Beriate 500 Ui Polvo Y Disolvente Para Solucion Inyectable O Perfusion
"I
an
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Beriate 250 U. I. polvo y disolvente para solución inyectable o perfusión Beriate 500 U. I. polvo y disolvente para solución inyectable o perfusión Beriate 1.000 U. I. polvo y disolvente para solución inyectable o perfusión Beriate 2.000 U. I. polvo y disolvente para solución inyectable o perfusión
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Beriate se presenta como polvo y disolvente para solución para inyección o perfusión que contiene por vial, nominalmente, 250 UI, 500 UI ó 1.000 UI de factor VIII de la coagulación humano.
Composición cualitativa y cuantitativa
Beriate 250/500/1000 reconstituido con 2,5 ml, 5 ml ó 10 ml de Agua para inyectables contiene, 100 UI de factor VIII de la coagulación sanguínea humano por ml, aproximadamente. Beriate 2000 se reconstituye con 10 ml de agua para inyectables y contiene aproximadamente 200 UI/ml de factor VIII de la coagulación sanguínea humano.
La potencia (UI) se determina usando el método del sustrato cromogénico de la Farmacopea Europea. La actividad específica media es de 270 UI/mg de proteína, aproximadamente.
Excipiente(s) con efecto conocido
Sodio.
Para consultar la lista completa de excipientes, ver Sección 6.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución para inyección o perfusión.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII).
Este producto puede usarse en la terapéutica de la deficiencia adquirida de factor VIII.
Este medicamento no contiene factor de von Willebrand en cantidades farmacológicamente activas, por lo que este producto no está indicado en la enfermedad de von Willebrand.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia.
Posología
La dosis y la duración de la terapia de sustitución dependen de la gravedad de la deficiencia de factor VIII, de la localización y gravedad de la hemorragia y de la condición clínica del paciente.
El número de unidades de factor VIII administradas se expresa en Unidades Internacionales (UI), que se corresponden con el estándar vigente de la OMS para productos que contienen factor VIII. La actividad
ÍTTI
plasmática de factor VIII puede expresarse, bien en porcentaje (referido al plasma humano normal) o bien, en Unidades Internacionales (referido a un estándar internacional para el factor VIII plasmático).
La actividad de una UI de factor VIII equivale a la cantidad de factor VIII presente en un ml de plasma humano normal.
El cálculo de la dosis requerida de factor VIII se basa en el hallazgo empírico de que:
1 UI de factor VIII por Kg de peso corporal aumenta la actividad plasmática de factor VIII en aproximadamente un 2% de la actividad normal (2 UI/dl). La dosis requerida se determina usando la siguiente fórmula:
Unidades requeridas = peso corporal [Kg] x aumento deseado de factor VIII [% o UI/dl] x 0,5.
La dosis y la frecuencia de administración deben estar siempre orientadas a conseguir la eficacia clínica en cada caso.
En los siguientes episodios hemorrágicos, la actividad de factor VIII no debe ser inferior al nivel plasmático de actividad que se indica (en % del nivel normal o UI/dl), durante el periodo correspondiente.
La tabla siguiente puede usarse como una guía posológica en episodios hemorrágicos y cirugía
|
Tipo de episodio hemorrágico/ Tipo de procedimiento quirúrgico |
Nivel de factor VIII requerido (% o UI/dl) |
Frecuencia de dosificación (horas) / Duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis precoz, sangrado |
20-40 |
Repetir la perfusión cada |
|
muscular o de la cavidad bucal |
12-24 horas. Al menos 1 día, hasta que la hemorragia se haya resuelto, en función del dolor, o hasta la cicatrización adecuada de la herida | |
|
Hemartrosis más extensa, sangrado |
Repetir la perfusión cada | |
|
muscular o hematoma |
30-60 |
12-24 horas, durante 3-4 días o más, hasta que el dolor y la discapacidad aguda se hayan resuelto |
|
Hemorragias con riesgo vital |
Repetir la perfusión cada | |
|
60-100 |
8-24 horas hasta que desaparezca el riesgo. |
|
Tipo de episodio hemorrágico/ Tipo de procedimiento quirúrgico |
Nivel de factor VIII requerido (% o UI/dl) |
Frecuencia de dosificación (horas) / Duración de la terapia (días) |
|
Cirugía Menor, incluyendo extracciones dentales |
30-60 |
Cada 24 horas, al menos 1 día, hasta la cicatrización de la herida. |
|
Mayor |
80-100 (pre y postoperatorio) |
Repetir la perfusión cada 8-24 horas hasta la cicatrización adecuada de la herida; continuar la terapia durante un mínimo de 7 días más para mantener una actividad de factor VIII del 30 al 60 % (UI/dl) |
Durante el tratamiento se recomienda controlar, adecuadamente, los niveles de factor VIII para determinar la dosis a administrar y la frecuencia de las perfusiones. En el caso especial de la cirugía mayor, es indispensable monitorizar con precisión la terapia de sustitución mediante pruebas de la coagulación (actividad plasmática del factor VIII). La respuesta de cada paciente frente al factor VIII puede variar y alcanzar distintos niveles de recuperación in vivo y demostrar semividas diferentes.
Para la profilaxis a largo plazo de las hemorragias, en pacientes con hemofilia A grave, las dosis usuales son de 20 a 40 UI de factor VIII por kg de peso corporal, cada 2 ó 3 días. En ciertos casos, especialmente en pacientes jóvenes, puede ser necesario acortar los intervalos entre administraciones, o utilizar dosis más elevadas.
La posología en pediatría se basa en el peso corporal y por lo tanto sigue, generalmente, las mismas directrices que se usan para los adultos. La frecuencia de administración debe estar siempre orientada a conseguir la eficacia clínica en cada caso. Existe alguna experiencia clínica en el tratamiento de niños menores de 6 años (Ver Sección 5.1).
En los pacientes deberá controlarse el desarrollo de inhibidores del factor VIII. Si no se alcanzan los niveles de actividad plasmática de factor VIII esperados, o si el sangrado no se controla con la dosis adecuada, deberá realizarse una prueba para determinar la presencia de inhibidores del factor VIIII. En pacientes con títulos altos de inhibidores, la terapia con factor VIII puede ser no efectiva, por lo que deberán considerarse otras opciones terapéuticas. El manejo de estos pacientes deberá estar dirigido por médicos con experiencia en el tratamiento de la hemofilia.
Véase también Sección 4.4.
Forma de administración
Reconstituir el preparado tal como se describe en el punto 6.6. El producto debe llevarse a temperatura ambiente o corporal antes de la administración. Inyectar o perfundir por vía intravenosa lenta, a una velocidad de administración que sea confortable para el paciente. La velocidad de inyección o perfusión no debe ser superior a los 2 ml por minuto.
Los pacientes deben mantenerse bajo observación por si se presenta alguna reacción inmediata. Si, se presentara alguna reacción que estuviera relacionada con la administración de Beriate, debe disminuirse la
ÍTTI
velocidad de perfusión o interrumpirse la administración del producto, si así lo requiriera la condición clínica del paciente (véase también Sección 4.4).
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a los excipientes del preparado.
4.4 Advertencias y precauciones especiales de empleo
Tal como sucede con cualquier otro producto proteico, que se administra por vía intravenosa, es posible que se presenten reacciones de hipersensibilidad de tipo alérgico. Los pacientes deben ser informados sobre la aparición de síntomas precoces de reacciones de hipersensibilidad incluyendo, habones, urticaria generalizada, opresión torácica, respiración dificultosa, hipotensión y anafilaxia. Si se presentan estos síntomas, debe informárseles que debe interrumpirse inmediatamente la administración del producto y comunicarlo a su médico.
En caso de shock, se seguirán las pautas médicas vigentes para su tratamiento.
Beriate 1000 UI contiene hasta 28 mg de sodio. Beriate 500 UI contiene hasta 14 mg de sodio y Beriate 250 UI contiene hasta 7 mg de sodio. Esto debe tenerse en cuenta por aquellos pacientes con una dieta limitada en sodio.
Para prevenir la transmisión de enfermedades infecciosas cuando se administran medicamentos derivados de la sangre o plasma humanos, se toman medidas estándares como la selección de donantes, análisis de marcadores específicos de infecciones en las donaciones individuales y en las mezclas de plasma, así como la inclusión de etapas en el proceso de fabricación para eliminar / inactivar virus. A pesar de esto, cuando se administran medicamentos derivados de la sangre o plasma humanos, la posibilidad de transmisión de agentes infecciosos no se puede excluir totalmente. Esto también se refiere a virus y agentes infecciosos emergentes de naturaleza desconocida.
Estas medidas se consideran efectivas para virus envueltos como VIH, VHB y VHC y para los virus no envueltos VHA y parvovirus B19.
Se recomienda la vacunación apropiada (hepatitis A y B) para los pacientes que reciban regularmente concentrados plasmáticos de factor VIII derivados de plasma humano.
La formación de anticuerpos neutralizantes (inhibidores) es una complicación bien conocida en el tratamiento de pacientes con hemofilia A. Estos inhibidores son generalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII. Esta actividad se cuantifica en Unidades Bethesda (UB) por ml de plasma utilizando el método modificado. El riesgo de desarrollar inhibidores se correlaciona con la exposición a factor VIII antihemofílico, siendo este riesgo mayor en los primeros 20 días de exposición. De manera poco frecuente, pueden desarrollarse inhibidores tras los primeros 100 días de exposición. En los pacientes tratados con el factor VIII humano de la coagulación se debe controlar, cuidadosamente, el desarrollo de inhibidores mediante observación clínica adecuada y pruebas de laboratorio. Ver también “Sección 4.8 Reacciones adversas”.
A fin de mantener la trazabilidad del producto y en beneficio de los pacientes se recomienda encarecidamente que, siempre que sea posible, cada vez que se les administre Beriate se deje constancia del nombre del medicamento y número de lote administrado.
4 de 1 1 MINISTWODE
SANIDAD, POLITICA SOCIAL E IGUALDAD Agencia es pañosa de medicamentos y productos san-lanos
ÍTTI
4.5 Interacción con otros medicamentos y otras formas de interacción
No se conocen interacciones de productos que contienen el factor VIII de la coagulación sanguínea humano con otros medicamentos.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción animal con factor VIII.
Ya que la hemofilia A es excepcional en mujeres, no se dispone de experiencia clínica sobre el uso de factor VIII durante el embarazo y la lactancia.
Por lo tanto, el factor VIII sólo debe ser usado en el embarazo y la lactancia, en el caso de que esté claramente indicado.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han observado efectos sobre la capacidad de conducir vehículos o utilizar maquinaria.
4.8 Reacciones adversas
Las reacciones adversas siguientes se referencian en la literatura científica y en la experiencia postcomercialización de Beriate. Se usan las siguientes categorías estándares según su frecuencia:
-Muy frecuente > 1/10
-Frecuente > 1/100 y < 1/10
-Infrecuente >1/1.000 y < 1/100
-Rara >1/10.000 y < 1/1.000
-Muy rara < 1/10.0000 (incluyendo los casos individuales reportados)
• Trastornos del Sistema Inmunitario
Muy raramente se han observado reacciones alérgicas o de hipersensibilidad (que pueden incluir angioedema, sensación de quemazón y picor en el lugar de la perfusión, escalofríos, enrojecimiento, urticaria generalizada, cefalea, urticaria, hipotensión, letargia, náuseas, intranquilidad, taquicardia, opresión torácica, hormigueo, vómitos, dificultad respiratoria) que en algunos casos puede evolucionar a anafilaxia grave (incluido el shock).
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes del factor VIII (inhibidores).
La aparición de estos inhibidores se manifiesta como una respuesta clínica insuficiente. En estos casos se recomienda contactar con un centro especializado en hemofilia.
La experiencia clínica obtenida de los ensayos clínicos con Beriate en pacientes previamente no tratados (PUPs) es muy limitada. Por consiguiente, no es posible aportar cifras válidas sobre la incidencia de inhibidores específicos clínicamente relevantes.
• Trastornos generales
En muy raras ocasiones se ha observado fiebre.
Para información sobre seguridad viral, ver Sección 4.4.
4.9 Sobredosis
Hasta ahora no se conocen síntomas de sobredosificación con el factor VIII de la coagulación humano.
"I
an
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico Antihemorrágicos: factor VIII de la coagulación Código ATC: B02BD02
El complejo de Factor VIII/Factor de von Willebrand consiste en dos moléculas (factor VIII y factor de von Willebrand) con diferentes funciones fisiológicas.
Cuando se perfunde el factor VIII a pacientes hemofílicos, éste se une al factor von Willenbrand presente en la circulación sanguínea del paciente.
El factor VIII activado, actúa como cofactor del factor IX activado, acelerando la conversión de factor X en factor X activado. Éste convierte la protrombina en trombina. La trombina convierte a su vez el fibrinógeno en fibrina, con lo que puede formarse el coágulo sanguíneo. La hemofilia A es una alteración de la coagulación sanguínea hereditaria ligada al sexo y se debe a una disminución de los niveles de factor VIII:
C que da lugar a un sangrado profuso en las articulaciones, músculos u órganos internos, ya sea de forma espontánea o a causa de un traumatismo accidental o quirúrgico. La terapia de sustitución aumenta los niveles plasmáticos de factor VIII, obteniéndose una restauración temporal de la deficiencia de este factor y una corrección de la tendencia al sangrado.
Además de su función protectora del factor VIII, el factor de von Willebrand facilita la adhesión de las plaquetas en los sitios con una herida vascular y juega un papel en la agregación plaquetaria.
Se dispone de datos sobre el tratamiento de 16 niños menores de 6 años y los resultados obtenidos de eficacia y seguridad clínica son consistentes con los datos obtenidos en pacientes con mas edad.
5.2 Propiedades farmacocinéticas
Tras la administración intravenosa del producto, la actividad del factor VIII disminuye siguiendo un modelo mono o biexponencial. La semivida terminal varía entre 5 y 22 horas, con un valor medio de 12 horas, aproximadamente. El incremento de la actividad de factor VIII tras la administración de 1 UI de factor VIII por Kg de peso corporal (incremento de la recuperación) fue de un 2% aproximadamente, con una variabilidad interindividual del 1,5 al 3%. El tiempo medio de residencia (TMR) fue de 17 horas (desviación estándar 5,5 horas); la media del área bajo la curva obtenida por extrapolación (AUC) fue de 0,4 h x Kg/ml (desviación estándar 0,2) y la media del aclaramiento fue de 3 ml/h/Kg (desviación estándar 1,5 ml/h/Kg).
5.3 Datos preclínicos sobre seguridad Toxicidad General:
No se han realizado pruebas de toxicidad con dosis repetidas en animales, debido al desarrollo de anticuerpos contra las proteínas heterólogas.
No se han observado efectos tóxicos en los animales de laboratorio, ni siquiera con dosis que suponen varias veces la dosis humana recomendada por kilogramo de peso corporal.
Las pruebas llevadas a cabo en preparados de factor VIII sometidos a tratamiento térmico de precipitación con anticuerpos policlonales (de conejo) mediante el test de Ouchterlony y la prueba anafilaxia cutánea
ÍTTI
pasiva en cobayas, no muestran cambios en estas reacciones inmunológicas, cuando se comparan con proteína no tratada.
Mutagenicidad:
Dado que la experiencia clínica no proporciona ningún indicio de efectos carcinogénicos o mutagénicos del factor VIII de coagulación sanguínea humano, no se considera necesario realizar estudios experimentales, especialmente en especies heterólogas.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Glicina, cloruro cálcico, hidróxido sódico (en pequeñas cantidades para ajustar el pH), sacarosa y cloruro sódico.
Disolvente que se suministra: 2,5 ml, 5ml y 10 ml de Agua para preparaciones inyectables.
6.2 Incompatibilidades
Este producto no debe mezclarse con otros medicamentos, disolventes o diluyentes.
6.3 Periodo de validez 3 años.
Después de la reconstitución, la estabilidad físico-química ha sido demostrada para un tiempo de 8 horas a temperatura ambiente (máx. 25° C). Desde un punto de vista microbiológico la solución reconstituida debe usarse inmediatamente. Si ello no es posible, no almacenar más de 8 horas a temperatura ambiente.
6.4 Precauciones especiales de conservación
Almacenar en nevera entre 2° C y 8° C. No congelar. Mantener el producto en su envase original, para protegerlo de la luz.
Durante su periodo de validez Beriate puede almacenarse hasta 25° C, durante un periodo de 1 mes como máximo. Este periodo de almacenamiento a temperatura ambiente debe apuntarse en su diario de tratamiento a fin de cumplir el periodo de 1 mes en su totalidad.
No exponer los viales al calor directo. Los viales no deben calentarse por encima de la temperatura corporal (37° C).
Mantener fuera de la vista y del alcance de los niños.
6.5 Naturaleza y contenido del envase Envase primario:
Vial para inyección de vidrio incoloro Tipo I según Farmacopea Europea, cerrados al vacío, provisto de tapón de goma, disco de plástico y cápsula de aluminio.
"I ■"
an
Presentaciones:
Un envase con 250 UI que contiene:
- 1 vial con polvo
- 1 vial con 2,5 ml de Agua para preparaciones inyectables Un envase con equipo de administración que contiene:
- 1 trasvasador con filtro 20/20
- 1 jeringa de 5 ml de un solo uso
- 1 equipo de punción venosa
- 2 toallitas con alcohol
- 1 apósito adhesivo no estéril
Un envase con 500 UI que contiene:
- 1 vial con polvo
- 1 vial con 5 ml de Agua para preparaciones inyectables
Un envase con equipo de administración que contiene:
- 1 trasvasador con filtro 20/20
- 1 jeringa de 5 ml de un solo uso
- 1 equipo de punción venosa
- 2 toallitas con alcohol
- 1 apósito adhesivo no estéril.
Un envase con 1.000 UI que contiene:
- 1 vial con polvo
- 1 vial con 10 ml de Agua para preparaciones inyectables Un envase con equipo de administración que contiene:
- 1 trasvasador con filtro 20/20
- 1 jeringa de 10 ml de un solo uso
- 1 equipo de punción venosa
- 2 toallitas con alcohol
- 1 apósito adhesivo no estéril
Un envase con 2.000 UI que contiene:
- 1 vial con polvo
- 1 vial con 10 ml de Agua para preparaciones inyectables Un envase con equipo de administración que contiene:
- 1 trasvasador con filtro 20/20
- 1 jeringa de 10 ml de un solo uso
- 1 equipo de punción venosa
- 2 toallitas con alcohol
- 1 apósito adhesivo no estéril
6.6 Precauciones especiales de eliminación y otras manipulaciones Instrucciones Generales
- La solución debe ser clara o ligeramente opalescente. Después de extraer y filtrar el producto reconstituido (véase más adelante) debe revisarse visualmente, antes de la administración, para detectar la presencia de partículas extrañas y decoloraciones. No debe usarse soluciones turbias o que presenten sedimentos (precipitados/partículas).
- La reconstitución y la extracción deben realizarse bajo condiciones asépticas.
- Tras la administración, cualquier producto no utilizado o del material empleado se eliminarán de acuerdo con los requerimientos de la legislación local
"I
Atemperar el disolvente a temperatura ambiente. Retirar las cápsulas de los viales, desinfectar la superficie expuesta de los tapones de goma, dejándolos secar antes de proceder a abrir el envase que contiene el Mix2Vial.
1
1. Abrir el envase que contiene el Mix2Vial, desprendiendo el precinto. No retire el Mix2Vial del blister.

2
2. Coloque el vial del disolvente sobre una superficie limpia y plana y sujételo con firmeza. Sujete el Mix2Vial junto con el blister y empuje el terminal azul hacia abajo encajándolo en el tapón del vial del disolvente.

3


4
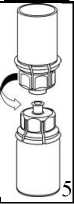

6
3. Retire, con cuidado, el blister del Mix2Vial sujetando el borde y tirando verticalmente hacia arriba. Asegúrese de que sólo retira el blister y no el Mix2Vial.
4. Coloque el vial del polvo liofilizado sobre una
superficie plana y firme. Invierta el vial del disolvente con el Mix2Vial acoplado y empuje el terminal del adaptador transparente hacia abajo encajándolo en el tapón del vial con el polvo. El disolvente se transferirá automáticamente al vial del polvo liofilizado._
5. Con una mano, sujete el vial con el producto con el Mix2Vial y, con la otra mano, sujete el vial del disolvente y desenrosque con cuidado el sistema separándolo en dos piezas.
Deseche el vial del disolvente con el adaptador azul del Mix2Vial acoplado
6. Someta el vial de la solución reconstituida con el adaptador transparente acoplado a movimientos de rotación suaves hasta que la sustancia se haya disuelto por completo. No lo agite.
.<5^.
"I

7
7. Llene de aire una jeringa vacía y estéril. Manteniendo el vial con la solución en posición vertical, conecte la jeringa al adaptador Luer Lock del Mix2Vial. Inyecte el aire en el vial de la solución.
Aplicación y retirada:
|
ú¡ |
8. Manteniendo presionado el émbolo de la jeringa, invertir el sistema, aspirar la solución al interior de la jeringa haciendo retroceder lentamente el émbolo de la jeringa. | ||
|
3Í 9 |
9. Una vez que la solución ha sido transferida a la jeringa, sujetar firmemente el cuerpo de la jeringa (manteniendo el émbolo de la jeringa mirando hacia abajo) y desconectar el adaptador transparente del Mix2Vial de la jeringa. |
Para la inyección de Beriate se recomienda el uso de jeringas de plástico de un solo uso, ya que la superficie de todas las jeringas de vidrio tiene tendencia a adherirse a este tipo de soluciones.
Perfundir o inyectar inmediatamente por vía intravenosa lenta (ver Sección 4.2. Método de administración), teniendo cuidado de que no entre sangre en la jeringa. Usar el equipo de venopunción que se suministra con este medicamento. Insertar la aguja en la vena. Dejar que entre la sangre hasta el final del tubo. Acoplar la jeringa al final roscado y con cierre del equipo de venopunción.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
CSL Behring GmbH Emil-von-Behring-Str. 76 35041 Marburg Germany
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
Beriate 250 UI. N° de Registro: 63.010 Beriate 500 UI. N° de Registro: 63.023 Beriate 1.000 UI. N° de Registro: 63.009 Beriate 2.000 UI. N° de Registro: xx.xxx
MINISrSUODE SANIDAD, POLITICA SOCIAL E IGUALDAD Agencia es paño» de medicamentos y proouctos sabíanos
ílMt
ÍTTI
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Marzo 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
Fecha última revisión: Mayo 2012
La información detallada y actualizada de este medicamento está disponible en la página Web de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) http://www.aemps.gob.es