Anoro 55Mcg/22Mcg Polvo Para Inhalacion Unidosis
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
ANORO 55 microgramos/22 microgramos polvo para inhalación (unidosis)
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada inhalación proporciona una dosis liberada (la dosis que sale por la boquilla) de 65 microgramos de bromuro de umeclidinio equivalente a 55 microgramos de umeclidinio y 22 microgramos de vilanterol (como trifenatato). Esto se corresponde con una dosis pre-dispensada de 74,2 microgramos de bromuro de umeclidinio equivalente a 62,5 microgramos de umeclidinio y 25 microgramos de vilanterol (como trifenatato).
Excipiente con efecto conocido
Cada dosis liberada contiene aproximadamente 25 mg de lactosa (como monohidrato).
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo para inhalación, unidosis (polvo para inhalación).
Polvo blanco en un inhalador (Ellipta) de color gris claro con una boquilla protectora de color rojo y un contador de dosis.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
ANORO está indicado como tratamiento broncodilatador de mantenimiento para aliviar los síntomas de la enfermedad pulmonar obstructiva crónica (EPOC) en pacientes adultos.
4.2 Posología y forma de administración
Posología
Adultos
La dosis recomendada es una inhalación de ANORO 55/22 microgramos una vez al día.
Para mantener la broncodilatación, ANORO debe administrarse una vez al día, a la misma hora del día, cada día. La dosis máxima es una inhalación de ANORO 55/22 microgramos una vez al día.
Poblaciones especiales Pacientes de edad avanzada
No se requiere ajuste de dosis en pacientes mayores de 65 años.
Insuficiencia renal
No se requiere ajuste de dosis en pacientes con insuficiencia renal.
Insuficiencia hepática
No se requiere ajuste de dosis en pacientes con insuficiencia hepática leve o moderada. No se ha estudiado el uso de ANORO en pacientes con insuficiencia hepática grave y se debe usar con precaución en esta población.
Población pediátrica
No existe una recomendación de uso específica para ANORO en la población pediátrica (menores de 18 años de edad) para la indicación de EPOC.
Forma de administración
ANORO se administra solo por vía inhalatoria.
Instrucciones de uso:
Las instrucciones de uso que se muestran a continuación para el inhalador de 30 dosis también son aplicables al inhalador de 7 dosis.
El inhalador Ellipta contiene unidosis y está listo para usar.
El inhalador está envasado en una bandeja que contiene una bolsa con desecante para reducir la humedad. La bolsa de desecante debe desecharse y no debe comerse o inhalarse. Se debe advertir al paciente de que no abra la bandeja hasta que esté preparado para inhalar la dosis.
Cuando se saca el inhalador por primera vez de la bandeja sellada, estará en la posición “cerrado”. La fecha de “desechar el” se debe escribir en el espacio designado para ello en la etiqueta del inhalador. La fecha de “desechar el” es de 6 semanas desde la fecha de apertura de la bandeja. Después de esta fecha, el inhalador se debe desechar. La bandeja se puede desechar después de la primera apertura.
Si la tapa del inhalador se abre y se cierra sin que se inhale el medicamento, se perderá la dosis. La dosis perdida quedará retenida de forma segura dentro del inhalador, pero no estará disponible para ser inhalada.
No es posible administrar de forma accidental una dosis adicional o una dosis doble mediante una inhalación.
a) Preparar una dosis
Cuando esté preparado, abrir la tapa para inhalar una dosis. No debe agitar el inhalador.
Deslizar la tapa hacia abajo hasta oír un ‘clic’. Ahora, el medicamento está preparado para poder inhalarlo.
Como confirmación, el contador de dosis disminuye en 1 unidad. Si el contador de dosis no disminuye al oír el ‘clic’, el inhalador no liberará la dosis del medicamento y debe llevarlo al farmacéutico para solicitar ayuda.
b) Cómo inhalar el medicamento
Mantener el inhalador alejado de la boca y espirar tanto como le sea posible. No espirar dentro del inhalador.
Colocar la boquilla entre los labios y cerrarlos firmemente alrededor de la boquilla. Las ranuras de ventilación no deben bloquearse con los dedos durante su uso.
• Realizar una inspiración prolongada, continua y profunda. Mantener la inspiración tanto tiempo como sea posible (al menos 3-4 segundos).
• Retirar el inhalador de la boca.
• Espirar suave y lentamente.
Puede que no distinga el sabor o note el medicamento, incluso cuando utiliza el inhalador de forma correcta.
c) Cerrar el inhalador
Antes de cerrar la tapa, la boquilla del inhalador puede limpiarse utilizando un pañuelo seco.
Deslizar la tapa hacia arriba hasta el tope para proteger la boquilla.
4.3 Contraindicaciones
Hipersensibilidad a los principios activos o a alguno de los excipientes incluidos en la sección 6.1.
4.4 Advertencias y precauciones especiales de empleo
Asma
Umeclidinio/vilanterol no se debe utilizar en pacientes con asma, ya que no se ha estudiado en esta población de pacientes.
Broncoespasmo paradójico
Al igual que en otros tratamientos inhalados, la administración de umeclidinio/vilanterol puede causar broncoespasmo paradójico que puede poner en peligro la vida. Si se produce broncoespasmo paradójico, se debe interrumpir el tratamiento con umeclidinio/vilanterol inmediatamente y si es necesario, instaurar un tratamiento alternativo.
No para uso agudo
Umeclidinio/vilanterol no está indicado como tratamiento para los episodios agudos de broncoespasmo.
Empeoramiento de la enfermedad
El aumento del uso de broncodilatadores de acción corta para aliviar los síntomas, indica un empeoramiento en el control de la enfermedad. En el caso de empeoramiento de la EPOC durante el tratamiento con umeclidinio/vilanterol, se debe realizar una reevaluación del paciente y de la pauta posológica del tratamiento para la EPOC.
Efectos cardiovasculares
Tras la administración de antagonistas de receptores muscarínicos y simpaticomiméticos, incluyendo umeclidinio/vilanterol, se pueden observar efectos cardiovasculares, como arritmias cardiacas, por ejemplo fibrilación auricular y taquicardia. Los pacientes con enfermedad cardiovascular clínicamente relevante no controlada fueron excluidos de los estudios clínicos. Por lo tanto, umeclidinio/vilanterol, se debe utilizar con precaución en pacientes con alteraciones cardiovasculares graves.
Actividad antimuscarínica
Debido a su actividad antimuscarínica, umeclidinio/vilanterol se debe emplear con precaución en pacientes con retención urinaria o con glaucoma de ángulo cerrado.
Hipocaliemia
Los agonistas_p2-adrenérgicos pueden producir hipocaliemia significativa en algunos pacientes, siendo posible que se produzcan efectos cardiovasculares. La disminución del potasio sérico suele ser transitoria y no requiere suplementos.
En estudios clínicos con umeclidinio/vilanterol, a la dosis terapéutica recomendada, no se observaron efectos clínicamente relevantes de hipocaliemia. Se debe tener precaución cuando umeclidinio/vilanterol se utilice con otros medicamentos que también puedan causar hipocaliemia (ver sección 4.5).
Hiperglucemia
Los agonistas p2-adrenérgicos pueden producir hiperglucemia transitoria en algunos pacientes.
En estudios clínicos con umeclidinio/vilanterol, a la dosis terapéutica recomendada, no se observaron efectos clínicamente relevantes de hiperglucemia. En pacientes diabéticos, se debe monitorizar estrechamente los niveles de glucosa en plasma al inicio del tratamiento con umeclidinio/vilanterol.
Afecciones coexistentes
Umeclidinio/vilanterol se debe utilizar con precaución en pacientes con trastornos convulsivos o tirotoxicosis, y en pacientes que son inusualmente sensibles a los agonistas p2-adrenérgicos.
Excipientes
Este medicamento contiene lactosa. Los pacientes con alteraciones hereditarias poco frecuentes de intolerancia a la galactosa, insuficiencia de lactasa de Lapp o malabsorción de glucosa o galactosa no deben utilizar este medicamento.
4.5 Interacción con otros medicamentos y otras formas de interacción
Bloqueantes p-adrenérgicos
Los medicamentos que contienen bloqueantes p-adrenérgicos pueden disminuir o antagonizar el efecto de los agonistas p2-adrenérgicos, como vilanterol. Se debe evitar el uso concomitante de bloqueantes p-adrenérgicos no selectivos o selectivos, a menos que existan razones de peso para su uso.
Interacciones metabólicas e interacciones basadas en transportadores
Vilanterol es un sustrato del citocromo P450 3A4 (CYP3A4). La administración concomitante de inhibidores potentes del CYP3A4 (por ejemplo ketoconazol, claritromicina, itraconazol, ritonavir, telitromicina) puede inhibir el metabolismo de vilanterol, y aumentar la exposición sistémica al mismo. La administración junto con ketoconazol (400 mg) en voluntarios sanos aumentó la media del AUC(0-t) y Cmax, de vilanterol en un 65% y 22% respectivamente. El incremento en la exposición a vilanterol no se asoció con un aumento de los efectos sistémicos sobre el ritmo cardiaco, los niveles de potasio en sangre o el intervalo QT (corregido mediante el método Fridericia) relacionados con los agonistas p-adrenérgicos. Se recomienda tener precaución cuando se administren conjuntamente umeclidinio/vilanterol con ketoconazol y otros inhibidores potentes conocidos del CYP3A4 ya que existe la posibilidad de un aumento de la exposición sistémica a vilanterol, que puede dar lugar a un posible aumento de reacciones adversas. Verapamilo, un inhibidor moderado del CYP3A4, no afectó de forma significativa a la farmacocinética de vilanterol.
Umeclidinio es un sustrato del citocromo P450 2D6 (CYP2D6). La farmacocinética de umeclidinio, en estado estacionario, se evaluó en voluntarios sanos que carecían de CYP2D6 (metabolizadores lentos). A una dosis 8 veces superior a la dosis terapéutica, no se observaron efectos en el AUC o en la Cmax de umeclidinio. A dosis 16 veces superiores a la dosis terapéutica, se observó un aumento en el AUC de
umeclidinio de aproximadamente 1,3 sin verse afectada la Cmax del mismo. En base a la magnitud de estos cambios, no se esperan interacciones medicamentosas clínicamente relevantes cuando se administre conjuntamente umeclidinio/vilanterol junto a inhibidores del CYP2D6 o cuando se administre a pacientes genéticamente deficientes en la actividad del CYP2D6 (metabolizadores lentos).
Tanto umeclidinio como vilanterol son sustratos de la glicoproteína-P transportadora (P-gp). Se evaluó el efecto de verapamilo (240 mg una vez al día), un inhibidor moderado de la P-gp, sobre la farmacocinética en estado estacionario de umeclidinio y vilanterol, en voluntarios sanos. No se observaron efectos causados por verapamilo en la Cmax de umeclidinio o vilanterol. Se observó un aumento en el AUC de umeclidinio de aproximadamente 1,4 veces sin efectos en el AUC de vilanterol. En base a la magnitud de estos cambios, no se esperan interacciones medicamentosas clínicamente relevantes cuando se administre conjuntamente umeclidinio/vilanterol con inhibidores de la P-gp.
Otros agentes antimuscarínicos y simpaticomiméticos
No se ha estudiado la administración conjunta de umeclidinio/vilanterol con otros antagonistas muscarínicos de acción prolongada, agonistas p2-adrenérgicos de acción prolongada u otros medicamentos que contengan alguno de estos componentes, y no se recomienda su uso ya que se pueden potenciar reacciones adversas conocidas de antagonistas muscarínicos inhalados o agonistas p2-adrenérgicos (ver sección 4.4 y sección 4.9).
Hipocaliemia
El tratamiento hipocaliémico concomitante con derivados de metilxantina, esteroides, o diuréticos no ahorradores de potasio puede potenciar el posible efecto hipocalémico de los agonistas p2-adrenérgicos, y por lo tanto, de deben utilizar con precaución (ver sección 4.4)
Otros medicamentos para la EPOC
Aunque no se han realizado estudios formales in vivo de interacción de medicamentos, umeclidinio/vilanterol inhalado se ha utilizado junto con otros medicamentos para la EPOC, incluyendo broncodilatadores simpaticomiméticos de acción corta y corticosteroides inhalados sin evidencias clínicas de interacción medicamentosa.
4.6 Fertilidad, embarazo y lactancia
Embarazo
No hay datos relativos al uso de umeclidinio/vilanterol en mujeres embarazadas. Los estudios realizados en animales tras la administración de vilanterol han mostrado toxicidad para la reproducción a niveles de exposición que no son clínicamente relevantes (ver sección 5.3).
Solo se debe considerar la administración de umeclidinio/vilanterol durante el embarazo si el beneficio esperado para la madre justifica el posible riesgo para el feto.
Lactancia
Se desconoce si umeclidinio o vilanterol se excretan en la leche materna. Sin embargo, otros agonistas p2-adrenérgicos son detectados en la leche materna. No se puede descartar el riesgo en recién nacidos/lactantes. Se debe decidir si es necesario interrumpir la lactancia o interrumpir el tratamiento con umeclidinio/vilanterol tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre.
Fertilidad
No hay datos sobre los efectos de umeclidinio/vilanterol sobre la fertilidad humana. Los estudios realizados en animales no muestran efectos de umeclidinio o vilanterol sobre la fertilidad.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de umeclidinio/vilanterol sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
4.8 Reacciones adversas
Resumen del perfil de seguridad
La reacción adversa notificada con mayor frecuencia en umeclidinio/vilanterol fue nasofaringitis (9%). Tabla de reacciones adversas
El perfil de seguridad de ANORO está basado en los datos de seguridad procedentes del programa de desarrollo clínico de umeclidinio/vilanterol y sus componentes por separado, que comprendía 6.855 pacientes con EPOC y de la notificación espontánea. El programa de desarrollo clínico incluyó 2.354 pacientes que recibieron umeclidinio/vilanterol una vez al día, en los estudios clínicos de Fase III de 24 semanas de duración o de mayor duración, de los cuales 1.296 pacientes recibieron la dosis recomendada de 55/22 microgramos en los estudios de 24 semanas, 832 pacientes recibieron una dosis mayor 113/22 microgramos en los estudios de 24 semanas y 226 pacientes recibieron 113/22 microgramos en un estudio de 12 meses de duración.
Las frecuencias asignadas a las reacciones adversas que se identifican en la siguiente tabla, incluyen las tasas de incidencia bruta observadas en la integración de cinco estudios de 24 semanas y en el estudio de seguridad de 12 meses de duración.
Para la clasificación de frecuencias se utiliza el siguiente convenio: muy frecuentes (>1/10); frecuentes (>1/100 a <1/10); poco frecuentes (>1/1.000 a <1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
|
Sistema de clasificación de órganos |
Reacciones adversas |
Frecuencia |
|
Infecciones e infestaciones |
Infección del tracto urinario |
Frecuentes |
|
Sinusitis |
Frecuentes | |
|
Nasofaringitis |
Frecuentes | |
|
Faringitis |
Frecuentes | |
|
Infecciones del tracto respiratorio superior |
Frecuentes | |
|
Trastornos del sistema |
Reacciones de hipersensibilidad que | |
|
inmunológico |
incluyen: | |
|
Erupción cutánea |
Poco frecuentes | |
|
Anafilaxia, angioedema y urticaria |
Raras | |
|
Trastornos del sistema |
Cefalea |
Frecuente |
|
nervioso |
Temblor |
Poco frecuentes |
|
Disgeusia |
Poco frecuentes | |
|
Trastornos oculares |
Glaucoma |
Frecuencia no conocida |
|
Trastornos cardiacos |
Fibrilación auricular |
Poco frecuentes |
|
Taquicardia supraventricular |
Poco frecuentes | |
|
Ritmo idioventricular |
Poco frecuentes | |
|
Taquicardia |
Poco frecuentes | |
|
Extrasístoles supraventricular |
Poco frecuentes | |
|
Palpitaciones |
Poco frecuentes | |
|
Trastornos respiratorios, |
Tos |
Frecuentes |
|
torácicos y mediastínicos |
Dolor orofaríngeo |
Frecuentes |
|
Trastornos gastrointestinales |
Estreñimiento |
Frecuentes |
|
Boca seca |
Frecuentes |
|
Sistema de clasificación de órganos |
Reacciones adversas |
Frecuencia |
|
Trastornos de la piel y del tejido subcutáneo |
Erupción cutánea |
Poco frecuentes |
|
Trastornos renales y |
Retención urinaria |
Raras |
|
urinarios |
Disuria |
Raras |
|
Obstrucción de la vejiga |
Raras |
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Es probable que una sobredosis de umeclidinio/vilanterol pueda producir signos y síntomas debidos a la acción de los componentes por separado, consistentes con los efectos adversos conocidos de los antagonistas muscarínicos inhalados (por ejemplo, boca seca, alteraciones en la acomodación visual y taquicardia) o con la sobredosis de otros agonistas p2-adrenérgicos (por ejemplo, arritmias, temblor, cefalea, palpitaciones, náuseas, hiperglucemia e hipocaliemia).
En caso de sobredosis, el paciente debe recibir tratamiento de soporte y, si es necesario, un seguimiento apropiado.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agentes para las enfermedades obstructivas de las vías respiratorias, adrenérgicos en combinación con anticolinérgicos, código ATC: R03AL03.
Mecanismo de acción
Umeclidinio/vilanterol es una combinación inhalada de un antagonista de receptores muscarínicos de acción prolongada/agonista p2-adrenérgico de acción prolongada (LAMA/LABA). Tras su inhalación oral, ambos compuestos actúan localmente en las vías respiratorias produciendo broncodilatación mediante mecanismos distintos.
Umeclidinio
Umeclidinio es un antagonista de receptores muscarínicos de acción prolongada (también conocido como anticolinérgico). Es un derivado de la quinuclidina con actividad sobre múltiples subtipos de receptores muscarínicos. Umeclidinio ejerce su actividad broncodilatadora inhibiendo competitivamente la unión de acetilcolina a los receptores muscarínicos del músculo liso bronquial. En los modelos pre-clínicos, demuestra tener reversibilidad lenta en los subtipos de receptores muscarínicos humanos M3 in vitro, y una duración de acción prolongada in vivo cuando se administra directamente en los pulmones.
Vilanterol
Vilanterol es un agonista selectivo de los receptores p2-adrenérgicos de acción prolongada (agonista p2-adrenérgico). Los efectos farmacológicos de los agonistas p2-adrenérgicos, incluido vilanterol, son atribuibles, al menos en parte, a la estimulación de la adenilato ciclasa intracelular, enzima que cataliza la conversión de adenosín trifosfato (ATP) a adenosín monofosfato-3’, 5’ cíclico (AMP cíclico). El aumento de los niveles del AMP cíclico produce relajación del músculo liso bronquial e inhibe la liberación de mediadores de la hipersensibilidad inmediata de las células, especialmente de los mastocitos.
Efectos farmacodinámicos
En los estudios de Fase III, de 6 meses de duración, umeclidinio/vilanterol, demostró una mejoría clínicamente relevante frente a placebo en la función pulmonar (medida mediante el volumen espiratorio forzado en el primer segundo [FEV1]) durante más de 24 horas tras la administración una vez al día, que fue evidente a los 15 minutos tras la administración de la primera dosis (mejoría frente a placebo de 102 ml (p<0,001 ). El promedio máximo de mejoría en el FEV1 en las primeras 6 horas tras administrar la dosis con respecto a placebo fue de 130 ml (p<0,001 ) en la semana 24. No hubo evidencia de taquifilaxia en el efecto de ANORO a lo largo del tiempo.
Electrofisiología cardiaca
Se evaluó el efecto de umeclidinio/vilanterol sobre el intervalo QT en un estudio QT controlado con placebo y activo (moxifloxacino) en el que se administró umeclidinio/vilanterol 113/22 microgramos o 500/100 microgramos (dosis pre-dispensada con umeclidinio a ocho veces la dosis recomendada y vilanterol a cuatro veces la dosis recomendada) una vez al día, durante 10 días, en 103 voluntarios sanos. La media de la diferencia máxima en la prolongación del intervalo QT (corregido mediante el método Fridericia, QTcF) respecto a placebo tras la corrección basal fue de 4,3 milisegundos (IC 90% =2,2 a 6,4) observados 10 minutos detrás la administración de umeclidinio/vilanterol 113/22 microgramos y 8,2 milisegundos (IC 90% =6,2 a 10,2) observados 30 minutos tras la administración de umeclidinio/vilanterol 500/100 microgramos. Por tanto, no se observó ningún posible efecto arritmógeno clínicamente relevante relacionado con la prolongación del intervalo QT con umeclidinio/vilanterol 113/22 microgramos.
También se observó un aumento en la frecuencia cardiaca dosis-dependiente. La diferencia máxima media en la frecuencia cardiaca respecto a placebo tras la corrección basal fue 8,4 pulsaciones/minuto (IC 90% =7,0 a 9,8) y 20,3 pulsaciones/minuto (IC 90% =18,9 a 21,7) observados 10 minutos después de la administración de umeclidinio/vilanterol 113/22 microgramos y 500/100 microgramos respectivamente.
Además, no se observaron efectos clínicamente relevantes sobre el ritmo cardiaco monitorizado con Holter durante 24 horas en el grupo de 53 pacientes con EPOC que fueron tratados con umeclidinio/vilanterol 55/22 microgramos una vez al día en un estudio de 6 meses de duración, en el grupo de 55 pacientes que recibieron umeclidinio/vilanterol 113/22 microgramos una vez al día en otro estudio de 6 meses de duración ni en el grupo de 226 pacientes que recibieron 113/22 microgramos una vez al día en el estudio de 12 meses de duración.
Eficacia clínica
La eficacia clínica de umeclidinio/vilanterol administrado una vez al día fue evaluada en ocho ensayos clínicos de Fase III en el que participaron 6.835 pacientes adultos con diagnóstico clínico de EPOC.
Un total de 5.618 pacientes procedentes de cinco estudios de 6 meses de duración (dos estudios controlados por placebo y tres estudios controlados con el comparador activo [tiotropio]), 655 pacientes procedentes de dos estudios de 3 meses de duración sobre la resistencia al ejercicio/función pulmonar, y 562 pacientes procedentes del estudio complementario de 12 meses de duración.
Efectos sobre la función pulmonar
ANORO demostró mejoras en la función pulmonar (definida por el cambio respecto a los valores basales en el FEVi valle) en diversos estudios. En un estudio de Fase III de 6 meses de duración, ANORO demostró mejoras estadísticamente significativas en el FEV1 valle (variable principal) en la semana 24 en comparación con placebo y con cada brazo de tratamiento de los componentes en monoterapia. Además, ANORO demostró mejoras clínicamente relevantes y estadísticamente significativas en el FEV1 valle en frente a tiotropio en dos de los tres estudios de 6 meses de duración con comparador activo, y mayores mejoras numéricas con respecto a tiotropio en el tercer estudio con comparador activo (ver Tabla 1). No hubo atenuación del efecto broncodilatador con el tiempo.
Resultados sobre los síntomas Dificultad para respirar:
ANORO demostró una reducción estadísticamente significativa y clínicamente relevante en la dificultad para respirar evaluada por un aumento en la puntuación focal del Índice Transicional de Disnea (ITD) en la semana 24 (variable secundaria principal) en comparación con placebo (ver Tabla 1). Las mejoras en la puntuación focal del ITD en comparación con cada componente en monoterapia y tiotropio no fueron estadísticamente significativas (ver Tabla 1).
El porcentaje de pacientes que respondieron al menos con la Diferencia Mínima Clínicamente Importante (DMCI) de 1 unidad de puntuación focal del ITD en la semana 24 fue mayor para ANORO (58%) en comparación con placebo (41%) y con cada componente en monoterapia (53% para umeclidinio y 51% para vilanterol).
Calidad de vida relacionada con la salud:
ANORO ha mostrado también una mejora en la calidad de vida relacionada con la salud evaluada mediante el cuestionario St. George’s Respiratory Questionnaire (SGRQ) tal como se indica en la reducción de la puntuación total del SGRQ en la semana 24 en comparación con placebo y con cada componente en monoterapia (ver Tabla 1). ANORO mostró una reducción estadísticamente significativa en la puntuación total del SGRQ en comparación con tiotropio en uno de los tres estudios con comparador activo (ver Tabla 1).
El porcentaje de pacientes que respondieron con al menos la DMCI en la puntuación de SGRQ (definida como una disminución de 4 unidades respecto a los valores basales) en la semana 24 fue mayor para ANORO (49%) en comparación con placebo (34%) y con cada componente en monoterapia (44% para umeclidinio y 48% para vilanterol). En un estudio comparador de activo, un porcentaje más elevado de pacientes tratados con ANORO respondieron con una mejora clínicamente relevante en la puntuación SGRQ en la semana 24 (53%) en comparación con tiotropio (46%). En los otros dos estudios comparadores de activo, una proporción similar de pacientes alcanzó al menos la DMCI con ANORO y tiotropio; 49% y 54% para ANORO 55/22 microgramos y 52% y 55% para tiotropio.
Uso de medicación de rescate
ANORO redujo el uso de medicación de rescate con salbutamol durante las semanas 1-24 en comparación con placebo y umeclidinio (ver Tabla 1), y demostró un aumento desde valores basales en la proporción de días en los que no se necesitó medicación de rescate (una media de 11,1%) en comparación con una disminución desde valores basales para placebo (una media de 0,9%).
En los tres estudios controlados con comparador activo de 6 meses de duración, ANORO redujo el uso de medicación de rescate con salbutamol en comparación con tiotropio, con reducciones estadísticamente significativas observadas en dos de los tres estudios (ver Tabla 1). En los tres estudios clínicos, ANORO también demostró un mayor aumento desde valores basales en la proporción de días en los que no se necesitó medicación de rescate (promedio entre 17,6% y 21,5%) en comparación con tiotropio (promedio entre 11,7% y 13,4%).
Tabla 1. Resultados sobre la función pulmonar, sintomáticos y de calidad de vida relacionada con la salud en la semana 24
|
Comparación de tratamientos con ANORO 55/22 mcg |
Diferencia de tratamiento1 (Intervalo de confianza 95%, valor de p) | |||
|
FEV1 valle (ml) |
Puntuación Focal del ITD |
Puntuación Total SGRQ |
Uso de medicación de rescate3 | |
|
ANORO (N = 413) versus Placebo (N = 280) |
167 (128, 207) <0,001 |
1,2 (0,7; 1,7) <0,001 |
-5,51 (-7,88; -3,13) <0,001* |
-0,8 (-1,3; -0,3) 0,001* |
|
ANORO (N = 413) versus Umeclidinio 55 mcg (N = 418) |
52 (17, 87) 0,004 |
0,3 (-0,2; 0,7) 0,244 |
-0,82 (-2,90; 1,27) 0,441 |
-0,6 (-1,0; -0,1) 0,014* |
|
ANORO (N = 413) versus Vilanterol 22 mcg (N = 421) |
95 (60, 130) <0,001 |
0,4 (-0,1; 0,8) 0,117 |
-0,32 (-2,41; 1,78) 0,767 |
0,1 (-0,3; 0,5) 0,675 |
|
ANORO (N = 454) versus Tiotropio 18 mcg (N = 451) (Estudio ZEP117115) |
112 (81, 144) <0,001 |
n/e |
-2,10 (-3,61; -0,59) 0,006 |
-0,5 (-0,7; -0,2) <0,001 |
|
ANORO (N = 207) versus Tiotropio 18 mcg (N = 203) (Estudio DB2113360) |
90 (39, 141) <0,001 |
0,12 (-0,4; 0,5) 0,817 |
0,75 (-2,12; 3,63) 0,607 |
-0,7 (-1,2; -0,1) 0,022 |
|
ANORO (N = 217) versus Tiotropio 18 mcg (N = 215) (Estudio DB2113374) |
60 (10, 109) 0,018* |
-0,17 (-2,85; 2,52) 0,904 |
-0,6 (-1,2; 0.0) 0,069 | |
N= número de la población por intención de tratar mcg = microgramos n/e = no evaluado
1. Media por mínimos cuadrados
2. Datos agrupados del estudio DB2113360 y el estudio DB2113374
3. Diferencia entre la media de número de inhalaciones al día durante las semanas 1-24
También se estudió una dosis mayor de umeclidinio/vilanterol (113/22 microgramos), en un estudio clínico controlado con placebo de 24 semanas y en dos de los tres estudios controlados con comparador activo de 24 semanas. Los resultados fueron similares a los obtenidos para la dosis de ANORO y proporcionaron evidencias adicionales de la eficacia de ANORO.
Exacerbaciones de EPOC
ANORO redujo el riesgo de exacerbaciones en EPOC en un 50% en comparación con placebo (análisis basado en el tiempo de la primera exacerbación: Hazard Ratio (HR) 0,5; p=0,004*); en un 20% en comparación con umeclidinio (HR 0,8; p=0,391); y en un 30% en comparación con vilanterol (HR 0,7; p=0,121). De los tres estudios con comparador activo, el riesgo de padecer exacerbaciones en EPOC en comparación con tiotropio se redujo en un 50% en un estudio (HR 0,5; p=0,044) y se incrementó en un 20% y un 90% en dos estudios (HR 1,2; p=0,709 y HR 1,9; p=0,062 respectivamente). Estos estudios no fueron específicamente diseñados para evaluar el efecto de los tratamientos en exacerbaciones de la EPOC y los pacientes con exacerbaciones fueron retirados del estudio.
Resistencia al ejercicio y volúmenes pulmonares
En uno de los dos estudios, ANORO 55/22 microgramos mejoró el tiempo de resistencia al ejercicio en comparación con placebo, tal como se evaluó con el test de marcha de carga constante (endurance shuttle walk test, ESWT), y en ambos estudios mejoró las medidas de volumen pulmonar en comparación con placebo en pacientes adultos con EPOC con hiperinflación (capacidad funcional residual [CFR] >120%). En el primer estudio, ANORO 55/22 microgramos demostró una mejora estadísticamente significativa y una mejora clínicamente relevante (basada en la diferencia mínima clínicamente importante (DMCI) entre los 45 y 85 segundos) frente a placebo en el tiempo de resistencia al ejercicio (exercise endurance time, EET) obtenido 3 horas después de la administración en la semana 12 (69,4 segundos [p=0,003]). La mejora en el EET en comparación con placebo se observó en el día 2 y se mantuvo en las semanas 6 y 12. En el segundo estudio, la diferencia de tratamiento en el EET entre ANORO 55/22 microgramos y placebo fué 21,9 segundos (p=0,234) en la semana 12.
ANORO 55/22 microgramos también mostró mejoras estadísticamente significativas en comparación con placebo en el cambio respecto a los valores basales de las medidas del volumen pulmonar en el punto valle y a las 3 horas tras la administración de la dosis en la semana 12 del primer estudio (capacidad inspiratoria: de 237 ml y 316 ml respectivamente, volumen residual: de -466 ml y -643 ml respectivamente y una capacidad funcional residual: de -351 ml y -522 ml respectivamente; todos p<0,001). En el segundo estudio, ANORO 55/22 microgramos mostró mejoras en comparación con placebo en el cambio respecto a los valores basales de las medidas del volumen pulmonar en el punto valle y a las 3 horas tras la administración de la dosis en la semana 12 estudio (capacidad inspiratoria: de 198 ml y 238 ml respectivamente, volumen residual: de -295 ml y -351 ml respectivamente y una capacidad funcional residual: de -238 ml y -302 ml respectivamente); todos los valores de p fueron <0,001*.
Población pediátrica
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con ANORO en los diferentes grupos de la población pediátrica en EPOC (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Cuando umeclidinio y vilanterol fueron administrados en combinación por vía inhalatoria, la farmacocinética de cada componente fue similar a la observada cuando se administra cada principio activo por separado. Por tanto, para los efectos farmacocinéticos se puede considerar cada componente por separado.
Absorción
Umeclidinio
Tras la administración por vía inhalatoria de umeclidinio a voluntarios sanos, la Cmax se alcanzó a los 5-15 minutos. La biodisponibilidad absoluta de umeclidinio administrado por vía inhalatoria fue de promedio el 13% de la dosis, con una contribución inapreciable de la absorción oral. Después de la administración de dosis repetidas de umeclidinio inhalado, el estado estacionario se alcanzó entre los días del 7 al 10, con una acumulación de 1,5 a 1,8 veces.
Vilanterol
Tras la administración por vía inhalatoria de vilanterol en voluntarios sanos, la Cmax se alcanzó a los 515 minutos. La biodisponibilidad absoluta de vilanterol administrado por vía inhalatoria fue del 27%, con una contribución inapreciable de la absorción oral. Después de la administración de dosis repetidas de vilanterol inhalado, el estado estacionario se alcanzó en los 6 días, con una acumulación de hasta 2,4 veces.
Distribución
Umeclidinio
Tras la administración intravenosa a sujetos sanos, el volumen medio de distribución fue de 86 litros. In vitro, el promedio de unión a proteínas plasmáticas en plasma humano fue el 89%.
Vilanterol
Tras la administración intravenosa a voluntarios sanos, el volumen medio de distribución en estado estacionario fue de 165 litros. In vitro, el promedio de unión a proteínas plasmáticas en plasma humano fue el 94%.
Biotransformación
Umeclidinio
En estudios in vitro se observó que umeclidinio se metaboliza principalmente por el citocromo P450 2D6 (CYP2D6) y que es sustrato de la glicoproteína-P (P-gp) transportadora. Las principales rutas metabólicas de umeclidinio son la oxidativa (hidroxilación, O-desalquilación) seguida de la conjugación (glucuronidación, etc.), dando lugar a una variedad de metabolitos con actividad farmacológica reducida o metabolitos para los que la actividad farmacológica no se ha establecido. La exposición sistémica a los metabolitos es baja.
Vilanterol
En estudios in vitro se observó que vilanterol se metaboliza principalmente por el citocromo P450 3A4 (CYP3A4) y que es sustrato de la P-gp transportadora. La principal ruta metabólica para el vilanterol es la O-desalquilación que da lugar a una variedad de metabolitos con actividad agonista p1 y p2 adrenérgica significativamente reducida. Los perfiles metabólicos plasmáticos tras la administración oral de vilanterol en un estudio con radiomarcador en humanos fueron consistentes con un elevado metabolismo de primer paso. La exposición sistémica de los metabolitos es baja.
Eliminación
Umeclidinio
Tras la administración intravenosa, el aclaramiento plasmático fue de 151 litros/hora. Tras la administración intravenosa, aproximadamente el 58% de la dosis administrada marcada con radiomarcadores (o el 73% de la dosis radiomarcada recuperada) fue excretada en las heces en las 192 horas después de la dosis. La eliminación urinaria representó el 22% de la dosis administrada con radiomarcadores en las 168 horas después de la dosis (27% de la dosis radiomarcada recuperada). La excreción de la materia relacionada con el fármaco en las heces tras la dosis administrada por vía intravenosa indica secreción biliar. Tras la administración oral a sujetos varones sanos, la radiactividad
total fue eliminada principalmente por las heces (92% de la dosis administrada con radiomarcadores o 99% de la dosis radiactiva recuperada) en las 168 horas después de la dosis. Menos del 1% de la dosis oral administrada (1% de la dosis radiactiva recuperada) fue excretada en orina, lo que sugiere una absorción inapreciable tras administración oral. El promedio de la vida media de eliminación plasmática de umeclidinio tras la administración por vía inhalatoria durante 10 días fue de 19 horas, con un 3% a 4% de excreción de sustancia activa inalterada en orina en el estado estacionario.
Vilanterol
Tras la administración por vía intravenosa, el aclaramiento plasmático fue de 108 litros/hora. Después de la administración oral de vilanterol radiomarcado, el balance de masa mostró el 70% del compuesto marcado radiactivamente en orina y el 30% en heces. La eliminación primaria de vilanterol fue mediante metabolismo seguida por la excreción de metabolitos en orina y heces. La vida media de eliminación plasmática de vilanterol tras la administración por vía inhalatoria durante 10 días fue de un promedio de 11 horas.
Características en poblaciones especiales de voluntarios sanos o pacientes
Pacientes de edad avanzada
El análisis farmacocinético poblacional mostró que la farmacocinética de umeclidinio y vilanterol fue similar en los pacientes de 65 años de edad y mayores de 65 años con EPOC, y para aquellos menores de 65 años.
Insuficiencia renal
Tras la administración de dos veces la dosis recomendada de umeclidinio y la dosis recomendada de vilanterol a pacientes con insuficiencia renal grave, no se observaron evidencias de un aumento en la exposición sistémica a umeclidinio o vilanterol (Cmax y AUC). Tampoco hubo evidencia de alteración en la unión a proteínas entre pacientes con insuficiencia renal grave y voluntarios sanos.
Insuficiencia hepática
Tras la administración de dos veces la dosis recomendada de umeclidinio y la dosis recomendada de vilanterol a pacientes con insuficiencia hepática moderada (clasificación B Child-Pugh) no se observaron evidencias de un aumento en la exposición sistémica a umeclidinio o vilanterol (Cmax y AUC). Tampoco hubo evidencia de alteración en la unión a proteínas entre pacientes con insuficiencia hepática moderada y voluntarios sanos. Umeclidinio/vilanterol no ha sido evaluado en pacientes con insuficiencia hepática grave.
Otras poblaciones especiales
El análisis farmacocinético poblacional mostró que no se requiere ajuste en la dosis de bromuro de umeclidinio en base al efecto de la edad, la raza, y el género, el uso de corticosteroides inhalados o el peso. Un estudio realizado en metabolizadores lentos de CYP2D6 no mostró evidencia de un efecto clínicamente relevante del polimorfismo genético de CYP2D6 sobre la exposición sistémica a bromuro de umeclidinio.
5.3 Datos preclínicos sobre seguridad
En estudios no clínicos con umeclidinio y vilanterol, en monoterapia y en combinación, los resultados fueron los típicamente asociados con la farmacología primaria tanto de los antagonistas del receptor muscarínico o agonistas p2-adrenérgicos respectivamente y/o irritación local. A continuación se muestran los resultados de los estudios realizados sobre los componentes por separado.
Genotoxicidad y carcinogénesis
Umeclidinio no resultó genotóxico en una batería estándar de estudios, ni resultó carcinogénico en estudios de inhalación a tiempo real realizados en ratas o ratones a exposiciones > 26 o > 22 veces la exposición clínica en humanos de umeclidinio 55 microgramos, respectivamente, en base al AUC.
En estudios de toxicidad genética, vilanterol (como a-fenilcinamato) y el ácido trifenilacético no resultaron genotóxicos, lo cual indica que vilanterol (como trifenatato) no representa un peligro genotóxico para humanos. De acuerdo con los resultados identificados en otros agonistas p2 adrenérgicos, en los estudios de inhalación a tiempo real, vilanterol trifenatato produjo efectos proliferativos en el aparato reproductor de ratas y ratones hembras y en la glándula pituitaria de las ratas. No hubo un aumento en la incidencia de tumores en ratas o ratones a exposiciones 0,5 o 13 veces la exposición clínica en humanos de vilanterol 22 microgramos, respectivamente, en base al AUC.
Toxicidad reproductiva
Umeclidinio no fue teratogénico ni en ratas ni en conejos. En estudios pre y post-natales, la administración subcutánea de umeclidinio en ratas dio como resultado un menor incremento en el peso corporal de la madre y en el consumo de alimentos y disminuyó ligeramente el peso corporal antes del destete de las crías en madres que recibieron dosis de 180 microgramos/kg/día (aproximadamente 80 veces la dosis clínica en humanos a umeclidinio 55 microgramos, en base al AUC).
Vilanterol no fue teratogénico en ratas. En estudios de inhalación en conejos, el vilanterol produjo efectos similares a los que se observaban con otros p2-agonistas adrenérgicos (paladar hendido, párpados abiertos, fusión esternebral y malrotación/flexión de extremidades) a 6 veces la dosis clínica en humanos en base al AUC. No hubo efectos a 36 veces la dosis clínica en humanos de vilanterol 22 microgramos, en base al AUC.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Lactosa monohidrato,
Estearato de magnesio.
6.2 Incompatibilidades
No procede.
6.3 Periodo de validez
2 años.
Periodo de validez una vez abierta la bandeja: 6 semanas.
6.4 Precauciones especiales de conservación
No conservar a temperatura superior a 30°C. Si se conserva en nevera, se debe permitir que el inhalador alcance la temperatura ambiente durante al menos una hora antes de su uso.
Mantener el inhalador dentro de la bandeja sellada para protegerlo de la humedad y retirar la tapa inmediatamente antes de su primer uso.
Se debe utilizar en un plazo de 6 semanas tras la apertura de la bandeja.
Escribir la fecha en la que el inhalador se debe desechar en el espacio designado para ello, que aparece en la etiqueta del inhalador. La fecha se debe anotar tan pronto como el inhalador se saque de la bandeja.
6.5 Naturaleza y contenido del envase
El inhalador Ellipta está formado por un cuerpo gris claro, un protector de la boquilla rojo y un contador de dosis, envasado en una bandeja de aluminio laminada que contiene una bolsa desecante. La bandeja está sellada con una tapa de aluminio desplegable.
El inhalador contiene dos blísters de aluminio laminado de 7 o 30 dosis.
El inhalador es un dispositivo multi-componente compuesto de polipropileno, polietileno de alta densidad, polioximetileno, polibutileno tereftalato, acrilonitrilo butadieno estireno, policarbonato y acero inoxidable.
Tamaño de los envases de 7 o 30 dosis por inhalador.
Envase clínico de 3 x 30 dosis por inhalador.
Puede que solamente estén comercializados algunos tamaños de envases.
6.6 Precauciones especiales de eliminación y otras eliminaciones
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Para instrucciones de uso, ver sección 4.2.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Glaxo Group Limited
980 Great West Road
Brentford
Middlesex
TW8 9GS
Reino Unido
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/14/898/001
EU/1/14/898/002
EU/1/14/898/003
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 08 Mayo 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección del fabricante responsable de la liberación de los lotes Glaxo Operations UK Ltd. (operando como Glaxo Wellcome Operations)
Priory Street
Ware, Hertfordshire SG12 0DJ Reino Unido
Glaxo Operations UK Ltd. (operando como Glaxo Wellcome Operations)
Harmire Road
Barnard Castle, County Durham DL12 8DT Reino Unido
El prospecto impreso del medicamento debe especificar el nombre y dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica.
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107 quarter, apartado 7, de la Directiva 2001/83/CE y cualquier actualización posterior publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo
acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de
Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
• Obligación de llevar a cabo medidas post-autorización
El TAC deberá llevar a cabo, dentro del plazo establecido, las siguientes medidas:
|
Descripción |
Fecha límite |
|
Presentación del informe final del estudio clínico observacional post-autorización de seguridad, cuyo protocolo fue acordado con el PRAC, donde se comparaba una cohorte de pacientes en tratamiento con Anoro frente a tiotropio (estudio 201038), con |
Q3 2024 |
el objetivo de cuantificar la incidencia de eventos cardiovasculares y cerebrovasculares seleccionados en pacientes con EPOC._
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
55 microgramos/22 microgramos
1. NOMBRE DEL MEDICAMENTO
ANORO 55 microgramos/22 microgramos polvo para inhalación (unidosis) umeclidinio/vilanterol
2. PRINCIPIO(S) ACTIVO(S)
Cada dosis liberada contiene 55 microgramos de umeclidinio (equivalente a 65 microgramos de bromuro de umeclidinio) y 22 microgramos de vilanterol (como trifenatato).
3. LISTA DE EXCIPIENTES
También contiene lactosa y estearato de magnesio. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo para inhalación (unidosis), Ellipta 1 inhalador Ellipta de 7 dosis 1 inhalador Ellipta de 30 dosis
Envase clínico: 90 dosis (3 inhaladores Ellipta de 30 dosis) - 3 x 30 dosis
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía inhalatoria, una vez al día
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
No conservar a temperatura superior a 30°C.
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Glaxo Group Limited, 980 Great West Road, Brentford, Middlesex, TW8 9GS, Reino Unido. Logo Glaxo Group Ltd
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
issi s
EU/1/14/898/001 1 inhalador Ellipta de 7 dosis EU/1/14/898/002 1 inhalador Ellipta de 30 dosis EU/1/14/898/003 Envase Clínico: 90 dosis (3 inhaladores Ellipta de 30 dosis)
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
anoro ellipta
55 microgramos/22 microgramos
1. NOMBRE DEL MEDICAMENTO
ANORO 55 microgramos/22 microgramos polvo para inhalación (unidosis) umeclidinio/vilanterol
2. PRINCIPIO(S) ACTIVO(S)
Cada dosis liberada contiene 55 microgramos de umeclidinio (equivalente a 65 microgramos de bromuro de umeclidinio) y 22 microgramos de vilanterol (como trifenatato).
3. LISTA DE EXCIPIENTES
También contiene lactosa y estearato de magnesio. Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
1 inhalador de 30 dosis Ellipta
Envase clínico. Prohibida su venta al detalle.
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Vía inhalatoria, una vez al día
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
No conservar a temperatura superior a 30°C.
Conservar en el embalaje original para protegerlo de la humedad.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE
COMERCIALIZACIÓN_
Glaxo Group Limited, 980 Great West Road, Brentford, Middlesex, TW8 9GS, Reino Unido. Logo Glaxo Group Ltd
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN_
EU/1/14/898/003
13. NÚMERO DE LOTE_
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN_
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
anoro ellipta
INFORMACIÓN MÍNIMA A INCLUIR EN BLÍSTERS O TIRAS ETIQUETA DE LA BANDEJA DE ALUMINIO 55 microgramos/22 microgramos
1. NOMBRE DEL MEDICAMENTO
ANORO 55/22 mcg polvo para inhalación umeclidinio/vilanterol
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Logo Glaxo Group Ltd
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. OTROS
No abrir hasta que esté preparado para inhalar. Periodo de validez tras la apertura: 6 semanas. 7 dosis 30 dosis Ellipta
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETA DEL DISPOSITIVO
55 microgramos/22 microgramos
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ANORO 55/22 mcg polvo para inhalación umeclidinio/vilanterol
Vía inhalatoria
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
7 dosis 30 dosis
6. OTROS
Periodo de validez tras la apertura: 6 semanas. Desechar el:
Ellipta
B. PROSPECTO
Prospecto: información para el usuario
ANORO 55 microgramos/22 microgramos polvo para inhalación (unidosis)
umeclidinio/vilanterol
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado solamente a usted, y no debe dárselo a otras personas aunque tengan los mismos síntomas que usted, ya que puede perjudicarles.
- Si experimenta efectos adversos, consulte a su médico, farmacéutico o enfermero, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es ANORO y para qué se utiliza
2. Qué necesita saber antes de empezar a usar ANORO
3. Cómo usar ANORO
4. Posibles efectos adversos
5. Conservación de ANORO
6. Contenido del envase e información adicional Instrucciones de uso paso a paso
1. Qué es ANORO y para qué se utiliza Qué es ANORO
ANORO contiene dos principios activos llamados umeclidinio y vilanterol. Estos pertenecen al grupo de medicamentos llamados broncodilatadores.
Para qué se utiliza ANORO
ANORO se utiliza para tratar la enfermedad pulmonar obstructiva crónica (EPOC) en adultos. La EPOC es una enfermedad crónica, que empeora lentamente y se caracteriza por provocar dificultad para respirar.
En la EPOC los músculos que rodean las vías aéreas se contraen. Este medicamento impide la contracción de estos músculos en los pulmones, facilitando la entrada y salida de aire de los pulmones. Cuando se utiliza de forma regular, ayuda a controlar las dificultades para respirar y reduce los efectos de la EPOC en su vida cotidiana.
ANORO no se debe utilizar para aliviar un ataque repentino de ahogo o sibilancias (sonidos silbantes al respirar).
Si tiene este tipo de ataques debe utilizar un inhalador de “rescate” de acción rápida (como salbutamol).
2. Qué necesita saber antes de empezar a usar ANORO No use ANORO:
- si es alérgico a umeclidinio, vilanterol o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Si piensa que lo anterior le aplica, no use este medicamento hasta haber consultado con su médico. Advertencias y precauciones
Consulte a su médico antes de empezar a usar este medicamento:
- si tiene asma (No use ANORO para el tratamiento del asma)
- si tiene problemas cardiacos o tensión arterial alta
- si tiene un problema ocular llamado glaucoma de ángulo cerrado
- si tiene próstata agrandada, dificultad para orinar o una obstrucción en la vejiga
- si tiene epilepsia
- si tiene problemas de tiroides
- si tiene diabetes
- si tiene problemas de hígado graves.
Consulte con su médico si piensa que cualquiera de las condiciones anteriores le aplican. Dificultades respiratorias urgentes
Si tiene opresión en el pecho, tos, sibilancias o dificultad para respirar inmediatamente después de utilizar su inhalador ANORO:
Deje de usar este medicamento y busque atención médica inmediatamente, ya que puede tener una afección grave llamada broncoespasmo paradójico.
Problemas oculares durante el tratamiento con ANORO
Si tiene dolor ocular o molestias, visión borrosa durante un tiempo, halos visuales o imágenes coloreadas asociadas a enrojecimiento de los ojos durante el tratamiento con ANORO:
Deje de usar este medicamento y busque ayuda médica inmediatamente, ya que estos signos pueden deberse a un ataque agudo de glaucoma de ángulo cerrado.
Niños y adolescentes
Este medicamento no se debe administrar a niños o adolescentes menores de 18 años.
Uso de ANORO con otros medicamentos
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento.
Algunos medicamentos pueden afectar a la forma de actuar de este medicamento, o hacer que sea más probable que sufra efectos adversos. Estos incluyen:
- medicamentos llamados betabloqueantes (como propranolol), utilizados en el tratamiento de la tensión arterial alta u otras enfermedades del corazón
- ketoconazol o itraconazol, para tratar infecciones por hongos
- claritromicina o telitromicina, para tratar infecciones bacterianas
- ritonavir, para tratar el VIH
- medicamentos que disminuyen los niveles de potasio en sangre, como por ejemplo algunos diuréticos.
- otros medicamentos de acción prolongada similares a este medicamento utilizados en el tratamiento de problemas respiratorios, por ejemplo tiotropio, indacaterol. No use ANORO si está tomando estos medicamentos.
Consulte con su médico o farmacéutico si está tomando alguno de estos medicamentos. Embarazo, lactancia y fertilidad
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico antes de utilizar este medicamento. Si está embarazada, no utilice este medicamento a menos que su médico le indique que puede hacerlo.
Se desconoce si los componentes de ANORO se excretan en la leche materna. Si está en periodo de lactancia, consulte con su médico antes de utilizar ANORO. Si está dando el pecho, no utilice este medicamento a menos que su médico le indique que puede hacerlo.
Conducción y uso de máquinas
Es poco probable que ANORO afecte a su capacidad para conducir o utilizar máquinas.
ANORO contiene lactosa
Si su médico le ha diagnosticado una intolerancia a ciertos azúcares, consulte con su médico antes de utilizar este medicamento.
3. Cómo usar ANORO
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte con su médico o farmacéutico.
La dosis recomendada es una inhalación todos los días, a la misma hora cada día. Solo necesita una inhalación al día, ya que el efecto de este medicamento dura 24 horas.
No utilice más dosis de las que su médico le haya indicado.
Use ANORO con regularidad
Es muy importante que utilice ANORO todos los días, como le haya indicado su médico. Esto le ayudará a no tener síntomas a lo largo del día y la noche.
No utilice este medicamento para aliviar un ataque repentino de ahogo o sibilancias. Si tiene este tipo de ataque debe utilizar un inhalador de “rescate” de acción rápida (como salbutamol).
Cómo usar el inhalador
Para obtener la información completa lea las “Instrucciones de uso paso a paso” incluidas en este prospecto.
Para usar ANORO, inspírelo hacia sus pulmones a través de la boca utilizando el inhalador Ellipta.
Si los síntomas no mejoran
Si sus síntomas de EPOC (ahogo, sibilancias, tos) no mejoran o empeoran, o si está utilizando su inhalador de “rescate” de acción rápida más a menudo de lo habitual:
Contacte con su médico lo antes posible.
Si usa más ANORO del que debe
Si accidentalmente toma demasiado medicamento, contacte con su médico o farmacéutico inmediatamente, ya que puede necesitar atención médica. Si es posible, muéstreles el inhalador, el envase o su prospecto. Podría notar que su corazón late más rápido de lo normal, tener alteraciones visuales, la boca seca o dolor de cabeza.
Si olvidó usar ANORO
No tome una dosis doble para compensar las dosis olvidadas. Tome la siguiente dosis a su hora habitual. Si tiene sibilancias o ahogo, utilice su inhalador de “rescate” de acción rápida (como salbutamol) y busque asesoramiento médico.
Si interrumpe el tratamiento con ANORO
Utilice ANORO durante el tiempo que le haya recomendado su médico. Solo será eficaz durante el tiempo que siga utilizándolo. No deje de utilizarlo hasta que su médico se lo indique, aunque se encuentre mejor, ya que sus síntomas pueden empeorar.
Si tiene alguna duda sobre el uso de este medicamento, pregunte a su médico, farmacéutico o enfermero.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Las reacciones alérgicas a ANORO son poco frecuentes (afectan a menos de 1 de cada 100 personas). Si tiene alguno de los síntomas descritos a continuación después de tomar ANORO, deje de tomar este medicamento e informe inmediatamente a su médico:
• erupción en la piel (habones) o enrojecimiento
• hinchazón, algunas veces de la cara o de la boca (angioedema)
• aumento de las sibilancias (sonido silbante que se produce al respirar), tos o tener dificultad para respirar
• sensación de debilidad repentina o mareo (que puede provocar colapso o pérdida de la consciencia).
Dificultades respiratorias urgentes
Si tiene opresión en el pecho, tos, sibilancias o dificultad para respirar inmediatamente después de utilizar este medicamento:
Deje de usar este medicamento y busque atención médica inmediatamente, ya que puede tener una afección grave llamada broncoespasmo paradójico.
Efectos adversos frecuentes
Estos pueden afectar hasta de 1 de cada 10 personas:
• orina frecuente y dolorosa (puede ser síntoma de infección del tracto urinario)
• combinación de dolor de garganta y moqueo
• dolor de garganta
• sensación de presión o dolor en las mejillas y la frente (puede ser síntoma de inflamación de los senos llamado sinusitis)
• dolor de cabeza
• tos
• dolor e irritación en la parte posterior de la boca y garganta
• estreñimiento
• boca seca
• infección de las vías aéreas superiores.
Efectos adversos poco frecuentes
Estos pueden afectar hasta 1 de cada 100 personas:
• latido del corazón irregular
• latido del corazón más rápido
• sentir los latidos del corazón (palpitaciones)
• erupción cutánea
• temblor
• alteración del gusto.
Efectos adversos raros
Estos pueden afectar hasta 1 de cada 1.000 personas:
• dificultad y dolor al orinar, estos pueden ser signos de obstrucción de la vejiga o retención urinaria.
Otros efectos adversos
En un número muy reducido de personas han ocurrido otros efectos adversos pero su frecuencia exacta se desconoce:
• disminución en la visión o dolor en los ojos debido a una presión ocular elevada (posibles signos de glaucoma).
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico o farmacéutico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de ANORO
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase, bandeja e inhalador, después de CAD. La fecha de caducidad es el último día del mes que se indica.
Conservar en el embalaje original para protegerlo de la humedad y no abrir la tapa de aluminio hasta que esté preparado para inhalar. Una vez abierta la bandeja, el inhalador se puede usar durante un plazo de 6 semanas, contando desde la fecha de apertura de la bandeja. Escribir la fecha en la que se debe desechar el inhalador en el espacio designado para ello en la etiqueta del inhalador. La fecha se debe anotar tan pronto como el inhalador se saque de la bandeja.
No conservar a temperatura superior a 30°C.
Si lo conserva en la nevera, deje que el inhalador vuelva a la temperatura ambiente al menos una hora antes de utilizarlo.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de ANORO
Los principios activos son bromuro de umeclidinio y vilanterol.
Cada inhalación proporciona una dosis liberada (dosis que sale por la boquilla) de 55 microgramos de umeclidinio (equivalente a 65 microgramos de bromuro de umeclidinio) y 22 microgramos de vilanterol (como trifenatato).
Los demás componentes son lactosa monohidrato y estearato de magnesio.
Aspecto del producto y contenido del envase
El dispositivo inhalador está formado por un cuerpo gris claro de plástico, un protector de la boquilla rojo y un contador de dosis. Está envasado en una bandeja de aluminio laminada. La bandeja contiene una bolsa desecante para reducir la humedad del envase.
Las sustancias activas se presentan como un polvo blanco en blísters separados dentro del inhalador. Cada inhalador contiene 7 o 30 dosis. También están disponibles los envases clínicos que contienen 90 (3 inhaladores de 30 dosis). Puede que solamente estén comercializados algunos tamaños de envases en su país.
Titular de la autorización de comercialización y responsable de la fabricación
Titular de la autorización de comercialización:
Glaxo Group Limited
980 Great West Road
Brentford
Middlesex
TW8 9GS
Reino Unido
Fabricante:
Glaxo Operations UK Limited (operando como Glaxo Wellcome Operations)
Priory Street Ware
Hertfordshire SG12 0DJ Reino Unido
Glaxo Operations UK Limited (operando como Glaxo Wellcome Operations)
Harmire Road Barnard Castle County Durham DL12 8DT Reino Unido
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
Belgie/Belgique/Belgien
GlaxoSmithKline Pharmaceuticals s.a./n.v. Tél/Tel: + 32 (0) 10 85 52 00
EtarapHH
LaaKCoCMHTKaaHH EOOfl Tea.: + 359 2 953 10 34
Ceská republika
GlaxoSmithKline, s.r.o.
Tel: + 420 222 001 111 cz.info@gsk.com
Danmark
GlaxoSmithKline Pharma A/S Tlf: + 45 36 35 91 00 dk-info@gsk.com
Deutschland
GlaxoSmithKline GmbH & Co. KG Tel.: + 49 (0)89 36044 8701 produkt.info@gsk.com
Eesti
GlaxoSmithKline Eesti OÜ
Lietuva
GlaxoSmithKline Lietuva UAB Tel: + 370 5 264 90 00 info.lt@gsk.com
Luxembourg/Luxemburg
GlaxoSmithKline Pharmaceuticals s.a./n.v.
Belgique/Belgien
Tél/Tel: + 32 (0) 10 85 52 00
Magyarország GlaxoSmithKline Kft.
Tel.: + 36 1 225 5300
Malta
GlaxoSmithKline (Malta) Limited Tel: + 356 21 238131
Nederland
GlaxoSmithKline BV Tel: + 31 (0)30 6938100 nlinfo@gsk.com
Norge
GlaxoSmithKline AS
|
Tel: + 372 6676 900 estonia@gsk.com |
Tlf: + 47 22 70 20 00 firmapost@gsk.no |
|
ELXáóa GlaxoSmithKline A.E.B.E. Tnk: + 30 210 68 82 100 |
Osterreich GlaxoSmithKline Pharma GmbH Tel: + 43 (0)1 97075 0 at.info@gsk.com |
|
España GlaxoSmithKline, S.A. Tel: + 34 902 202 700 es-ci@gsk.com |
Polska GSK Services Sp. z o.o. Tel.: + 48 (0)22 576 9000 |
|
France Laboratoire GlaxoSmithKline Tél: + 33 (0)1 39 17 84 44 diam@gsk.com |
Portugal GlaxoSmithKline - Produtos Farmacéuticos, Lda. Tel: + 351 21 412 95 00 FI.PT@gsk.com |
|
Hrvatska GlaxoSmithKline d.o.o. Tel: +385 1 6051999 |
Romania GlaxoSmithKline (GSK) S.R.L. Tel: + 4021 3028 208 |
|
Ireland GlaxoSmithKline (Ireland) Limited Tel: + 353 (0)1 4955000 |
Slovenija GlaxoSmithKline d.o.o. Tel: + 386 (0)1 280 25 00 medical.x.si@gsk.com |
|
Ísland Vistor hf. Sími: + 354 535 7000 |
Slovenská republika GlaxoSmithKline Slovakia s. r. o. Tel: + 421 (0)2 48 26 11 11 recepcia.sk@gsk.com |
|
Italia GlaxoSmithKline S.p.A. Tel: + 39 (0)45 9218 111 |
Suomi/Finland GlaxoSmithKline Oy Puh/Tel: + 358 (0)10 30 30 30 Finland.tuoteinfo@gsk.com |
|
Kúnpoq GlaxoSmithKline (Cyprus) Ltd Tnk: + 357 22 39 70 00 gskcyprus@gsk.com |
Sverige GlaxoSmithKline AB Tel: + 46 (0)8 638 93 00 info.produkt@gsk.com |
|
Latvija GlaxoSmithKline Latvia SIA Tel: + 371 67312687 lv-epasts@gsk.com |
United Kingdom GlaxoSmithKline UK Ltd Tel: + 44 (0)800 221441 customercontactuk@gsk.com |
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http: //www .ema.europa.eu.
¿Qué es el inhalador?
La primera vez que utilice ANORO, no necesita asegurarse de que el inhalador está funcionando correctamente, ya que contiene dosis previamente medidas y está listo para utilizarse directamente.
El inhalador está envasado en una bandeja que contiene una bolsa desecante, para reducir la humedad. Tire la bolsa del desecante, no lo ingiera o inhale.
Cuando saque el inhalador de su caja (bandeja sellada), estará en la posición de “cerrado”. No lo abra hasta que esté preparado para inhalar una dosis del medicamento. Cuando se abre la bandeja, se debe anotar la fecha de “desechar el” en el espacio designado para ello que aparece en la etiqueta del inhalador. La fecha de “desechar el” es de 6 semanas desde la fecha de apertura de la bandeja.
Después de esta fecha el inhalador no se debe utilizar más. La bandeja se puede desechar después de la primera apertura.
Las instrucciones de uso del inhalador proporcionadas a continuación pueden ser usadas tanto para el inhalador de 30 dosis como para el inhalador de 7 dosis.
Lea esta información antes de comenzar
Si la tapa del inhalador se abre y se cierra sin que se inhale el medicamento, se perderá la dosis. La dosis perdida quedará retenida de forma segura dentro del inhalador, pero no estará disponible para ser inhalada.
No es posible administrar de forma accidental una dosis adicional o una dosis doble mediante una inhalación.
Contador de dosis
El contador de dosis indica cuántas dosis de medicamento quedan en el dispositivo. Antes de usar el inhalador, debe indicar exactamente 30 dosis.
Cada vez que se abre la tapa, el contador disminuye en 1 unidad.
Cuando quedan menos de 10 dosis, la mitad del contador de dosis se pone de color rojo.
Una vez se utiliza la última dosis, la mitad del contador de dosis se pone de color rojo e indica el número 0. El inhalador ahora está vacío.
Si se abre la tapa cuando el inhalador está vacío, el contador de dosis pasa de estar la mitad de color rojo a estarlo completamente.
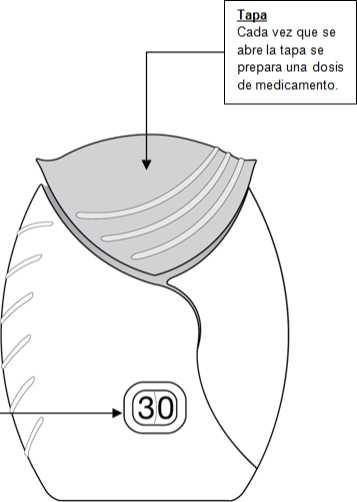
1) Preparar una dosis
Cuando esté preparado para inhalar una dosis, abrir la tapa del inhalador. No agite el inhalador.
• Deslizar la tapa hacia abaj o hasta oír un ‘ clic’.
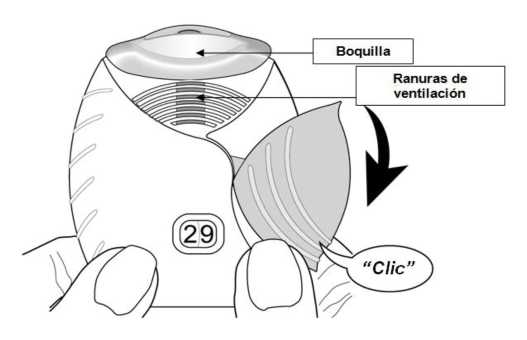
Ahora, el medicamento está preparado para poder inhalarlo.
Como confirmación, el contador de dosis disminuye en 1 unidad.
• Si el contador de dosis no disminuye al oír el ‘clic’, el inhalador no liberará la dosis del medicamento.
Llévelo al farmacéutico y solicite ayuda.
2) Inhale su medicamento
• Mientras mantiene el inhalador alejado de la boca, espire tanto como le sea posible.
No espire dentro del inhalador.
• Coloque la boquilla entre los labios, y ciérrelos firmemente alrededor de la boquilla.
No bloquee las ranuras de ventilación con los dedos.
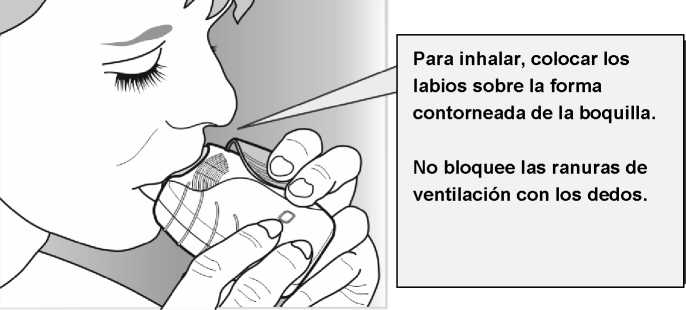
• Realice una inspiración larga, continua y profunda. Mantenga la inspiración tanto tiempo como sea posible (al menos 3-4 segundos).
• Retire el inhalador de la boca.
Espire suave y lentamente.
Puede que no sea capaz de distinguir el sabor o notar el medicamento, incluso cuando utiliza el inhalador de forma correcta.
3) Cerrar el inhalador
Antes de cerrar la tapa, la boquilla del inhalador puede limpiarse utilizando un pañuelo seco.
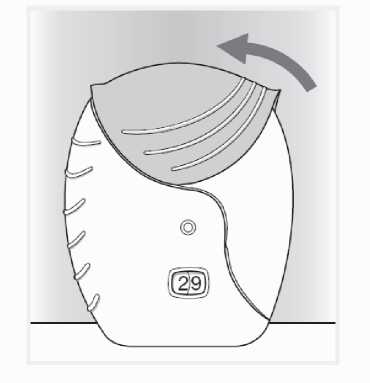
• Deslice la tapa hacia arriba, hasta el tope, para cubrir la boquilla.
ANEXO IV
CONCLUSIONES CIENTÍFICAS Y MOTIVOS PARA LA MODIFICACIÓN DE LAS CONDICIONES DE LAS AUTORIZACIONES DE COMERCIALIZACIÓN
Teniendo en cuenta lo dispuesto en el Informe de Evaluación del Comité para la Evaluación de Riesgos en Farmacovigilancia (PRAC) sobre los informes periódicos de seguridad (IPS) para bromuro de umeclidinio/vilanterol, las conclusiones científicas del Comité de Medicamentos de Uso Humano (CHMP) son las siguientes:
Se recibieron un total de 30 casos espontáneos, en los que se describen 31 reacciones adversas según el término preferente PT para reacciones adversas urinarias asociados con bromuro de umeclidinio/vilanterol (UMEC/VI).
De aquellos, se consideraron un total de 12 casos espontáneos suficientemente detallados como para realizar una evaluación completa, que incluyeron 13 acontecimientos adversos urinarios: 8 acontecimientos graves de retención urinaria, 2 acontecimientos graves de obstrucción del tracto urinario, 2 casos no graves de disminución del flujo urinario y 1 caso de disminución de la frecuencia de micción. Se ha notificado un tiempo hasta su aparición (< 2 semanas) después del inicio del tratamiento con UMEC/VI en 7/12 casos evaluables. En 8 casos, hubo evidencia positiva de la retirada después de la interrupción del tratamiento con UMEC/VI.
"La retención urinaria" es un efecto de clase conocido de los medicamentos anticolinérgicos (Halpin, 2015 Stephenson, 2011; Afonso, 2011) y ya ha sido incluida una advertencia en la ficha técnica recomendando el uso de UMEC/VI con precaución en los pacientes con retención urinaria.
De acuerdo a los resultados de las evaluaciones acumuladas y considerando la plausibilidad farmacológica del efecto anticolinérgico sobre la función urinaria, hay evidencias suficientemente robustas para garantizar una actualización de la Información de Producto con “retención urinaria, obstrucción de la vejiga y disuria” como reacciones adversas asociados al tratamiento con UMEC/VI.
Por tanto, en vista de los datos presentados en los IPS revisados, el PRAC considera que los cambios en la Información de Producto de los medicamentos que contienen bromuro de umeclidinio/vilanterol están j ustificados.
El CHMP está de acuerdo con las conclusiones científicas del PRAC.
Motivos para la modificación de las condiciones de la autorización de comercialización
De acuerdo con las conclusiones científicas para bromuro de umeclidinio/vilanterol, el CHMP considera que el balance beneficio-riesgo del medicamento o medicamentos que contienen bromuro de umeclidinio/vilanterol no se modifica sujeto a los cambios propuestos en la información del producto.
El CHMP recomienda que se modifiquen las condiciones de la autorización de comercialización.