Benefix 3000 Ui Polvo Y Disolvente Para Solucion Inyectable
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
BeneFIX 250 UI polvo y disolvente para solución inyectable BeneFIX 500 UI polvo y disolvente para solución inyectable BeneFIX 1000 UI polvo y disolvente para solución inyectable BeneFIX 2000 UI polvo y disolvente para solución inyectable BeneFIX 3000 UI polvo y disolvente para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
BeneFIX 250 UI polvo y disolvente para solución inyectable
Cada vial contiene nominalmente 250 UI de nonacog alfa (factor IX de coagulación recombinante). Después de la reconstitución con los 5 ml de solución de cloruro de sodio para inyección que acompañan (al 0,234%), cada ml de la solución contiene aproximadamente 50 UI de nonacog alfa.
BeneFIX 500 UI polvo y disolvente para solución inyectable
Cada vial contiene nominalmente 500 UI de nonacog alfa (factor IX de coagulación recombinante). Después de la reconstitución con los 5 ml de solución de cloruro de sodio para inyección que acompañan (al 0,234%), cada ml de la solución contiene aproximadamente 100 UI de nonacog alfa.
BeneFIX 1000 UI polvo y disolvente para solución inyectable
Cada vial contiene nominalmente 1000 UI de nonacog alfa (factor IX de coagulación recombinante). Después de la reconstitución con los 5 ml de solución de cloruro de sodio para inyección que acompañan (al 0,234%), cada ml de la solución contiene aproximadamente 200 UI de nonacog alfa.
BeneFIX 2000 UI polvo y disolvente para solución inyectable
Cada vial contiene nominalmente 2000 UI de nonacog alfa (factor IX de coagulación recombinante). Después de la reconstitución con los 5 ml de solución de cloruro de sodio para inyección que acompañan (al 0,234%), cada ml de la solución contiene aproximadamente 400 UI de nonacog alfa.
BeneFIX 3000 UI polvo y disolvente para solución inyectable
Cada vial contiene nominalmente 3000 UI de nonacog alfa (factor IX de coagulación recombinante). Después de la reconstitución con los 5 ml de solución de cloruro de sodio para inyección que acompañan (al 0,234%), cada ml de la solución contiene aproximadamente 600 UI de nonacog alfa.
La potencia (UI) se determina utilizando el ensayo de coagulación en una etapa de la Farmacopea Europea. La actividad específica de BeneFIX, es al menos de 200 UI/mg de proteína.
BeneFIX contiene factor IX de coagulación recombinante, (DCI = nonacog alfa). El factor IX de coagulación recombinante es una proteína purificada que posee 415 aminoácidos en una cadena única. Tiene una secuencia primaria de aminoácidos comparable a la forma alélica Ala148 del factor IX plasmático, y algunas de las modificaciones post-translacionales de la molécula recombinante son diferentes a las de la molécula plasmática. El factor IX de coagulación recombinante es una glucoproteína secretada por células de mamífero sometidas a ingeniería genética, derivadas de una línea celular de ovario de hámster chino (CHO).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
BeneFIX 250 UI, 500 UI, 1000 UI, 2000 UI o 3000 UI polvo y disolvente para solución inyectable Polvo y disolvente para solución inyectable.
Polvo blanco o blanquecino y disolvente transparente e incoloro.
DATOS CLÍNICOS
4.
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia B (deficiencia congénita de factor IX).
BeneFIX se puede utilizar en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe realizarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia.
Seguimiento del tratamiento
En el transcurso del tratamiento, es recomendable la determinación adecuada de las concentraciones de factor IX ya que puede servir de orientación para establecer la dosis que se debe administrar y la frecuencia de las perfusiones repetidas. La respuesta de cada paciente al factor IX puede ser distinta, con diferentes semividas y recuperaciones. La dosis basada en el peso corporal puede precisar ajuste en el caso de los pacientes con bajo peso o sobrepeso. En concreto, en el caso de intervenciones quirúrgicas mayores, es imprescindible un seguimiento preciso del tratamiento sustitutivo mediante el análisis de la coagulación (la actividad plasmática del factor IX).
Cuando se utilice un ensayo de coagulación en una etapa basado en el tiempo de tromboplastina (TTPa) in vitro para la determinación de la actividad de factor IX en las muestras de sangre de los pacientes, los resultados de la actividad de factor IX pueden verse afectados considerablemente tanto por el tipo de reactivo de TTP como por el patrón de referencia usado en el ensayo. Esto reviste importancia sobre todo al cambiar el laboratorio o los reactivos usados en el ensayo.
Posología
La dosis y duración de la terapia de sustitución dependen de la gravedad de la deficiencia del factor IX, de la localización y el grado de la hemorragia, y del estado clínico del paciente.
El número de unidades de factor IX administradas se expresa en Unidades Internacionales (UI) que se relacionan con el estándar actual de la OMS para productos de factor IX. La actividad de factor IX en plasma se expresa bien como porcentaje (referido a plasma humano normal) o en Unidades Internacionales (relativo a un Estándar Internacional de factor IX en plasma).
Una unidad internacional (UI) de actividad de factor IX es equivalente a la cantidad de factor IX presente en un mililitro de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis necesaria de BeneFIX puede basarse en el hallazgo de que cabe esperar que una unidad de actividad de factor IX por kg de peso corporal aumente el nivel circulante de factor IX una media de 0,8 UI/dl (rango de 0,4 a 1,4 UI/dl) en pacientes > 12 años (ver más información en la sección 5.2).
La dosis requerida se determina usando la siguiente fórmula:
|
U.I. de factor |
= peso corporal (en |
x aumento deseado de |
x reciproca de la |
|
IX requeridas |
Kg) |
factor IX (%) o (UI/dl) |
recuperación observada |
Ejemplo: para una recuperación de 0,8 UI/dl, la fórmula es:
|
U.I. de factor |
= peso corporal (en |
x aumento deseado de x 1,3 U.I./Kg. |
|
IX requeridas |
Kg) |
factor IX (%) o (UI/dl) |
La cantidad que se debe administrar y la frecuencia de administración se debe orientar siempre a la eficacia clínica de cada caso en particular.
En el caso de los episodios hemorrágicos siguientes, la actividad de factor IX no debe dejarse caer por debajo de los niveles de actividad de plasma dados (en % de normal o en UI/dl) en el periodo correspondiente. Puede emplearse la siguiente tabla como guía para la dosificación en episodios hemorrágicos y cirugía:
|
Grado de hemorragia / tipo de |
Nivel de Factor IX |
Frecuencia de dosis (horas) / |
|
proceso quirúrgico |
requerido (%) o (IU/dl) |
Duración de terapia (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente, |
O 1 o <N |
Repetir cada 24 horas. Al menos 1 día, |
|
hemorragia muscular u oral |
hasta que el episodio hemorrágico según indique el dolor se resuelva o se logre la curación | |
|
Hemartrosis más extensa, |
O 1 o en |
Repetir la perfusión cada 24 horas |
|
hemorragia muscular o |
durante 3 - 4 días o más, hasta que | |
|
hematoma |
cese el dolor y la incapacidad. | |
|
Hemorragias con riesgo vital |
o o i o |
Repetir la perfusión cada 8 a 24 horas hasta superar el peligro |
|
Cirugía | ||
|
Menor: |
o 1 o en |
Cada 24 horas, al menos un día, hasta |
|
Incluyendo extracción dental |
que se logre la curación. | |
|
Mayor |
00 0 1 o o |
Repetir la perfusión cada 8-24 horas |
|
(pre y post operatorio) |
hasta la curación adecuada de la herida, luego seguir la terapia durante al menos otros 7 días para mantener la actividad de factor IX entre un 30% y un 60% (UI/dl) |
Profilaxis
Se puede administrar BeneFIX para la profilaxis de larga duración, frente a hemorragias en pacientes con hemofilia B grave. En un estudio clínico para profilaxis secundaria rutinaria, la dosis media para pacientes previamente tratados (PTP) fue 40 UI/kg (rango entre 13 y 78 UI/kg) en intervalos de 3 a 4 días.
En algunos casos, especialmente en los pacientes más jóvenes, se pueden precisar intervalos de administración más cortos o dosis más altas.
Población pediátrica
Hay poca documentación sobre el tratamiento a demanda y la cirugía en pacientes pediátricos menores de 6 años tratados con BeneFIX.
La dosis media (± desviación estándar) para la profilaxis fue de 63,7 (±19,1) UI/kg en intervalos de 3 a 7 días. Es posible que en pacientes más jóvenes se necesiten dosis más elevadas o intervalos más cortos entre las dosis. El consumo de FIX para la profilaxis rutinaria en 22 pacientes evaluables fue de 4.607 (± 1.849) UI/kg al año y 378 (±152) UI/kg al mes.
Se debe realizar un control estricto de la actividad de factor IX en plasma según esté indicado clínicamente, así como calcular los parámetros farmacocinéticos tales como la recuperación y la vida media, con objeto de ajustar la dosis adecuadamente.
Población de edad avanzada
En los estudios de BeneFIX no se incluyeron suficientes sujetos de 65 años de edad y mayores, que permitiera determinar si respondían de forma diferente que los sujetos más jóvenes. Al igual que con cualquier paciente que recibe BeneFIX, se debe individualizar la selección de la dosis para un paciente de edad avanzada.
Forma de administración
BeneFIX se administra en perfusión intravenosa tras la reconstitución del polvo liofilizado para solución inyectable con solución de cloruro de sodio al 0,234% (ver sección 6.6).
BeneFIX debe administrarse a una velocidad de perfusión lenta. En la mayoría de los casos, se utilizó una velocidad de perfusión de hasta 4 ml por minuto. La velocidad de administración deberá determinarla el grado de comodidad del paciente.
Si se produce alguna sospecha de reacción de hipersensibilidad que se considere relacionada con la administración de BeneFIX, se debe reducir la velocidad de perfusión o se debe detener la perfusión (ver secciones 4.4 y 4.8).
Aglutinación de hematíes en el tubo o jeringa
Se han comunicado casos de aglutinación de hematíes en el tubo o en la jeringa durante la administración de BeneFIX. No se ha notificado ninguna reacción adversa en relación con esta observación. Para reducir al mínimo la posibilidad de aglutinación, es importante limitar la cantidad de sangre que entra en el tubo. La sangre no debe entrar en la jeringa. En caso de observar aglutinación de hematíes en el tubo o en la jeringa, se debe desechar todo este material (tubo, jeringa y solución de BeneFIX) y se debe reanudar la administración con un envase nuevo.
Perfusión continua
No se ha aprobado la administración mediante perfusión continua y no se recomienda (ver también las secciones 4.4 y 6.6).
Para las instrucciones sobre la reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1. Antecedentes conocidos de reacción alérgica a las proteínas de hámster.
4.4 Advertencias y precauciones especiales de empleo Hipersensibilidad
Es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico con BeneFIX. El producto contiene trazas de proteínas de hámster. Con productos de factor IX, incluyendo BeneFIX se han producido reacciones anafilácticas/anafilactoides que potencialmente pueden poner en riesgo la vida. Si se producen síntomas de hipersensibilidad, se debe aconsejar a los pacientes que suspendan inmediatamente el uso del medicamento y que consulten al médico. Los pacientes deben ser informados acerca de los signos iniciales de las reacciones de hipersensibilidad, que incluyen dificultad de respirar, respiración entrecortada, tumefacción, urticaria, urticaria generalizada, prurito, opresión en el pecho, broncoespasmo, laringoespasmo, jadeo, hipotensión, visión borrosa y anafilaxia.
En algunos casos, estas reacciones han progresado hasta anafilaxis grave. En el caso de shock, se deben observar los estándares médicos actuales para tratamientos de shock. En el caso de reacciones alérgicas graves, deben considerarse medidas hemostáticas alternativas.
Inhibidores
La aparición de inhibidores es un evento poco frecuente en pacientes previamente tratados (PTP) que reciben productos que contienen factor IX. Dado que durante los estudios clínicos un PTP con BeneFIX desarrolló un inhibidor de baja respuesta, clínicamente relevante, y que la experiencia acerca de la antigenicidad con factor IX recombinante es aún limitada, debe vigilarse cuidadosamente a los pacientes tratados con BeneFIX por el desarrollo de inhibidores del factor IX, que deben ser titulados en Unidades Bethesda mediante test biológicos adecuados.
Hay informes en la literatura que muestran una correlación entre la aparición de un inhibidor de factor IX y reacciones alérgicas. Por consiguiente, los pacientes que experimentan reacciones alérgicas deben ser evaluados por si existe presencia de inhibidor. Debe advertirse que los pacientes que desarrollan inhibidores de factor IX pueden tener un riesgo aumentado de anafilaxis con la estimulación subsiguiente con factor IX. La información preliminar indica que podría haber una relación entre la presencia de mutaciones de deleción importantes en el gen del factor IX de un paciente y un aumento del riesgo de formación de inhibidores y de reacciones de hipersensibilidad agudas. Los pacientes que se sabe que tienen mutaciones de deleción importantes en el gen del factor IX deberán ser observados de cerca por si hubiera signos y síntomas de reacciones de hipersensibilidad agudas, especialmente durante las fases tempranas de exposición inicial al producto.
Dado el riesgo de reacciones alérgicas con concentrados de factor IX, las administraciones iniciales de factor IX deben, según el juicio del médico que hace el tratamiento, realizarse bajo supervisión médica donde pueda proporcionarse un cuidado médico adecuado para las reacciones alérgicas.
Trombosis
Aunque BeneFIX contiene únicamente factor IX, el riesgo de trombosis y coagulación intravascular diseminada (CID) debe tenerse en cuenta. Como el uso de concentrados de complejo de factor IX ha estado históricamente asociado con el desarrollo de complicaciones tromboembólicas, el uso de productos que contienen factor IX puede ser potencialmente peligroso en pacientes con signos de fibrinolisis y en pacientes con coagulación intravascular diseminada (CID). Debido al riesgo potencial de complicaciones trombóticas, cuando el producto se administre a pacientes con enfermedad hepática, a pacientes en post-operatorio, a recién nacidos o a pacientes con riesgo de fenómenos trombóticos o CID, debe iniciarse un seguimiento clínico con los ensayos biológicos apropiados para detectar signos precoces de trombosis y coagulopatía de consumo. En cada una de estas situaciones, debe evaluarse el beneficio del tratamiento con BeneFIX, frente al riesgo de esas complicaciones.
No se ha establecido la seguridad y eficacia de la administración de BeneFIX mediante perfusión continua (ver también las secciones 4.2 y 4.8). Tras la comercialización, se han notificado casos de acontecimientos trombóticos, incluyendo el síndrome de la vena cava superior (SVC) potencialmente mortal en neonatos críticamente enfermos, mientras recibían BeneFIX en perfusión continua a través de un catéter venoso central (ver también la sección 4.8).
Acontecimientos cardiovasculares
En los pacientes con factores de riesgo cardiovascular, el tratamiento sustitutivo con FIX puede aumentar el riesgo cardiovascular.
Síndrome nefrótico
Se han notificado casos de síndrome nefrótico después de intentar la inducción de inmunotolerancia en pacientes con hemofilia B con inhibidores de factor IX y antecedentes de reacciones alérgicas. No se ha establecido la seguridad y eficacia del uso de BeneFIX para la inducción de inmunotolerancia.
Poblaciones especiales
En los ensayos clínicos, no se han obtenido datos suficientes sobre el tratamiento de los pacientes no tratados previamente (PUPs) con BeneFIX.
Registro de uso
Se recomienda encarecidamente que, cada vez que se administre BeneFIX a un paciente, se tome nota del nombre y del número de lote del producto con el fin de mantener un vínculo entre el paciente y el lote del medicamento. Los pacientes pueden pegar una de las etiquetas despegables que se encuentran en el vial, para dejar constancia del número de lote en su diario o para notificar cualquier reacción adversa.
4.5 Interacción con otros medicamentos y otras formas de interacción
No se han notificado interacciones de los productos de factor IX de la coagulación humana (ADNr) con otros medicamentos.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción en animales con factor IX. Dados los raros casos de hemofilia B en mujeres, no se dispone de experiencia sobre el uso del factor IX durante el embarazo y la lactancia. Por lo tanto, la administración de factor IX durante el embarazo y la lactancia, debe estar claramente indicada.
No se ha establecido el efecto de BeneFIX en la fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de BeneFIX sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Se han observado hipersensibilidad o reacciones alérgicas (que pueden consistir en angioedema, escozor y picor en el lugar de perfusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habón urticarial, hipotensión, letargia, náuseas, inquietud, taquicardia, opresión en el pecho, hormigueo, vómitos, sibilancias) y, en algunos casos, pueden progresar a anafilaxia grave (incluido shock). En algunos casos, estas reacciones han progresado a anafilaxia grave y se han producido en estrecha asociación en el tiempo con la aparición de anticuerpos inhibidores del factor IX (ver también sección 4.4). Se ha descrito el síndrome nefrótico después de intentar la inducción de la inmunotolerancia en pacientes afectados con hemofilia B y con anticuerpos inhibidores del factor IX, y con antecedentes de reacción alérgica.
En muy raras ocasiones, se ha observado la aparición de anticuerpos frente a proteínas del hámster con reacciones de hipersensibilidad relacionadas.
Los pacientes que padecen hemofilia B pueden presentar anticuerpos neutralizantes (inhibidores) frente al factor IX. Si se producen tales inhibidores, el evento se manifestará como una respuesta clínica insuficiente. En esos casos, se recomienda ponerse en contacto con un centro especializado en hemofilia.
Existe el posible riesgo de sufrir episodios tromboembólicos después de la administración de productos del factor IX; ver sección 4.4.
Tabla de reacciones adversas
En la siguiente tabla se presenta la clasificación de órganos del sistema MedDRA (COS y nivel de términos preferidos). Se han evaluado las frecuencias según la siguiente clasificación: muy frecuentes (> 1/10); frecuentes (> 1/100 a < 1/10); poco frecuentes (> 1/1.000 a < 1/100), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). En la tabla se enumeran las reacciones adversas notificadas en los ensayos clínicos de pacientes previamente tratados e identificados en el uso poscomercialización. Las frecuencias se basan en los acontecimientos adversos surgidos durante el tratamiento debidos a todas las causas en ensayos clínicos agrupados con 224 sujetos.
Dentro de cada grupo de frecuencia, las reacciones adversas se enumeran en orden decreciente de gravedad.
|
Sistema de clasificación de órganos |
Muy frecuentes >1/10 |
Frecuentes > 1/100 a < 1/10 |
Poco frecuentes > 1/1.000 a < 1/100 |
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles) |
|
Infecciones e infestaciones |
Celulitis en el lugar de perfusión3 | |||
|
Trastornos de la sangre y del sistema linfático |
Inhibición del factor IXb | |||
|
Trastornos del sistema inmunitario |
Hipersensibilidadc |
Reacción anafiláctica* | ||
|
Trastornos del sistema nervioso |
Cefalead |
Mareo; disgeusia |
Somnolencia; temblores | |
|
Trastornos oculares |
Alteración visuale | |||
|
Trastornos cardiacos |
Taquicardiaf | |||
|
Trastornos vasculares |
Flebitis; rubefaccióng |
Hipotensión11 |
Síndrome de la vena cava superior^*; trombosis venosa profunda*; trombosis*; tromboflebitis* | |
|
Trastornos respiratorios,torácicos y mediastínicos |
Tosj | |||
|
Trastornos gastrointestinales |
Vómitos; náuseas | |||
|
Trastornos de la piel y del tejido subcutáneo |
Erupción cutánea ; urticaria | |||
|
Trastornos renales y urinarios |
Infarto renall | |||
|
Trastornos generales y alteraciones en el lugar de administración |
Pirexia |
Molestia torácica13; reacción en el lugar de perfusión; dolor en el lugar de perfusión |
Respuesta terapéutica insuficiente* | |
|
Exploraciones complementarias |
Recuperación insuficiente del factor IXp, * | |||
|
* Reacción adversa al medicamento identificada en el periodo poscomercialización. a Incluida celulitis. b Formación de inhibidores transitorios de bajo título. c Incluidos hipersensibilidad al fármaco, angioedema, broncoespasmo, sibilancias, disnea y laringoespasmo. d Incluidas migraña, cefalea sinusal. | ||||
|
Sistema de |
Muy |
Frecuentes |
Poco |
Frecuencia no |
|
clasificación de |
frecuentes |
> 1/100 |
frecuentes |
conocida |
|
órganos |
>1/10 |
a < 1/10 |
> 1/1.000 a |
(no puede estimarse |
|
< 1/100 |
a partir de los datos | |||
|
disponibles) |
e Incluidos escotoma centelleante y visión borrosa. f Incluidos aumento de la frecuencia cardiaca, taquicardia sinusal. g Incluidos sofocos, sensación de calor, calor en la piel. h Incluida disminución de la presión arterial.
i Síndrome de la vena cava superior (VCS) en recién nacidos críticamente enfermos, mientras recibián una perfusión continua de BeneFIX mediante un catéter venoso central. j Incluida tos productiva.
k Incluidas erupción cutánea macular, erupción cutánea papular, erupción cutánea maculopapular.
l Aparecido en un paciente con un resultado positivo para los anticuerpos de la hepatitis C, 12 días después de recibir una dosis de BeneFIX por un episodio hemorrágico. m Incluidos dolor en el lugar de perfusión, molestia en el lugar de perfusión. n Incluidos prurito en el lugar de perfusión, eritema en el lugar de perfusión. o Incluidos dolor torácico y tirantez de pecho.
p Este es un término textual. No se recuperó ningún término preferido de MedDRA 17.1._
Descripción de reacciones adversas seleccionadas
Hipersensibilidad / reacciones alérgicas
Si aparece cualquier sospecha de reacción adversa que se considere relacionada con la administración de BeneFIX, ver secciones 4.2 y 4.4.
Desarrollo de inhibidor
Se detectó la aparición de un inhibidor de baja respuesta, clínicamente relevante, en 1 de 65 pacientes tratados con BeneFIX que habían recibido previamente productos derivados de plasma (incluyendo 9 pacientes que participaban únicamente en el estudio quirúrgico). Este paciente pudo continuar el tratamiento con BeneFIX sin aumento anamnéstico del inhibidor ni anafilaxia (ver sección 4.4).
Población pediátrica
Es posible que se den reacciones alérgicas en niños con una mayor frecuencia que en adultos.
No existen datos suficientes para dar información sobre la incidencia de inhibidores en PUPs (ver sección 5.1).
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
No se han notificado síntomas de sobredosis con los productos de factor IX de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágico, factor IX de coagulación sanguínea; Código ATC: B02BD09
Mecanismo de acción
BeneFIX contiene de factor IX de coagulación recombinante, (nonacog alfa). El factor IX de coagulación recombinante es una glucoproteína de cadena única con un peso molecular de 55.000 Daltons aproximadamente, que forma parte de la familia de serina proteasas de los factores de la coagulación dependientes de la vitamina K. El factor IX de coagulación recombinante es una proteína recombinante basada en el ADN, de carácter terapéutico que posee características estructurales y funcionales comparables a las del factor IX endógeno. El factor IX es activado por el complejo de factor VII/factor tisular en la vía extrínseca, así como por el factor XIa en la vía intrínseca de la coagulación. El factor IX activado, en combinación con el factor VIII activado, activa al factor X. El resultado final es la conversión de la protrombina en trombina. La trombina convierte luego el fibrinógeno en fibrina, con lo que puede formarse un coágulo. La actividad del factor IX es nula o está muy reducida en los pacientes con hemofilia B, y puede ser necesario el tratamiento de reposición.
Efectos farmacodinámicos
La hemofilia B es una alteración hereditaria de la coagulación sanguínea ligada al sexo y que es debida a la disminución de los niveles de factor IX que da como resultado una hemorragia profusa en las articulaciones, músculos u órganos internos, bien de forma espontánea o bien como resultado de un trauma accidental o quirúrgico. Mediante terapia de sustitución, los niveles plasmáticos de factor IX aumentan, consiguiendo una corrección temporal de la deficiencia de factor y una corrección de las tendencias hemorrágicas.
Población pediátrica
El análisis de eficacia del estudio 3090A1-301-WW se basó en 22 sujetos pediátricos evaluables en régimen de profilaxis, incluidos 4 pacientes a demanda que en poco tiempo cambiaron a profilaxis. Dos pacientes se sometieron a operaciones quirúrgicas (circuncisión e inserción de un sistema Port-a-cath). Los análisis de seguridad de 25 pacientes evaluables mostraron el perfil de seguridad esperado. El único efecto adverso grave reportado relacionado con BeneFIX se dio en el único PUP participante, que experimentó hipersensibilidad y desarrollo de inhibidor.
En dos estudios abiertos se comprobó que BeneFIX puede ser administrado de forma segura con una dosis de 100 UI/kg una vez por semana. Sin embargo, la vida media del medicamento (ver sección 5.2) y los escasos datos del estudio farmacocinético con el régimen de una vez por semana no permiten en general recomendar este régimen para la profilaxis a largo plazo en pacientes con hemofilia B grave.
5.2 Propiedades farmacocinéticas
En un estudio farmacocinético aleatorizado y cruzado se comprobó que BeneFIX reconstituido al 0,234% con cloruro de sodio es farmacocinéticamente equivalente al BeneFIX previamente comercializado (reconstituido con agua esterilizada) en 24 pacientes tratados previamente (> 12 años) con una dosis de 75 UI/kg. Además, los parámetros farmacocinéticos fueron monitorizados en 23 de los mismos pacientes después de la administración repetida de BeneFIX durante seis meses y se comprobó que no había cambios en comparación con los obtenidos en la evaluación inicial. Un resumen de los datos farmacocinéticos se muestra en la Tabla 1.
|
Tabla 1. Valores calculados de los parámetros farmacocinéticos de BeneFIX (75 UI/kg) basales y al mes 6 en pacientes con hemofilia B tratados previamente | ||
|
Parámetro |
Valor basal n = 24 |
Mes 6 n = 23 |
|
Media ± DE |
Media ± DE | |
|
Cm4x (UI/dl) |
54,5 ± 15,0 |
57,3 ± 13,2 |
|
AUC» (UI-h/dl) |
940 ± 237 |
923 ± 205 |
|
11/2 (h) |
22,4 ± 5,3 |
23,8 ± 6,5 |
|
CL (ml/h/kg) |
8,47 ± 2,12 |
8,54 ± 2,04 |
|
Recuperación |
0,73 ± 0,20 |
0,76 ± 0,18 |
|
(UI/dl por UI/kg) | ||
Abreviaturas: AUCm = área bajo la curva de concentración plasmática y tiempo desde tiempo cero hasta infinito; Cmáx = concentración máxima; t1/2 = vida media de eliminación del plasma; CL = aclaramiento; DE = desviación estándar.
Se creó un modelo farmacocinético de población con datos recopilados en 73 pacientes de edades comprendidas entre los 7 meses y los 60 años. Los parámetros calculados mediante el modelo final de dos compartimentos se muestran en la Tabla 2. Los bebés y los niños presentaron un aclaramiento más elevado, un volumen mayor de distribución, una vida media menor y una recuperación menor que los adolescentes y los adultos. La fase terminal no se ha cubierto explícitamente, debido a la falta de datos después de transcurridas 24 horas en sujetos pediátricos < 6 años de edad.
|
Tabla 2. Valor promedio (± DE) de parámetros farmacocinéticos en funciói individuales de Bayes a partir del análisis farmacocinético de la pob |
i de cálculos ación | ||||
|
Grupo de edad (años) |
Bebés <2 |
Niños pequeños 2 a < 6 |
Niños 6 a < 12 |
Adolescentes 12 a < 18 |
Adultos 18 a 60 |
|
Número de sujetos |
7 |
16 |
1 |
19 |
30 |
|
Aclaramiento (ml/h/kg) |
13,1 ± 2,1 |
13,1 ± 2,9 |
15,5 |
9,2 ± 2,3 |
8,0 ± 0,6 |
|
Vss (ml/kg) |
252 ± 35 |
257 ± 25 |
303 |
234 ± 49 |
225 ± 59 |
|
Vida media de eliminación (h) |
15,6 ± 1,2 |
16,7 ± 1,9 |
16,3 |
21,5 ± 5,0 |
23,9 ± 4,5 |
|
Recuperación (UI/dl por UI/kg) |
0,61 ± 0,10 |
0,60 ±0,08 |
0,47 |
0,69 ± 0,16 |
0,74 ± 0,20 |
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de genotoxicidad.
No se han realizado investigaciones sobre carcinogenicidad, alteraciones de la fertilidad ni desarrollo fetal.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo Sacarosa Glicina L-Histidina Polisorbato 80
Disolvente
Solución de cloruro de sodio
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros. Sólo deben utilizarse los sistemas de perfusión que se suministran. El tratamiento puede fallar como consecuencia de la adsorción del factor IX humano de la coagulación a las superficies internas de algunos sistemas de perfusión.
6.3 Período de validez 2 años.
El producto reconstituido no contiene conservante y debe ser utilizado inmediatamente, o no más tarde de 3 horas después de la reconstitución. Se ha demostrado su estabilidad química y física durante 3 horas a temperaturas de hasta 25°C.
6.4 Precauciones especiales de conservación
Conservar por debajo de 30 °C. No congelar.
6.5 Naturaleza y contenido del envase
BeneFIX 250 UI, 500 UI, 1000 UI, 2000 UI o 3000 UI polvo y disolvente para solución inyectable Un vial de 10 ml (de vidrio tipo I) con BeneFIX 250 UI, 500 UI, 1000 UI, 2000 UI o 3000 UI de polvo, con un tapón (de clorobutilo) y una cápsula “flip-off’ (de aluminio) y 5 ml de disolvente transparente e incoloro en una jeringa precargada (de vidrio tipo I) con un tapón del émbolo (de bromobutilo), un tapón-filtro (de bromobutilo), un adaptador estéril para la reconstitución, un sistema de perfusión estéril, dos torundas con alcohol, un apósito adhesivo y una compresa de gasa.
6.6 Precauciones especiales de eliminación y otras manipulaciones
BeneFIX se administra mediante perfusión intravenosa después de reconstituir el polvo liofilizado para inyección con el disolvente (solución de cloruro de sodio al 0,234 % p/v) proporcionado en la jeringa precargada (ver sección 3 del prospecto para las instrucciones de reconstitución).
Una vez reconstituido, BeneFIX contiene polisorbato 80, que aumenta el índice de extracción de di-(2-etilhexil) ftalato (DEHP) del cloruro de polivinilo (PVC). Esto se debe tener en cuenta durante la preparación y administración de BeneFIX. Es importante que se sigan estrictamente las recomendaciones indicadas en la sección 4.2.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con las normativas locales.
Dado que no se ha evaluado el uso de BeneFIX mediante perfusión continua, no debe mezclarse BeneFIX con soluciones para perfusión ni administrarse en un goteo.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
8. NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/047/004
EU/1/97/047/005
EU/1/97/047/006
EU/1/97/047/007
EU/1/97/047/008
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 27 de Agosto de 1997 Fecha de la última renovación: 20 de julio de 2012
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea del Medicamento http://www.ema.europa.eu
A. FABRICANTE(S) DEL (DE LOS) PRINCIPIO(S) ACTIVO(S) BIOLÓGICO(S) Y FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE(S) DEL (DE LOS) PRINCIPIO(S) ACTIVO(S) BIOLÓGICO(S) Y FABRICANTE(S) RESPONSABLE(S) DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante del principio activo biológico Wyeth BioPharma
Division of Wyeth Pharmaceuticals Inc., a subsidiary of Pfizer Inc.
One Burtt Road Andover MA 01810 EEUU
Nombre y dirección del fabricante responsable de la liberación de los lotes Wyeth Farma, S.A.
Autovía del Norte. A-1, Km 23. Desvío Algete, Km 1, 28700, San Sebastián de los Reyes, Madrid España
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo
acordado en la versión del PGR incluido en el Módulo 1.8.2. de la Autorización de
Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR ESTUCHE
|
1. |
NOMBRE DEL MEDICAMENTO |
BeneFIX 250 UI polvo y disolvente para solución inyectable BeneFIX 500 UI polvo y disolvente para solución inyectable BeneFIX 1000 UI polvo y disolvente para solución inyectable BeneFIX 2000 UI polvo y disolvente para solución inyectable BeneFIX 3000 UI polvo y disolvente para solución inyectable Nonacog alfa (factor IX de coagulación recombinante)
2. PRINCIPIO(S) ACTIVO(S)_
1 vial: 250 UI de nonacog alfa (aprox. 50 UI/ml tras la reconstitución).
1 vial: 500 UI de nonacog alfa (aprox. 100 UI/ml tras la reconstitución).
1 vial: 1000 UI de nonacog alfa (aprox. 200 UI/ml tras la reconstitución).
1 vial: 2000 UI de nonacog alfa (aprox. 400 UI/ml tras la reconstitución).
1 vial: 3000 UI de nonacog alfa (aprox. 600 UI/ml tras la reconstitución).
3. LISTA DE EXCIPIENTES
Sacarosa, glicina, L-histidina, cloruro de sodio, polisorbato 80.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo y disolvente para solución inyectable
1 vial con 250 UI de nonacog alfa 1 vial con 500 UI de nonacog alfa 1 vial con 1000 UI de nonacog alfa 1 vial con 2000 UI de nonacog alfa 1 vial con 3000 UI de nonacog alfa
1 jeringa precargada con 5 ml de disolvente 1 adaptador estéril para la reconstitución
1 sistema de perfusión estéril
2 torundas con alcohol 1 apósito adhesivo
1 compresa de gasa
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Vía intravenosa. Administrar en un único uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
Para la reconstitución, emplear sólo la jeringa precargada de disolvente incluida en el estuche.
8. FECHA DE CADUCIDAD
CAD
Utilizar inmediatamente o en un plazo de 3 horas desde la reconstitución.
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar por debajo de 30 °C. No congelar.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/97/047/004
EU/1/97/047/005
EU/1/97/047/006
EU/1/97/047/007
EU/1/97/047/008
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
BeneFIX 250 BeneFIX 500 BeneFIX 1000 BeneFIX 2000 BeneFIX 3000
ETIQUETA DEL VIAL
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
BeneFIX 250 UI polvo para solución inyectable
BeneFIX 500 UI polvo para solución inyectable
BeneFIX 1000 UI polvo para solución inyectable
BeneFIX 2000 UI polvo para solución inyectable
BeneFIX 3000 UI polvo para solución inyectable
Nonacog alfa (factor IX de coagulación recombinante)
Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
Inyección de un solo uso.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
Ver parte frontal de la etiqueta (Lote, Cad)
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
250 UI 500 UI 1000 UI 2000 UI 3000 UI
6. OTROS
Conservar por debajo de 30 °C. No congelar.
Para la reconstitución, emplear sólo la jeringa precargada incluida en el estuche.
ETIQUETA DE LA JERINGA PRECARGADA DE DISOLVENTE
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Disolvente para BeneFIX Vía intravenosa.
2. FORMA DE ADMINISTRACIÓN
Usar el contenido completo.
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
Pfizer Limited
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
Contiene 5 ml de solución de cloruro de sodio al 0,234% para inyección.
6. OTROS
B. PROSPECTO
Prospecto: información para el usuario
BeneFIX 250 UI polvo y disolvente para solución inyectable BeneFIX 500 UI polvo y disolvente para solución inyectable BeneFIX 1000 UI polvo y disolvente para solución inyectable BeneFIX 2000 UI polvo y disolvente para solución inyectable BeneFIX 3000 UI polvo y disolvente para solución inyectable Nonacog alfa (factor IX de coagulación recombinante)
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es BeneFIX y para qué se utiliza
2. Qué necesita saber antes de empezar a usar BeneFIX
3. Cómo usar BeneFIX
4. Posibles efectos adversos
5. Conservación de BeneFIX
6. Contenido del envase e información adicional
1. Qué es BeneFIX y para qué se utiliza
BeneFIX es un producto de factor IX de la coagulación inyectable obtenido por tecnología de ADN recombinante. La sustancia activa de BeneFIX es nonacog alfa. Las personas que nacen con hemofilia B (enfermedad de Christmas) no tienen suficiente factor IX para controlar las hemorragias. BeneFIX, actúa reponiendo el factor IX en los pacientes con hemofilia B para permitir que su sangre pueda coagularse.
BeneFIX se utiliza para el tratamiento y prevención de hemorragias en pacientes con hemofilia B (deficiencia de factor IX congénita) en todos los grupos de edad.
2. Qué necesita saber antes de empezar a usar BeneFIX No use BeneFIX:
- Si es alérgico a nonacog alfa o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- Si es alérgico a las proteínas de hámster.
Advertencias y precauciones
- Consulte a su médico inmediatamente si su hemorragia no se detiene como cabría esperar.
Se pueden producir reacciones alérgicas. El producto puede contener trazas de proteínas de hámster (ver No use BeneFIX). Con productos de factor IX, incluyendo BeneFIX se han producido reacciones anafilácticas (reacciones alérgicas graves) que potencialmente pueden poner en riesgo la vida. Los signos iniciales de las reacciones de alergia incluyen dificultad para respirar, respiración entrecortada, tumefacción o hinchazón, habón urticarial, picor, urticaria generalizada, opresión en el pecho, jadeo, presión arterial baja, visión borrosa y anafilaxis (reacción alérgica grave que puede causar dificultad al tragar y/o al respirar, enrojecimiento o hinchazón de la cara y/o de las manos).
Si se produce una reacción de tipo alérgico o anafiláctico detenga inmediatamente la perfusión y contacte con un médico o busque inmediatamente ayuda médica de urgencia.
En caso de reacciones alérgicas graves, debe considerarse la utilización de una terapia alternativa.
La aparición de anticuerpos neutralizantes (inhibidores) es un acontecimiento poco frecuente en pacientes que han recibido tratamiento previo con productos que contienen factor IX. No obstante, como con todos los productos que contienen factor IX, debe ser vigilado cuidadosamente por si desarrolla inhibidores del factor IX mientras está en tratamiento con BeneFIX.
Estudios han mostrado una relación entre la aparición de un inhibidor del factor IX y reacciones alérgicas. Por ello, si experimenta reacciones alérgicas como las descritas arriba, deberá ser examinado para determinar la presencia de un inhibidor. Se debe tener en cuenta que los pacientes con un inhibidor del factor IX pueden presentar un riesgo aumentado de anafilaxis en un futuro tratamiento con BeneFIX.
La producción de factor IX en el organismo está controlada por el gen del factor IX. Los pacientes con mutaciones específicas del gen del factor IX como una delección importante, podrán tener más probabilidades de desarrollar un inhibidor del factor IX y/o padecer reacciones alérgicas. Por lo tanto, si se sabe que tiene dicha mutación, su médico puede monitorizarle más de cerca por si hubiera signos de una reacción alérgica, especialmente cuando empiece a tomar BeneFIX por primera vez.
Dado el riesgo de reacciones alérgicas con factor IX, las administraciones iniciales de BeneFIX deben realizarse bajo supervisión médica, donde pueda proporcionarse un cuidado médico adecuado para las reacciones alérgicas.
Aún en ausencia de inhibidor del factor IX, pueden ser necesarias dosis mayores de BeneFIX que las que se precisan para otros de factor IX derivados de plasma que haya recibido anteriormente. Por consiguiente, se tiene que realizar una estrecha monitorización de la actividad de factor IX en plasma (que mide la capacidad de su sangre para formar coágulos) con objeto de ajustar la dosis adecuadamente. Si la hemorragia no se controla con la dosis recomendada contacte con su médico.
Si sufre de enfermedad del hígado o del corazón o si ha sufrido recientemente una intervención quirúrgica, existe un mayor riesgo de complicaciones de coagulación de la sangre.
Se ha comunicado una alteración de los riñones (síndrome nefrótico) tras la administración de dosis altas de factor IX derivado del plasma en pacientes con hemofilia B con inhibidores del factor IX y antecedentes de reacciones alérgicas.
No se han obtenido datos suficientes de los ensayos clínicos de pacientes no tratados previamente (pacientes que nunca han recibido una perfusión con factor IX) con BeneFIX.
- Se recomienda que cada vez que use BeneFIX registre el nombre y el número de lote del
producto. Puede usar una de las etiquetas despegables que se encuentran en el vial para dejar constancia del número de lote en su diario o para notificar cualquier reacción adversa.
Uso de BeneFIX con otros medicamentos
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Embarazo, lactancia y fertilidad
Si está usted embarazada o en período de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, sólo debe recibir BeneFIX por indicación específica de su médico. Se desconoce si BeneFIX puede causar daño al feto cuando se administra a mujeres embarazadas. Si está en período de lactancia o se queda embarazada, el médico puede aconsejarle que suspenda el tratamiento con BeneFIX.
Consulte a su médico o farmacéutico antes de utilizar este medicamento.
Conducción y uso de máquinas
La influencia de BeneFIX sobre la capacidad para conducir y utilizar máquinas es nula.
3. Cómo usar BeneFIX
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte de nuevo a su médico o farmacéutico.
Su médico decidirá la dosis de BeneFIX que recibirá. Esta dosis y su duración dependerán de sus necesidades individuales de tratamiento sustitutivo con factor IX y de la rapidez con que su organismo utilice el factor IX lo que será comprobado regularmente. Podrá apreciar una diferencia en la dosis que recibe si cambia de un producto de factor IX derivado de plasma a BeneFIX.
Su médico puede modificar la dosis de BeneFIX que reciba a lo largo del tratamiento.
Reconstitución y administración
Las instrucciones facilitadas a continuación son una guía para la reconstitución y administración de BeneFIX. Los pacientes deben seguir las instrucciones de venopunción específicas indicadas por su médico.
BeneFIX se administra mediante perfusión intravenosa (IV) después de la reconstitución del polvo para inyección con el disolvente incluido en la jeringa precargada (solución de cloruro de sodio).
Lávese siempre las manos antes de realizar los procedimientos siguientes. Durante el procedimiento de reconstitución debe seguirse una técnica aséptica (que significa limpio y libre de gérmenes).
Reconstitución:
BeneFIX se administrará por perfusión intravenosa (IV) después de la reconstitución con disolvente estéril para inyección.
1. Permita que el vial liofilizado de BeneFIX y la jeringa precargada de disolvente alcancen la temperatura ambiente.
Retire la cápsula de cierre del vial de BeneFIX para que quede visible la parte central del tapón de goma.
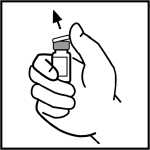
3. Limpie la parte superior del vial con la torunda empapada en alcohol que se proporciona o utilice otra solución antiséptica y espere a que se seque. Después de limpiar, no toque el tapón de goma con su mano y evite que toque ninguna superficie.
4. Retire la tapa protectora del envase de plástico transparente del adaptador. No saque el adaptador del envase.
5. Coloque el vial sobre una superficie plana. Mientras sujeta el adaptador en su envase, colóquelo sobre el vial. Presiónelo firmemente hasta que el adaptador encaje en lo alto del vial, con la punta del adaptador penetrando en el tapón del vial.
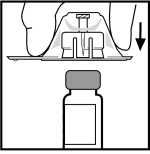
6. Retire el envase del adaptador y deséchelo.
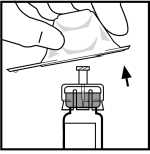
7. Encaje la barra del émbolo a la jeringa del disolvente empujándola y girándola firmemente.
8. Rompa la tapa de plástico resistente a la manipulación partiendo la perforación del capuchón. Esto se realiza doblando el capuchón hacia arriba y abajo hasta que se rompa la perforación. No toque el interior del capuchón ni la punta de la jeringa. El capuchón puede necesitar ser reemplazado (si BeneFIX reconstituido no es administrado inmediatamente), por tanto deséchelo colocándolo en su parte superior.
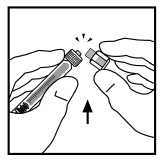
Coloque el vial en una superficie plana. Conecte la jeringa del disolvente al adaptador del vial insertando la punta de la jeringa en la apertura del adaptador mientras empuja y gira firmemente la jeringa en el sentido de las agujas del reloj hasta que encaje perfectamente.
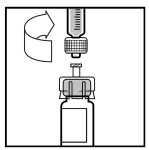
10.
Haga descender lentamente el émbolo para inyectar todo el disolvente en el vial de BeneFIX.
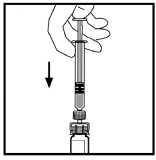
11.
Con la jeringa aún conectada al adaptador, gire suavemente el vial hasta que se disuelva el polvo.
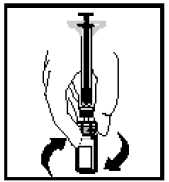
12.
La solución final debe ser examinada visualmente en busca de partículas finas antes de la administración. La solución debe aparecer transparente e incolora.
Nota: Si utiliza más de un vial de BeneFIX para perfusión, cada vial debe ser reconstituido de acuerdo con las instrucciones anteriores. La jeringa de disolvente deberá desecharse, dejando el adaptador del vial en su sitio y a continuación se utilizará una jeringa grande tipo Luer Lock (un dispositivo que conecta la jeringa al vial) para retirar el contenido reconstituido de cada vial individual.
13.
Gire el vial asegurándose de que el émbolo de la jeringa ha descendido por completo. Aspire lentamente toda la solución al interior de la jeringa.
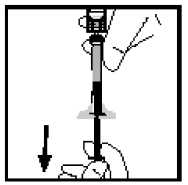
Libere la jeringa del adaptador empujando y girando lentamente la jeringa en el sentido de las agujas del reloj. Deseche el vial unido al adaptador.
Nota: Si la solución no va a ser utilizada inmediatamente, el capuchón debe ser reemplazado cuidadosamente. No toque la punta de la jeringa ni el interior del capuchón.
BeneFIX, debe administrarse inmediatamente después de su reconstitución, o bien dentro de las 3 horas siguientes. La solución reconstituida puede almacenarse a temperatura ambiente antes de la administración.
Administración (Inyección intravenosa):
BeneFIX debe administrarse mediante la jeringa precargada de disolvente que se suministra o mediante una jeringa de plástico desechable estéril tipo Luer Lock. Además, la solución debe extraerse del vial utilizando el adaptador del vial.
BeneFIX debe ser inyectado intravenosamente durante varios minutos. El médico le podrá cambiar la velocidad de la perfusión recomendada a fin de que le resulte más cómoda.
Se han notificado casos de aglutinación de hematíes en el tubo o en la jeringa durante la administración de BeneFIX. No se han comunicado reacciones adversas en relación con esta observación. Para reducir al mínimo la posibilidad de aglutinación, es importante limitar la cantidad de sangre que entra en el tubo. La sangre no debe entrar en la jeringa. Si se observa aglutinación de hematíes en el tubo o en la jeringa, deseche todo este material (tubo, jeringa y solución de BeneFIX) y reanude la administración con un nuevo envase.
Dado que el uso de BeneFIX en perfusión continua (gota a gota) no ha sido evaluado, BeneFIX no debe mezclarse con soluciones para perfusión o administrarse gota a gota.
Deseche la solución no utilizada, los viales vacíos y las agujas y jeringas utilizadas en un recipiente adecuado para eliminar desechos que puedan provocar lesiones, si no se manejan adecuadamente.
Si usa más BeneFIX del que debiera
Si se inyecta una cantidad de BeneFIX, superior a la recomendada por el médico, póngase en contacto con él inmediatamente.
Si interrumpe el tratamiento con BeneFIX
No interrumpa el tratamiento con BeneFIX sin consultar con su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermera.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede tener efectos adversos, aunque no todas las personas los sufran.
Reacciones de hipersensibilidad/alergia
Es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico con BeneFIX. Tales reacciones pueden consistir en hinchazón de la cara o la garganta, escozor y picor en el lugar de perfusión, escalofríos, rubefacción (enrojecimiento de la piel), prurito, dolor de cabeza, habón urticarial, disminución de la presión arterial, letargo, náuseas, inquietud, aumento de la frecuencia cardiaca, opresión en el pecho, hormigueo, vómitos, sibilancias (ruidos al respirar). En algunos casos, estas reacciones han progresado hasta convertirse en anafilaxias graves. Pueden aparecer reacciones alérgicas junto con el desarrollo de inhibidores del factor IX (ver también “Advertencias y precauciones”).
Estas reacciones pueden potencialmente poner en riesgo la vida. Si se producen reacciones anafilácticas/alérgicas, detenga inmediatamente la perfusión y contacte con un médico o busque inmediatamente ayuda médica de urgencia. El tratamiento requerido dependerá de la naturaleza y gravedad de los efectos adversos (ver también “Advertencias y precauciones”).
Desarrollo de inhibidores
Los pacientes con hemofilia B pueden desarrollar anticuerpos neutralizantes (inhibidores) al factor IX. Si esto ocurriera, un signo puede ser un incremento en la cantidad de BeneFIX que normalmente se requiere para tratar una hemorragia, y/o que la hemorragia continúe después del tratamiento. En estos casos, se recomienda contactar con un centro especializado en hemofilia. Su médico puede monitorizarle para controlar el desarrollo de inhibidores (ver “Advertencias y precauciones”).
Se ha notificado una alteración de los riñones tras la administración de dosis altas de factor IX derivado del plasma para la inducción de inmunotolerancia en pacientes con hemofilia B con inhibidores de factor IX y antecedentes de reacciones alérgicas (ver también “Advertencias y precauciones”).
Acontecimientos trombóticos
BeneFIX puede aumentar el riesgo de trombosis (coágulos de sangre anormales) en su organismo, si tiene factores de riesgo para el desarrollo de coágulos de sangre, incluyendo un catéter venoso permanente. Se han notificado casos graves de coágulos de sangre, incluyendo coágulos de sangre con riesgo para la vida en bebés críticamente enfermos, mientras recibían BeneFIX en perfusión continua a través de un catéter venoso central. También se han notificado casos de tromboflebitis periférica (dolor y enrojecimiento de las venas) y trombosis venosa profunda (coágulos de sangre en las extremidades); en la mayoría de estos casos, BeneFIX se había administrado por medio de perfusión continua, que es un método de administración no aprobado.
Efectos adversos muy frecuentes (pueden afectar a más de 1 de cada 10 personas)
• Dolor de cabeza
• Tos
• Fiebre
Efectos adversos frecuentes (pueden afectar hasta a 1 de cada 10 personas)
• Reacciones de hipersensibilidad o alergia
• Mareos, alteración del sentido del gusto
• Flebitis (dolor y enrojecimiento de las venas), rubefacción (enrojecimiento de la piel)
• Vómitos, náuseas
• Erupción cutánea, habón urticarial
• Molestia en el pecho (incluido dolor en el pecho)
• Reacción en el lugar de perfusión (incluidos picor y enrojecimiento en el lugar de perfusión), dolor y molestia en el lugar de perfusión
Efectos adversos poco frecuentes (pueden afectar hasta a 1 de cada 100 personas)
• Aparición de anticuerpos neutralizantes (inhibidores)
• Celulitis en el lugar de perfusión (dolor y enrojecimiento de la piel)
• Somnolencia, temblores
• Alteración de la visión (incluidas visión borrosa, aparición de manchas o luces)
• Aumento de la frecuencia cardiaca, disminución de la presión arterial
• Infarto renal (interrupción del suministro sanguíneo al riñón)
Efectos adversos de frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
• Reacción anafiláctica
• Acontecimientos trombóticos (coágulos de sangre anormales)
• Ausencia de respuesta al tratamiento (los episodios de sangrado no se pueden detener ni evitar) Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto.También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de BeneFIX
Mantener fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el estuche y en la etiqueta del vial. La fecha de caducidad es el último día del mes que se indica.
BeneFIX, debe conservarse por debajo de 30 °C y debe ser usado antes de la fecha de caducidad que aparece en la etiqueta.
No congelar el producto para evitar que se dañe la jeringa precargada.
El producto reconstituido debe utilizarse inmediatamente o en un plazo de 3 horas.
No utilice este medicamento si observa que la solución no es transparente o incolora.
Para la reconstitución sólo debe utilizarse la jeringa precargada que se incluye en el estuche. Para la administración, pueden emplearse otras jeringas desechables estériles.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional
- El principio activo es nonacog alfa (factor IX de coagulación recombinante). Cada vial de BeneFIX contiene nominalmente 250, 500, 1000, 2000 ó 3000 UI de nonacog alfa.
- Los demás ingredientes son sacarosa, glicina, L-histidina, polisorbato 80. Se incluye un disolvente para la reconstitución (solución de cloruro de sodio al 0,234%).
- Después de la reconstitución con el disolvente incluido, (solución de cloruro de sodio al 0,234%) cada vial contiene 50, 100, 200, 400 ó 600 UI respectivamente (ver tabla 1) de nonacog alfa por 1 ml de la solución para inyección preparada.
Tabla 1. Concentración de BeneFIX por ml de solución preparada
|
Cantidad de BeneFIX por Vial |
Cantidad de BeneFIX por 1 ml de solución preparada para inyección |
|
250 UI |
50 UI |
|
500 UI |
100 UI |
|
1000 UI |
200 UI |
|
2000 UI |
400 UI |
|
3000 UI |
600 UI |
Aspecto del producto y contenido del envase
BeneFIX se suministra como un polvo para inyección en un vial de vidrio y un disolvente suministrado en una jeringa precargada.
El pack contiene:
• un vial con BeneFIX 250, 500, 1000, 2000 o 3000 UI polvo
• una jeringa precargada con disolvente, 5 ml de solución de cloruro de sodio al 0,234% para la reconstitución
• un adaptador estéril para la reconstitución
• un sistema de perfusión estéril
• dos torundas con alcohol
• un apósito adhesivo
• una compresa de gasa
Titular de la autorización de comercialización
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Reino Unido
Responsable de la fabricación
Wyeth Farma S.A
Autovía del Norte.A-1, Km. 23. Desvío Algete, Km. 1, 28700 San Sebastián de los Reyes, Madrid España
Pueden solicitar más información respecto a este medicamento, dirigiéndose al representante local del titular de la autorización de comercialización.
Belgie /Belgique / Belgien
Pfizer S.A./N.V.
Tél/Tel: +32 (0)2 554 62 11
Lietuva
Pfizer Luxembourg SARL fili Lietuvoje
Tel. +3705 2514000
Bt^rapna
n$au3ep HroKceMÓypr CAPH, KaoH Buarapua
Tea.: +359 2 970 4333
Luxembourg/Luxemburg
Pfizer S.A.
Tél/Tel: +32 (0)2 554 62 11
|
Ceská Republika Pfizer PFE, spol. s.r.o. Tel: +420-283-004-111 |
Magyarország Pfizer Kft. Tel.: + 36 1 488 37 00 |
|
Danmark |
Malta |
|
Pfizer ApS Tlf: +45 44 20 11 00 |
Vivian Corporation Ltd. Tel : + 35621 344610 |
|
Deutschland Pfizer Pharma GmbH Tel: +49 (0)30 550055 51000 |
Nederland Pfizer bv Tel: +31 (0)10 406 43 01 |
|
Eesti Pfizer Luxembourg SARL Eesti Filial Tel: +372 666 7500 |
Norge Pfizer Norge AS Tlf: +47 67 52 61 00 |
|
EXXáSa PFIZER EAAAI A.E. Tp^: +30 210 678 5800 |
Osterreich Pfizer Corporation Austria Ges.m.b.H Tel: +43 (0)1 521 15-0 |
|
España Pfizer S.L. Tel: +34 91 490 99 00 |
Polska Pfizer Polska Sp. z o.o., Tel.: +48 22 335 61 00 |
|
France Pfizer |
Portugal Pfizer Biofarmaceutica, Sociedade Unipessoal Lda |
|
Tél: +33 (0)1 58 07 34 40 |
Tel: +351 21 423 5500 |
|
Hrvatska Pfizer Croatia d.o.o. Tel: + 385 1 3908 777 |
Romanía Pfizer Romania S.R.L. Tel: +40 21 207 28 00 |
|
Ireland Pfizer Healthcare Ireland Tel: 1800 633 363 (toll free) +44 (0)1304 616161 |
Slovenija Pfizer Luxembourg SARL Pfizer, podruznica za svetovanje s podrocja farmacevtske dejavnosti, Ljubljana Tel: + 386 (0) 1 52 11 400 |
|
Ísland Icepharma hf. Sími: + 354 540 8000 |
Slovenská republika Pfizer Luxembourg SARL, organizacná zlozka Tel: +421-2-3355 5500 |
|
Italia Pfizer S.r.l. Tel: +39 06 33 18 21 |
Suomi/Finland Pfizer Oy Puh/Tel: +358 (0)9 43 00 40 |
Kúrcpog Sverige
PFIZER EAAAE A.E. Pfizer Innovations AB
(CYPRUS BRANCH) Tel: + 46 (0)8 550 520 00
Tqk +357 22 817690
United Kingdom
Pfizer Limited
Tel: +44 (0)1304 616161
Latvija
Pfizer Luxembourg SARL filiale Latvija
Tel: +371 670 35 775 Fecha de la última revisión de este prospecto:
La información detallada de este medicamento está disponible en la página web de la Agencia Europea del Medicamento http://www.ema.europa.eu. También presenta enlaces con otras páginas web sobre enfermedades raras y medicamentos huérfanos.
35