náuseas intensas o vómitos, dificultad al tragar. Puedes tener una inflamación intensa, posiblemente con sensación de dolor o constricción, en garganta/ tubo por donde pasan los alimentos.
Raras (pueden afectar hasta 1 de cada 1.000 personas)
• picor, hinchazón de la cara, labios, lengua y garganta, con dificultad para respirar.
• dolor de ojo persistente e inflamación
• dolor nuevo, debilidad o molestias en el muslo, la cadera o la ingle. Pueden ser síntomas precoces de una posible fractura inusual del hueso del muslo
Muy raras (pueden afectra hasta 1 de cada 10.000 personas)
• dolor o sensación de dolor en la boca o mandíbula. Pueden ser síntomas precoces de problemas graves de mandíbula [necrosis (muerte del tejido óseo) del hueso de la mandíbula] reacción alérgica grave que puede suponer una amenaza para la vida
Otros efectos adversos posibles
Frecuentes (pueden afectar hasta 1 de cada 10 personas):
• dolor de cabeza
• ardor de estómago, molestia al tragar, dolor de estómago o tripa (debido a una inflamación del estómago), indigestión, nausea, diarrea (pérdidas intestinales)
• calambres musculares, rigidez de articulaciones y extremidades
Poco frecuentes (pueden afectar hasta 1 de cada 100 personas)
• mareos
• flatulencia (ventoseo, sensación de hinchada)
• dolor de espalda
• sensación de fatiga y agotamiento
Raras (pueden afectar hasta 1 de cada 1.000 personas)
• Inflamación del duodeno (primera sección del intestino) que causa dolor de estómago.
Urticaria
<<>
si se trata de efectos
Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso adversos que no aparecen en este prospecto.
5. Conservación de Bondenza
Mantener este medicamento fuera de la vista y del alcance de los niños No requiere condiciones especiales de conservación.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase después de CAD. La fecha de caducidad es el último día del mes que se indica.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medic amentos que no necesita. De esta forma ayudará a proteger el
medio ambiente.
6
Composición
El principio activo es el ácido ibandrónico. Cada comprimido contiene 150 mg de ácido ibandrónico (en forma de monohidrato sódico).
Los demás componentes son:
núcleo del comprimido: lactosa monohidrato, povidona, celulosa microcristalina, crospovidona, ácido esteárico, sílice coloidal anhidra cubierta del comprimido: hipromelosa, dióxido de titanio (E 171), talco, macrogol 6000 Aspecto del producto y contenido del envase
Los comprimidos de Bondenza son de color blanco o blanquecino, tienen forma oblonga y la inscripción “BNVA” en una cara, y “150” en la otra cara. Se pueden suministrar en envases de 1 o 3 comprimidos.
Puede que solamente estén comercializados algunos tamaños de envases.
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
Responsable de la fabricación
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Alemania
Puede solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Luxembourg/Luxemburg
(Voir/siehe Belgique/Belgien)
Magyarország
Roche (Magyarország) Kft.
Tel: +36 - 23 446 800
Malta
(See United Kingdom)
Belgie/Belgique/Belgien
N.V. Roche S.A.
Tél/Tel: +32 (0) 2 525 82 11
EtarapHH
Pom Etarapna EOOfl Tea: +359 2 818 44 44
O*
V
Nederland
Roche Nederland B.V. Tel: +31 (0) 348 438050
Norge
Roche Norge AS Tlf: +47 - 22 78 90 00
Osterreich
Roche Austria GmbH Tel: +43 (0) 1 27739
Ekláóa
Roche (Hellas) A.E.
Ttfk: +30 210 61 66 100
Polska
Roche Polska Sp.z o.o. Tel: +48 - 22 345 18 88
España
Faes Farma, S.A.
Tel: +34 - 94 481 83 00
Portugal
Roche Farmacéutica Química, Lda Tel: +351 - 21 425 70 00
France
Roche
Tél: +33 (0) 1 47 61 40 00
Romania
Roche Romania S.R.L. Tel: +40 21 206 47 01
Ireland
Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700
Ísland
Roche a/s
c/o Icepharma hf
Tel: +354 540 8000
Slovenija
Roche farmacevtska druzba d.o.o Tel: +386 - 1 360 26 00
Slovenská republika
Roche Slovensko, s.r.o.
Tel: +421 - 2 52638201
Kúrcpog
G.A Stamatis & Co.(Cyprus) Ltd Tr|k: +357 - 22 76 62 76
Latvija
Roche Latvija SIA Tel: +371 - 6 7 039831
Sverige
Roche AB
Tel: +46 (0) 8 726 1200
United Kingdom Roche Products Ltd. Tel: +44 (0) 1707 3660
000
La información detallada de este medicame: de Medicamentos http://www.ema.europa.

tá disponible en la página web de la Agencia Europea

INFORMACIÓN INCLUIDA EN PEGATINAS AUTO-ADHESIVAS A USAR EN SU CALENDARIO PERSONAL
PLANIFICACIÓN DE LA TOMA DE BONDENZA
La dosis de Bondenza es de un comprimido al mes. Elija un día del mes que le sea fácil de recordar:
- bien sea la misma fecha (como el primer día de cada mes)
- o siempre el mismo día (como el primer domingo de cada mes).
Use las pegatinas auto-adhesivas para marcar los días en su calendario.
Una vez haya tomado su comprimido, ponga una señal en la pegatina de la caja.
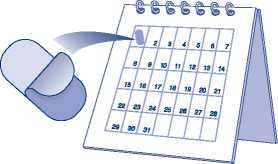
PEGATINAS AUTO-ADHESIVAS PARA SU CALENDARIO PERSONAL.
Comprimido mensual
Comprimido mensual
Comprimido mensual Bondenza
Es importante tomar Bondenza todos los meses.
./! C
Bondenza Bondenza

Prospecto: información para el usuario
Bondenza 3 mg solución inyectable
Ácido ibandrónico
Lea todo el prospecto detenidamente antes de empezar a usar el medicamento porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte con su médico, farmacéutico o enfermera.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermera, incluso si se trata de efectos adversos que no aparecen en este prospecto
1.
2.
3.
4.
5.
6.
Qué es Bondenza y para qué se utiliza
Qué necesita saber antes de empezar a recibir Bondenza
Cómo recibir Bondenza
Posibles efectos adversos
Conservación de Bondenza
Contenido del envase e información adicional
1.
Qué es Bondenza y para qué se utiliza
inados b
Bondenza pertenece a un grupo de medicamentos denominados bifosfonatos . Contiene el principio activo ácido ibandrónico.
Bondenza puede invertir la pérdida de hueso ya que impide que se pierda más hueso y aumenta la masa de hueso en la mayoría de las mujeres que lo toman, aún incluso cuando éstas no son capaces de ver o apreciar la diferencia. Bondenza puede ayudar a reducir los casos de rotura de huesos (fracturas). Esta reducción ha sido demostrada en fracturas vertebrales pero no en las de cadera.
Bondenza 3 mg solución inyectable en jeringas precargadas es una solución intravenosa para ser administrada por un profesional sanitario. No se inyecte Bondenza usted mismo.
Se le ha recetado Bondenza para tratar su osteoporosis posmenopáusica porque tiene un riesgo elevado de sufrir fracturas. La osteoporosis consiste en un adelgazamiento y debilitamiento de los huesos, hecho frecuente entre las mujeres después de la menopausia. En la menopausia, los ovarios dejan de producir la hormona femenina —los estrógenos— que ayuda a conservar la salud del esqueleto.
Cuanto antes llegue una mujer a la menopausia, mayor es el riesgo de que sufra fracturas por osteoporosis.
Otros factores que aumentan el riesgo de sufrir fracturas son:
aporte insuficiente de calcio y de vitamina D en la dieta tabaquismo o consumo excesivo de alcohol pocos paseos u otros ejercicios con carga de peso antecedentes familiares de osteoporosis
Los hábitos saludables de vida también facilitan los efectos favorables del tratamiento. Entre éstas se encuentran:
- una alimentación equilibrada, rica en calcio y en vitamina D
- ; los paseos o cualquier otro ejercicio con carga
- ; no fumar y un consumo no excesivo de alcohol.
2. Qué necesita saber antes de empezar a recibir Bondenza
No reciba Bondenza:
- si tiene o ha tenido valores bajos de calcio en sangre. Por favor consulte a su médico
- si es alérgico al ácido ibandrónico o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Advertencias y precauciones
Algunas personas precisan cuidados especiales durante el tratamiento con Bondenza. Consulte a su médico antes de empezar a recibir Bondenza:
- Si usted tiene o ha tenido problemas de riñón, fallo renal o ha necesitado diálisis, o si usted tiene cualquier otra enfermedad que pueda afectar a sus riñones.
Si padece algún trastorno del metabolismo mineral (por ejemplo, carencia de vitamina D). Usted debería tomar suplementos de calcio y vitamina D durante el tratamiento con Bondenza. Si no pudiera, debe informar a su médico.
Si se encuentra bajo tratamiento dental o tiene prevista alguna cirugía dental, comunique a su dentista que está en tratamiento con Bondenza.
tom
Si tiene problemas de corazón y su médico le ha recomendado una toma limitada diaria de líquidos.
do una
Se han notificado casos graves, algunas veces mortales, de reacción ac. ibandrónico intravenoso.
alérgica en pacientes tratados con
Debería avisar inmediatamente a su médico o enfermera si experimenta uno de los siguientes
i — ----- o
sación de opresión en la garganta, hinchazón de o, rojez o hinchazón de la cara, sarpullido
síntomas, como falta de aire/dificultad respiratoria, s la lengua, mareo, sensación de pérdida del conoci corporal, nausea y vómito (ver sección 4).
Niños y adolescentes
No se debe usar Bondenza en niños o adolescentes menores de 18 años.
Otros medicamentos y Bonden
Informe a su médico o farmacéutico si está utilizando o ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento..
Embarazo y lactancia

No debe usar Bo Si se encuentra e Consulte a su

si está embarazada, o si cree que pudiera estarlo. período de lactancia, usted tendrá que suspenderla para usar Bondenza. ico o farmacéutico antes de utilizar este medicamento.
Conducción y uso de máquinas
Puede conducir y utilizar máquinas ya que se espera que Bondenza no tenga efecto o éste sea despreciable sobre su capacidad para conducir y utilizar máquinas.
Bondenza contiene menos de 1 mmol de sodio (23 mg) por dosis (3 ml), es decir, está esencialmente “exento de sodio”.
3. Cómo recibir Bondenza
La dosis recomendada de Bondenza inyectable intravenoso es de 3 mg (1 jeringa precargada) cada 3 meses.
La inyección debe administrarse por vía intravenosa por un médico o personal sanitario cualificado/entrenado. No se administre la inyección usted mismo.
Bondenza inyectable debe administrase en vena y no por cualquier otra vía.
Continuación del tratamiento con Bondenza
Para conseguir el máximo beneficio del tratamiento, es importante continuar recibiendo las inyecciones cada 3 meses durante el periodo prescrito por su médico.
Bondenza trata la osteoporosis sólo mientras usted recibe el tratamiento, incluso si usted no viera o sintiese la diferencia.
Usted deberá tomar suplementos de calcio y vitamina D, como su médico le indique.
Si usted usa más Bondenza del que debiera
Usted puede sufrir disminución de los niveles de calcio, fósforo o magnesio en sangre. Su médico puede tomar medidas para corregir estas alteraciones, administrándole una inyección de estos minerales.
Si olvidó usar Bondenza
Usted deberá pedir una cita a su médico para que le administre la próxima inyección lo antes posible. A partir de esa fecha continúe recibiendo las inyecciones cada 3 meses.
&
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede tener efectos adversos aunque no todas
las personas lo sufran.
Informe a su médico o enfermera inmediatamente si nota cualquiera de los siguientes efectos
adversos graves ya que podría necesitar tratamiento médico urgente:
Frecuentes (pueden afectar hasta 1 de cada 10 personas):
• síntomas seudo-gripales, incluyendo fiebre, escalofríos y tiritona, sensación de malestar, fatiga, dolor de huesos y de músculos y articulaciones. Consulte a su enfermera o médico si cualquier efecto llega a ser molesto o dura más de un par de días.
• erupción cutánea. Puede que esté teniendo una reacción alérgica al medicamento
Raras (pueden afectar hasta 1 de cada 1.000 personas)
• picor, hinchazón de la cara, labios, lengua y garganta, con dificultad para respirar.
• dolor de ojo persistente e inflamación (si se prolonga)
• dolor nuevo, debilidad o molestias en el muslo, la cadera o la ingle. Pueden ser síntomas precoces de una posible fractura inusual del hueso del muslo
Muy raras (pueden afectra hasta 1 de cada 10.000 personas)
• dolor o sensación de dolor en la boca o mandíbula. Pueden ser síntomas precoces de problemas graves de mandíbula [necrosis (muerte del tejido óseo) del hueso de la mandíbula]
• reaccion alérgica grave que puede suponer una amenaza para la vida (ver sección 2)
Otros efectos adversos posibles
Frecuentes (pueden afectar hasta 1 de cada 10 personas):
• dolor de cabeza
• dolor de estómago (como gastritis) o dolor de tripa, indigestión, nausea, diarrea (perdidas intestinales) o estreñimiento.
• dolor en músculos, articulaciones y espalda.
Poco frecuentes (pueden afectar hasta 1 de cada 100 personas)
• inflamación de la vena
• dolor o lesión en el lugar de la inyección
• dolor de huesos
• sensación de debilidad
Raras (pueden afectar a hasta 1 de cada 1.000 personas)
• urticaria
Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluso si se trata de efectos adversos que no aparecen en este prospecto.
5. CONSERVACION DE BONDENZA
Mantener este medicamento fuera de la vista y del alcance de los niños Este medicamento no requiere condiciones especiales de conservación.
&
No utilice este medicamento después de la fecha de caducidad que aparece en el envase y en la jeringa después de CAD. La fecha de caducidad es el último día del mes que se indica.
La persona que administra la inyección, debe desechar cualquier resto de solución no utilizado y depositar la jeringa y la aguja para inyección usadas en el recipiente destinado a tal efecto.
6. Contenido del envase e información adicional Composición de Bondenza
- El principio activo es ácido ibandrónico. Una jeringa precargada contiene 3 mg de ácido ibandrónico en 3 ml de solución (como monohidrato sódico).
- Los demás componentes son cloruro de sodio, ácido acético, acetato de sodio trihidrato, agua para preparaciones inyectables.
Aspecto del producto y contenido del envase
Bondenza 3 mg solución inyectable en jeringas precargadas es una solución clara e incolora. Cada jeringa precargada contiene 3ml de sol ución.
Bondenza está disponible en envases de 1 jeringa precargada y 1 aguja para inyección ó 4 jeringas precargadas y 4 agujas para inyección.
Puede que solamente estén comercializados algunos tamaños de envases.
jp
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Reino Unido
Responsable de la fabricación
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Alemania
Puede solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización.
Belgie/Belgique/Belgien Luxembourg/Luxemburg
N.V. Roche S.A. (Voir/siehe Belgique/Belgien)
Tél/Tel: +32 (0) 2 525 82 11
Etarapun
Pom Etarapna EOOfl
Tea: +359 2 818 44 44
Magyarország
Roche (Magyarország) Kft. Tel: +36 - 23 446 800
España
Faes Farma, S.A.
Tel: +34 - 94 481 83 00
V
France
Roche
Tél: +33 (0) 1 47 61 40 00 Ireland
&
^oche Polska Sp.z o.o.
Tel: +48 - 22 345 18 88
Portugal
Roche Farmacéutica Química, Lda Tel: +351 - 21 425 70 00
Danmark
Roche a/s
Tlf: +45 - 36 39 99 99
Deutschland
Roche Pharma AG Tel: +49 (0) 7624 140
Eesti
Roche Eesti OÜ Tel: + 372 - 6 112 401
Eklába
Roche (Hellas) A.E.
Ttfk: +30 210 61 66 100
Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700
Nederland
Roche Nederland B.V. Tel: +31 (0) 348 438050
Norge
Roche Norge AS Tlf: +47 - 22 78 90 00
- .¿f0
Osterreich
Roche Austria GmbH Tel: +43 (0) 1 27739
Ísland
Roche a/s c/o Icepharma hf
Tel: +354 540 8000
Slovenská republika
Roche Slovensko, s.r.o. Tel: +421 - 2 52638201
United Kingdom
Latvija
Roche Latvija SIA Tel: +371 - 6 7 039831
Roche Products Ltd.
Tel: +44 (0) 1707 366000
Lietuva
UAB “Roche Lietuva”
Tel: +370 5 2546799
Fecha de la última revisión de este prospecto MM/AAAA}
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.

Esta información está destinada únicamente a profesionales del sector sanitario:
INFORMACION PARA EL PROFESIONAL SANITARIO
Para más información por favor consulte la Ficha Técnica del producto:
Administración de Bondenza 3 mg solución inyectable en jeringa precargada:
Bondenza 3 mg solución inyectable en jeringa precargada debe administrarse de forma intravenosa durante un periodo de 15 - 30 segundos.
La solución es irritante, por lo tanto es importante respetar la vía de administración. Si de forma accidental se inyecta en los tejidos alrededor de la vena, las pacientes pueden experimentar irritación local, dolor e inflamación en el lugar de la inyección.
Bondenza 3 mg solución inyectable en jeringa precargada no debe mezclarse con soluciones que contengan calcio (tales como solución Lactato de Ringer, heparina cálcica) u otros medicamentos de administración intravenosa. Cuando Bondenza se administra utilizando una vía intravenosa existente, la infusión intravenosa debe restringirse a solución isotónica salina o solución de 50mg/ml de glucosa al 5%.
Olvido de dosis:
Si se olvida una dosis, la inyección debe administrarse tan pronto como sea posible. A partir de ahí, las inyecciones deberían programarse cada tres meses desde la fecha de la última inyección.
Sobredosis:
No se dispone de información específica so bre el tratamiento de sobredosis con Bondenza.
Basándose en el conocimiento de esta clase de fármacos, la sobredosificación intravenosa puede provocar hipocalcemia, hipofosfatemia e hipomagnesemia, que puede causar parestesia. En casos graves puede ser necesaria la infusión intravenosa de dosis apropiadas de gluconato cálcico, fosfato sódico o potásico y sulfato magnésico.
Advertencias generales:
NT
Bondenza 3 mg solución inyectable en jeringa precargada al igual que otros bifosfonatos administrados por vía intravenosa, puede provocar un descenso transitorio de los valores de calcio en suero.
Antes de iniciar el tratamiento con Bondenza inyectable, se debe valorar y tratar de manera eficaz la hipocalcemia, así como otros trastornos del metabolismo óseo y mineral. Es importante en todas las pacientes una ingesta adecuada de calcio y vitamina D. Todas las pacientes deben recibir suplementos de calcio y vitamina D.
Las pacientes que presenten enfermedades concomitantes o que utilicen medicamentos con potenciales reacciones adversas sobre el riñón deben ser estrechamente vigiladas durante el tratamiento de acuerdo a la práctica clínica habitual. La solución que no se use en la inyección, así como la jeringa y la aguja para inyección deberían desecharse de acuerdo con los requerimientos locales.
56