Azzalure 10 Unidades Speywood/0,05 Ml Polvo Para Solucion Inyectable
FICHA TÉCNICA
1. NOMBRE DEL MEDICAMENTO
Azzalure, 10 unidades Speywood/0,05 ml, polvo para solución inyectable
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Toxina botulínica tipo A* 10 unidades Speywood**/0,05 ml de solución reconstituida.
Vial de 125 unidades.
*Toxina botulínica tipo A de Clostridium botulinum - complejo hemaglutinina.
**Una unidad Speywood (U) se define como la dosis mediana letal intraperitoneal en el ratón (DL50).
Las unidades Speywood de Azzalure son específicas de este producto y no son intercambiables con otros preparados de toxina botulínica.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo para solución inyectable.
Polvo blanco.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Azzalure está indicado para la mejoría temporal en el aspecto de las líneas glabelares (líneas verticales entre las cejas) del entrecejo de intensidad de moderada a grave, en pacientes adultos menores de 65 años, cuando la severidad de estas líneas tiene un impacto psicológico importante para el paciente.
4.2 Posología y forma de administración
4.2.1 Posología
Las unidades de toxina botulínica son distintas dependiendo del medicamento. Las unidades Speywood de Azzalure son específicas de este producto y no son intercambiables con otros preparados de toxina botulínica.
Población pediátrica
La seguridad y la efectividad de Azzalure en individuos menores de 18 años de edad no han sido demostradas.
4.2.2 Forma de administración
Azzalure sólo puede ser administrado por médicos con la cualificación y experiencia adecuadas en este tratamiento y que dispongan del equipo adecuado.
Una vez reconstituido, Azzalure sólo debe utilizarse para tratar un único paciente y durante una única sesión de tratamiento.
Antes de la inyección, el producto debe ser reconstituido. Las instrucciones para la reconstitución se describen en la Sección 6.6.
Retirar cualquier resto de maquillaje y desinfectar la piel con un antiséptico tópico.
Se deben aplicar inyecciones intramusculares en ángulo recto respecto a la piel con una aguja estéril de calibre 29-30 G.
Instrucciones de administración:
La dosis recomendada es de 50 unidades Speywood (0,25 ml de solución reconstituida) de Azzalure administrada en 5 lugares de inyección, se administran 10 unidades Speywood (0,05 ml de solución reconstituida) por vía intramuscular en cada uno de los cinco lugares previstos: 2 inyecciones en cada músculo corrugador y 1 en el músculo prócero cerca del ángulo nasofrontal, como se muestra en el dibujo:
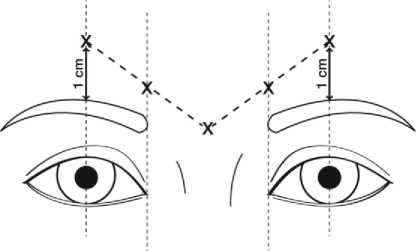
Los puntos anatómicos de referencia se identifican fácilmente al observar y palpar con el entrecejo fruncido al máximo. Antes de aplicar la inyección, coloque el dedo pulgar o el índice firmemente debajo del borde orbital para evitar la extravasación por debajo del mismo. Durante la inyección, la aguja se debe orientar superiormente y medialmente. Para reducir el riesgo de ptosis, no aplicar la inyección cerca del músculo elevador del párpado superior, especialmente, en pacientes con complejos ceja-depresor (depresor superciliar) más grandes. Las inyecciones en el músculo corrugador se deben realizar en la parte central de dicho músculo, al menos 1 cm por encima del borde orbital.
El intervalo entre cada tratamiento depende de la respuesta individual de cada paciente tras su valoración. En estudios clínicos, el efecto óptimo se mantiene hasta 4 meses después de la inyección. Algunos pacientes siguieron respondiendo a los 5 meses (ver sección 5.1). El intervalo entre cada tratamiento no debe ser más frecuente que de cada 3 meses.
En caso de que falle el tratamiento o disminuya el efecto después de inyecciones repetidas, se deberán utilizar métodos de tratamiento alternativos. En caso de que el tratamiento no sea eficaz después de la primera sesión, se deberán tomar las siguientes medidas:
• Analizar las causas del fallo, por ejemplo, inyección en músculos erróneos, técnica de inyección y formación de anticuerpos frente a la toxina;
• Evaluar nuevamente si es adecuado el tratamiento con toxina botulínica tipo A.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1
- En caso de presencia de infección en los puntos en que se pretende inyectar;
- En caso de miastenia grave, Síndrome de Eaton Lambert o esclerosis lateral amiotrófica.
4.4 Advertencias y precauciones especiales de empleo
Azzalure se debe utilizar con precaución en pacientes con riesgo, o signos clínicos, de trastornos importantes de la transmisión neuromuscular. Es posible que estos pacientes tengan una mayor sensibilidad a los fármacos del tipo de Azzalure, lo que puede provocar debilidad muscular acusada.
En muy raras ocasiones se han observado efectos adversos debidos a la diseminación de los efectos de la toxina a lugares alejados del lugar de administración. Los pacientes tratados con dosis terapéuticas pueden experimentar debilidad muscular exagerada.
No se recomienda la administración de Azzalure en pacientes con antecedentes de disfagia y aspiración.
En caso de dificultad respiratoria, de deglución o del habla, el paciente o el cuidador deben consultar a su médico inmediatamente.
No se deben sobrepasar ni la dosis ni la frecuencia de administración recomendadas de Azzalure.
Antes de administrar Azzalure es muy importante estudiar la anatomía facial del paciente. Se debe tener en cuenta la existencia de asimetría facial, ptosis, dermatocalasia excesiva, cicatrices o cualquier alteración de la anatomía como resultado de alguna intervención quirúrgica facial previa.
Se deberá tener cuidado al utilizar Azzalure en caso de inflamación de la zona en que se pretende inyectar o cuando el músculo a tratar esté excesivamente debilitado o atrofiado.
Como ocurre con cualquier inyección intramuscular, no se recomienda la administración de Azzalure en pacientes con un tiempo de hemorragia prolongado.
La administración de inyecciones a intervalos más frecuentes o a dosis más elevadas puede incrementar el riesgo de formación de anticuerpos contra la toxina botulínica. Clínicamente, la formación de anticuerpos neutralizantes puede reducir la eficacia del tratamiento.
Se desconoce el efecto producido por la administración de diferentes toxinas botulínicas durante el tratamiento con Azzalure y debe evitarse.
Es preceptivo que Azzalure séa usado para un solo tratamiento y para un solo paciente durante una única sesión. El producto sobrante sin usar debe ser eliminado tal y como se detalla en la sección 6.6.
Deberían tomarse precauciones especiales a la hora de la preparación y la administración, al igual que para la inactivación y la eliminación de la solución sobrante sin usar (ver sección 6.6).
4.5 Interacción con otros medicamentos y otras formas de interacción
Azzalure se debe utilizar con precaución a la hora de tratamiento concomitante con aminoglucósidos u otros fármacos que afecten a la transmisión neuromuscular (p. ej. fármacos curariformes), en estos casos es posible que se potencie el efecto de la toxina botulínica tipo A.
No se han realizado estudios de interacciones. No se han observado otras interacciones de interés clínico.
4.6 Fertilidad, embarazo y lactancia
4.6.1 Embarazo
Azzalure no debe utilizarse durante el embarazo a menos que sea claramente necesario. No existen datos suficientes sobre la utilización de la toxina botulínica tipo A en mujeres embarazadas. Los estudios en animales han mostrado toxicidad reproductiva a altas dosis (ver Sección 5.3). Se desconoce el riesgo en seres humanos.
4.6.2 Lactancia
Se desconoce si Azzalure se excreta en la leche humana. No se puede recomendar el uso de Azzalure durante la lactancia materna.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Existe un posible riesgo de debilidad muscular localizada o alteraciones visuales debidas al uso de este medicamento, que pueden afectar de manera transitoria la capacidad para conducir y utilizar máquinas.
4.8 Reacciones adversas
En diferentes ensayos clínicos, más de 1900 pacientes fueron expuestos a Azzalure.
En estudios clínicos pivotales, estudios a doble ciego, controlados con placebo, y estudios de larga duración, abiertos, se trataron alrededor de 1500 pacientes con líneas glabelares moderadas a graves con la dosis recomendada de 50 unidades
En estudios a doble ciego, controlados con placebo, con dosis únicas, el 22,3% de los pacientes tratados con la dosis de Azzalure recomendada (50 U) y el 16,6% de los pacientes tratados con placebo presentaron una reacción provocada por el tratamiento, por la técnica de inyección o por ambos. En el estudio en fase III, abierto y a largo plazo, donde los pacientes recibieron múltiples ciclos de inyecciones, el 26% de los pacientes presentaron, al menos, una reacción adversa provocada por el tratamiento después de la primera inyección. La incidencia de reacciones adversas debidas a la técnica de inyección/tratamiento se redujo a medida que se repitieron los ciclos.
Las reacciones adversas más frecuentes fueron cefalea y las reacciones en el lugar de inyección. Generalmente, las reacciones debidas a la técnica de inyección/tratamiento aparecen en la primera semana después de la inyección y suelen ser transitorias. La mayoría de las reacciones observadas fueron de gravedad leve o moderada y fueron reversibles.
Clasificación de la frecuencia de las reacciones adversas:
Muy frecuentes (> 1/10); frecuentes (> 1/100 a < 1/10); poco frecuentes (> 1/1.000 a < 1/100); raras (> 1/10.000 a < 1/1.000); muy raras (< 1/10.000); frecuencia no conocida ( no puede estimarse a partir de los datos disponibles).
|
Trastornos del sistema nervioso |
Muy frecuentes Cefalea Frecuentes Paresia facial (generalmente descrita como paresia de la frente) Poco frecuentes Mareos |
|
Trastornos oculares |
Frecuentes Astenopía, ptosis, edema palpebral, lagrimeo, sequedad ocular, fasciculación (contracciones musculares alrededor de los ojos) Poco frecuentes Trastornos visuales, visión borrosa, diplopía, movimiento involuntario de los ojos |
|
Trastornos de la piel y del tejido |
Poco frecuentes |
|
subcutáneo |
Prurito, eritema Raras Urticaria |
|
Trastornos generales y |
Muy frecuentes |
|
alteraciones en el lugar de |
Reacciones en el lugar de inyección (por ejemplo: |
|
administración |
eritema, edema, irritación, erupción, prurito, parestesia, dolor, molestias, escozor y hematoma) |
|
Trastornos del sistema |
Poco frecuentes |
|
inmunológico |
Hipersensibilidad |
Muy raramente se han comunicado efectos adversos con toxina botulínica producidos por una distribución de los efectos de la toxina a zonas alejadas del lugar de inyección (debilidad muscular excesiva, disfagia, neumonía por aspiración con consecuencias fatales en algún caso) (ver sección 4.4).
Notificación de sospechas de reacciones adversas:
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales
sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Español de Farmacovigilancia de medicamentos de Uso Humano: https://www.notificaRAM.es
4.9 Sobredosis
No se observaron casos de sobredosis durante los estudios clínicos.
Se espera que una dosis excesiva de toxina botulínica provoque debilidad neuromuscular junto con otros síntomas. En caso de sobredosis, puede ser necesaria la instauración de respiración asistida debido a la parálisis de los músculos respiratorios. Asimismo, se deberá vigilar al paciente por si presenta debilidad muscular excesiva o parálisis muscular. Si fuera necesario se instaurará un tratamiento sintomático.
Es posible que los síntomas de sobredosis no aparezcan inmediatamente después de la inyección.
En pacientes que muestren síntomas de intoxicación por toxina botulinica tipo A, (por ejemplo: una combinación de debilidad muscular, ptosis, diplopia, dificultad en la deglución y al hablar, o paresias de los músculos respiratorios), debe considerarse la posibilidad de hospitalización.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Otros agentes relajantes musculares de acción periférica.
Código ATC: M03AX01.
El efecto farmacodinámico principal de la toxina botulínica tipo A de Clostridium botulinum se debe a la denervación química del músculo en tratamiento. Este fenómeno, produce una reducción cuantificable del potencial de acción muscular compuesto, lo que provoca una disminución localizada, o una parálisis, de la actividad muscular.
Datos clínicos
Durante el desarrollo clínico de Azzalure, se realizaron diferentes ensayos clínicos donde participaron más de 2600 pacientes.
En los estudios clínicos, 1907 pacientes que presentaban líneas glabelares moderadas a graves, se trataron con la dosis recomendada de 50 unidades Speywood. Entre ellos, 305 fueron tratados con 50 unidades en dos estudios pivotales de fase III, a doble ciego y controlados con placebo, y 1200 con 50 unidades en un estudio de fase III de dosis repetidas, abierto y a largo plazo. El resto de los pacientes se trataron en estudios de apoyo y de determinación de dosis.
La mediana del tiempo hasta el inicio de la respuesta fue de 2 o 3 días después del tratamiento, observándose el efecto máximo el día 30. En ambos estudios pivotales controlados con placebo de Fase III, las inyecciones de Azzalure redujeron significativamente la intensidad de las líneas glabelares hasta 4 meses después de la inyección. El efecto duró significativamente tras 5 meses en uno de los 2 estudiospivotales.
Treinta días después de la inyección, la evaluación de los investigadores indicó que el 90% (273/305) de los pacientes respondió al tratamiento (no presentaba líneas glabelares o éstas eran leves en fruncimiento máximo), mientras que en el grupo que recibió placebo, la proporción fue del 3% (4/153). Cinco meses después de la inyección, el 17% (32/190) de los pacientes tratados con Azzalure continuaban respondiendo al tratamiento en comparación con el 1% (1/92) de los pacientes tratados con placebo en este estudio. La evaluación del propio paciente a los 30 días y en fruncimiento máximo indica un índice de respuesta del 82% (251/305) con Azzalure y del 6% (9/153) en el grupo que recibió placebo. En el estudio clínico pivotal de fase III en el que se llevó a cabo esta evaluación, la proporción de pacientes que, según la valoración del investigador, mostraron una mejoría de 2 grados en máximo fruncimiento fue del 77% (79/103).
Un subgrupo de 177 pacientes presentaba líneas glabelares moderadas o graves en reposo antes del tratamiento. La evaluación del investigador para este grupo de pacientes, 30 días después del tratamiento,
indicó que el 71% (125/177) de los pacientes tratados con Azzalure respondieron al tratamiento frente al 10% (8/78) de los tratados con placebo.
El estudio abierto, con dosis repetidas y a largo plazo mostró que la mediana del tiempo hasta el inicio de la respuesta de 3 días se mantuvo en los sucesivos ciclos de tratamiento. Asimismo, se mantuvo, tras sucesivos ciclos, la tasa de respuesta en máximo fruncimiento determinada por el investigador en el día 30 (varió a lo largo de los 5 ciclos entre el 80% y el 91%). La tasa de respuesta en reposo en los sucesivos ciclos también fue coherente con los estudios de dosis única, con el 56%-74% de los pacientes tratados con Azzalure que respondieron 30 días después del tratamiento según la valoración de los investigadores. Ninguna de las variables clínicas incluyó una evaluación objetiva del impacto psicológico.
5.2 Propiedades farmacocinéticas
No se prevé encontrar Azzalure en cantidades cuantificables en la sangre periférica tras la inyección i.m. de las dosis recomendadas. Por lo tanto, no se han realizado estudios farmacocinéticos con Azzalure.
5.3 Datos preclínicos sobre seguridad
En los estudios para la reproducción en ratas y conejos, a altas dosis se ha observado toxicidad materna grave asociada a pérdida en la implantación. A dosis entre 60 y 100 veces la recomendada en humanos (50 U) en ratas y conejos, respectivamente, no se observó toxicidad embriofetal. En ambas especies no se observaron efectos teratogénicos. A altas dosis se observó en ratas una reducción de la fertilidad en machos y hembras debida a una falta de emparejamiento provocada por la parálisis muscular.
En un estudio de toxicidad crónica realizado en ratas, no se observó toxicidad sistémica a dosis 75 veces mayores que las recomendadas para los humanos (50 U), divididas por igual entre los músculos de los glúteos derecho e izquierdo.
En los estudios de toxicidad aguda, crónica y tolerancia local en el lugar de inyección no se observaron reacciones adversas inusuales, locales o sistémicas, a dosis clinicamente relevantes.
6 . DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Albúmina humana 200g/L Lactosa monohidrato.
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros excepto con los mencionados en la sección 6.6.
6.3 Periodo de validez 2 años.
Solución reconstituida:
Una vez reconstituido, se ha demostrado una estabilidad química y física durante 24 horas entre 2-8°C. Desde un punto de vista microbiológico, salvo que el método de reconstitución excluya el riesgo de contaminación microbiana, el medicamento se debe utilizar inmediatamente. En el caso que no se utilice de forma inmediata, el tiempo y las condiciones de almacenamiento una vez reconstituido, son de responsabilidad del usuario.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Para las condiciones de conservación del medicamento reconstituido, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Vial (vidrio tipo I) con polvo de 125 unidades Speywood, con un tapón (halobutilo) y un precinto (aluminio).
Envase con 1 o 2 viales.
Puede que solamente estén comercializados algunos tamaños de envase.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Se deben seguir estrictamente las instrucciones de uso, manipulación y eliminación.
La reconstitución se debe realizar de acuerdo con las instrucciones de buenas prácticas, especialmente respetando las condiciones asépticas.
Azzalure se debe reconstituir con una solución inyectable de cloruro de sodio de 9 mg/ml (0,9%).
En la tabla de dilución a continuación, se indica la cantidad requerida de solución inyectable de cloruro de sodio de 9 mg/ml (0,9%) que se debe disponer en una jeringa para obtener una solución reconstituida incolora con una concentración de 10 U/0,05 ml:
|
Cantidad de disolvente añadido (solución de cloruro de sodio 0.9%) para un vial de 125 U |
Dosis resultante (Unidades para 0.05 ml) |
|
0.63 ml |
10 U |
Para medir con exactitud los 0,63 mlse puede utilizar una jeringa de 1 ml graduada en incrementos de 0,1 ml y 0,01ml.
RECOMENDACIONES PARA LA ELIMINACIÓN DE MATERIALES CONTAMINADOS Inmediatamente después de su uso y antes de su eliminación, la solución de Azzalure reconstituida no utilizada (en el vial o en la jeringa) debe ser inactivada con 2 ml de solución de hipoclorito sódico diluida al 0,55 o al 1% (solución de Dakin).
Los viales, jeringas y materiales utilizados no se deben vaciar, sino que se deben depositar en contenedores adecuados y ser eliminados según los procedimientos locales establecidos.
RECOMENDACIONES EN CASO DE ACCIDENTE DURANTE LA MANIPULACIÓN DELA TOXINA BOTULÍNICA
• Se debe limpiar cualquier resto de producto, bien con un material absorbente empapado en una solución de hipoclorito sódico (lejía) si se trata del polvo, bien con un material absorbente seco, si se trata del producto reconstituido.
• Las superficies contaminadas se deben limpiar con un material absorbente empapado en una solución de hipoclorito sódico (lejía) y luego secar.
• Si se rompe un vial, se debe proceder como se indica anteriormente, recogiendo con cuidado los fragmentos de cristal y limpiando el producto, evitando cortarse con los cristales rotos.
• Si el producto entra en contacto con la piel, lávese la zona afectada con una solución de hipoclorito sódico (lejía) y aclárese con abundante agua.
• Si el producto entra en contacto con los ojos, límpiese cuidadosamente con abundante agua o con una solución de limpieza oftálmica.
• Si el producto entra en contacto con una herida, corte o piel levantada, límpiese cuidadosamente con abundante agua y tome las medidas médicas oportunas según la dosis inyectada.
Se deberán seguir estrictamente estas instrucciones de uso, manipulación y eliminación.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Ipsen Pharma, S.A. Torre Realia, Plaza de Europa, 41-43. 08908 L’Hospitalet de Llobregat, Barcelona -España
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
71.300
9. FECHA DE LA PRIMERA AUTORIZACIÓN/ RENOVACIÓN DE LA AUTORIZACIÓN
Agosto 2009 / Mayo 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
Noviembre 2015
¡y
taños
8 de 8