Arzerra 100 Mg Concentrado Para Solucion Para Perfusion
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
Arzerra 100 mg concentrado para solución para perfusión.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un ml de concentrado contiene 20 mg de ofatumumab.
Cada vial contiene 100 mg de ofatumumab en 5 ml.
Ofatumumab es un anticuerpo monoclonal humano producido en una línea celular recombinante murina (NS0).
Excipiente(s) con efecto conocido:
Este medicamento contiene 34,8 mg de sodio por dosis de 300 mg, 116 mg de sodio por dosis de 1.000 mg y 232 mg de sodio por dosis de 2.000 mg.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Concentrado para solución para perfusión (concentrado estéril).
Líquido de transparente a opalescente, de incoloro a color amarillo pálido.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Leucemia linfocítica crónica (LLC) no tratada previamente:
Arzerra en combinación con clorambucilo o bendamustina está indicado para el tratamiento de pacientes con LLC que no han recibido tratamiento previo y que no son adecuados para un tratamiento basado en fludarabina.
Para mayor información, ver sección 5.1.
LLC refractaria:
Arzerra está indicado para el tratamiento de pacientes con LLC que son refractarios a fludarabina y alemtuzumab.
Para mayor información, ver sección 5.1.
4.2 Posología y forma de administración
Arzerra se debe administrar bajo la supervisión de un médico experimentado en el uso de tratamientos para el cáncer y en un entorno donde se disponga de instalaciones para la reanimación completa de forma inmediata.
Monitorización
Durante la administración de ofatumumab, especialmente durante la primera perfusión, se debe monitorizar estrechamente a los pacientes ante la aparición de reacciones debidas a la perfusión, incluyendo el síndrome de liberación de citoquinas.
Premedicación
Los pacientes siempre deben ser premedicados de 30 minutos a 2 horas antes de la perfusión de Arzerra, de acuerdo a los siguientes esquemas de dosificación:
LLC no tratada previamente:
• 1.000 mg de paracetamol por vía oral (acetaminofeno) o equivalente, más
• antihistamínico por vía oral o intravenosa (50 mg de difenhidramina o 10 mg de cetirizina o equivalente), más
• corticosteroides por vía intravenosa (50 mg de prednisolona u otro equivalente)
Después de la primera y la segunda perfusión, si el paciente no experimenta una reacción adversa grave al medicamento (RAM), se puede reducir u omitir la premedicación con corticosteroides en las perfusiones posteriores, según el criterio del médico.
LLC refractaria:
• 1.000 mg de paracetamol (acetaminofeno) por vía oral o equivalente, más
• antihistamínico por vía oral o intravenosa (50 mg de difenhidramina o 10 mg de cetirizina u otro equivalente), más
• corticosteroides por vía intravenosa (100 mg de prednisolona o equivalente)
Si la segunda perfusión semanal se ha completado sin RAM graves, se puede reducir la dosis de corticosteroides en las perfusiones de la 3 a la 8, según el criterio del médico.
Antes de la novena perfusión (primera perfusión mensual), los pacientes deben recibir la dosis total de medicamentos utilizados en la premedicación tal y como se describe anteriormente. Si la novena perfusión se completa sin RAM graves, la dosis puede ser reducida al equivalente de 50 mg de prednisolona en las perfusiones posteriores, según el criterio del médico.
Posología
LLC no tratada previamente:
La dosis recomendada y programada es de 300 mg en el día 1, seguido de 1.000 mg una semana después en el día 8 (ciclo 1), seguido de 1.000 mg en el día 1 de los ciclos posteriores, durante un mínimo de 3 ciclos, hasta obtener la mejor respuesta o un máximo de 12 ciclos (cada 28 días).
La mejor respuesta, fue aquella respuesta clínica que no mejoró tras recibir 3 ciclos adicionales de tratamiento.
Primera perfusión
La velocidad inicial de la primera perfusión de Arzerra debe ser de 12 ml/hora. Durante la perfusión, la velocidad se debe incrementar cada 30 minutos hasta un máximo de 400 ml/hora (ver sección 6.6).
Perfusiones posteriores
Si la primera perfusión se ha completado sin RAM graves relacionadas con la perfusión, las perfusiones posteriores se pueden iniciar a una velocidad de 25 ml/hora que se debe incrementar cada 30 minutos hasta un máximo de 400 ml/hora (ver sección 6.6).
LLC refractaria:
La dosis recomendada es de 300 mg para la primera perfusión y de 2.000 mg para todas las perfusiones posteriores. El esquema de perfusión es de 8 semanas consecutivas, y transcurrido un periodo de interrupción de 4-5 semanas, dicho esquema pasará a ser de 4 meses consecutivos (por ej. una perfusión cada 4 semanas).
Primera y segunda perfusión
La velocidad inicial de la primera y la segunda perfusión de Arzerra debe ser de 12 ml/hora. Durante la perfusión, la velocidad se debe incrementar cada 30 minutos hasta un máximo de 200 ml/hora (ver sección 6.6).
Perfusiones posteriores
Si la segunda perfusión se ha completado sin RAM graves relacionadas con la perfusión, las perfusiones restantes se pueden iniciar a una velocidad de 25 ml/hora y esta se debe incrementar cada 30 minutos hasta un máximo de 400 ml/hora (ver sección 6.6).
Modificación de la dosis y reinicio del tratamiento para RAM relacionadas con la perfusión - en pacientes con LLC no tratados previamente y pacientes con LLC refractaria.
Si aparecen RAM relacionadas con la perfusión de cualquier gravedad, interrumpir la perfusión. El tratamiento se puede retomar según el criterio del médico responsable del tratamiento. A modo de guía, se pueden utilizar las siguientes modificaciones de la velocidad de perfusión:
• En el caso de que se produzca una RAM de leve a moderada, la perfusión se debe interrumpir y reiniciarse cuando el estado del paciente sea estable, a la mitad de la velocidad de perfusión que se aplicaba en el momento de la interrupción. Antes de interrumpir la perfusión debido a una RAM, si la velocidad de perfusión no se ha incrementado desde la velocidad inicial de 12 ml/hora, la perfusión debe reiniciarse a una velocidad de 12 ml/hora, velocidad de perfusión estándar. La velocidad de perfusión se puede seguir incrementando de acuerdo a los procedimientos estándar, de acuerdo al criterio médico y a la tolerancia del paciente (no exceder el incremento de la velocidad de cada 30 minutos).
• En el caso de una reacción adversa grave, la perfusión se debe interrumpir y reiniciarse a una velocidad de perfusión de 12 ml/hora, cuando el estado del paciente sea estable. La velocidad de perfusión se puede seguir incrementando de acuerdo a los procedimientos estándar, de acuerdo al criterio del médico y a la tolerancia del paciente (no exceder el incremento de la velocidad de cada 30 minutos).
Población pediátrica
Arzerra no está recomendado para el uso en niños menores de 18 años debido a datos insuficientes en la seguridad y/o eficacia.
Pacientes de edad avanzada
No se observaron diferencias sustanciales en la seguridad y la eficacia relacionadas con la edad (ver sección 5.1). En base a los datos de seguridad y eficacia en pacientes de edad avanzada, no se requieren ajustes de la dosis (ver sección 5.2).
Insuficiencia renal
No se han realizado estudios formales con Arzerra en pacientes con insuficiencia renal. No se recomiendan ajustes de la dosis para insuficiencia renal de leve a moderada (aclaramiento de creatinina > 30 ml/min) (ver sección 5.2).
Insuficiencia hepática
No se han realizado estudios formales con Arzerra en pacientes con insuficiencia hepática. Sin embargo, es improbable que los pacientes con insuficiencia hepática requieran una modificación de la dosis (ver sección 5.2).
Forma de administración
Arzerra se administra en perfusión intravenosa y se debe diluir antes de la administración. Para consultar las instrucciones de dilución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad a ofatumumab o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Reacciones debidas a la perfusión
La administración intravenosa de ofatumumab se ha asociado con reacciones debidas a la perfusión. Estas reacciones pueden provocar la interrupción temporal del tratamiento, o la retirada del mismo. Las premedicaciones atenúan las reacciones debidas a la perfusión, pero éstas pueden seguir apareciendo, predominantemente durante la primera perfusión. Las reacciones debidas a la perfusión pueden incluir, pero no estar limitadas a, acontecimientos anafilactoides, broncoespasmo, acontecimientos cardiacos (por ejemplo, isquemia miocárdica / infarto, bradicardia), escalofríos, tos, síndrome de liberación de citoquinas, diarrea, disnea, fatiga, rubefacción, hipertensión, hipotensión, náuseas, dolor, edema pulmonar, prurito, pirexia, erupción y urticaria. En raras ocasiones, estas reacciones pueden provocar la muerte. Incluso con la premedicación, tras el uso de ofatumumab se han notificado reacciones graves, incluyendo síndrome de liberación de citoquinas. En los casos de reacción grave debida a la perfusión, la perfusión de Arzerra se debe interrumpir inmediatamente e iniciar el tratamiento sintomático (ver sección 4.2).
Las reacciones debidas a la perfusión aparecen con mayor frecuencia en el primer día de perfusión y tienden a disminuir con las perfusiones posteriores. Los pacientes con un historial de función pulmonar disminuida, pueden tener mayor riesgo de complicaciones pulmonares debido a reacciones graves y se deben monitorizar estrechamente durante la perfusión de ofatumumab.
Síndrome de lisis tumoral
En pacientes con LLC, puede aparecer síndrome de lisis tumoral (SLT) con el uso de ofatumumab. Los factores de riesgo del SLT incluyen una mayor carga tumoral, concentraciones elevadas de células circulantes (> 25.000/mm3), hipovolemia, insuficiencia renal, niveles elevados de ácido úrico antes del tratamiento y niveles elevados de lactato deshidrogenasa. El manejo del SLT incluye la corrección de las alteraciones electrolíticas, monitorización de la función renal, mantenimiento del balance de fluidos y tratamiento de soporte.
Leucoencefalopatía multifocal progresiva
Se ha notificado leucoencefalopatía multifocal progresiva (LMP) y muerte en pacientes con LLC que reciben farmacoterapia citotóxica, incluyendo ofatumumab. Se debe valorar la realización de un diagnóstico de LMP en cualquier paciente en tratamiento con Arzerra que notifique la aparicición de nuevos signos y síntomas neurológicos o cambios en los signos y síntomas neurológicos preexistentes. Si se sospecha de un diagnóstico de LMP, se debe interrumpir el tratamiento con Arzerra y remitir al paciente a un neurólogo.
Inmunizaciones
No se ha estudiado la seguridad y la capacidad para generar una respuesta primaria o anamnésica a la inmunización con vacunas vivas atenuadas o inactivadas durante el tratamiento con ofatumumab. La respuesta a la vacunación podría verse afectada cuando se agotan las células B. Debido al riesgo de infección, la administración de vacunas vivas atenuadas se debe evitar durante y después del tratamiento con ofatumumab, hasta que los recuentos de células B se normalicen. Se deben valorar los riesgos y los beneficios de la vacunación de pacientes durante el tratamiento con ofatumumab.
Hepatitis B
En pacientes tratados con medicamentos clasificados como anticuerpos citolíticos de acción directa sobre CD20, entre los que se incluye Arzerra, puede aparecer infección y reactivación del virus de la hepatitis B (VHB), que en algunos casos da lugar a hepatitis fulminante. Se han notificado casos en pacientes con resultados positivos del antígeno de superficie de la hepatitis B (HBsAg), y también en aquellos pacientes con resultados positivos de anticuerpos frente al antigeno core de la hepatitis B (anti-HBc) y con HBsAg negativo. La reactivación del VHB también ha aparecido en pacientes que aparentemente ya no presentaban infección por hepatitis B (por ejemplo, HBsAG negativo, anti-HBc positivo, y anti-HBs positivo).
La reactivación del VHB se define como un incremento brusco en la replicación del VHB, que se manifiesta con un aumento rápido de los niveles de ADN del VHB en suero, o mediante la detección del HBsAg en una persona que previamente presentó resultados HBsAg negativo y anti-HBc positivo. Generalmente, la reactivación de la replicación del VHB va seguida de hepatitis, es decir: aumento de los niveles de transaminasas, y en casos graves, incremento de los niveles de bilirrubina, fallo hepático, y muerte.
Antes de iniciar el tratamiento con Arzerra, todos los pacientes deben ser monitorizados para descartar una posible infección por el VHB, mediante la medición de HBsAg y anti-HBc. Los pacientes que presenten evidencias de infección previa por hepatitis B (HBsAg negativo, anti-HBc positivo), deben consultar a médicos con experiencia en el manejo de la hepatitis B en relación a la monitorización e inicio del tratamiento antiviral del VHB. No se debe iniciar el tratamiento con Arzerra en pacientes con evidencias recientes de infección por hepatitis B (HbsAg positivo), hasta que la infección se haya tratado adecuadamente.
Los pacientes que presenten evidencias de infección previa por el VHB, deben ser monitorizados ante cualquier signo clínico o de laboratorio relacionado con la hepatitis o la reactivación del VHB, durante el tratamiento con Arzerra y durante los 6-12 meses siguientes a la última perfusión de Arzerra. Se han notificados casos de reactivación del VHB hasta 12 meses después de finalizar el tratamiento. La interrupción del tratamiento antiviral del VHB se debe consultar con un médico con experiencia en el manejo de la hepatitis B.
Los pacientes que desarrollen una reactivación del VHB durante el tratamiento con Arzerra, deben de interrumpir inmediatamente el tratamiento con Arzerra y cualquier otra quimioterapia concomitante, e instaurar un tratamiento adecuado. Existen pocos datos de seguridad sobre la reanudación del tratamiento con Arzerra en pacientes que desarrollan una reactivación del VHB. La decisión de reanudar el tratamiento con Arzerra en pacientes que presenten una reactivación del VHB se debe consultar con un médico con experiencia en el manejor de la hepatitis B.
Cardiovascular
Se debe monitorizar estrechamente a los pacientes con antecedentes de enfermedad cardiaca. Se debe interrumpir el tratamiento con Arzerra en los pacientes que experimentan arritmias cardiacas graves o potencialmente mortales.
En un análisis agrupado de tres estudios abiertos en pacientes con LLC (N = 85), se evaluó el efecto de múltiples dosis de Arzerra sobre el intervalo QTc. En el análisis agrupado, se observaron incrementos por encima de 5 milisegundos en la mediana/media de los intervalos QT/QTc. No se detectaron grandes cambios en la media del intervalo QTc (esto es, > 20 milisegundos). Ningún paciente presentó un incremento en el intervalo QTc > 500 milisegundos. No se detectaron incrementos en el intervalo QTc dependientes de la concentración. Antes y durante la administración de ofatumumab, se recomienda medir los niveles de electrolitos, como el potasio y el magnesio, a los pacientes. Las anomalías en los niveles de electrolitos deben ser corregidas. Se desconoce el efecto de ofatumumab en pacientes con prolongación del intervalo QT (por ejemplo, prolongación adquirida o congénita).
Obstrucción intestinal
Se ha notificado obstrucción intestinal en pacientes que reciben tratamiento con anticuerpos monoclonales anti CD20, incluyendo ofatumumab. Se debe evaluar a los pacientes que presentan dolor abdominal, especialmente al principio del ciclo de tratamiento con ofatumumab, e iniciar el tratamiento adecuado.
Monitorización de valores analíticos de laboratorio
Durante el tratamiento con ofatumumab se han notificado citopenias, incluyendo neutropenia prolongada y neutropenia tardía. Durante el tratamiento con ofatumumab, se deben realizar recuentos sanguíneos completos, incluyendo recuento de neutrófilos y de plaquetas a intervalos regulares, y con mayor frecuencia en pacientes que desarrollan citopenias.
Contenido en sodio
Este medicamento contiene 34,8 mg de sodio por dosis de 300 mg, 116 mg por dosis de 1.000 mg y 232 mg de sodio por dosis de 2.000 mg. Esto debe tenerse en cuenta en pacientes con una dieta controlada en sodio.
4.5 Interacción con otros medicamentos y otras formas de interacción
Aunque existen datos limitados de interacción de ofatumumab con otros fármacos, no se conocen interacciones clínicamente significativas con otros medicamentos.
Ofatumumab no tiene un efecto clínicamente relevante sobre la farmacocinética de clorambucilo o su metabolito activo, la mostaza del ácido fenilacético.
La eficacia de vacunas vivas atenuadas o inactivadas puede verse alterada con ofatumumab. Por tanto, debe evitarse el uso concomitante de estos agentes con ofatumumab. Si la coadminsitración se considera inevitable, se deben valorar los riesgos y los beneficios de vacunar a los pacientes durante el tratamiento con ofatumumab (ver sección 4.4).
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos sobre el uso de ofatumumab en mujeres embarazadas. Los estudios en animales no indican efectos dañinos directos o indirectos en relación con la toxicidad reproductiva (ver sección 5.3). Ofatumumab no debe adminsitrase a mujeres embarazadas a menos que el posible beneficio para la madre supere el posible riesgo para el feto.
Las mujeres en edad fértil tienen que utilizar métodos anticonceptivos eficaces durante y hasta 12 meses depués del tratamiento con ofatumumab.
Lactancia
Se desconoce si ofatumumab se excreta en la leche materna, no obstante la IgG humana se secreta en la leche materna. No se ha establecido el uso seguro de ofatumumab en humanos durante la lactancia. La excrección de ofatumumab en la leche no ha sido estudiada en animales. Los datos publicados sugieren que el consumo de leche materna en recien nacidos y lactantes no da lugar a una absorción importante de éstos anticuerpos maternos en circulación. No se puede descartar el riesgo en recien nacidos/lactantes. La lactancia debe suspenderse durante el tratamiento con ofatumumab y durante los 12 meses posteriores al tratamiento.
Fertilidad
No hay datos sobre los efectos de ofatumumab en la fertilidad humana. Los efectos en la fertilidad masculina y femenina no han sido evaluados en estudios con animales.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas.
En base a la farmacología de ofatumumab, no se esperan efectos perjudiciales en estas actividades. Al considerar la capacidad del paciente para realizar tareas que requieran juicio, capacidades motoras o cognitivas, se debe tener en cuenta el estado clínico del paciente y el perfil de reacciones adversas de ofatumumab (ver sección 4.8)
4.8 Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad global de ofatumumab en pacientes con LLC (no tratados previamente y en recaída o refractarios) está basado en los datos procedentes de 511 pacientes incluidos en los ensayos clínicos (ver sección 5.1). Aquí se incluyen los 250 pacientes tratados con ofatumumab en monoterapia (en pacientes con LLC en recaída o refractarios) y 261 pacientes tratados en combinación con un agente alquilante (en pacientes que no han recibido tratamiento previo para la LLC y que no son adecuados para recibir un tratamiento a base de fludarabina).
El perfil de efectos adversos de ofatumumab en pacientes con LLC refractarios a fludarabina que no respondieron al menos a dos tratamientos previos y que presentaban enfermedad voluminosa, fue consistente con el perfil de seguridad global establecido a partir de otros estudios en LLC, tal y como se describe a continuación, en la tabla.
Tabla de reacciones adversas
Las reacciones adversas notificadas con ofatumumab en pacientes con LLC no tratados previamente y en recaída o refractarios, ya sea solo o en combinación con un agente alquilante, se incluyen a continuación por la clasificación de órganos del sistema MedDRA y por frecuencia. Muy frecuentes (> 1/10), Frecuentes (> 1/100 a < 1/10), Poco frecuentes (> 1/1.000 a < 1/100), Raras (> 1/10.000 a < 1/1.000),
Muy raras (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Clasificación de órganos del sistema MedDRA |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
|
Infecciones e infestaciones |
Infección del tracto respiratorio inferior, incluyendo neumonía, infección del tracto respiratorio superior |
Sepsis, incluyendo sepsis neutropénica y shock séptico, infección por virus del herpes, infección del tracto urinario |
Infección de Hepatitis B y reactivación | |
|
Trastornos de la sangre y del sistema linfático |
Neutropenia, anemia |
Neutropenia febril, trombocitopenia, leucopenia |
Agranulocitosis, coagulopatía, aplasia eritrocitaria, linfopenia | |
|
Trastornos del sistema inmunológico |
Reacciones anafilactoides*, hipersensibilidad * |
Shock anafiláctico* | ||
|
Trastornos del metabolismo y de la nutrición |
Síndrome de lisis tumoral | |||
|
Trastornos cardiacos |
Taquicardia* |
Bradicardia* | ||
|
Trastornos vasculares |
Hipotensión*, hipertensión* | |||
|
Trastornos respiratorios, torácicos y mediastínicos |
Broncoespasmo*, hipoxia*, disnea*, malestar torácico*, dolor faringolaríngeo*, tos*, congestión nasal* |
Edema pulmonar* | ||
|
Trastornos gastrointestinales |
Náusea* |
Diarrea* |
Obstrucción en el intestino delgado | |
|
Trastornos de la piel y del tejido subcutáneo |
Erupción* |
Urticaria*, prurito*, rubefacción* | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor de espalda* | |||
|
Trastornos generales y alteraciones en el lugar de administración |
Pirexia* |
Síndrome de liberación de citoquinas*, sensación de frío*, escalofríos*, hiperhidrosis*, fatiga* | ||
|
*Es probable que estos eventos sean atribuibles |
a ofatumumab al producirse una reacción a |
la perfusión, y | ||
éstos normalmente aparecieron después de iniciar la perfusión y en las 24 horas siguientes a la finalización de la perfusión (ver sección 4.4).
Descripción de las reacciones adversas seleccionadas
Reacciones relacionadas con la perfusión
En los ensayos clínicos en pacientes que recibieron ofatumumab, las reacciones adversas observadas con mayor frecuencia fueron reacciones relacionadas con la perfusión, que ocurrieron en un 68 % (348/511) de los pacientes, durante cualquier momento del tratamiento. La mayoria de las reacciones relacionadas con la perfusión fueron de gravedad Grado 1 o Grado 2. El ocho por ciento de los pacientes presentaron reacciones relacionadas con la perfusión de Grado > 3, durante cualquier momento del tratamiento. El dos por ciento de las reacciones relacionas con la perfusión condujeron a la suspensión del tratamiento. No hubo reacciones mortales relacionadas con la perfusión (ver sección 4.4).
Infecciones
De los 511 pacientes que recibieron ofatumumab en los ensayos clínicos, 300 pacientes (59 %) experimentaron una infección. Estas fueron infecciones bacterianas, virales o fúngicas. Ciento cuatro (20 %) de los 511 pacientes experimentaron infecciones > Grado 3. Veintiocho (5 %) de los 511 pacientes experimentaron infecciones mortales.
Neutropenia
De los 511 pacientes que recibieron ofatumumab en los ensayos clínicos, 139 pacientes (27 %) experimentaron un evento adverso asociado con un descenso en el recuento de neutrófilos; 118 (23 %) de los 511 pacientes experimentaron eventos adversos > Grado 3 asociados con un descenso en el recuento de neutrófilos. Cuarenta y dos (8 %) pacientes experimentaron eventos adversos graves asociados con un descenso en el recuento de neutrófilos.
En el estudio pivotal en pacientes con CLL no tratados previamente (OMB110911), se notificó neutropenia prolongada (definida como neutropenia de Grado 3 o 4, que no se resolvió entre los 24 y 42 días desde el último tratamiento) en 41 pacientes (23 pacientes tratados con ofatumumab y clorambucilo, 18 pacientes tratados con clorambucilo en monoterapia). Nueve pacientes tratados con ofatumumab y clorambucilo, y tres pacientes tratados solamente con clorambucilo presentaron neutropenia tardía, definida como neutropenia de Grado 3 o 4 que comienza al menos 42 días después del último tratamiento.
Cardiovascular
En un análisis agrupado de tres estudios abiertos en pacientes con LLC (N = 85), se evaluó el efecto de multiples dosis de Arzerra sobre el intervalo QTc. En el análisis agrupado, se observaron incrementos por encima de 5 milisegundos en la mediana/media de los intervalos QT/QTc. No se detectaron grandes cambios en la media del intervalo QTc (esto es, > 20 milisegundos). Ningún paciente presentó incrementos en el intervalo QTc > 500 milisegundos. No se detectaron incrementos en el intervalo QTc dependientes de la concentración.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se han notificado casos de sobredosis.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: anticuerpos monoclonales, código ATC: L01XC10 Mecanismo de acción
Ofatumumab es un anticuerpo monoclonal humano (IgG1) que se une específicamente a un epítopo bien diferenciado que abarca los bucles extracelulares grande y pequeño de la molécula CD20. La molécula CD20 es una fosfoproteína transmembrana expresada en los linfocitos B desde el estadío pre-B hasta linfocito B maduro y en tumores de células B. Los tumores de células B incluyen LLC (generalmente asociados con niveles más bajos de expresión de CD20) y linfomas no Hodgkin (donde > 90 % de los tumores presentan niveles elevados de expresión de CD20). La molécula CD20 no se desprende de la superficie celular y no se interioriza tras la unión del anticuerpo.
La unión de ofatumumab al epítopo proximal a la membrana de la molécula CD20 induce el reclutamiento y la activación de la vía del complemento en la superficie celular, que origina citotoxicidad dependiente del complemento y consiguiente lisis de las células tumorales. Se ha demostrado que ofatumumab induce de manera importante la lisis de las células con elevados niveles de expresión de las moléculas de defensa del complemento. También se ha demostrado que ofatumumab induce la lisis celular de células con alto y bajo grado de expresión de CD20 y en células resistentes a rituximab. Además, la unión de ofatumumab permite el reclutamiento de las células natural killer, que permiten la inducción de la muerte celular a través de la citotoxicidad celular anticuerpo dependiente.
Efectos farmacodinámicos
Los recuentos de células B periféricas disminuyeron tras la primera perfusión de ofatumumab en pacientes con neoplasias hematológicas. En pacientes con LLC refractaria, la mediana del descenso en el recuento de células B fue de un 22 % después de la primera perfusión y de un 92 % en la octava perfusión semanal. En la mayoría de los pacientes el recuento de células B periféricas se mantuvo bajo durante el resto del tratamiento, y en los pacientes que respondieron al tratamiento permaneció por debajo de los valores de referencia hasta 15 meses después de la última dosis.
En pacientes con LLC no tratados previamente, la mediana de las reducciones del recuento de células B después del primer ciclo de tratamiento y antes del sexto ciclo mensual fue de un 94 % y > 99 % respectivamente para la combinación de ofatumumab con clorambucilo, y de un 73 % y un 97 % respectivamente para clorambucilo en monoterapia. A los 6 meses después de la última dosis, la mediana de las reducciones del recuento de células B fue > 99 % para la combinación de ofatumumab y clorambucilo, y de un 94 % para clorambucilo en monoterapia.
Inmunogenicidad
Es posible que aparezca inmunogenicidad con proteínas terapéuticas como ofatumumab. A lo largo del programa clínico en LLC, se analizaron muestras de suero de más de 440 pacientes para detectar la presencia de anticuerpos anti-ofatumumab (bien mediante un ensayo inmunoabsorbente ligado a enzimas [ELISA] o por electroquimioluminiscencia) durante y después de periodos de tratamiento de 4 a 45 semanas. No hubo formación de anticuerpos anti-ofatumumab en pacientes con LLC después del tratamiento con ofatumumab.
Eficacia clínica y seguridad
Se ha evaluado la eficacia de Arzerra en dos estudios clínicos (OMB110911 y OMB115991) en pacientes con LLC no tratados previamente y considerados no adecuados para recibir un tratamiento basado en fludarabina, y en dos estudios clínicos (Hx-CD20-406 y Hx-CD20-402) en pacientes con LLC en recaída o refractaria.
LLC no tratada previamente:
El estudio OMB110911 (aleatorizado, abierto, con un brazo en paralelo, multicéntrico) evaluó la eficacia de Arzerra en combinación con clorambucilo frente a clorambucilo en monoterapia en 447 pacientes con LLC no tratados previamente, y considerados no adecuados para recibir un tratamiento basado en fludarabina (por ejemplo, debido a una edad avanzada o a la presencia de comorbilidades), con enfermedad activa e indicados para el tratamiento. Los pacientes recibieron Arzerra mediante perfusiones intravenosas mensuales (Ciclo 1: 300 mg en el día 1, y 1.000 mg en el día 8. Ciclos posteriores: 1.000 mg en el día 1, cada 28 días) en combinación con clorambucilo (10 mg/m2 administrados por vía oral en los días 1-7, cada 28 días) o clorambucilo en monoterapia (10 mg/m2 administrados por vía oral en los días 1-7, cada 28 días). Los pacientes recibieron el tratamiento durante un mínimo de 3 meses hasta obtener la mejor respuesta o hasta un máximo de 12 ciclos. La mediana de la edad fue de 69 años (rango: 35 a 92 años), el 27 % de los pacientes eran > 75 años de edad, el 63 % eran hombres y el 89 % eran de raza blanca. La mediana de la escala de valoración acumulativa de enfermedades en geriatría (Cumulative Illness Rating Score for Geriatrics, CIRS-G) fue 9, y el 31 % de los pacientes presentaban un CIRS-G > 10. La mediana del aclaramiento de creatinina (Creatinine Clearance, CrCl), evaluado mediante la fórmula de Cockroft-Gault, fue de 70 ml/min, y el 48 % de los pacientes presentaron un CrCl de < 70 ml/min. Los pacientes con un estado funcional de 0 a 2 según la Eastern Cooperative Oncology Group (ECOG) fueron reclutados en el estudio, y el 91 % presentaban una estado funcional ECOG de 0 ó 1. Aproximadamente el 60 % de los pacientes recibieron de 3-6 ciclos de Arzerra, y un 32 % recibieron de 7-12 ciclos. La mediana del número de ciclos completos en pacientes fue de 6 (una dosis total de Arzerra de 6.300 mg).
La variable principal fue la mediana de la supervivencia libre de progresión (SLP) evaluada por un Comité de Revisión Independiente (Independent Review Committee, IRC) ciego, y se utilizaron las guías International Workshop for Chronic Lymphocytic Leukaemia (IWCLL) (año 2008), actualizada por el Working Group esponsorizado por el National Cancer Institute (NCI-WG). La tasa global de respuesta (Overall Response Rate, ORR), incluyendo respuestas completas (RC) fueron también evaluadas por un IRC utilizando las guías del IWCLL del año 2008.
Arzerra en combinación con clorambucilo, mostró una mejora estadísticamente significativa de un 71 % en la mediana de la SLP en comparación con clorambucilo en monoterapia (HR: 0,57; IC 95 %: 0,45; 0,72) (ver Tabla 1, Figura 1). Los beneficios en la SLP con la incorporación de Arzerra se observaron en todos los pacientes, incluyendo aquellos con características biológicas de peor pronóstico (como las deleciones 17p o 11q, IGHV no mutado, P2M > 3.500 pg/l, y expresión ZAP-70).
Tabla 1. Resumen de la SLP de Arzerra en combinación con Clorambucilo en comparación con Clorambucilo en monoterapia, en pacientes con LLC no tratados previamente
|
Evaluación primaria por el IRC y Análisis de Subgrupos para la SLP, Meses |
Clorambucilo (N = 226) |
Arzerra y Clorambucilo (N = 221) |
|
Mediana, todos los pacientes |
13,1 |
22,4 |
|
IC 95 % |
(10,6; 13,8) |
(19,0; 25,2) |
|
Hazard Ratio |
0,57 (0,45; 0,72) | |
|
Valor de P |
p < 0,001 | |
|
Edad > 75 años (n = 119) |
12,2 |
23,8 |
|
Comorbilidad 0 ó 1 (n = 126) |
10,9 |
23,0 |
|
Comorbilidad 2 ó mayor (n = 321) |
13,3 |
21,9 |
|
ECOG 0, 1 (n = 411) |
13,3 |
23,0 |
|
ECOG 2 (n = 35) |
7,9 |
20,9 |
|
CIRS-G < 10 (n = 310) |
13,1 |
21,7 |
|
CIRS-G > 10 (n = 137) |
12,2 |
23,2 |
|
CrCl < 70 mL/min (n = 214) |
10,9 |
23,1 |
|
CrCl > 70 mL/min (n = 227) |
14,5 |
22,1 |
|
17p or 11q deletion (n = 90) |
7,9 |
13,6 |
|
IGHV mutado (< 98 %) (n = 177) |
12,2 |
30,5 |
|
IGHV no mutado (> 98 %) (n = 227) |
11,7 |
17,3 |
|
P2M < 3.500 pg/l (n = 109) |
13,8 |
25,5 |
|
P2M > 3.500 pg/l (n = 322) |
11,6 |
19,6 |
|
ZAP-70 positivo (n = 161) |
9,7 |
17,7 |
|
ZAP-70 intermedio (n = 160) |
13,6 |
25,3 |
|
ZAP-70 negativo (n = 100) |
13,8 |
25,6 |
|
IGHV mutado & ZAP-70 negativo (n = 60) |
10,5 |
NR |
|
IGHV mutado & ZAP-70 positivo |
7,9 |
27,2 |
|
(n = 35) | ||
|
IGHV no mutado & ZAP-70 negativo |
16,7 |
16,2 |
|
(n = 27) | ||
|
IGHV no mutado & ZAP-70 positivo |
11,2 |
16,2 |
|
(n = 122) | ||
Abreviaturas: P2M= Beta-2-microglobulina, IC= intervalo de confianza; CIRS-G= Cumulative Illness Rating Scale for Geriatrics (escala de valoración acumulativa de enfermedades en geriatría); LLC= Leucemia Linfocítica Crónica, CrCl= Creatinine Clearance (aclaramiento de creatinina), ECOG= Eastern Cooperative Oncology Group, IGHV= Immunoglobulin Heavy Chain Variable Region (región variable de la cadena pesada de inmunoglobulina); IRC= Independent Review Committee (comité de evaluación independiente); N= número; NR= Not reached (no alcanzado); SLP= Supervivencia Libre de Progresión, ZAP-70= Zeta-Chain-associated protein kinase 70 (cadena Zeta asociada a la proteína quinasa 70).
Los datos disponibles en población heterogénea que no es de raza blanca y en pacientes con un estado funcional ECOG = 2, son limitados.
Figura 1. Estimaciones de Kaplan-Meier por el IRC-evaluación de la SLP
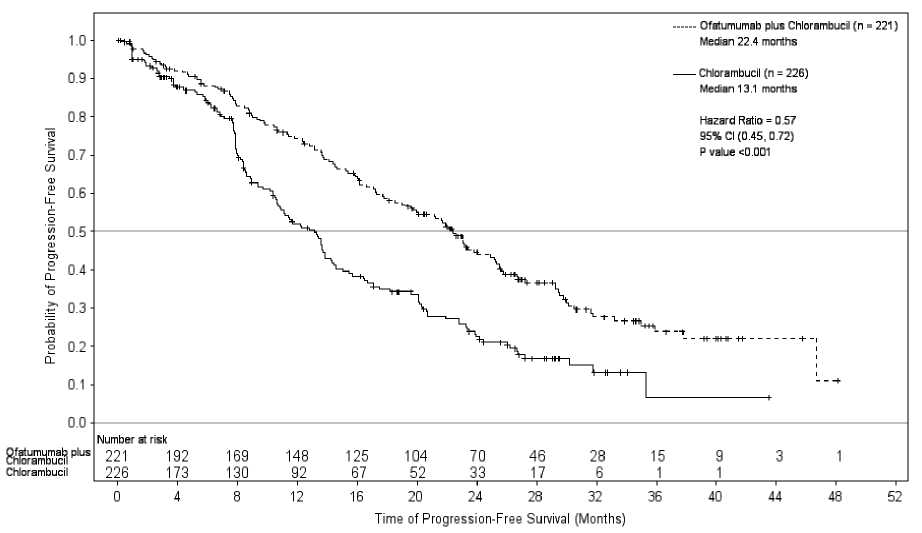
Tabla 2. Resumen de las Variables Secundarias de Arzerra en Combinación con Clorambucilo Comparada con Clorambucilo en monoterapia, en pacientes con LLC No Tratados Previamente
|
Evaluación de las variables secundarias por un IRC |
Clorambucilo (N = 226) |
Arzerra y Clorambucilo (N = 221) |
|
ORR (%) |
69 |
82 |
|
IC 95 % |
(62,1; 74,6) |
(76,7; 87,1) |
|
Valor de P |
p < 0,001 | |
|
RC (%) |
1 |
12 |
|
RC con MRD negativa (% of CR) |
0 |
37 |
|
Mediana duración de la respuesta, todos os |
13 2 |
22 1 |
|
pacientes, meses | ||
|
IC 95 % |
(10,8; 16,4) |
(19,1; 24,6) |
|
Valor de P |
p < 0,001 | |
Abreviaturas: IC= intervalo de confianza; LLC= Leucemia Linfocítica Crónica, RC= Respuesta Completa; IRC= Independent Review Committee (comité de revisión independiente), MRD= Minimal Residue Disease (enfermedad residual mínima), N= número; ORR= Overall Response Rate (tasa de respuesta global).
El estudio OMB115991 evaluó la eficacia de Arzerra en combinación con bendamustina en 44 pacientes con LLC no tratados previamente, y considerados no adecuados para recibir tratamiento basado en fludarabina. Los pacientes recibieron Arzerra mediante perfusiones intravenosas mensuales (Ciclo 1:
300 mg en el día 1, y 1.000 mg en el día 8. Ciclos posteriores: 1.000 mg en el día 1, cada 28 días) en combinación con bendamustina administrada por vía intravenosa (90 mg/m2 en los días 1 y 2, cada 28 días). Los pacientes recibieron el tratamiento durante un mínimo de 3 ciclos, y los pacientes con enfermedad estable o que respondieron después de 3 ciclos, continuaron el tratamiento durante otros 3 ciclos hasta un máximo de 6 ciclos. La mediana del número de ciclos completados en pacientes fue de 6 (una dosis total de Arzerra de 6.300 mg).
La variable principal fue la ORR evaluada por el investigador de acuerdo con las guías IWCLL del año 2008.
Los resultados de este estudio mostraron que Arzerra en combinación con bendamustina es un tratamiento efectivo, proporcionando una ORR del 95 % (IC 95 %: 85, 99) y una RC del 43 %. Más de la mitad de los pacientes (56 %) con RC eran MRD negativos tras completar el tratamiento en estudio.
No hay datos disponibles que comparen Arzerra en combinación con bendamustina o con clorambucilo, frente a un régimen de tratamiento basado en rituximab, tal como rituximab con clorambucilo. Por consiguiente, el beneficio de la nueva combinación frente a un régimen basado en rituximab se desconoce.
LLC refractaria:
Arzerra fue administrado en monoterapia a 223 pacientes con LLC refractaria (estudio Hx-CD20-406). La mediana de edad de los pacientes fue de 64 años (intervalo: de 41 a 87 años), y la mayoría fueron hombres (73 %) y de raza blanca (96 %). Los pacientes habían recibido una media de 5 tratamientos previos, incluyendo rituximab (57 %). De estos 223 pacientes, 95 pacientes fueron refractarios al tratamiento con fludarabina y alemtuzumab (definido como la incapacidad de alcanzar al menos una respuesta parcial con el tratamiento con fludarabina o alemtuzumab, o progresión de la enfermedad dentro de los 6 meses desde la última dosis de fludarabina o alemtuzumab). Los datos citogenéticos basales (FISH) estuvieron disponibles para 209 pacientes. Se detectaron aberraciones cromosómicas en 174 pacientes y 36 pacientes presentaron un cariotipo normal; hubo 47 pacientes con deleción 17p, 73 pacientes con deleción 11q,
23 pacientes con trisomía 12q y 31 pacientes con deleción 13q como única aberración.
La tasa de respuesta global (ORR) fue del 49 % en pacientes refractarios a fludarabina y alemtuzumab (ver en la Tabla 3 un resumen de los datos de eficacia del estudio). Los pacientes que recibieron tratamiento previo con rituximab, ya sea como monoterapia o en combinación con otros medicamentos, respondieron al tratamiento con ofatumumab en una tasa similar a aquellos que no recibieron tratamiento previo con rituximab.
Tabla 3. Resumen de la respuesta a Arzerra en pacientes con LLC refractaria
|
Variable (Primaria) 1 |
Pacientes refractarios a fludarabina y alemtuzumab n = 95 |
|
Tasa de respuesta global Respondedores, n (%) |
47 (49) |
|
IC (%) 95,3 % |
39; 60 |
|
Tasa de respuesta en pacientes con tratamiento previo con rituximab Respondedores, n (%) |
25/56 (45) |
|
IC (%) 95 % |
31; 59 |
|
Tasa de respuesta en pacientes con anomalías cromosómicas Deleción de 17p Respondedores, n (%) |
10/27 (37) |
|
IC (%) 95 % |
19; 58 |
|
Deleción de 11q Respondedores, n (%) |
15/32 (47) |
|
IC (%) 95 % |
29; 65 |
|
Mediana de supervivencia global Meses |
13,9 |
|
IC 95 % |
9,9; 18,6 |
|
Supervivencia libre de progresión Meses |
4,6 |
|
IC 95 % |
3,9; 6,3 |
|
Mediana de duración de la respuesta Meses |
5,5 |
|
IC 95 % |
3,7; 7,2 |
|
Mediana del tiempo hasta el siguiente tratamiento de LLC Meses |
8,5 |
|
IC 95 % |
7,2; 9,9 |
|
1 La respuesta global fue evaluada por un Comité de Respuesta Independiente utilizando las guías del | |
|
NCIWG para LLC del año 1996. | |
También se demostraron mejoras en los componentes de los criterios de respuesta del NCI-WG. Estos incluyeron mejoras asociadas con síntomas constitucionales, linfoadenopatía, organomegalia, o citopenias (ver Tabla 4).
Tabla 4. Resumen de mejora clínica con una duración mínima de 2 meses en pacientes refractarios con anomalías en situación basal
|
Variable de eficacia o parámetro hematológicoa |
Pacientes con beneficios/pacientes con anomalías en situación basal (%) |
|
Pacientes refractarios a fludarabina y alemtuzumab | |
|
Recuento de linfocitos Disminución > 50 % Normalización (< 4 x 109/l) |
49/71 (69) 36/71 (51) |
|
Resolución completa de los síntomas constitucionalesb |
21/47 (45) |
|
Linfoadenopatía0 Mejoría > 50 % Resolución completa |
51/88 (58) 17/88 (19) |
|
Esplenomegalia Mejoría > 50 % Resolución completa |
27/47 (57) 23/47 (49) |
|
Hepatomegalia | |
|
Mejoría > 50% |
14/24 (58) |
|
Resolución completa |
11/24 (46) |
|
Hemoglobina < 11 g/dl en situación basal hasta > 11 g/dl post basal |
12/49 (24) |
|
Recuento de plaquetas < 100 x 109/l en situación basal hasta incremento > 50 % o > 100 x 109/l post basal |
19/50 (38) |
|
Neutrófilos < 1 x 109/l en situación basal hasta > 1,5 x 109/l |
1/17 (6) |
|
a Excluye las visitas de pacientes desde la fecha de la primera transfusión, tratamiento con eritropoyetina, o tratamiento con factores de crecimiento. Para los pacientes con ausencia de datos en situación basal, la última exploración/no programada de datos se tomará como situación basal. b Resolución completa de los síntomas constitucionales (fiebre, sudores nocturnos, fatiga, pérdida de peso) definido como la presencia de cualquier síntoma en situación basal, seguido de ausencia de síntomas. c Linfoadenopatía medida por la suma de los productos de los diámetros mayores (SPD) evaluados mediante exploración física. | |
Arzerra fue también administrado a un grupo de pacientes (n = 112) con linfoadenopatía voluminosa (definida como al menos un nódulo linfático > 5 cm) que eran además refractarios a fludarabina. La tasa de respuesta global (ORR) en este grupo fue del 43 % (IC 95,3 %: 33, 53). La mediana de supervivencia libre de progresión fue de 5,5 meses (IC 95 %: 4,6; 6,4) y la mediana de supervivencia global fue de 17,4 meses (IC 95 %: 15,0; 24,0). La tasa de respuesta en pacientes tratados previamente con rituximab fue del 38 % (IC 95 %: 23; 61). Estos pacientes también experimentaron una mejoría clínica comparable, en términos de variables de eficacia y parámetros hematológicos detallados anteriormente, con los pacientes refractarios a fludarabina y alemtuzumab
Adicionalmente, un grupo de pacientes (n=16) que eran intolerantes/inelegibles al tratamiento con fludarabina y/o intolerantes al tratamiento con alemtuzumab, fueron tratados con Arzerra. La tasa de respuesta global en este grupo fue del 63 % (IC 95,3 %: 35, 85).
Se realizó un estudio abierto, de dos brazos de tratamiento, aleatorizado (OMB114242) en pacientes con LLC refractarios a fludarabina que no respondieron al menos a dos tratamientos previos (n = 122) y que presentaban enfermedad voluminosa, en el que se comparó Arzerra en monoterapia (n = 79) frente a un tratamiento elegido por el médico (n = 43). No hubo diferencia estadísticamente significativa en la variable principal de la SLP evaluada por un Comité de Revisión Independiente (5,4 vs. 3,6 meses,
HR = 0,79, p = 0,27). Los resultados de la SLP en el brazo de Arzerra en monoterapia fueron comparables a los resultados observados en el tratamiento de Arzerra en monoterapia del estudio Hx-CD20-406.
Se realizó un estudio (Hx-CD20-402) de escalado de dosis en 33 pacientes con LLC en recaída o refractaria. La mediana de la edad de los pacientes fue de 61 años (intervalo: de 27 a 82 años), la mayoría fueron hombres (58%) y todos eran blancos. El tratamiento con ofatumumab (cuando se administran 4 perfusiones semanales), obtuvo una tasa de respuesta objetiva del 50 % en el grupo de dosis más alta (1a dosis: 500 mg; 2a, 3a y 4a dosis: 2.000 mg) e incluyó 12 remisiones parciales y una remisión parcial nodular. Para el grupo de dosis más alta, la mediana de tiempo a progresión fue de 15,6 semanas (95 %
IC: 15-22,6) en el análisis completo de la población, y de 23 semanas (IC: 20-31,4) en los respondedores. La duración de la respuesta fue de 16 semanas (IC: 13,3 - 19,0) y el tiempo hasta el próximo tratamiento para LLC fue de 52,4 semanas (IC: 36,9 - no estimable).
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Arzerra en los diferentes grupos de la población pediátrica en Leucemia Linfocítica Crónica (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Ofatumumab se administra mediante perfusión intravenosa; por lo tanto, la absorción no es aplicable. Las concentraciones plasmáticas máximas de ofatumumab se observaron generalmente al final o inmediatamente después de la perfusión. Los datos farmacocinéticos procedentes de 215 pacientes con LLC refractaria estuvieron disponibles. El valor de la media geométrica de Cmax fue 61 pg/ml tras la primera perfusión (300 mg); después de la octava perfusión semanal (siete perfusiones de 2.000 mg), el valor de la media geométrica de Cmax fue de 1.391 pg/ml y el valor de la media geométrica del AUC^.^ fue de 463.418 pg.h/ml; después de la duodécima perfusión, (cuatro perfusiones mensuales de 2.000 mg), el valor de la media geométrica de Cmax fue 827 pg/ml y el valor de la media geométrica del AUC(0.^) fue 203.536 pg.h/ml. En pacientes con LLC no tratados previamente que recibieron ofatumumab y clorambucilo, la media geométrica de los valores de Cmax tras la primera perfusión (300 mg), la perfusión de 1.000 mg en el día 8 y la perfusión de 1.000 mg en el cuarto ciclo mensual, fue de 52 pg/ml, 241 pg/ml, y 285 pg/ml respectivamente; la media geométrica del valor de AUC(0-X) en el cuarto ciclo fue de 65.100 pg.h/ml.
Distribución
Ofatumumab tiene un volumen de distribución pequeño, con valores medios de Vss que van desde 1,7 hasta 8,1 l a través de los estudios, niveles de dosis y número de perfusión.
Biotransformación
Ofatumumab es una proteína para la cual se espera que la ruta metabólica sea la degradación en pequeños péptidos y aminoácidos individuales mediante enzimas proteolíticas omnipresentes. No se han realizado estudios clásicos de biotransformación.
Eliminación
Ofatumumab es eliminado de dos formas: una ruta independiente de la diana como otras moléculas IgG y una ruta mediada por dianas que está relacionada con la unión a células B. Hubo una reducción rápida y sostenida de las células CD20+ B después de la primera perfusión de ofatumumab, dejando un número reducido de células CD20+ disponibles para que el anticuerpo se una en las perfusiones posteriores. Como resultado, la tasa de aclaramiento de ofatumumab fue menor y los valores de t./2 fueron significativamente mayores tras las perfusiones posteriores que tras la perfusión inicial; durante las perfusiones semanales repetidas, los valores del AUC y Cmax de ofatumumab aumentaron más que la acumulación esperada basándose en los datos de la primera perfusión.
En los estudios en pacientes con LLC en recaída o refractarios, los valores de la media geométrica para CL y t/ fueron 64 ml/h (intervalo 4,3-1.122 ml/h) y 1,3 días (intervalo 0,2-6,0 días) después de la primera perfusión, 8,5 ml/h (intervalo 1,3-41,5 ml/h) y 11,5 días (intervalo 2,3-30,6 días) después de la cuarta perfusión, 11,7 ml/h (intervalo 3,9-54,2 ml/h) y 13,6 días (intervalo 2,4-36,0 días) después de la octava perfusión, y 12,1 ml/h (intervalo 3,0-233 ml/h) y 11,5 días (intervalo 1,8-36,4 días) después de la duodécima perfusión.
En pacientes con LLC no tratados previamente que recibieron ofatumumab y clorambucilo, la media geométrica de CL y los valores de t/ fueron de 15,4 ml/h (intervalo 4,1-146 ml/h) y 18,5 días (intervalo 2,7-82,6 días) después de la cuarta perfusión.
Pacientes de edad avanzada (65 años y mayores)
En el análisis farmacocinético de un estudio cruzado de una población de pacientes con edades comprendidas entre 21 y 87 años, la edad no resultó ser un factor significativo en la farmacocinética de ofatumumab.
Niños y adolescentes
No hay datos farmacocinéticos disponible en pacientes pediátricos.
Género
En el análisis de población de un estudio cruzado, el género tuvo un efecto modesto (12 %) en el volumen de distribución central de ofatumumab, con valores de Cmax y AUC mayores observados en mujeres (el 48 % de los pacientes del análisis fueron hombres y el 52 % fueron mujeres); estos efectos no se consideran clínicamente relevantes y no se recomiendan ajustes de la dosis.
Insuficiencia renal
En el análisis de un estudio cruzado de una población de pacientes con un aclaramiento de creatinina calculado en un rango de valores de 26 a 287 ml/min, el aclaramiento de creatinina calculado en situación basal no resultó ser un factor significativo en la farmacocinética de ofatumumab. No se recomienda el ajuste de la dosis para la insuficiencia renal de leve a moderada (aclaramiento de creatinina > 30 ml/min). Los datos farmacocinéticos en pacientes con insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min), son limitados.
Insuficiencia hepática
No se han llevado a cabo estudios formales para examinar el efecto de la insuficiencia hepática. Las moléculas IgG1 como ofatumumab son catabolizadas por enzimas proteolíticas omnipresentes, que no están restringidas al tejido hepático; por lo tanto, es improbable que los cambios en la función hepática tengan algún efecto en la eliminación de ofatumumab.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos.
La administración intravenosa y subcutánea en monos trajo como consecuencia una reducción esperada en el recuento de células B linfoides y periféricas, sin hallazagos toxicológicos asociados. Como se preveía, se observó una reducción en la respuesta inmune humoral de IgG en la hemocianina de lapa, pero no hubo efectos en las respuestas de hipersensibilidad de tipo retardado. En unos pocos animales, se produjo un aumento de la destrucción de glóbulos rojos como resultado de los anticuerpos antifármaco que recubren los glóbulos rojos de los monos. Se observó el correspondiente aumento en los recuentos de reticulocitos en estos monos, que fue indicativo de una respuesta regenerativa en médula ósea.
La administración intravenosa de 100 mg/kg de ofatumumab una vez a la semana en monas cinomolgus preñadas, entre los días 20 y 50 de gestación, no provocó toxicidad fetal o maternal o teratogenicidad. En el día 100 de la gestación, se observó una reducción de células B relacionada con la actividad farmacológica de ofatumumab en la sangre del cordón fetal y en los tejidos esplénicos fetales. No se han realizado estudios de desarrollo pre y postnatal. Por lo tanto, la recuperación postnatal no ha sido demostrada.
Dado que ofatumumab es un anticuerpo monoclonal, no se han realizado estudios de genotoxicidad y carcinogenicidad con ofatumumab.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Arginina
Acetato sódico (E262)
Cloruro sódico Polisorbato 80 (E433)
Edetato disódico (E386)
Ácido clorhídrico (E507) (para ajustar el pH)
Agua para preparaciones inyectables
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros excepto con los mencionados en la sección 6.6.
6.3 Periodo de validez Vial
3 años.
Perfusión diluida
Se ha demostrado estabilidad en uso química y física durante 48 horas en condiciones ambientales (menos de 25°C).
Desde el punto de vista microbiológico, el medicamento se debe utilizar inmediatamente. Si no se utiliza inmediatamente, los tiempos y condiciones de almacenamiento en uso previos a la utilización, son responsabilidad del usuario y no deben ser mayores de 24 horas a 2-8°C, a menos que la reconstitución/dilución se hayan realizado en condiciones asépticas controladas y validadas.
6.4 Precauciones especiales de conservación
Conservar y transportar refrigerado (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Para las condiciones de conservación tras la dilución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Vial de vidrio claro Tipo I con tapón de goma de bromobutilo libre de látex y lámina de cierre de aluminio, conteniendo 5 ml de concentrado para solución para perfusión.
Arzerra está disponible en envases de 3 viales.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Arzerra concentrado para solución para perfusión no contiene conservantes; por consiguiente la dilución debe llevarse a cabo bajo condiciones asépticas. La solución diluida para la perfusión debe utilizarse en las 24 horas siguientes a la preparación. Transcurrido ese tiempo, se debe desechar la solución no utilizada.
• Antes de diluir Arzerra
Antes de la dilución se debe comprobar el concentrado de Arzerra para detectar cualquier partícula o decoloración. Ofatumumab debe ser una solución de incolora a color amarillo pálido. No utilizar el concentrado de Arzerra si presenta decoloración.
No se debe agitar el vial de ofatumumab para esta inspección.
• Cómo se debe diluir la solución para perfusión
Antes de la administración, el concentrado de Arzerra debe diluirse en una solución para inyección de cloruro sódico 9 mg/ml (0,9 %), utilizando técnicas asépticas.
Dosis de 300 mg - Utilizar 3 viales (15 ml en total, 5 ml por vial):
- extraer y desechar 15 ml de una bolsa con 1.000 ml de solución para inyección de cloruro sódico 9 mg/ml (0,9 %);
- extraer 5 ml de ofatumumab de cada uno de los 3 viales e inyectarlos en la bolsa de 1.000 ml;
- no se debe agitar, mezclar la solución diluida mediante una inversión suave.
• Cómo se debe administrar la solución diluida
Arzerra no se debe administrar como una inyección intravenosa o en bolo. Se debe administrar utilizando una bomba para perfusión intravenosa.
La perfusión debe ser completada en un plazo de 24 horas tras su preparación. Transcurrido ese tiempo desechar la solución no utilizada.
Arzerra no se debe mezclar o administrar como una perfusión con otros medicamentos o soluciones intravenosas. Para evitar esto, limpiar la vía con una solución para inyección de cloruro sódico 9 mg/ml (0,9 %) antes y después de la administración de ofatumumab.
LLC no tratada previamente:
En la primera perfusión, administrar durante más de 4,5 horas (ver sección 4.2), a través de una vía periférica o un catéter permanente, de acuerdo con el siguiente esquema:
Perfusión 1: esquema
|
Tiempo (minutos) |
ml/hora |
|
0 - 30 |
12 |
|
31 - 60 |
25 |
|
61 - 90 |
50 |
|
91 - 120 |
100 |
|
121 - 150 |
200 |
|
151 - 180 |
300 |
|
180 + |
400 |
Si la primera perfusión se ha completado sin una reacción adversa grave, las perfusiones restantes (2-13) de 1.000 mg se deben administrar durante más de 4 horas (ver sección 4.2), a través de una vía periférica o un catéter permanente, de acuerdo con el siguiente esquema:
Perfusiones de la 2 a la 13: esquema
|
Tiempo (minutos) |
ml/hora |
|
0 - 30 |
25 |
|
31 - 60 |
50 |
|
61 - 90 |
100 |
|
91 - 120 |
200 |
|
121 + |
400 |
LLC refractaria:
La primera y la segunda perfusión se deben administrar durante más de 6,5 horas (ver sección 4.2), a través de una vía periférica o un catéter permanente, de acuerdo con el siguiente esquema:
Perfusiones 1 y 2: esquema
|
Tiempo (minutos) |
ml/hora |
|
0 - 30 |
12 |
|
31 - 60 |
25 |
|
61 - 90 |
50 |
|
91 - 120 |
100 |
|
121 + |
200 |
Si la segunda perfusión se ha completado sin ninguna reacción adversa grave, las perfusiones restantes (3-12) deben ser administradas durante más de 4 horas (ver sección 4.2), a través de una vía periférica o un catéter permanente, de acuerdo con el siguiente esquema:
Perfusiones 3 a 12: esquema
|
Tiempo (minutos) |
ml/hora |
|
0 - 30 |
25 |
|
31 - 60 |
50 |
|
61 - 90 |
100 |
|
91 - 120 |
200 |
|
121 + |
400 |
Si se observa cualquier reacción adversa, la velocidad de perfusión se debe reducir (ver sección 4.2).
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/10/625/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 19/04/2010 Fecha de la última renovación: 16/01/2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
1. NOMBRE DEL MEDICAMENTO
Arzerra 1.000 mg concentrado para solución para perfusión
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un ml de concentrado contiene 20 mg de ofatumumab.
Cada vial contiene 1.000 mg de ofatumumab en 50 ml.
Ofatumumab es un anticuerpo monoclonal humano producido en una línea celular recombinante murina (NS0).
Excipiente(s) con efecto conocido:
Este medicamento contiene 34,8 mg de sodio por dosis de 300 mg, 116 mg de sodio por dosis de 1.000 mg y 232 mg de sodio por dosis de 2.000 mg.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Concentrado para solución para perfusión (concentrado estéril).
Líquido de transparente a opalescente, de incoloro a color amarillo pálido.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Leucemia linfocítica crónica (LLC) no tratada previamente:
Arzerra en combinación con clorambucilo o bendamustina está indicado para el tratamiento de pacientes con LLC que no han recibido tratamiento previo y que no son adecuados para un tratamiento basado en fludarabina.
Para mayor información, ver sección 5.1.
LLC refractaria:
Arzerra está indicado para el tratamiento de pacientes con LLC que son refractarios a fludarabina y alemtuzumab.
Para mayor información, ver sección 5.1.
4.2 Posología y forma de administración
Arzerra se debe administrar bajo la supervisión de un médico experimentado en el uso de tratamientos para el cáncer y en un entorno donde se disponga de instalaciones para la reanimación completa de forma inmediata.
Monitorización
Durante la administración de ofatumumab, especialmente durante la primera perfusión, se debe monitorizar estrechamente a los pacientes ante la aparición de reacciones debidas a la perfusión, incluyendo el síndrome de liberación de citoquinas.
Premedicación
Los pacientes siempre deben ser premedicados de 30 minutos a 2 horas antes de la perfusión de Arzerra, de acuerdo a los siguientes esquemas de dosificación:
LLC no tratada previamente:
• 1.000 mg de paracetamol por vía oral (acetaminofeno) o equivalente, más
• antihistamínico por vía oral o intravenosa (50 mg de difenhidramina o 10 mg de cetirizina o equivalente), más
• corticosteroides por vía intravenosa (50 mg de prednisolona u otro equivalente)
Después de la primera y la segunda perfusión, si el paciente no experimenta una reacción adversa grave al medicamento (RAM), se puede reducir u omitir la premedicación con corticosteroides en las perfusiones posteriores, según el criterio del médico.
LLC refractaria:
• 1.000 mg de paracetamol (acetaminofeno) por vía oral o equivalente, más
• antihistamínico por vía oral o intravenosa (50 mg de difenhidramina o 10 mg de cetirizina u otro equivalente), más
• corticosteroides por vía intravenosa (100 mg de prednisolona o equivalente)
Si la segunda perfusión semanal se ha completado sin RAM graves, se puede reducir la dosis de corticosteroides en las perfusiones de la 3 a la 8, según el criterio del médico.
Antes de la novena perfusión (primera perfusión mensual), los pacientes deben recibir la dosis total de medicamentos utilizados en la premedicación tal y como se describe anteriormente. Si la novena perfusión se completa sin RAM graves, la dosis puede ser reducida al equivalente de 50 mg de prednisolona en las perfusiones posteriores, según el criterio del médico.
Posología
LLC no tratada previamente:
La dosis recomendada y programada es de 300 mg en el día 1, seguido de 1.000 mg una semana después en el día 8 (ciclo 1), seguido de 1.000 mg en el día 1 de los ciclos posteriores, durante un mínimo de 3 ciclos, hasta obtener la mejor respuesta o un máximo de 12 ciclos (cada 28 días).
La mejor respuesta, fue aquella respuesta clínica que no mejoró tras recibir 3 ciclos adicionales de tratamiento.
Primera perfusión
La velocidad inicial de la primera perfusión de Arzerra debe ser de 12 ml/hora. Durante la perfusión, la velocidad se debe incrementar cada 30 minutos hasta un máximo de 400 ml/hora (ver sección 6.6).
Perfusiones posteriores
Si la primera perfusión se ha completado sin RAM graves relacionadas con la perfusión, las perfusiones posteriores se pueden iniciar a una velocidad de 25 ml/hora que se debe incrementar cada 30 minutos hasta un máximo de 400 ml/hora (ver sección 6.6).
LLC refractaria:
La dosis recomendada es de 300 mg para la primera perfusión y de 2.000 mg para todas las perfusiones posteriores. El esquema de perfusión es de 8 semanas consecutivas, y transcurrido un periodo de interrupción de 4-5 semanas, dicho esquema pasará a ser de 4 meses consecutivos (por ej. una perfusión cada 4 semanas).
Primera y segunda perfusión
La velocidad inicial de la primera y la segunda perfusión de Arzerra debe ser de 12 ml/hora. Durante la perfusión, la velocidad se debe incrementar cada 30 minutos hasta un máximo de 200 ml/hora (ver sección 6.6).
Perfusiones posteriores
Si la segunda perfusión se ha completado sin RAM graves relacionadas con la perfusión, las perfusiones restantes se pueden iniciar a una velocidad de 25 ml/hora y esta se debe incrementar cada 30 minutos hasta un máximo de 400 ml/hora (ver sección 6.6).
Modificación de la dosis y reinicio del tratamiento para RAM relacionadas con la perfusión - en pacientes con LLC no tratados previamente y pacientes con LLC refractaria.
Si aparecen RAM relacionadas con la perfusión de cualquier gravedad, interrumpir la perfusión. El tratamiento se puede retomar según el criterio del médico responsable del tratamiento. A modo de guía, se pueden utilizar las siguientes modificaciones de la velocidad de perfusión:
• En el caso de que se produzca una RAM de leve a moderada, la perfusión se debe interrumpir y reiniciarse cuando el estado del paciente sea estable, a la mitad de la velocidad de perfusión que se aplicaba en el momento de la interrupción. Antes de interrumpir la perfusión debido a una RAM, si la velocidad de perfusión no se ha incrementado desde la velocidad inicial de 12 ml/hora, la perfusión debe reiniciarse a una velocidad de 12 ml/hora, velocidad de perfusión estándar. La velocidad de perfusión se puede seguir incrementando de acuerdo a los procedimientos estándar, de acuerdo al criterio médico y a la tolerancia del paciente (no exceder el incremento de la velocidad de cada 30 minutos).
• En el caso de una reacción adversa grave, la perfusión se debe interrumpir y reiniciarse a una velocidad de perfusión de 12 ml/hora, cuando el estado del paciente sea estable. La velocidad de perfusión se puede seguir incrementando de acuerdo a los procedimientos estándar, de acuerdo al criterio del médico y a la tolerancia del paciente (no exceder el incremento de la velocidad de cada 30 minutos).
Población pediátrica
Arzerra no está recomendado para el uso en niños menores de 18 años debido a datos insuficientes en la seguridad y/o eficacia.
Pacientes de edad avanzada
No se observaron diferencias sustanciales en la seguridad y la eficacia relacionadas con la edad (ver sección 5.1). En base a los datos de seguridad y eficacia en pacientes de edad avanzada, no se requieren ajustes de la dosis (ver sección 5.2).
Insuficiencia renal
No se han realizado estudios formales con Arzerra en pacientes con insuficiencia renal. No se recomiendan ajustes de la dosis para insuficiencia renal de leve a moderada (aclaramiento de creatinina > 30 ml/min) (ver sección 5.2).
Insuficiencia hepática
No se han realizado estudios formales con Arzerra en pacientes con insuficiencia hepática. Sin embargo, es improbable que los pacientes con insuficiencia hepática requieran una modificación de la dosis (ver sección 5.2).
Forma de administración
Arzerra se administra en perfusión intravenosa y se debe diluir antes de la administración. Para consultar las instrucciones de dilución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad a ofatumumab o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Reacciones debidas a la perfusión
La administración intravenosa de ofatumumab se ha asociado con reacciones debidas a la perfusión. Estas reacciones pueden provocar la interrupción temporal del tratamiento, o la retirada del mismo. Las premedicaciones atenúan las reacciones debidas a la perfusión, pero éstas pueden seguir apareciendo, predominantemente durante la primera perfusión. Las reacciones debidas a la perfusión pueden incluir, pero no estar limitadas a, acontecimientos anafilactoides, broncoespasmo, acontecimientos cardiacos (por ejemplo, isquemia miocárdica / infarto, bradicardia), escalofríos, tos, síndrome de liberación de citoquinas, diarrea, disnea, fatiga, rubefacción, hipertensión, hipotensión, náuseas, dolor, edema pulmonar, prurito, pirexia, erupción y urticaria. En raras ocasiones, estas reacciones pueden provocar la muerte. Incluso con la premedicación, tras el uso de ofatumumab se han notificado reacciones graves, incluyendo síndrome de liberación de citoquinas. En los casos de reacción grave debida a la perfusión, la perfusión de Arzerra se debe interrumpir inmediatamente e iniciar el tratamiento sintomático (ver sección 4.2).
Las reacciones debidas a la perfusión aparecen con mayor frecuencia en el primer día de perfusión y tienden a disminuir con las perfusiones posteriores. Los pacientes con un historial de función pulmonar disminuida, pueden tener mayor riesgo de complicaciones pulmonares debido a reacciones graves y se deben monitorizar estrechamente durante la perfusión de ofatumumab.
Síndrome de lisis tumoral
En pacientes con LLC, puede aparecer síndrome de lisis tumoral (SLT) con el uso de ofatumumab. Los factores de riesgo del SLT incluyen una mayor carga tumoral, concentraciones elevadas de células circulantes (> 25.000/mm3), hipovolemia, insuficiencia renal, niveles elevados de ácido úrico antes del tratamiento y niveles elevados de lactato deshidrogenasa. El manejo del SLT incluye la corrección de las alteraciones electrolíticas, monitorización de la función renal, mantenimiento del balance de fluidos y tratamiento de soporte.
Leucoencefalopatía multifocal progresiva
Se ha notificado leucoencefalopatía multifocal progresiva (LMP) y muerte en pacientes con LLC que reciben farmacoterapia citotóxica, incluyendo ofatumumab. Se debe valorar la realización de un diagnóstico de LMP en cualquier paciente en tratamiento con Arzerra que notifique la aparicición de nuevos signos y síntomas neurológicos o cambios en los signos y síntomas neurológicos preexistentes. Si se sospecha de un diagnóstico de LMP, se debe interrumpir el tratamiento con Arzerra y remitir al paciente a un neurólogo.
Inmunizaciones
No se ha estudiado la seguridad y la capacidad para generar una respuesta primaria o anamnésica a la inmunización con vacunas vivas atenuadas o inactivadas durante el tratamiento con ofatumumab. La respuesta a la vacunación podría verse afectada cuando se agotan las células B. Debido al riesgo de infección, la administración de vacunas vivas atenuadas se debe evitar durante y después del tratamiento con ofatumumab, hasta que los recuentos de células B se normalicen. Se deben valorar los riesgos y los beneficios de la vacunación de pacientes durante el tratamiento con ofatumumab.
Hepatitis B
En pacientes tratados con medicamentos clasificados como anticuerpos citolíticos de acción directa sobre CD20, entre los que se incluye Arzerra, puede aparecer infección y reactivación del virus de la hepatitis B (VHB), que en algunos casos da lugar a hepatitis fulminante. Se han notificado casos en pacientes con resultados positivos del antígeno de superficie de la hepatitis B (HBsAg), y también en aquellos pacientes con resultados positivos de anticuerpos frente al antigeno core de la hepatitis B (anti-HBc) y con HBsAg negativo. La reactivación del VHB también ha aparecido en pacientes que aparentemente ya no presentaban infección por hepatitis B (por ejemplo, HBsAG negativo, anti-HBc positivo, y anti-HBs positivo).
La reactivación del VHB se define como un incremento brusco en la replicación del VHB, que se manifiesta con un aumento rápido de los niveles de ADN del VHB en suero, o mediante la detección del HBsAg en una persona que previamente presentó resultados HBsAg negativo y anti-HBc positivo. Generalmente, la reactivación de la replicación del VHB va seguida de hepatitis, es decir: aumento de los niveles de transaminasas, y en casos graves, incremento de los niveles de bilirrubina, fallo hepático, y muerte.
Antes de iniciar el tratamiento con Arzerra, todos los pacientes deben ser monitorizados para descartar una posible infección por el VHB, mediante la medición de HBsAg y anti-HBc. Los pacientes que presenten evidencias de infección previa por hepatitis B (HBsAg negativo, anti-HBc positivo), deben consultar a médicos con experiencia en el manejo de la hepatitis B en relación a la monitorización e inicio del tratamiento antiviral del VHB. No se debe iniciar el tratamiento con Arzerra en pacientes con evidencias recientes de infección por hepatitis B (HbsAg positivo), hasta que la infección se haya tratado adecuadamente.
Los pacientes que presenten evidencias de infección previa por el VHB, deben ser monitorizados ante cualquier signo clínico o de laboratorio relacionado con la hepatitis o la reactivación del VHB, durante el tratamiento con Arzerra y durante los 6-12 meses siguientes a la última perfusión de Arzerra. Se han notificados casos de reactivación del VHB hasta 12 meses después de finalizar el tratamiento. La interrupción del tratamiento antiviral del VHB se debe consultar con un médico con experiencia en el manejo de la hepatitis B.
Los pacientes que desarrollen una reactivación del VHB durante el tratamiento con Arzerra, deben de interrumpir inmediatamente el tratamiento con Arzerra y cualquier otra quimioterapia concomitante, e instaurar un tratamiento adecuado. Existen pocos datos de seguridad sobre la reanudación del tratamiento con Arzerra en pacientes que desarrollan una reactivación del VHB. La decisión de reanudar el tratamiento con Arzerra en pacientes que presenten una reactivación del VHB se debe consultar con un médico con experiencia en el manejor de la hepatitis B.
Cardiovascular
Se debe monitorizar estrechamente a los pacientes con antecedentes de enfermedad cardiaca. Se debe interrumpir el tratamiento con Arzerra en los pacientes que experimentan arritmias cardiacas graves o potencialmente mortales.
En un análisis agrupado de tres estudios abiertos en pacientes con LLC (N = 85), se evaluó el efecto de múltiples dosis de Arzerra sobre el intervalo QTc. En el análisis agrupado, se observaron incrementos por encima de 5 milisegundos en la mediana/media de los intervalos QT/QTc. No se detectaron grandes cambios en la media del intervalo QTc (esto es, > 20 milisegundos). Ningún paciente presentó un incremento en el intervalo QTc > 500 milisegundos. No se detectaron incrementos en el intervalo QTc dependientes de la concentración. Antes y durante la administración de ofatumumab, se recomienda medir los niveles de electrolitos, como el potasio y el magnesio, a los pacientes. Las anomalías en los niveles de electrolitos deben ser corregidas. Se desconoce el efecto de ofatumumab en pacientes con prolongación del intervalo QT (por ejemplo, prolongación adquirida o congénita).
Obstrucción intestinal
Se ha notificado obstrucción intestinal en pacientes que reciben tratamiento con anticuerpos monoclonales anti CD20, incluyendo ofatumumab. Se debe evaluar a los pacientes que presentan dolor abdominal, especialmente al principio del ciclo de tratamiento con ofatumumab, e iniciar el tratamiento adecuado.
Monitorización de valores analíticos de laboratorio
Durante el tratamiento con ofatumumab se han notificado citopenias, incluyendo neutropenia prolongada y neutropenia tardía. Durante el tratamiento con ofatumumab, se deben realizar recuentos sanguíneos completos, incluyendo recuento de neutrófilos y de plaquetas a intervalos regulares, y con mayor frecuencia en pacientes que desarrollan citopenias.
Contenido en sodio
Este medicamento contiene 34,8 mg de sodio por dosis de 300 mg, 116 mg por dosis de 1.000 mg y 232 mg de sodio por dosis de 2.000 mg. Esto debe tenerse en cuenta en pacientes con una dieta controlada en sodio.
4.5 Interacción con otros medicamentos y otras formas de interacción
Aunque existen datos limitados de interacción de ofatumumab con otros fármacos, no se conocen interacciones clínicamente significativas con otros medicamentos.
Ofatumumab no tiene un efecto clínicamente relevante sobre la farmacocinética de clorambucilo o su metabolito activo, la mostaza del ácido fenilacético.
La eficacia de vacunas vivas atenuadas o inactivadas puede verse alterada con ofatumumab. Por tanto, debe evitarse el uso concomitante de estos agentes con ofatumumab. Si la coadminsitración se considera inevitable, se deben valorar los riesgos y los beneficios de vacunar a los pacientes durante el tratamiento con ofatumumab (ver sección 4.4).
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos sobre el uso de ofatumumab en mujeres embarazadas. Los estudios en animales no indican efectos dañinos directos o indirectos en relación con la toxicidad reproductiva (ver sección 5.3). Ofatumumab no debe adminsitrase a mujeres embarazadas a menos que el posible beneficio para la madre supere el posible riesgo para el feto.
Las mujeres en edad fértil tienen que utilizar métodos anticonceptivos eficaces durante y hasta 12 meses depués del tratamiento con ofatumumab.
Lactancia
Se desconoce si ofatumumab se excreta en la leche materna, no obstante la IgG humana se secreta en la leche materna. No se ha establecido el uso seguro de ofatumumab en humanos durante la lactancia. La excrección de ofatumumab en la leche no ha sido estudiada en animales. Los datos publicados sugieren que el consumo de leche materna en recien nacidos y lactantes no da lugar a una absorción importante de éstos anticuerpos maternos en circulación. No se puede descartar el riesgo en recien nacidos/lactantes. La lactancia debe suspenderse durante el tratamiento con ofatumumab y durante los 12 meses posteriores al tratamiento.
Fertilidad
No hay datos sobre los efectos de ofatumumab en la fertilidad humana. Los efectos en la fertilidad masculina y femenina no han sido evaluados en estudios con animales.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas.
En base a la farmacología de ofatumumab, no se esperan efectos perjudiciales en estas actividades. Al considerar la capacidad del paciente para realizar tareas que requieran juicio, capacidades motoras o cognitivas, se debe tener en cuenta el estado clínico del paciente y el perfil de reacciones adversas de ofatumumab (ver sección 4.8)
4.8 Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad global de ofatumumab en pacientes con LLC (no tratados previamente y en recaída o refractarios) está basado en los datos procedentes de 511 pacientes incluidos en los ensayos clínicos (ver sección 5.1). Aquí se incluyen los 250 pacientes tratados con ofatumumab en monoterapia (en pacientes con LLC en recaída o refractarios) y 261 pacientes tratados en combinación con un agente alquilante (en pacientes que no han recibido tratamiento previo para la LLC y que no son adecuados para recibir un tratamiento a base de fludarabina).
El perfil de efectos adversos de ofatumumab en pacientes con LLC refractarios a fludarabina que no respondieron al menos a dos tratamientos previos y que presentaban enfermedad voluminosa, fue consistente con el perfil de seguridad global establecido a partir de otros estudios en LLC, tal y como se describe a continuación, en la tabla.
Tabla de reacciones adversas
Las reacciones adversas notificadas con ofatumumab en pacientes con LLC no tratados previamente y en recaída o refractarios, ya sea solo o en combinación con un agente alquilante, se incluyen a continuación por la clasificación de órganos del sistema MedDRA y por frecuencia. Muy frecuentes (> 1/10), Frecuentes (> 1/100 a < 1/10), Poco frecuentes (> 1/1.000 a < 1/100), Raras (> 1/10.000 a < 1/1.000),
Muy raras (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Clasificación de órganos del sistema MedDRA |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
|
Infecciones e infestaciones |
Infección del tracto respiratorio inferior, incluyendo neumonía, infección del tracto respiratorio superior |
Sepsis, incluyendo sepsis neutropénica y shock séptico, infección por virus del herpes, infección del tracto urinario |
Infección de Hepatitis B y reactivación | |
|
Trastornos de la sangre y del sistema linfático |
Neutropenia, anemia |
Neutropenia febril, trombocitopenia, leucopenia |
Agranulocitosis, coagulopatía, aplasia eritrocitaria, linfopenia | |
|
Trastornos del sistema inmunológico |
Reacciones anafilactoides*, hipersensibilidad * |
Shock anafiláctico* | ||
|
Trastornos del metabolismo y de la nutrición |
Síndrome de lisis tumoral | |||
|
Trastornos cardiacos |
Taquicardia* |
Bradicardia* | ||
|
Trastornos vasculares |
Hipotensión*, hipertensión* | |||
|
Trastornos respiratorios, torácicos y mediastínicos |
Broncoespasmo*, hipoxia*, disnea*, malestar torácico*, dolor faringolaríngeo*, tos*, congestión nasal* |
Edema pulmonar* | ||
|
Trastornos gastrointestinales |
Náusea* |
Diarrea* |
Obstrucción en el intestino delgado | |
|
Trastornos de la piel y del tejido subcutáneo |
Erupción* |
Urticaria*, prurito*, rubefacción* | ||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor de espalda* | |||
|
Trastornos generales y alteraciones en el lugar de administración |
Pirexia* |
Síndrome de liberación de citoquinas*, sensación de frío*, escalofríos*, hiperhidrosis*, fatiga* | ||
|
*Es probable que estos eventos sean atribuibles a ofatumumab al producirse una reacción a |
la perfusión, y | |||
éstos normalmente aparecieron después de iniciar la perfusión y en las 24 horas siguientes a la finalización de la perfusión (ver sección 4.4).
Descripción de las reacciones adversas seleccionadas
Reacciones relacionadas con la perfusión
En los ensayos clínicos en pacientes que recibieron ofatumumab, las reacciones adversas observadas con mayor frecuencia fueron reacciones relacionadas con la perfusión, que ocurrieron en un 68 % (348/511) de los pacientes, durante cualquier momento del tratamiento. La mayoria de las reacciones relacionadas con la perfusión fueron de gravedad Grado 1 o Grado 2. El ocho por ciento de los pacientes presentaron reacciones relacionadas con la perfusión de Grado > 3, durante cualquier momento del tratamiento. El dos por ciento de las reacciones relacionas con la perfusión condujeron a la suspensión del tratamiento. No hubo reacciones mortales relacionadas con la perfusión (ver sección 4.4).
Infecciones
De los 511 pacientes que recibieron ofatumumab en los ensayos clínicos, 300 pacientes (59 %) experimentaron una infección. Estas fueron infecciones bacterianas, virales o fúngicas. Ciento cuatro (20 %) de los 511 pacientes experimentaron infecciones > Grado 3. Veintiocho (5 %) de los 511 pacientes experimentaron infecciones mortales.
Neutropenia
De los 511 pacientes que recibieron ofatumumab en los ensayos clínicos, 139 pacientes (27 %) experimentaron un evento adverso asociado con un descenso en el recuento de neutrófilos; 118 (23 %) de los 511 pacientes experimentaron eventos adversos > Grado 3 asociados con un descenso en el recuento de neutrófilos. Cuarenta y dos (8 %) pacientes experimentaron eventos adversos graves asociados con un descenso en el recuento de neutrófilos.
En el estudio pivotal en pacientes con CLL no tratados previamente (OMB110911), se notificó neutropenia prolongada (definida como neutropenia de Grado 3 o 4, que no se resolvió entre los 24 y 42 días desde el último tratamiento) en 41 pacientes (23 pacientes tratados con ofatumumab y clorambucilo, 18 pacientes tratados con clorambucilo en monoterapia). Nueve pacientes tratados con ofatumumab y clorambucilo, y tres pacientes tratados solamente con clorambucilo presentaron neutropenia tardía, definida como neutropenia de Grado 3 o 4 que comienza al menos 42 días después del último tratamiento.
Cardiovascular
En un análisis agrupado de tres estudios abiertos en pacientes con LLC (N = 85), se evaluó el efecto de multiples dosis de Arzerra sobre el intervalo QTc. En el análisis agrupado, se observaron incrementos por encima de 5 milisegundos en la mediana/media de los intervalos QT/QTc. No se detectaron grandes cambios en la media del intervalo QTc (esto es, > 20 milisegundos). Ningún paciente presentó incrementos en el intervalo QTc > 500 milisegundos. No se detectaron incrementos en el intervalo QTc dependientes de la concentración.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Anexo V.
4.9 Sobredosis
No se han notificado casos de sobredosis.
PROPIEDADES FARMACOLÓGICAS
5.
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: anticuerpos monoclonales, código ATC: L01XC10 Mecanismo de acción
Ofatumumab es un anticuerpo monoclonal humano (IgG1) que se une específicamente a un epítopo bien diferenciado que abarca los bucles extracelulares grande y pequeño de la molécula CD20. La molécula CD20 es una fosfoproteína transmembrana expresada en los linfocitos B desde el estadío pre-B hasta linfocito B maduro y en tumores de células B. Los tumores de células B incluyen LLC (generalmente asociados con niveles más bajos de expresión de CD20) y linfomas no Hodgkin (donde > 90 % de los tumores presentan niveles elevados de expresión de CD20). La molécula CD20 no se desprende de la superficie celular y no se interioriza tras la unión del anticuerpo.
La unión de ofatumumab al epítopo proximal a la membrana de la molécula CD20 induce el reclutamiento y la activación de la vía del complemento en la superficie celular, que origina citotoxicidad dependiente del complemento y consiguiente lisis de las células tumorales. Se ha demostrado que ofatumumab induce de manera importante la lisis de las células con elevados niveles de expresión de las moléculas de defensa del complemento. También se ha demostrado que ofatumumab induce la lisis celular de células con alto y bajo grado de expresión de CD20 y en células resistentes a rituximab. Además, la unión de ofatumumab permite el reclutamiento de las células natural killer, que permiten la inducción de la muerte celular a través de la citotoxicidad celular anticuerpo dependiente.
Efectos farmacodinámicos
Los recuentos de células B periféricas disminuyeron tras la primera perfusión de ofatumumab en pacientes con neoplasias hematológicas. En pacientes con LLC refractaria, la mediana del descenso en el recuento de células B fue de un 22 % después de la primera perfusión y de un 92 % en la octava perfusión semanal. En la mayoría de los pacientes el recuento de células B periféricas se mantuvo bajo durante el resto del tratamiento, y en los pacientes que respondieron al tratamiento permaneció por debajo de los valores de referencia hasta 15 meses después de la última dosis.
En pacientes con LLC no tratados previamente, la mediana de las reducciones del recuento de células B después del primer ciclo de tratamiento y antes del sexto ciclo mensual fue de un 94 % y > 99 % respectivamente para la combinación de ofatumumab con clorambucilo, y de un 73 % y un 97 % respectivamente para clorambucilo en monoterapia. A los 6 meses después de la última dosis, la mediana de las reducciones del recuento de células B fue > 99 % para la combinación de ofatumumab y clorambucilo, y de un 94 % para clorambucilo en monoterapia.
Inmunogenicidad
Es posible que aparezca inmunogenicidad con proteínas terapéuticas como ofatumumab. A lo largo del programa clínico en LLC, se analizaron muestras de suero de más de 440 pacientes para detectar la presencia de anticuerpos anti-ofatumumab (bien mediante un ensayo inmunoabsorbente ligado a enzimas [ELISA] o por electroquimioluminiscencia) durante y después de periodos de tratamiento de 4 a 45 semanas. No hubo formación de anticuerpos anti-ofatumumab en pacientes con LLC después del tratamiento con ofatumumab.
Eficacia clínica y seguridad
Se ha evaluado la eficacia de Arzerra en dos estudios clínicos (OMB110911 y OMB115991) en pacientes con LLC no tratados previamente y considerados no adecuados para recibir un tratamiento basado en fludarabina, y en dos estudios clínicos (Hx-CD20-406 y Hx-CD20-402) en pacientes con LLC en recaída o refractaria.
LLC no tratada previamente:
El estudio OMB110911 (aleatorizado, abierto, con un brazo en paralelo, multicéntrico) evaluó la eficacia de Arzerra en combinación con clorambucilo frente a clorambucilo en monoterapia en 447 pacientes con LLC no tratados previamente, y considerados no adecuados para recibir un tratamiento basado en fludarabina (por ejemplo, debido a una edad avanzada o a la presencia de comorbilidades), con enfermedad activa e indicados para el tratamiento. Los pacientes recibieron Arzerra mediante perfusiones intravenosas mensuales (Ciclo 1: 300 mg en el día 1, y 1.000 mg en el día 8. Ciclos posteriores: 1.000 mg en el día 1, cada 28 días) en combinación con clorambucilo (10 mg/m2 administrados por vía oral en los días 1-7, cada 28 días) o clorambucilo en monoterapia (10 mg/m2 administrados por vía oral en los días 1-7, cada 28 días). Los pacientes recibieron el tratamiento durante un mínimo de 3 meses hasta obtener la mejor respuesta o hasta un máximo de 12 ciclos. La mediana de la edad fue de 69 años (rango: 35 a 92 años), el 27 % de los pacientes eran > 75 años de edad, el 63 % eran hombres y el 89 % eran de raza blanca. La mediana de la escala de valoración acumulativa de enfermedades en geriatría (Cumulative Illness Rating Score for Geriatrics, CIRS-G) fue 9, y el 31 % de los pacientes presentaban un CIRS-G > 10. La mediana del aclaramiento de creatinina (Creatinine Clearance, CrCl), evaluado mediante la fórmula de Cockroft-Gault, fue de 70 ml/min, y el 48 % de los pacientes presentaron un CrCl de < 70 ml/min. Los pacientes con un estado funcional de 0 a 2 según la Eastern Cooperative Oncology Group (ECOG) fueron reclutados en el estudio, y el 91 % presentaban una estado funcional ECOG de 0 ó 1. Aproximadamente el 60 % de los pacientes recibieron de 3-6 ciclos de Arzerra, y un 32 % recibieron de 7-12 ciclos. La mediana del número de ciclos completos en pacientes fue de 6 (una dosis total de Arzerra de 6.300 mg).
La variable principal fue la mediana de la supervivencia libre de progresión (SLP) evaluada por un Comité de Revisión Independiente (Independent Review Committee, IRC) ciego, y se utilizaron las guías International Workshop for Chronic Lymphocytic Leukaemia (IWCLL) (año 2008), actualizada por el Working Group esponsorizado por el National Cancer Institute (NCI-WG). La tasa global de respuesta (Overall Response Rate, ORR), incluyendo respuestas completas (RC) fueron también evaluadas por un IRC utilizando las guías del IWCLL del año 2008.
Arzerra en combinación con clorambucilo, mostró una mejora estadísticamente significativa de un 71 % en la mediana de la SLP en comparación con clorambucilo en monoterapia (HR: 0,57; IC 95 %: 0,45; 0,72) (ver Tabla 1, Figura 1). Los beneficios en la SLP con la incorporación de Arzerra se observaron en todos los pacientes, incluyendo aquellos con características biológicas de peor pronóstico (como las deleciones 17p o 11q, IGHV no mutado, P2M > 3.500 pg/l, y expresión ZAP-70).
Tabla 1. Resumen de la SLP de Arzerra en combinación con Clorambucilo en comparación con Clorambucilo en monoterapia, en pacientes con LLC no tratados previamente
|
Evaluación primaria por el IRC y Análisis de Subgrupos para la SLP, Meses |
Clorambucilo (N = 226) |
Arzerra y Clorambucilo (N = 221) |
|
Mediana, todos los pacientes |
13,1 |
22,4 |
|
IC 95 % |
(10,6; 13,8) |
(19,0; 25,2) |
|
Hazard Ratio |
0,57 (0,45; 0,72) | |
|
Valor de P |
p < 0,001 | |
|
Edad >75 años (n = 119) |
12,2 |
23,8 |
|
Comorbilidad 0 ó 1 (n = 126) |
10,9 |
23,0 |
|
Comorbilidad 2 ó mayor (n = 321) |
13,3 |
21,9 |
|
ECOG 0, 1 (n = 411) |
13,3 |
23,0 |
|
ECOG 2 (n = 35) |
7,9 |
20,9 |
|
CIRS-G < 10 (n = 310) |
13,1 |
21,7 |
|
CIRS-G > 10 (n = 137) |
12,2 |
23,2 |
|
CrCl < 70 mL/min (n = 214) |
10,9 |
23,1 |
|
CrCl > 70 mL/min (n = 227) |
14,5 |
22,1 |
|
17p or 11q deletion (n = 90) |
7,9 |
13,6 |
|
IGHV mutado (< 98 %) (n = 177) |
12,2 |
30,5 |
|
IGHV no mutado (> 98 %) (n = 227) |
11,7 |
17,3 |
|
P2M < 3.500 pg/l (n = 109) |
13,8 |
25,5 |
|
P2M > 3.500 pg/l (n = 322) |
11,6 |
19,6 |
|
ZAP-70 positivo (n = 161) |
9,7 |
17,7 |
|
ZAP-70 intermedio (n = 160) |
13,6 |
25,3 |
|
ZAP-70 negativo (n = 100) |
13,8 |
25,6 |
|
IGHV mutado & ZAP-70 negativo (n = 60) |
10,5 |
NR |
|
IGHV mutado & ZAP-70 positivo |
7,9 |
27,2 |
|
(n = 35) | ||
|
IGHV no mutado & ZAP-70 negativo |
16,7 |
16,2 |
|
(n = 27) | ||
|
IGHV no mutado & ZAP-70 positivo |
11,2 |
16,2 |
|
(n = 122) | ||
|
Abreviaturas: P2M= Beta-2-microglobu |
ina, IC= intervalo de confianza; CIRS-G= Cumulative Illi | |
Rating Scale for Geriatrics (escala de valoración acumulativa de enfermedades en geriatría); LLC= Leucemia Linfocítica Crónica, CrCl= Creatinine Clearance (aclaramiento de creatinina), ECOG= Eastern Cooperative Oncology Group, IGHV= Immunoglobulin Heavy Chain Variable Region (región variable de la cadena pesada de inmunoglobulina); IRC= Independent Review Committee (comité de evaluación independiente); N= número; NR= Not reached (no alcanzado); SLP= Supervivencia Libre de Progresión, ZAP-70= Zeta-Chain-associated protein kinase 70 (cadena Zeta asociada a la proteína quinasa 70).
Los datos disponibles en población heterogénea que no es de raza blanca y en pacientes con un estado funcional ECOG = 2, son limitados.
Figura 1. Estimaciones de Kaplan-Meier por el IRC-evaluación de la SLP
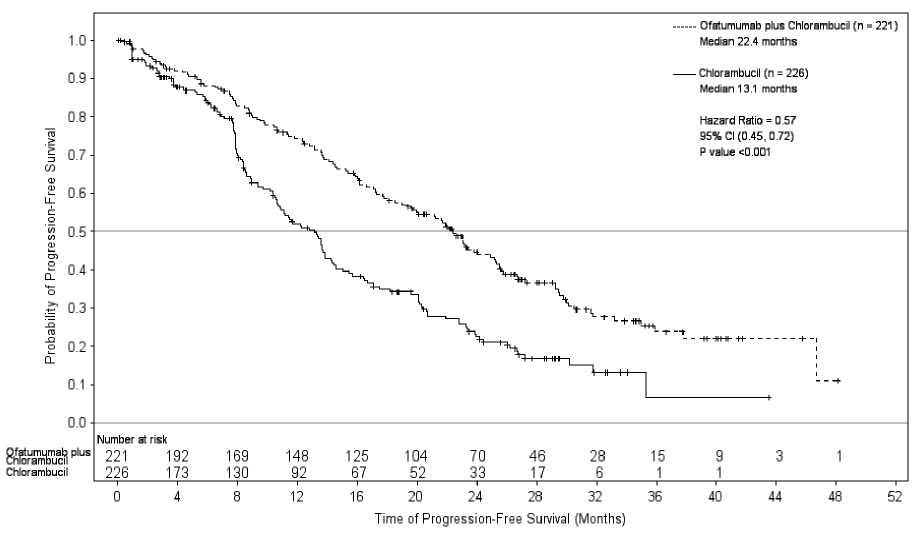
Tabla 2. Resumen de las Variables Secundarias de Arzerra en Combinación con Clorambucilo Comparada con Clorambucilo en monoterapia, en pacientes con LLC No Tratados Previamente
|
Evaluación de las variables secundarias por un IRC |
Clorambucilo (N = 226) |
Arzerra y Clorambucilo (N = 221) |
|
ORR (%) |
69 |
82 |
|
IC 95 % |
(62,1; 74,6) |
(76,7; 87,1) |
|
Valor de P |
p < 0,001 | |
|
RC (%) |
1 |
12 |
|
RC con MRD negativa (% of CR) |
0 |
37 |
|
Mediana duración de la respuesta, todos os |
13 2 |
22 1 |
|
pacientes, meses | ||
|
IC 95 % |
(10,8; 16,4) |
(19,1; 24,6) |
|
Valor de P |
p < 0,001 | |
Abreviaturas: IC= intervalo de confianza; LLC= Leucemia Linfocítica Crónica, RC= Respuesta Completa; IRC= Independent Review Committee (comité de revisión independiente), MRD= Minimal Residue Disease (enfermedad residual mínima), N= número; ORR= Overall Response Rate (tasa de respuesta global).
El estudio OMB115991 evaluó la eficacia de Arzerra en combinación con bendamustina en 44 pacientes con LLC no tratados previamente, y considerados no adecuados para recibir tratamiento basado en fludarabina. Los pacientes recibieron Arzerra mediante perfusiones intravenosas mensuales (Ciclo 1:
300 mg en el día 1, y 1.000 mg en el día 8. Ciclos posteriores: 1.000 mg en el día 1, cada 28 días) en combinación con bendamustina administrada por vía intravenosa (90 mg/m2 en los días 1 y 2, cada 28 días). Los pacientes recibieron el tratamiento durante un mínimo de 3 ciclos, y los pacientes con enfermedad estable o que respondieron después de 3 ciclos, continuaron el tratamiento durante otros 3 ciclos hasta un máximo de 6 ciclos. La mediana del número de ciclos completados en pacientes fue de 6 (una dosis total de Arzerra de 6.300 mg).
La variable principal fue la ORR evaluada por el investigador de acuerdo con las guías IWCLL del año 2008.
Los resultados de este estudio mostraron que Arzerra en combinación con bendamustina es un tratamiento efectivo, proporcionando una ORR del 95 % (IC 95 %: 85, 99) y una RC del 43 %. Más de la mitad de los pacientes (56 %) con RC eran MRD negativos tras completar el tratamiento en estudio.
No hay datos disponibles que comparen Arzerra en combinación con bendamustina o con clorambucilo, frente a un régimen de tratamiento basado en rituximab, tal como rituximab con clorambucilo. Por consiguiente, el beneficio de la nueva combinación frente a un régimen basado en rituximab se desconoce.
LLC refractaria:
Arzerra fue administrado en monoterapia a 223 pacientes con LLC refractaria (estudio Hx-CD20-406). La mediana de edad de los pacientes fue de 64 años (intervalo: de 41 a 87 años), y la mayoría fueron hombres (73 %) y de raza blanca (96 %). Los pacientes habían recibido una media de 5 tratamientos previos, incluyendo rituximab (57 %). De estos 223 pacientes, 95 pacientes fueron refractarios al tratamiento con fludarabina y alemtuzumab (definido como la incapacidad de alcanzar al menos una respuesta parcial con el tratamiento con fludarabina o alemtuzumab, o progresión de la enfermedad dentro de los 6 meses desde la última dosis de fludarabina o alemtuzumab). Los datos citogenéticos basales (FISH) estuvieron disponibles para 209 pacientes. Se detectaron aberraciones cromosómicas en 174 pacientes y 36 pacientes presentaron un cariotipo normal; hubo 47 pacientes con deleción 17p, 73 pacientes con deleción 11q,
23 pacientes con trisomía 12q y 31 pacientes con deleción 13q como única aberración.
La tasa de respuesta global (ORR) fue del 49 % en pacientes refractarios a fludarabina y alemtuzumab (ver en la Tabla 3 un resumen de los datos de eficacia del estudio). Los pacientes que recibieron tratamiento previo con rituximab, ya sea como monoterapia o en combinación con otros medicamentos, respondieron al tratamiento con ofatumumab en una tasa similar a aquellos que no recibieron tratamiento previo con rituximab.
Tabla 3. Resumen de la respuesta a Arzerra en pacientes con LLC refractaria
|
Variable (Primaria) 1 |
Pacientes refractarios a fludarabina y alemtuzumab n = 95 |
|
Tasa de respuesta global Respondedores, n (%) |
47 (49) |
|
IC (%) 95,3 % |
39; 60 |
|
Tasa de respuesta en pacientes con tratamiento previo con rituximab Respondedores, n (%) |
25/56 (45) |
|
IC (%) 95 % |
31; 59 |
|
Tasa de respuesta en pacientes con anomalías cromosómicas Deleción de 17p Respondedores, n (%) |
10/27 (37) |
|
IC (%) 95 % |
19; 58 |
|
Deleción de 11q Respondedores, n (%) |
15/32 (47) |
|
IC (%) 95 % |
29; 65 |
|
Mediana de supervivencia global Meses |
13,9 |
|
IC 95 % |
9,9; 18,6 |
|
Supervivencia libre de progresión Meses |
4,6 |
|
IC 95 % |
3,9; 6,3 |
|
Mediana de duración de la respuesta Meses |
5,5 |
|
IC 95 % |
3,7; 7,2 |
|
Mediana del tiempo hasta el siguiente tratamiento de LLC Meses |
8,5 |
|
IC 95 % |
7,2; 9,9 |
|
1 La respuesta global fue evaluada por un Comité de Respuesta Independiente utilizando las guías del | |
|
NCIWG para LLC del año 1996. | |
También se demostraron mejoras en los componentes de los criterios de respuesta del NCI-WG. Estos incluyeron mejoras asociadas con síntomas constitucionales, linfoadenopatía, organomegalia, o citopenias (ver Tabla 4).
Tabla 4. Resumen de mejora clínica con una duración mínima de 2 meses en pacientes refractarios con anomalías en situación basal
|
Variable de eficacia o parámetro hematológicoa |
Pacientes con beneficios/pacientes con anomalías en situación basal (%) |
|
Pacientes refractarios a fludarabina y alemtuzumab | |
|
Recuento de linfocitos Disminución > 50 % Normalización (< 4 x 109/l) |
49/71 (69) 36/71 (51) |
|
Resolución completa de los síntomas constitucionalesb |
21/47 (45) |
|
Linfoadenopatía0 Mejoría > 50 % Resolución completa |
51/88 (58) 17/88 (19) |
|
Esplenomegalia Mejoría > 50 % Resolución completa |
27/47 (57) 23/47 (49) |
|
Hepatomegalia | |
|
Mejoría > 50 % |
14/24 (58) |
|
Resolución completa |
11/24 (46) |
|
Hemoglobina < 11 g/dl en situación basal hasta > 11 g/dl post basal |
12/49 (24) |
|
Recuento de plaquetas < 100 x 109/l en situación basal hasta incremento > 50 % o > 100 x 109/l post basal |
19/50 (38) |
|
Neutrófilos < 1 x 109/l en situación basal hasta > 1,5 x 109/l |
1/17 (6) |
|
a Excluye las visitas de pacientes desde la fecha de la primera transfusión, tratamiento con eritropoyetina, o tratamiento con factores de crecimiento. Para los pacientes con ausencia de datos en situación basal, la última exploración/no programada de datos se tomará como situación basal. b Resolución completa de los síntomas constitucionales (fiebre, sudores nocturnos, fatiga, pérdida de peso) definido como la presencia de cualquier síntoma en situación basal, seguido de ausencia de síntomas. c Linfoadenopatía medida por la suma de los productos de los diámetros mayores (SPD) evaluados mediante exploración física. | |
Arzerra fue también administrado a un grupo de pacientes (n = 112) con linfoadenopatía voluminosa (definida como al menos un nódulo linfático > 5 cm) que eran además refractarios a fludarabina. La tasa de respuesta global (ORR) en este grupo fue del 43 % (IC 95,3 %: 33, 53). La mediana de supervivencia libre de progresión fue de 5,5 meses (IC 95 %: 4,6; 6,4) y la mediana de supervivencia global fue de 17,4 meses (IC 95 %: 15,0; 24,0). La tasa de respuesta en pacientes tratados previamente con rituximab fue del 38 % (IC 95 %: 23; 61). Estos pacientes también experimentaron una mejoría clínica comparable, en términos de variables de eficacia y parámetros hematológicos detallados anteriormente, con los pacientes refractarios a fludarabina y alemtuzumab
Adicionalmente, un grupo de pacientes (n = 16) que eran intolerantes/inelegibles al tratamiento con fludarabina y/o intolerantes al tratamiento con alemtuzumab, fueron tratados con Arzerra. La tasa de respuesta global en este grupo fue del 63 % (IC 95,3 %: 35, 85).
Se realizó un estudio abierto, de dos brazos de tratamiento, aleatorizado (OMB114242) en pacientes con LLC refractarios a fludarabina que no respondieron al menos a dos tratamientos previos (n = 122) y que presentaban enfermedad voluminosa, en el que se comparó Arzerra en monoterapia (n = 79) frente a un tratamiento elegido por el médico (n = 43). No hubo diferencia estadísticamente significativa en la variable principal de la SLP evaluada por un Comité de Revisión Independiente (5,4 vs. 3,6 meses,
HR = 0,79, p = 0,27). Los resultados de la SLP en el brazo de Arzerra en monoterapia fueron comparables a los resultados observados en el tratamiento de Arzerra en monoterapia del estudio Hx-CD20-406.
Se realizó un estudio (Hx-CD20-402) de escalado de dosis en 33 pacientes con LLC en recaída o refractaria. La mediana de la edad de los pacientes fue de 61 años (intervalo: de 27 a 82 años), la mayoría fueron hombres (58 %) y todos eran blancos. El tratamiento con ofatumumab (cuando se administran 4 perfusiones semanales), obtuvo una tasa de respuesta objetiva del 50 % en el grupo de dosis más alta (1a dosis: 500 mg; 2a, 3a y 4a dosis: 2.000 mg) e incluyó 12 remisiones parciales y una remisión parcial nodular. Para el grupo de dosis más alta, la mediana de tiempo a progresión fue de 15,6 semanas (95 %
IC: 15-22,6) en el análisis completo de la población, y de 23 semanas (IC: 20-31,4) en los respondedores. La duración de la respuesta fue de 16 semanas (IC: 13,3 - 19,0) y el tiempo hasta el próximo tratamiento para LLC fue de 52,4 semanas (IC: 36,9 - no estimable).
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Arzerra en los diferentes grupos de la población pediátrica en Leucemia Linfocítica Crónica (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
5.2 Propiedades farmacocinéticas
Absorción
Ofatumumab se administra mediante perfusión intravenosa; por lo tanto, la absorción no es aplicable. Las concentraciones plasmáticas máximas de ofatumumab se observaron generalmente al final o inmediatamente después de la perfusión. Los datos farmacocinéticos procedentes de 215 pacientes con LLC refractaria estuvieron disponibles. El valor de la media geométrica de Cmax fue 61 pg/ml tras la primera perfusión (300 mg); después de la octava perfusión semanal (siete perfusiones de 2.000 mg), el valor de la media geométrica de Cmax fue de 1.391 pg/ml y el valor de la media geométrica del AUC(0_«,) fue de 463.418 pg.h/ml; después de la duodécima perfusión, (cuatro perfusiones mensuales de 2.000 mg), el valor de la media geométrica de Cmax fue 827 pg/ml y el valor de la media geométrica del AUC(0.^) fue 203.536 pg.h/ml. En pacientes con LLC no tratados previamente que recibieron ofatumumab y clorambucilo, la media geométrica de los valores de Cmax tras la primera perfusión (300 mg), la perfusión de 1.000 mg en el día 8 y la perfusión de 1.000 mg en el cuarto ciclo mensual, fue de 52 pg/ml, 241 pg/ml, y 285 pg/ml respectivamente; la media geométrica del valor de AUC(0.X) en el cuarto ciclo fue de 65.100 pg.h/ml.
Distribución
Ofatumumab tiene un volumen de distribución pequeño, con valores medios de Vss que van desde 1,7 hasta 8,1 l a través de los estudios, niveles de dosis y número de perfusión.
Biotransformación
Ofatumumab es una proteína para la cual se espera que la ruta metabólica sea la degradación en pequeños péptidos y aminoácidos individuales mediante enzimas proteolíticas omnipresentes. No se han realizado estudios clásicos de biotransformación.
Ofatumumab es eliminado de dos formas: una ruta independiente de la diana como otras moléculas IgG y una ruta mediada por dianas que está relacionada con la unión a células B. Hubo una reducción rápida y sostenida de las células CD20+ B después de la primera perfusión de ofatumumab, dejando un número reducido de células CD20+ disponibles para que el anticuerpo se una en las perfusiones posteriores. Como resultado, la tasa de aclaramiento de ofatumumab fue menor y los valores de t/2 fueron significativamente mayores tras las perfusiones posteriores que tras la perfusión inicial; durante las perfusiones semanales repetidas, los valores del AUC y Cmax de ofatumumab aumentaron más que la acumulación esperada basándose en los datos de la primera perfusión.
En los estudios en pacientes con LLC en recaída o refractarios, los valores de la media geométrica para CL y t/ fueron 64 ml/h (intervalo 4,3-1.122 ml/h) y 1,3 días (intervalo 0,2-6,0 días) después de la primera perfusión, 8,5 ml/h (intervalo 1,3-41,5 ml/h) y 11,5 días (intervalo 2,3-30,6 días) después de la cuarta perfusión, 11,7 ml/h (intervalo 3,9-54,2 ml/h) y 13,6 días (intervalo 2,4-36,0 días) después de la octava perfusión, y 12,1 ml/h (intervalo 3,0-233 ml/h) y 11,5 días (intervalo 1,8-36,4 días) después de la duodécima perfusión.
En pacientes con LLC no tratados previamente que recibieron ofatumumab y clorambucilo, la media geométrica de CL y los valores de t/ fueron de 15,4 ml/h (intervalo 4,1-146 ml/h) y 18,5 días (intervalo 2,7-82,6 días) después de la cuarta perfusión.
Pacientes de edad avanzada (65 años y mayores)
En el análisis farmacocinético de un estudio cruzado de una población de pacientes con edades comprendidas entre 21 y 87 años, la edad no resultó ser un factor significativo en la farmacocinética de ofatumumab.
Niños y adolescentes
No hay datos farmacocinéticos disponible en pacientes pediátricos.
Género
En el análisis de población de un estudio cruzado, el género tuvo un efecto modesto (12 %) en el volumen de distribución central de ofatumumab, con valores de Cmax y AUC mayores observados en mujeres (el 48 % de los pacientes del análisis fueron hombres y el 52 % fueron mujeres); estos efectos no se consideran clínicamente relevantes y no se recomiendan ajustes de la dosis.
Insuficiencia renal
En el análisis de un estudio cruzado de una población de pacientes con un aclaramiento de creatinina calculado en un rango de valores de 26 a 287 ml/min, el aclaramiento de creatinina calculado en situación basal no resultó ser un factor significativo en la farmacocinética de ofatumumab. No se recomienda el ajuste de la dosis para la insuficiencia renal de leve a moderada (aclaramiento de creatinina > 30 ml/min). Los datos farmacocinéticos en pacientes con insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min), son limitados.
Insuficiencia hepática
No se han llevado a cabo estudios formales para examinar el efecto de la insuficiencia hepática. Las moléculas IgG1 como ofatumumab son catabolizadas por enzimas proteolíticas omnipresentes, que no están restringidas al tejido hepático; por lo tanto, es improbable que los cambios en la función hepática tengan algún efecto en la eliminación de ofatumumab.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos.
La administración intravenosa y subcutánea en monos trajo como consecuencia una reducción esperada en el recuento de células B linfoides y periféricas, sin hallazagos toxicológicos asociados. Como se preveía, se observó una reducción en la respuesta inmune humoral de IgG en la hemocianina de lapa, pero no hubo efectos en las respuestas de hipersensibilidad de tipo retardado. En unos pocos animales, se produjo un aumento de la destrucción de glóbulos rojos como resultado de los anticuerpos antifármaco que recubren los glóbulos rojos de los monos. Se observó el correspondiente aumento en los recuentos de reticulocitos en estos monos, que fue indicativo de una respuesta regenerativa en médula ósea.
La administración intravenosa de 100 mg/kg de ofatumumab una vez a la semana en monas cinomolgus preñadas, entre los días 20 y 50 de gestación, no provocó toxicidad fetal o maternal o teratogenicidad. En el día 100 de la gestación, se observó una reducción de células B relacionada con la actividad farmacológica de ofatumumab en la sangre del cordón fetal y en los tejidos esplénicos fetales. No se han realizado estudios de desarrollo pre y postnatal. Por lo tanto, la recuperación postnatal no ha sido demostrada.
Dado que ofatumumab es un anticuerpo monoclonal, no se han realizado estudios de genotoxicidad y carcinogenicidad con ofatumumab.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Arginina
Acetato sódico (E262)
Cloruro sódico Polisorbato 80 (E433)
Edetato disódico (E386)
Ácido clorhídrico (E507) (para ajustar el pH)
Agua para preparaciones inyectables
6.2 Incompatibilidades
Este medicamento no debe mezclarse con otros excepto con los mencionados en la sección 6.6.
6.3 Periodo de validez Vial
3 años.
Perfusión diluida
Se ha demostrado estabilidad en uso química y física durante 48 horas en condiciones ambientales (menos de 25°C).
Desde el punto de vista microbiológico, el medicamento se debe utilizar inmediatamente. Si no se utiliza inmediatamente, los tiempos y condiciones de almacenamiento en uso previos a la utilización, son responsabilidad del usuario y no deben ser mayores de 24 horas a 2-8°C, a menos que la reconstitución/dilución se hayan realizado en condiciones asépticas controladas y validadas.
6.4 Precauciones especiales de conservación
Conservar y transportar refrigerado (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Para las condiciones de conservación tras la dilución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Vial de vidrio claro Tipo I con tapón de goma de bromobutilo libre de látex y lámina de cierre de aluminio, conteniendo 5 ml de concentrado para solución para perfusión.
Arzerra está disponible en envases de 1 vial.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Arzerra concentrado para solución para perfusión no contiene conservantes; por consiguiente la dilución debe llevarse a cabo bajo condiciones asépticas. La solución diluida para la perfusión debe utilizarse en las 24 horas siguientes a la preparación. Transcurrido ese tiempo, se debe desechar la solución no utilizada.
• Antes de diluir Arzerra
Antes de la dilución se debe comprobar el concentrado de Arzerra para detectar cualquier partícula o decoloración. Ofatumumab debe ser una solución de incolora a color amarillo pálido. No utilizar el concentrado de Arzerra si presenta decoloración.
No se debe agitar el vial de ofatumumab para esta inspección.
• Cómo se debe diluir la solución para perfusión
Antes de la administración, el concentrado de Arzerra debe diluirse en una solución para inyección de cloruro sódico 9 mg/ml (0,9 %), utilizando técnicas asépticas.
Dosis de 1.000 mg - Utilizar 1 vial (50 ml en total, 50 ml por vial):
- extraer y desechar 50 ml de una bolsa con 1.000 ml de solución para inyección de cloruro sódico 9 mg/ml (0,9 %);
- extraer 50 ml de ofatumumab del vial e inyectarlos en la bolsa de 1.000 ml;
- no se debe agitar, mezclar la solución diluida mediante una inversión suave.
Dosis de 2.000 mg - Utilizar 2 viales (100 ml en total, 50 ml por vial):
- extraer y desechar 100 ml de una bolsa con 1.000 ml de solución para inyección de cloruro sódico 9 mg/ml (0,9 %);
- extraer 50 ml de ofatumumab de cada uno de los 2 viales e inyectarlos en la bolsa de 1.000 ml;
- no se debe agitar, mezclar la solución diluida mediante una inversión suave.
• Cómo se debe administrar la solución diluida
Arzerra no se debe administrar como una inyección intravenosa o en bolo. Se debe administrar utilizando una bomba para perfusión intravenosa.
La perfusión debe ser completada en un plazo de 24 horas tras su preparación. Transcurrido ese tiempo desechar la solución no utilizada.
Arzerra no se debe mezclar o administrar como una perfusión con otros medicamentos o soluciones intravenosas. Para evitar esto, limpiar la vía con una solución para inyección de cloruro sódico 9 mg/ml (0,9 %) antes y después de la administración de ofatumumab.
LLC no tratada previamente:
En la primera perfusión, administrar durante más de 4,5 horas (ver sección 4.2), a través de una vía periférica o un catéter permanente, de acuerdo con el siguiente esquema:
Perfusión 1: esquema
|
Tiempo (minutos) |
ml/hora |
|
0 - 30 |
12 |
|
31 - 60 |
25 |
|
61 - 90 |
50 |
|
91 - 120 |
100 |
|
121 - 150 |
200 |
|
151 - 180 |
300 |
|
180 + |
400 |
Si la primera perfusión se ha completado sin una reacción adversa grave, las perfusiones restantes (2-13) de 1.000 mg se deben administrar durante más de 4 horas (ver sección 4.2), a través de una vía periférica o un catéter permanente, de acuerdo con el siguiente esquema:
|
Perfusiones de la 2 a |
a 13: esquema |
|
Tiempo (minutos) |
ml/hora |
|
0 - 30 |
25 |
|
31 - 60 |
50 |
|
61 - 90 |
100 |
|
91 - 120 |
200 |
|
121 + |
400 |
LLC refractaria:
La primera y la segunda perfusión se deben administrar durante más de 6,5 horas (ver sección 4.2), a través de una vía periférica o un catéter permanente, de acuerdo con el siguiente esquema:
Perfusiones 1 y 2: esquema
|
Tiempo (minutos) |
ml/hora |
|
0 - 30 |
12 |
|
31 - 60 |
25 |
|
61 - 90 |
50 |
|
91 - 120 |
100 |
|
121 + |
200 |
Si la segunda perfusión se ha completado sin ninguna reacción adversa grave, las perfusiones restantes (3-12) deben ser administradas durante más de 4 horas (ver sección 4.2), a través de una vía periférica o un catéter permanente, de acuerdo con el siguiente esquema:
Perfusiones 3 a 12: esquema
|
Tiempo (minutos) |
ml/hora |
|
0 - 30 |
25 |
|
31 - 60 |
50 |
|
61 - 90 |
100 |
|
91 - 120 |
200 |
|
121 + |
400 |
Si se observa cualquier reacción adversa, la velocidad de perfusión se debe reducir (ver sección 4.2).
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/10/625/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 19/04/2010 Fecha de la última renovación: 16/01/2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu/.
A. FABRICANTES DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIÓNES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
FABRICANTES DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
A.
Nombre y dirección de los fabricantes del principio activo biológico.
Lonza Biologics plc 228 Bath Road Slough, Berks SL1 4DX Reino Unido
Lonza Biologics, Inc.
101 International Drive Portsmouth, NH 03801-2815 Estados Unidos
Nombre y dirección de los fabricantes responsables de la liberación de los lotes
Glaxo Operations UK Ltd.
Harmire Road Barnard Castle Durham, DL12 8DT Reino Unido
Novartis Pharmaceuticals UK Limited
Frimley Business Park
Frimley
Camberley, Surrey GU16 7SR Reino Unido
Novartis Pharma GmbH RoonstraBe 25 D-90429 Nuremberg Alemania
El prospecto impreso del medicamento debe especificar el nombre y dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD), prevista en el artículo 107 ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
D.
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2. de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de una nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
1. NOMBRE DEL MEDICAMENTO
Arzerra 100 mg concentrado para solución para perfusión Ofatumumab
2. PRINCIPIO ACTIVO
Un ml contiene 20 mg de ofatumumab.
Cada vial contiene 100 mg de ofatumumab en 5 ml.
3. LISTA DE EXCIPIENTES
Arginina, acetato sódico (E262), cloruro sódico, polisorbato 80 (E433), edetato disódico (E386), ácido clorhídrico (E507), agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Concentrado para solución para perfusión.
100 mg/5 ml 3 viales
5. FORMA Y VÍA DE ADMINISTRACIÓN
Vía intravenosa.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar y transportar refrigerado.
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/10/625/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille
ETIQUETA DEL VIAL
|
1. |
NOMBRE DEL MEDICAMENTO |
Arzerra 100 mg concentrado estéril
Ofatumumab
IV
|
2. |
MÉTODO DE ADMINISTRACIÓN |
|
3. |
FECHA DE CADUCIDAD |
|
EXP | |
|
4. |
NÚMERO DE LOTE |
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
100 mg/5 ml
6. OTROS
1. NOMBRE DEL MEDICAMENTO
Arzerra 1.000 mg concentrado para solución para perfusión Ofatumumab
2. PRINCIPIO ACTIVO
Un ml contiene 20 mg de ofatumumab.
Cada vial contiene 1.000 mg de ofatumumab en 50 ml.
3. LISTA DE EXCIPIENTES
Arginina, acetato sódico (E262), cloruro sódico, polisorbato 80 (E433), edetato disódico (E386), ácido clorhídrico (E507), agua para preparaciones inyectables.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Concentrado para solución para perfusión.
1.000 mg/50 ml 1 vial
5. FORMA Y VÍA DE ADMINISTRACIÓN
Vía intravenosa.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar y transportar refrigerado.
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO
UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/10/625/003
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille
ETIQUETA DEL VIAL
|
1. |
NOMBRE DEL MEDICAMENTO |
Arzerra 1.000 mg concentrado estéril
Ofatumumab
IV
|
2. |
MÉTODO DE ADMINISTRACIÓN |
|
3. |
FECHA DE CADUCIDAD |
|
EXP | |
|
4. |
NÚMERO DE LOTE |
Lot
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
1.000 mg/50 ml
6. OTROS
B. PROSPECTO
Prospecto: información para el paciente
Arzerra 100 mg concentrado para solución para perfusión Arzerra 1.000 mg concentrado para solución para perfusión
Ofatumumab
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o enfermero.
- Si experimenta efectos adversos, consulte a su médico, o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Arzerra y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Arzerra
3. Cómo usar Arzerra
4. Posibles efectos adversos
5 Conservación de Arzerra
6. Contenido del envase e información adicional
1. Qué es Arzerra y para qué se utiliza
Arzerra contiene ofatumumab, que pertenece a un grupo de medicamentos denominados anticuerpos monoclonales.
Arzerra se utiliza para el tratamiento de la leucemia linfocítica crónica (LLC). La LLC es un cáncer de la sangre que afecta a un tipo de glóbulos blancos llamados linfocitos. Los linfocitos se multiplican muy rápidamente y viven mucho tiempo, por eso hay demasiados circulando en su sangre. La enfermedad también puede afectar a otros órganos en su cuerpo. El anticuerpo en Arzerra reconoce una sustancia de la superficie de los linfocitos y causa la muerte de los linfocitos.
2. Qué necesita saber antes de empezar a usar Arzerra No use Arzerra:
• si es alérgico (hipersensible) a ofatumumab o a cualquiera de los demás componentes de Arzerra
(incluidos en la sección 6 “Contenido del envase e información adicional”).
Consulte con su médico si cree que esto puede sucederle a usted.
Advertencias y precauciones
Consulte a su médico o enfermero antes de emperzar a utilizar Arzerra.
• si ha tenido problemas de corazón,
• si tiene enfermedad pulmonar,
Consulte con su médico si piensa que puede tener cualquiera de estos problemas. Puede que necesite revisiones adicionales mientras esté en tratamiento con Arzerra.
Su médico puede que compruebe la cantidad de sales que tiene en sangre, como el magnesio y el potasio (electrolitos), antes y durante el tratamiento con Arzerra. Su médico puede ponerle un tratamiento para los desequilibrios en las sales.
Vacunación y Arzerra
Si se está poniendo alguna vacuna, informe a su médico o a la persona que le esté poniendo la vacuna, de que está en tratamiento con Arzerra. Su respuesta a la vacuna puede debilitarse y puede que no esté protegido totalmente.
Hepatitis B
Antes de iniciar el tratamiento con Arzerra, deben realizarle pruebas para comprobar si tiene hepatitis B (una enfermedad del hígado). Si ha tenido hepatitis B, Arzerra podría hacer que su hepatitis B vuelva a activarse de nuevo. Su médico puede tratarle con un medicamento antiviral adecuado para ayudarle a prevenir esto.
Si tiene o ha tenido hepatitis B, informe a su médico antes de que le administren Arzerra.
Reacciones debidas a la perfusión
Los medicamentos de este tipo (anticuerpos monoclonales) pueden provocar reacciones debidas a la perfusión cuando se inyectan en el organismo. Le administrarán medicamentos como antihistamínicos, esteroides o analgésicos para ayudarle a reducir cualquier reacción. Véase también el apartado 4,
“Posibles efectos secundarios”.
Si cree que ha tenido una reacción similar anteriormente, informe a su médico antes de que le administren Arzerra.
Leucoencefalopatía multifocal progresiva (LEMP)
Se han comunicado casos de Leucoencefalopatía multifocal progresiva (LEMP), una enfermedad grave del cerebro que pone en peligro la vida, con el uso de medicamentos como Arzerra. Informe a su médico inmediatamente si tiene pérdida de memoria, dificultad para pensar, dificultad para caminar o pérdida de visión. Si experimentó estos síntomas antes del tratamiento con Arzerra, informe a su médico inmediatamente sobre cualquier cambio en estos síntomas.
Niños y adolescentes
Se desconoce si Arzerra actúa en niños y adolescentes. Por lo tanto, Arzerra no está recomendado para su uso en niños y adolescentes.
Uso de otros medicamentos y Arzerra
Informe a su médico o farmacéutico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento. Entre estos se incluyen las plantas medicinales y otros medicamentos adquiridos sin receta.
Embarazo, lactancia y fertilidad
Generalmente no se recomienda utilizar Arzerra durante el embarazo. No hay información sobre la seguridad de Arzerra en mujeres embarazadas.
• Informe a su médico si está embarazada o está planeando quedarse embarazada. Su médico valorará el beneficio de administrarle Arzerra durante el embarazo frente al riesgo para su bebé.
• Use un método anticonceptivo fiable para evitar quedarse embarazada mientras esté en tratamiento con Arzerra, y durante los 12 meses después de su último tratamiento.
• Si se queda embarazada durante el tratamiento con Arzerra, informe a su médico.
Se desconoce si los componentes de Arzerra pasan a la leche materna. La lactancia materna no está recomendada durante el tratamiento con Arzerra ni durante los 12 meses después desde la última vez que fue tratado con Arzerra.
Conducción y uso de máquinas
Es poco probable que Arzerra afecte a su capacidad para conducir o utilizar máquinas.
Arzerra contiene sodio
Arzerra contiene 34,8 mg de sodio en cada dosis de 300 mg, 116 mg de sodio en cada dosis de 1.000 mg y 232 mg de sodio en cada dosis de 2.000 mg. Es necesario tener esto en cuenta si está siguiendo una dieta baja en sodio.
3. Cómo usar Arzerra
Si tiene cualquier duda sobre la administración de Arzerra, consulte con el médico que le está administrando la perfusión.
La dosis normal
La dosis normal de Arzerra para la primera perfusión es de 300 mg. Esta dosis se incrementará, normalmente a 1.000 mg ó 2.000 mg, para las perfusiones restantes.
Cómo se debe administrar
Arzerra se administra en una vena (intravenosamente) como una perfusión (un goteo) durante varias horas.
Si no ha recibido tratamiento previo para la LLC, recibirá un máximo de 13 perfusiones. Se le
administrará una primera perfusión, seguida de una segunda perfusión 7 días después. Las perfusiones restantes le serán administradas una vez al mes durante un máximo de 11 meses.
Si ha recibido tratamiento previo para la LLC, recibirá por lo general un ciclo de 12 perfusiones. Se
le administrará una perfusión una vez a la semana durante ocho semanas, seguido de un periodo de interrupción de cuatro a cinco semanas. Las perfusiones restantes se administrarán una vez al mes durante cuatro meses.
Medicamentos administrados antes de cada perfusión
Antes de cada perfusión de Arzerra, se le administrarán medicamentos como premedicación que ayudan a reducir cualquier reacción debida a la perfusión. Entre estos se pueden incluir antihistamínicos, esteroides y analgésicos. Se le vigilará estrechamente y si presenta cualquier reacción, estas serán tratadas.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Reacciones debidas a la perfusión
Los medicamentos de este tipo (anticuerpos monoclonales) pueden causar reacciones debidas a la perfusión, que en ocasiones son graves, y pueden causar la muerte. Las reacciones aparecen con mayor probabilidad durante el primer tratamiento.
Efectos adversos muy frecuentes de una reacción debida a la perfusión (pueden afectar a más de 1 de
cada 10 personas):
• náuseas
• fiebre
• erupción cutánea
Efectos adversos frecuentes de una reacción debida a la perfusión (pueden afectar hasta 1 de cada 10 personas):
• reacciones alérgicas, algunas veces graves, con hinchazón de la cara o boca que causan dificultad para respirar (reacciones anafilácticas)
• dificultad para respirar, respiración difícil, opresión en el pecho, tos
• presión sanguínea baja (puede causar ligeros mareos al ponerse en pie)
• rubefacción
• sudoración excesiva
• temblores o tiritona
• latidos rápidos del corazón
• diarrea
• dolor de espalda
• presión sanguínea alta
• picor, erupción papulosa (habones)
• dolor de garganta o irritación
• falta de energía
• nariz taponada
Síntomas poco frecuentes de una reacción debida a la perfusión (pueden afectar hasta 1 de cada 100 personas).
• líquido en los pulmones (edema pulmonar) que causa dificultad para respirar
• ritmo cardiaco lento.
Informe inmediatamente a su médico o enfermero si presenta cualquiera de estos síntomas.
Efectos adversos muy frecuentes
Pueden afectar a más de 1 de cada 10 personas:
• infección en los pulmones o en las vías respiratorias (tracto respiratorio), como neumonía
• infección de oido, nariz o garganta
Efectos adversos muy frecuentes que pueden aparecer en sus análisis de sangre:
• niveles bajos de glóbulos blancos (neutropenia)
• niveles bajos de glóbulos rojos (anemia)
Efectos adversos frecuentes
Pueden afectar hasta 1 de cada 10 personas:
• fiebre debida a una infección y niveles bajos de glóbulos blancos
• infecciones sanguíneas
• infecciones del tracto urinario
• herpes zoster
• herpes labial
Efectos adversos frecuentes que pueden aparecer en sus análisis de sangre:
• niveles bajos de plaquetas en la sangre (células de la sangre que ayudan a la coagulación)
Efectos adversos poco frecuentes Pueden afectar hasta 1 de cada 100 personas:
• obstrucción en el intestino, que se puede sentir como un dolor de estómago
Si usted tiene dolor de estómago persistente, consulte a su médico tan pronto como le sea posible.
• aumentos de potasio, fosfato y ácido úrico en la sangre que puede causar problemas renales (síndrome de lisis tumoral)
Los síntomas de esta enfermedad incluyen:
• menos producción de orina de lo normal
• espasmos musculares.
Si nota estos síntomas, contacte con su médico tan pronto como sea posible.
Efectos adversos poco frecuentes que pueden aparecer en sus análisis de sangre:
• problemas de coagulación de la sangre
• incapacidad de la médula ósea para producir suficientes glóbulos blancos o rojos
Efectos adversos raros
Pueden afectar hasta 1 de cada 1.000 personas:
• Infección o reactivación del virus de la hepatitis B.
Si usted presenta efectos adversos
Informe a su médico o enfermero si considera que alguno de los efectos adversos que sufre es grave o molesto, o si aprecia cualquier efecto adverso no mencionado en este prospecto.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
Conservación de Arzerra
5.
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase y en la etiqueta del vial. La fecha de caducidad es el último día del mes que se indica.
Conservar y transportar refrigerado (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Conservar la solución diluida para perfusión entre 2°C y 8°C y utilizar durante las 24 horas siguientes. La solución para perfusión no utilizada debe ser desechada a las 24 horas después de su preparación.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Arzerra
- El principio activo es ofatumumab. Un ml del concentrado contiene 20 mg de ofatumumab.
- Los demás componentes son arginina, acetato sódico (E262), cloruro sódico, polisorbato 80 (E433), edetato disódico (E386), ácido clorhídrico (E507) (para ajustar el pH), agua para preparaciones inyectables.
Aspecto del producto y contenido del envase
Arzerra es un concentrado para solución para perfusión de incoloro a color amarillo pálido.
Arzerra 100 mg está disponible en un envase que contiene 3 viales. Cada vial de vidrio está sellado con un tapón de goma libre de látex y una lámina de cierre de aluminio, y contiene 5 ml de concentrado (100 mg de ofatumumab).
Arzerra 1.000 mg está disponible en un envase que contiene 1 vial. Cada vial de vidrio está sellado con un tapón de goma libre de látex y una lámina de cierre de aluminio, y contiene 50 ml de concentrado (1.000 mg de ofatumumab).
Titular de la autorización de comercialización
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Reino Unido
Responsable de la fabricación
Glaxo Operations UK Limited (Trading as Glaxo Wellcome Operations), Harmire Road, Barnard Castle, County Durham, DL12 8DT, Reino Unido.
Novartis Pharmaceuticals UK Limited, Frimley Business Park, Frimley, Camberley, Surrey GU16 7SR, Reino Unido
Novartis Pharma GmbH, RoonstraBe 25, D-90429 Nuremberg, Alemania
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Etarapun Novartis Pharma Services Inc. Tea: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
|
Ceská republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 555 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EXXáda Novartis (Hellas) A.E.B.E. T^k: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
España Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmacéuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
Ísland
Vistor hf.
Sími: +354 535 7000 Italia
Novartis Farma S.p.A.
Tel: +39 02 96 54 1
Kúrcpog
Novartis Pharma Services Inc. Tr(k: +357 22 690 690
Latvija
Novartis Pharma Services Inc. Tel: +371 67 887 070
Slovenská republika
Novartis Slovakia s.r.o.
Tel: +421 2 5542 5439
Suomi/Finland
Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200
Sverige
Novartis Sverige AB Tel: +46 8 732 32 00
United Kingdom
Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370
Fecha de la última revisión de este prospecto:
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu/.
La siguiente información está destinada únicamente a médicos o profesionales del sector sanitario:
1) Antes de diluir Arzerra
Antes de la dilución se debe comprobar el concentrado de Arzerra para detectar cualquier partícula o decoloración. Ofatumumab debe ser una solución de incolora a color amarillo pálido. No utilizar el concentrado de Arzerra si presenta decoloración.
No agite el vial de ofatumumab para esta inspección.
2) Cómo se debe diluir la solución para perfusión
Antes de la administración, el concentrado de Arzerra debe diluirse en una solución para inyección de cloruro sódico 9 mg/ml (0,9 %), utilizando técnicas de asepsia adecuadas.
Dosis de 300 mg - Utilizar 3 viales de 100 mg/5 ml (15 ml en total, 5 ml por vial):
• extraer y desechar 15 ml de una bolsa con 1.000 ml de solución para inyección de cloruro sódico 9 mg/ml (0,9 %);
• extraer 5 ml de ofatumumab de cada uno de los 3 viales de 100 mg e inyectarlos en la bolsa de
1.000 ml;
• no agitar, mezclar la solución diluida mediante una inversión suave.
Dosis de 1.000 mg - Utilizar 1 vial de 1.000 mg/50 ml (50 ml en total, 50 ml por vial):
• extraer y desechar 50 ml de una bolsa con 1.000 ml de solución para inyección de cloruro sódico 9 mg/ml (0,9 %);
• extraer 50 ml de ofatumumab del vial de 1.000 mg e inyectarlos en la bolsa de 1.000 ml;
• no agitar, mezclar la solución diluida mediante una inversión suave.
Dosis de 2.000 mg - Utilizar 2 viales de 1.000 mg/50 ml (100 ml en total, 50 ml por vial):
• extraer y desechar 100 ml de una bolsa con 1.000 ml de solución para inyección de cloruro sódico 9 mg/ml (0,9 %);
• extraer 50 ml de ofatumumab de cada uno de los 2 viales de 1.000 mg e inyectarlos en la bolsa de
1.000 ml;
• no agitar, mezclar la solución diluida mediante una inversión suave.
3) Cómo se debe administrar la solución diluida
Arzerra no se debe administrar por vía intravenosa o bolo. Se debe administrar utilizando una bomba para perfusión intravenosa.
La perfusión debe ser completada en un plazo de 24 horas tras su preparación. Transcurrido ese tiempo desechar la solución no utilizada.
Arzerra no se debe mezclar o administrar como una perfusión con otros medicamentos o soluciones intravenosas. Para evitar esto, limpiar la vía con una solución para inyección de cloruro sódico 9 mg/ml (0,9 %) antes y después de la administración de ofatumumab.
LLC no tratada previamente:
En la primera perfusión, administrar durante más de 4,5 horas (ver sección 4.2 de la Ficha técnica), a través de una vía periférica o un catéter permanente, de acuerdo con el siguiente esquema:
Perfusión 1: esquema
|
Tiempo (minutos) |
ml/hora |
|
0 - 30 |
12 |
|
31 - 60 |
25 |
|
61 - 90 |
50 |
|
91 - 120 |
100 |
|
121 - 150 |
200 |
|
151 - 180 |
300 |
|
180 + |
400 |
Si la primera perfusión se ha completado sin ninguna reacción adversa grave, las perfusiones restantes (2-13) de 1.000 mg se deben administrar durante más de 4 horas (ver sección 4.2 de la Ficha técnica), a través de una vía periférica o un catéter permanente, de acuerdo con el siguiente esquema:
Perfusiones de la 2 a la 13: esquema
|
Tiempo (minutos) |
ml/hora |
|
0 - 30 |
25 |
|
31 - 60 |
50 |
|
61 - 90 |
100 |
|
91 - 120 |
200 |
|
121 + |
400 |
LLC refractaria:
La primera y la segunda perfusión se deben administrar durante más de 6,5 horas (ver la sección 4.2 de la Ficha técnica), a través de una vía periférica o un catéter permanente, de acuerdo con el siguiente esquema:
Perfusiones 1 y 2: esquema
|
Tiempo (minutos) |
ml/hora |
|
0 - 30 |
12 |
|
31 - 60 |
25 |
|
61 - 90 |
50 |
|
91 - 120 |
100 |
|
121 + |
200 |
Si la segunda perfusión se ha completado sin ninguna reacción adversa grave, las perfusiones restantes (3-12) deben ser administradas durante más de 4 horas (ver la sección 4.2 de la Ficha técnica), a través de una vía periférica o un catéter permanente, de acuerdo con el siguiente esquema:
Perfusiones 3 a 12: esquema
|
Tiempo (minutos) |
ml/hora |
|
0 - 30 |
25 |
|
31 - 60 |
50 |
|
61 - 90 |
100 |
|
91 - 120 |
200 |
|
121 + |
400 |
Si se observa cualquier reacción adversa, la velocidad de perfusión se debe reducir de acuerdo con la sección 4.2 de la Ficha técnica.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
69