Advate 250 Ui Polvo Y Disolvente Para Solucion Inyectable
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
1. NOMBRE DEL MEDICAMENTO
ADVATE 250 UI polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 250 UI de factor VIII humano de coagulación (ADNr), octocog alfa. Después de la reconstitución, ADVATE contiene aproximadamente 50 UI por ml de factor VIII humano de coagulación (ADNr), octocog alfa.
La potencia (Unidades Internacionales) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de ADVATE es aproximadamente 4.000 -10.000 UI/mg/proteína.
El octocog alfa (factor VIII humano de coagulación (ADNr)) es una proteína purificada con 2332 aminoácidos. Está producida por tecnología de ADN recombinante en células de ovario de hámster chino (CHO). Preparado sin la adición de ninguna proteína (exógena) de origen humano o animal en el proceso del cultivo celular, purificación o formulación final.
Excipientes con efectos conocidos:
0,45 mmol de sodio (10 mg) por vial.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: polvo friable de color blanco a blanquecino. Disolvente: solución transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII). ADVATE está indicado en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia y con apoyo de resucitación disponible inmediatamente en caso de anafilaxia.
Posología
La dosis y duración de la terapia de sustitución depende de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII se expresa en Unidades Internacionales (UI), que se relacionan con el estándar de la OMS para medicamentos con factor VIII. La actividad de Factor VIII en plasma se expresa bien como porcentaje (referido a plasma humano normal) o en UI (referidas al Estándar Internacional de factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII es equivalente a la cantidad de factor VIII existente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis de factor VIII requerida se basa en el hallazgo empírico de que 1 UI de factor VIII por kg de peso corporal incrementa la actividad de factor VIII del plasma en 2 UI/dl. La dosis requerida se determina mediante la siguiente fórmula:
Unidades requeridas (UI) = peso corporal (kg) x aumento de factor VIII deseado (%) x 0,5
En el caso de los episodios hemorrágicos siguientes, la actividad de factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. En cirugía y en los episodios hemorrágicos, puede utilizarse la siguiente tabla 1 como guía de dosificación:
|
Tabla 1 Guía de dosificación en episodios hemorrágicos y cirugía | ||
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (% o UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente o hemorragia muscular u oral. |
20 - 40 |
Repetir la inyección cada 12 a 24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma. |
30 - 60 |
Repetir la inyección cada 12-24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) durante 3 a 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir la inyección cada 8 a 24 horas (cada 6 a 12 horas en los pacientes menores de 6 años) hasta superar el peligro. |
|
Cirugía | ||
|
Menor Incluyendo extracción dental. |
30 - 60 |
Cada 24 horas (cada 12 a 24 horas en los pacientes menores de 6 años), al menos 1 día, hasta lograr la curación. |
|
Mayor |
80 - 100 (pre y postoperatorio) |
Repetir la inyección cada 8-24 horas (cada 6 a 24 horas en los pacientes menores de 6 años) hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor VIII del 30% al 60% (UI/dl). |
La dosis y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual. En determinadas circunstancias (ej.: presencia de un título bajo de inhibidor) pueden ser necesarias dosis mayores a las calculadas mediante la fórmula.
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis que se debe administrar y la frecuencia con la que se debe repetir la inyección. En el caso especial de las intervenciones de cirugía mayor, es indispensable controlar con precisión el tratamiento de sustitución mediante pruebas de la coagulación (actividad del factor VIII plasmático). La respuesta individual de cada paciente frente al factor VIII puede variar, alcanzar distintos niveles de recuperación in vivo y presentar semividas diferentes.
Profilaxis
Para la profilaxis de larga duración frente a hemorragias en pacientes con hemofilia A grave, las dosis normales son de 20 a 40 UI de factor VIII por kg de peso corporal a intervalos de 2 a 3 días.
Población _ pediátrica
La dosis de tratamiento a demanda en pacientes pediátricos (0 a 18 años de edad) no difiere de la de los pacientes adultos. En pacientes menores de 6 años, se recomiendan dosis de 20 a 50 UI de factor VIII por kg de peso corporal de 3 a 4 veces a la semana como terapia profiláctica.
Forma de administración
ADVATE se debe administrar por vía intravenosa. En caso de ser administrado por un profesional no sanitario, es necesario realizar un entrenamiento adecuado.
Se debe determinar la velocidad de administración para asegurar la comodidad del paciente, hasta un máximo de 10 ml/min.
Después de la reconstitución, la solución es transparente, incolora y libre de partículas extrañas y tiene un pH de entre 6,7 y 7,3.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1 o a las proteínas de ratón o hámster.
4.4 Advertencias y precauciones especiales de empleo
Hipersensibilidad
Con ADVATE se han notificado reacciones de hipersensibilidad de tipo alérgico, incluyendo anafilaxis. El medicamento contiene trazas de proteínas de ratón y de hámster. Se debe informar a los pacientes de que en caso de que ocurran síntomas de hipersensibilidad, deben interrumpir el tratamiento y consultar con su médico de forma inmediata. Se debe informar a los pacientes de los signos de las reacciones de hipersensibilidad de tipo inmediato incluyendo habón urticarial, urticaria generalizada, tirantez en el pecho, sibilancia, hipotensión y anafilaxia.
En caso de que se produzca un shock, se deben seguir las pautas médicas estándar para su tratamiento.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) frente al factor VIII es una complicación conocida en el tratamiento de individuos con hemofilia A. Estos inhibidores son habitualmente inmunoglobulinas IgGs dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en unidades Bethesda (UB) por ml de plasma empleando el ensayo modificado. En pacientes que desarrollan anticuerpos neutralizantes (inhibidores) al Factor VIII, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia. El riesgo de desarrollar inhibidores se relaciona con el grado de exposición al factor VIII, siendo mayor durante los primeros 20 días de exposición, y con otros factores ambientales y genéticos. Raramente se desarrollan inhibidores después de los 100 primeros días de exposición.
Se han observado casos recurrentes de aparición de inhibidores (a títulos bajos), después de cambiar de un factor VIII a otro en los pacientes con un tratamiento previo de más de 100 días de exposición y con antecedentes de desarrollo de inhibidores. Así, se recomienda monitorizar cuidadosamente la aparición de inhibidores en todos los pacientes después de cualquier cambio de producto.
En general, en todos los pacientes tratados con un factor VIII de coagulación se debe monitorizar cuidadosamente la aparición de inhibidores mediante la realización de observaciones clínicas y de las pruebas de laboratorio apropiadas. Si no se obtienen los niveles de actividad de factor VIII en plasma esperados, o si no se controla la hemorragia con una dosis adecuada, se debe realizar una prueba de detección de inhibidor de factor VIII. En pacientes con niveles altos de inhibidor, la terapia de sustitución de factor VIII puede no ser efectiva y se deben considerar otras opciones terapéuticas.
El tratamiento de tales pacientes debe estar dirigido por médicos con experiencia en el cuidado de pacientes con hemofilia e inhibidores del factor VIII.
Complicación relacionada con catéter en el tratamiento
Si se requiere un dispositivo de acceso venoso (CVAD) debe considerarse el riesgo de complicaciones relacionadas con CVAD incluyendo infecciones locales, bacteremia y trombosis del lado del catéter.
Consideraciones relativas al excipiente
Después de la reconstitución, este medicamento contiene 0,45 mmol de sodio (10 mg) por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
Se recomienda encarecidamente registrar el nombre y el número de lote del producto cada vez que se administre ADVATE a un paciente para poder mantener una relación entre el paciente y el lote del medicamento.
Población pediátrica:
Las advertencias y precauciones incluidas se aplican tanto a adultos como a niños.
4.5 Interacción con otros medicamentos y otras formas de interacción.
No se han realizado estudios de interacciones con ADVATE.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción en animales con factor VIII. Dados los raros casos de hemofilia A en mujeres, no se dispone de experiencia relacionada con el uso de factor VIII durante el embarazo y el periodo de lactancia. Por lo tanto, sólo debe utilizarse factor VIII durante el embarazo y el periodo de lactancia si está claramente indicado.
4.7 Efectos sobre la capacidad de conducir y utilizar máquinas
La influencia de ADVATE sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los ensayos clínicos con ADVATE incluyeron 418 sujetos con al menos una exposición a ADVATE y registraron un total de 93 reacciones adversas al medicamento (RAM). Las RAM que aparecieron con mayor frecuencia fueron el desarrollo de anticuerpos neutralizantes frente al factor VIII (inhibidores), cefalea y fiebre.
La hipersensibilidad y las reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el lugar de infusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habón urticarial, hipotensión, letargia, náuseas, inquietud, taquicardia, tirantez en el pecho, cosquilleo, vómitos, sibilancia) se observaron con poca frecuencia pero, en algunos casos, pueden progresar a anafilaxia grave (incluyendo shock).
Se puede observar desarrollo de anticuerpos a la proteína de ratón y/o hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) frente al factor VIII. Si aparecen tales inhibidores, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia.
Resumen tabulado de reacciones adversas
La siguiente tabla 2 indica la frecuencia de las reacciones adversas en los ensayos clínicos y procedente de informes espontáneos. La tabla sigue la clasificación de órganos del sistema MedDRA (COS y Nivel de término preferido).
Las frecuencias se han clasificado utilizando el siguiente criterio: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100), raros (> 1/10.000 a < 1/1.000), muy raros (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de informes espontáneos | ||
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Infecciones e infestaciones |
Gripe |
Poco frecuentes |
|
Laringitis |
Poco frecuentes | |
|
Linfangitis |
Poco frecuentes | |
|
Trastornos de la sangre y del sistema linfático |
Inhibición del factor VIII Linfangitis |
Frecuentes Poco frecuentes |
|
Trastornos del sistema inmunológico |
Reacción anafiláctica |
Frecuencia no conocida |
|
Hipersensibilidadc |
Frecuencia no conocida | |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuentes |
|
Mareos |
Poco frecuentes | |
|
Problemas de memoria Síncope |
Poco frecuentes Poco frecuentes | |
|
Temblores |
Poco frecuentes | |
|
Migraña |
Poco frecuentes | |
|
Disgeusia |
Poco frecuentes | |
|
Trastornos oculares |
Inflamación ocular |
Poco frecuentes |
|
Trastornos cardiacos |
Palpitaciones |
Poco frecuentes |
|
Trastornos vasculares |
Hematoma |
Poco frecuentes |
|
Sofocos |
Poco frecuentes | |
|
Palidez |
Poco frecuentes | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
Poco frecuentes |
|
Trastornos gastrointestinales |
Diarrea |
Poco frecuentes |
|
Dolor en el abdomen superior |
Poco frecuentes | |
|
Nauseas |
Poco frecuentes | |
|
Vómitos |
Poco frecuentes | |
|
Trastornos de la piel y del tejido subcutáneo |
Prurito |
Poco frecuentes |
|
Sarpullido |
Poco frecuentes | |
|
Hiperhidrosis |
Poco frecuentes | |
|
Urticaria |
Poco frecuentes | |
|
Trastornos generales y alteraciones en el lugar de administración |
Fiebre |
Frecuentes |
|
Edema periférico |
Poco frecuentes | |
|
Dolor torácico Malestar torácico |
Poco frecuentes Poco frecuentes | |
|
Escalofríos |
Poco frecuentes | |
|
Sensación anormal Hematoma en el lugar de punción de un vaso sanguíneo |
Poco frecuentes Poco frecuentes | |
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de __informes espontáneos__
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Fatiga Reacción en la zona de inyección |
Frecuencia no conocida Frecuencia no conocida | |
|
Malestar general |
Frecuencia no conocida | |
|
Exploraciones complementarias | ||
|
Recuento elevado de monocitos |
Poco frecuentes | |
|
Nivel reducido de factor de coagulación VIIIb |
Poco frecuentes | |
|
Descenso del hematocrito |
Poco frecuentes | |
|
Test de laboratorio anormal |
Poco frecuentes | |
|
Lesiones, intoxicaciones y complicaciones de procedimientos terapéuticos |
Complicaciones tras el procedimiento |
Poco frecuentes |
|
Hemorragia tras el procedimiento |
Poco frecuentes | |
|
Reacción en el lugar del procedimiento |
Poco frecuentes |
a) Calculado según un número total único de pacientes que recibieron ADVATE (418)
b) El descenso inesperado de los niveles de factor VIII de coagulación se produjo en un paciente durante la perfusión continua de ADVATE después de cirugía (días 10-14 de postoperatorio). Se mantuvo la hemostasis durante todo este periodo y tanto los niveles de factor VIII de plasma como los niveles de aclaramiento volvieron a los niveles adecuados el día 15 de postoperatorio. Los ensayos de inhibidores del factor VIII realizados tras la finalización de la perfusión continua y la terminación del estudio fueron negativos.
c) RAM explicadas en la sección siguiente.
Descripción de reacciones adversas seleccionadas Desarrollo de inhibidor
Se ha notificado un desarrollo de inhibidor en pacientes con tratamientos previos y en pacientes no tratados previamente. Para información detallada ver secciones 5.1 (Propiedades farmacológicas) y 4.4 (Advertencias y precauciones especiales de empleo).
RAM específicas _para residuos del _proceso de _ fabricación
De los 229 pacientes en tratamiento en los que se evaluaron los anticuerpos antiproteínas de células de ovario de hámster chino (CHO), 3 mostraron una tendencia ascendente estadísticamente significativa en los títulos, 4 mostraron picos sostenidos o picos momentáneos y un paciente mostró ambos, pero no tuvo ningún otro síntoma clínico. De los 229 pacientes tratados a los que se realizó una evaluación de los anticuerpos anti-IgG murina, 10 mostraron una tendencia ascendente, dos mostraron un pico sostenido o pico momentáneo y un paciente mostró ambos. Cuatro de estos pacientes sufrieron episodios aislados de urticaria, prurito, sarpullido y recuentos ligeramente elevados de eosinófilos entre exposiciones repetidas al medicamento del estudio.
Hipersensibilidad
Reacciones de tipo alérgico incluyen anafilaxis y se han manifestado como mareos, parestesias, erupción, soplado, tumefacción facial, urticaria y prurito.
Población _ pediátrica
En los estudios clínicos no se han notado diferencias específicas por edad en RAM excepto el desarrollo de inhibidores en pacientes pediátricos no tratados previamente (PUP) y complicaciones relacionadas con catéter.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación pertinente incluido en el Anexo V.
4.9 Sobredosis
No se han notificado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos: factor VIII de coagulación. Código ATC: B02BD02.
El complejo factor VIII /Factor von Willebrand consta de dos moléculas (factor VIII y factor von Willebrand) con diferentes funciones fisiológicas. ADVATE contiene factor VIII de coagulación recombinante (octocog alfa), una glicoproteína que es biológicamente equivalente a la glicoproteína de factor VIII del plasma humano.
Octocog alfa es una glicoproteína que consta de 2332 aminoácidos con un peso molecular aproximado de 280 kD. Cuando se perfunde en un paciente hemofílico, octocog alfa se enlaza al Factor von Willebrand endógeno en la circulación del paciente. El factor VIII activado actúa como un Cofactor para el Factor IX activado, acelerando la conversión del Factor X a Factor X activado. El Factor X activado convierte la protrombina en trombina. La trombina convierte el fibrinógeno en fibrina, con la que se puede formar el coágulo. La hemofilia A es una alteración hereditaria de la coagulación sanguínea ligada al sexo y producida por la disminución de los niveles de factor VIII que da como resultado una hemorragia profusa en las articulaciones, músculos u órganos internos, bien de forma espontánea o bien como resultado de un trauma accidental o quirúrgico. Mediante terapia de sustitución, los niveles plasmáticos de factor VIII aumentan, consiguiendo una corrección temporal de la deficiencia de factor VIII y una corrección de las tendencias hemorrágicas.
Desarrollo del inhibidor
Se evaluó la inmunogenicidad de ADVATE en pacientes con tratamientos previos. Durante los ensayos clínicos con ADVATE en 233 pacientes pediátricos y adultos [pacientes pediátricos (0 a 16 años de edad) y pacientes adultos (más de 16 años de edad)] diagnosticados con hemofilia A grave (factor VIII < 1%), con exposición previa a concentrados de factor VIII durante un periodo igual o superior a 150 días para adultos y niños mayores e igual o superior a 50 días para niños menores de 6 años, un paciente desarrolló un título bajo de inhibidor (2,4 BU en el ensayo Bethesda modificado) tras 26 días de exposición a ADVATE. Las pruebas de seguimiento de inhibidores en este paciente tras abandonar el ensayo fueron negativas. En todos los estudios, la exposición media a ADVATE fue de 97,0 días de exposición por sujeto (intervalo 1 a 709) para pacientes previamente tratados. La incidencia global de cualquier desarrollo de inhibidor del factor VIII (alto o bajo) fue de 0,4% (1 de 233).
En el ensayo completado no controlado 060103, 16 de los 45 (35,6%) pacientes sin tratamiento previo con hemofilia A grave (FVIII < 1%) con al menos 25 días de exposición a FVIII desarrollaron inhibidores del FVIII: 7 (15,6%) sujetos desarrollaron inhibidores de título alto y 9 (20%) de los sujetos desarrollaron inhibidores de título bajo, 1 de los cuales también se clasificó como inhibidor temporal.
Los factores de riesgo relacionados con el desarrollo del inhibidor en este ensayo incluyeron etnicidad no caucásica, antecedentes familiares de inhibidores y tratamiento intensivo a dosis altas en los primeros 20 días de exposición. En los 20 sujetos que no tenían ninguno de estos factores de riesgo no se produjo el desarrollo del inhibidor.
Se han recogido datos sobre Inducción de tolerancia inmunitaria (ITI) en pacientes con inhibidores.
En un subestudio del estudio PUP 060103, se documentaron tratamientos ITI en 11 PUP. Se hizo un análisis de cuadro retrospective de 30 materias sobre ITI (estudio 060703) y recogida de datos de registro está en curso.
En el estudio 060201, se compararon dos esquemas de tratamiento de profilaxis a largo plazo en 53 sujetos previamente tratados: un régimen individualizado de dosificación guiada farmacocinética (dentro de un rango de 20 a 80 UI de factor VIII por kg de peso corporal a intervalos de 72 ± 6 horas, n = 23) con un régimen de dosificación profiláctica estándar (de 20 a 40 UI/kg cada 48 ± 6 horas, n = 30). El régimen de dosificación guiada farmacocinética (de acuerdo con una fórmula específica) tenía como objetivo mantener el factor VIII en niveles > 1% en el intervalo entre dosis de 72 horas. Los datos de este estudio demuestran que los dos regímenes de dosificación profiláctica son comparables en lo que se refiere a la reducción de la frecuencia de hemorragias.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con ADVATE en los diferentes grupos de la población pediátrica con hemofilia A (deficiencia congénita de factor VIII) en “Inducción de tolerancia inmunitaria (ITI) en pacientes con hemofilia A (deficiencia congénita de factor VIII) que han desarrollado inhibidores al factor VIII” y “tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII)” (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Todos los estudios farmacocinéticos con ADVATE se realizaron en pacientes previamente tratados con hemofilia A grave o moderadamente grave (valor basal del factor VIII < 2%). El análisis de las muestras de plasma se realizó en un laboratorio central utilizando el ensayo de coagulación en una etapa.
Un total de 195 sujetos con hemofilia A grave (valor basal del factor VIII < 1%) proporcionaron parámetros PK que se incluyeron en el conjunto de análisis de PK según el protocolo. Las categorías de estos análisis para lactantes (1 mes a <2 años), niños (2 a <5 años), niños de mayor edad (5 a <12 años), adolescentes (12 a <18 años) y adultos (18 años y mayores) se utilizaron para resumir los parámetros PK, definiéndose la edad como la edad en el momento de la perfusión de PK.
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Área bajo la curva total (UI*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Recuperación incremental ajustada a Cmáx (UI/dl por UI/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Semivida (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Concentración máxima en plasma tras la perfusión (UI/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Tiempo de residencia medio (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Volumen de distribución en estado estacionario (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Aclaramiento (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Calculada como (Cmáx - valor basal del factor VI la medida máxima de factor VIII tras la perfusión.
I) dividida por la dosis en UI/kg, donde Cmáx es
La seguridad y la eficacia hemostática de ADVATE en la población pediátrica son similares a las de los pacientes adultos. La recuperación y la semivida terminal (t/2) ajustadas fue aproximadamente un 20% inferior en niños pequeños (menores de 6 años) que en adultos, lo que lo que puede deberse en parte a un volumen mayor conocido de plasma por kg de peso corporal en los pacientes más jóvenes.
No se dispone de datos farmacocinéticos de ADVATE en pacientes sin tratamiento previo.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de la seguridad, toxicología aguda, toxicidad en dosis repetida, toxicidad local y genotoxicidad.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo
Manitol
Cloruro de sodio Histidina Trealosa Cloruro de calcio Trometamol Polisorbato 80 Glutatión (reducido)
Disolvente
Agua esterilizada para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
2 años.
Después de la reconstitución, desde un punto de vista microbiológico, el medicamento debe utilizarse de inmediato. No obstante, se ha demostrado una estabilidad física y química en uso durante 3 horas a 25 °C.
Durante el periodo de validez, el medicamento puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único no superior a 6 meses. Se debe anotar la fecha del final del periodo de conservación a temperatura ambiente de 6 meses en el embalaje del medicamento. El medicamento no puede refrigerarse de nuevo.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
ADVATE con el equipo BAXJECT II: Conservar el vial del producto en el embalaje exterior para protegerlo de la luz.
ADVATE en el sistema BAXJECT III: Conservar el blister sellado en el embalaje exterior para protegerlo de la luz.
Para consultar las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Tanto el vial de polvo como el vial de disolvente de 5 ml son de vidrio tipo I cerrado con tapones de caucho de clorobutilo. El medicamento se proporciona con una de las configuraciones siguientes:
- ADVATE con el equipo BAXJECT II: Cada envase contiene un vial de polvo, un vial de disolvente de 5 ml y un equipo para reconstitución (BAXJECT II).
- ADVATE en el sistema BAXJECT III: Cada envase contiene un sistema BAXJECT III listo para usar en un blister sellado (el vial de polvo y el vial de disolvente de 5 ml están precargados con el sistema para su reconstitución).
6.6 Precauciones especiales de eliminación y otras manipulaciones
ADVATE debe administrarse intravenosamente después de la reconstitución del producto.
La solución reconstituida se debe inspeccionar visualmente para comprobar que no presenta partículas extrañas y/o descoloración.
Después de la reconstitución la solución debe ser transparente, incolora y no presentar partículas extrañas.
No utilizar soluciones turbias o con depósitos.
- Para la administración se requiere el uso de una jeringa luer-lock.
- Utilizar dentro de las tres horas después de la reconstitución.
- No refrigerar la preparación después de la reconstitución.
- La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Reconstitución con el equipo BAXJECT II:
- Para la reconstitución utilizar sólo el agua esterilizada para preparaciones inyectables y el equipo de reconstitución incluido en el envase.
- No utilizar si el equipo BAXJECT II, el sistema estéril de protección o su envase está dañado o muestra algún signo de deterioro.
- Se debe utilizar una técnica aséptica:
1. Si el medicamento se encuentra en la nevera, coger de la nevera los viales de polvo y de disolvente de ADVATE y dejarlos a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Quitar los protectores de los viales de polvo y disolvente.
4. Limpiar los tapones con las toallitas impregnadas de alcohol. Colocar los viales en una superficie plana y limpia.
5. Abrir el envoltorio del equipo BAXJECT II quitando la tapa de papel sin tocar el interior (Fig. a). No sacar el equipo del envoltorio. No utilizar si el equipo BAXJECT II, su sistema de barrera estéril o su acondicionamiento están dañados o muestran algún signo de deterioro.
6. Dar la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente. Coger el envoltorio por su extremo y sacar el equipo BAXJECT II de su envoltorio (Fig. b). No quitar el protector azul del equipo BAXJECT II.
7. Para la reconstitución, utilizar solamente el agua esterilizada para preparaciones inyectables y el equipo para reconstitución incluidos en el envase. Con el equipo BAXJECT II unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico blanca dentro del tapón del vial de polvo ADVATE. El vacío hará que el disolvente penetre en el vial de polvo ADVATE (Fig. c).
8. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará
a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig. a
Fig. b
Fig. c
Reconstitución con el sistema BAXJECT III
- No lo utilice si la tapa del blister no está perfectamente sellada.
1. Si el medicamento se encuentra en la nevera, coger de la nevera el blister sellado (contiene viales de polvo y disolvente precargados con el sistema para su reconstitución) y dejarlo a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Abrir el envoltorio de ADVATE quitando la tapa. Extraer el sistema BAXJECT III del blister.
4. Colocar ADVATE en una superficie plana con el vial de disolvente encima (Fig. 1). El vial de disolvente tiene una raya azul. No quitar el protector azul hasta que se le indique más adelante.
5. Mientras sujeta ADVATE con una mano en el sistema BAXJECT III, presionar con fuerza el vial de disolvente con la otra mano hasta que el sistema quede totalmente contraído y el disolvente entre dentro del vial de ADVATE (Fig. 2). No inclinar el sistema hasta que haya finalizado la transferencia.
6. Comprobar que ha finalizado la transferencia del disolvente. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig.l
Fig.2
Fig. 3
Administración:
Utilizar técnica aséptica:
Los medicamentos para administración parenteral deben inspeccionarse visualmente antes de su
administración para verificar la ausencia de partículas, cuando la solución y el envase lo permitan.
Sólo se debe utilizar una solución transparente y sin coloración.
1. Quitar el protector azul del equipo BAXJECT II / al sistema BAXJECT III. No introducir aire en la jeringa. Conectar la jeringa al equipo BAXJECT II / al sistema BAXJECT III.
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente.
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar intravenosamente. La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de ADVATE. Si se observa una elevación significativa del pulso, reducir la velocidad de administración o interrumpir temporalmente la inyección suele hacer que los síntomas normalmente desaparezcan pronto (ver secciones 4.4 y 4.8).
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
Industriestrasse 67
A-1221 Viena
Austria
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/001
EU/1/03/271/011
9. FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 2 de marzo de 2004 Fecha de la última renovación: 2 de marzo de 2014
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
ADVATE 500 UI polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 500 UI de factor VIII humano de coagulación (ADNr), octocog alfa. Después de la reconstitución, ADVATE contiene aproximadamente 100 UI por ml de factor VIII humano de coagulación (ADNr), octocog alfa.
La potencia (Unidades Internacionales) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de ADVATE es aproximadamente 4.000 -10.000 UI/mg/proteína.
El octocog alfa (factor VIII humano de coagulación (ADNr)) es una proteína purificada con 2332 aminoácidos. Está producida por tecnología de ADN recombinante en células de ovario de hámster chino (CHO). Preparado sin la adición de ninguna proteína (exógena) de origen humano o animal en el proceso del cultivo celular, purificación o formulación final.
Excipientes con efectos conocidos:
0,45 mmol de sodio (10 mg) por vial.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: polvo friable de color blanco a blanquecino. Disolvente: solución transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII). ADVATE está indicado en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia y con apoyo de resucitación disponible inmediatamente en caso de anafilaxia.
Posología
La dosis y duración de la terapia de sustitución depende de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII se expresa en Unidades Internacionales (UI), que se relacionan con el estándar de la OMS para medicamentos con factor VIII. La actividad de Factor VIII en plasma se expresa bien como porcentaje (referido a plasma humano normal) o en UI (referidas al Estándar Internacional de factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII es equivalente a la cantidad de factor VIII existente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis de factor VIII requerida se basa en el hallazgo empírico de que 1 UI de factor VIII por kg de peso corporal incrementa la actividad de factor VIII del plasma en 2 UI/dl. La dosis requerida se determina mediante la siguiente fórmula:
Unidades requeridas (UI) = peso corporal (kg) x aumento de factor VIII deseado (%) x 0,5
En el caso de los episodios hemorrágicos siguientes, la actividad de factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. En cirugía y en los episodios hemorrágicos, puede utilizarse la siguiente tabla 1 como guía de dosificación:
|
Tabla 1 Guía de dosificación en episodios hemorrágicos y cirugía | ||
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (% o UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente o hemorragia muscular u oral. |
20 - 40 |
Repetir la inyección cada 12 a 24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma. |
30 - 60 |
Repetir la inyección cada 12-24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) durante 3 a 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir la inyección cada 8 a 24 horas (cada 6 a 12 horas en los pacientes menores de 6 años) hasta superar el peligro. |
|
Cirugía | ||
|
Menor Incluyendo extracción dental. |
30 - 60 |
Cada 24 horas (cada 12 a 24 horas en los pacientes menores de 6 años), al menos 1 día, hasta lograr la curación. |
|
Mayor |
80 - 100 (pre y postoperatorio) |
Repetir la inyección cada 8-24 horas (cada 6 a 24 horas en los pacientes menores de 6 años) hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor VIII del 30% al 60% (UI/dl). |
La dosis y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual. En determinadas circunstancias (ej.: presencia de un título bajo de inhibidor) pueden ser necesarias dosis mayores a las calculadas mediante la fórmula.
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis que se debe administrar y la frecuencia con la que se debe repetir la inyección. En el caso especial de las intervenciones de cirugía mayor, es indispensable controlar con precisión el tratamiento de sustitución mediante pruebas de la coagulación (actividad del factor VIII plasmático). La respuesta individual de cada paciente frente al factor VIII puede variar, alcanzar distintos niveles de recuperación in vivo y presentar semividas diferentes.
Profilaxis
Para la profilaxis de larga duración frente a hemorragias en pacientes con hemofilia A grave, las dosis normales son de 20 a 40 UI de factor VIII por kg de peso corporal a intervalos de 2 a 3 días.
Población _ pediátrica
La dosis de tratamiento a demanda en pacientes pediátricos (0 a 18 años de edad) no difiere de la de los pacientes adultos. En pacientes menores de 6 años, se recomiendan dosis de 20 a 50 UI de factor VIII por kg de peso corporal de 3 a 4 veces a la semana como terapia profiláctica.
Forma de administración
ADVATE se debe administrar por vía intravenosa. En caso de ser administrado por un profesional no sanitario, es necesario realizar un entrenamiento adecuado.
Se debe determinar la velocidad de administración para asegurar la comodidad del paciente, hasta un máximo de 10 ml/min.
Después de la reconstitución, la solución es transparente, incolora y libre de partículas extrañas y tiene un pH de entre 6,7 y 7,3.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1 o a las proteínas de ratón o hámster.
4.4 Advertencias y precauciones especiales de empleo
Hipersensibilidad
Con ADVATE se han notificado reacciones de hipersensibilidad de tipo alérgico, incluyendo anafilaxis. El medicamento contiene trazas de proteínas de ratón y de hámster. Se debe informar a los pacientes de que en caso de que ocurran síntomas de hipersensibilidad, deben interrumpir el tratamiento y consultar con su médico de forma inmediata. Se debe informar a los pacientes de los signos de las reacciones de hipersensibilidad de tipo inmediato incluyendo habón urticarial, urticaria generalizada, tirantez en el pecho, sibilancia, hipotensión y anafilaxia.
En caso de que se produzca un shock, se deben seguir las pautas médicas estándar para su tratamiento.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) frente al factor VIII es una complicación conocida en el tratamiento de individuos con hemofilia A. Estos inhibidores son habitualmente inmunoglobulinas IgGs dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en unidades Bethesda (UB) por ml de plasma empleando el ensayo modificado. En pacientes que desarrollan anticuerpos neutralizantes (inhibidores) al Factor VIII, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia. El riesgo de desarrollar inhibidores se relaciona con el grado de exposición al factor VIII, siendo mayor durante los primeros 20 días de exposición, y con otros factores ambientales y genéticos. Raramente se desarrollan inhibidores después de los 100 primeros días de exposición.
Se han observado casos recurrentes de aparición de inhibidores (a títulos bajos), después de cambiar de un factor VIII a otro en los pacientes con un tratamiento previo de más de 100 días de exposición y con antecedentes de desarrollo de inhibidores. Así, se recomienda monitorizar cuidadosamente la aparición de inhibidores en todos los pacientes después de cualquier cambio de producto.
En general, en todos los pacientes tratados con un factor VIII de coagulación se debe monitorizar cuidadosamente la aparición de inhibidores mediante la realización de observaciones clínicas y de las pruebas de laboratorio apropiadas. Si no se obtienen los niveles de actividad de factor VIII en plasma esperados, o si no se controla la hemorragia con una dosis adecuada, se debe realizar una prueba de detección de inhibidor de factor VIII. En pacientes con niveles altos de inhibidor, la terapia de sustitución de factor VIII puede no ser efectiva y se deben considerar otras opciones terapéuticas.
El tratamiento de tales pacientes debe estar dirigido por médicos con experiencia en el cuidado de pacientes con hemofilia e inhibidores del factor VIII.
Complicación relacionada con catéter en el tratamiento
Si se requiere un dispositivo de acceso venoso (CVAD) debe considerarse el riesgo de complicaciones relacionadas con CVAD incluyendo infecciones locales, bacteremia y trombosis del lado del catéter.
Consideraciones relativas al excipiente
Después de la reconstitución, este medicamento contiene 0,45 mmol de sodio (10 mg) por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
Se recomienda encarecidamente registrar el nombre y el número de lote del producto cada vez que se administre ADVATE a un paciente para poder mantener una relación entre el paciente y el lote del medicamento.
Población pediátrica:
Las advertencias y precauciones incluidas se aplican tanto a adultos como a niños.
4.5 Interacción con otros medicamentos y otras formas de interacción.
No se han realizado estudios de interacciones con ADVATE.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción en animales con factor VIII. Dados los raros casos de hemofilia A en mujeres, no se dispone de experiencia relacionada con el uso de factor VIII durante el embarazo y el periodo de lactancia. Por lo tanto, sólo debe utilizarse factor VIII durante el embarazo y el periodo de lactancia si está claramente indicado.
4.7 Efectos sobre la capacidad de conducir y utilizar máquinas
La influencia de ADVATE sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los ensayos clínicos con ADVATE incluyeron 418 sujetos con al menos una exposición a ADVATE y registraron un total de 93 reacciones adversas al medicamento (RAM). Las RAM que aparecieron con mayor frecuencia fueron el desarrollo de anticuerpos neutralizantes frente al factor VIII (inhibidores), cefalea y fiebre.
La hipersensibilidad y las reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el lugar de infusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habón urticarial, hipotensión, letargia, náuseas, inquietud, taquicardia, tirantez en el pecho, cosquilleo, vómitos, sibilancia) se observaron con poca frecuencia pero, en algunos casos, pueden progresar a anafilaxia grave (incluyendo shock).
Se puede observar desarrollo de anticuerpos a la proteína de ratón y/o hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) frente al factor VIII. Si aparecen tales inhibidores, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia.
Resumen tabulado de reacciones adversas
La siguiente tabla 2 indica la frecuencia de las reacciones adversas en los ensayos clínicos y procedente de informes espontáneos. La tabla sigue la clasificación de órganos del sistema MedDRA (COS y Nivel de término preferido).
Las frecuencias se han clasificado utilizando el siguiente criterio: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100), raros (> 1/10.000 a < 1/1.000), muy raros (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de informes espontáneos | ||
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Infecciones e infestaciones |
Gripe |
Poco frecuentes |
|
Laringitis |
Poco frecuentes | |
|
Linfangitis |
Poco frecuentes | |
|
Trastornos de la sangre y del sistema linfático |
Inhibición del factor VIII Linfangitis |
Frecuentes Poco frecuentes |
|
Trastornos del sistema inmunológico |
Reacción anafiláctica |
Frecuencia no conocida |
|
Hipersensibilidadc |
Frecuencia no conocida | |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuentes |
|
Mareos |
Poco frecuentes | |
|
Problemas de memoria Síncope |
Poco frecuentes Poco frecuentes | |
|
Temblores |
Poco frecuentes | |
|
Migraña |
Poco frecuentes | |
|
Disgeusia |
Poco frecuentes | |
|
Trastornos oculares |
Inflamación ocular |
Poco frecuentes |
|
Trastornos cardiacos |
Palpitaciones |
Poco frecuentes |
|
Trastornos vasculares |
Hematoma |
Poco frecuentes |
|
Sofocos |
Poco frecuentes | |
|
Palidez |
Poco frecuentes | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
Poco frecuentes |
|
Trastornos gastrointestinales |
Diarrea |
Poco frecuentes |
|
Dolor en el abdomen superior |
Poco frecuentes | |
|
Nauseas |
Poco frecuentes | |
|
Vómitos |
Poco frecuentes | |
|
Trastornos de la piel y del tejido subcutáneo |
Prurito |
Poco frecuentes |
|
Sarpullido |
Poco frecuentes | |
|
Hiperhidrosis |
Poco frecuentes | |
|
Urticaria |
Poco frecuentes | |
|
Trastornos generales y alteraciones en el lugar de administración |
Fiebre |
Frecuentes |
|
Edema periférico |
Poco frecuentes | |
|
Dolor torácico Malestar torácico |
Poco frecuentes Poco frecuentes | |
|
Escalofríos |
Poco frecuentes | |
|
Sensación anormal Hematoma en el lugar de punción de un vaso sanguíneo |
Poco frecuentes Poco frecuentes | |
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de __informes espontáneos__
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Fatiga Reacción en la zona de inyección |
Frecuencia no conocida Frecuencia no conocida | |
|
Malestar general |
Frecuencia no conocida | |
|
Exploraciones complementarias | ||
|
Recuento elevado de monocitos |
Poco frecuentes | |
|
Nivel reducido de factor de coagulación VIIIb |
Poco frecuentes | |
|
Descenso del hematocrito |
Poco frecuentes | |
|
Test de laboratorio anormal |
Poco frecuentes | |
|
Lesiones, intoxicaciones y complicaciones de procedimientos terapéuticos |
Complicaciones tras el procedimiento |
Poco frecuentes |
|
Hemorragia tras el procedimiento |
Poco frecuentes | |
|
Reacción en el lugar del procedimiento |
Poco frecuentes |
a) Calculado según un número total único de pacientes que recibieron ADVATE (418)
b) El descenso inesperado de los niveles de factor VIII de coagulación se produjo en un paciente durante la perfusión continua de ADVATE después de cirugía (días 10-14 de postoperatorio).
Se mantuvo la hemostasis durante todo este periodo y tanto los niveles de factor VIII de plasma como los niveles de aclaramiento volvieron a los niveles adecuados el día 15 de postoperatorio. Los ensayos de inhibidores del factor VIII realizados tras la finalización de la perfusión continua y la terminación del estudio fueron negativos.
c) RAM explicadas en la sección siguiente.
Descripción de reacciones adversas seleccionadas Desarrollo de inhibidor
Se ha notificado un desarrollo de inhibidor en pacientes con tratamientos previos y en pacientes no tratados previamente. Para información detallada ver secciones 5.1 (Propiedades farmacológicas) y 4.4 (Advertencias y precauciones especiales de empleo).
RAM específicas para residuos del proceso de fabricación
De los 229 pacientes en tratamiento en los que se evaluaron los anticuerpos antiproteínas de células de ovario de hámster chino (CHO), 3 mostraron una tendencia ascendente estadísticamente significativa en los títulos, 4 mostraron picos sostenidos o picos momentáneos y un paciente mostró ambos, pero no tuvo ningún otro síntoma clínico. De los 229 pacientes tratados a los que se realizó una evaluación de los anticuerpos anti-IgG murina, 10 mostraron una tendencia ascendente, dos mostraron un pico sostenido o pico momentáneo y un paciente mostró ambos. Cuatro de estos pacientes sufrieron episodios aislados de urticaria, prurito, sarpullido y recuentos ligeramente elevados de eosinófilos entre exposiciones repetidas al medicamento del estudio.
Hipersensibilidad
Reacciones de tipo alérgico incluyen anafilaxis y se han manifestado como mareos, parestesias, erupción, soplado, tumefacción facial, urticaria y prurito.
Población pediátrica
En los estudios clínicos no se han notado diferencias específicas por edad en RAM excepto el desarrollo de inhibidores en pacientes pediátricos no tratados previamente (PUP) y complicaciones relacionadas con catéter.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación pertinente incluido en el Anexo V.
4.9 Sobredosis
No se han notificado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos: factor VIII de coagulación. Código ATC: B02BD02.
El complejo factor VIII /Factor von Willebrand consta de dos moléculas (factor VIII y factor von Willebrand) con diferentes funciones fisiológicas. ADVATE contiene factor VIII de coagulación recombinante (octocog alfa), una glicoproteína que es biológicamente equivalente a la glicoproteína de factor VIII del plasma humano.
Octocog alfa es una glicoproteína que consta de 2332 aminoácidos con un peso molecular aproximado de 280 kD. Cuando se perfunde en un paciente hemofílico, octocog alfa se enlaza al Factor von Willebrand endógeno en la circulación del paciente. El factor VIII activado actúa como un Cofactor para el Factor IX activado, acelerando la conversión del Factor X a Factor X activado. El Factor X activado convierte la protrombina en trombina. La trombina convierte el fibrinógeno en fibrina, con la que se puede formar el coágulo. La hemofilia A es una alteración hereditaria de la coagulación sanguínea ligada al sexo y producida por la disminución de los niveles de factor VIII que da como resultado una hemorragia profusa en las articulaciones, músculos u órganos internos, bien de forma espontánea o bien como resultado de un trauma accidental o quirúrgico. Mediante terapia de sustitución, los niveles plasmáticos de factor VIII aumentan, consiguiendo una corrección temporal de la deficiencia de factor VIII y una corrección de las tendencias hemorrágicas.
Desarrollo del inhibidor
Se evaluó la inmunogenicidad de ADVATE en pacientes con tratamientos previos. Durante los ensayos clínicos con ADVATE en 233 pacientes pediátricos y adultos [pacientes pediátricos (0 a 16 años de edad) y pacientes adultos (más de 16 años de edad)] diagnosticados con hemofilia A grave (factor VIII < 1%), con exposición previa a concentrados de factor VIII durante un periodo igual o superior a 150 días para adultos y niños mayores e igual o superior a 50 días para niños menores de 6 años, un paciente desarrolló un título bajo de inhibidor (2,4 BU en el ensayo Bethesda modificado) tras 26 días de exposición a ADVATE. Las pruebas de seguimiento de inhibidores en este paciente tras abandonar el ensayo fueron negativas. En todos los estudios, la exposición media a ADVATE fue de 97,0 días de exposición por sujeto (intervalo 1 a 709) para pacientes previamente tratados. La incidencia global de cualquier desarrollo de inhibidor del factor VIII (alto o bajo) fue de 0,4% (1 de 233).
En el ensayo completado no controlado 060103, 16 de los 45 (35,6%) pacientes sin tratamiento previo con hemofilia A grave (FVIII < 1%) con al menos 25 días de exposición a FVIII desarrollaron inhibidores del FVIII: 7 (15,6%) sujetos desarrollaron inhibidores de título alto y 9 (20%) de los sujetos desarrollaron inhibidores de título bajo, 1 de los cuales también se clasificó como inhibidor temporal.
Los factores de riesgo relacionados con el desarrollo del inhibidor en este ensayo incluyeron etnicidad no caucásica, antecedentes familiares de inhibidores y tratamiento intensivo a dosis altas en los primeros 20 días de exposición. En los 20 sujetos que no tenían ninguno de estos factores de riesgo no se produjo el desarrollo del inhibidor.
Se han recogido datos sobre Inducción de tolerancia inmunitaria (ITI) en pacientes con inhibidores.
En un subestudio del estudio PUP 060103, se documentaron tratamientos ITI en 11 PUP. Se hizo un análisis de cuadro retrospective de 30 materias sobre ITI (estudio 060703) y recogida de datos de registro está en curso.
En el estudio 060201, se compararon dos esquemas de tratamiento de profilaxis a largo plazo en 53 sujetos previamente tratados: un régimen individualizado de dosificación guiada farmacocinética (dentro de un rango de 20 a 80 UI de factor VIII por kg de peso corporal a intervalos de 72 ± 6 horas, n = 23) con un régimen de dosificación profiláctica estándar (de 20 a 40 UI/kg cada 48 ± 6 horas, n = 30). El régimen de dosificación guiada farmacocinética (de acuerdo con una fórmula específica) tenía como objetivo mantener el factor VIII en niveles > 1% en el intervalo entre dosis de 72 horas. Los datos de este estudio demuestran que los dos regímenes de dosificación profiláctica son comparables en lo que se refiere a la reducción de la frecuencia de hemorragias.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con ADVATE en los diferentes grupos de la población pediátrica con hemofilia A (deficiencia congénita de factor VIII) en “Inducción de tolerancia inmunitaria (ITI) en pacientes con hemofilia A (deficiencia congénita de factor VIII) que han desarrollado inhibidores al factor VIII” y “tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII)” (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Todos los estudios farmacocinéticos con ADVATE se realizaron en pacientes previamente tratados con hemofilia A grave o moderadamente grave (valor basal del factor VIII < 2%). El análisis de las muestras de plasma se realizó en un laboratorio central utilizando el ensayo de coagulación en una etapa.
Un total de 195 sujetos con hemofilia A grave (valor basal del factor VIII < 1%) proporcionaron parámetros PK que se incluyeron en el conjunto de análisis de PK según el protocolo. Las categorías de estos análisis para lactantes (1 mes a <2 años), niños (2 a <5 años), niños de mayor edad (5 a <12 años), adolescentes (12 a <18 años) y adultos (18 años y mayores) se utilizaron para resumir los parámetros PK, definiéndose la edad como la edad en el momento de la perfusión de PK.
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Área bajo la curva total (UI*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Recuperación incremental ajustada a Cmáx (UI/dl por UI/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Semivida (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Concentración máxima en plasma tras la perfusión (UI/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Tiempo de residencia medio (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Volumen de distribución en estado estacionario (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Aclaramiento (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Calculada como (Cmáx - valor basal del factor VIII) dividida por la dosis en Ul/kg, donde Cmáx es la medida máxima de factor VIII tras la perfusión.
La seguridad y la eficacia hemostática de ADVATE en la población pediátrica son similares a las de los pacientes adultos. La recuperación y la semivida terminal (t/2) ajustadas fUe aproximadamente un 20% inferior en niños pequeños (menores de 6 años) que en adultos, lo que lo que puede deberse en parte a un volumen mayor conocido de plasma por kg de peso corporal en los pacientes más jóvenes.
No se dispone de datos farmacocinéticos de ADVATE en pacientes sin tratamiento previo.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de la seguridad, toxicología aguda, toxicidad en dosis repetida, toxicidad local y genotoxicidad.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo
Manitol
Cloruro de sodio
Histidina
Trealosa
Cloruro de calcio Trometamol Polisorbato 80 Glutatión (reducido)
Disolvente
Agua esterilizada para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
2 años.
Después de la reconstitución, desde un punto de vista microbiológico, el medicamento debe utilizarse de inmediato. No obstante, se ha demostrado una estabilidad física y química en uso durante 3 horas a 25 °C.
Durante el periodo de validez, el medicamento puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único no superior a 6 meses. Se debe anotar la fecha del final del periodo de conservación a temperatura ambiente de 6 meses en el embalaje del medicamento. El medicamento no puede refrigerarse de nuevo.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
ADVATE con el equipo BAXJECT II: Conservar el vial del producto en el embalaje exterior para protegerlo de la luz.
ADVATE en el sistema BAXJECT III: Conservar el blister sellado en el embalaje exterior para protegerlo de la luz.
Para consultar las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Tanto el vial de polvo como el vial de disolvente de 5 ml son de vidrio tipo I cerrado con tapones de caucho de clorobutilo. El medicamento se proporciona con una de las configuraciones siguientes:
- ADVATE con el equipo BAXJECT II: Cada envase contiene un vial de polvo, un vial de disolvente de 5 ml y un equipo para reconstitución (BAXJECT II).
- ADVATE en el sistema BAXJECT III: Cada envase contiene un sistema BAXJECT III listo para usar en un blister sellado (el vial de polvo y el vial de disolvente de 5 ml están precargados con el sistema para su reconstitución).
6.6 Precauciones especiales de eliminación y otras manipulaciones
ADVATE debe administrarse intravenosamente después de la reconstitución del producto.
La solución reconstituida se debe inspeccionar visualmente para comprobar que no presenta partículas extrañas y/o descoloración.
Después de la reconstitución la solución debe ser transparente, incolora y no presentar partículas extrañas.
No utilizar soluciones turbias o con depósitos.
- Para la administración se requiere el uso de una jeringa luer-lock.
- Utilizar dentro de las tres horas después de la reconstitución.
- No refrigerar la preparación después de la reconstitución.
- La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Reconstitución con el equipo BAXJECT II:
- Para la reconstitución utilizar sólo el agua esterilizada para preparaciones inyectables y el equipo de reconstitución incluido en el envase.
- No utilizar si el equipo BAXJECT II, el sistema estéril de protección o su envase está dañado o muestra algún signo de deterioro.
- Se debe utilizar una técnica aséptica:
1. Si el medicamento se encuentra en la nevera, coger de la nevera los viales de polvo y de disolvente de ADVATE y dejarlos a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Quitar los protectores de los viales de polvo y disolvente.
4. Limpiar los tapones con las toallitas impregnadas de alcohol. Colocar los viales en una superficie plana y limpia.
5. Abrir el envoltorio del equipo BAXJECT II quitando la tapa de papel sin tocar el interior (Fig. a). No sacar el equipo del envoltorio. No utilizar si el equipo BAXJECT II, su sistema de barrera estéril o su acondicionamiento están dañados o muestran algún signo de deterioro.
6. Dar la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente. Coger el envoltorio por su extremo y sacar el equipo BAXJECT II de su envoltorio (Fig. b). No quitar el protector azul del equipo BAXJECT II.
7. Para la reconstitución, utilizar solamente el agua esterilizada para preparaciones inyectables y el equipo para reconstitución incluidos en el envase. Con el equipo BAXJECT II unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico blanca dentro del tapón del vial de polvo ADVATE. El vacío hará que el disolvente penetre en el vial de polvo ADVATE (Fig. c).
8. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará
a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig. a
Fig. b
Fig. c
Reconstitución con el sistema BAXJECT III
- No lo utilice si la tapa del blister no está perfectamente sellada.
1. Si el medicamento se encuentra en la nevera, coger de la nevera el blister sellado (contiene viales de polvo y disolvente precargados con el sistema para su reconstitución) y dejarlo a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Abrir el envoltorio de ADVATE quitando la tapa. Extraer el sistema BAXJECT III del blister.
4. Colocar ADVATE en una superficie plana con el vial de disolvente encima (Fig. 1). El vial de disolvente tiene una raya azul. No quitar el protector azul hasta que se le indique más adelante.
5. Mientras sujeta ADVATE con una mano en el sistema BAXJECT III, presionar con fuerza el vial de disolvente con la otra mano hasta que el sistema quede totalmente contraído y el disolvente entre dentro del vial de ADVATE (Fig. 2). No inclinar el sistema hasta que haya finalizado la transferencia.
6. Comprobar que ha finalizado la transferencia del disolvente. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig.l
Fig.2
Fig. 3
Administración:
Utilizar técnica aséptica:
Los medicamentos para administración parenteral deben inspeccionarse visualmente antes de su
administración para verificar la ausencia de partículas, cuando la solución y el envase lo permitan.
Sólo se debe utilizar una solución transparente y sin coloración.
1. Quitar el protector azul del equipo BAXJECT II / al sistema BAXJECT III. No introducir aire en la jeringa. Conectar la jeringa al equipo BAXJECT II / al sistema BAXJECT III.
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente.
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar intravenosamente. La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de ADVATE. Si se observa una elevación significativa del pulso, reducir la velocidad de administración o interrumpir temporalmente la inyección suele hacer que los síntomas normalmente desaparezcan pronto (ver secciones 4.4 y 4.8).
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
Industriestrasse 67
A-1221 Viena
Austria
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/002
EU/1/03/271/012
9. FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 2 de marzo de 2004 Fecha de la última renovación: 2 de marzo de 2014
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
ADVATE 1000 UI polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 1000 UI de factor VIII humano de coagulación (ADNr), octocog alfa. Después de la reconstitución, ADVATE contiene aproximadamente 200 UI por ml de factor VIII humano de coagulación (ADNr), octocog alfa.
La potencia (Unidades Internacionales) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de ADVATE es aproximadamente 4.000 -10.000 UI/mg/proteína.
El octocog alfa (factor VIII humano de coagulación (ADNr)) es una proteína purificada con 2332 aminoácidos. Está producida por tecnología de ADN recombinante en células de ovario de hámster chino (CHO). Preparado sin la adición de ninguna proteína (exógena) de origen humano o animal en el proceso del cultivo celular, purificación o formulación final.
Excipientes con efectos conocidos:
0,45 mmol de sodio (10 mg) por vial.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: polvo friable de color blanco a blanquecino. Disolvente: solución transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII). ADVATE está indicado en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia y con apoyo de resucitación disponible inmediatamente en caso de anafilaxia.
Posología
La dosis y duración de la terapia de sustitución depende de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII se expresa en Unidades Internacionales (UI), que se relacionan con el estándar de la OMS para medicamentos con factor VIII. La actividad de Factor VIII en plasma se expresa bien como porcentaje (referido a plasma humano normal) o en UI (referidas al Estándar Internacional de factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII es equivalente a la cantidad de factor VIII existente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis de factor VIII requerida se basa en el hallazgo empírico de que 1 UI de factor VIII por kg de peso corporal incrementa la actividad de factor VIII del plasma en 2 UI/dl. La dosis requerida se determina mediante la siguiente fórmula:
Unidades requeridas (UI) = peso corporal (kg) x aumento de factor VIII deseado (%) x 0,5
En el caso de los episodios hemorrágicos siguientes, la actividad de factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. En cirugía y en los episodios hemorrágicos, puede utilizarse la siguiente tabla 1 como guía de dosificación:
|
Tabla 1 Guía de dosificación en episodios hemorrágicos y cirugía | ||
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (% o UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente o hemorragia muscular u oral. |
20 - 40 |
Repetir la inyección cada 12 a 24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma. |
30 - 60 |
Repetir la inyección cada 12-24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) durante 3 a 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir la inyección cada 8 a 24 horas (cada 6 a 12 horas en los pacientes menores de 6 años) hasta superar el peligro. |
|
Cirugía | ||
|
Menor Incluyendo extracción dental. |
30 - 60 |
Cada 24 horas (cada 12 a 24 horas en los pacientes menores de 6 años), al menos 1 día, hasta lograr la curación. |
|
Mayor |
80 - 100 (pre y postoperatorio) |
Repetir la inyección cada 8-24 horas (cada 6 a 24 horas en los pacientes menores de 6 años) hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor VIII del 30% al 60% (UI/dl). |
La dosis y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual. En determinadas circunstancias (ej.: presencia de un título bajo de inhibidor) pueden ser necesarias dosis mayores a las calculadas mediante la fórmula.
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis que se debe administrar y la frecuencia con la que se debe repetir la inyección. En el caso especial de las intervenciones de cirugía mayor, es indispensable controlar con precisión el tratamiento de sustitución mediante pruebas de la coagulación (actividad del factor VIII plasmático). La respuesta individual de cada paciente frente al factor VIII puede variar, alcanzar distintos niveles de recuperación in vivo y presentar semividas diferentes.
Profilaxis
Para la profilaxis de larga duración frente a hemorragias en pacientes con hemofilia A grave, las dosis normales son de 20 a 40 UI de factor VIII por kg de peso corporal a intervalos de 2 a 3 días.
Población _ pediátrica
La dosis de tratamiento a demanda en pacientes pediátricos (0 a 18 años de edad) no difiere de la de los pacientes adultos. En pacientes menores de 6 años, se recomiendan dosis de 20 a 50 UI de factor VIII por kg de peso corporal de 3 a 4 veces a la semana como terapia profiláctica.
Forma de administración
ADVATE se debe administrar por vía intravenosa. En caso de ser administrado por un profesional no sanitario, es necesario realizar un entrenamiento adecuado.
Se debe determinar la velocidad de administración para asegurar la comodidad del paciente, hasta un máximo de 10 ml/min.
Después de la reconstitución, la solución es transparente, incolora y libre de partículas extrañas y tiene un pH de entre 6,7 y 7,3.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1 o a las proteínas de ratón o hámster.
4.4 Advertencias y precauciones especiales de empleo
Hipersensibilidad
Con ADVATE se han notificado reacciones de hipersensibilidad de tipo alérgico, incluyendo anafilaxis. El medicamento contiene trazas de proteínas de ratón y de hámster. Se debe informar a los pacientes de que en caso de que ocurran síntomas de hipersensibilidad, deben interrumpir el tratamiento y consultar con su médico de forma inmediata. Se debe informar a los pacientes de los signos de las reacciones de hipersensibilidad de tipo inmediato incluyendo habón urticarial, urticaria generalizada, tirantez en el pecho, sibilancia, hipotensión y anafilaxia.
En caso de que se produzca un shock, se deben seguir las pautas médicas estándar para su tratamiento.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) frente al factor VIII es una complicación conocida en el tratamiento de individuos con hemofilia A. Estos inhibidores son habitualmente inmunoglobulinas IgGs dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en unidades Bethesda (UB) por ml de plasma empleando el ensayo modificado. En pacientes que desarrollan anticuerpos neutralizantes (inhibidores) al Factor VIII, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia. El riesgo de desarrollar inhibidores se relaciona con el grado de exposición al factor VIII, siendo mayor durante los primeros 20 días de exposición, y con otros factores ambientales y genéticos. Raramente se desarrollan inhibidores después de los 100 primeros días de exposición.
Se han observado casos recurrentes de aparición de inhibidores (a títulos bajos), después de cambiar de un factor VIII a otro en los pacientes con un tratamiento previo de más de 100 días de exposición y con antecedentes de desarrollo de inhibidores. Así, se recomienda monitorizar cuidadosamente la aparición de inhibidores en todos los pacientes después de cualquier cambio de producto.
En general, en todos los pacientes tratados con un factor VIII de coagulación se debe monitorizar cuidadosamente la aparición de inhibidores mediante la realización de observaciones clínicas y de las pruebas de laboratorio apropiadas. Si no se obtienen los niveles de actividad de factor VIII en plasma esperados, o si no se controla la hemorragia con una dosis adecuada, se debe realizar una prueba de detección de inhibidor de factor VIII. En pacientes con niveles altos de inhibidor, la terapia de sustitución de factor VIII puede no ser efectiva y se deben considerar otras opciones terapéuticas.
El tratamiento de tales pacientes debe estar dirigido por médicos con experiencia en el cuidado de pacientes con hemofilia e inhibidores del factor VIII.
Complicación relacionada con catéter en el tratamiento
Si se requiere un dispositivo de acceso venoso (CVAD) debe considerarse el riesgo de complicaciones relacionadas con CVAD incluyendo infecciones locales, bacteremia y trombosis del lado del catéter.
Consideraciones relativas al excipiente
Después de la reconstitución, este medicamento contiene 0,45 mmol de sodio (10 mg) por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
Se recomienda encarecidamente registrar el nombre y el número de lote del producto cada vez que se administre ADVATE a un paciente para poder mantener una relación entre el paciente y el lote del medicamento.
Población pediátrica:
Las advertencias y precauciones incluidas se aplican tanto a adultos como a niños.
4.5 Interacción con otros medicamentos y otras formas de interacción.
No se han realizado estudios de interacciones con ADVATE.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción en animales con factor VIII. Dados los raros casos de hemofilia A en mujeres, no se dispone de experiencia relacionada con el uso de factor VIII durante el embarazo y el periodo de lactancia. Por lo tanto, sólo debe utilizarse factor VIII durante el embarazo y el periodo de lactancia si está claramente indicado.
4.7 Efectos sobre la capacidad de conducir y utilizar máquinas
La influencia de ADVATE sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los ensayos clínicos con ADVATE incluyeron 418 sujetos con al menos una exposición a ADVATE y registraron un total de 93 reacciones adversas al medicamento (RAM). Las RAM que aparecieron con mayor frecuencia fueron el desarrollo de anticuerpos neutralizantes frente al factor VIII (inhibidores), cefalea y fiebre.
La hipersensibilidad y las reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el lugar de infusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habón urticarial, hipotensión, letargia, náuseas, inquietud, taquicardia, tirantez en el pecho, cosquilleo, vómitos, sibilancia) se observaron con poca frecuencia pero, en algunos casos, pueden progresar a anafilaxia grave (incluyendo shock).
Se puede observar desarrollo de anticuerpos a la proteína de ratón y/o hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) frente al factor VIII. Si aparecen tales inhibidores, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia.
Resumen tabulado de reacciones adversas
La siguiente tabla 2 indica la frecuencia de las reacciones adversas en los ensayos clínicos y procedente de informes espontáneos. La tabla sigue la clasificación de órganos del sistema MedDRA (COS y Nivel de término preferido).
Las frecuencias se han clasificado utilizando el siguiente criterio: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100), raros (> 1/10.000 a < 1/1.000), muy raros (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de informes espontáneos | ||
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Infecciones e infestaciones |
Gripe |
Poco frecuentes |
|
Laringitis |
Poco frecuentes | |
|
Linfangitis |
Poco frecuentes | |
|
Trastornos de la sangre y del sistema linfático |
Inhibición del factor VIII Linfangitis |
Frecuentes Poco frecuentes |
|
Trastornos del sistema inmunológico |
Reacción anafiláctica |
Frecuencia no conocida |
|
Hipersensibilidadc |
Frecuencia no conocida | |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuentes |
|
Mareos |
Poco frecuentes | |
|
Problemas de memoria Síncope |
Poco frecuentes Poco frecuentes | |
|
Temblores |
Poco frecuentes | |
|
Migraña |
Poco frecuentes | |
|
Disgeusia |
Poco frecuentes | |
|
Trastornos oculares |
Inflamación ocular |
Poco frecuentes |
|
Trastornos cardiacos |
Palpitaciones |
Poco frecuentes |
|
Trastornos vasculares |
Hematoma |
Poco frecuentes |
|
Sofocos |
Poco frecuentes | |
|
Palidez |
Poco frecuentes | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
Poco frecuentes |
|
Trastornos gastrointestinales |
Diarrea |
Poco frecuentes |
|
Dolor en el abdomen superior |
Poco frecuentes | |
|
Nauseas |
Poco frecuentes | |
|
Vómitos |
Poco frecuentes | |
|
Trastornos de la piel y del tejido subcutáneo |
Prurito |
Poco frecuentes |
|
Sarpullido |
Poco frecuentes | |
|
Hiperhidrosis |
Poco frecuentes | |
|
Urticaria |
Poco frecuentes | |
|
Trastornos generales y alteraciones en el lugar de administración |
Fiebre |
Frecuentes |
|
Edema periférico |
Poco frecuentes | |
|
Dolor torácico Malestar torácico |
Poco frecuentes Poco frecuentes | |
|
Escalofríos |
Poco frecuentes | |
|
Sensación anormal Hematoma en el lugar de punción de un vaso sanguíneo |
Poco frecuentes Poco frecuentes | |
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de __informes espontáneos__
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Fatiga Reacción en la zona de inyección |
Frecuencia no conocida Frecuencia no conocida | |
|
Malestar general |
Frecuencia no conocida | |
|
Exploraciones complementarias | ||
|
Recuento elevado de monocitos |
Poco frecuentes | |
|
Nivel reducido de factor de coagulación VIIIb |
Poco frecuentes | |
|
Descenso del hematocrito |
Poco frecuentes | |
|
Test de laboratorio anormal |
Poco frecuentes | |
|
Lesiones, intoxicaciones y complicaciones de procedimientos terapéuticos |
Complicaciones tras el procedimiento |
Poco frecuentes |
|
Hemorragia tras el procedimiento |
Poco frecuentes | |
|
Reacción en el lugar del procedimiento |
Poco frecuentes |
a) Calculado según un número total único de pacientes que recibieron ADVATE (418)
b) El descenso inesperado de los niveles de factor VIII de coagulación se produjo en un paciente durante la perfusión continua de ADVATE después de cirugía (días 10-14 de postoperatorio).
Se mantuvo la hemostasis durante todo este periodo y tanto los niveles de factor VIII de plasma como los niveles de aclaramiento volvieron a los niveles adecuados el día 15 de postoperatorio. Los ensayos de inhibidores del factor VIII realizados tras la finalización de la perfusión continua y la terminación del estudio fueron negativos.
c) RAM explicadas en la sección siguiente.
Descripción de reacciones adversas seleccionadas Desarrollo de inhibidor
Se ha notificado un desarrollo de inhibidor en pacientes con tratamientos previos y en pacientes no tratados previamente. Para información detallada ver secciones 5.1 (Propiedades farmacológicas) y 4.4 (Advertencias y precauciones especiales de empleo).
RAM específicas para residuos del proceso de fabricación
De los 229 pacientes en tratamiento en los que se evaluaron los anticuerpos antiproteínas de células de ovario de hámster chino (CHO), 3 mostraron una tendencia ascendente estadísticamente significativa en los títulos, 4 mostraron picos sostenidos o picos momentáneos y un paciente mostró ambos, pero no tuvo ningún otro síntoma clínico. De los 229 pacientes tratados a los que se realizó una evaluación de los anticuerpos anti-IgG murina, 10 mostraron una tendencia ascendente, dos mostraron un pico sostenido o pico momentáneo y un paciente mostró ambos. Cuatro de estos pacientes sufrieron episodios aislados de urticaria, prurito, sarpullido y recuentos ligeramente elevados de eosinófilos entre exposiciones repetidas al medicamento del estudio.
Hipersensibilidad
Reacciones de tipo alérgico incluyen anafilaxis y se han manifestado como mareos, parestesias, erupción, soplado, tumefacción facial, urticaria y prurito.
Población pediátrica
En los estudios clínicos no se han notado diferencias específicas por edad en RAM excepto el desarrollo de inhibidores en pacientes pediátricos no tratados previamente (PUP) y complicaciones relacionadas con catéter.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación pertinente incluido en el Anexo V.
4.9 Sobredosis
No se han notificado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos: factor VIII de coagulación. Código ATC: B02BD02.
El complejo factor VIII /Factor von Willebrand consta de dos moléculas (factor VIII y factor von Willebrand) con diferentes funciones fisiológicas. ADVATE contiene factor VIII de coagulación recombinante (octocog alfa), una glicoproteína que es biológicamente equivalente a la glicoproteína de factor VIII del plasma humano.
Octocog alfa es una glicoproteína que consta de 2332 aminoácidos con un peso molecular aproximado de 280 kD. Cuando se perfunde en un paciente hemofílico, octocog alfa se enlaza al Factor von Willebrand endógeno en la circulación del paciente. El factor VIII activado actúa como un Cofactor para el Factor IX activado, acelerando la conversión del Factor X a Factor X activado. El Factor X activado convierte la protrombina en trombina. La trombina convierte el fibrinógeno en fibrina, con la que se puede formar el coágulo. La hemofilia A es una alteración hereditaria de la coagulación sanguínea ligada al sexo y producida por la disminución de los niveles de factor VIII que da como resultado una hemorragia profusa en las articulaciones, músculos u órganos internos, bien de forma espontánea o bien como resultado de un trauma accidental o quirúrgico. Mediante terapia de sustitución, los niveles plasmáticos de factor VIII aumentan, consiguiendo una corrección temporal de la deficiencia de factor VIII y una corrección de las tendencias hemorrágicas.
Desarrollo del inhibidor
Se evaluó la inmunogenicidad de ADVATE en pacientes con tratamientos previos. Durante los ensayos clínicos con ADVATE en 233 pacientes pediátricos y adultos [pacientes pediátricos (0 a 16 años de edad) y pacientes adultos (más de 16 años de edad)] diagnosticados con hemofilia A grave (factor VIII < 1%), con exposición previa a concentrados de factor VIII durante un periodo igual o superior a 150 días para adultos y niños mayores e igual o superior a 50 días para niños menores de 6 años, un paciente desarrolló un título bajo de inhibidor (2,4 BU en el ensayo Bethesda modificado) tras 26 días de exposición a ADVATE. Las pruebas de seguimiento de inhibidores en este paciente tras abandonar el ensayo fueron negativas. En todos los estudios, la exposición media a ADVATE fue de 97,0 días de exposición por sujeto (intervalo 1 a 709) para pacientes previamente tratados. La incidencia global de cualquier desarrollo de inhibidor del factor VIII (alto o bajo) fue de 0,4% (1 de 233).
En el ensayo completado no controlado 060103, 16 de los 45 (35,6%) pacientes sin tratamiento previo con hemofilia A grave (FVIII < 1%) con al menos 25 días de exposición a FVIII desarrollaron inhibidores del FVIII: 7 (15,6%) sujetos desarrollaron inhibidores de título alto y 9 (20%) de los sujetos desarrollaron inhibidores de título bajo, 1 de los cuales también se clasificó como inhibidor temporal.
Los factores de riesgo relacionados con el desarrollo del inhibidor en este ensayo incluyeron etnicidad no caucásica, antecedentes familiares de inhibidores y tratamiento intensivo a dosis altas en los primeros 20 días de exposición. En los 20 sujetos que no tenían ninguno de estos factores de riesgo no se produjo el desarrollo del inhibidor.
Se han recogido datos sobre Inducción de tolerancia inmunitaria (ITI) en pacientes con inhibidores.
En un subestudio del estudio PUP 060103, se documentaron tratamientos ITI en 11 PUP. Se hizo un análisis de cuadro retrospective de 30 materias sobre ITI (estudio 060703) y recogida de datos de registro está en curso.
En el estudio 060201, se compararon dos esquemas de tratamiento de profilaxis a largo plazo en 53 sujetos previamente tratados: un régimen individualizado de dosificación guiada farmacocinética (dentro de un rango de 20 a 80 UI de factor VIII por kg de peso corporal a intervalos de 72 ± 6 horas, n = 23) con un régimen de dosificación profiláctica estándar (de 20 a 40 UI/kg cada 48 ± 6 horas, n = 30). El régimen de dosificación guiada farmacocinética (de acuerdo con una fórmula específica) tenía como objetivo mantener el factor VIII en niveles > 1% en el intervalo entre dosis de 72 horas. Los datos de este estudio demuestran que los dos regímenes de dosificación profiláctica son comparables en lo que se refiere a la reducción de la frecuencia de hemorragias.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con ADVATE en los diferentes grupos de la población pediátrica con hemofilia A (deficiencia congénita de factor VIII) en “Inducción de tolerancia inmunitaria (ITI) en pacientes con hemofilia A (deficiencia congénita de factor VIII) que han desarrollado inhibidores al factor VIII” y “tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII)” (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Todos los estudios farmacocinéticos con ADVATE se realizaron en pacientes previamente tratados con hemofilia A grave o moderadamente grave (valor basal del factor VIII < 2%). El análisis de las muestras de plasma se realizó en un laboratorio central utilizando el ensayo de coagulación en una etapa.
Un total de 195 sujetos con hemofilia A grave (valor basal del factor VIII < 1%) proporcionaron parámetros PK que se incluyeron en el conjunto de análisis de PK según el protocolo. Las categorías de estos análisis para lactantes (1 mes a <2 años), niños (2 a <5 años), niños de mayor edad (5 a <12 años), adolescentes (12 a <18 años) y adultos (18 años y mayores) se utilizaron para resumir los parámetros PK, definiéndose la edad como la edad en el momento de la perfusión de PK.
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Área bajo la curva total (UI*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Recuperación incremental ajustada a Cmáx (UI/dl por UI/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Semivida (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Concentración máxima en plasma tras la perfusión (UI/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Tiempo de residencia medio (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Volumen de distribución en estado estacionario (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Aclaramiento (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Calculada como (Cmáx - valor basal del factor VIII) dividida por la dosis en Ul/kg, donde Cmáx es la medida máxima de factor VIII tras la perfusión.
La seguridad y la eficacia hemostática de ADVATE en la población pediátrica son similares a las de los pacientes adultos. La recuperación y la semivida terminal (t/2) ajustadas fUe aproximadamente un 20% inferior en niños pequeños (menores de 6 años) que en adultos, lo que lo que puede deberse en parte a un volumen mayor conocido de plasma por kg de peso corporal en los pacientes más jóvenes.
No se dispone de datos farmacocinéticos de ADVATE en pacientes sin tratamiento previo.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de la seguridad, toxicología aguda, toxicidad en dosis repetida, toxicidad local y genotoxicidad.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo
Manitol
Cloruro de sodio
Histidina
Trealosa
Cloruro de calcio Trometamol Polisorbato 80 Glutatión (reducido)
Disolvente
Agua esterilizada para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
2 años.
Después de la reconstitución, desde un punto de vista microbiológico, el medicamento debe utilizarse de inmediato. No obstante, se ha demostrado una estabilidad física y química en uso durante 3 horas a 25 °C.
Durante el periodo de validez, el medicamento puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único no superior a 6 meses. Se debe anotar la fecha del final del periodo de conservación a temperatura ambiente de 6 meses en el embalaje del medicamento. El medicamento no puede refrigerarse de nuevo.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
ADVATE con el equipo BAXJECT II: Conservar el vial del producto en el embalaje exterior para protegerlo de la luz.
ADVATE en el sistema BAXJECT III: Conservar el blister sellado en el embalaje exterior para protegerlo de la luz.
Para consultar las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Tanto el vial de polvo como el vial de disolvente de 5 ml son de vidrio tipo I cerrado con tapones de caucho de clorobutilo. El medicamento se proporciona con una de las configuraciones siguientes:
- ADVATE con el equipo BAXJECT II: Cada envase contiene un vial de polvo, un vial de disolvente de 5 ml y un equipo para reconstitución (BAXJECT II).
- ADVATE en el sistema BAXJECT III: Cada envase contiene un sistema BAXJECT III listo para usar en un blister sellado (el vial de polvo y el vial de disolvente de 5 ml están precargados con el sistema para su reconstitución).
6.6 Precauciones especiales de eliminación y otras manipulaciones
ADVATE debe administrarse intravenosamente después de la reconstitución del producto.
La solución reconstituida se debe inspeccionar visualmente para comprobar que no presenta partículas extrañas y/o descoloración.
Después de la reconstitución la solución debe ser transparente, incolora y no presentar partículas extrañas.
No utilizar soluciones turbias o con depósitos.
- Para la administración se requiere el uso de una jeringa luer-lock.
- Utilizar dentro de las tres horas después de la reconstitución.
- No refrigerar la preparación después de la reconstitución.
- La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Reconstitución con el equipo BAXJECT II:
- Para la reconstitución utilizar sólo el agua esterilizada para preparaciones inyectables y el equipo de reconstitución incluido en el envase.
- No utilizar si el equipo BAXJECT II, el sistema estéril de protección o su envase está dañado o muestra algún signo de deterioro.
- Se debe utilizar una técnica aséptica:
1. Si el medicamento se encuentra en la nevera, coger de la nevera los viales de polvo y de disolvente de ADVATE y dejarlos a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Quitar los protectores de los viales de polvo y disolvente.
4. Limpiar los tapones con las toallitas impregnadas de alcohol. Colocar los viales en una superficie plana y limpia.
5. Abrir el envoltorio del equipo BAXJECT II quitando la tapa de papel sin tocar el interior (Fig. a). No sacar el equipo del envoltorio. No utilizar si el equipo BAXJECT II, su sistema de barrera estéril o su acondicionamiento están dañados o muestran algún signo de deterioro.
6. Dar la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente. Coger el envoltorio por su extremo y sacar el equipo BAXJECT II de su envoltorio (Fig. b). No quitar el protector azul del equipo BAXJECT II.
7. Para la reconstitución, utilizar solamente el agua esterilizada para preparaciones inyectables y el equipo para reconstitución incluidos en el envase. Con el equipo BAXJECT II unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico blanca dentro del tapón del vial de polvo ADVATE. El vacío hará que el disolvente penetre en el vial de polvo ADVATE (Fig. c).
8. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará
a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig. a
Fig. b
Fig. c
Reconstitución con el sistema BAXJECT III
- No lo utilice si la tapa del blister no está perfectamente sellada.
1. Si el medicamento se encuentra en la nevera, coger de la nevera el blister sellado (contiene viales de polvo y disolvente precargados con el sistema para su reconstitución) y dejarlo a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Abrir el envoltorio de ADVATE quitando la tapa. Extraer el sistema BAXJECT III del blister.
4. Colocar ADVATE en una superficie plana con el vial de disolvente encima (Fig. 1). El vial de disolvente tiene una raya azul. No quitar el protector azul hasta que se le indique más adelante.
5. Mientras sujeta ADVATE con una mano en el sistema BAXJECT III, presionar con fuerza el vial de disolvente con la otra mano hasta que el sistema quede totalmente contraído y el disolvente entre dentro del vial de ADVATE (Fig. 2). No inclinar el sistema hasta que haya finalizado la transferencia.
6. Comprobar que ha finalizado la transferencia del disolvente. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig.l
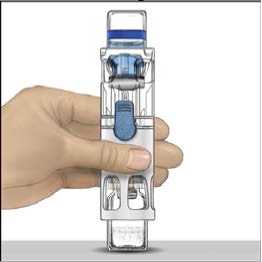
Fig.2
Fig. 3
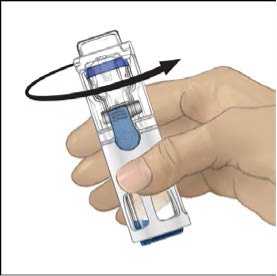
Administración:
Utilizar técnica aséptica:
Los medicamentos para administración parenteral deben inspeccionarse visualmente antes de su
administración para verificar la ausencia de partículas, cuando la solución y el envase lo permitan.
Sólo se debe utilizar una solución transparente y sin coloración.
1. Quitar el protector azul del equipo BAXJECT II / al sistema BAXJECT III. No introducir aire en la jeringa. Conectar la jeringa al equipo BAXJECT II / al sistema BAXJECT III.
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente.
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar intravenosamente. La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de ADVATE. Si se observa una elevación significativa del pulso, reducir la velocidad de administración o interrumpir temporalmente la inyección suele hacer que los síntomas normalmente desaparezcan pronto (ver secciones 4.4 y 4.8).
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
Industriestrasse 67
A-1221 Viena
Austria
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/003
EU/1/03/271/013
9. FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 2 de marzo de 2004 Fecha de la última renovación: 2 de marzo de 2014
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
ADVATE 1500 UI polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 1500 UI de factor VIII humano de coagulación (ADNr), octocog alfa. Después de la reconstitución, ADVATE contiene aproximadamente 300 UI por ml de factor VIII humano de coagulación (ADNr), octocog alfa.
La potencia (Unidades Internacionales) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de ADVATE es aproximadamente 4.000 -10.000 UI/mg/proteína.
El octocog alfa (factor VIII humano de coagulación (ADNr)) es una proteína purificada con 2332 aminoácidos. Está producida por tecnología de ADN recombinante en células de ovario de hámster chino (CHO). Preparado sin la adición de ninguna proteína (exógena) de origen humano o animal en el proceso del cultivo celular, purificación o formulación final.
Excipientes con efectos conocidos:
0,45 mmol de sodio (10 mg) por vial.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: polvo friable de color blanco a blanquecino. Disolvente: solución transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII). ADVATE está indicado en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia y con apoyo de resucitación disponible inmediatamente en caso de anafilaxia.
Posología
La dosis y duración de la terapia de sustitución depende de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII se expresa en Unidades Internacionales (UI), que se relacionan con el estándar de la OMS para medicamentos con factor VIII. La actividad de Factor VIII en plasma se expresa bien como porcentaje (referido a plasma humano normal) o en UI (referidas al Estándar Internacional de factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII es equivalente a la cantidad de factor VIII existente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis de factor VIII requerida se basa en el hallazgo empírico de que 1 UI de factor VIII por kg de peso corporal incrementa la actividad de factor VIII del plasma en 2 UI/dl. La dosis requerida se determina mediante la siguiente fórmula:
Unidades requeridas (UI) = peso corporal (kg) x aumento de factor VIII deseado (%) x 0,5
En el caso de los episodios hemorrágicos siguientes, la actividad de factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. En cirugía y en los episodios hemorrágicos, puede utilizarse la siguiente tabla 1 como guía de dosificación:
|
Tabla 1 Guía de dosificación en episodios hemorrágicos y cirugía | ||
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (% o UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente o hemorragia muscular u oral. |
20 - 40 |
Repetir la inyección cada 12 a 24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma. |
30 - 60 |
Repetir la inyección cada 12-24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) durante 3 a 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir la inyección cada 8 a 24 horas (cada 6 a 12 horas en los pacientes menores de 6 años) hasta superar el peligro. |
|
Cirugía | ||
|
Menor Incluyendo extracción dental. |
30 - 60 |
Cada 24 horas (cada 12 a 24 horas en los pacientes menores de 6 años), al menos 1 día, hasta lograr la curación. |
|
Mayor |
80 - 100 (pre y postoperatorio) |
Repetir la inyección cada 8-24 horas (cada 6 a 24 horas en los pacientes menores de 6 años) hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor VIII del 30% al 60% (UI/dl). |
La dosis y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual. En determinadas circunstancias (ej.: presencia de un título bajo de inhibidor) pueden ser necesarias dosis mayores a las calculadas mediante la fórmula.
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis que se debe administrar y la frecuencia con la que se debe repetir la inyección. En el caso especial de las intervenciones de cirugía mayor, es indispensable controlar con precisión el tratamiento de sustitución mediante pruebas de la coagulación (actividad del factor VIII plasmático). La respuesta individual de cada paciente frente al factor VIII puede variar, alcanzar distintos niveles de recuperación in vivo y presentar semividas diferentes.
Profilaxis
Para la profilaxis de larga duración frente a hemorragias en pacientes con hemofilia A grave, las dosis normales son de 20 a 40 UI de factor VIII por kg de peso corporal a intervalos de 2 a 3 días.
Población _ pediátrica
La dosis de tratamiento a demanda en pacientes pediátricos (0 a 18 años de edad) no difiere de la de los pacientes adultos. En pacientes menores de 6 años, se recomiendan dosis de 20 a 50 UI de factor VIII por kg de peso corporal de 3 a 4 veces a la semana como terapia profiláctica.
Forma de administración
ADVATE se debe administrar por vía intravenosa. En caso de ser administrado por un profesional no sanitario, es necesario realizar un entrenamiento adecuado.
Se debe determinar la velocidad de administración para asegurar la comodidad del paciente, hasta un máximo de 10 ml/min.
Después de la reconstitución, la solución es transparente, incolora y libre de partículas extrañas y tiene un pH de entre 6,7 y 7,3.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1 o a las proteínas de ratón o hámster.
4.4 Advertencias y precauciones especiales de empleo
Hipersensibilidad
Con ADVATE se han notificado reacciones de hipersensibilidad de tipo alérgico, incluyendo anafilaxis. El medicamento contiene trazas de proteínas de ratón y de hámster. Se debe informar a los pacientes de que en caso de que ocurran síntomas de hipersensibilidad, deben interrumpir el tratamiento y consultar con su médico de forma inmediata. Se debe informar a los pacientes de los signos de las reacciones de hipersensibilidad de tipo inmediato incluyendo habón urticarial, urticaria generalizada, tirantez en el pecho, sibilancia, hipotensión y anafilaxia.
En caso de que se produzca un shock, se deben seguir las pautas médicas estándar para su tratamiento.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) frente al factor VIII es una complicación conocida en el tratamiento de individuos con hemofilia A. Estos inhibidores son habitualmente inmunoglobulinas IgGs dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en unidades Bethesda (UB) por ml de plasma empleando el ensayo modificado. En pacientes que desarrollan anticuerpos neutralizantes (inhibidores) al Factor VIII, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia. El riesgo de desarrollar inhibidores se relaciona con el grado de exposición al factor VIII, siendo mayor durante los primeros 20 días de exposición, y con otros factores ambientales y genéticos. Raramente se desarrollan inhibidores después de los 100 primeros días de exposición.
Se han observado casos recurrentes de aparición de inhibidores (a títulos bajos), después de cambiar de un factor VIII a otro en los pacientes con un tratamiento previo de más de 100 días de exposición y con antecedentes de desarrollo de inhibidores. Así, se recomienda monitorizar cuidadosamente la aparición de inhibidores en todos los pacientes después de cualquier cambio de producto.
En general, en todos los pacientes tratados con un factor VIII de coagulación se debe monitorizar cuidadosamente la aparición de inhibidores mediante la realización de observaciones clínicas y de las pruebas de laboratorio apropiadas. Si no se obtienen los niveles de actividad de factor VIII en plasma esperados, o si no se controla la hemorragia con una dosis adecuada, se debe realizar una prueba de detección de inhibidor de factor VIII. En pacientes con niveles altos de inhibidor, la terapia de sustitución de factor VIII puede no ser efectiva y se deben considerar otras opciones terapéuticas.
El tratamiento de tales pacientes debe estar dirigido por médicos con experiencia en el cuidado de pacientes con hemofilia e inhibidores del factor VIII.
Complicación relacionada con catéter en el tratamiento
Si se requiere un dispositivo de acceso venoso (CVAD) debe considerarse el riesgo de complicaciones relacionadas con CVAD incluyendo infecciones locales, bacteremia y trombosis del lado del catéter.
Consideraciones relativas al excipiente
Después de la reconstitución, este medicamento contiene 0,45 mmol de sodio (10 mg) por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
Se recomienda encarecidamente registrar el nombre y el número de lote del producto cada vez que se administre ADVATE a un paciente para poder mantener una relación entre el paciente y el lote del medicamento.
Población pediátrica:
Las advertencias y precauciones incluidas se aplican tanto a adultos como a niños.
4.5 Interacción con otros medicamentos y otras formas de interacción.
No se han realizado estudios de interacciones con ADVATE.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción en animales con factor VIII. Dados los raros casos de hemofilia A en mujeres, no se dispone de experiencia relacionada con el uso de factor VIII durante el embarazo y el periodo de lactancia. Por lo tanto, sólo debe utilizarse factor VIII durante el embarazo y el periodo de lactancia si está claramente indicado.
4.7 Efectos sobre la capacidad de conducir y utilizar máquinas
La influencia de ADVATE sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los ensayos clínicos con ADVATE incluyeron 418 sujetos con al menos una exposición a ADVATE y registraron un total de 93 reacciones adversas al medicamento (RAM). Las RAM que aparecieron con mayor frecuencia fueron el desarrollo de anticuerpos neutralizantes frente al factor VIII (inhibidores), cefalea y fiebre.
La hipersensibilidad y las reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el lugar de infusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habón urticarial, hipotensión, letargia, náuseas, inquietud, taquicardia, tirantez en el pecho, cosquilleo, vómitos, sibilancia) se observaron con poca frecuencia pero, en algunos casos, pueden progresar a anafilaxia grave (incluyendo shock).
Se puede observar desarrollo de anticuerpos a la proteína de ratón y/o hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) frente al factor VIII. Si aparecen tales inhibidores, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia.
Resumen tabulado de reacciones adversas
La siguiente tabla 2 indica la frecuencia de las reacciones adversas en los ensayos clínicos y procedente de informes espontáneos. La tabla sigue la clasificación de órganos del sistema MedDRA (COS y Nivel de término preferido).
Las frecuencias se han clasificado utilizando el siguiente criterio: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100), raros (> 1/10.000 a < 1/1.000), muy raros (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de informes espontáneos | ||
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Infecciones e infestaciones |
Gripe |
Poco frecuentes |
|
Laringitis |
Poco frecuentes | |
|
Linfangitis |
Poco frecuentes | |
|
Trastornos de la sangre y del sistema linfático |
Inhibición del factor VIII Linfangitis |
Frecuentes Poco frecuentes |
|
Trastornos del sistema inmunológico |
Reacción anafiláctica |
Frecuencia no conocida |
|
Hipersensibilidadc |
Frecuencia no conocida | |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuentes |
|
Mareos |
Poco frecuentes | |
|
Problemas de memoria Síncope |
Poco frecuentes Poco frecuentes | |
|
Temblores |
Poco frecuentes | |
|
Migraña |
Poco frecuentes | |
|
Disgeusia |
Poco frecuentes | |
|
Trastornos oculares |
Inflamación ocular |
Poco frecuentes |
|
Trastornos cardiacos |
Palpitaciones |
Poco frecuentes |
|
Trastornos vasculares |
Hematoma |
Poco frecuentes |
|
Sofocos |
Poco frecuentes | |
|
Palidez |
Poco frecuentes | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
Poco frecuentes |
|
Trastornos gastrointestinales |
Diarrea |
Poco frecuentes |
|
Dolor en el abdomen superior |
Poco frecuentes | |
|
Nauseas |
Poco frecuentes | |
|
Vómitos |
Poco frecuentes | |
|
Trastornos de la piel y del tejido subcutáneo |
Prurito |
Poco frecuentes |
|
Sarpullido |
Poco frecuentes | |
|
Hiperhidrosis |
Poco frecuentes | |
|
Urticaria |
Poco frecuentes | |
|
Trastornos generales y alteraciones en el lugar de administración |
Fiebre |
Frecuentes |
|
Edema periférico |
Poco frecuentes | |
|
Dolor torácico Malestar torácico |
Poco frecuentes Poco frecuentes | |
|
Escalofríos |
Poco frecuentes | |
|
Sensación anormal Hematoma en el lugar de punción de un vaso sanguíneo |
Poco frecuentes Poco frecuentes | |
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de __informes espontáneos__
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Fatiga Reacción en la zona de inyección |
Frecuencia no conocida Frecuencia no conocida | |
|
Malestar general |
Frecuencia no conocida | |
|
Exploraciones complementarias | ||
|
Recuento elevado de monocitos |
Poco frecuentes | |
|
Nivel reducido de factor de coagulación VIIIb |
Poco frecuentes | |
|
Descenso del hematocrito |
Poco frecuentes | |
|
Test de laboratorio anormal |
Poco frecuentes | |
|
Lesiones, intoxicaciones y complicaciones de procedimientos terapéuticos |
Complicaciones tras el procedimiento |
Poco frecuentes |
|
Hemorragia tras el procedimiento |
Poco frecuentes | |
|
Reacción en el lugar del procedimiento |
Poco frecuentes |
a) Calculado según un número total único de pacientes que recibieron ADVATE (418)
b) El descenso inesperado de los niveles de factor VIII de coagulación se produjo en un paciente durante la perfusión continua de ADVATE después de cirugía (días 10-14 de postoperatorio).
Se mantuvo la hemostasis durante todo este periodo y tanto los niveles de factor VIII de plasma como los niveles de aclaramiento volvieron a los niveles adecuados el día 15 de postoperatorio. Los ensayos de inhibidores del factor VIII realizados tras la finalización de la perfusión continua y la terminación del estudio fueron negativos.
c) RAM explicadas en la sección siguiente.
Descripción de reacciones adversas seleccionadas Desarrollo de inhibidor
Se ha notificado un desarrollo de inhibidor en pacientes con tratamientos previos y en pacientes no tratados previamente. Para información detallada ver secciones 5.1 (Propiedades farmacológicas) y 4.4 (Advertencias y precauciones especiales de empleo).
RAM específicas para residuos del proceso de fabricación
De los 229 pacientes en tratamiento en los que se evaluaron los anticuerpos antiproteínas de células de ovario de hámster chino (CHO), 3 mostraron una tendencia ascendente estadísticamente significativa en los títulos, 4 mostraron picos sostenidos o picos momentáneos y un paciente mostró ambos, pero no tuvo ningún otro síntoma clínico. De los 229 pacientes tratados a los que se realizó una evaluación de los anticuerpos anti-IgG murina, 10 mostraron una tendencia ascendente, dos mostraron un pico sostenido o pico momentáneo y un paciente mostró ambos. Cuatro de estos pacientes sufrieron episodios aislados de urticaria, prurito, sarpullido y recuentos ligeramente elevados de eosinófilos entre exposiciones repetidas al medicamento del estudio.
Hipersensibilidad
Reacciones de tipo alérgico incluyen anafilaxis y se han manifestado como mareos, parestesias, erupción, soplado, tumefacción facial, urticaria y prurito.
Población pediátrica
En los estudios clínicos no se han notado diferencias específicas por edad en RAM excepto el desarrollo de inhibidores en pacientes pediátricos no tratados previamente (PUP) y complicaciones relacionadas con catéter.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación pertinente incluido en el Anexo V.
4.9 Sobredosis
No se han notificado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos: factor VIII de coagulación. Código ATC: B02BD02.
El complejo factor VIII /Factor von Willebrand consta de dos moléculas (factor VIII y factor von Willebrand) con diferentes funciones fisiológicas. ADVATE contiene factor VIII de coagulación recombinante (octocog alfa), una glicoproteína que es biológicamente equivalente a la glicoproteína de factor VIII del plasma humano.
Octocog alfa es una glicoproteína que consta de 2332 aminoácidos con un peso molecular aproximado de 280 kD. Cuando se perfunde en un paciente hemofílico, octocog alfa se enlaza al Factor von Willebrand endógeno en la circulación del paciente. El factor VIII activado actúa como un Cofactor para el Factor IX activado, acelerando la conversión del Factor X a Factor X activado. El Factor X activado convierte la protrombina en trombina. La trombina convierte el fibrinógeno en fibrina, con la que se puede formar el coágulo. La hemofilia A es una alteración hereditaria de la coagulación sanguínea ligada al sexo y producida por la disminución de los niveles de factor VIII que da como resultado una hemorragia profusa en las articulaciones, músculos u órganos internos, bien de forma espontánea o bien como resultado de un trauma accidental o quirúrgico. Mediante terapia de sustitución, los niveles plasmáticos de factor VIII aumentan, consiguiendo una corrección temporal de la deficiencia de factor VIII y una corrección de las tendencias hemorrágicas.
Desarrollo del inhibidor
Se evaluó la inmunogenicidad de ADVATE en pacientes con tratamientos previos. Durante los ensayos clínicos con ADVATE en 233 pacientes pediátricos y adultos [pacientes pediátricos (0 a 16 años de edad) y pacientes adultos (más de 16 años de edad)] diagnosticados con hemofilia A grave (factor VIII < 1%), con exposición previa a concentrados de factor VIII durante un periodo igual o superior a 150 días para adultos y niños mayores e igual o superior a 50 días para niños menores de 6 años, un paciente desarrolló un título bajo de inhibidor (2,4 BU en el ensayo Bethesda modificado) tras 26 días de exposición a ADVATE. Las pruebas de seguimiento de inhibidores en este paciente tras abandonar el ensayo fueron negativas. En todos los estudios, la exposición media a ADVATE fue de 97,0 días de exposición por sujeto (intervalo 1 a 709) para pacientes previamente tratados. La incidencia global de cualquier desarrollo de inhibidor del factor VIII (alto o bajo) fue de 0,4% (1 de 233).
En el ensayo completado no controlado 060103, 16 de los 45 (35,6%) pacientes sin tratamiento previo con hemofilia A grave (FVIII < 1%) con al menos 25 días de exposición a FVIII desarrollaron inhibidores del FVIII: 7 (15,6%) sujetos desarrollaron inhibidores de título alto y 9 (20%) de los sujetos desarrollaron inhibidores de título bajo, 1 de los cuales también se clasificó como inhibidor temporal.
Los factores de riesgo relacionados con el desarrollo del inhibidor en este ensayo incluyeron etnicidad no caucásica, antecedentes familiares de inhibidores y tratamiento intensivo a dosis altas en los primeros 20 días de exposición. En los 20 sujetos que no tenían ninguno de estos factores de riesgo no se produjo el desarrollo del inhibidor.
Se han recogido datos sobre Inducción de tolerancia inmunitaria (ITI) en pacientes con inhibidores.
En un subestudio del estudio PUP 060103, se documentaron tratamientos ITI en 11 PUP. Se hizo un análisis de cuadro retrospective de 30 materias sobre ITI (estudio 060703) y recogida de datos de registro está en curso.
En el estudio 060201, se compararon dos esquemas de tratamiento de profilaxis a largo plazo en 53 sujetos previamente tratados: un régimen individualizado de dosificación guiada farmacocinética (dentro de un rango de 20 a 80 UI de factor VIII por kg de peso corporal a intervalos de 72 ± 6 horas, n = 23) con un régimen de dosificación profiláctica estándar (de 20 a 40 UI/kg cada 48 ± 6 horas, n = 30). El régimen de dosificación guiada farmacocinética (de acuerdo con una fórmula específica) tenía como objetivo mantener el factor VIII en niveles > 1% en el intervalo entre dosis de 72 horas. Los datos de este estudio demuestran que los dos regímenes de dosificación profiláctica son comparables en lo que se refiere a la reducción de la frecuencia de hemorragias.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con ADVATE en los diferentes grupos de la población pediátrica con hemofilia A (deficiencia congénita de factor VIII) en “Inducción de tolerancia inmunitaria (ITI) en pacientes con hemofilia A (deficiencia congénita de factor VIII) que han desarrollado inhibidores al factor VIII” y “tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII)” (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Todos los estudios farmacocinéticos con ADVATE se realizaron en pacientes previamente tratados con hemofilia A grave o moderadamente grave (valor basal del factor VIII < 2%). El análisis de las muestras de plasma se realizó en un laboratorio central utilizando el ensayo de coagulación en una etapa.
Un total de 195 sujetos con hemofilia A grave (valor basal del factor VIII < 1%) proporcionaron parámetros PK que se incluyeron en el conjunto de análisis de PK según el protocolo. Las categorías de estos análisis para lactantes (1 mes a <2 años), niños (2 a <5 años), niños de mayor edad (5 a <12 años), adolescentes (12 a <18 años) y adultos (18 años y mayores) se utilizaron para resumir los parámetros PK, definiéndose la edad como la edad en el momento de la perfusión de PK.
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Área bajo la curva total (UI*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Recuperación incremental ajustada a Cmáx (UI/dl por UI/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Semivida (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Concentración máxima en plasma tras la perfusión (UI/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Tiempo de residencia medio (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Volumen de distribución en estado estacionario (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Aclaramiento (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Calculada como (Cmáx - valor basal del factor VIII) dividida por la dosis en Ul/kg, donde Cmáx es la medida máxima de factor VIII tras la perfusión.
La seguridad y la eficacia hemostática de ADVATE en la población pediátrica son similares a las de los pacientes adultos. La recuperación y la semivida terminal (t/2) ajustadas fUe aproximadamente un 20% inferior en niños pequeños (menores de 6 años) que en adultos, lo que lo que puede deberse en parte a un volumen mayor conocido de plasma por kg de peso corporal en los pacientes más jóvenes.
No se dispone de datos farmacocinéticos de ADVATE en pacientes sin tratamiento previo.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de la seguridad, toxicología aguda, toxicidad en dosis repetida, toxicidad local y genotoxicidad.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo
Manitol
Cloruro de sodio
Histidina
Trealosa
Cloruro de calcio Trometamol Polisorbato 80 Glutatión (reducido)
Disolvente
Agua esterilizada para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
2 años.
Después de la reconstitución, desde un punto de vista microbiológico, el medicamento debe utilizarse de inmediato. No obstante, se ha demostrado una estabilidad física y química en uso durante 3 horas a 25 °C.
Durante el periodo de validez, el medicamento puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único no superior a 6 meses. Se debe anotar la fecha del final del periodo de conservación a temperatura ambiente de 6 meses en el embalaje del medicamento. El medicamento no puede refrigerarse de nuevo.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
ADVATE con el equipo BAXJECT II: Conservar el vial en el embalaje exterior para protegerlo de la luz.
ADVATE en el sistema BAXJECT III: Conservar el blister sellado en el embalaje exterior para protegerlo de la luz.
Para consultar las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Tanto el vial de polvo como el vial de disolvente de 5 ml son de vidrio tipo I cerrado con tapones de caucho de clorobutilo. El medicamento se proporciona con una de las configuraciones siguientes:
- ADVATE con el equipo BAXJECT II: Cada envase contiene un vial de polvo, un vial de disolvente de 5 ml y un equipo para reconstitución (BAXJECT II).
- ADVATE en el sistema BAXJECT III: Cada envase contiene un sistema BAXJECT III listo para usar en un blister sellado (el vial de polvo y el vial de disolvente de 5 ml están precargados con el sistema para su reconstitución).
6.6 Precauciones especiales de eliminación y otras manipulaciones
ADVATE debe administrarse intravenosamente después de la reconstitución del producto.
La solución reconstituida se debe inspeccionar visualmente para comprobar que no presenta partículas extrañas y/o descoloración.
Después de la reconstitución la solución debe ser transparente, incolora y no presentar partículas extrañas.
No utilizar soluciones turbias o con depósitos.
- Para la administración se requiere el uso de una jeringa luer-lock.
- Utilizar dentro de las tres horas después de la reconstitución.
- No refrigerar la preparación después de la reconstitución.
- La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Reconstitución con el equipo BAXJECT II:
- Para la reconstitución utilizar sólo el agua esterilizada para preparaciones inyectables y el equipo de reconstitución incluido en el envase.
- No utilizar si el equipo BAXJECT II, el sistema estéril de protección o su envase está dañado o muestra algún signo de deterioro.
- Se debe utilizar una técnica aséptica:
1. Si el medicamento se encuentra en la nevera, coger de la nevera los viales de polvo y de disolvente de ADVATE y dejarlos a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Quitar los protectores de los viales de polvo y disolvente.
4. Limpiar los tapones con las toallitas impregnadas de alcohol. Colocar los viales en una superficie plana y limpia.
5. Abrir el envoltorio del equipo BAXJECT II quitando la tapa de papel sin tocar el interior (Fig. a). No sacar el equipo del envoltorio. No utilizar si el equipo BAXJECT II, su sistema de barrera estéril o su acondicionamiento están dañados o muestran algún signo de deterioro.
6. Dar la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente. Coger el envoltorio por su extremo y sacar el equipo BAXJECT II de su envoltorio (Fig. b). No quitar el protector azul del equipo BAXJECT II.
7. Para la reconstitución, utilizar solamente el agua esterilizada para preparaciones inyectables y el equipo para reconstitución incluidos en el envase. Con el equipo BAXJECT II unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico blanca dentro del tapón del vial de polvo ADVATE. El vacío hará que el disolvente penetre en el vial de polvo ADVATE (Fig. c).
8. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará
a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig. a
Fig. b
Fig. c
Reconstitución con el sistema BAXJECT III
- No lo utilice si la tapa del blister no está perfectamente sellada.
1. Si el medicamento se encuentra en la nevera, coger de la nevera el blister sellado (contiene viales de polvo y disolvente precargados con el sistema para su reconstitución) y dejarlo a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Abrir el envoltorio de ADVATE quitando la tapa. Extraer el sistema BAXJECT III del blister
4. Colocar ADVATE en una superficie plana con el vial de disolvente encima (Fig. 1). El vial de disolvente tiene una raya azul. No quitar el protector azul hasta que se le indique más adelante.
5. Mientras sujeta ADVATE con una mano en el sistema BAXJECT III, presionar con fuerza el vial de disolvente con la otra mano hasta que el sistema quede totalmente contraído y el disolvente entre dentro del vial de ADVATE (Fig. 2). No inclinar el sistema hasta que haya finalizado la transferencia.
6. Comprobar que ha finalizado la transferencia del disolvente. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig.l
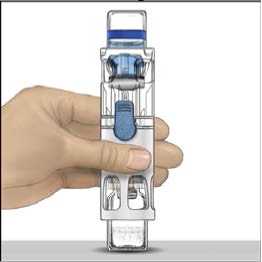
Fig.2
Fig. 3
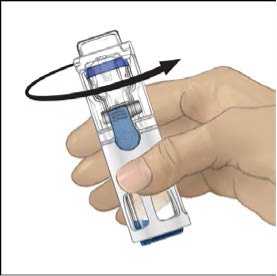
Administración:
Utilizar técnica aséptica:
Los medicamentos para administración parenteral deben inspeccionarse visualmente antes de su
administración para verificar la ausencia de partículas, cuando la solución y el envase lo permitan.
Sólo se debe utilizar una solución transparente y sin coloración.
1. Quitar el protector azul del equipo BAXJECT II / sistema BAXJECT III. No introducir aire en la jeringa. Conectar la jeringa al equipo BAXJECT II / sistema BAXJECT III.
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente.
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar intravenosamente. La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de ADVATE. Si se observa una elevación significativa del pulso, reducir la velocidad de administración o interrumpir temporalmente la inyección suele hacer que los síntomas normalmente desaparezcan pronto (ver secciones 4.4 y 4.8).
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
Industriestrasse 67
A-1221 Viena
Austria
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/004
EU/1/03/271/014
9. FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 2 de marzo de 2004 Fecha de la última renovación: 2 de marzo de 2014
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
ADVATE 2000 UI polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 2000 UI de factor VIII humano de coagulación (ADNr), octocog alfa. Después de la reconstitución, ADVATE contiene aproximadamente 400 UI por ml de factor VIII humano de coagulación (ADNr), octocog alfa.
La potencia (Unidades Internacionales) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de ADVATE es aproximadamente 4.000 -10.000 UI/mg/proteína.
El octocog alfa (factor VIII humano de coagulación (ADNr)) es una proteína purificada con 2332 aminoácidos. Está producida por tecnología de ADN recombinante en células de ovario de hámster chino (CHO). Preparado sin la adición de ninguna proteína (exógena) de origen humano o animal en el proceso del cultivo celular, purificación o formulación final.
Excipientes con efectos conocidos:
0,45 mmol de sodio (10 mg) por vial.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: polvo friable de color blanco a blanquecino. Disolvente: solución transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII). ADVATE está indicado en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia y con apoyo de resucitación disponible inmediatamente en caso de anafilaxia.
Posología
La dosis y duración de la terapia de sustitución depende de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII se expresa en Unidades Internacionales (UI), que se relacionan con el estándar de la OMS para medicamentos con factor VIII. La actividad de Factor VIII en plasma se expresa bien como porcentaje (referido a plasma humano normal) o en UI (referidas al Estándar Internacional de factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII es equivalente a la cantidad de factor VIII existente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis de factor VIII requerida se basa en el hallazgo empírico de que 1 UI de factor VIII por kg de peso corporal incrementa la actividad de factor VIII del plasma en 2 UI/dl. La dosis requerida se determina mediante la siguiente fórmula:
Unidades requeridas (UI) = peso corporal (kg) x aumento de factor VIII deseado (%) x 0,5
En el caso de los episodios hemorrágicos siguientes, la actividad de factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. En cirugía y en los episodios hemorrágicos, puede utilizarse la siguiente tabla 1 como guía de dosificación:
|
Tabla 1 Guía de dosificación en episodios hemorrágicos y cirugía | ||
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (% o UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente o hemorragia muscular u oral. |
20 - 40 |
Repetir la inyección cada 12 a 24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma. |
30 - 60 |
Repetir la inyección cada 12-24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) durante 3 a 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir la inyección cada 8 a 24 horas (cada 6 a 12 horas en los pacientes menores de 6 años) hasta superar el peligro. |
|
Cirugía | ||
|
Menor Incluyendo extracción dental. |
30 - 60 |
Cada 24 horas (cada 12 a 24 horas en los pacientes menores de 6 años), al menos 1 día, hasta lograr la curación. |
|
Mayor |
80 - 100 (pre y postoperatorio) |
Repetir la inyección cada 8-24 horas (cada 6 a 24 horas en los pacientes menores de 6 años) hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor VIII del 30% al 60% (UI/dl). |
La dosis y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual. En determinadas circunstancias (ej.: presencia de un título bajo de inhibidor) pueden ser necesarias dosis mayores a las calculadas mediante la fórmula.
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis que se debe administrar y la frecuencia con la que se debe repetir la inyección. En el caso especial de las intervenciones de cirugía mayor, es indispensable controlar con precisión el tratamiento de sustitución mediante pruebas de la coagulación (actividad del factor VIII plasmático). La respuesta individual de cada paciente frente al factor VIII puede variar, alcanzar distintos niveles de recuperación in vivo y presentar semividas diferentes.
Profilaxis
Para la profilaxis de larga duración frente a hemorragias en pacientes con hemofilia A grave, las dosis normales son de 20 a 40 UI de factor VIII por kg de peso corporal a intervalos de 2 a 3 días.
Población _ pediátrica
La dosis de tratamiento a demanda en pacientes pediátricos (0 a 18 años de edad) no difiere de la de los pacientes adultos. En pacientes menores de 6 años, se recomiendan dosis de 20 a 50 UI de factor VIII por kg de peso corporal de 3 a 4 veces a la semana como terapia profiláctica.
Forma de administración
ADVATE se debe administrar por vía intravenosa. En caso de ser administrado por un profesional no sanitario, es necesario realizar un entrenamiento adecuado.
Se debe determinar la velocidad de administración para asegurar la comodidad del paciente, hasta un máximo de 10 ml/min.
Después de la reconstitución, la solución es transparente, incolora y libre de partículas extrañas y tiene un pH de entre 6,7 y 7,3.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1 o a las proteínas de ratón o hámster.
4.4 Advertencias y precauciones especiales de empleo
Hipersensibilidad
Con ADVATE se han notificado reacciones de hipersensibilidad de tipo alérgico, incluyendo anafilaxis. El medicamento contiene trazas de proteínas de ratón y de hámster. Se debe informar a los pacientes de que en caso de que ocurran síntomas de hipersensibilidad, deben interrumpir el tratamiento y consultar con su médico de forma inmediata. Se debe informar a los pacientes de los signos de las reacciones de hipersensibilidad de tipo inmediato incluyendo habón urticarial, urticaria generalizada, tirantez en el pecho, sibilancia, hipotensión y anafilaxia.
En caso de que se produzca un shock, se deben seguir las pautas médicas estándar para su tratamiento.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) frente al factor VIII es una complicación conocida en el tratamiento de individuos con hemofilia A. Estos inhibidores son habitualmente inmunoglobulinas IgGs dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en unidades Bethesda (UB) por ml de plasma empleando el ensayo modificado. En pacientes que desarrollan anticuerpos neutralizantes (inhibidores) al Factor VIII, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia. El riesgo de desarrollar inhibidores se relaciona con el grado de exposición al factor VIII, siendo mayor durante los primeros 20 días de exposición, y con otros factores ambientales y genéticos. Raramente se desarrollan inhibidores después de los 100 primeros días de exposición.
Se han observado casos recurrentes de aparición de inhibidores (a títulos bajos), después de cambiar de un factor VIII a otro en los pacientes con un tratamiento previo de más de 100 días de exposición y con antecedentes de desarrollo de inhibidores. Así, se recomienda monitorizar cuidadosamente la aparición de inhibidores en todos los pacientes después de cualquier cambio de producto.
En general, en todos los pacientes tratados con un factor VIII de coagulación se debe monitorizar cuidadosamente la aparición de inhibidores mediante la realización de observaciones clínicas y de las pruebas de laboratorio apropiadas. Si no se obtienen los niveles de actividad de factor VIII en plasma esperados, o si no se controla la hemorragia con una dosis adecuada, se debe realizar una prueba de detección de inhibidor de factor VIII. En pacientes con niveles altos de inhibidor, la terapia de sustitución de factor VIII puede no ser efectiva y se deben considerar otras opciones terapéuticas.
El tratamiento de tales pacientes debe estar dirigido por médicos con experiencia en el cuidado de pacientes con hemofilia e inhibidores del factor VIII.
Complicación relacionada con catéter en el tratamiento
Si se requiere un dispositivo de acceso venoso (CVAD) debe considerarse el riesgo de complicaciones relacionadas con CVAD incluyendo infecciones locales, bacteremia y trombosis del lado del catéter.
Consideraciones relativas al excipiente
Después de la reconstitución, este medicamento contiene 0,45 mmol de sodio (10 mg) por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
Se recomienda encarecidamente registrar el nombre y el número de lote del producto cada vez que se administre ADVATE a un paciente para poder mantener una relación entre el paciente y el lote del medicamento.
Población pediátrica:
Las advertencias y precauciones incluidas se aplican tanto a adultos como a niños.
4.5 Interacción con otros medicamentos y otras formas de interacción.
No se han realizado estudios de interacciones con ADVATE.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción en animales con factor VIII. Dados los raros casos de hemofilia A en mujeres, no se dispone de experiencia relacionada con el uso de factor VIII durante el embarazo y el periodo de lactancia. Por lo tanto, sólo debe utilizarse factor VIII durante el embarazo y el periodo de lactancia si está claramente indicado.
4.7 Efectos sobre la capacidad de conducir y utilizar máquinas
La influencia de ADVATE sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los ensayos clínicos con ADVATE incluyeron 418 sujetos con al menos una exposición a ADVATE y registraron un total de 93 reacciones adversas al medicamento (RAM). Las RAM que aparecieron con mayor frecuencia fueron el desarrollo de anticuerpos neutralizantes frente al factor VIII (inhibidores), cefalea y fiebre.
La hipersensibilidad y las reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el lugar de infusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habón urticarial, hipotensión, letargia, náuseas, inquietud, taquicardia, tirantez en el pecho, cosquilleo, vómitos, sibilancia) se observaron con poca frecuencia pero, en algunos casos, pueden progresar a anafilaxia grave (incluyendo shock).
Se puede observar desarrollo de anticuerpos a la proteína de ratón y/o hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) frente al factor VIII. Si aparecen tales inhibidores, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia.
Resumen tabulado de reacciones adversas
La siguiente tabla 2 indica la frecuencia de las reacciones adversas en los ensayos clínicos y procedente de informes espontáneos. La tabla sigue la clasificación de órganos del sistema MedDRA (COS y Nivel de término preferido).
Las frecuencias se han clasificado utilizando el siguiente criterio: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100), raros (> 1/10.000 a < 1/1.000), muy raros (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de informes espontáneos | ||
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Infecciones e infestaciones |
Gripe |
Poco frecuentes |
|
Laringitis |
Poco frecuentes | |
|
Linfangitis |
Poco frecuentes | |
|
Trastornos de la sangre y del sistema linfático |
Inhibición del factor VIII Linfangitis |
Frecuentes Poco frecuentes |
|
Trastornos del sistema inmunológico |
Reacción anafiláctica |
Frecuencia no conocida |
|
Hipersensibilidadc |
Frecuencia no conocida | |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuentes |
|
Mareos |
Poco frecuentes | |
|
Problemas de memoria Síncope |
Poco frecuentes Poco frecuentes | |
|
Temblores |
Poco frecuentes | |
|
Migraña |
Poco frecuentes | |
|
Disgeusia |
Poco frecuentes | |
|
Trastornos oculares |
Inflamación ocular |
Poco frecuentes |
|
Trastornos cardiacos |
Palpitaciones |
Poco frecuentes |
|
Trastornos vasculares |
Hematoma |
Poco frecuentes |
|
Sofocos |
Poco frecuentes | |
|
Palidez |
Poco frecuentes | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
Poco frecuentes |
|
Trastornos gastrointestinales |
Diarrea |
Poco frecuentes |
|
Dolor en el abdomen superior |
Poco frecuentes | |
|
Nauseas |
Poco frecuentes | |
|
Vómitos |
Poco frecuentes | |
|
Trastornos de la piel y del tejido subcutáneo |
Prurito |
Poco frecuentes |
|
Sarpullido |
Poco frecuentes | |
|
Hiperhidrosis |
Poco frecuentes | |
|
Urticaria |
Poco frecuentes | |
|
Trastornos generales y alteraciones en el lugar de administración |
Fiebre |
Frecuentes |
|
Edema periférico |
Poco frecuentes | |
|
Dolor torácico Malestar torácico |
Poco frecuentes Poco frecuentes | |
|
Escalofríos |
Poco frecuentes | |
|
Sensación anormal Hematoma en el lugar de punción de un vaso sanguíneo |
Poco frecuentes Poco frecuentes | |
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de __informes espontáneos__
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Fatiga Reacción en la zona de inyección |
Frecuencia no conocida Frecuencia no conocida | |
|
Malestar general |
Frecuencia no conocida | |
|
Exploraciones complementarias | ||
|
Recuento elevado de monocitos |
Poco frecuentes | |
|
Nivel reducido de factor de coagulación VIIIb |
Poco frecuentes | |
|
Descenso del hematocrito |
Poco frecuentes | |
|
Test de laboratorio anormal |
Poco frecuentes | |
|
Lesiones, intoxicaciones y complicaciones de procedimientos terapéuticos |
Complicaciones tras el procedimiento |
Poco frecuentes |
|
Hemorragia tras el procedimiento |
Poco frecuentes | |
|
Reacción en el lugar del procedimiento |
Poco frecuentes |
a) Calculado según un número total único de pacientes que recibieron ADVATE (418)
b) El descenso inesperado de los niveles de factor VIII de coagulación se produjo en un paciente durante la perfusión continua de ADVATE después de cirugía (días 10-14 de postoperatorio).
Se mantuvo la hemostasis durante todo este periodo y tanto los niveles de factor VIII de plasma como los niveles de aclaramiento volvieron a los niveles adecuados el día 15 de postoperatorio. Los ensayos de inhibidores del factor VIII realizados tras la finalización de la perfusión continua y la terminación del estudio fueron negativos.
c) RAM explicadas en la sección siguiente.
Descripción de reacciones adversas seleccionadas Desarrollo de inhibidor
Se ha notificado un desarrollo de inhibidor en pacientes con tratamientos previos y en pacientes no tratados previamente. Para información detallada ver secciones 5.1 (Propiedades farmacológicas) y 4.4 (Advertencias y precauciones especiales de empleo).
RAM específicas para residuos del proceso de fabricación
De los 229 pacientes en tratamiento en los que se evaluaron los anticuerpos antiproteínas de células de ovario de hámster chino (CHO), 3 mostraron una tendencia ascendente estadísticamente significativa en los títulos, 4 mostraron picos sostenidos o picos momentáneos y un paciente mostró ambos, pero no tuvo ningún otro síntoma clínico. De los 229 pacientes tratados a los que se realizó una evaluación de los anticuerpos anti-IgG murina, 10 mostraron una tendencia ascendente, dos mostraron un pico sostenido o pico momentáneo y un paciente mostró ambos. Cuatro de estos pacientes sufrieron episodios aislados de urticaria, prurito, sarpullido y recuentos ligeramente elevados de eosinófilos entre exposiciones repetidas al medicamento del estudio.
Hipersensibilidad
Reacciones de tipo alérgico incluyen anafilaxis y se han manifestado como mareos, parestesias, erupción, soplado, tumefacción facial, urticaria y prurito.
Población pediátrica
En los estudios clínicos no se han notado diferencias específicas por edad en RAM excepto el desarrollo de inhibidores en pacientes pediátricos no tratados previamente (PUP) y complicaciones relacionadas con catéter.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación pertinente incluido en el Anexo V.
4.9 Sobredosis
No se han notificado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos: factor VIII de coagulación. Código ATC: B02BD02.
El complejo factor VIII /Factor von Willebrand consta de dos moléculas (factor VIII y factor von Willebrand) con diferentes funciones fisiológicas. ADVATE contiene factor VIII de coagulación recombinante (octocog alfa), una glicoproteína que es biológicamente equivalente a la glicoproteína de factor VIII del plasma humano.
Octocog alfa es una glicoproteína que consta de 2332 aminoácidos con un peso molecular aproximado de 280 kD. Cuando se perfunde en un paciente hemofílico, octocog alfa se enlaza al Factor von Willebrand endógeno en la circulación del paciente. El factor VIII activado actúa como un Cofactor para el Factor IX activado, acelerando la conversión del Factor X a Factor X activado. El Factor X activado convierte la protrombina en trombina. La trombina convierte el fibrinógeno en fibrina, con la que se puede formar el coágulo. La hemofilia A es una alteración hereditaria de la coagulación sanguínea ligada al sexo y producida por la disminución de los niveles de factor VIII que da como resultado una hemorragia profusa en las articulaciones, músculos u órganos internos, bien de forma espontánea o bien como resultado de un trauma accidental o quirúrgico. Mediante terapia de sustitución, los niveles plasmáticos de factor VIII aumentan, consiguiendo una corrección temporal de la deficiencia de factor VIII y una corrección de las tendencias hemorrágicas.
Desarrollo del inhibidor
Se evaluó la inmunogenicidad de ADVATE en pacientes con tratamientos previos. Durante los ensayos clínicos con ADVATE en 233 pacientes pediátricos y adultos [pacientes pediátricos (0 a 16 años de edad) y pacientes adultos (más de 16 años de edad)] diagnosticados con hemofilia A grave (factor VIII < 1%), con exposición previa a concentrados de factor VIII durante un periodo igual o superior a 150 días para adultos y niños mayores e igual o superior a 50 días para niños menores de 6 años, un paciente desarrolló un título bajo de inhibidor (2,4 BU en el ensayo Bethesda modificado) tras 26 días de exposición a ADVATE. Las pruebas de seguimiento de inhibidores en este paciente tras abandonar el ensayo fueron negativas. En todos los estudios, la exposición media a ADVATE fue de 97,0 días de exposición por sujeto (intervalo 1 a 709) para pacientes previamente tratados. La incidencia global de cualquier desarrollo de inhibidor del factor VIII (alto o bajo) fue de 0,4% (1 de 233).
En el ensayo completado no controlado 060103, 16 de los 45 (35,6%) pacientes sin tratamiento previo con hemofilia A grave (FVIII < 1%) con al menos 25 días de exposición a FVIII desarrollaron inhibidores del FVIII: 7 (15,6%) sujetos desarrollaron inhibidores de título alto y 9 (20%) de los sujetos desarrollaron inhibidores de título bajo, 1 de los cuales también se clasificó como inhibidor temporal.
Los factores de riesgo relacionados con el desarrollo del inhibidor en este ensayo incluyeron etnicidad no caucásica, antecedentes familiares de inhibidores y tratamiento intensivo a dosis altas en los primeros 20 días de exposición. En los 20 sujetos que no tenían ninguno de estos factores de riesgo no se produjo el desarrollo del inhibidor.
Se han recogido datos sobre Inducción de tolerancia inmunitaria (ITI) en pacientes con inhibidores.
En un subestudio del estudio PUP 060103, se documentaron tratamientos ITI en 11 PUP. Se hizo un análisis de cuadro retrospective de 30 materias sobre ITI (estudio 060703) y recogida de datos de registro está en curso.
En el estudio 060201, se compararon dos esquemas de tratamiento de profilaxis a largo plazo en 53 sujetos previamente tratados: un régimen individualizado de dosificación guiada farmacocinética (dentro de un rango de 20 a 80 UI de factor VIII por kg de peso corporal a intervalos de 72 ± 6 horas, n = 23) con un régimen de dosificación profiláctica estándar (de 20 a 40 UI/kg cada 48 ± 6 horas, n = 30). El régimen de dosificación guiada farmacocinética (de acuerdo con una fórmula específica) tenía como objetivo mantener el factor VIII en niveles > 1% en el intervalo entre dosis de 72 horas. Los datos de este estudio demuestran que los dos regímenes de dosificación profiláctica son comparables en lo que se refiere a la reducción de la frecuencia de hemorragias.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con ADVATE en los diferentes grupos de la población pediátrica con hemofilia A (deficiencia congénita de factor VIII) en “Inducción de tolerancia inmunitaria (ITI) en pacientes con hemofilia A (deficiencia congénita de factor VIII) que han desarrollado inhibidores al factor VIII” y “tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII)” (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Todos los estudios farmacocinéticos con ADVATE se realizaron en pacientes previamente tratados con hemofilia A grave o moderadamente grave (valor basal del factor VIII < 2%). El análisis de las muestras de plasma se realizó en un laboratorio central utilizando el ensayo de coagulación en una etapa.
Un total de 195 sujetos con hemofilia A grave (valor basal del factor VIII < 1%) proporcionaron parámetros PK que se incluyeron en el conjunto de análisis de PK según el protocolo. Las categorías de estos análisis para lactantes (1 mes a <2 años), niños (2 a <5 años), niños de mayor edad (5 a <12 años), adolescentes (12 a <18 años) y adultos (18 años y mayores) se utilizaron para resumir los parámetros PK, definiéndose la edad como la edad en el momento de la perfusión de PK.
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Área bajo la curva total (UI*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Recuperación incremental ajustada a Cmáx (UI/dl por UI/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Semivida (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Concentración máxima en plasma tras la perfusión (UI/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Tiempo de residencia medio (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Volumen de distribución en estado estacionario (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Aclaramiento (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Calculada como (Cmáx - valor basal del factor VIII) dividida por la dosis en Ul/kg, donde Cmáx es la medida máxima de factor VIII tras la perfusión.
La seguridad y la eficacia hemostática de ADVATE en la población pediátrica son similares a las de los pacientes adultos. La recuperación y la semivida terminal (t/2) ajustadas fUe aproximadamente un 20% inferior en niños pequeños (menores de 6 años) que en adultos, lo que lo que puede deberse en parte a un volumen mayor conocido de plasma por kg de peso corporal en los pacientes más jóvenes.
No se dispone de datos farmacocinéticos de ADVATE en pacientes sin tratamiento previo.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de la seguridad, toxicología aguda, toxicidad en dosis repetida, toxicidad local y genotoxicidad.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo
Manitol
Cloruro de sodio
Histidina
Trealosa
Cloruro de calcio Trometamol Polisorbato 80 Glutatión (reducido)
Disolvente
Agua esterilizada para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
2 años.
Después de la reconstitución, desde un punto de vista microbiológico, el medicamento debe utilizarse de inmediato. No obstante, se ha demostrado una estabilidad física y química en uso durante 3 horas a 25 °C.
Durante el periodo de validez, el medicamento puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único no superior a 6 meses. Se debe anotar la fecha del final del periodo de conservación a temperatura ambiente de 6 meses en el embalaje del medicamento. El medicamento no puede refrigerarse de nuevo.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
ADVATE con el equipo BAXJECT II: Conservar el vial del producto en el embalaje exterior para protegerlo de la luz.
ADVATE en el sistema BAXJECT III: Conservar el blister sellado en el embalaje exterior para protegerlo de la luz.
Para consultar las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Tanto el vial de polvo como el vial de disolvente de 5 ml son de vidrio tipo I cerrado con tapones de caucho de clorobutilo. El medicamento se proporciona con una de las configuraciones siguientes:
- ADVATE con el equipo BAXJECT II: Cada envase contiene un vial de polvo, un vial de disolvente de 5 ml y un equipo para reconstitución (BAXJECT II).
- ADVATE en el sistema BAXJECT III: Cada envase contiene un sistema BAXJECT III listo para usar en un blister sellado (el vial de polvo y el vial de disolvente de 5 ml están precargados con el sistema para su reconstitución).
6.6 Precauciones especiales de eliminación y otras manipulaciones
ADVATE debe administrarse intravenosamente después de la reconstitución del producto.
La solución reconstituida se debe inspeccionar visualmente para comprobar que no presenta partículas extrañas y/o descoloración.
Después de la reconstitución la solución debe ser transparente, incolora y no presentar partículas extrañas.
No utilizar soluciones turbias o con depósitos.
- Para la administración se requiere el uso de una jeringa luer-lock.
- Utilizar dentro de las tres horas después de la reconstitución.
- No refrigerar la preparación después de la reconstitución.
- La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Reconstitución con el equipo BAXJECT II:
- Para la reconstitución utilizar sólo el agua esterilizada para preparaciones inyectables y el equipo de reconstitución incluido en el envase.
- No utilizar si el equipo BAXJECT II, el sistema estéril de protección o su envase está dañado o muestra algún signo de deterioro.
- Se debe utilizar una técnica aséptica:
1. Si el medicamento se encuentra en la nevera, coger de la nevera los viales de polvo y de disolvente de ADVATE y dejarlos a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Quitar los protectores de los viales de polvo y disolvente.
4. Limpiar los tapones con las toallitas impregnadas de alcohol. Colocar los viales en una superficie plana y limpia.
5. Abrir el envoltorio del equipo BAXJECT II quitando la tapa de papel sin tocar el interior (Fig. a). No sacar el equipo del envoltorio. No utilizar si el equipo BAXJECT II, su sistema de barrera estéril o su acondicionamiento están dañados o muestran algún signo de deterioro.
6. Dar la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente. Coger el envoltorio por su extremo y sacar el equipo BAXJECT II de su envoltorio (Fig. b). No quitar el protector azul del equipo BAXJECT II.
7. Para la reconstitución, utilizar solamente el agua esterilizada para preparaciones inyectables y el equipo para reconstitución incluidos en el envase. Con el equipo BAXJECT II unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico blanca dentro del tapón del vial de polvo ADVATE. El vacío hará que el disolvente penetre en el vial de polvo ADVATE (Fig. c).
8. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará
a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig. a
Fig. b
Fig. c
Reconstitución con el sistema BAXJECT III:
- No lo utilice si la tapa del blister no está perfectamente sellada.
1. Si el medicamento se encuentra en la nevera, coger de la nevera el blister sellado (contiene viales de polvo y disolvente precargados con el sistema para su reconstitución) y dejarlo a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Abrir el envoltorio de ADVATE quitando la tapa.Extraer el sistema BAXJECT III del blister.
4. Colocar ADVATE en una superficie plana con el vial de disolvente encima (Fig. 1). El vial de disolvente tiene una raya azul. No quitar el protector azul hasta que se le indique más adelante.
5. Mientras sujeta ADVATE con una mano en el sistema BAXJECT III, presionar con fuerza el vial de disolvente con la otra mano hasta que el sistema quede totalmente contraído y el disolvente entre dentro del vial de ADVATE (Fig. 2). No inclinar el sistema hasta que haya finalizado la transferencia.
6. Comprobar que ha finalizado la transferencia del disolvente. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.

Fig. 3

Administración:
Utilizar técnica aséptica:
Los medicamentos para administración parenteral deben inspeccionarse visualmente antes de su
administración para verificar la ausencia de partículas, cuando la solución y el envase lo permitan.
Sólo se debe utilizar una solución transparente y sin coloración.
1. Quitar el protector azul del equipo BAXJECT II / al sistema BAXJECT III. No introducir aire en la jeringa. Conectar la jeringa al equipo BAXJECT II / al sistema BAXJECT III (Fig. d).
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente (Fig. e).
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar intravenosamente. La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de ADVATE. Si se observa una elevación significativa del pulso, reducir la velocidad de administración o interrumpir temporalmente la inyección suele hacer que los síntomas normalmente desaparezcan pronto (ver secciones 4.4 y 4.8).
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
Industriestrasse 67
A-1221 Viena
Austria
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/005
EU/1/03/271/015
9. FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 2 de marzo de 2004 Fecha de la última renovación: 2 de marzo de 2014
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
ADVATE 3000 UI polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 3000 UI de factor VIII humano de coagulación (ADNr), octocog alfa. Después de la reconstitución, ADVATE contiene aproximadamente 600 UI por ml de factor VIII humano de coagulación (ADNr), octocog alfa.
La potencia (Unidades Internacionales) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de ADVATE es aproximadamente 4.000 -10.000 UI/mg/proteína.
El octocog alfa (factor VIII humano de coagulación (ADNr)) es una proteína purificada con 2332 aminoácidos. Está producida por tecnología de ADN recombinante en células de ovario de hámster chino (CHO). Preparado sin la adición de ninguna proteína (exógena) de origen humano o animal en el proceso del cultivo celular, purificación o formulación final.
Excipientes con efectos conocidos:
0,45 mmol de sodio (10 mg) por vial.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: polvo friable de color blanco a blanquecino. Disolvente: solución transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII). ADVATE está indicado en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia y con apoyo de resucitación disponible inmediatamente en caso de anafilaxia.
Posología
La dosis y duración de la terapia de sustitución depende de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII se expresa en Unidades Internacionales (UI), que se relacionan con el estándar de la OMS para medicamentos con factor VIII. La actividad de Factor VIII en plasma se expresa bien como porcentaje (referido a plasma humano normal) o en UI (referidas al Estándar Internacional de factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII es equivalente a la cantidad de factor VIII existente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis de factor VIII requerida se basa en el hallazgo empírico de que 1 UI de factor VIII por kg de peso corporal incrementa la actividad de factor VIII del plasma en 2 UI/dl. La dosis requerida se determina mediante la siguiente fórmula:
Unidades requeridas (UI) = peso corporal (kg) x aumento de factor VIII deseado (%) x 0,5
En el caso de los episodios hemorrágicos siguientes, la actividad de factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. En cirugía y en los episodios hemorrágicos, puede utilizarse la siguiente tabla 1 como guía de dosificación:
|
Tabla 1 Guía de dosificación en episodios hemorrágicos y cirugía | ||
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (% o UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente o hemorragia muscular u oral. |
20 - 40 |
Repetir la inyección cada 12 a 24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma. |
30 - 60 |
Repetir la inyección cada 12-24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) durante 3 a 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir la inyección cada 8 a 24 horas (cada 6 a 12 horas en los pacientes menores de 6 años) hasta superar el peligro. |
|
Cirugía | ||
|
Menor Incluyendo extracción dental. |
30 - 60 |
Cada 24 horas (cada 12 a 24 horas en los pacientes menores de 6 años), al menos 1 día, hasta lograr la curación. |
|
Mayor |
80 - 100 (pre y postoperatorio) |
Repetir la inyección cada 8-24 horas (cada 6 a 24 horas en los pacientes menores de 6 años) hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor VIII del 30% al 60% (UI/dl). |
La dosis y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual. En determinadas circunstancias (ej.: presencia de un título bajo de inhibidor) pueden ser necesarias dosis mayores a las calculadas mediante la fórmula.
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis que se debe administrar y la frecuencia con la que se debe repetir la inyección. En el caso especial de las intervenciones de cirugía mayor, es indispensable controlar con precisión el tratamiento de sustitución mediante pruebas de la coagulación (actividad del factor VIII plasmático). La respuesta individual de cada paciente frente al factor VIII puede variar, alcanzar distintos niveles de recuperación in vivo y presentar semividas diferentes.
Profilaxis
Para la profilaxis de larga duración frente a hemorragias en pacientes con hemofilia A grave, las dosis normales son de 20 a 40 UI de factor VIII por kg de peso corporal a intervalos de 2 a 3 días.
Población _ pediátrica
La dosis de tratamiento a demanda en pacientes pediátricos (0 a 18 años de edad) no difiere de la de los pacientes adultos. En pacientes menores de 6 años, se recomiendan dosis de 20 a 50 UI de factor VIII por kg de peso corporal de 3 a 4 veces a la semana como terapia profiláctica.
Forma de administración
ADVATE se debe administrar por vía intravenosa. En caso de ser administrado por un profesional no sanitario, es necesario realizar un entrenamiento adecuado.
Se debe determinar la velocidad de administración para asegurar la comodidad del paciente, hasta un máximo de 10 ml/min.
Después de la reconstitución, la solución es transparente, incolora y libre de partículas extrañas y tiene un pH de entre 6,7 y 7,3.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1 o a las proteínas de ratón o hámster.
4.4 Advertencias y precauciones especiales de empleo
Hipersensibilidad
Con ADVATE se han notificado reacciones de hipersensibilidad de tipo alérgico, incluyendo anafilaxis. El medicamento contiene trazas de proteínas de ratón y de hámster. Se debe informar a los pacientes de que en caso de que ocurran síntomas de hipersensibilidad, deben interrumpir el tratamiento y consultar con su médico de forma inmediata. Se debe informar a los pacientes de los signos de las reacciones de hipersensibilidad de tipo inmediato incluyendo habón urticarial, urticaria generalizada, tirantez en el pecho, sibilancia, hipotensión y anafilaxia.
En caso de que se produzca un shock, se deben seguir las pautas médicas estándar para su tratamiento.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) frente al factor VIII es una complicación conocida en el tratamiento de individuos con hemofilia A. Estos inhibidores son habitualmente inmunoglobulinas IgGs dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en unidades Bethesda (UB) por ml de plasma empleando el ensayo modificado. En pacientes que desarrollan anticuerpos neutralizantes (inhibidores) al Factor VIII, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia. El riesgo de desarrollar inhibidores se relaciona con el grado de exposición al factor VIII, siendo mayor durante los primeros 20 días de exposición, y con otros factores ambientales y genéticos. Raramente se desarrollan inhibidores después de los 100 primeros días de exposición.
Se han observado casos recurrentes de aparición de inhibidores (a títulos bajos), después de cambiar de un factor VIII a otro en los pacientes con un tratamiento previo de más de 100 días de exposición y con antecedentes de desarrollo de inhibidores. Así, se recomienda monitorizar cuidadosamente la aparición de inhibidores en todos los pacientes después de cualquier cambio de producto.
En general, en todos los pacientes tratados con un factor VIII de coagulación se debe monitorizar cuidadosamente la aparición de inhibidores mediante la realización de observaciones clínicas y de las pruebas de laboratorio apropiadas. Si no se obtienen los niveles de actividad de factor VIII en plasma esperados, o si no se controla la hemorragia con una dosis adecuada, se debe realizar una prueba de detección de inhibidor de factor VIII. En pacientes con niveles altos de inhibidor, la terapia de sustitución de factor VIII puede no ser efectiva y se deben considerar otras opciones terapéuticas.
El tratamiento de tales pacientes debe estar dirigido por médicos con experiencia en el cuidado de pacientes con hemofilia e inhibidores del factor VIII.
Complicación relacionada con catéter en el tratamiento
Si se requiere un dispositivo de acceso venoso (CVAD) debe considerarse el riesgo de complicaciones relacionadas con CVAD incluyendo infecciones locales, bacteremia y trombosis del lado del catéter.
Consideraciones relativas al excipiente
Después de la reconstitución, este medicamento contiene 0,45 mmol de sodio (10 mg) por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
Se recomienda encarecidamente registrar el nombre y el número de lote del producto cada vez que se administre ADVATE a un paciente para poder mantener una relación entre el paciente y el lote del medicamento.
Población pediátrica:
Las advertencias y precauciones incluidas se aplican tanto a adultos como a niños.
4.5 Interacción con otros medicamentos y otras formas de interacción.
No se han realizado estudios de interacciones con ADVATE.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción en animales con factor VIII. Dados los raros casos de hemofilia A en mujeres, no se dispone de experiencia relacionada con el uso de factor VIII durante el embarazo y el periodo de lactancia. Por lo tanto, sólo debe utilizarse factor VIII durante el embarazo y el periodo de lactancia si está claramente indicado.
4.7 Efectos sobre la capacidad de conducir y utilizar máquinas
La influencia de ADVATE sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del perfil de seguridad
Los ensayos clínicos con ADVATE incluyeron 418 sujetos con al menos una exposición a ADVATE y registraron un total de 93 reacciones adversas al medicamento (RAM). Las RAM que aparecieron con mayor frecuencia fueron el desarrollo de anticuerpos neutralizantes frente al factor VIII (inhibidores), cefalea y fiebre.
La hipersensibilidad y las reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el lugar de infusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habón urticarial, hipotensión, letargia, náuseas, inquietud, taquicardia, tirantez en el pecho, cosquilleo, vómitos, sibilancia) se observaron con poca frecuencia pero, en algunos casos, pueden progresar a anafilaxia grave (incluyendo shock).
Se puede observar desarrollo de anticuerpos a la proteína de ratón y/o hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) frente al factor VIII. Si aparecen tales inhibidores, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia.
Resumen tabulado de reacciones adversas
La siguiente tabla 2 indica la frecuencia de las reacciones adversas en los ensayos clínicos y procedente de informes espontáneos. La tabla sigue la clasificación de órganos del sistema MedDRA (COS y Nivel de término preferido).
Las frecuencias se han clasificado utilizando el siguiente criterio: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100), raros (> 1/10.000 a < 1/1.000), muy raros (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de informes espontáneos | ||
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Infecciones e infestaciones |
Gripe |
Poco frecuentes |
|
Laringitis |
Poco frecuentes | |
|
Linfangitis |
Poco frecuentes | |
|
Trastornos de la sangre y del sistema linfático |
Inhibición del factor VIII Linfangitis |
Frecuentes Poco frecuentes |
|
Trastornos del sistema inmunológico |
Reacción anafiláctica |
Frecuencia no conocida |
|
Hipersensibilidadc |
Frecuencia no conocida | |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
Frecuentes |
|
Mareos |
Poco frecuentes | |
|
Problemas de memoria Síncope |
Poco frecuentes Poco frecuentes | |
|
Temblores |
Poco frecuentes | |
|
Migraña |
Poco frecuentes | |
|
Disgeusia |
Poco frecuentes | |
|
Trastornos oculares |
Inflamación ocular |
Poco frecuentes |
|
Trastornos cardiacos |
Palpitaciones |
Poco frecuentes |
|
Trastornos vasculares |
Hematoma |
Poco frecuentes |
|
Sofocos |
Poco frecuentes | |
|
Palidez |
Poco frecuentes | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
Poco frecuentes |
|
Trastornos gastrointestinales |
Diarrea |
Poco frecuentes |
|
Dolor en el abdomen superior |
Poco frecuentes | |
|
Nauseas |
Poco frecuentes | |
|
Vómitos |
Poco frecuentes | |
|
Trastornos de la piel y del tejido subcutáneo |
Prurito |
Poco frecuentes |
|
Sarpullido |
Poco frecuentes | |
|
Hiperhidrosis |
Poco frecuentes | |
|
Urticaria |
Poco frecuentes | |
|
Trastornos generales y alteraciones en el lugar de administración |
Fiebre |
Frecuentes |
|
Edema periférico |
Poco frecuentes | |
|
Dolor torácico Malestar torácico |
Poco frecuentes Poco frecuentes | |
|
Escalofríos |
Poco frecuentes | |
|
Sensación anormal Hematoma en el lugar de punción de un vaso sanguíneo |
Poco frecuentes Poco frecuentes | |
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de __informes espontáneos__
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Fatiga Reacción en la zona de inyección |
Frecuencia no conocida Frecuencia no conocida | |
|
Malestar general |
Frecuencia no conocida | |
|
Exploraciones complementarias | ||
|
Recuento elevado de monocitos |
Poco frecuentes | |
|
Nivel reducido de factor de coagulación VIIIb |
Poco frecuentes | |
|
Descenso del hematocrito |
Poco frecuentes | |
|
Test de laboratorio anormal |
Poco frecuentes | |
|
Lesiones, intoxicaciones y complicaciones de procedimientos terapéuticos |
Complicaciones tras el procedimiento |
Poco frecuentes |
|
Hemorragia tras el procedimiento |
Poco frecuentes | |
|
Reacción en el lugar del procedimiento |
Poco frecuentes |
a) Calculado según un número total único de pacientes que recibieron ADVATE (418)
b) El descenso inesperado de los niveles de factor VIII de coagulación se produjo en un paciente durante la perfusión continua de ADVATE después de cirugía (días 10-14 de postoperatorio).
Se mantuvo la hemostasis durante todo este periodo y tanto los niveles de factor VIII de plasma como los niveles de aclaramiento volvieron a los niveles adecuados el día 15 de postoperatorio. Los ensayos de inhibidores del factor VIII realizados tras la finalización de la perfusión continua y la terminación del estudio fueron negativos.
c) RAM explicadas en la sección siguiente.
Descripción de reacciones adversas seleccionadas Desarrollo de inhibidor
Se ha notificado un desarrollo de inhibidor en pacientes con tratamientos previos y en pacientes no tratados previamente. Para información detallada ver secciones 5.1 (Propiedades farmacológicas) y 4.4 (Advertencias y precauciones especiales de empleo).
RAM específicas para residuos del proceso de fabricación
De los 229 pacientes en tratamiento en los que se evaluaron los anticuerpos antiproteínas de células de ovario de hámster chino (CHO), 3 mostraron una tendencia ascendente estadísticamente significativa en los títulos, 4 mostraron picos sostenidos o picos momentáneos y un paciente mostró ambos, pero no tuvo ningún otro síntoma clínico. De los 229 pacientes tratados a los que se realizó una evaluación de los anticuerpos anti-IgG murina, 10 mostraron una tendencia ascendente, dos mostraron un pico sostenido o pico momentáneo y un paciente mostró ambos. Cuatro de estos pacientes sufrieron episodios aislados de urticaria, prurito, sarpullido y recuentos ligeramente elevados de eosinófilos entre exposiciones repetidas al medicamento del estudio.
Hipersensibilidad
Reacciones de tipo alérgico incluyen anafilaxis y se han manifestado como mareos, parestesias, erupción, soplado, tumefacción facial, urticaria y prurito.
Población pediátrica
En los estudios clínicos no se han notado diferencias específicas por edad en RAM excepto el desarrollo de inhibidores en pacientes pediátricos no tratados previamente (PUP) y complicaciones relacionadas con catéter.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación pertinente incluido en el Anexo V.
4.9 Sobredosis
No se han notificado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos: factor VIII de coagulación. Código ATC: B02BD02.
El complejo factor VIII /Factor von Willebrand consta de dos moléculas (factor VIII y factor von Willebrand) con diferentes funciones fisiológicas. ADVATE contiene factor VIII de coagulación recombinante (octocog alfa), una glicoproteína que es biológicamente equivalente a la glicoproteína de factor VIII del plasma humano.
Octocog alfa es una glicoproteína que consta de 2332 aminoácidos con un peso molecular aproximado de 280 kD. Cuando se perfunde en un paciente hemofílico, octocog alfa se enlaza al Factor von Willebrand endógeno en la circulación del paciente. El factor VIII activado actúa como un Cofactor para el Factor IX activado, acelerando la conversión del Factor X a Factor X activado. El Factor X activado convierte la protrombina en trombina. La trombina convierte el fibrinógeno en fibrina, con la que se puede formar el coágulo. La hemofilia A es una alteración hereditaria de la coagulación sanguínea ligada al sexo y producida por la disminución de los niveles de factor VIII que da como resultado una hemorragia profusa en las articulaciones, músculos u órganos internos, bien de forma espontánea o bien como resultado de un trauma accidental o quirúrgico. Mediante terapia de sustitución, los niveles plasmáticos de factor VIII aumentan, consiguiendo una corrección temporal de la deficiencia de factor VIII y una corrección de las tendencias hemorrágicas.
Desarrollo del inhibidor
Se evaluó la inmunogenicidad de ADVATE en pacientes con tratamientos previos. Durante los ensayos clínicos con ADVATE en 233 pacientes pediátricos y adultos [pacientes pediátricos (0 a 16 años de edad) y pacientes adultos (más de 16 años de edad)] diagnosticados con hemofilia A grave (factor VIII < 1%), con exposición previa a concentrados de factor VIII durante un periodo igual o superior a 150 días para adultos y niños mayores e igual o superior a 50 días para niños menores de 6 años, un paciente desarrolló un título bajo de inhibidor (2,4 BU en el ensayo Bethesda modificado) tras 26 días de exposición a ADVATE. Las pruebas de seguimiento de inhibidores en este paciente tras abandonar el ensayo fueron negativas. En todos los estudios, la exposición media a ADVATE fue de 97,0 días de exposición por sujeto (intervalo 1 a 709) para pacientes previamente tratados. La incidencia global de cualquier desarrollo de inhibidor del factor VIII (alto o bajo) fue de 0,4% (1 de 233).
En el ensayo completado no controlado 060103, 16 de los 45 (35,6%) pacientes sin tratamiento previo con hemofilia A grave (FVIII < 1%) con al menos 25 días de exposición a FVIII desarrollaron inhibidores del FVIII: 7 (15,6%) sujetos desarrollaron inhibidores de título alto y 9 (20%) de los sujetos desarrollaron inhibidores de título bajo, 1 de los cuales también se clasificó como inhibidor temporal.
Los factores de riesgo relacionados con el desarrollo del inhibidor en este ensayo incluyeron etnicidad no caucásica, antecedentes familiares de inhibidores y tratamiento intensivo a dosis altas en los primeros 20 días de exposición. En los 20 sujetos que no tenían ninguno de estos factores de riesgo no se produjo el desarrollo del inhibidor.
Se han recogido datos sobre Inducción de tolerancia inmunitaria (ITI) en pacientes con inhibidores.
En un subestudio del estudio PUP 060103, se documentaron tratamientos ITI en 11 PUP. Se hizo un análisis de cuadro retrospective de 30 materias sobre ITI (estudio 060703) y recogida de datos de registro está en curso.
En el estudio 060201, se compararon dos esquemas de tratamiento de profilaxis a largo plazo en 53 sujetos previamente tratados: un régimen individualizado de dosificación guiada farmacocinética (dentro de un rango de 20 a 80 UI de factor VIII por kg de peso corporal a intervalos de 72 ± 6 horas, n = 23) con un régimen de dosificación profiláctica estándar (de 20 a 40 UI/kg cada 48 ± 6 horas, n = 30). El régimen de dosificación guiada farmacocinética (de acuerdo con una fórmula específica) tenía como objetivo mantener el factor VIII en niveles > 1% en el intervalo entre dosis de 72 horas. Los datos de este estudio demuestran que los dos regímenes de dosificación profiláctica son comparables en lo que se refiere a la reducción de la frecuencia de hemorragias.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con ADVATE en los diferentes grupos de la población pediátrica con hemofilia A (deficiencia congénita de factor VIII) en “Inducción de tolerancia inmunitaria (ITI) en pacientes con hemofilia A (deficiencia congénita de factor VIII) que han desarrollado inhibidores al factor VIII” y “tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII)” (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Todos los estudios farmacocinéticos con ADVATE se realizaron en pacientes previamente tratados con hemofilia A grave o moderadamente grave (valor basal del factor VIII < 2%). El análisis de las muestras de plasma se realizó en un laboratorio central utilizando el ensayo de coagulación en una etapa.
Un total de 195 sujetos con hemofilia A grave (valor basal del factor VIII < 1%) proporcionaron parámetros PK que se incluyeron en el conjunto de análisis de PK según el protocolo. Las categorías de estos análisis para lactantes (1 mes a <2 años), niños (2 a <5 años), niños de mayor edad (5 a <12 años), adolescentes (12 a <18 años) y adultos (18 años y mayores) se utilizaron para resumir los parámetros PK, definiéndose la edad como la edad en el momento de la perfusión de PK.
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Área bajo la curva total (UI*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Recuperación incremental ajustada a Cmáx (UI/dl por UI/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Semivida (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Concentración máxima en plasma tras la perfusión (UI/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Tiempo de residencia medio (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Volumen de distribución en estado estacionario (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Aclaramiento (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Calculada como (Cmáx - valor basal del factor VIII) dividida por la dosis en Ul/kg, donde Cmáx es la medida máxima de factor VIII tras la perfusión.
La seguridad y la eficacia hemostática de ADVATE en la población pediátrica son similares a las de los pacientes adultos. La recuperación y la semivida terminal (t/2) ajustadas fUe aproximadamente un 20% inferior en niños pequeños (menores de 6 años) que en adultos, lo que lo que puede deberse en parte a un volumen mayor conocido de plasma por kg de peso corporal en los pacientes más jóvenes.
No se dispone de datos farmacocinéticos de ADVATE en pacientes sin tratamiento previo.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de la seguridad, toxicología aguda, toxicidad en dosis repetida, toxicidad local y genotoxicidad.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Polvo
Manitol
Cloruro de sodio
Histidina
Trealosa
Cloruro de calcio Trometamol Polisorbato 80 Glutatión (reducido)
Disolvente
Agua esterilizada para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad este medicamento no debe mezclarse con otros.
6.3 Periodo de validez
2 años.
Después de la reconstitución, desde un punto de vista microbiológico, el medicamento debe utilizarse de inmediato. No obstante, se ha demostrado una estabilidad física y química en uso durante 3 horas a 25 °C.
Durante el periodo de validez, el medicamento puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único no superior a 6 meses. Se debe anotar la fecha del final del periodo de conservación a temperatura ambiente de 6 meses en el embalaje del medicamento. El medicamento no puede refrigerarse de nuevo.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
ADVATE con el equipo BAXJECT II: Conservar el vial del producto en el embalaje exterior para protegerlo de la luz.
ADVATE en el sistema BAXJECT III: Conservar el blister sellado en el embalaje exterior para protegerlo de la luz.
Para consultar las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Tanto el vial de polvo como el vial de disolvente de 5 ml son de vidrio tipo I cerrado con tapones de caucho de clorobutilo. El medicamento se proporciona con una de las configuraciones siguientes:
- ADVATE con el equipo BAXJECT II: Cada envase contiene un vial de polvo, un vial de disolvente de 5 ml y un equipo para reconstitución (BAXJECT II).
- ADVATE en el sistema BAXJECT III: Cada envase contiene un sistema BAXJECT III listo para usar en un blister sellado (el vial de polvo y el vial de disolvente de 5 ml están precargados con el sistema para su reconstitución).
6.6 Precauciones especiales de eliminación y otras manipulaciones
ADVATE debe administrarse intravenosamente después de la reconstitución del producto.
La solución reconstituida se debe inspeccionar visualmente para comprobar que no presenta partículas extrañas y/o descoloración.
Después de la reconstitución la solución debe ser transparente, incolora y no presentar partículas extrañas.
No utilizar soluciones turbias o con depósitos.
- Para la administración se requiere el uso de una jeringa luer-lock.
- Utilizar dentro de las tres horas después de la reconstitución.
- No refrigerar la preparación después de la reconstitución.
- La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Reconstitución con el equipo BAXJECT II:
- Para la reconstitución utilizar sólo el agua esterilizada para preparaciones inyectables y el equipo de reconstitución incluido en el envase.
- No utilizar si el equipo BAXJECT II, el sistema estéril de protección o su envase está dañado o muestra algún signo de deterioro.
- Se debe utilizar una técnica aséptica:
1. Si el medicamento se encuentra en la nevera, coger de la nevera los viales de polvo y de disolvente de ADVATE y dejarlos a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Quitar los protectores de los viales de polvo y disolvente.
4. Limpiar los tapones con las toallitas impregnadas de alcohol. Colocar los viales en una superficie plana y limpia.
5. Abrir el envoltorio del equipo BAXJECT II quitando la tapa de papel sin tocar el interior (Fig. a). No sacar el equipo del envoltorio. No utilizar si el equipo BAXJECT II, su sistema de barrera estéril o su acondicionamiento están dañados o muestran algún signo de deterioro.
6. Dar la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente. Coger el envoltorio por su extremo y sacar el equipo BAXJECT II de su envoltorio (Fig. b). No quitar el protector azul del equipo BAXJECT II.
7. Para la reconstitución, utilizar solamente el agua esterilizada para preparaciones inyectables y el equipo para reconstitución incluidos en el envase. Con el equipo BAXJECT II unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico blanca dentro del tapón del vial de polvo ADVATE. El vacío hará que el disolvente penetre en el vial de polvo ADVATE (Fig. c).
8. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará
a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig. a Fig. b Fig. c

Reconstitución con el sistema BAXJECT III:
- No lo utilice si la tapa del blister no está perfectamente sellada.
1. Si el medicamento se encuentra en la nevera, coger de la nevera el blister sellado (contiene viales de polvo y disolvente precargados con el sistema para su reconstitución) y dejarlo a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Abrir el envoltorio de ADVATE quitando la tapa. Extraer el sistema BAXJECT III del blister.
4. Colocar ADVATE en una superficie plana con el vial de disolvente encima (Fig. 1). El vial de disolvente tiene una raya azul. No quitar el protector azul hasta que se le indique más adelante.
5. Mientras sujeta ADVATE con una mano en el sistema BAXJECT III, presionar con fuerza el vial de disolvente con la otra mano hasta que el sistema quede totalmente contraído y el disolvente entre dentro del vial de ADVATE (Fig. 2). No inclinar el sistema hasta que haya finalizado la transferencia.
6. Comprobar que ha finalizado la transferencia del disolvente. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig.l

Fig.2
Fig. 3

Administración:
Utilizar técnica aséptica:
Los medicamentos para administración parenteral deben inspeccionarse visualmente antes de su
administración para verificar la ausencia de partículas, cuando la solución y el envase lo permitan.
Sólo se debe utilizar una solución transparente y sin coloración.
1. Quitar el protector azul del equipo BAXJECT II / al sistema BAXJECT III. No introducir aire en la jeringa. Conectar la jeringa al equipo BAXJECT II / al sistema BAXJECT III.
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente.
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar intravenosamente. La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de ADVATE. Si se observa una elevación significativa del pulso, reducir la velocidad de administración o interrumpir temporalmente la inyección suele hacer que los síntomas normalmente desaparezcan pronto (ver secciones 4.4 y 4.8).
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
Industriestrasse 67
A-1221 Viena
Austria
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/006
EU/1/03/271/016
9. FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 2 de marzo de 2004 Fecha de la última renovación: 2 de marzo de 2014
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
ADVATE 250 UI polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 250 UI de factor VIII humano de coagulación (ADNr), octocog alfa. Después de la reconstitución, ADVATE contiene aproximadamente 125 UI por ml de factor VIII humano de coagulación (ADNr), octocog alfa.
La potencia (Unidades Internacionales) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de ADVATE es aproximadamente 4.000 -10.000 UI/mg/proteína.
El octocog alfa (factor VIII humano de coagulación (ADNr)) es una proteína purificada con 2332 aminoácidos. Está producida por tecnología de ADN recombinante en células de ovario de hámster chino (CHO). Preparado sin la adición de ninguna proteína (exógena) de origen humano o animal en el proceso del cultivo celular, purificación o formulación final.
Excipientes con efectos conocidos:
0,45 mmol de sodio (10 mg) por vial.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: polvo friable de color blanco a blanquecino. Disolvente: solución transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII). ADVATE está indicado en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia y con apoyo de resucitación disponible inmediatamente en caso de anafilaxia.
Posología
La dosis y duración de la terapia de sustitución depende de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII se expresa en Unidades Internacionales (UI), que se relacionan con el estándar de la OMS para medicamentos con factor VIII. La actividad de Factor VIII en plasma se expresa bien como porcentaje (referido a plasma humano normal) o en UI (referidas al Estándar Internacional de factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII es equivalente a la cantidad de factor VIII existente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis de factor VIII requerida se basa en el hallazgo empírico de que 1 UI de factor VIII por kg de peso corporal incrementa la actividad de factor VIII del plasma en 2 UI/dl. La dosis requerida se determina mediante la siguiente fórmula:
Unidades requeridas (UI) = peso corporal (kg) x aumento de factor VIII deseado (%) x 0,5
En el caso de los episodios hemorrágicos siguientes, la actividad de factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. En cirugía y en los episodios hemorrágicos, puede utilizarse la siguiente tabla 1 como guía de dosificación:
|
Tabla 1 Guía de dosificación en episodios hemorrágicos y cirugía | ||
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (% o UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente o hemorragia muscular u oral. |
20 - 40 |
Repetir la inyección cada 12 a 24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma. |
30 - 60 |
Repetir la inyección cada 12-24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) durante 3 a 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir la inyección cada 8 a 24 horas (cada 6 a 12 horas en los pacientes menores de 6 años) hasta superar el peligro. |
|
Cirugía | ||
|
Menor Incluyendo extracción dental. |
30 - 60 |
Cada 24 horas (cada 12 a 24 horas en los pacientes menores de 6 años), al menos 1 día, hasta lograr la curación. |
|
Mayor |
80 - 100 (pre y postoperatorio) |
Repetir la inyección cada 8-24 horas (cada 6 a 24 horas en los pacientes menores de 6 años) hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor VIII del 30% al 60% (UI/dl). |
La dosis y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual. En determinadas circunstancias (ej.: presencia de un título bajo de inhibidor) pueden ser necesarias dosis mayores a las calculadas mediante la fórmula.
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis que se debe administrar y la frecuencia con la que se debe repetir la inyección. En el caso especial de las intervenciones de cirugía mayor, es indispensable controlar con precisión el tratamiento de sustitución mediante pruebas de la coagulación (actividad del factor VIII plasmático). La respuesta individual de cada paciente frente al factor VIII puede variar, alcanzar distintos niveles de recuperación in vivo y presentar semividas diferentes.
Profilaxis
Para la profilaxis de larga duración frente a hemorragias en pacientes con hemofilia A grave, las dosis normales son de 20 a 40 UI de factor VIII por kg de peso corporal a intervalos de 2 a 3 días.
Población _ pediátrica
La dosis de tratamiento a demanda en pacientes pediátricos (0 a 18 años de edad) no difiere de la de los pacientes adultos. En pacientes menores de 6 años, se recomiendan dosis de 20 a 50 UI de factor VIII por kg de peso corporal de 3 a 4 veces a la semana como terapia profiláctica.
No se ha descrito el uso de las presentaciones de 2 ml para sujetos pediátricos de menos de 2 años.
Forma de administración
ADVATE se debe administrar por vía intravenosa. En caso de ser administrado por un profesional no sanitario, es necesario realizar un entrenamiento adecuado.
Se debe determinar la velocidad de administración para asegurar la comodidad del paciente, hasta un máximo de 10 ml/min.
Después de la reconstitución, la solución es transparente, incolora y libre de partículas extrañas y tiene un pH de entre 6,7 y 7,3.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1 o a las proteínas de ratón o hámster.
4.4 Advertencias y precauciones especiales de empleo
Hipersensibilidad
Con ADVATE se han notificado reacciones de hipersensibilidad de tipo alérgico, incluyendo anafilaxis. El medicamento contiene trazas de proteínas de ratón y de hámster. Se debe informar a los pacientes de que en caso de que ocurran síntomas de hipersensibilidad, deben interrumpir el tratamiento y consultar con su médico de forma inmediata. Se debe informar a los pacientes de los signos de las reacciones de hipersensibilidad de tipo inmediato incluyendo habón urticarial, urticaria generalizada, tirantez en el pecho, sibilancia, hipotensión y anafilaxia.
En caso de que se produzca un shock, se deben seguir las pautas médicas estándar para su tratamiento.
Debido a la disminución del volumen de inyección de ADVATE reconstituido en 2 ml de agua esterilizada para preparaciones inyectables, si se producen reacciones de hipersensibilidad hay menos tiempo para reaccionar deteniendo la inyección. Por tanto, se aconseja tener cuidado durante la inyección de ADVATE reconstituido en 2 ml de agua esterilizada para preparaciones inyectables, especialmente en niños.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) frente al factor VIII es una complicación conocida en el tratamiento de individuos con hemofilia A. Estos inhibidores son habitualmente inmunoglobulinas IgGs dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en unidades Bethesda (UB) por ml de plasma empleando el ensayo modificado. En pacientes que desarrollan anticuerpos neutralizantes (inhibidores) al Factor VIII, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia. El riesgo de desarrollar inhibidores se relaciona con el grado de exposición al factor
VIII, siendo mayor durante los primeros 20 días de exposición, y con otros factores ambientales y genéticos. Raramente se desarrollan inhibidores después de los 100 primeros días de exposición.
Se han observado casos recurrentes de aparición de inhibidores (a títulos bajos), después de cambiar de un factor VIII a otro en los pacientes con un tratamiento previo de más de 100 días de exposición y con antecedentes de desarrollo de inhibidores. Así, se recomienda monitorizar cuidadosamente la aparición de inhibidores en todos los pacientes después de cualquier cambio de producto.
En general, en todos los pacientes tratados con un factor VIII de coagulación se debe monitorizar cuidadosamente la aparición de inhibidores mediante la realización de observaciones clínicas y de las pruebas de laboratorio apropiadas. Si no se obtienen los niveles de actividad de factor VIII en plasma esperados, o si no se controla la hemorragia con una dosis adecuada, se debe realizar una prueba de detección de inhibidor de factor VIII. En pacientes con niveles altos de inhibidor, la terapia de sustitución de factor VIII puede no ser efectiva y se deben considerar otras opciones terapéuticas.
El tratamiento de tales pacientes debe estar dirigido por médicos con experiencia en el cuidado de pacientes con hemofilia e inhibidores del factor VIII.
Aplicación incorrecta de ADVATE
Para ADVATE reconstituido con 2 ml de agua esterilizada para preparaciones inyectables, la aplicación incorrecta (vía intraarterial o paravenosa) puede producir reacciones en el lugar de inyección leves y a corto plazo, como cardenales y eritema.
Complicación relacionada con catéter en el tratamiento
Si se requiere un dispositivo de acceso venoso (CVAD) debe considerarse el riesgo de complicaciones relacionadas con CVAD incluyendo infecciones locales, bacteremia y trombosis del lado del catéter.
Consideraciones relativas al excipiente
Después de la reconstitución, este medicamento contiene 0,45 mmol de sodio (10 mg) por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
Se recomienda encarecidamente registrar el nombre y el número de lote del producto cada vez que se administre ADVATE a un paciente para poder mantener una relación entre el paciente y el lote del medicamento.
Población _ pediátrica:
Las advertencias y precauciones incluidas se aplican tanto a adultos como a niños.
4.5 Interacción con otros medicamentos y otras formas de interacción.
No se han realizado estudios de interacciones con ADVATE.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción en animales con factor VIII. Dados los raros casos de hemofilia A en mujeres, no se dispone de experiencia relacionada con el uso de factor VIII durante el embarazo y el periodo de lactancia. Por lo tanto, sólo debe utilizarse factor VIII durante el embarazo y el periodo de lactancia si está claramente indicado.
4.7 Efectos sobre la capacidad de conducir y utilizar máquinas
La influencia de ADVATE sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del_perfil de seguridad
Los ensayos clínicos con ADVATE incluyeron 418 sujetos con al menos una exposición a ADVATE y registraron un total de 93 reacciones adversas al medicamento (RAM). Las RAM que aparecieron con mayor frecuencia fueron el desarrollo de anticuerpos neutralizantes frente al factor VIII (inhibidores), cefalea y fiebre.
La hipersensibilidad y las reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el lugar de infusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habón urticarial, hipotensión, letargia, náuseas, inquietud, taquicardia, tirantez en el pecho, cosquilleo, vómitos, sibilancia) se observaron con poca frecuencia pero, en algunos casos, pueden progresar a anafilaxia grave (incluyendo shock).
Se puede observar desarrollo de anticuerpos a la proteína de ratón y/o hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) frente al factor VIII. Si aparecen tales inhibidores, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia.
Resumen tabulado de reacciones adversas
La siguiente tabla 2 indica la frecuencia de las reacciones adversas en los ensayos clínicos y procedente de informes espontáneos. La tabla sigue la clasificación de órganos del sistema MedDRA (COS y Nivel de término preferido).
Las frecuencias se han clasificado utilizando el siguiente criterio: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100), raros (> 1/10.000 a < 1/1.000), muy raros (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de __informes espontáneos__
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Infecciones e infestaciones |
Gripe |
Poco frecuentes |
|
Laringitis |
Poco frecuentes | |
|
Linfangitis |
Poco frecuentes | |
|
Trastornos de la sangre y |
Inhibición del factor VIII |
Frecuentes |
|
del sistema linfático |
Linfangitis |
Poco frecuentes |
|
Trastornos del sistema |
Reacción anafiláctica |
Frecuencia no conocida |
|
inmunológico |
Hipersensibilidad0 |
Frecuencia no conocida |
|
Trastornos del sistema |
Dolor de cabeza |
Frecuentes |
|
nervioso |
Mareos |
Poco frecuentes |
|
Problemas de memoria |
Poco frecuentes | |
|
Síncope |
Poco frecuentes | |
|
Temblores |
Poco frecuentes | |
|
Migraña |
Poco frecuentes | |
|
Disgeusia |
Poco frecuentes | |
|
Trastornos oculares |
Inflamación ocular |
Poco frecuentes |
|
Trastornos cardiacos |
Palpitaciones |
Poco frecuentes |
|
Trastornos vasculares |
Hematoma |
Poco frecuentes |
|
Sofocos |
Poco frecuentes | |
|
Palidez |
Poco frecuentes | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
Poco frecuentes |
|
Trastornos |
Diarrea |
Poco frecuentes |
|
gastrointestinales |
Dolor en el abdomen superior |
Poco frecuentes |
|
Nauseas |
Poco frecuentes | |
|
Vómitos |
Poco frecuentes |
|
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de informes espontáneos | ||
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Trastornos de la piel y del tejido subcutáneo |
Prurito |
Poco frecuentes |
|
Sarpullido |
Poco frecuentes | |
|
Hiperhidrosis |
Poco frecuentes | |
|
Urticaria |
Poco frecuentes | |
|
Trastornos generales y alteraciones en el lugar de administración |
Fiebre |
Frecuentes |
|
Edema periférico |
Poco frecuentes | |
|
Dolor torácico Malestar torácico |
Poco frecuentes Poco frecuentes | |
|
Escalofríos |
Poco frecuentes | |
|
Sensación anormal Hematoma en el lugar de punción de un vaso sanguíneo |
Poco frecuentes Poco frecuentes | |
|
Fatiga Reacción en la zona de inyección |
Frecuencia no conocida Frecuencia no conocida | |
|
Malestar general |
Frecuencia no conocida | |
|
Exploraciones complementarias | ||
|
Recuento elevado de monocitos |
Poco frecuentes | |
|
Nivel reducido de factor de coagulación VIIIb |
Poco frecuentes | |
|
Descenso del hematocrito |
Poco frecuentes | |
|
Test de laboratorio anormal |
Poco frecuentes | |
|
Lesiones, intoxicaciones y complicaciones de procedimientos terapéuticos |
Complicaciones tras el procedimiento |
Poco frecuentes |
|
Hemorragia tras el procedimiento |
Poco frecuentes | |
|
Reacción en el lugar del procedimiento |
Poco frecuentes | |
a) Calculado según un número total único de pacientes que recibieron ADVATE (418)
b) El descenso inesperado de los niveles de factor VIII de coagulación se produjo en un paciente durante la perfusión continua de ADVATE después de cirugía (días 10-14 de postoperatorio).
Se mantuvo la hemostasis durante todo este periodo y tanto los niveles de factor VIII de plasma como los niveles de aclaramiento volvieron a los niveles adecuados el día 15 de postoperatorio. Los ensayos de inhibidores del factor VIII realizados tras la finalización de la perfusión continua y la terminación del estudio füeron negativos.
c) RAM explicadas en la sección siguiente.
Descripción de reacciones adversas seleccionadas Desarrollo de inhibidor
Se ha notificado un desarrollo de inhibidor en pacientes con tratamientos previos y en pacientes no tratados previamente. Para información detallada ver secciones 5.1 (Propiedades farmacológicas) y 4.4 (Advertencias y precauciones especiales de empleo).
RAM específicas para residuos del proceso de fabricación
De los 229 pacientes en tratamiento en los que se evaluaron los anticuerpos antiproteínas de células de ovario de hámster chino (CHO), 3 mostraron una tendencia ascendente estadísticamente significativa en los títulos, 4 mostraron picos sostenidos o picos momentáneos y un paciente mostró ambos, pero no tuvo ningún otro síntoma clínico. De los 229 pacientes tratados a los que se realizó una evaluación de los anticuerpos anti-IgG murina, 10 mostraron una tendencia ascendente, dos mostraron un pico sostenido o pico momentáneo y un paciente mostró ambos. Cuatro de estos pacientes sufrieron episodios aislados de urticaria, prurito, sarpullido y recuentos ligeramente elevados de eosinófilos entre exposiciones repetidas al medicamento del estudio.
Hipersensibilidad
Reacciones de tipo alérgico incluyen anafilaxis y se han manifestado como mareos, parestesias, erupción, soplado, tumefacción facial, urticaria y prurito.
Población pediátrica
En los estudios clínicos no se han notado diferencias específicas por edad en RAM excepto el desarrollo de inhibidores en pacientes pediátricos no tratados previamente (PUP) y complicaciones relacionadas con catéter.
Notificación de sospechas de reacciones adversas
. Se invita a los sistema nacional
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento profesionales sanitarios a notificar las sospechas de reacciones adversas a través del de notificación pertinente incluido en el Anexo V.
4.9 Sobredosis
No se han notificado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos: factor VIII de coagulación. Código ATC: B02BD02.
El complejo factor VIII /Factor von Willebrand consta de dos moléculas (factor VIII y factor von Willebrand) con diferentes funciones fisiológicas. ADVATE contiene factor VIII de coagulación recombinante (octocog alfa), una glicoproteína que es biológicamente equivalente a la glicoproteína de factor VIII del plasma humano.
Octocog alfa es una glicoproteína que consta de 2332 aminoácidos con un peso molecular aproximado de 280 kD. Cuando se perfunde en un paciente hemofílico, octocog alfa se enlaza al Factor von Willebrand endógeno en la circulación del paciente. El factor VIII activado actúa como un Cofactor para el Factor IX activado, acelerando la conversión del Factor X a Factor X activado. El Factor X activado convierte la protrombina en trombina. La trombina convierte el fibrinógeno en fibrina, con la que se puede formar el coágulo. La hemofilia A es una alteración hereditaria de la coagulación sanguínea ligada al sexo y producida por la disminución de los niveles de factor VIII que da como resultado una hemorragia profusa en las articulaciones, músculos u órganos internos, bien de forma espontánea o bien como resultado de un trauma accidental o quirúrgico. Mediante terapia de sustitución, los niveles plasmáticos de factor VIII aumentan, consiguiendo una corrección temporal de la deficiencia de factor VIII y una corrección de las tendencias hemorrágicas.
Desarrollo del inhibidor
Se evaluó la inmunogenicidad de ADVATE en pacientes con tratamientos previos. Durante los ensayos clínicos con ADVATE en 233 pacientes pediátricos y adultos [pacientes pediátricos (0 a 16 años de edad) y pacientes adultos (más de 16 años de edad)] diagnosticados con hemofilia A grave (factor VIII < 1%), con exposición previa a concentrados de factor VIII durante un periodo igual o superior a 150 días para adultos y niños mayores e igual o superior a 50 días para niños menores de 6 años, un paciente desarrolló un título bajo de inhibidor (2,4 BU en el ensayo Bethesda modificado) tras 26 días de exposición a ADVATE. Las pruebas de seguimiento de inhibidores en este paciente tras abandonar el ensayo fueron negativas. En todos los estudios, la exposición media a ADVATE fue de 97,0 días de exposición por sujeto (intervalo 1 a 709) para pacientes previamente tratados. La incidencia global de cualquier desarrollo de inhibidor del factor VIII (alto o bajo) fue de 0,4% (1 de 233).
En el ensayo completado no controlado 060103, 16 de los 45 (35,6%) pacientes sin tratamiento previo con hemofilia A grave (FVIII < 1%) con al menos 25 días de exposición a FVIII desarrollaron inhibidores del FVIII: 7 (15,6%) sujetos desarrollaron inhibidores de título alto y 9 (20%) de los sujetos desarrollaron inhibidores de título bajo, 1 de los cuales también se clasificó como inhibidor temporal.
Los factores de riesgo relacionados con el desarrollo del inhibidor en este ensayo incluyeron etnicidad no caucásica, antecedentes familiares de inhibidores y tratamiento intensivo a dosis altas en los primeros 20 días de exposición. En los 20 sujetos que no tenían ninguno de estos factores de riesgo no se produjo el desarrollo del inhibidor.
Se han recogido datos sobre Inducción de tolerancia inmunitaria (ITI) en pacientes con inhibidores.
En un subestudio del estudio PUP 060103, se documentaron tratamientos ITI en 11 PUP. Se hizo un análisis de cuadro retrospective de 30 materias sobre ITI (estudio 060703) y recogida de datos de registro está en curso.
En el estudio 060201, se compararon dos esquemas de tratamiento de profilaxis a largo plazo en 53 sujetos previamente tratados: un régimen individualizado de dosificación guiada farmacocinética (dentro de un rango de 20 a 80 UI de factor VIII por kg de peso corporal a intervalos de 72 ± 6 horas, n = 23) con un régimen de dosificación profiláctica estándar (de 20 a 40 UI/kg cada 48 ± 6 horas, n = 30). El régimen de dosificación guiada farmacocinética (de acuerdo con una fórmula específica) tenía como objetivo mantener el factor VIII en niveles > 1% en el intervalo entre dosis de 72 horas. Los datos de este estudio demuestran que los dos regímenes de dosificación profiláctica son comparables en lo que se refiere a la reducción de la frecuencia de hemorragias.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con ADVATE en los diferentes grupos de la población pediátrica con hemofilia A (deficiencia congénita de factor VIII) en “Inducción de tolerancia inmunitaria (ITI) en pacientes con hemofilia A (deficiencia congénita de factor VIII) que han desarrollado inhibidores al factor VIII” y “tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII)” (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Todos los estudios farmacocinéticos con ADVATE se realizaron en pacientes previamente tratados con hemofilia A grave o moderadamente grave (valor basal del factor VIII < 2%). El análisis de las muestras de plasma se realizó en un laboratorio central utilizando el ensayo de coagulación en una etapa.
Un total de 195 sujetos con hemofilia A grave (valor basal del factor VIII < 1%) proporcionaron parámetros PK que se incluyeron en el conjunto de análisis de PK según el protocolo. Las categorías de estos análisis para lactantes (1 mes a <2 años), niños (2 a <5 años), niños de mayor edad (5 a <12 años), adolescentes (12 a <18 años) y adultos (18 años y mayores) se utilizaron para resumir los parámetros PK, definiéndose la edad como la edad en el momento de la perfusión de PK.
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Área bajo la curva total (UI*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Recuperación incremental ajustada a Cmáx (UI/dl por UI/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Semivida (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Concentración máxima en plasma tras la perfusión (UI/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Tiempo de residencia medio (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Volumen de distribución en estado estacionario (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Aclaramiento (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Calculada como (Cmáx - valor basal del factor VIII) dividida por la dosis en Ul/kg, donde Cmáx es la medida máxima de factor VIII tras la perfusión.
La seguridad y la eficacia hemostática de ADVATE en la población pediátrica son similares a las de los pacientes adultos. La recuperación y la semivida terminal (t./2) ajustadas fUe aproximadamente un 20% inferior en niños pequeños (menores de 6 años) que en adultos, lo que lo que puede deberse en parte a un volumen mayor conocido de plasma por kg de peso corporal en los pacientes más jóvenes.
No se dispone de datos farmacocinéticos de ADVATE en pacientes sin tratamiento previo.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de la seguridad, toxicología aguda, toxicidad en dosis repetida, toxicidad local y genotoxicidad.
Un estudio de tolerancia local con conejos demostró que ADVATE reconstituido con 2 ml de agua esterilizada para preparaciones inyectables se tolera bien tras una administración intravenosa.
Se observó un leve enrojecimiento temporal en el lugar de la administración después de la aplicación intraarterial y después de la aplicación paravenosa. No obstante, no se observaron cambios histopatológicos adversos correlacionados que indiquen una naturaleza temporal de esta observación.
DATOS FARMACÉUTICOS
6.
6.1 Lista de excipientes
Polvo
Manitol
Cloruro de sodio Histidina Trealosa Cloruro de calcio Trometamol Polisorbato 80 Glutatión (reducido)
Disolvente
Agua esterilizada para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
Después de la reconstitución, desde un punto de vista microbiológico, el medicamento debe utilizarse de inmediato. No obstante, se ha demostrado una estabilidad física y química en uso durante 3 horas a 25 °C.
Durante el periodo de validez, el medicamento puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único no superior a 6 meses. Se debe anotar la fecha del final del periodo de conservación a temperatura ambiente de 6 meses en el embalaje del medicamento. El medicamento no puede refrigerarse de nuevo.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
ADVATE con el equipo BAXJECT II: Conservar el vial del producto en el embalaje exterior para protegerlo de la luz.
ADVATE en el sistema BAXJECT III: Conservar el blister sellado en el embalaje exterior para protegerlo de la luz.
Para consultar las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Tanto el vial de polvo como el vial de disolvente de 2 ml son de vidrio tipo I cerrado con tapones de caucho de clorobutilo. El medicamento se proporciona con una de las configuraciones siguientes:
- ADVATE con el equipo BAXJECT II: Cada envase contiene un vial de polvo, un vial de disolvente de 2 ml y un equipo para reconstitución (BAXJECT II).
- ADVATE en el sistema BAXJECT III: Cada envase contiene un sistema BAXJECT III listo para usar en un blister sellado (el vial de polvo y el vial de disolvente de 2 ml están precargados con el sistema para su reconstitución).
6.6 Precauciones especiales de eliminación y otras manipulaciones
ADVATE debe administrarse intravenosamente después de la reconstitución del producto.
La solución reconstituida se debe inspeccionar visualmente para comprobar que no presenta partículas
extrañas y/o descoloración.
Después de la reconstitución la solución debe ser transparente, incolora y no presentar partículas
extrañas.
No utilizar soluciones turbias o con depósitos.
- Para la administración se requiere el uso de una jeringa luer-lock.
- Utilizar dentro de las tres horas después de la reconstitución.
- No refrigerar la preparación después de la reconstitución.
- La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Reconstitución con el equipo BAXJECT II:
- Para la reconstitución utilizar sólo el agua esterilizada para preparaciones inyectables y el equipo de reconstitución incluido en el envase.
- No utilizar si el equipo BAXJECT II, el sistema estéril de protección o su envase está dañado o muestra algún signo de deterioro.
- Se debe utilizar una técnica aséptica:
1. Si el medicamento se encuentra en la nevera, coger de la nevera los viales de polvo y de disolvente de ADVATE y dejarlos a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Quitar los protectores de los viales de polvo y disolvente.
4. Limpiar los tapones con las toallitas impregnadas de alcohol. Colocar los viales en una superficie plana y limpia.
5. Abrir el envoltorio del equipo BAXJECT II quitando la tapa de papel sin tocar el interior (Fig. a). No sacar el equipo del envoltorio. No utilizar si el equipo BAXJECT II, su sistema de barrera estéril o su acondicionamiento están dañados o muestran algún signo de deterioro.
6. Dar la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente. Coger el envoltorio por su extremo y sacar el equipo BAXJECT II de su envoltorio (Fig. b). No quitar el protector azul del equipo BAXJECT II.
7. Para la reconstitución, utilizar solamente el agua esterilizada para preparaciones inyectables y el equipo para reconstitución incluidos en el envase. Con el equipo BAXJECT II unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico blanca dentro del tapón del vial de polvo ADVATE. El vacío hará que el disolvente penetre en el vial de polvo ADVATE (Fig. c).
8. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará
a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig. a
Fig. c

Reconstitución con el sistema BAXJECT III:
- No lo utilice si la tapa del blister no está perfectamente sellada.
1. Si el medicamento se encuentra en la nevera, coger de la nevera el blister sellado (contiene viales de polvo y disolvente precargados con el sistema para su reconstitución) y dejarlo a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Abrir el envoltorio de ADVATE quitando la tapa. Extraer el sistema BAXJECT III del blister.
4. Colocar ADVATE en una superficie plana con el vial de disolvente encima (Fig. 1). El vial de disolvente tiene una raya azul. No quitar el protector azul hasta que se le indique más adelante.
5. Mientras sujeta ADVATE con una mano en el sistema BAXJECT III, presionar con fuerza el vial de disolvente con la otra mano hasta que el sistema quede totalmente contraído y el disolvente entre dentro del vial de ADVATE (Fig. 2). No inclinar el sistema hasta que haya finalizado la transferencia.
6. Comprobar que ha finalizado la transferencia del disolvente. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig- 1

Fig. 2

Fig. 3

Administración:
Utilizar técnica aséptica:
Los medicamentos para administración parenteral deben inspeccionarse visualmente antes de su
administración para verificar la ausencia de partículas, cuando la solución y el envase lo permitan.
Sólo se debe utilizar una solución transparente y sin coloración.
1. Quitar el protector azul del equipo BAXJECT II / al sistema BAXJECT III. No introducir aire en la jeringa. Conectar la jeringa al equipo BAXJECT II / al sistemaBAXJECT III.
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente.
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar intravenosamente. La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de ADVATE. Si se observa una elevación significativa del pulso, reducir la velocidad de administración o interrumpir temporalmente la inyección suele hacer que los síntomas normalmente desaparezcan pronto (ver secciones 4.4 y 4.8).
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG Industriestrasse 67 A-1221 Viena Austria
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/007
EU/1/03/271/017
9. FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA
AUTORIZACIÓN
Fecha de la primera autorización: 2 de marzo de 2004 Fecha de la última renovación: 2 de marzo de 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
1. NOMBRE DEL MEDICAMENTO
ADVATE 500 UI polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 500 UI de factor VIII humano de coagulación (ADNr), octocog alfa. Después de la reconstitución, ADVATE contiene aproximadamente 250 UI por ml de factor VIII humano de coagulación (ADNr), octocog alfa.
La potencia (Unidades Internacionales) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de ADVATE es aproximadamente 4.000 -10.000 UI/mg/proteína.
El octocog alfa (factor VIII humano de coagulación (ADNr)) es una proteína purificada con 2332 aminoácidos. Está producida por tecnología de ADN recombinante en células de ovario de hámster chino (CHO). Preparado sin la adición de ninguna proteína (exógena) de origen humano o animal en el proceso del cultivo celular, purificación o formulación final.
Excipientes con efectos conocidos:
0,45 mmol de sodio (10 mg) por vial.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: polvo friable de color blanco a blanquecino. Disolvente: solución transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII). ADVATE está indicado en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia y con apoyo de resucitación disponible inmediatamente en caso de anafilaxia.
Posología
La dosis y duración de la terapia de sustitución depende de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII se expresa en Unidades Internacionales (UI), que se relacionan con el estándar de la OMS para medicamentos con factor VIII. La actividad de Factor VIII en plasma se expresa bien como porcentaje (referido a plasma humano normal) o en UI (referidas al Estándar Internacional de factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII es equivalente a la cantidad de factor VIII existente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis de factor VIII requerida se basa en el hallazgo empírico de que 1 UI de factor VIII por kg de peso corporal incrementa la actividad de factor VIII del plasma en 2 UI/dl. La dosis requerida se determina mediante la siguiente fórmula:
Unidades requeridas (UI) = peso corporal (kg) x aumento de factor VIII deseado (%) x 0,5
En el caso de los episodios hemorrágicos siguientes, la actividad de factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. En cirugía y en los episodios hemorrágicos, puede utilizarse la siguiente tabla 1 como guía de dosificación:
|
Tabla 1 Guía de dosificación en episodios hemorrágicos y cirugía | ||
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (% o UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente o hemorragia muscular u oral. |
20 - 40 |
Repetir la inyección cada 12 a 24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma. |
30 - 60 |
Repetir la inyección cada 12-24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) durante 3 a 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir la inyección cada 8 a 24 horas (cada 6 a 12 horas en los pacientes menores de 6 años) hasta superar el peligro. |
|
Cirugía | ||
|
Menor Incluyendo extracción dental. |
30 - 60 |
Cada 24 horas (cada 12 a 24 horas en los pacientes menores de 6 años), al menos 1 día, hasta lograr la curación. |
|
Mayor |
80 - 100 (pre y postoperatorio) |
Repetir la inyección cada 8-24 horas (cada 6 a 24 horas en los pacientes menores de 6 años) hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor VIII del 30% al 60% (UI/dl). |
La dosis y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual. En determinadas circunstancias (ej.: presencia de un título bajo de inhibidor) pueden ser necesarias dosis mayores a las calculadas mediante la fórmula.
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis que se debe administrar y la frecuencia con la que se debe repetir la inyección. En el caso especial de las intervenciones de cirugía mayor, es indispensable controlar con precisión el tratamiento de sustitución mediante pruebas de la coagulación (actividad del factor VIII plasmático). La respuesta individual de cada paciente frente al factor VIII puede variar, alcanzar distintos niveles de recuperación in vivo y presentar semividas diferentes.
Profilaxis
Para la profilaxis de larga duración frente a hemorragias en pacientes con hemofilia A grave, las dosis normales son de 20 a 40 UI de factor VIII por kg de peso corporal a intervalos de 2 a 3 días.
Población _ pediátrica
La dosis de tratamiento a demanda en pacientes pediátricos (0 a 18 años de edad) no difiere de la de los pacientes adultos. En pacientes menores de 6 años, se recomiendan dosis de 20 a 50 UI de factor VIII por kg de peso corporal de 3 a 4 veces a la semana como terapia profiláctica.
No se ha descrito el uso de las presentaciones de 2 ml para sujetos pediátricos de menos de 2 años.
Forma de administración
ADVATE se debe administrar por vía intravenosa. En caso de ser administrado por un profesional no sanitario, es necesario realizar un entrenamiento adecuado.
Se debe determinar la velocidad de administración para asegurar la comodidad del paciente, hasta un máximo de 10 ml/min.
Después de la reconstitución, la solución es transparente, incolora y libre de partículas extrañas y tiene un pH de entre 6,7 y 7,3.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1 o a las proteínas de ratón o hámster.
4.4 Advertencias y precauciones especiales de empleo
Hipersensibilidad
Con ADVATE se han notificado reacciones de hipersensibilidad de tipo alérgico, incluyendo anafilaxis. El medicamento contiene trazas de proteínas de ratón y de hámster. Se debe informar a los pacientes de que en caso de que ocurran síntomas de hipersensibilidad, deben interrumpir el tratamiento y consultar con su médico de forma inmediata. Se debe informar a los pacientes de los signos de las reacciones de hipersensibilidad de tipo inmediato incluyendo habón urticarial, urticaria generalizada, tirantez en el pecho, sibilancia, hipotensión y anafilaxia.
En caso de que se produzca un shock, se deben seguir las pautas médicas estándar para su tratamiento.
Debido a la disminución del volumen de inyección de ADVATE reconstituido en 2 ml de agua esterilizada para preparaciones inyectables, si se producen reacciones de hipersensibilidad hay menos tiempo para reaccionar deteniendo la inyección. Por tanto, se aconseja tener cuidado durante la inyección de ADVATE reconstituido en 2 ml de agua esterilizada para preparaciones inyectables, especialmente en niños.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) frente al factor VIII es una complicación conocida en el tratamiento de individuos con hemofilia A. Estos inhibidores son habitualmente inmunoglobulinas IgGs dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en unidades Bethesda (UB) por ml de plasma empleando el ensayo modificado. En pacientes que desarrollan anticuerpos neutralizantes (inhibidores) al Factor VIII, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia. El riesgo de desarrollar inhibidores se relaciona con el grado de exposición al factor
VIII, siendo mayor durante los primeros 20 días de exposición, y con otros factores ambientales y genéticos. Raramente se desarrollan inhibidores después de los 100 primeros días de exposición.
Se han observado casos recurrentes de aparición de inhibidores (a títulos bajos), después de cambiar de un factor VIII a otro en los pacientes con un tratamiento previo de más de 100 días de exposición y con antecedentes de desarrollo de inhibidores. Así, se recomienda monitorizar cuidadosamente la aparición de inhibidores en todos los pacientes después de cualquier cambio de producto.
En general, en todos los pacientes tratados con un factor VIII de coagulación se debe monitorizar cuidadosamente la aparición de inhibidores mediante la realización de observaciones clínicas y de las pruebas de laboratorio apropiadas. Si no se obtienen los niveles de actividad de factor VIII en plasma esperados, o si no se controla la hemorragia con una dosis adecuada, se debe realizar una prueba de detección de inhibidor de factor VIII. En pacientes con niveles altos de inhibidor, la terapia de sustitución de factor VIII puede no ser efectiva y se deben considerar otras opciones terapéuticas.
El tratamiento de tales pacientes debe estar dirigido por médicos con experiencia en el cuidado de pacientes con hemofilia e inhibidores del factor VIII.
Aplicación incorrecta de ADVATE
Para ADVATE reconstituido con 2 ml de agua esterilizada para preparaciones inyectables, la aplicación incorrecta (vía intraarterial o paravenosa) puede producir reacciones en el lugar de inyección leves y a corto plazo, como cardenales y eritema.
Complicación relacionada con catéter en el tratamiento
Si se requiere un dispositivo de acceso venoso (CVAD) debe considerarse el riesgo de complicaciones relacionadas con CVAD incluyendo infecciones locales, bacteremia y trombosis del lado del catéter.
Consideraciones relativas al excipiente
Después de la reconstitución, este medicamento contiene 0,45 mmol de sodio (10 mg) por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
Se recomienda encarecidamente registrar el nombre y el número de lote del producto cada vez que se administre ADVATE a un paciente para poder mantener una relación entre el paciente y el lote del medicamento.
Población _ pediátrica:
Las advertencias y precauciones incluidas se aplican tanto a adultos como a niños.
4.5 Interacción con otros medicamentos y otras formas de interacción.
No se han realizado estudios de interacciones con ADVATE.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción en animales con factor VIII. Dados los raros casos de hemofilia A en mujeres, no se dispone de experiencia relacionada con el uso de factor VIII durante el embarazo y el periodo de lactancia. Por lo tanto, sólo debe utilizarse factor VIII durante el embarazo y el periodo de lactancia si está claramente indicado.
4.7 Efectos sobre la capacidad de conducir y utilizar máquinas
La influencia de ADVATE sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del_perfil de seguridad
Los ensayos clínicos con ADVATE incluyeron 418 sujetos con al menos una exposición a ADVATE y registraron un total de 93 reacciones adversas al medicamento (RAM). Las RAM que aparecieron
con mayor frecuencia fueron el desarrollo de anticuerpos neutralizantes frente al factor VIII (inhibidores), cefalea y fiebre.
La hipersensibilidad y las reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el lugar de infusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habón urticarial, hipotensión, letargia, náuseas, inquietud, taquicardia, tirantez en el pecho, cosquilleo, vómitos, sibilancia) se observaron con poca frecuencia pero, en algunos casos, pueden progresar a anafilaxia grave (incluyendo shock).
Se puede observar desarrollo de anticuerpos a la proteína de ratón y/o hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) frente al factor VIII. Si aparecen tales inhibidores, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia.
Resumen tabulado de reacciones adversas
La siguiente tabla 2 indica la frecuencia de las reacciones adversas en los ensayos clínicos y procedente de informes espontáneos. La tabla sigue la clasificación de órganos del sistema MedDRA (COS y Nivel de término preferido).
Las frecuencias se han clasificado utilizando el siguiente criterio: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100), raros (> 1/10.000 a < 1/1.000), muy raros (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de __informes espontáneos__
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Infecciones e infestaciones |
Gripe |
Poco frecuentes |
|
Laringitis |
Poco frecuentes | |
|
Linfangitis |
Poco frecuentes | |
|
Trastornos de la sangre y |
Inhibición del factor VIII |
Frecuentes |
|
del sistema linfático |
Linfangitis |
Poco frecuentes |
|
Trastornos del sistema |
Reacción anafiláctica |
Frecuencia no conocida |
|
inmunológico |
Hipersensibilidad0 |
Frecuencia no conocida |
|
Trastornos del sistema |
Dolor de cabeza |
Frecuentes |
|
nervioso |
Mareos |
Poco frecuentes |
|
Problemas de memoria |
Poco frecuentes | |
|
Síncope |
Poco frecuentes | |
|
Temblores |
Poco frecuentes | |
|
Migraña |
Poco frecuentes | |
|
Disgeusia |
Poco frecuentes | |
|
Trastornos oculares |
Inflamación ocular |
Poco frecuentes |
|
Trastornos cardiacos |
Palpitaciones |
Poco frecuentes |
|
Trastornos vasculares |
Hematoma |
Poco frecuentes |
|
Sofocos |
Poco frecuentes | |
|
Palidez |
Poco frecuentes | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
Poco frecuentes |
|
Trastornos |
Diarrea |
Poco frecuentes |
|
gastrointestinales |
Dolor en el abdomen superior |
Poco frecuentes |
|
Nauseas |
Poco frecuentes | |
|
Vómitos |
Poco frecuentes |
|
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de informes espontáneos | ||
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Trastornos de la piel y del tejido subcutáneo |
Prurito |
Poco frecuentes |
|
Sarpullido |
Poco frecuentes | |
|
Hiperhidrosis |
Poco frecuentes | |
|
Urticaria |
Poco frecuentes | |
|
Trastornos generales y alteraciones en el lugar de administración |
Fiebre |
Frecuentes |
|
Edema periférico |
Poco frecuentes | |
|
Dolor torácico Malestar torácico |
Poco frecuentes Poco frecuentes | |
|
Escalofríos |
Poco frecuentes | |
|
Sensación anormal Hematoma en el lugar de punción de un vaso sanguíneo |
Poco frecuentes Poco frecuentes | |
|
Fatiga Reacción en la zona de inyección |
Frecuencia no conocida Frecuencia no conocida | |
|
Malestar general |
Frecuencia no conocida | |
|
Exploraciones complementarias | ||
|
Recuento elevado de monocitos |
Poco frecuentes | |
|
Nivel reducido de factor de coagulación VIIIb |
Poco frecuentes | |
|
Descenso del hematocrito |
Poco frecuentes | |
|
Test de laboratorio anormal |
Poco frecuentes | |
|
Lesiones, intoxicaciones y complicaciones de procedimientos terapéuticos |
Complicaciones tras el procedimiento |
Poco frecuentes |
|
Hemorragia tras el procedimiento |
Poco frecuentes | |
|
Reacción en el lugar del procedimiento |
Poco frecuentes | |
a) Calculado según un número total único de pacientes que recibieron ADVATE (418)
b) El descenso inesperado de los niveles de factor VIII de coagulación se produjo en un paciente durante la perfusión continua de ADVATE después de cirugía (días 10-14 de postoperatorio).
Se mantuvo la hemostasis durante todo este periodo y tanto los niveles de factor VIII de plasma como los niveles de aclaramiento volvieron a los niveles adecuados el día 15 de postoperatorio. Los ensayos de inhibidores del factor VIII realizados tras la finalización de la perfusión continua y la terminación del estudio füeron negativos.
c) RAM explicadas en la sección siguiente.
Descripción de reacciones adversas seleccionadas Desarrollo de inhibidor
Se ha notificado un desarrollo de inhibidor en pacientes con tratamientos previos y en pacientes no tratados previamente. Para información detallada ver secciones 5.1 (Propiedades farmacológicas) y 4.4 (Advertencias y precauciones especiales de empleo).
RAM específicas para residuos del proceso de fabricación
De los 229 pacientes en tratamiento en los que se evaluaron los anticuerpos antiproteínas de células de ovario de hámster chino (CHO), 3 mostraron una tendencia ascendente estadísticamente significativa en los títulos, 4 mostraron picos sostenidos o picos momentáneos y un paciente mostró ambos, pero no tuvo ningún otro síntoma clínico. De los 229 pacientes tratados a los que se realizó una evaluación de los anticuerpos anti-IgG murina, 10 mostraron una tendencia ascendente, dos mostraron un pico sostenido o pico momentáneo y un paciente mostró ambos. Cuatro de estos pacientes sufrieron episodios aislados de urticaria, prurito, sarpullido y recuentos ligeramente elevados de eosinófilos entre exposiciones repetidas al medicamento del estudio.
Hipersensibilidad
Reacciones de tipo alérgico incluyen anafilaxis y se han manifestado como mareos, parestesias, erupción, soplado, tumefacción facial, urticaria y prurito.
Población pediátrica
En los estudios clínicos no se han notado diferencias específicas por edad en RAM excepto el desarrollo de inhibidores en pacientes pediátricos no tratados previamente (PUP) y complicaciones relacionadas con catéter.
Notificación de sospechas de reacciones adversas
. Se invita a los sistema nacional
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento profesionales sanitarios a notificar las sospechas de reacciones adversas a través del de notificación pertinente incluido en el Anexo V.
4.9 Sobredosis
No se han notificado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos: factor VIII de coagulación. Código ATC: B02BD02.
El complejo factor VIII /Factor von Willebrand consta de dos moléculas (factor VIII y factor von Willebrand) con diferentes funciones fisiológicas. ADVATE contiene factor VIII de coagulación recombinante (octocog alfa), una glicoproteína que es biológicamente equivalente a la glicoproteína de factor VIII del plasma humano.
Octocog alfa es una glicoproteína que consta de 2332 aminoácidos con un peso molecular aproximado de 280 kD. Cuando se perfunde en un paciente hemofílico, octocog alfa se enlaza al Factor von Willebrand endógeno en la circulación del paciente. El factor VIII activado actúa como un Cofactor para el Factor IX activado, acelerando la conversión del Factor X a Factor X activado. El Factor X activado convierte la protrombina en trombina. La trombina convierte el fibrinógeno en fibrina, con la que se puede formar el coágulo. La hemofilia A es una alteración hereditaria de la coagulación sanguínea ligada al sexo y producida por la disminución de los niveles de factor VIII que da como resultado una hemorragia profusa en las articulaciones, músculos u órganos internos, bien de forma espontánea o bien como resultado de un trauma accidental o quirúrgico. Mediante terapia de sustitución, los niveles plasmáticos de factor VIII aumentan, consiguiendo una corrección temporal de la deficiencia de factor VIII y una corrección de las tendencias hemorrágicas.
Desarrollo del inhibidor
Se evaluó la inmunogenicidad de ADVATE en pacientes con tratamientos previos. Durante los ensayos clínicos con ADVATE en 233 pacientes pediátricos y adultos [pacientes pediátricos (0 a 16 años de edad) y pacientes adultos (más de 16 años de edad)] diagnosticados con hemofilia A grave (factor VIII < 1%), con exposición previa a concentrados de factor VIII durante un periodo igual o superior a 150 días para adultos y niños mayores e igual o superior a 50 días para niños menores de 6 años, un paciente desarrolló un título bajo de inhibidor (2,4 BU en el ensayo Bethesda modificado) tras 26 días de exposición a ADVATE. Las pruebas de seguimiento de inhibidores en este paciente tras abandonar el ensayo fueron negativas. En todos los estudios, la exposición media a ADVATE fue de 97,0 días de exposición por sujeto (intervalo 1 a 709) para pacientes previamente tratados. La incidencia global de cualquier desarrollo de inhibidor del factor VIII (alto o bajo) fue de 0,4% (1 de 233).
En el ensayo completado no controlado 060103, 16 de los 45 (35,6%) pacientes sin tratamiento previo con hemofilia A grave (FVIII < 1%) con al menos 25 días de exposición a FVIII desarrollaron inhibidores del FVIII: 7 (15,6%) sujetos desarrollaron inhibidores de título alto y 9 (20%) de los sujetos desarrollaron inhibidores de título bajo, 1 de los cuales también se clasificó como inhibidor temporal.
Los factores de riesgo relacionados con el desarrollo del inhibidor en este ensayo incluyeron etnicidad no caucásica, antecedentes familiares de inhibidores y tratamiento intensivo a dosis altas en los primeros 20 días de exposición. En los 20 sujetos que no tenían ninguno de estos factores de riesgo no se produjo el desarrollo del inhibidor.
Se han recogido datos sobre Inducción de tolerancia inmunitaria (ITI) en pacientes con inhibidores.
En un subestudio del estudio PUP 060103, se documentaron tratamientos ITI en 11 PUP. Se hizo un análisis de cuadro retrospective de 30 materias sobre ITI (estudio 060703) y recogida de datos de registro está en curso.
En el estudio 060201, se compararon dos esquemas de tratamiento de profilaxis a largo plazo en 53 sujetos previamente tratados: un régimen individualizado de dosificación guiada farmacocinética (dentro de un rango de 20 a 80 UI de factor VIII por kg de peso corporal a intervalos de 72 ± 6 horas, n = 23) con un régimen de dosificación profiláctica estándar (de 20 a 40 UI/kg cada 48 ± 6 horas, n = 30). El régimen de dosificación guiada farmacocinética (de acuerdo con una fórmula específica) tenía como objetivo mantener el factor VIII en niveles > 1% en el intervalo entre dosis de 72 horas. Los datos de este estudio demuestran que los dos regímenes de dosificación profiláctica son comparables en lo que se refiere a la reducción de la frecuencia de hemorragias.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con ADVATE en los diferentes grupos de la población pediátrica con hemofilia A (deficiencia congénita de factor VIII) en “Inducción de tolerancia inmunitaria (ITI) en pacientes con hemofilia A (deficiencia congénita de factor VIII) que han desarrollado inhibidores al factor VIII” y “tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII)” (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Todos los estudios farmacocinéticos con ADVATE se realizaron en pacientes previamente tratados con hemofilia A grave o moderadamente grave (valor basal del factor VIII < 2%). El análisis de las muestras de plasma se realizó en un laboratorio central utilizando el ensayo de coagulación en una etapa.
Un total de 195 sujetos con hemofilia A grave (valor basal del factor VIII < 1%) proporcionaron parámetros PK que se incluyeron en el conjunto de análisis de PK según el protocolo. Las categorías de estos análisis para lactantes (1 mes a <2 años), niños (2 a <5 años), niños de mayor edad (5 a <12 años), adolescentes (12 a <18 años) y adultos (18 años y mayores) se utilizaron para resumir los parámetros PK, definiéndose la edad como la edad en el momento de la perfusión de PK.
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Área bajo la curva total (UI*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Recuperación incremental ajustada a Cmáx (UI/dl por UI/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Semivida (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Concentración máxima en plasma tras la perfusión (UI/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Tiempo de residencia medio (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Volumen de distribución en estado estacionario (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Aclaramiento (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Calculada como (Cmáx - valor basal del factor VIII) dividida por la dosis en Ul/kg, donde Cmáx es la medida máxima de factor VIII tras la perfusión.
La seguridad y la eficacia hemostática de ADVATE en la población pediátrica son similares a las de los pacientes adultos. La recuperación y la semivida terminal (t./2) ajustadas fUe aproximadamente un 20% inferior en niños pequeños (menores de 6 años) que en adultos, lo que lo que puede deberse en parte a un volumen mayor conocido de plasma por kg de peso corporal en los pacientes más jóvenes.
No se dispone de datos farmacocinéticos de ADVATE en pacientes sin tratamiento previo.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de la seguridad, toxicología aguda, toxicidad en dosis repetida, toxicidad local y genotoxicidad.
Un estudio de tolerancia local con conejos demostró que ADVATE reconstituido con 2 ml de agua esterilizada para preparaciones inyectables se tolera bien tras una administración intravenosa.
Se observó un leve enrojecimiento temporal en el lugar de la administración después de la aplicación intraarterial y después de la aplicación paravenosa. No obstante, no se observaron cambios histopatológicos adversos correlacionados que indiquen una naturaleza temporal de esta observación.
DATOS FARMACÉUTICOS
6.
6.1 Lista de excipientes
Polvo
Manitol
Cloruro de sodio Histidina Trealosa Cloruro de calcio Trometamol Polisorbato 80 Glutatión (reducido)
Disolvente
Agua esterilizada para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
Después de la reconstitución, desde un punto de vista microbiológico, el medicamento debe utilizarse de inmediato. No obstante, se ha demostrado una estabilidad física y química en uso durante 3 horas a 25 °C.
Durante el periodo de validez, el medicamento puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único no superior a 6 meses. Se debe anotar la fecha del final del periodo de conservación a temperatura ambiente de 6 meses en el embalaje del medicamento. El medicamento no puede refrigerarse de nuevo.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
ADVATE con el equipo BAXJECT II: Conservar el vial en el embalaje exterior para protegerlo de la luz.
ADVATE en el sistema BAXJECT III: Conservar el blister sellado en el embalaje exterior para protegerlo de la luz.
Para consultar las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Tanto el vial de polvo como el vial de disolvente de 2 ml son de vidrio tipo I cerrado con tapones de caucho de clorobutilo. El medicamento se proporciona con una de las configuraciones siguientes:
- ADVATE con el equipo BAXJECT II: Cada envase contiene un vial de polvo, un vial de disolvente de 2 ml y un equipo para reconstitución (BAXJECT II).
- ADVATE en el sistema BAXJECT III: Cada envase contiene un sistema BAXJECT III listo para usar en un blister sellado (el vial de polvo y el vial de disolvente de 2 ml están precargados con el sistema para su reconstitución).
6.6 Precauciones especiales de eliminación y otras manipulaciones
ADVATE debe administrarse intravenosamente después de la reconstitución del producto.
La solución reconstituida se debe inspeccionar visualmente para comprobar que no presenta partículas
extrañas y/o descoloración.
Después de la reconstitución la solución debe ser transparente, incolora y no presentar partículas
extrañas.
No utilizar soluciones turbias o con depósitos.
- Para la administración se requiere el uso de una jeringa luer-lock.
- Utilizar dentro de las tres horas después de la reconstitución.
- No refrigerar la preparación después de la reconstitución.
- La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Reconstitución con el equipo BAXJECT II:
- Para la reconstitución utilizar sólo el agua esterilizada para preparaciones inyectables y el equipo de reconstitución incluido en el envase.
- No utilizar si el equipo BAXJECT II, el sistema estéril de protección o su envase está dañado o muestra algún signo de deterioro.
- Se debe utilizar una técnica aséptica:
1. Si el medicamento se encuentra en la nevera, coger de la nevera los viales de polvo y de disolvente de ADVATE y dejarlos a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Quitar los protectores de los viales de polvo y disolvente.
4. Limpiar los tapones con las toallitas impregnadas de alcohol. Colocar los viales en una superficie plana y limpia.
5. Abrir el envoltorio del equipo BAXJECT II quitando la tapa de papel sin tocar el interior (Fig. a). No sacar el equipo del envoltorio. No utilizar si el equipo BAXJECT II, su sistema de barrera estéril o su acondicionamiento están dañados o muestran algún signo de deterioro.
6. Dar la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente. Coger el envoltorio por su extremo y sacar el equipo BAXJECT II de su envoltorio (Fig. b). No quitar el protector azul del equipo BAXJECT II.
7. Para la reconstitución, utilizar solamente el agua esterilizada para preparaciones inyectables y el equipo para reconstitución incluidos en el envase. Con el equipo BAXJECT II unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico blanca dentro del tapón del vial de polvo ADVATE. El vacío hará que el disolvente penetre en el vial de polvo ADVATE (Fig. c).
8. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará
a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig. a
Fig. c

Reconstitución con el sistema BAXJECT III:
- No lo utilice si la tapa del blister no está perfectamente sellada.
1. Si el medicamento se encuentra en la nevera, coger de la nevera el blister sellado (contiene viales de polvo y disolvente precargados con el sistema para su reconstitución) y dejarlo a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Abrir el envoltorio de ADVATE quitando la tapa. Extraer el sistema BAXJECT III del blister.
4. Colocar ADVATE en una superficie plana con el vial de disolvente encima (Fig. 1). El vial de disolvente tiene una raya azul. No quitar el protector azul hasta que se le indique más adelante.
5. Mientras sujeta ADVATE con una mano en el sistema BAXJECT III, presionar con fuerza el vial de disolvente con la otra mano hasta que el sistema quede totalmente contraído y el disolvente entre dentro del vial de ADVATE (Fig. 2). No inclinar el sistema hasta que haya finalizado la transferencia.
6. Comprobar que ha finalizado la transferencia del disolvente. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig- 1

Fig. 2

Fig. 3

Administración:
Utilizar técnica aséptica:
Los medicamentos para administración parenteral deben inspeccionarse visualmente antes de su
administración para verificar la ausencia de partículas, cuando la solución y el envase lo permitan.
Sólo se debe utilizar una solución transparente y sin coloración.
1. Quitar el protector azul del equipo BAXJECT II / al sistema BAXJECT III. No introducir aire en la jeringa. Conectar la jeringa al equipo BAXJECT II / al sistema BAXJECT III.
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente.
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar intravenosamente. La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de ADVATE. Si se observa una elevación significativa del pulso, reducir la velocidad de administración o interrumpir temporalmente la inyección suele hacer que los síntomas normalmente desaparezcan pronto (ver secciones 4.4 y 4.8).
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG Industriestrasse 67 A-1221 Viena Austria
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/008
EU/1/03/271/018
9. FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 2 de marzo de 2004 Fecha de la última renovación: 2 de marzo de 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
1. NOMBRE DEL MEDICAMENTO
ADVATE 1000 UI polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 1000 UI de factor VIII humano de coagulación (ADNr), octocog alfa. Después de la reconstitución, ADVATE contiene aproximadamente 500 UI por ml de factor VIII humano de coagulación (ADNr), octocog alfa.
La potencia (Unidades Internacionales) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de ADVATE es aproximadamente 4.000 -10.000 UI/mg/proteína.
El octocog alfa (factor VIII humano de coagulación (ADNr)) es una proteína purificada con 2332 aminoácidos. Está producida por tecnología de ADN recombinante en células de ovario de hámster chino (CHO). Preparado sin la adición de ninguna proteína (exógena) de origen humano o animal en el proceso del cultivo celular, purificación o formulación final.
Excipientes con efectos conocidos:
0,45 mmol de sodio (10 mg) por vial.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: polvo friable de color blanco a blanquecino. Disolvente: solución transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII). ADVATE está indicado en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia y con apoyo de resucitación disponible inmediatamente en caso de anafilaxia.
Posología
La dosis y duración de la terapia de sustitución depende de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII se expresa en Unidades Internacionales (UI), que se relacionan con el estándar de la OMS para medicamentos con factor VIII. La actividad de Factor VIII en plasma se expresa bien como porcentaje (referido a plasma humano normal) o en UI (referidas al Estándar Internacional de factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII es equivalente a la cantidad de factor VIII existente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis de factor VIII requerida se basa en el hallazgo empírico de que 1 UI de factor VIII por kg de peso corporal incrementa la actividad de factor VIII del plasma en 2 UI/dl. La dosis requerida se determina mediante la siguiente fórmula:
Unidades requeridas (UI) = peso corporal (kg) x aumento de factor VIII deseado (%) x 0,5
En el caso de los episodios hemorrágicos siguientes, la actividad de factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. En cirugía y en los episodios hemorrágicos, puede utilizarse la siguiente tabla 1 como guía de dosificación:
|
Tabla 1 Guía de dosificación en episodios hemorrágicos y cirugía | ||
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (% o UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente o hemorragia muscular u oral. |
20 - 40 |
Repetir la inyección cada 12 a 24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma. |
30 - 60 |
Repetir la inyección cada 12-24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) durante 3 a 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir la inyección cada 8 a 24 horas (cada 6 a 12 horas en los pacientes menores de 6 años) hasta superar el peligro. |
|
Cirugía | ||
|
Menor Incluyendo extracción dental. |
30 - 60 |
Cada 24 horas (cada 12 a 24 horas en los pacientes menores de 6 años), al menos 1 día, hasta lograr la curación. |
|
Mayor |
80 - 100 (pre y postoperatorio) |
Repetir la inyección cada 8-24 horas (cada 6 a 24 horas en los pacientes menores de 6 años) hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor VIII del 30% al 60% (UI/dl). |
La dosis y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual. En determinadas circunstancias (ej.: presencia de un título bajo de inhibidor) pueden ser necesarias dosis mayores a las calculadas mediante la fórmula.
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis que se debe administrar y la frecuencia con la que se debe repetir la inyección. En el caso especial de las intervenciones de cirugía mayor, es indispensable controlar con precisión el tratamiento de sustitución mediante pruebas de la coagulación (actividad del factor VIII plasmático). La respuesta individual de cada paciente frente al factor VIII puede variar, alcanzar distintos niveles de recuperación in vivo y presentar semividas diferentes.
Profilaxis
Para la profilaxis de larga duración frente a hemorragias en pacientes con hemofilia A grave, las dosis normales son de 20 a 40 UI de factor VIII por kg de peso corporal a intervalos de 2 a 3 días.
Población _ pediátrica
La dosis de tratamiento a demanda en pacientes pediátricos (0 a 18 años de edad) no difiere de la de los pacientes adultos. En pacientes menores de 6 años, se recomiendan dosis de 20 a 50 UI de factor VIII por kg de peso corporal de 3 a 4 veces a la semana como terapia profiláctica.
No se ha descrito el uso de las presentaciones de 2 ml para sujetos pediátricos de menos de 2 años.
Forma de administración
ADVATE se debe administrar por vía intravenosa. En caso de ser administrado por un profesional no sanitario, es necesario realizar un entrenamiento adecuado.
Se debe determinar la velocidad de administración para asegurar la comodidad del paciente, hasta un máximo de 10 ml/min.
Después de la reconstitución, la solución es transparente, incolora y libre de partículas extrañas y tiene un pH de entre 6,7 y 7,3.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1 o a las proteínas de ratón o hámster.
4.4 Advertencias y precauciones especiales de empleo
Hipersensibilidad
Con ADVATE se han notificado reacciones de hipersensibilidad de tipo alérgico, incluyendo anafilaxis. El medicamento contiene trazas de proteínas de ratón y de hámster. Se debe informar a los pacientes de que en caso de que ocurran síntomas de hipersensibilidad, deben interrumpir el tratamiento y consultar con su médico de forma inmediata. Se debe informar a los pacientes de los signos de las reacciones de hipersensibilidad de tipo inmediato incluyendo habón urticarial, urticaria generalizada, tirantez en el pecho, sibilancia, hipotensión y anafilaxia.
En caso de que se produzca un shock, se deben seguir las pautas médicas estándar para su tratamiento.
Debido a la disminución del volumen de inyección de ADVATE reconstituido en 2 ml de agua esterilizada para preparaciones inyectables, si se producen reacciones de hipersensibilidad hay menos tiempo para reaccionar deteniendo la inyección. Por tanto, se aconseja tener cuidado durante la inyección de ADVATE reconstituido en 2 ml de agua esterilizada para preparaciones inyectables, especialmente en niños.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) frente al factor VIII es una complicación conocida en el tratamiento de individuos con hemofilia A. Estos inhibidores son habitualmente inmunoglobulinas IgGs dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en unidades Bethesda (UB) por ml de plasma empleando el ensayo modificado. En pacientes que desarrollan anticuerpos neutralizantes (inhibidores) al Factor VIII, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia. El riesgo de desarrollar inhibidores se relaciona con el grado de exposición al factor
VIII, siendo mayor durante los primeros 20 días de exposición, y con otros factores ambientales y genéticos. Raramente se desarrollan inhibidores después de los 100 primeros días de exposición.
Se han observado casos recurrentes de aparición de inhibidores (a títulos bajos), después de cambiar de un factor VIII a otro en los pacientes con un tratamiento previo de más de 100 días de exposición y con antecedentes de desarrollo de inhibidores. Así, se recomienda monitorizar cuidadosamente la aparición de inhibidores en todos los pacientes después de cualquier cambio de producto.
En general, en todos los pacientes tratados con un factor VIII de coagulación se debe monitorizar cuidadosamente la aparición de inhibidores mediante la realización de observaciones clínicas y de las pruebas de laboratorio apropiadas. Si no se obtienen los niveles de actividad de factor VIII en plasma esperados, o si no se controla la hemorragia con una dosis adecuada, se debe realizar una prueba de detección de inhibidor de factor VIII. En pacientes con niveles altos de inhibidor, la terapia de sustitución de factor VIII puede no ser efectiva y se deben considerar otras opciones terapéuticas.
El tratamiento de tales pacientes debe estar dirigido por médicos con experiencia en el cuidado de pacientes con hemofilia e inhibidores del factor VIII.
Aplicación incorrecta de ADVATE
Para ADVATE reconstituido con 2 ml de agua esterilizada para preparaciones inyectables, la aplicación incorrecta (vía intraarterial o paravenosa) puede producir reacciones en el lugar de inyección leves y a corto plazo, como cardenales y eritema.
Complicación relacionada con catéter en el tratamiento
Si se requiere un dispositivo de acceso venoso (CVAD) debe considerarse el riesgo de complicaciones relacionadas con CVAD incluyendo infecciones locales, bacteremia y trombosis del lado del catéter.
Consideraciones relativas al excipiente
Después de la reconstitución, este medicamento contiene 0,45 mmol de sodio (10 mg) por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
Se recomienda encarecidamente registrar el nombre y el número de lote del producto cada vez que se administre ADVATE a un paciente para poder mantener una relación entre el paciente y el lote del medicamento.
Población _ pediátrica:
Las advertencias y precauciones incluidas se aplican tanto a adultos como a niños.
4.5 Interacción con otros medicamentos y otras formas de interacción.
No se han realizado estudios de interacciones con ADVATE.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción en animales con factor VIII. Dados los raros casos de hemofilia A en mujeres, no se dispone de experiencia relacionada con el uso de factor VIII durante el embarazo y el periodo de lactancia. Por lo tanto, sólo debe utilizarse factor VIII durante el embarazo y el periodo de lactancia si está claramente indicado.
4.7 Efectos sobre la capacidad de conducir y utilizar máquinas
La influencia de ADVATE sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del_perfil de seguridad
Los ensayos clínicos con ADVATE incluyeron 418 sujetos con al menos una exposición a ADVATE y registraron un total de 93 reacciones adversas al medicamento (RAM). Las RAM que aparecieron con mayor frecuencia fueron el desarrollo de anticuerpos neutralizantes frente al factor VIII (inhibidores), cefalea y fiebre.
La hipersensibilidad y las reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el lugar de infusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habón urticarial, hipotensión, letargia, náuseas, inquietud, taquicardia, tirantez en el pecho, cosquilleo, vómitos, sibilancia) se observaron con poca frecuencia pero, en algunos casos, pueden progresar a anafilaxia grave (incluyendo shock).
Se puede observar desarrollo de anticuerpos a la proteína de ratón y/o hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) frente al factor VIII. Si aparecen tales inhibidores, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia.
Resumen tabulado de reacciones adversas
La siguiente tabla 2 indica la frecuencia de las reacciones adversas en los ensayos clínicos y procedente de informes espontáneos. La tabla sigue la clasificación de órganos del sistema MedDRA (COS y Nivel de término preferido).
Las frecuencias se han clasificado utilizando el siguiente criterio: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100), raros (> 1/10.000 a < 1/1.000), muy raros (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de __informes espontáneos__
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Infecciones e infestaciones |
Gripe |
Poco frecuentes |
|
Laringitis |
Poco frecuentes | |
|
Linfangitis |
Poco frecuentes | |
|
Trastornos de la sangre y |
Inhibición del factor VIII |
Frecuentes |
|
del sistema linfático |
Linfangitis |
Poco frecuentes |
|
Trastornos del sistema |
Reacción anafiláctica |
Frecuencia no conocida |
|
inmunológico |
Hipersensibilidad0 |
Frecuencia no conocida |
|
Trastornos del sistema |
Dolor de cabeza |
Frecuentes |
|
nervioso |
Mareos |
Poco frecuentes |
|
Problemas de memoria |
Poco frecuentes | |
|
Síncope |
Poco frecuentes | |
|
Temblores |
Poco frecuentes | |
|
Migraña |
Poco frecuentes | |
|
Disgeusia |
Poco frecuentes | |
|
Trastornos oculares |
Inflamación ocular |
Poco frecuentes |
|
Trastornos cardiacos |
Palpitaciones |
Poco frecuentes |
|
Trastornos vasculares |
Hematoma |
Poco frecuentes |
|
Sofocos |
Poco frecuentes | |
|
Palidez |
Poco frecuentes | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
Poco frecuentes |
|
Trastornos |
Diarrea |
Poco frecuentes |
|
gastrointestinales |
Dolor en el abdomen superior |
Poco frecuentes |
|
Nauseas |
Poco frecuentes | |
|
Vómitos |
Poco frecuentes |
|
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de informes espontáneos | ||
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Trastornos de la piel y del tejido subcutáneo |
Prurito |
Poco frecuentes |
|
Sarpullido |
Poco frecuentes | |
|
Hiperhidrosis |
Poco frecuentes | |
|
Urticaria |
Poco frecuentes | |
|
Trastornos generales y alteraciones en el lugar de administración |
Fiebre |
Frecuentes |
|
Edema periférico |
Poco frecuentes | |
|
Dolor torácico Malestar torácico |
Poco frecuentes Poco frecuentes | |
|
Escalofríos |
Poco frecuentes | |
|
Sensación anormal Hematoma en el lugar de punción de un vaso sanguíneo |
Poco frecuentes Poco frecuentes | |
|
Fatiga Reacción en la zona de inyección |
Frecuencia no conocida Frecuencia no conocida | |
|
Malestar general |
Frecuencia no conocida | |
|
Exploraciones complementarias | ||
|
Recuento elevado de monocitos |
Poco frecuentes | |
|
Nivel reducido de factor de coagulación VIIIb |
Poco frecuentes | |
|
Descenso del hematocrito |
Poco frecuentes | |
|
Test de laboratorio anormal |
Poco frecuentes | |
|
Lesiones, intoxicaciones y complicaciones de procedimientos terapéuticos |
Complicaciones tras el procedimiento |
Poco frecuentes |
|
Hemorragia tras el procedimiento |
Poco frecuentes | |
|
Reacción en el lugar del procedimiento |
Poco frecuentes | |
a) Calculado según un número total único de pacientes que recibieron ADVATE (418)
b) El descenso inesperado de los niveles de factor VIII de coagulación se produjo en un paciente durante la perfusión continua de ADVATE después de cirugía (días 10-14 de postoperatorio).
Se mantuvo la hemostasis durante todo este periodo y tanto los niveles de factor VIII de plasma como los niveles de aclaramiento volvieron a los niveles adecuados el día 15 de postoperatorio. Los ensayos de inhibidores del factor VIII realizados tras la finalización de la perfusión continua y la terminación del estudio füeron negativos.
c) RAM explicadas en la sección siguiente.
Descripción de reacciones adversas seleccionadas Desarrollo de inhibidor
Se ha notificado un desarrollo de inhibidor en pacientes con tratamientos previos y en pacientes no tratados previamente. Para información detallada ver secciones 5.1 (Propiedades farmacológicas) y 4.4 (Advertencias y precauciones especiales de empleo).
RAM específicas para residuos del proceso de fabricación
De los 229 pacientes en tratamiento en los que se evaluaron los anticuerpos antiproteínas de células de ovario de hámster chino (CHO), 3 mostraron una tendencia ascendente estadísticamente significativa en los títulos, 4 mostraron picos sostenidos o picos momentáneos y un paciente mostró ambos, pero no tuvo ningún otro síntoma clínico. De los 229 pacientes tratados a los que se realizó una evaluación de los anticuerpos anti-IgG murina, 10 mostraron una tendencia ascendente, dos mostraron un pico sostenido o pico momentáneo y un paciente mostró ambos. Cuatro de estos pacientes sufrieron episodios aislados de urticaria, prurito, sarpullido y recuentos ligeramente elevados de eosinófilos entre exposiciones repetidas al medicamento del estudio.
Hipersensibilidad
Reacciones de tipo alérgico incluyen anafilaxis y se han manifestado como mareos, parestesias, erupción, soplado, tumefacción facial, urticaria y prurito.
Población pediátrica
En los estudios clínicos no se han notado diferencias específicas por edad en RAM excepto el desarrollo de inhibidores en pacientes pediátricos no tratados previamente (PUP) y complicaciones relacionadas con catéter.
Notificación de sospechas de reacciones adversas
. Se invita a los sistema nacional
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento profesionales sanitarios a notificar las sospechas de reacciones adversas a través del de notificación pertinente incluido en el Anexo V.
4.9 Sobredosis
No se han notificado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos: factor VIII de coagulación. Código ATC: B02BD02.
El complejo factor VIII /Factor von Willebrand consta de dos moléculas (factor VIII y factor von Willebrand) con diferentes funciones fisiológicas. ADVATE contiene factor VIII de coagulación recombinante (octocog alfa), una glicoproteína que es biológicamente equivalente a la glicoproteína de factor VIII del plasma humano.
Octocog alfa es una glicoproteína que consta de 2332 aminoácidos con un peso molecular aproximado de 280 kD. Cuando se perfunde en un paciente hemofílico, octocog alfa se enlaza al Factor von Willebrand endógeno en la circulación del paciente. El factor VIII activado actúa como un Cofactor para el Factor IX activado, acelerando la conversión del Factor X a Factor X activado. El Factor X activado convierte la protrombina en trombina. La trombina convierte el fibrinógeno en fibrina, con la que se puede formar el coágulo. La hemofilia A es una alteración hereditaria de la coagulación sanguínea ligada al sexo y producida por la disminución de los niveles de factor VIII que da como resultado una hemorragia profusa en las articulaciones, músculos u órganos internos, bien de forma espontánea o bien como resultado de un trauma accidental o quirúrgico. Mediante terapia de sustitución, los niveles plasmáticos de factor VIII aumentan, consiguiendo una corrección temporal de la deficiencia de factor VIII y una corrección de las tendencias hemorrágicas.
Desarrollo del inhibidor
Se evaluó la inmunogenicidad de ADVATE en pacientes con tratamientos previos. Durante los ensayos clínicos con ADVATE en 233 pacientes pediátricos y adultos [pacientes pediátricos (0 a 16 años de edad) y pacientes adultos (más de 16 años de edad)] diagnosticados con hemofilia A grave (factor VIII < 1%), con exposición previa a concentrados de factor VIII durante un periodo igual o superior a 150 días para adultos y niños mayores e igual o superior a 50 días para niños menores de 6 años, un paciente desarrolló un título bajo de inhibidor (2,4 BU en el ensayo Bethesda modificado) tras 26 días de exposición a ADVATE. Las pruebas de seguimiento de inhibidores en este paciente tras abandonar el ensayo fueron negativas. En todos los estudios, la exposición media a ADVATE fue de 97,0 días de exposición por sujeto (intervalo 1 a 709) para pacientes previamente tratados. La incidencia global de cualquier desarrollo de inhibidor del factor VIII (alto o bajo) fue de 0,4% (1 de 233).
En el ensayo completado no controlado 060103, 16 de los 45 (35,6%) pacientes sin tratamiento previo con hemofilia A grave (FVIII < 1%) con al menos 25 días de exposición a FVIII desarrollaron inhibidores del FVIII: 7 (15,6%) sujetos desarrollaron inhibidores de título alto y 9 (20%) de los sujetos desarrollaron inhibidores de título bajo, 1 de los cuales también se clasificó como inhibidor temporal.
Los factores de riesgo relacionados con el desarrollo del inhibidor en este ensayo incluyeron etnicidad no caucásica, antecedentes familiares de inhibidores y tratamiento intensivo a dosis altas en los primeros 20 días de exposición. En los 20 sujetos que no tenían ninguno de estos factores de riesgo no se produjo el desarrollo del inhibidor.
Se han recogido datos sobre Inducción de tolerancia inmunitaria (ITI) en pacientes con inhibidores.
En un subestudio del estudio PUP 060103, se documentaron tratamientos ITI en 11 PUP. Se hizo un análisis de cuadro retrospective de 30 materias sobre ITI (estudio 060703) y recogida de datos de registro está en curso.
En el estudio 060201, se compararon dos esquemas de tratamiento de profilaxis a largo plazo en 53 sujetos previamente tratados: un régimen individualizado de dosificación guiada farmacocinética (dentro de un rango de 20 a 80 UI de factor VIII por kg de peso corporal a intervalos de 72 ± 6 horas, n = 23) con un régimen de dosificación profiláctica estándar (de 20 a 40 UI/kg cada 48 ± 6 horas, n = 30). El régimen de dosificación guiada farmacocinética (de acuerdo con una fórmula específica) tenía como objetivo mantener el factor VIII en niveles > 1% en el intervalo entre dosis de 72 horas. Los datos de este estudio demuestran que los dos regímenes de dosificación profiláctica son comparables en lo que se refiere a la reducción de la frecuencia de hemorragias.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con ADVATE en los diferentes grupos de la población pediátrica con hemofilia A (deficiencia congénita de factor VIII) en “Inducción de tolerancia inmunitaria (ITI) en pacientes con hemofilia A (deficiencia congénita de factor VIII) que han desarrollado inhibidores al factor VIII” y “tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII)“ (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Todos los estudios farmacocinéticos con ADVATE se realizaron en pacientes previamente tratados con hemofilia A grave o moderadamente grave (valor basal del factor VIII < 2%). El análisis de las muestras de plasma se realizó en un laboratorio central utilizando el ensayo de coagulación en una etapa.
Un total de 195 sujetos con hemofilia A grave (valor basal del factor VIII < 1%) proporcionaron parámetros PK que se incluyeron en el conjunto de análisis de PK según el protocolo. Las categorías de estos análisis para lactantes (1 mes a <2 años), niños (2 a <5 años), niños de mayor edad (5 a <12 años), adolescentes (12 a <18 años) y adultos (18 años y mayores) se utilizaron para resumir los parámetros PK, definiéndose la edad como la edad en el momento de la perfusión de PK.
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Área bajo la curva total (UI*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Recuperación incremental ajustada a Cmáx (UI/dl por UI/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Semivida (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Concentración máxima en plasma tras la perfusión (UI/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Tiempo de residencia medio (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Volumen de distribución en estado estacionario (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Aclaramiento (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Calculada como (Cmáx - valor basal del factor VIII) dividida por la dosis en UI/kg, donde Cmáx es la medida máxima de factor VIII tras la perfusión.
La seguridad y la eficacia hemostática de ADVATE en la población pediátrica son similares a las de los pacientes adultos. La recuperación y la semivida terminal (t/2) ajustadas fue aproximadamente un 20% inferior en niños pequeños (menores de 6 años) que en adultos, lo que lo que puede deberse en parte a un volumen mayor conocido de plasma por kg de peso corporal en los pacientes más jóvenes.
No se dispone de datos farmacocinéticos de ADVATE en pacientes sin tratamiento previo.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de la seguridad, toxicología aguda, toxicidad en dosis repetida, toxicidad local y genotoxicidad.
Un estudio de tolerancia local con conejos demostró que ADVATE reconstituido con 2 ml de agua esterilizada para preparaciones inyectables se tolera bien tras una administración intravenosa.
Se observó un leve enrojecimiento temporal en el lugar de la administración después de la aplicación intraarterial y después de la aplicación paravenosa. No obstante, no se observaron cambios histopatológicos adversos correlacionados que indiquen una naturaleza temporal de esta observación.
DATOS FARMACÉUTICOS
6.
6.1 Lista de excipientes
Polvo
Manitol
Cloruro de sodio Histidina Trealosa Cloruro de calcio Trometamol Polisorbato 80 Glutatión (reducido)
Disolvente
Agua esterilizada para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
Después de la reconstitución, desde un punto de vista microbiológico, el medicamento debe utilizarse de inmediato. No obstante, se ha demostrado una estabilidad física y química en uso durante 3 horas a 25 °C.
Durante el periodo de validez, el medicamento puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único no superior a 6 meses. Se debe anotar la fecha del final del periodo de conservación a temperatura ambiente de 6 meses en el embalaje del medicamento. El medicamento no puede refrigerarse de nuevo.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
ADVATE con el equipo BAXJECT II: Conservar el vial en el embalaje exterior para protegerlo de la luz.
ADVATE en el sistema BAXJECT III: Conservar el blister sellado en el embalaje exterior para protegerlo de la luz.
Para consultar las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Tanto el vial de polvo como el vial de disolvente de 2 ml son de vidrio tipo I cerrado con tapones de caucho de clorobutilo. El medicamento se proporciona con una de las configuraciones siguientes:
- ADVATE con el equipo BAXJECT II: Cada envase contiene un vial de polvo, un vial de disolvente de 2 ml y un equipo para reconstitución (BAXJECT II).
- ADVATE en el sistema BAXJECT III: Cada envase contiene un sistema BAXJECT III listo para usar en un blister sellado (el vial de polvo y el vial de disolvente de 2 ml están precargados con el sistema para su reconstitución).
6.6 Precauciones especiales de eliminación y otras manipulaciones
ADVATE debe administrarse intravenosamente después de la reconstitución del producto.
La solución reconstituida se debe inspeccionar visualmente para comprobar que no presenta partículas
extrañas y/o descoloración.
Después de la reconstitución la solución debe ser transparente, incolora y no presentar partículas
extrañas.
No utilizar soluciones turbias o con depósitos.
- Para la administración se requiere el uso de una jeringa luer-lock.
- Utilizar dentro de las tres horas después de la reconstitución.
- No refrigerar la preparación después de la reconstitución.
- La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Reconstitución con el equipo BAXJECT II:
- Para la reconstitución utilizar sólo el agua esterilizada para preparaciones inyectables y el equipo de reconstitución incluido en el envase.
- No utilizar si el equipo BAXJECT II, el sistema estéril de protección o su envase está dañado o muestra algún signo de deterioro.
- Se debe utilizar una técnica aséptica:
1. Si el medicamento se encuentra en la nevera, coger de la nevera los viales de polvo y de disolvente de ADVATE y dejarlos a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Quitar los protectores de los viales de polvo y disolvente.
4. Limpiar los tapones con las toallitas impregnadas de alcohol. Colocar los viales en una superficie plana y limpia.
5. Abrir el envoltorio del equipo BAXJECT II quitando la tapa de papel sin tocar el interior (Fig. a). No sacar el equipo del envoltorio. No utilizar si el equipo BAXJECT II, su sistema de barrera estéril o su acondicionamiento están dañados o muestran algún signo de deterioro.
6. Dar la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente. Coger el envoltorio por su extremo y sacar el equipo BAXJECT II de su envoltorio (Fig. b). No quitar el protector azul del equipo BAXJECT II.
7. Para la reconstitución, utilizar solamente el agua esterilizada para preparaciones inyectables y el equipo para reconstitución incluidos en el envase. Con el equipo BAXJECT II unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico blanca dentro del tapón del vial de polvo ADVATE. El vacío hará que el disolvente penetre en el vial de polvo ADVATE (Fig. c).
8. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará
a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.

Reconstitución con el sistema BAXJECT III:
- No lo utilice si la tapa del blister no está perfectamente sellada.
1. Si el medicamento se encuentra en la nevera, coger de la nevera el blister sellado (contiene viales de polvo y disolvente precargados con el sistema para su reconstitución) y dejarlo a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Abrir el envoltorio de ADVATE quitando la tapa. Extraer el sistema BAXJECT III del blister.
4. Colocar ADVATE en una superficie plana con el vial de disolvente encima (Fig. 1). El vial de disolvente tiene una raya azul. No quitar el protector azul hasta que se le indique más adelante.
5. Mientras sujeta ADVATE con una mano en el sistema BAXJECT III, presionar con fuerza el vial de disolvente con la otra mano hasta que el sistema quede totalmente contraído y el disolvente entre dentro del vial de ADVATE (Fig. 2). No inclinar el sistema hasta que haya finalizado la transferencia.
6. Comprobar que ha finalizado la transferencia del disolvente. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig- 1

Fig. 3

Fig. 2
Administración:
Utilizar técnica aséptica:
Los medicamentos para administración parenteral deben inspeccionarse visualmente antes de su administración para verificar la ausencia de partículas, cuando la solución y el envase lo permitan. Sólo se debe utilizar una solución transparente y sin coloración.
1. Quitar el protector azul del equipo BAXJECT II / al sistema BAXJECT III. No introducir aire en la jeringa. Conectar la jeringa al equipo BAXJECT II / al sistema BAXJECT III.
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente.
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar intravenosamente. La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de ADVATE. Si se observa una elevación significativa del pulso, reducir la velocidad de administración o interrumpir temporalmente la inyección suele hacer que los síntomas normalmente desaparezcan pronto (ver secciones 4.4 y 4.8).
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG Industriestrasse 67 A-1221 Viena Austria
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/009
EU/1/03/271/019
9. FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 2 de marzo de 2004 Fecha de la última renovación: 2 de marzo de 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
1. NOMBRE DEL MEDICAMENTO
ADVATE 1500 UI polvo y disolvente para solución inyectable.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene nominalmente 1500 UI de factor VIII humano de coagulación (ADNr), octocog alfa. Después de la reconstitución, ADVATE contiene aproximadamente 750 UI por ml de factor VIII humano de coagulación (ADNr), octocog alfa.
La potencia (Unidades Internacionales) se determina utilizando el ensayo cromogénico de la Farmacopea Europea. La actividad específica de ADVATE es aproximadamente 4.000 -10.000 UI/mg/proteína.
El octocog alfa (factor VIII humano de coagulación (ADNr)) es una proteína purificada con 2332 aminoácidos. Está producida por tecnología de ADN recombinante en células de ovario de hámster chino (CHO). Preparado sin la adición de ninguna proteína (exógena) de origen humano o animal en el proceso del cultivo celular, purificación o formulación final.
Excipientes con efectos conocidos:
0,45 mmol de sodio (10 mg) por vial.
Para consultar la lista completa de excipientes ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo y disolvente para solución inyectable.
Polvo: polvo friable de color blanco a blanquecino. Disolvente: solución transparente e incolora.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII). ADVATE está indicado en todos los grupos de edad.
4.2 Posología y forma de administración
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia y con apoyo de resucitación disponible inmediatamente en caso de anafilaxia.
Posología
La dosis y duración de la terapia de sustitución depende de la gravedad de la deficiencia de factor VIII, de la localización y extensión de la hemorragia y del estado clínico del paciente.
El número de unidades de factor VIII se expresa en Unidades Internacionales (UI), que se relacionan con el estándar de la OMS para medicamentos con factor VIII. La actividad de Factor VIII en plasma se expresa bien como porcentaje (referido a plasma humano normal) o en UI (referidas al Estándar Internacional de factor VIII en plasma).
Una Unidad Internacional (UI) de actividad de factor VIII es equivalente a la cantidad de factor VIII existente en un ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis de factor VIII requerida se basa en el hallazgo empírico de que 1 UI de factor VIII por kg de peso corporal incrementa la actividad de factor VIII del plasma en 2 UI/dl. La dosis requerida se determina mediante la siguiente fórmula:
Unidades requeridas (UI) = peso corporal (kg) x aumento de factor VIII deseado (%) x 0,5
En el caso de los episodios hemorrágicos siguientes, la actividad de factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. En cirugía y en los episodios hemorrágicos, puede utilizarse la siguiente tabla 1 como guía de dosificación:
|
Tabla 1 Guía de dosificación en episodios hemorrágicos y cirugía | ||
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (% o UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente o hemorragia muscular u oral. |
20 - 40 |
Repetir la inyección cada 12 a 24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma. |
30 - 60 |
Repetir la inyección cada 12-24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) durante 3 a 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir la inyección cada 8 a 24 horas (cada 6 a 12 horas en los pacientes menores de 6 años) hasta superar el peligro. |
|
Cirugía | ||
|
Menor Incluyendo extracción dental. |
30 - 60 |
Cada 24 horas (cada 12 a 24 horas en los pacientes menores de 6 años), al menos 1 día, hasta lograr la curación. |
|
Mayor |
80 - 100 (pre y postoperatorio) |
Repetir la inyección cada 8-24 horas (cada 6 a 24 horas en los pacientes menores de 6 años) hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor VIII del 30% al 60% (UI/dl). |
La dosis y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual. En determinadas circunstancias (ej.: presencia de un título bajo de inhibidor) pueden ser necesarias dosis mayores a las calculadas mediante la fórmula.
Durante el tratamiento se recomienda controlar el nivel de factor VIII para determinar la dosis que se debe administrar y la frecuencia con la que se debe repetir la inyección. En el caso especial de las intervenciones de cirugía mayor, es indispensable controlar con precisión el tratamiento de sustitución mediante pruebas de la coagulación (actividad del factor VIII plasmático). La respuesta individual de cada paciente frente al factor VIII puede variar, alcanzar distintos niveles de recuperación in vivo y presentar semividas diferentes.
Profilaxis
Para la profilaxis de larga duración frente a hemorragias en pacientes con hemofilia A grave, las dosis normales son de 20 a 40 UI de factor VIII por kg de peso corporal a intervalos de 2 a 3 días.
Población _ pediátrica
La dosis de tratamiento a demanda en pacientes pediátricos (0 a 18 años de edad) no difiere de la de los pacientes adultos. En pacientes menores de 6 años, se recomiendan dosis de 20 a 50 UI de factor VIII por kg de peso corporal de 3 a 4 veces a la semana como terapia profiláctica.
No se ha descrito el uso de las presentaciones de 2 ml para sujetos pediátricos de menos de 2 años.
Forma de administración
ADVATE se debe administrar por vía intravenosa. En caso de ser administrado por un profesional no sanitario, es necesario realizar un entrenamiento adecuado.
Se debe determinar la velocidad de administración para asegurar la comodidad del paciente, hasta un máximo de 10 ml/min.
Después de la reconstitución, la solución es transparente, incolora y libre de partículas extrañas y tiene un pH de entre 6,7 y 7,3.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección 6.6.
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1 o a las proteínas de ratón o hámster.
4.4 Advertencias y precauciones especiales de empleo
Hipersensibilidad
Con ADVATE se han notificado reacciones de hipersensibilidad de tipo alérgico, incluyendo anafilaxis. El medicamento contiene trazas de proteínas de ratón y de hámster. Se debe informar a los pacientes de que en caso de que ocurran síntomas de hipersensibilidad, deben interrumpir el tratamiento y consultar con su médico de forma inmediata. Se debe informar a los pacientes de los signos de las reacciones de hipersensibilidad de tipo inmediato incluyendo habón urticarial, urticaria generalizada, tirantez en el pecho, sibilancia, hipotensión y anafilaxia.
En caso de que se produzca un shock, se deben seguir las pautas médicas estándar para su tratamiento.
Debido a la disminución del volumen de inyección de ADVATE reconstituido en 2 ml de agua esterilizada para preparaciones inyectables, si se producen reacciones de hipersensibilidad hay menos tiempo para reaccionar deteniendo la inyección. Por tanto, se aconseja tener cuidado durante la inyección de ADVATE reconstituido en 2 ml de agua esterilizada para preparaciones inyectables, especialmente en niños.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) frente al factor VIII es una complicación conocida en el tratamiento de individuos con hemofilia A. Estos inhibidores son habitualmente inmunoglobulinas IgGs dirigidas contra la actividad procoagulante del factor VIII, que se cuantifican en unidades Bethesda (UB) por ml de plasma empleando el ensayo modificado. En pacientes que desarrollan anticuerpos neutralizantes (inhibidores) al Factor VIII, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia. El riesgo de desarrollar inhibidores se relaciona con el grado de exposición al factor
VIII, siendo mayor durante los primeros 20 días de exposición, y con otros factores ambientales y genéticos. Raramente se desarrollan inhibidores después de los 100 primeros días de exposición.
Se han observado casos recurrentes de aparición de inhibidores (a títulos bajos), después de cambiar de un factor VIII a otro en los pacientes con un tratamiento previo de más de 100 días de exposición y con antecedentes de desarrollo de inhibidores. Así, se recomienda monitorizar cuidadosamente la aparición de inhibidores en todos los pacientes después de cualquier cambio de producto.
En general, en todos los pacientes tratados con un factor VIII de coagulación se debe monitorizar cuidadosamente la aparición de inhibidores mediante la realización de observaciones clínicas y de las pruebas de laboratorio apropiadas. Si no se obtienen los niveles de actividad de factor VIII en plasma esperados, o si no se controla la hemorragia con una dosis adecuada, se debe realizar una prueba de detección de inhibidor de factor VIII. En pacientes con niveles altos de inhibidor, la terapia de sustitución de factor VIII puede no ser efectiva y se deben considerar otras opciones terapéuticas.
El tratamiento de tales pacientes debe estar dirigido por médicos con experiencia en el cuidado de pacientes con hemofilia e inhibidores del factor VIII.
Aplicación incorrecta de ADVATE
Para ADVATE reconstituido con 2 ml de agua esterilizada para preparaciones inyectables, la aplicación incorrecta (vía intraarterial o paravenosa) puede producir reacciones en el lugar de inyección leves y a corto plazo, como cardenales y eritema.
Complicación relacionada con catéter en el tratamiento
Si se requiere un dispositivo de acceso venoso (CVAD) debe considerarse el riesgo de complicaciones relacionadas con CVAD incluyendo infecciones locales, bacteremia y trombosis del lado del catéter.
Consideraciones relativas al excipiente
Después de la reconstitución, este medicamento contiene 0,45 mmol de sodio (10 mg) por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
Se recomienda encarecidamente registrar el nombre y el número de lote del producto cada vez que se administre ADVATE a un paciente para poder mantener una relación entre el paciente y el lote del medicamento.
Población _ pediátrica:
Las advertencias y precauciones incluidas se aplican tanto a adultos como a niños.
4.5 Interacción con otros medicamentos y otras formas de interacción.
No se han realizado estudios de interacciones con ADVATE.
4.6 Fertilidad, embarazo y lactancia
No se han realizado estudios de reproducción en animales con factor VIII. Dados los raros casos de hemofilia A en mujeres, no se dispone de experiencia relacionada con el uso de factor VIII durante el embarazo y el periodo de lactancia. Por lo tanto, sólo debe utilizarse factor VIII durante el embarazo y el periodo de lactancia si está claramente indicado.
4.7 Efectos sobre la capacidad de conducir y utilizar máquinas
La influencia de ADVATE sobre la capacidad para conducir y utilizar máquinas es nula.
4.8 Reacciones adversas
Resumen del_perfil de seguridad
Los ensayos clínicos con ADVATE incluyeron 418 sujetos con al menos una exposición a ADVATE y registraron un total de 93 reacciones adversas al medicamento (RAM). Las RAM que aparecieron con mayor frecuencia fueron el desarrollo de anticuerpos neutralizantes frente al factor VIII (inhibidores), cefalea y fiebre.
La hipersensibilidad y las reacciones alérgicas (que pueden incluir angioedema, escozor y punzadas en el lugar de infusión, escalofríos, rubefacción, urticaria generalizada, cefalea, habón urticarial, hipotensión, letargia, náuseas, inquietud, taquicardia, tirantez en el pecho, cosquilleo, vómitos, sibilancia) se observaron con poca frecuencia pero, en algunos casos, pueden progresar a anafilaxia grave (incluyendo shock).
Se puede observar desarrollo de anticuerpos a la proteína de ratón y/o hámster con reacciones de hipersensibilidad asociadas.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) frente al factor VIII. Si aparecen tales inhibidores, esta condición se manifestará con una respuesta clínica insuficiente. En esos casos, se recomienda contactar con un centro especializado en hemofilia.
Resumen tabulado de reacciones adversas
La siguiente tabla 2 indica la frecuencia de las reacciones adversas en los ensayos clínicos y procedente de informes espontáneos. La tabla sigue la clasificación de órganos del sistema MedDRA (COS y Nivel de término preferido).
Las frecuencias se han clasificado utilizando el siguiente criterio: muy frecuentes (> 1/10), frecuentes (> 1/100 a < 1/10), poco frecuentes (> 1/1.000 a < 1/100), raros (> 1/10.000 a < 1/1.000), muy raros (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de __informes espontáneos__
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Infecciones e infestaciones |
Gripe |
Poco frecuentes |
|
Laringitis |
Poco frecuentes | |
|
Linfangitis |
Poco frecuentes | |
|
Trastornos de la sangre y |
Inhibición del factor VIII |
Frecuentes |
|
del sistema linfático |
Linfangitis |
Poco frecuentes |
|
Trastornos del sistema |
Reacción anafiláctica |
Frecuencia no conocida |
|
inmunológico |
Hipersensibilidad0 |
Frecuencia no conocida |
|
Trastornos del sistema |
Dolor de cabeza |
Frecuentes |
|
nervioso |
Mareos |
Poco frecuentes |
|
Problemas de memoria |
Poco frecuentes | |
|
Síncope |
Poco frecuentes | |
|
Temblores |
Poco frecuentes | |
|
Migraña |
Poco frecuentes | |
|
Disgeusia |
Poco frecuentes | |
|
Trastornos oculares |
Inflamación ocular |
Poco frecuentes |
|
Trastornos cardiacos |
Palpitaciones |
Poco frecuentes |
|
Trastornos vasculares |
Hematoma |
Poco frecuentes |
|
Sofocos |
Poco frecuentes | |
|
Palidez |
Poco frecuentes | |
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
Poco frecuentes |
|
Trastornos |
Diarrea |
Poco frecuentes |
|
gastrointestinales |
Dolor en el abdomen superior |
Poco frecuentes |
|
Nauseas |
Poco frecuentes | |
|
Vómitos |
Poco frecuentes |
|
Tabla 2 Frecuencia de las reacciones adversas (RAM) en los ensayos clínicos y procedentes de informes espontáneos | ||
|
Clasificación de órganos del sistema MedDRA |
Reacción adversa |
Frecuencia3 |
|
Trastornos de la piel y del tejido subcutáneo |
Prurito |
Poco frecuentes |
|
Sarpullido |
Poco frecuentes | |
|
Hiperhidrosis |
Poco frecuentes | |
|
Urticaria |
Poco frecuentes | |
|
Trastornos generales y alteraciones en el lugar de administración |
Fiebre |
Frecuentes |
|
Edema periférico |
Poco frecuentes | |
|
Dolor torácico Malestar torácico |
Poco frecuentes Poco frecuentes | |
|
Escalofríos |
Poco frecuentes | |
|
Sensación anormal Hematoma en el lugar de punción de un vaso sanguíneo |
Poco frecuentes Poco frecuentes | |
|
Fatiga Reacción en la zona de inyección |
Frecuencia no conocida Frecuencia no conocida | |
|
Malestar general |
Frecuencia no conocida | |
|
Exploraciones complementarias | ||
|
Recuento elevado de monocitos |
Poco frecuentes | |
|
Nivel reducido de factor de coagulación VIIIb |
Poco frecuentes | |
|
Descenso del hematocrito |
Poco frecuentes | |
|
Test de laboratorio anormal |
Poco frecuentes | |
|
Lesiones, intoxicaciones y complicaciones de procedimientos terapéuticos |
Complicaciones tras el procedimiento |
Poco frecuentes |
|
Hemorragia tras el procedimiento |
Poco frecuentes | |
|
Reacción en el lugar del procedimiento |
Poco frecuentes | |
a) Calculado según un número total único de pacientes que recibieron ADVATE (418)
b) El descenso inesperado de los niveles de factor VIII de coagulación se produjo en un paciente durante la perfusión continua de ADVATE después de cirugía (días 10-14 de postoperatorio).
Se mantuvo la hemostasis durante todo este periodo y tanto los niveles de factor VIII de plasma como los niveles de aclaramiento volvieron a los niveles adecuados el día 15 de postoperatorio. Los ensayos de inhibidores del factor VIII realizados tras la finalización de la perfusión continua y la terminación del estudio füeron negativos.
c) RAM explicadas en la sección siguiente.
Descripción de reacciones adversas seleccionadas Desarrollo de inhibidor
Se ha notificado un desarrollo de inhibidor en pacientes con tratamientos previos y en pacientes no tratados previamente. Para información detallada ver secciones 5.1 (Propiedades farmacológicas) y 4.4 (Advertencias y precauciones especiales de empleo).
RAM específicas para residuos del proceso de fabricación
De los 229 pacientes en tratamiento en los que se evaluaron los anticuerpos antiproteínas de células de ovario de hámster chino (CHO), 3 mostraron una tendencia ascendente estadísticamente significativa en los títulos, 4 mostraron picos sostenidos o picos momentáneos y un paciente mostró ambos, pero no tuvo ningún otro síntoma clínico. De los 229 pacientes tratados a los que se realizó una evaluación de los anticuerpos anti-IgG murina, 10 mostraron una tendencia ascendente, dos mostraron un pico sostenido o pico momentáneo y un paciente mostró ambos. Cuatro de estos pacientes sufrieron episodios aislados de urticaria, prurito, sarpullido y recuentos ligeramente elevados de eosinófilos entre exposiciones repetidas al medicamento del estudio.
Hipersensibilidad
Reacciones de tipo alérgico incluyen anafilaxis y se han manifestado como mareos, parestesias, erupción, soplado, tumefacción facial, urticaria y prurito.
Población pediátrica
En los estudios clínicos no se han notado diferencias específicas por edad en RAM excepto el desarrollo de inhibidores en pacientes pediátricos no tratados previamente (PUP) y complicaciones relacionadas con catéter.
Notificación de sospechas de reacciones adversas
. Se invita a los sistema nacional
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento profesionales sanitarios a notificar las sospechas de reacciones adversas a través del de notificación pertinente incluido en el Anexo V.
4.9 Sobredosis
No se han notificado síntomas de sobredosis con factor VIII de coagulación recombinante.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: antihemorrágicos: factor VIII de coagulación. Código ATC: B02BD02.
El complejo factor VIII /Factor von Willebrand consta de dos moléculas (factor VIII y factor von Willebrand) con diferentes funciones fisiológicas. ADVATE contiene factor VIII de coagulación recombinante (octocog alfa), una glicoproteína que es biológicamente equivalente a la glicoproteína de factor VIII del plasma humano.
Octocog alfa es una glicoproteína que consta de 2332 aminoácidos con un peso molecular aproximado de 280 kD. Cuando se perfunde en un paciente hemofílico, octocog alfa se enlaza al Factor von Willebrand endógeno en la circulación del paciente. El factor VIII activado actúa como un Cofactor para el Factor IX activado, acelerando la conversión del Factor X a Factor X activado. El Factor X activado convierte la protrombina en trombina. La trombina convierte el fibrinógeno en fibrina, con la que se puede formar el coágulo. La hemofilia A es una alteración hereditaria de la coagulación sanguínea ligada al sexo y producida por la disminución de los niveles de factor VIII que da como resultado una hemorragia profusa en las articulaciones, músculos u órganos internos, bien de forma espontánea o bien como resultado de un trauma accidental o quirúrgico. Mediante terapia de sustitución, los niveles plasmáticos de factor VIII aumentan, consiguiendo una corrección temporal de la deficiencia de factor VIII y una corrección de las tendencias hemorrágicas.
Desarrollo del inhibidor
Se evaluó la inmunogenicidad de ADVATE en pacientes con tratamientos previos. Durante los ensayos clínicos con ADVATE en 233 pacientes pediátricos y adultos [pacientes pediátricos (0 a 16 años de edad) y pacientes adultos (más de 16 años de edad)] diagnosticados con hemofilia A grave (factor VIII < 1%), con exposición previa a concentrados de factor VIII durante un periodo igual o superior a 150 días para adultos y niños mayores e igual o superior a 50 días para niños menores de 6 años, un paciente desarrolló un título bajo de inhibidor (2,4 BU en el ensayo Bethesda modificado) tras 26 días de exposición a ADVATE. Las pruebas de seguimiento de inhibidores en este paciente tras abandonar el ensayo fueron negativas. En todos los estudios, la exposición media a ADVATE fue de 97,0 días de exposición por sujeto (intervalo 1 a 709) para pacientes previamente tratados. La incidencia global de cualquier desarrollo de inhibidor del factor VIII (alto o bajo) fue de 0,4% (1 de 233).
En el ensayo completado no controlado 060103, 16 de los 45 (35,6%) pacientes sin tratamiento previo con hemofilia A grave (FVIII < 1%) con al menos 25 días de exposición a FVIII desarrollaron inhibidores del FVIII: 7 (15,6%) sujetos desarrollaron inhibidores de título alto y 9 (20%) de los sujetos desarrollaron inhibidores de título bajo, 1 de los cuales también se clasificó como inhibidor temporal.
Los factores de riesgo relacionados con el desarrollo del inhibidor en este ensayo incluyeron etnicidad no caucásica, antecedentes familiares de inhibidores y tratamiento intensivo a dosis altas en los primeros 20 días de exposición. En los 20 sujetos que no tenían ninguno de estos factores de riesgo no se produjo el desarrollo del inhibidor.
Se han recogido datos sobre Inducción de tolerancia inmunitaria (ITI) en pacientes con inhibidores.
En un subestudio del estudio PUP 060103, se documentaron tratamientos ITI en 11 PUP. Se hizo un análisis de cuadro retrospective de 30 materias sobre ITI (estudio 060703) y recogida de datos de registro está en curso.
En el estudio 060201, se compararon dos esquemas de tratamiento de profilaxis a largo plazo en 53 sujetos previamente tratados: un régimen individualizado de dosificación guiada farmacocinética (dentro de un rango de 20 a 80 UI de factor VIII por kg de peso corporal a intervalos de 72 ± 6 horas, n = 23) con un régimen de dosificación profiláctica estándar (de 20 a 40 UI/kg cada 48 ± 6 horas, n = 30). El régimen de dosificación guiada farmacocinética (de acuerdo con una fórmula específica) tenía como objetivo mantener el factor VIII en niveles > 1% en el intervalo entre dosis de 72 horas. Los datos de este estudio demuestran que los dos regímenes de dosificación profiláctica son comparables en lo que se refiere a la reducción de la frecuencia de hemorragias.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con ADVATE en los diferentes grupos de la población pediátrica con hemofilia A (deficiencia congénita de factor VIII) en “Inducción de tolerancia inmunitaria (ITI) en pacientes con hemofilia A (deficiencia congénita de factor VIII) que han desarrollado inhibidores al factor VIII” y “tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita de factor VIII)” (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
5.2 Propiedades farmacocinéticas
Todos los estudios farmacocinéticos con ADVATE se realizaron en pacientes previamente tratados con hemofilia A grave o moderadamente grave (valor basal del factor VIII < 2%). El análisis de las muestras de plasma se realizó en un laboratorio central utilizando el ensayo de coagulación en una etapa.
Un total de 195 sujetos con hemofilia A grave (valor basal del factor VIII < 1%) proporcionaron parámetros PK que se incluyeron en el conjunto de análisis de PK según el protocolo. Las categorías de estos análisis para lactantes (1 mes a <2 años), niños (2 a <5 años), niños de mayor edad (5 a <12 años), adolescentes (12 a <18 años) y adultos (18 años y mayores) se utilizaron para resumir los parámetros PK, definiéndose la edad como la edad en el momento de la perfusión de PK.
|
Tabla 3 Resumen de parámetros farmacocinéticos de ADVATE por grupo de edad con hemofilia A grave (valor basal del factor VIII < 1%) | |||||
|
Parámetro (media ± desviación estándar) |
Lactantes (n = 5) |
Niños (n = 30) |
Niños de mayor edad (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
|
Área bajo la curva total (UI*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Recuperación incremental ajustada a Cmáx (UI/dl por UI/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Semivida (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Concentración máxima en plasma tras la perfusión (UI/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Tiempo de residencia medio (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Volumen de distribución en estado estacionario (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Aclaramiento (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Calculada como (Cmáx - valor basal del factor VIII) dividida por la dosis en UI/kg, donde Cmáx es la medida máxima de factor VIII tras la perfusión.
La seguridad y la eficacia hemostática de ADVATE en la población pediátrica son similares a las de los pacientes adultos. La recuperación y la semivida terminal (t/2) ajustadas fue aproximadamente un 20% inferior en niños pequeños (menores de 6 años) que en adultos, lo que lo que puede deberse en parte a un volumen mayor conocido de plasma por kg de peso corporal en los pacientes más jóvenes.
No se dispone de datos farmacocinéticos de ADVATE en pacientes sin tratamiento previo.
5.3 Datos preclínicos sobre seguridad
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de la seguridad, toxicología aguda, toxicidad en dosis repetida, toxicidad local y genotoxicidad.
Un estudio de tolerancia local con conejos demostró que ADVATE reconstituido con 2 ml de agua esterilizada para preparaciones inyectables se tolera bien tras una administración intravenosa.
Se observó un leve enrojecimiento temporal en el lugar de la administración después de la aplicación intraarterial y después de la aplicación paravenosa. No obstante, no se observaron cambios histopatológicos adversos correlacionados que indiquen una naturaleza temporal de esta observación.
DATOS FARMACÉUTICOS
6.
6.1 Lista de excipientes
Polvo
Manitol
Cloruro de sodio Histidina Trealosa Cloruro de calcio Trometamol Polisorbato 80 Glutatión (reducido)
Disolvente
Agua esterilizada para preparaciones inyectables.
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad este medicamento no debe mezclarse con otros.
6.3 Periodo de validez 2 años.
Después de la reconstitución, desde un punto de vista microbiológico, el medicamento debe utilizarse de inmediato. No obstante, se ha demostrado una estabilidad física y química en uso durante 3 horas a 25 °C.
Durante el periodo de validez, el medicamento puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único no superior a 6 meses. Se debe anotar la fecha del final del periodo de conservación a temperatura ambiente de 6 meses en el embalaje del medicamento. El medicamento no puede refrigerarse de nuevo.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
ADVATE con el equipo BAXJECT II: Conservar el vial del producto en el embalaje exterior para protegerlo de la luz.
ADVATE en el sistema BAXJECT III: Conservar el blister sellado en el embalaje exterior para protegerlo de la luz.
Para consultar las condiciones de conservación tras la reconstitución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Tanto el vial de polvo como el vial de disolvente de 2 ml son de vidrio tipo I cerrado con tapones de
caucho de clorobutilo. El medicamento se proporciona con una de las configuraciones siguientes:
- ADVATE con el equipo BAXJECT II: Cada envase contiene un vial de polvo, un vial de disolvente de 2 ml y un equipo para reconstitución (BAXJECT II).
- ADVATE en el sistema BAXJECT III: Cada envase contiene un sistema BAXJECT III listo para usar en un blister sellado (el vial de polvo y el vial de disolvente de 2 ml están precargados con el sistema para su reconstitución).
6.6 Precauciones especiales de eliminación y otras manipulaciones
ADVATE debe administrarse intravenosamente después de la reconstitución del producto.
La solución reconstituida se debe inspeccionar visualmente para comprobar que no presenta partículas
extrañas y/o descoloración.
Después de la reconstitución la solución debe ser transparente, incolora y no presentar partículas
extrañas.
No utilizar soluciones turbias o con depósitos.
- Para la administración se requiere el uso de una jeringa luer-lock.
- Utilizar dentro de las tres horas después de la reconstitución.
- No refrigerar la preparación después de la reconstitución.
- La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Reconstitución con el equipo BAXJECT II:
- Para la reconstitución utilizar sólo el agua esterilizada para preparaciones inyectables y el equipo de reconstitución incluido en el envase.
- No utilizar si el equipo BAXJECT II, el sistema estéril de protección o su envase está dañado o muestra algún signo de deterioro.
- Se debe utilizar una técnica aséptica:
1. Si el medicamento se encuentra en la nevera, coger de la nevera los viales de polvo y de disolvente de ADVATE y dejarlos a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Quitar los protectores de los viales de polvo y disolvente.
4. Limpiar los tapones con las toallitas impregnadas de alcohol. Colocar los viales en una superficie plana y limpia.
5. Abrir el envoltorio del equipo BAXJECT II quitando la tapa de papel sin tocar el interior (Fig. a). No sacar el equipo del envoltorio. No utilizar si el equipo BAXJECT II, su sistema de barrera estéril o su acondicionamiento están dañados o muestran algún signo de deterioro.
6. Dar la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente. Coger el envoltorio por su extremo y sacar el equipo BAXJECT II de su envoltorio (Fig. b). No quitar el protector azul del equipo BAXJECT II.
7. Para la reconstitución, utilizar solamente el agua esterilizada para preparaciones inyectables y el equipo para reconstitución incluidos en el envase. Con el equipo BAXJECT II unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico blanca dentro del tapón del vial de polvo ADVATE. El vacío hará que el disolvente penetre en el vial de polvo ADVATE (Fig. c).
8. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará
a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.

Reconstitución con el sistema BAXJECT III:
- No lo utilice si la tapa del blister no está perfectamente sellada.
1. Si el medicamento se encuentra en la nevera, coger de la nevera el blister sellado (contiene viales de polvo y disolvente precargados con el sistema para su reconstitución) y dejarlo a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Abrir el envoltorio de ADVATE quitando la tapa. Extraer el sistema BAXJECT III del blister.
4. Colocar ADVATE en una superficie plana con el vial de disolvente encima (Fig. 1). El vial de disolvente tiene una raya azul. No quitar el protector azul hasta que se le indique más adelante.
5. Mientras sujeta ADVATE con una mano en el sistema BAXJECT III, presionar con fuerza el vial de disolvente con la otra mano hasta que el sistema quede totalmente contraído y el disolvente entre dentro del vial de ADVATE (Fig. 2). No inclinar el sistema hasta que haya finalizado la transferencia.
6. Comprobar que ha finalizado la transferencia del disolvente. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig- 1

Fig. 3

Fig. 2
Administración:
Utilizar técnica aséptica:
Los medicamentos para administración parenteral deben inspeccionarse visualmente antes de su administración para verificar la ausencia de partículas, cuando la solución y el envase lo permitan. Sólo se debe utilizar una solución transparente y sin coloración.
1. Quitar el protector azul del equipo BAXJECT II / al sistema BAXJECT III. No introducir aire en la jeringa. Conectar la jeringa al equipo BAXJECT II / al sistema BAXJECT III.
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente.
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar intravenosamente. La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de ADVATE. Si se observa una elevación significativa del pulso, reducir la velocidad de administración o interrumpir temporalmente la inyección suele hacer que los síntomas normalmente desaparezcan pronto (ver secciones 4.4 y 4.8).
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG Industriestrasse 67 A-1221 Viena Austria
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/010
EU/1/03/271/020
9. FECHA DE LA PRIMERA AUTORIZACIÓN / RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 2 de marzo de 2004 Fecha de la última renovación: 2 de marzo de 2014
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos http://www.ema.europa.eu.
A. FABRICANTES DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
A. FABRICANTES DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTE RESPONSABLE DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección de los fabricantes del principio activo biológico
Baxalta Manufacturing Sárl Route de Pierre-á-Bot 111 CH-2000 Neuchatel Suiza
Baxalta Manufacturing SARL Singapore Branch
2A Woodlands Industrial Park D Street 2
Singapore 737779
Singapur
Nombre y dirección de los fabricantes responsables de la liberación de los lotes
Baxalta Belgium Manufacturing SA Boulevard René Branquart 80 B-7860 Lessines Bélgica
Baxter SA
Boulevard René Branquart 80
B-7860 Lessines
Bélgica
El prospecto impreso del medicamento debe especificar el nombre y dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes periódicos de seguridad (IPS)
El Titular de la Autorización de Comercialización (TAC) presentará los informes periódicos de seguridad para este medicamento de conformidad con las exigencias establecidas en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107ter, párrafo 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2 de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuando se modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo,
o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos).
• Si coincide la presentación de un IPS con la actualización del PGR, ambos documentos se pueden presentar conjuntamente.
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
ADVATE 250 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante).
1 vial: 250 UI octocog alfa, aproximadamente 50 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 250 UI de octocog alfa, 1 vial con 5 ml de agua esterilizada para preparaciones inyectables, 1 equipo para reconstitución BAXJECT II.
Vía intravenosa, después de la reconstitución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:
No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Viena Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/001
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
MEDICAMENTO SUJETO A PRESCRIPCIÓN MÉDICA
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 250
ETIQUETA DEL VIAL DE POLVO (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 250 UI polvo para solución inyectable. Octocog alfa Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Para un solo uso.
3. FECHA DE CADUCIDAD
EXP:
4. NÚMERO DE LOTE
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
250 UI octocog alfa
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
EXP:
4. NÚMERO DE LOTE_
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES_
5 ml
6. OTROS
ADVATE 250 UI polvo y disolvente para solución inyectable Octocog alfa (factor VIII humano de coagulación recombinante)
1 vial: 250 UI octocog alfa, aproximadamente 50 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 250 UI de octocog alfa y 1 vial con 5 ml de agua esterilizada para preparaciones inyectables, precargado en el sistema BAXJECT III.
Vía intravenosa, después de la reconstitución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:
No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Vienna Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/011
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 250
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN BLISTERS O TIRAS ETIQUETA DEL BLISTER (SISTEMA BAXJECT III)_
ADVATE 250 UI polvo y disolvente para solución inyectable Octocog alfa
Baxter AG
CAD:
Lote:
Vía intravenosa, después de la reconstitución.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
No utilizar si el embalaje está abierto o dañado.
Vial de polvo y 5 ml de disolvente precargado en un sistema BAXJECT III.
1. NOMBRE DEL MEDICAMENTO
ADVATE 250
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. OTROS


ETIQUETA DEL VIAL DE POLVO (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 250
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ADVATE 500 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante).
1 vial: 500 UI octocog alfa, aproximadamente 100 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 500 UI de octocog alfa, 1 vial con 5 ml de agua esterilizada para preparaciones inyectables, 1 equipo para reconstitución BAXJECT II.
Vía intravenosa, después de la reconstitución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:
No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Viena Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/002
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
MEDICAMENTO SUJETO A PRESCRIPCIÓN MÉDICA
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 500
ETIQUETA DEL VIAL DE POLVO (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 500 UI polvo para solución inyectable Octocog alfa Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Para un solo uso
3. FECHA DE CADUCIDAD
EXP:
4. NÚMERO DE LOTE
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
500 UI octocog alfa
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
EXP:
4. NÚMERO DE LOTE_
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES_
5 ml
6. OTROS
ADVATE 500 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante)
1 vial: 500 UI octocog alfa, aproximadamente 100 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 500 UI octocog alfa y 1 vial con 5 ml de agua esterilizada para preparaciones inyectables, precargado en un sistema BAXJECT III.
Vía intravenosa, después de la reconstitución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:
No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Vienna Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/012
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 500
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN BLISTERS O TIRAS ETIQUETA DEL BLISTER (SISTEMA BAXJECT III)_
ADVATE 500 UI polvo y disolvente para solución inyectable Octocog alfa
Baxter AG
CAD:
Lote:
Vía intravenosa, después de la reconstitución.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
No utilizar si el embalaje está abierto o dañado.
Vial de polvo y 5 ml de disolvente precargado en un sistema BAXJECT III.
1. NOMBRE DEL MEDICAMENTO
ADVATE 500
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. OTROS

ETIQUETA DEL VIAL DE POLVO (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 500
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
CAD:
4. NÚMERO DE LOTE_
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES_
6. OTROS
ADVATE 1000 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante).
1 vial: 1000 UI octocog alfa, aproximadamente 200 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 1000 UI de octocog alfa, 1 vial con 5 ml de agua esterilizada para preparaciones inyectables, 1 equipo para reconstitución BAXJECT II.
Vía intravenosa, después de la reconstitución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:
No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Viena Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/003
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
MEDICAMENTO SUJETO A PRESCRIPCIÓN MÉDICA
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 1000
ETIQUETA DEL VIAL DE POLVO (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 1000 UI polvo para solución inyectable Octocog alfa Vía intravenosa,
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Para un solo uso.
3. FECHA DE CADUCIDAD
EXP:
4. NÚMERO DE LOTE
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
1000 UI octocog alfa
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
EXP:
4. NÚMERO DE LOTE_
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES_
5 ml
6. OTROS
ADVATE 1000 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante).
1 vial: 1000 UI octocog alfa, aproximadamente 200 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, clorurode sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 1000 UI octocog alfa y 1 vial con 5 ml de agua esterilizada para preparaciones inyectables, precargado en el sistema BAXJECT III.
Vía intravenosa, después de la reconstitución.:
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Vienna Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/013
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 1000
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN BLISTERS O TIRAS ETIQUETA DEL BLISTER (SISTEMA BAXJECT III)_
ADVATE 1000 UI polvo y disolvente para solución inyectable Octocog alfa
Baxter AG
CAD:
Lote:
Vía intravenosa, después de la reconstitución.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
No utilizar si el embalaje está abierto o dañado.
Vial de polvo y 5 ml de disolvente precargado en un sistema BAXJECT III.
1. NOMBRE DEL MEDICAMENTO
ADVATE 1000
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. OTROS

ETIQUETA DEL VIAL DE POLVO (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 1000
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ADVATE 1500 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante).
1 vial: 1500 UI octocog alfa, aproximadamente 300 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 1500 UI de octocog alfa, 1 vial con 5 ml de agua esterilizada para preparaciones inyectables, 1 equipo para reconstitución BAXJECT II.
Vía intravenosa, después de la reconstitución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:
No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Viena Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/004
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 1500
ETIQUETA DEL VIAL DE POLVO (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 1500 UI polvo para solución inyectable. Octocog alfa Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Para un solo uso.
3. FECHA DE CADUCIDAD
EXP:
4. NÚMERO DE LOTE
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
1500 UI octocog alfa
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
EXP:
4. NÚMERO DE LOTE_
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES_
5 ml
6. OTROS
ADVATE 1500 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante)
1 vial: 1500 UI octocog alfa, aproximadamente 300 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 1500 UI octocog alfa y 1 vial con 5 ml de agua esterilizada para preparaciones inyectables, precargado en un sistema BAXJECT III.
Vía intravenosa, después de la reconstitución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:
No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Vienna Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/014
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 1500
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN BLISTERS O TIRAS ETIQUETA DEL BLISTER (SISTEMA BAXJECT III)_
ADVATE 1500 UI polvo y disolvente para solución inyectable Octocog alfa
Baxter AG
CAD:
Lote:
Vía intravenosa, después de la reconstitución.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
No utilizar si el embalaje está abierto o dañado.
Vial de polvo y 5 ml de disolvente precargado en un sistema BAXJECT III.
1. NOMBRE DEL MEDICAMENTO
ADVATE 1500
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. OTROS

ETIQUETA DEL VIAL DE POLVO (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 1500
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ADVATE 2000 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante).
1 vial: 2000 UI octocog alfa, aproximadamente 400 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, clorurode sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 2000 UI de octocog alfa, 1 vial con 5 ml de agua esterilizada para preparaciones inyectables, 1 equipo para reconstitución BAXJECT II.
Vía intravenosa, después de la reconstitución.:
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Viena Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/005
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 2000
ETIQUETA DEL VIAL DE POLVO (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 2000 UI polvo para solución inyectable. Octocog alfa Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Para un solo uso.
3. FECHA DE CADUCIDAD
EXP:
4. NÚMERO DE LOTE
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
2000 UI octocog alfa
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
EXP:
4. NÚMERO DE LOTE_
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES_
5 ml
6. OTROS
ADVATE 2000 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante).
1 vial: 2000 UI octocog alfa, aproximadamente 400 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 2000 UI octocog alfa y 1 vial con 5 ml de agua esterilizada para preparaciones inyectables, precargado en el sistema BAXJECT III.
Vía intravenosa, después de la reconstitución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:
No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Vienna Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/015
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 2000
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN BLISTERS O TIRAS ETIQUETA DEL BLISTER (SISTEMA BAXJECT III)_
ADVATE 2000 UI polvo y disolvente para solución inyectable Octocog alfa
Baxter AG
CAD:
Lote:
Vía intravenosa, después de la reconstitución.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
No utilizar si el embalaje está abierto o dañado.
Vial de polvo y 5 ml de disolvente precargado en el sistema BAXJECT III.
1. NOMBRE DEL MEDICAMENTO
ADVATE 2000
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. OTROS

ETIQUETA DEL VIAL DE POLVO (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 2000
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ADVATE 3000 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante).
1 vial: 3000 UI octocog alfa, aproximadamente 600 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 3000 UI de octocog alfa, 1 vial con 5 ml de agua esterilizada para preparaciones inyectables, 1 equipo para reconstitución BAXJECT II.
Vía intravenosa.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:
No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Viena Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/006
13. NÚMERO DE LOTE
Lot:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 3000
ETIQUETA DEL VIAL DE POLVO (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 3000 UI polvo para solución inyectable. Octocog alfa Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Para un solo uso.
3. FECHA DE CADUCIDAD
EXP:
4. NÚMERO DE LOTE
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
3000 UI octocog alfa
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
EXP:
4. NÚMERO DE LOTE_
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES_
5 ml
6. OTROS
ADVATE 3000 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante)
1 vial: 3000 UI octocog alfa, aproximadamente 600 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4,000 - 10,000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 3000 UI octocog alfa y 1 vial con 5 ml de agua esterilizada para preparaciones inyectables, precargado en un sistema BAXJECT III.
Vía intravenosa, después de la reconstitución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:
No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Vienna Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/016
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 3000
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN BLISTERS O TIRAS ETIQUETA DEL BLISTER (SISTEMA BAXJECT III)_
ADVATE 3000 UI polvo y disolvente para solución inyectable Octocog alfa
Baxter AG
CAD:
Lote:
Vía intravenosa, después de la reconstitución.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
No utilizar si el embalaje está abierto o dañado.
Vial de polvo y 5 ml de disolvente precargado en el sistema BAXJECT III.
1. NOMBRE DEL MEDICAMENTO
ADVATE 3000
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. OTROS

ETIQUETA DEL VIAL DE POLVO (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 3000
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
CAD:
4. NÚMERO DE LOTE_
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES_
6. OTROS
ADVATE 250 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante).
1 vial: 250 UI octocog alfa, aproximadamente 125 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 250 UI de octocog alfa, 1 vial con 2 ml de agua esterilizada para preparaciones inyectables, 1 equipo para reconstitución BAXJECT II.
Vía intravenosa, después de la reconstitución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Viena Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/007
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 250
ETIQUETA DEL VIAL DE POLVO (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 250 UI polvo para solución inyectable. Octocog alfa Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Para un solo uso.
3. FECHA DE CADUCIDAD
EXP:
4. NÚMERO DE LOTE
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
250 UI octocog alfa
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
EXP:
4. NÚMERO DE LOTE_
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES_
2 ml
6. OTROS
ADVATE 250 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante).
1 vial: 250 UI octocog alfa, aproximadamente 125 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 250 UI octocog alfay 1 vial con 2 ml de agua esterilizada para preparaciones inyectables precargado en el sistema BAXJECT III.
Vía intravenosa, después de la reconstitución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:
No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Vienna Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/017
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 250
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN BLISTERS O TIRAS ETIQUETA DEL BLISTER (SISTEMA BAXJECT III)_
ADVATE 250 UI polvo y disolvente para solución inyectable Octocog alfa
Baxter AG
CAD:
Lote:
Vía intravenosa, después de la reconstitución.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
No utilizar si el embalaje está abierto o dañado.
Vial de polvo y 2 ml de disolvente precargado en el sistema BAXJECT III.
1. NOMBRE DEL MEDICAMENTO
ADVATE 250
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. OTROS

ETIQUETA DEL VIAL DE POLVO (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 250
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ADVATE 500 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante).
1 vial: 500 UI octocog alfa, aproximadamente 250 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 500 UI de octocog alfa, 1 vial con 2 ml de agua esterilizada para preparaciones inyectables, 1 equipo para reconstitución BAXJECT II.
Vía intravenosa, después de la reconstitución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:
No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Viena Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/008
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 500
ETIQUETA DEL VIAL DE POLVO (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 500 UI polvo para solución inyectable. Octocog alfa Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Para un solo uso.
3. FECHA DE CADUCIDAD
EXP:
4. NÚMERO DE LOTE
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
500 UI octocog alfa
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
EXP:
4. NÚMERO DE LOTE_
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES_
2 ml
6. OTROS
ADVATE 500 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante)
1 vial: 500 UI octocog alfa, aproximadamente 250 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, clorurode sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 500 UI octocog alfa y 1 vial con 2 ml de agua esterilizada para preparaciones inyectables precargado en el sistema BAXJECT III.
Vía intravenosa, después de la reconstitución.:
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Vienna Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/018
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 500
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN BLISTERS O TIRAS ETIQUETA DEL BLISTER (SISTEMA BAXJECT III)_
ADVATE 500 UI polvo y disolvente para solución inyectable Octocog alfa
Baxter AG
CAD:
Lote:
Vía intravenosa, después de la reconstitución.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
No utilizar si el embalaje está abierto o dañado.
Vial de polvo y 2 ml de disolvente precargado en el sistema BAXJECT III.
1. NOMBRE DEL MEDICAMENTO
ADVATE 500
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. OTROS

ETIQUETA DEL VIAL DE POLVO (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 500
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ADVATE 1000 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante).
1 vial: 1000 UI octocog alfa, aproximadamente 500 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 1000 UI de octocog alfa, y 1 vial con 2 ml de agua esterilizada para preparaciones inyectables, 1 equipo para reconstitución BAXJECT II.
Vía intravenosa, después de la reconstitución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Viena Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/009
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 1000
ETIQUETA DEL VIAL DE POLVO (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 1000 UI polvo para solución inyectable. Octocog alfa Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Para un solo uso.
3. FECHA DE CADUCIDAD
EXP:
4. NÚMERO DE LOTE
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
1000 UI octocog alfa
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
EXP:
4. NÚMERO DE LOTE_
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES_
2 ml
6. OTROS
ADVATE 1000 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante)
1 vial: 1000 UI octocog alfa, aproximadamente 500 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 1000 UI octocog alfa y1 vial con 2 ml de agua esterilizada para preparaciones inyectablesprecargado en el sistema BAXJECT III.
Vía intravenosa.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:
No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Vienna Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/019
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 1000
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN BLISTERS O TIRAS ETIQUETA DEL BLISTER (SISTEMA BAXJECT III)_
ADVATE 1000 UI polvo y disolvente para solución inyectable Octocog alfa
Baxter AG
CAD:
Lote:
Vía intravenosa, después de la reconstitución.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
No utilizar si el embalaje está abierto o dañado.
Vial de polvo y 2 ml de disolvente precargado en el sistema BAXJECT III.
1. NOMBRE DEL MEDICAMENTO
ADVATE 1000
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. OTROS

ETIQUETA DEL VIAL DE POLVO (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 1000_
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ADVATE 1500 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante).
1 vial: 1500 UI octocog alfa, aproximadamente 750 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 1500 UI de octocog alfa, 1 vial con 2 ml de agua esterilizada para preparaciones inyectables, 1 equipo para reconstitución BAXJECT II.
Vía intravenosa, después de la reconstitución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Viena Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/010
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 1500
ETIQUETA DEL VIAL DE POLVO (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 1500 UI polvo para solución inyectable. Octocog alfa Vía intravenosa
2. FORMA DE ADMINISTRACIÓN
Leer el prospecto antes de utilizar este medicamento. Para un solo uso.
3. FECHA DE CADUCIDAD
EXP:
4. NÚMERO DE LOTE
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
1500 UI octocog alfa
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (EQUIPO BAXJECT II)_
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN_
3. FECHA DE CADUCIDAD_
EXP:
4. NÚMERO DE LOTE_
Lot:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES_
2 ml
6. OTROS
ADVATE 1500 UI polvo y disolvente para solución inyectable. Octocog alfa (factor VIII humano de coagulación recombinante)
1 vial: 1500 UI octocog alfa, aproximadamente 750 UI/ml después de la reconstitución. Actividad específica: aproximadamente 4.000 - 10.000 UI/mg proteína.
Excipientes: manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80, glutatión (reducido).
Polvo y disolvente para solución inyectable
Contenido: 1 vial con 1500 UI octocog alfa y 1 vial con 2 ml de agua esterilizada para preparaciones inyectables precargado en el sistema BAXJECT III.
Vía intravenosa, después de la reconstitución.
Para un solo uso.
Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
CAD:
Fin del periodo de 6 meses, si se conserva a temperatura ambiente:No utilizar después de esta fecha de caducidad.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
Conservar en nevera.
No congelar.
Mantener en el envase original para protegerlo de la luz.
Puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único hasta 6 meses.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)_
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG A-1221 Vienna Austria
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/03/271/020
13. NÚMERO DE LOTE
Lote:
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica.
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
ADVATE 1500
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN BLISTERS O TIRAS ETIQUETA DEL BLISTER (SISTEMA BAXJECT III)_
ADVATE 1500 UI polvo y disolvente para solución inyectable Octocog alfa
Baxter AG
CAD:
Lote:
Vía intravenosa, después de la reconstitución.
Utilizar inmediatamente o en las 3 horas siguientes a la reconstitución.
No utilizar si el embalaje está abierto o dañado.
Vial de polvo y 2 ml de disolvente precargado en el sistema BAXJECT III.
1. NOMBRE DEL MEDICAMENTO
ADVATE 1500
2. NOMBRE DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Baxter AG
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. OTROS

ETIQUETA DEL VIAL DE POLVO (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADVATE 1500
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
ETIQUETA DEL VIAL DE DISOLVENTE (SISTEMA BAXJECT III)
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
Agua esterilizada para preparaciones inyectables.
2. FORMA DE ADMINISTRACIÓN
3. FECHA DE CADUCIDAD
CAD:
4. NÚMERO DE LOTE
Lote:
5. CONTENIDO EN PESO, VOLUMEN O EN UNIDADES
6. OTROS
B. PROSPECTO
Prospecto: Información para el usuario
ADVATE 250 UI polvo y disolvente para solución inyectable ADVATE 500 UI polvo y disolvente para solución inyectable ADVATE 1000 UI polvo y disolvente para solución inyectable ADVATE 1500 UI polvo y disolvente para solución inyectable ADVATE 2000 UI polvo y disolvente para solución inyectable ADVATE 3000 UI polvo y disolvente para solución inyectable
Octocog alfa (factor VIII humano de coagulación recombinante)
Lea todo el prospecto detenidamente antes de empezar a usar el medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Este medicamento se le ha recetado solamente a usted y no debe dárselo a otras personas, aunque tengan los mismos sintomas, ya que puede perjudicarles.
- Si experimenta cualquier tipo de efecto adverso, consulte a su medico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es ADVATE y para qué se utiliza
2. Qué necesita saber antes de empezar a usar ADVATE
3. Cómo usar ADVATE
4. Posibles efectos adversos
5. Conservación de ADVATE
6. Contenido del envase e nformación adicional
1. Qué es ADVATE y para qué se utiliza
ADVATE contiene el principio activo octocog alfa, factor VIII humano de coagulación producido por tecnología de ADN recombinante. El factor VIII es necesario para coagular la sangre y parar las hemorragias. En pacientes con hemofilia A el factor VIII falta o no funciona correctamente (ausencia hereditaria de factor VIII).
ADVATE se utiliza para el tratamiento y la prevención de hemorragias en pacientes de todos los grupos de edad que padecen hemofilia A (un trastorno hemorrágico hereditario provocado por la ausencia de factor VIII).
ADVATE se prepara sin añadir ninguna proteína derivada humana o animal en todo el proceso de fabricación.
2. Qué necesita saber antes de empezar a usar ADVATE No use ADVATE
- si es alérgico (hipersensible) al octocog alfa o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- si es alérgico a las proteínas del ratón o del hámster
Si no está seguro si es alérgico, consulte con su médico.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar ADVATE. Informe a su médico si ha sido tratado previamente con medicamentos que contienen factor VIII, especialmente si desarrolló inhibidores ya que puede existir un riesgo elevado de que esto vuelva a suceder. Los inhibidores son anticuerpos que bloquean el factor VIII que reducen la eficacia de ADVATE para prevenir el control del sangrado. El desarrollo de inhibidores es una complicación conocida en el tratamiento de la hemofilia A. Si su hemorragia no llega a controlarse con ADVATE, consulte a su médico inmediatamente.
Existe un riesgo escasode que pueda experimentar una reacción anafiláctica (una reacción alérgica repentina grave) a ADVATE. Debe ser consciente de que los primeros síntomas de una reacción alérgica son sarpullido, picor, ronchas, picor generalizado, hinchazón de los labios y lengua, dificultad para respirar, jadeos, opresión en el pecho, sensación de malestar general y mareos. Estos síntomas pueden ser un aviso de shock anafiláctico, que también puede producir mareos graves, pérdida de consciencia y dificultades graves para respirar.
Si experimenta cualquiera de estos síntomas, interrumpa inmediatamente la administración del medicamento y consulte con un médico. Los síntomas graves, incluyendo la dificultad para respirar y (casi) desmayos requieren un tratamiento de emergencia rápido.
Pacientes que desarrollan inhibidores del Factor VIII
Si el nivel de factor VIII en el plasma no alcanza los valores esperados con ADVATE, o si no se controla adecuadamente la hemorragia, puede deberse al desarrollo de inhibidores del factor VIII. Esto se comprobará por su médico.Usted puede necesitar una dosis mayor de ADVATE o, incluso, otro medicamento para controlar las hemorragias. No aumente la dosis total de ADVATE que utiliza para controlar su hemorragia sin consultar a su médico.
Niños y adolescentes
Las advertencias y precauciones indicadas se aplican a adultos y niños (de 0 a 18 años de edad).
Uso de ADVATE con otros medicamentos
Informe a su médico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su medico antes de utilizar este medicamento.
Conducción y uso de máquinas
El tratamiento con ADVATE no tiene influencia sobre la capacidad para conducir y utilizar maquinaria.
ADVATE contiene sodio
Este medicamento contiene 0,45 mmol de sodio (10 mg) por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
3. Cómo usar ADVATE
El tratamiento con ADVATE se iniciará por un médico con experiencia en el tratamiento de pacientes con hemofilia A.
Su médico calculará su dosis de ADVATE (en unidades internacionales o UI) en función de su estado, su peso corporal y de si va a ser utilizado para la prevención o para el tratamiento de hemorragias.
La frecuencia de administración dependerá de cómo actúe ADVATE. La terapia sustitutiva con ADVATE es normalmente un tratamiento de por vida.
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico.
Prevención de hemorragias
La dosis habitual de octocog alfa es de 20 a 40 UI por kg de peso corporal, administrado
cada 2 ó 3 días. Sin embargo, en algunos casos, especialmente en los pacientes más jóvenes, pueden
requerirse la administración más frecuente de las inyecciones o dosis mayores.
Tratamiento de hemorragias
La dosis de octocog alfa se calcula en función de su peso corporal y de los niveles de factor VIII que se quieran conseguir. Los niveles de factor VIII a alcanzar dependerán de la gravedad y de la localización de la hemorragia.
Dosis (UI) = peso corporal (kg) x aumento de Factor VIII deseado (% del normal) x 0,5
Si tiene la impresión de que el efecto de ADVATE es insuficiente, consulte con su médico.
Su médico realizará los análisis de laboratorio apropiados para asegurarse de que tiene los niveles adecuados de factor VIII. Esto es especialmente importante si se va a someter a una operación importante.
Uso en niños y adolescentes (de 0 a 18 años de edad)
Para el tratamiento de hemorragias, la dosis en niños no difiere de la de los pacientes adultos. Para evitar hemorragias en niños menores de 6 años, se recomiendan dosis de 20 a 50 UI por kg de peso corporal de 3 a 4 veces por semana. La administración de ADVATE en niños (intravenosa) no difiere de la administración en adultos. Puede ser necesario un dispositivo de acceso venoso central (CVAD) para permitir infusiones frecuentes de productos de factor VIII.
Cómo se administra ADVATE
ADVATE normalmente se inyecta en vena (vía intravenosa) por su médico o enfermera. Usted o cualquier otra persona pueden también administrar la inyección de ADVATE pero únicamente después de recibir la formación adecuada. Las instrucciones detalladas para su administración se describen al final de este prospecto.
Si usa más ADVATE del que debiera
Siga exactamente las instrucciones de administración de ADVATE indicadas por su médico. Consulte a su médico si tiene dudas.Si se inyecta una dosis mayor de ADVATE de la recomendada, consulte con su médico lo antes posible.
Si olvidó usar ADVATE
No se inyecte una dosis doble para compensar las dosis olvidadas. Adminístrese la siguiente inyección como está establecido y continúe como le había indicado su médico.
Si interrumpe el tratamiento con ADVATE
No deje de usar ADVATE sin consultarlo con su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Si se producen reacciones alérgicas (anafilácticas) repentinas y graves, se debe parar
inmediatamente la inyección. Contacte con su médico inmediatamente si tiene alguno de los siguientes síntomas iniciales de las reacciones alérgicas:
- erupción, sarpullido, ronchas y picor generalizado,
- hinchazón de los labios y la lengua,
- dificultad para respirar, jadeos, opresión en el pecho,
- sensación de malestar general,
- mareos y pérdida de consciencia.
Los síntomas graves, como dificultad para respirar y (casi) síncope, requieren un tratamiento temprano de emergencia.
Efectos adversos frecuentes (pueden afectar hasta a 1 de cada 10 pacientes) inhibidores del factor VIII, dolor de cabeza y fiebre
Efectos adversos poco frecuentes (pueden afectar hasta a 1 de cada 100 pacientes) mareos, gripe, desfallecimiento, pulsaciones anormales, manchas rojas asociadas a picor, molestias en el pecho, cardenal en la zona de inyección, reacción en la zona de inyección, picor, aumento de la sudoración, sabor extraño en la boca, sofocos, migrañas, pérdidas de memoria, escalofríos, diarrea, náusea, vómitos, dificultad para respirar, dolor de garganta, infección de los vasos linfáticos, palidez, inflamación ocular, sarpullidos, sudoración excesiva, inflamación de pies y piernas, disminución del número de glóbulos rojos, aumento de un tipo de leucocitos (monocitos) y dolor en la parte superior del abdomen o parte inferior del pecho.
Relacionados con la cirugía
infección relacionada con el catéter, disminución del número de glóbulos rojos, hinchazón de las extremidades y las articulaciones, sangrado prolongado después de la retirada del drenaje, disminución de los niveles de factor VIII y hematoma postoperatorio
Relacionados con los dispositivos de acceso venoso central (CVAD)
Infección relacionada con catéter, infección sistémica y coágulo de sangre local en el lado del catéter.
Efectos adversos con frecuencia desconocida (no se puede estimar la frecuencia a partir de los datos disponibles)
Reacciones que pueden ser mortales (anafilaxis) y otras reacciones alérgicas (hipersensibilidad), trastornos generales (cansancio, falta de energía).
Efectos adversos adicionales en niños
Además del desarrollo de inhibidores en pacientes pediátricos sin tratamiento previo y las complicaciones asociadas al catéter, en los ensayos clínicos no se observaron diferencias específicas de la edad en los efectos adversos.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de ADVATE
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase después de CAD. La fecha de caducidad se refiere al último día del mes que se indica.
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
Hasta la fecha de caducidad, el vial de polvo puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único no superior a 6 meses. En este caso, este medicamento caduca al final de este periodo de 6 meses o en la fecha de caducidad impresa en el vial del producto, lo que ocurra antes. Por favor, anote en el envase del medicamento la fecha de finalización del periodo de conservación a temperatura ambiente de 6 meses. El medicamento no puede refrigerarse de nuevo después de conservarse a temperatura ambiente.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Este medicamento es para un solo uso. Elimine la solución no utilizada de forma apropiada.
Utilizar el medicamento inmediatamente tras la disolución completa del polvo.
No refrigerar el medicamento después de la preparación.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de ADVATE
- El principio activo es octocog alfa (factor VIII humano de coagulación producido por tecnología de ADN recombinante). Cada vial de polvo contiene
nominalmente 250, 500, 1000, 1500, 2000, o 3000 UI de octocog alfa.
- Los demás componentes son manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80 y glutatión (reducido)
Vial de disolvente: 5 ml de agua esterilizada para preparaciones inyectables
Aspecto del producto y contenido del envase
ADVATE es un polvo desmenuzable de color blanco a blanquecino.
Después de su reconstitución, la solución es transparente, incolora y libre de partículas extrañas.
Cada envase contiene también un equipo para reconstitución (BAXJECT II).
Titular de la autorización de comercialización
Baxter AG Industriestrasse 67 A-1221 Viena
Responsables de la fabricación
Baxalta Belgium Manufacturing SA Boulevard René Branquart 80 B-7860 Lessines Bélgica
Baxter SA
Boulevard René Branquart 80
B-7860 Lessines
Bélgica
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00 |
Lietuva UAB "Baxter Lithuania'' Tel: +370 5 269 16 90 / +370 5 252 71 00 |
|
Etarapun EaKCTep Etarapna EOOfl Tea.: +359 2 9808482 |
Luxembourg/Luxemburg Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00 |
|
Ceská republika Baxter Czech spol.s.r.o. Tel.: +420 225774111 |
Magyarország Baxter Hungary Kft. Tel.: +36 1 202 1980 |
|
Danmark Baxalta Denmark A/S Tlf: +45 32 70 12 00 |
Malta Baxalta UK Limited Tel.: +44 1 635 798 777 |
|
Deutschland Baxalta Deutschland GmbH Tel: +49 89 262077-011 |
Nederland Baxalta Netherlands B.V. Tel: +31 30 799 27 77 |
|
Eesti OÜ Baxter Estonia Tel.: +372 6 515 120 |
Norge Baxalta Norway AS Tlf: +47 22 585 000 |
|
EXláda Baxter (Hellas) E.n.E. TpT,: +30 210 28 80 000 |
Osterreich Baxalta Osterreich GmbH Tel.: +43 1 20100-0 |
|
España Baxalta Spain S.L. Tel: +34 91 790 42 22 |
Polska Baxter Polska Sp. z o.o. Tel.: +48 22 4883 777 |
|
France Baxalta France S.A.S. Tél: +33 1 70 96 06 00 |
Portugal Baxalta Portugal, Unipessoal, Lda. Tel: +351 21 122 03 00 |
|
Hrvatska Baxter d.o.o. Tel: +386 1 420 16 80 |
Romania FARMACEUTICA REMEDIA SA Tel.: +40 21 321 16 40 |
|
Ireland Baxalta UK Limited Tel: +44 1 635 798 777 |
Slovenija Baxter d.o.o. Tel.: +386 1 420 16 80 |
|
Ísland Lyfjaver ehf. Sími: +354 533 6100 |
Slovenská republika Baxter Slovakia s.r.o. Tel: +421 2 3210 1150 |
|
Italia Baxalta Italy S.r.l. Tel: +39 06 45224 600 |
Suomi/Finland Baxalta Finland Oy Puh/Tel: +358 201478200 |
Fecha de la última revisión de este prospecto
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu/.
Instrucciones para la preparación y administración
Se requiere una técnica aséptica para la preparación de la solución y su administración.
Utilizar sólo el agua esterilizada para preparaciones inyectables y el equipo de reconstitución que se incluyen en cada envase de ADVATE. ADVATE no se debe mezclar con otros medicamentos o disolventes.
Se recomienda encarecidamente registrar el nombre y el número de lote del producto cada vez que se administre ADVATE.
Instrucciones para la reconstitución
• No usar después de la fecha de caducidad que aparece en las etiquetas y en el envase.
• No utilizar si el equipo BAXJECTII, el sistema estéril de protección o su envase está dañado

Kúrcpog
Baxter (Helias) E.n.E. TqX: +30 210 28 80 000
Sverige
Baxalta Sweden AB Tel: +46 8 50 53 26 00
Latvija
SIA BAXTER Latvia Tel.: +371 67 784 784
United Kingdom
Baxalta UK Limited Tel: +44 1 635 798 777
o muestra algún signo de deterioro, como indica el símbolo: No refrigerar la preparación después de la reconstitución.
1. Si el medicamento se encuentra en la nevera, coger de la nevera los viales de polvo y de disolvente de ADVATE y dejarlos a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Quitar los protectores de los viales de polvo y disolvente.
4. Limpiar los tapones con toallitas impregnadas de alcohol. Colocar los viales en una superficie plana y limpia.
5. Abrir el envoltorio del equipo BAXJECT II quitando la tapa de papel sin tocar el interior (Fig. a). No sacar el equipo del envoltorio. No utilizar si el equipo BAXJECT II, su sistema de barrera estéril o su acondicionamiento están dañados o muestran algún signo de deterioro.
6. Dar la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente. Coger el envoltorio por su extremo y sacar el equipo BAXJECT II de su envoltorio (Fig. b). No quitar el protector azul del equipo BAXJECT II.
7. Para la reconstitución, utilizar solamente el agua esterilizada para preparaciones inyectables y el equipo para reconstitución incluidos en el envase. Con el equipo BAXJECT II unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico blanca dentro del tapón del vial de polvo ADVATE. El vacío hará que el disolvente penetre en el vial de polvo ADVATE (Fig. c).
8. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará
a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presentar partículas extrañas.
Fig. a Fig. b Fig. c

Instrucciones para la inyección
Para la administración se requiere el uso de una jeringa luer-lock.
Nota importante:
• No intente administrar la inyección a menos que haya recibido una formación especial de su médico o enfermera.
• Inspeccione la solución preparada para detectar partículas o decoloración antes de la administración (la solución debe ser transparente, incolora y libre de partículas extrañas).
No utilice ADVATE si la solución no está totalmente transparente o no está disuelta completamente.
1. Quitar el protector azul del equipo BAXJECT II. No introducir aire en la jeringa. Conectar la jeringa al equipo BAXJECT II (Fig. d).
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente (Fig. e).
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar intravenosamente. La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de ADVATE. Si observa un aumento significativo del pulso, debe reducir la velocidad de administración o interrumpir temporalmente la inyección, normalmente esto permite que los síntomas desaparezcan en seguida (ver sección 4 “Posibles efectos adversos”).
5. Elimine la solución no utilizada de forma apropiada.
Fig. d

Fig. e

Esta información está destinada únicamente a profesionales del sector sanitario:
Tratamiento bajo demanda
En el caso de los episodios hemorrágicos posteriores, la actividad de factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. Se puede utilizar la siguiente tabla como guía de dosificación en episodios hemorrágicos y cirugía.
La dosis y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual. En determinadas circunstancias (ej.: presencia de un título bajo de inhibidor) pueden ser necesarias dosis mayores a las calculadas mediante la fórmula.
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (% o UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente o hemorragia muscular u oral. |
20 - 40 |
Repetir la inyección cada 12 a 24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma. |
30 - 60 |
Repetir la inyección cada 12-24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) durante 3 a 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir la inyección cada 8 a 24 horas (cada 6 a 12 horas en los pacientes menores de 6 años) hasta superar el peligro. |
|
Cirugía | ||
|
Menor Incluyendo extracción dental. |
30 - 60 |
Cada 24 horas (cada 12 a 24 horas en los pacientes menores de 6 años), al menos 1 día, hasta lograr la curación. |
|
Mayor |
80 - 100 (pre y postoperatorio) |
Repetir la inyección cada 8-24 horas (cada 6 a 24 horas en los pacientes menores de 6 años) hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor VIII del 30% al 60% (UI/dl). |
Prospecto: Información para el usuario
ADVATE 250 UI polvo y disolvente para solución inyectable ADVATE 500 UI polvo y disolvente para solución inyectable ADVATE 1000 UI polvo y disolvente para solución inyectable ADVATE 1500 UI polvo y disolvente para solución inyectable ADVATE 2000 UI polvo y disolvente para solución inyectable ADVATE 3000 UI polvo y disolvente para solución inyectable
Octocog alfa (factor VIII humano de coagulación recombinante)
Lea todo el prospecto detenidamente antes de empezar a usar el medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Este medicamento se le ha recetado solamente a usted y no debe dárselo a otras personas, aunque tengan los mismos sintomas, ya que puede perjudicarles.
- Si experimenta cualquier tipo de efecto adverso, consulte a su medico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es ADVATE y para qué se utiliza
2. Qué necesita saber antes de empezar a usar ADVATE
3. Cómo usar ADVATE
4. Posibles efectos adversos
5. Conservación de ADVATE
6. Contenido del envase e nformación adicional
1. Qué es ADVATE y para qué se utiliza
ADVATE contiene el principio activo octocog alfa, factor VIII humano de coagulación producido por tecnología de ADN recombinante. El factor VIII es necesario para coagular la sangre y parar las hemorragias. En pacientes con hemofilia A el factor VIII falta o no funciona correctamente (ausencia hereditaria de factor VIII).
ADVATE se utiliza para el tratamiento y la prevención de hemorragias en pacientes de todos los grupos de edad que padecen hemofilia A (un trastorno hemorrágico hereditario provocado por la ausencia de factor VIII).
ADVATE se prepara sin añadir ninguna proteína derivada humana o animal en todo el proceso de fabricación.
2. Qué necesita saber antes de empezar a usar ADVATE No use ADVATE
- si es alérgico (hipersensible) al octocog alfa o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- si es alérgico a las proteínas del ratón o del hámster
Si no está seguro si es alérgico, consulte con su médico.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar ADVATE. Informe a su médico si ha sido tratado previamente con medicamentos que contienen factor VIII, especialmente si desarrolló inhibidores ya que puede existir un riesgo elevado de que esto vuelva a suceder. Los inhibidores son anticuerpos que bloquean el factor VIII que reducen la eficacia de ADVATE para prevenir el control del sangrado. El desarrollo de inhibidores es una complicación conocida en el tratamiento de la hemofilia A. Si su hemorragia no llega a controlarse con ADVATE, consulte a su médico inmediatamente.
Existe un riesgo escasode que pueda experimentar una reacción anafiláctica (una reacción alérgica repentina grave) a ADVATE. Debe ser consciente de que los primeros síntomas de una reacción alérgica son sarpullido, picor, ronchas, picor generalizado, hinchazón de los labios y lengua, dificultad para respirar, jadeos, opresión en el pecho, sensación de malestar general y mareos. Estos síntomas pueden ser un aviso de shock anafiláctico, que también puede producir mareos graves, pérdida de consciencia y dificultades graves para respirar.
Si experimenta cualquiera de estos síntomas, interrumpa inmediatamente la administración del medicamento y consulte con un médico. Los síntomas graves, incluyendo la dificultad para respirar y (casi) desmayos requieren un tratamiento de emergencia rápido.
Pacientes que desarrollan inhibidores del Factor VIII
Si el nivel de factor VIII en el plasma no alcanza los valores esperados con ADVATE, o si no se controla adecuadamente la hemorragia, puede deberse al desarrollo de inhibidores del factor VIII. Esto se comprobará por su médico.Usted puede necesitar una dosis mayor de ADVATE o, incluso, otro medicamento para controlar las hemorragias. No aumente la dosis total de ADVATE que utiliza para controlar su hemorragia sin consultar a su médico.
Niños y adolescentes
Las advertencias y precauciones indicadas se aplican a adultos y niños (de 0 a 18 años de edad).
Uso de ADVATE con otros medicamentos
Informe a su médico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su medico antes de utilizar este medicamento.
Conducción y uso de máquinas
El tratamiento con ADVATE no tiene influencia sobre la capacidad para conducir y utilizar maquinaria.
ADVATE contiene sodio
Este medicamento contiene 0,45 mmol de sodio (10 mg) por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
3. Cómo usar ADVATE
El tratamiento con ADVATE se iniciará por un médico con experiencia en el tratamiento de pacientes con hemofilia A.
Su médico calculará su dosis de ADVATE (en unidades internacionales o UI) en función de su estado, su peso corporal y de si va a ser utilizado para la prevención o para el tratamiento de hemorragias.
La frecuencia de administración dependerá de cómo actúe ADVATE. La terapia sustitutiva con ADVATE es normalmente un tratamiento de por vida.
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico.
Prevención de hemorragias
La dosis habitual de octocog alfa es de 20 a 40 UI por kg de peso corporal, administrado
cada 2 ó 3 días. Sin embargo, en algunos casos, especialmente en los pacientes más jóvenes, pueden
requerirse la administración más frecuente de las inyecciones o dosis mayores.
Tratamiento de hemorragias
La dosis de octocog alfa se calcula en función de su peso corporal y de los niveles de factor VIII que se quieran conseguir. Los niveles de factor VIII a alcanzar dependerán de la gravedad y de la localización de la hemorragia.
Dosis (UI) = peso corporal (kg) x aumento de Factor VIII deseado (% del normal) x 0,5
Si tiene la impresión de que el efecto de ADVATE es insuficiente, consulte con su médico.
Su médico realizará los análisis de laboratorio apropiados para asegurarse de que tiene los niveles adecuados de factor VIII. Esto es especialmente importante si se va a someter a una operación importante.
Uso en niños y adolescentes (de 0 a 18 años de edad)
Para el tratamiento de hemorragias, la dosis en niños no difiere de la de los pacientes adultos. Para evitar hemorragias en niños menores de 6 años, se recomiendan dosis de 20 a 50 UI por kg de peso corporal de 3 a 4 veces por semana. La administración de ADVATE en niños (intravenosa) no difiere de la administración en adultos. Puede ser necesario un dispositivo de acceso venoso central (CVAD) para permitir infusiones frecuentes de productos de factor VIII.
Cómo se administra ADVATE
ADVATE normalmente se inyecta en vena (vía intravenosa) por su médico o enfermera. Usted o cualquier otra persona pueden también administrar la inyección de ADVATE pero únicamente después de recibir la formación adecuada. Las instrucciones detalladas para su administración se describen al final de este prospecto.
Si usa más ADVATE del que debiera
Siga exactamente las instrucciones de administración de ADVATE indicadas por su médico. Consulte a su médico si tiene dudas.Si se inyecta una dosis mayor de ADVATE de la recomendada, consulte con su médico lo antes posible.
Si olvidó usar ADVATE
No se inyecte una dosis doble para compensar las dosis olvidadas. Adminístrese la siguiente inyección como está establecido y continúe como le había indicado su médico.
Si interrumpe el tratamiento con ADVATE
No deje de usar ADVATE sin consultarlo con su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Si se producen reacciones alérgicas (anafilácticas) repentinas y graves, se debe parar
inmediatamente la inyección. Contacte con su médico inmediatamente si tiene alguno de los siguientes síntomas iniciales de las reacciones alérgicas:
- erupción, sarpullido, ronchas y picor generalizado,
- hinchazón de los labios y la lengua,
- dificultad para respirar, jadeos, opresión en el pecho,
- sensación de malestar general,
- mareos y pérdida de consciencia.
Los síntomas graves, como dificultad para respirar y (casi) síncope, requieren un tratamiento temprano de emergencia.
Efectos adversos frecuentes (pueden afectar hasta a 1 de cada 10 pacientes) inhibidores del factor VIII, dolor de cabeza y fiebre
Efectos adversos poco frecuentes (pueden afectar hasta a 1 de cada 100 pacientes) mareos, gripe, desfallecimiento, pulsaciones anormales, manchas rojas asociadas a picor, molestias en el pecho, cardenal en la zona de inyección, reacción en la zona de inyección, picor, aumento de la sudoración, sabor extraño en la boca, sofocos, migrañas, pérdidas de memoria, escalofríos, diarrea, náusea, vómitos, dificultad para respirar, dolor de garganta, infección de los vasos linfáticos, palidez, inflamación ocular, sarpullidos, sudoración excesiva, inflamación de pies y piernas, disminución del número de glóbulos rojos, aumento de un tipo de leucocitos (monocitos) y dolor en la parte superior del abdomen o parte inferior del pecho.
Relacionados con la cirugía
infección relacionada con el catéter, disminución del número de glóbulos rojos, hinchazón de las extremidades y las articulaciones, sangrado prolongado después de la retirada del drenaje, disminución de los niveles de factor VIII y hematoma postoperatorio
Relacionados con los dispositivos de acceso venoso central (CVAD)
Infección relacionada con catéter, infección sistémica y coágulo de sangre local en el lado del catéter.
Efectos adversos con frecuencia desconocida (no se puede estimar la frecuencia a partir de los datos disponibles)
Reacciones que pueden ser mortales (anafilaxis) y otras reacciones alérgicas (hipersensibilidad), trastornos generales (cansancio, falta de energía).
Efectos adversos adicionales en niños
Además del desarrollo de inhibidores en pacientes pediátricos sin tratamiento previo y las complicaciones asociadas al catéter, en los ensayos clínicos no se observaron diferencias específicas de la edad en los efectos adversos.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
Conservación de ADVATE
5.
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase después de CAD. La fecha de caducidad se refiere al último día del mes que se indica.
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
Hasta la fecha de caducidad, el blister con el medicamento puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único no superior a 6 meses. En este caso, este medicamento caduca al final de este periodo de 6 meses o en la fecha de caducidad impresa en el blister, lo que ocurra antes. Por favor, anote en el envase del medicamento la fecha de finalización del periodo de conservación a temperatura ambiente de 6 meses. El medicamento no puede refrigerarse de nuevo después de conservarse a temperatura ambiente.
Conservar el blister con el medicamento en el embalaje exterior para protegerlo de la luz.
Este medicamento es para un solo uso. Elimine la solución no utilizada de forma apropiada.
Utilizar el medicamento inmediatamente tras la disolución completa del polvo.
No refrigerar el medicamento después de la preparación.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de ADVATE
- El principio activo es octocog alfa (factor VIII humano de coagulación producido por tecnología de ADN recombinante). Cada vial de polvo contiene nominalmente 250, 500, 1000, 1500, 2000, o 3000 UI de octocog alfa.
- Los demás componentes son manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80 y glutatión (reducido)
Vial de disolvente: 5 ml de agua esterilizada para preparaciones inyectables
Aspecto del producto y contenido del envase
ADVATE es un polvo desmenuzable de color blanco a blanquecino.
Después de su reconstitución, la solución es transparente, incolora y libre de partículas extrañas.
Titular de la autorización de comercialización
Baxter AG Industriestrasse 67 A-1221 Viena
Responsables de la fabricación
Baxalta Belgium Manufacturing SA Boulevard René Branquart 80 B-7860 Lessines
Bélgica
Baxter SA
Boulevard René Branquart 80
B-7860 Lessines
Bélgica
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00 |
Lietuva UAB "Baxter Lithuania'' Tel: +370 5 269 16 90 / +370 5 252 71 00 |
|
Etarapna EaKCTep Etarapna EOOfl Tea.: +359 2 9808482 |
Luxembourg/Luxemburg Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00 |
|
Ceská republika Baxter Czech spol.s.r.o. Tel.: +420 225774111 |
Magyarország Baxter Hungary Kft. Tel.: +36 1 202 1980 |
|
Danmark Baxalta Denmark A/S Tlf: +45 32 70 12 00 |
Malta Baxalta UK Limited Tel.: +44 1 635 798 777 |
|
Deutschland Baxalta Deutschland GmbH Tel: +49 89 262077-011 |
Nederland Baxalta Netherlands B.V. Tel: +31 30 799 27 77 |
|
Eesti OÜ Baxter Estonia Tel.: +372 6 515 120 |
Norge Baxalta Norway AS Tlf: +47 22 585 000 |
|
EXlába Baxter (Hellas) E.n.E. TqL: +30 210 28 80 000 |
Osterreich Baxalta Osterreich GmbH Tel.: +43 1 20100-0 |
|
España Baxalta Spain S.L. Tel: +34 91 790 42 22 |
Polska Baxter Polska Sp. z o.o. Tel.: +48 22 4883 777 |
|
France Baxalta France S.A.S. Tél: +33 1 70 96 06 00 |
Portugal Baxalta Portugal, Unipessoal, Lda. Tel: +351 21 122 03 00 |
|
Hrvatska Baxter d.o.o. Tel: +386 1 420 16 80 |
Romania FARMACEUTICA REMEDIA SA Tel.: +40 21 321 16 40 |
|
Ireland Baxalta UK Limited Tel: +44 1 635 798 777 |
Slovenija Baxter d.o.o. Tel.: +386 1 420 16 80 |
Suomi/Finland
Italia
Baxalta Italy S.r.l.
Tel: +39 06 45224 600
Baxalta Finland Oy Puh/Tel: +358 201478200
Sverige
Baxalta Sweden AB Tel: +46 8 50 53 26 00
United Kingdom
Baxalta UK Limited Tel: +44 1 635 798 777
Fecha de la última revisión de este prospecto
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu/.
Instrucciones para la preparación y administración
ADVATE no se debe mezclar con otros medicamentos o disolventes.
Se recomienda encarecidamente registrar el nombre y el número de lote del producto cada vez que se
administre ADVATE.
Instrucciones para la reconstitución
• No usar después de la fecha de caducidad que aparece en las etiquetas y en el envase.
• No lo utilice si la tapa del blister no está perfectamente sellada.
• No refrigerar la preparación después de la reconstitución.
1. Si el medicamento se encuentra en la nevera, coger de la nevera el blister sellado (contiene viales de polvo y disolvente precargados con el sistema para su reconstitución) y dejarlo a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Abrir el envoltorio de ADVATE quitando la tapa. Extraer el sistema BAXJECT III del blister.
4. Colocar ADVATE en una superficie plana con el vial de disolvente encima (Fig. 1). El vial de disolvente tiene una raya azul. No quitar el protector azul hasta que se le indique más adelante.
5. Mientras sujeta ADVATE con una mano en el sistema BAXJECT III, presionar con fuerza el vial de disolvente con la otra mano hasta que el sistema quede totalmente contraído y el disolvente entre dentro del vial de ADVATE (Fig. 2). No inclinar el sistema hasta que haya finalizado la transferencia.
6. Comprobar que ha finalizado la transferencia del disolvente. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig. 1 Fig. 2 Fig. 3

Instrucciones para la inyección
Se requiere una técnica aséptica durante la administración.
Para la administración se requiere el uso de una jeringa luer-lock.
Nota importante:
• No intente administrar la inyección a menos que haya recibido una formación especial de su médico o enfermera.
• Inspeccione la solución preparada para detectar partículas o decoloración antes de la administración (la solución debe ser transparente, incolora y libre de partículas extrañas).
No utilice ADVATE si la solución no está totalmente transparente o no está disuelta completamente.
1. Quitar el protector azul del sistema BAXJECT III. No introducir aire en la jeringa. Conectar la jeringa al sistema BAXJECT III.
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente.
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar intravenosamente. La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de ADVATE. Si observa un aumento significativo del pulso, debe reducir la velocidad de administración o interrumpir temporalmente la inyección, normalmente esto permite que los síntomas desaparezcan en seguida (ver sección 4 “Posibles efectos adversos”).
5. Elimine la solución no utilizada de forma apropiada.
Esta información está destinada únicamente a profesionales del sector sanitario:
Tratamiento bajo demanda
En el caso de los episodios hemorrágicos posteriores, la actividad de factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. Se puede utilizar la siguiente tabla como guía de dosificación en episodios hemorrágicos y cirugía.
La dosis y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual.
En determinadas circunstancias (ej.: presencia de un título bajo de inhibidor) pueden ser necesarias dosis mayores a las calculadas mediante la fórmula.
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (% o UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente o hemorragia muscular u oral. |
20 - 40 |
Repetir la inyección cada 12 a 24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma. |
30 - 60 |
Repetir la inyección cada 12-24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) durante 3 a 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir la inyección cada 8 a 24 horas (cada 6 a 12 horas en los pacientes menores de 6 años) hasta superar el peligro. |
|
Cirugía | ||
|
Menor Incluyendo extracción dental. |
30 - 60 |
Cada 24 horas (cada 12 a 24 horas en los pacientes menores de 6 años), al menos 1 día, hasta lograr la curación. |
|
Mayor |
80 - 100 (pre y postoperatorio) |
Repetir la inyección cada 8-24 horas (cada 6 a 24 horas en los pacientes menores de 6 años) hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor VIII del 30% al 60% (UI/dl). |
Prospecto: Información para el usuario
ADVATE 250 UI polvo y disolvente para solución inyectable ADVATE 500 UI polvo y disolvente para solución inyectable ADVATE 1000 UI polvo y disolvente para solución inyectable ADVATE 1500 UI polvo y disolvente para solución inyectable
Octocog alfa (factor VIII humano de coagulación recombinante)
Lea todo el prospecto detenidamente antes de empezar a usar el medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Este medicamento se le ha recetado solamente a usted y no debe dárselo a otras personas, aunque tengan los mismos sintomas, ya que puede perjudicarles.
- Si experimenta cualquier tipo de efecto adverso, consulte a su medico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es ADVATE y para qué se utiliza
2. Qué necesita saber antes de empezar a usar ADVATE
3. Cómo usar ADVATE
4. Posibles efectos adversos
5. Conservación de ADVATE
6. Contenido del envase e nformación adicional
1. Qué es ADVATE y para qué se utiliza
ADVATE contiene el principio activo octocog alfa, factor VIII humano de coagulación producido por tecnología de ADN recombinante. El factor VIII es necesario para coagular la sangre y parar las hemorragias. En pacientes con hemofilia A el factor VIII falta o no funciona correctamente (ausencia hereditaria de factor VIII).
ADVATE se utiliza para el tratamiento y la prevención de hemorragias en pacientes de todos los grupos de edad que padecen hemofilia A (un trastorno hemorrágico hereditario provocado por la ausencia de factor VIII).
ADVATE se prepara sin añadir ninguna proteína derivada humana o animal en todo el proceso de fabricación.
2. Qué necesita saber antes de empezar a usar ADVATE No use ADVATE
- si es alérgico (hipersensible) al octocog alfa o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- si es alérgico a las proteínas del ratón o del hámster
Si no está seguro si es alérgico, consulte con su médico.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar ADVATE. Informe a su médico si ha sido tratado previamente con medicamentos que contienen factor VIII, especialmente si desarrolló inhibidores ya que puede existir un riesgo elevado de que esto vuelva a suceder. Los inhibidores son anticuerpos que
bloquean el factor VIII que reducen la eficacia de ADVATE para prevenir el control del sangrado. El desarrollo de inhibidores es una complicación conocida en el tratamiento de la hemofilia A. Si su hemorragia no llega a controlarse con ADVATE, consulte a su médico inmediatamente.
Existe un riesgo escasode que pueda experimentar una reacción anafiláctica (una reacción alérgica repentina grave) a ADVATE. Debe ser consciente de que los primeros síntomas de una reacción alérgica son sarpullido, picor, ronchas, picor generalizado, hinchazón de los labios y lengua, dificultad para respirar, jadeos, opresión en el pecho, sensación de malestar general y mareos. Estos síntomas pueden ser un aviso de shock anafiláctico, que también puede producir mareos graves, pérdida de consciencia y dificultades graves para respirar.
Si experimenta cualquiera de estos síntomas, interrumpa inmediatamente la administración del medicamento y consulte con un médico. Los síntomas graves, incluyendo la dificultad para respirar y (casi) desmayos requieren un tratamiento de emergencia rápido.
Pacientes que desarrollan inhibidores del Factor VIII
Si el nivel de factor VIII en el plasma no alcanza los valores esperados con ADVATE, o si no se controla adecuadamente la hemorragia, puede deberse al desarrollo de inhibidores del factor VIII. Esto se comprobará por su médico.Usted puede necesitar una dosis mayor de ADVATE o, incluso, otro medicamento para controlar las hemorragias. No aumente la dosis total de ADVATE que utiliza para controlar su hemorragia sin consultar a su médico.
Niños y adolescentes
Las advertencias y precauciones indicadas se aplican a adultos y niños (de 0 a 18 años de edad).
Uso de ADVATE con otros medicamentos
Informe a su médico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su medico antes de utilizar este medicamento.
Conducción y uso de máquinas
El tratamiento con ADVATE no tiene influencia sobre la capacidad para conducir y utilizar maquinaria.
ADVATE contiene sodio
Este medicamento contiene 0,45 mmol de sodio (10 mg) por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
Aplicación incorrecta de ADVATE
La aplicación incorrecta (la inyección en la arteria o fuera de la vena) se debe evitar ya que pueden producirse reacciones leves y a corto plazo en el lugar de la inyección, como cardenales y enrojecimiento.
3. Cómo usar ADVATE
El tratamiento con ADVATE se iniciará por un médico con experiencia en el tratamiento de pacientes con hemofilia A.
Su médico calculará su dosis de ADVATE (en unidades internacionales o UI) en función de su estado, su peso corporal y de si va a ser utilizado para la prevención o para el tratamiento de hemorragias.
La frecuencia de administración dependerá de cómo actúe ADVATE. La terapia sustitutiva con ADVATE es normalmente un tratamiento de por vida.
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico.
Prevención de hemorragias
La dosis habitual de octocog alfa es de 20 a 40 UI por kg de peso corporal, administrado
cada 2 ó 3 días. Sin embargo, en algunos casos, especialmente en los pacientes más jóvenes, pueden
requerirse la administración más frecuente de las inyecciones o dosis mayores.
Tratamiento de hemorragias
La dosis de octocog alfa se calcula en función de su peso corporal y de los niveles de factor VIII que se quieran conseguir. Los niveles de factor VIII a alcanzar dependerán de la gravedad y de la localización de la hemorragia.
Dosis (UI) = peso corporal (kg) x aumento de Factor VIII deseado (% del normal) x 0,5
Si tiene la impresión de que el efecto de ADVATE es insuficiente, consulte con su médico.
Su médico realizará los análisis de laboratorio apropiados para asegurarse de que tiene los niveles adecuados de factor VIII. Esto es especialmente importante si se va a someter a una operación importante.
Uso en niños y adolescentes (de 0 a 18 años de edad)
Para el tratamiento de hemorragias, la dosis en niños no difiere de la de los pacientes adultos. Para evitar hemorragias en niños menores de 6 años, se recomiendan dosis de 20 a 50 UI por kg de peso corporal de 3 a 4 veces por semana. La administración de ADVATE en niños (intravenosa) no difiere de la administración en adultos. Puede ser necesario un dispositivo de acceso venoso central (CVAD) para permitir infusiones frecuentes de productos de factor VIII. Esta presentación está disponible con 5 ml de disolvente y 2 ml de disolvente. No obstante, para la presentación de 2 ml no se dispone información en niños de menos de 2 años.
Debido a la disminución del volumen de inyección de ADVATE reconstituido en 2 ml, el tiempo para reaccionar a las reacciones de hipersensibilidad durante una inyección se reduce aún más. Por tanto, se aconseja tener precaución durante la inyección de ADVATE reconstituido en 2 ml, especialmente en niños.
Cómo se administra ADVATE
ADVATE normalmente se inyecta en vena (vía intravenosa) por su médico o enfermera. Usted o cualquier otra persona pueden también administrar la inyección de ADVATE pero únicamente después de recibir la formación adecuada. Las instrucciones detalladas para su administración se describen al final de este prospecto.
Si usa más ADVATE del que debiera
Siga exactamente las instrucciones de administración de ADVATE indicadas por su médico. Consulte a su médico si tiene dudas.Si se inyecta una dosis mayor de ADVATE de la recomendada, consulte con su médico lo antes posible.
Si olvidó usar ADVATE
No se inyecte una dosis doble para compensar las dosis olvidadas. Adminístrese la siguiente inyección como está establecido y continúe como le había indicado su médico.
Si interrumpe el tratamiento con ADVATE
No deje de usar ADVATE sin consultarlo con su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Si se producen reacciones alérgicas (anafilácticas) repentinas y graves, se debe parar
inmediatamente la inyección. Contacte con su médico inmediatamente si tiene alguno de los siguientes síntomas iniciales de las reacciones alérgicas:
- erupción, sarpullido, ronchas y picor generalizado,
- hinchazón de los labios y la lengua,
- dificultad para respirar, jadeos, opresión en el pecho,
- sensación de malestar general,
- mareos y pérdida de consciencia.
Los síntomas graves, como dificultad para respirar y (casi) síncope, requieren un tratamiento temprano de emergencia.
Efectos adversos frecuentes (pueden afectar hasta a 1 de cada 10 pacientes) inhibidores del factor VIII, dolor de cabeza y fiebre
Efectos adversos poco frecuentes (pueden afectar hasta a 1 de cada 100 pacientes) mareos, gripe, desfallecimiento, pulsaciones anormales, manchas rojas asociadas a picor, molestias en el pecho, cardenal en la zona de inyección, reacción en la zona de inyección, picor, aumento de la sudoración, sabor extraño en la boca, sofocos, migrañas, pérdidas de memoria, escalofríos, diarrea, náusea, vómitos, dificultad para respirar, dolor de garganta, infección de los vasos linfáticos, palidez, inflamación ocular, sarpullidos, sudoración excesiva, inflamación de pies y piernas, disminución del número de glóbulos rojos, aumento de un tipo de leucocitos (monocitos) y dolor en la parte superior del abdomen o parte inferior del pecho.
Relacionados con la cirugía
infección relacionada con el catéter, disminución del número de glóbulos rojos, hinchazón de las extremidades y las articulaciones, sangrado prolongado después de la retirada del drenaje, disminución de los niveles de factor VIII y hematoma postoperatorio
Relacionados con los dispositivos de acceso venoso central (CVAD)
Infección relacionada con catéter, infección sistémica y coágulo de sangre local en el lado del catéter.
Efectos adversos con frecuencia desconocida (no se puede estimar la frecuencia a partir de los datos disponibles)
Reacciones que pueden ser mortales (anafilaxis) y otras reacciones alérgicas (hipersensibilidad), trastornos generales (cansancio, falta de energía).
Efectos adversos adicionales en niños
Además del desarrollo de inhibidores en pacientes pediátricos sin tratamiento previo y las complicaciones asociadas al catéter, en los ensayos clínicos no se observaron diferencias específicas de la edad en los efectos adversos.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
Conservación de ADVATE
5.
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase después de CAD. La fecha de caducidad se refiere al último día del mes que se indica.
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
Hasta la fecha de caducidad, el vial de polvo puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único no superior a 6 meses. En este caso, este medicamento caduca al final de este periodo de 6 meses o en la fecha de caducidad impresa en el vial del producto, lo que ocurra antes. Por favor, anote en el envase del medicamento la fecha de finalización del periodo de conservación a temperatura ambiente de 6 meses. El medicamento no puede refrigerarse de nuevo después de conservarse a temperatura ambiente.
Conservar el vial en el embalaje exterior para protegerlo de la luz.
Este medicamento es para un solo uso. Elimine la solución no utilizada de forma apropiada.
Utilizar el medicamento inmediatamente tras la disolución completa del polvo.
No refrigerar el medicamento después de la preparación.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de ADVATE
- El principio activo es octocog alfa (factor VIII humano de coagulación producido por tecnología de ADN recombinante). Cada vial de polvo contiene nominalmente 250, 500, 1000 o 1500 UI de octocog alfa.
- Los demás componentes son manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80 y glutatión (reducido)
Vial de disolvente: 2 ml de agua esterilizada para preparaciones inyectables
Aspecto del producto y contenido del envase
ADVATE es un polvo desmenuzable de color blanco a blanquecino.
Después de su reconstitución, la solución es transparente, incolora y libre de partículas extrañas.
Cada envase contiene también un equipo para reconstitución (BAXJECT II).
Titular de la autorización de comercialización
Baxter AG Industriestrasse 67 A-1221 Viena
Responsables de la fabricación
Baxalta Belgium Manufacturing SA Boulevard René Branquart 80 B-7860 Lessines Bélgica
Baxter SA
Boulevard René Branquart 80
B-7860 Lessines
Bélgica
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00 |
Lietuva UAB "Baxter Lithuania'' Tel: +370 5 269 16 90 / +370 5 252 71 00 |
|
Etarapna EaKCTep Etarapna EOOfl Tea.: +359 2 9808482 |
Luxembourg/Luxemburg Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00 |
|
Ceská republika Baxter Czech spol.s.r.o. Tel.: +420 225774111 |
Magyarország Baxter Hungary Kft. Tel.: +36 1 202 1980 |
|
Danmark Baxalta Denmark A/S Tlf: +45 32 70 12 00 |
Malta Baxalta UK Limited Tel.: +44 1 635 798 777 |
|
Deutschland Baxalta Deutschland GmbH Tel: +49 89 262077-011 |
Nederland Baxalta Netherlands B.V. Tel: +31 30 799 27 77 |
|
Eesti OÜ Baxter Estonia Tel.: +372 6 515 120 |
Norge Baxalta Norway AS Tlf: +47 22 585 000 |
|
EXláóa Baxter (Hellas) E.n.E. TqL: +30 210 28 80 000 |
Osterreich Baxalta Osterreich GmbH Tel.: +43 1 20100-0 |
|
España Baxalta Spain S.L. Tel: +34 91 790 42 22 |
Polska Baxter Polska Sp. z o.o. Tel.: +48 22 4883 777 |
|
France Baxalta France S.A.S. Tél: +33 1 70 96 06 00 |
Portugal Baxalta Portugal, Unipessoal, Lda. Tel: +351 21 122 03 00 |
|
Hrvatska Baxter d.o.o. Tel: +386 1 420 16 80 |
Romania FARMACEUTICA REMEDIA SA Tel.: +40 21 321 16 40 |
|
Ireland Baxalta UK Limited Tel: +44 1 635 798 777 |
Slovenija Baxter d.o.o. Tel.: +386 1 420 16 80 |
|
Ísland Lyfjaver ehf. Sími: +354 533 6100 |
Slovenská republika Baxter Slovakia s.r.o. Tel: +421 2 3210 1150 |
|
Italia Baxalta Italy S.r.l. Tel: +39 06 45224 600 |
Suomi/Finland Baxalta Finland Oy Puh/Tel: +358 201478200 |
|
Kúnpoq Baxter (Hellas) E.n.E. TqL: +30 210 28 80 000 |
Sverige Baxalta Sweden AB Tel: +46 8 50 53 26 00 |
|
Latvija SIA BAXTER Latvia Tel.: +371 67 784 784 |
United Kingdom Baxalta UK Limited Tel: +44 1 635 798 777 |
Fecha de la última revisión de este prospecto
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu/.
Instrucciones para la preparación y administración
Se requiere una técnica aséptica para la preparación de la solución y su administración.
Utilizar sólo el agua esterilizada para preparaciones inyectables y el equipo de reconstitución que se incluyen en cada envase de ADVATE. ADVATE no se debe mezclar con otros medicamentos o disolventes.
Se recomienda encarecidamente registrar el nombre y el número de lote del producto cada vez que se administre ADVATE.
Instrucciones para la reconstitución
• No usar después de la fecha de caducidad que aparece en las etiquetas y en el envase.
• No utilizar si el equipo BAXJECTII, el sistema estéril de protección o su envase está dañado

o muestra algún signo de deterioro, como indica el símbolo: No refrigerar la preparación después de la reconstitución.
1. Si el medicamento se encuentra en la nevera, coger de la nevera los viales de polvo y de disolvente de ADVATE y dejarlos a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Quitar los protectores de los viales de polvo y disolvente.
4. Limpiar los tapones con toallitas impregnadas de alcohol. Colocar los viales en una superficie plana y limpia.
5. Abrir el envoltorio del equipo BAXJECT II quitando la tapa de papel sin tocar el interior (Fig. a). No sacar el equipo del envoltorio. No utilizar si el equipo BAXJECT II, su sistema de barrera estéril o su acondicionamiento están dañados o muestran algún signo de deterioro.
6. Dar la vuelta al envoltorio e insertar la punta de plástico a través del tapón del disolvente. Coger el envoltorio por su extremo y sacar el equipo BAXJECT II de su envoltorio (Fig. b). No quitar el protector azul del equipo BAXJECT II.
7. Para la reconstitución, utilizar solamente el agua esterilizada para preparaciones inyectables y el equipo para reconstitución incluidos en el envase. Con el equipo BAXJECT II unido al vial de disolvente, invertir el sistema de tal forma que el vial de disolvente esté en la parte superior del equipo. Insertar la punta de plástico blanca dentro del tapón del vial de polvo ADVATE. El vacío hará que el disolvente penetre en el vial de polvo ADVATE (Fig. c).
8. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará
a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presentar partículas extrañas.
Fig. a
Fig. b
Fig. c
Instrucciones para la inyección
Para la administración se requiere el uso de una jeringa luer-lock.
Nota importante:
• No intente administrar la inyección a menos que haya recibido una formación especial de su médico o enfermera.
• Inspeccione la solución preparada para detectar partículas o decoloración antes de la administración (la solución debe ser transparente, incolora y libre de partículas extrañas).
No utilice ADVATE si la solución no está totalmente transparente o no está disuelta completamente.
1. Quitar el protector azul del equipo BAXJECT II. No introducir aire en la jeringa. Conectar la jeringa al equipo BAXJECT II (Fig. d).
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente (Fig. e).
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar intravenosamente. La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de ADVATE. Si observa un aumento significativo del pulso, debe reducir la velocidad de administración o interrumpir temporalmente la inyección, normalmente esto permite que los síntomas desaparezcan en seguida (ver sección 4 “Posibles efectos adversos”).
5. Elimine la solución no utilizada de forma apropiada.
Fig. d
Fig. e

Esta información está destinada únicamente a profesionales del sector sanitario:
Tratamiento bajo demanda
En el caso de los episodios hemorrágicos posteriores, la actividad de factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. Se puede utilizar la siguiente tabla como guía de dosificación en episodios hemorrágicos y cirugía.
La dosis y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual.
En determinadas circunstancias (ej.: presencia de un título bajo de inhibidor) pueden ser necesarias dosis mayores a las calculadas mediante la fórmula.
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (% o UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente o hemorragia muscular u oral. |
20 - 40 |
Repetir la inyección cada 12 a 24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma. |
30 - 60 |
Repetir la inyección cada 12-24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) durante 3 a 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir la inyección cada 8 a 24 horas (cada 6 a 12 horas en los pacientes menores de 6 años) hasta superar el peligro. |
|
Cirugía | ||
|
Menor |
30 - 60 |
Cada 24 horas (cada 12 a 24 horas |
|
Incluyendo extracción dental. |
en los pacientes menores de 6 años), al menos 1 día, hasta lograr la curación. | |
|
Mayor |
80 - 100 |
Repetir la inyección cada 8-24 horas |
|
(pre y postoperatorio) |
(cada 6 a 24 horas en los pacientes menores de 6 años) hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor VIII del 30% al 60% (UI/dl). |
Prospecto: Información para el usuario
ADVATE 250 UI polvo y disolvente para solución inyectable ADVATE 500 UI polvo y disolvente para solución inyectable ADVATE 1000 UI polvo y disolvente para solución inyectable ADVATE 1500 UI polvo y disolvente para solución inyectable
Octocog alfa (factor VIII humano de coagulación recombinante)
Lea todo el prospecto detenidamente antes de empezar a usar el medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Este medicamento se le ha recetado solamente a usted y no debe dárselo a otras personas, aunque tengan los mismos sintomas, ya que puede perjudicarles.
- Si experimenta cualquier tipo de efecto adverso, consulte a su medico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto:
1. Qué es ADVATE y para qué se utiliza
2. Qué necesita saber antes de empezar a usar ADVATE
3. Cómo usar ADVATE
4. Posibles efectos adversos
5. Conservación de ADVATE
6. Contenido del envase e nformación adicional
1. Qué es ADVATE y para qué se utiliza
ADVATE contiene el principio activo octocog alfa, factor VIII humano de coagulación producido por tecnología de ADN recombinante. El factor VIII es necesario para coagular la sangre y parar las hemorragias. En pacientes con hemofilia A el factor VIII falta o no funciona correctamente (ausencia hereditaria de factor VIII).
ADVATE se utiliza para el tratamiento y la prevención de hemorragias en pacientes de todos los grupos de edad que padecen hemofilia A (un trastorno hemorrágico hereditario provocado por la ausencia de factor VIII).
ADVATE se prepara sin añadir ninguna proteína derivada humana o animal en todo el proceso de fabricación.
2. Qué necesita saber antes de empezar a usar ADVATE No use ADVATE
- si es alérgico (hipersensible) al octocog alfa o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- si es alérgico a las proteínas del ratón o del hámster
Si no está seguro si es alérgico, consulte con su médico.
Advertencias y precauciones
Consulte a su médico antes de empezar a usar ADVATE. Informe a su médico si ha sido tratado previamente con medicamentos que contienen factor VIII, especialmente si desarrolló inhibidores ya que puede existir un riesgo elevado de que esto vuelva a suceder. Los inhibidores son anticuerpos que bloquean el factor VIII que reducen la eficacia de ADVATE para prevenir el control del sangrado. El desarrollo de inhibidores es una complicación conocida en el tratamiento de la hemofilia A. Si su hemorragia no llega a controlarse con ADVATE, consulte a su médico inmediatamente.
Existe un riesgo escasode que pueda experimentar una reacción anafiláctica (una reacción alérgica repentina grave) a ADVATE. Debe ser consciente de que los primeros síntomas de una reacción alérgica son sarpullido, picor, ronchas, picor generalizado, hinchazón de los labios y lengua, dificultad para respirar, jadeos, opresión en el pecho, sensación de malestar general y mareos. Estos síntomas pueden ser un aviso de shock anafiláctico, que también puede producir mareos graves, pérdida de consciencia y dificultades graves para respirar.
Si experimenta cualquiera de estos síntomas, interrumpa inmediatamente la administración del medicamento y consulte con un médico. Los síntomas graves, incluyendo la dificultad para respirar y (casi) desmayos requieren un tratamiento de emergencia rápido.
Pacientes que desarrollan inhibidores del Factor VIII
Si el nivel de factor VIII en el plasma no alcanza los valores esperados con ADVATE, o si no se controla adecuadamente la hemorragia, puede deberse al desarrollo de inhibidores del factor VIII. Esto se comprobará por su médico.Usted puede necesitar una dosis mayor de ADVATE o, incluso, otro medicamento para controlar las hemorragias. No aumente la dosis total de ADVATE que utiliza para controlar su hemorragia sin consultar a su médico.
Niños y adolescentes
Las advertencias y precauciones indicadas se aplican a adultos y niños (de 0 a 18 años de edad).
Uso de ADVATE con otros medicamentos
Informe a su médico si está utilizando, ha utilizado recientemente o podría tener que utilizar cualquier otro medicamento.
Embarazo y lactancia
Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su medico antes de utilizar este medicamento.
Conducción y uso de máquinas
El tratamiento con ADVATE no tiene influencia sobre la capacidad para conducir y utilizar maquinaria.
ADVATE contiene sodio
Este medicamento contiene 0,45 mmol de sodio (10 mg) por vial, lo que se debe tener en cuenta en pacientes con dietas pobres en sodio.
Aplicación incorrecta de ADVATE
La aplicación incorrecta (la inyección en la arteria o fuera de la vena) se debe evitar ya que pueden producirse reacciones leves y a corto plazo en el lugar de la inyección, como cardenales y enrojecimiento.
3. Cómo usar ADVATE
El tratamiento con ADVATE se iniciará por un médico con experiencia en el tratamiento de pacientes con hemofilia A.
Su médico calculará su dosis de ADVATE (en unidades internacionales o UI) en función de su estado, su peso corporal y de si va a ser utilizado para la prevención o para el tratamiento de hemorragias.
La frecuencia de administración dependerá de cómo actúe ADVATE. La terapia sustitutiva con ADVATE es normalmente un tratamiento de por vida.
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte de nuevo a su médico.
Prevención de hemorragias
La dosis habitual de octocog alfa es de 20 a 40 UI por kg de peso corporal, administrado
cada 2 ó 3 días. Sin embargo, en algunos casos, especialmente en los pacientes más jóvenes, pueden
requerirse la administración más frecuente de las inyecciones o dosis mayores.
Tratamiento de hemorragias
La dosis de octocog alfa se calcula en función de su peso corporal y de los niveles de factor VIII que se quieran conseguir. Los niveles de factor VIII a alcanzar dependerán de la gravedad y de la localización de la hemorragia.
Dosis (UI) = peso corporal (kg) x aumento de Factor VIII deseado (% del normal) x 0,5
Si tiene la impresión de que el efecto de ADVATE es insuficiente, consulte con su médico.
Su médico realizará los análisis de laboratorio apropiados para asegurarse de que tiene los niveles adecuados de factor VIII. Esto es especialmente importante si se va a someter a una operación importante.
Uso en niños y adolescentes (de 0 a 18 años de edad)
Para el tratamiento de hemorragias, la dosis en niños no difiere de la de los pacientes adultos. Para evitar hemorragias en niños menores de 6 años, se recomiendan dosis de 20 a 50 UI por kg de peso corporal de 3 a 4 veces por semana. La administración de ADVATE en niños (intravenosa) no difiere de la administración en adultos. Puede ser necesario un dispositivo de acceso venoso central (CVAD) para permitir infusiones frecuentes de productos de factor VIII. Esta presentación está disponible con 5 ml de disolvente y 2 ml de disolvente. No obstante, para la presentación de 2 ml no se dispone información en niños de menos de 2 años.
Debido a la disminución del volumen de inyección de ADVATE reconstituido en 2 ml, el tiempo para reaccionar a las reacciones de hipersensibilidad durante una inyección se reduce aún más. Por tanto, se aconseja tener precaución durante la inyección de ADVATE reconstituido en 2 ml, especialmente en niños.
Cómo se administra ADVATE
ADVATE normalmente se inyecta en vena (vía intravenosa) por su médico o enfermera. Usted o cualquier otra persona pueden también administrar la inyección de ADVATE pero únicamente después de recibir la formación adecuada. Las instrucciones detalladas para su administración se describen al final de este prospecto.
Si usa más ADVATE del que debiera
Siga exactamente las instrucciones de administración de ADVATE indicadas por su médico. Consulte a su médico si tiene dudas.Si se inyecta una dosis mayor de ADVATE de la recomendada, consulte con su médico lo antes posible.
Si olvidó usar ADVATE
No se inyecte una dosis doble para compensar las dosis olvidadas. Adminístrese la siguiente inyección como está establecido y continúe como le había indicado su médico.
Si interrumpe el tratamiento con ADVATE
No deje de usar ADVATE sin consultarlo con su médico.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Si se producen reacciones alérgicas (anafilácticas) repentinas y graves, se debe parar
inmediatamente la inyección. Contacte con su médico inmediatamente si tiene alguno de los siguientes síntomas iniciales de las reacciones alérgicas:
- erupción, sarpullido, ronchas y picor generalizado,
- hinchazón de los labios y la lengua,
- dificultad para respirar, jadeos, opresión en el pecho,
- sensación de malestar general,
- mareos y pérdida de consciencia.
Los síntomas graves, como dificultad para respirar y (casi) síncope, requieren un tratamiento temprano de emergencia.
Efectos adversos frecuentes (pueden afectar hasta a 1 de cada 10 pacientes) inhibidores del factor VIII, dolor de cabeza y fiebre
Efectos adversos poco frecuentes (pueden afectar hasta a 1 de cada 100 pacientes) mareos, gripe, desfallecimiento, pulsaciones anormales, manchas rojas asociadas a picor, molestias en el pecho, cardenal en la zona de inyección, reacción en la zona de inyección, picor, aumento de la sudoración, sabor extraño en la boca, sofocos, migrañas, pérdidas de memoria, escalofríos, diarrea, náusea, vómitos, dificultad para respirar, dolor de garganta, infección de los vasos linfáticos, palidez, inflamación ocular, sarpullidos, sudoración excesiva, inflamación de pies y piernas, disminución del número de glóbulos rojos, aumento de un tipo de leucocitos (monocitos) y dolor en la parte superior del abdomen o parte inferior del pecho.
Relacionados con la cirugía
infección relacionada con el catéter, disminución del número de glóbulos rojos, hinchazón de las extremidades y las articulaciones, sangrado prolongado después de la retirada del drenaje, disminución de los niveles de factor VIII y hematoma postoperatorio
Relacionados con los dispositivos de acceso venoso central (CVAD)
Infección relacionada con catéter, infección sistémica y coágulo de sangre local en el lado del catéter.
Efectos adversos con frecuencia desconocida (no se puede estimar la frecuencia a partir de los datos disponibles)
Reacciones que pueden ser mortales (anafilaxis) y otras reacciones alérgicas (hipersensibilidad), trastornos generales (cansancio, falta de energía).
Efectos adversos adicionales en niños
Además del desarrollo de inhibidores en pacientes pediátricos sin tratamiento previo y las complicaciones asociadas al catéter, en los ensayos clínicos no se observaron diferencias específicas de la edad en los efectos adversos.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Anexo V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
Conservación de ADVATE
5.
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase después de CAD. La fecha de caducidad se refiere al último día del mes que se indica.
Conservar en nevera (entre 2 °C y 8 °C).
No congelar.
Hasta la fecha de caducidad, el blister con el medicamento puede conservarse a temperatura ambiente (hasta 25 °C) por un periodo único no superior a 6 meses. En este caso, este medicamento caduca al final de este periodo de 6 meses o en la fecha de caducidad impresa en el blister, lo que ocurra antes. Por favor, anote en el envase del medicamento la fecha de finalización del periodo de conservación a temperatura ambiente de 6 meses. El medicamento no puede refrigerarse de nuevo después de conservarse a temperatura ambiente.
Conservar el blister con el medicamento en el embalaje exterior para protegerlo de la luz.
Este medicamento es para un solo uso. Elimine la solución no utilizada de forma apropiada.
Utilizar el medicamento inmediatamente tras la disolución completa del polvo.
No refrigerar el medicamento después de la preparación.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de ADVATE
- El principio activo es octocog alfa (factor VIII humano de coagulación producido por tecnología de ADN recombinante). Cada vial de polvo contiene nominalmente 250, 500, 1000 o 1500 UI de octocog alfa.
- Los demás componentes son manitol, cloruro de sodio, histidina, trealosa, cloruro de calcio, trometamol, polisorbato 80 y glutatión (reducido)
Vial de disolvente: 2 ml de agua esterilizada para preparaciones inyectables
Aspecto del producto y contenido del envase
ADVATE es un polvo desmenuzable de color blanco a blanquecino.
Después de su reconstitución, la solución es transparente, incolora y libre de partículas extrañas.
Titular de la autorización de comercialización
Baxter AG Industriestrasse 67 A-1221 Viena
Responsables de la fabricación
Baxalta Belgium Manufacturing SA Boulevard René Branquart 80 B-7860 Lessines
Bélgica
Baxter SA
Boulevard René Branquart 80
B-7860 Lessines
Bélgica
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00 |
Lietuva UAB "Baxter Lithuania'' Tel: +370 5 269 16 90 / +370 5 252 71 00 |
|
Etarapna EaKCTep Etarapna EOOfl Tea.: +359 2 9808482 |
Luxembourg/Luxemburg Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00 |
|
Ceská republika Baxter Czech spol.s.r.o. Tel.: +420 225774111 |
Magyarország Baxter Hungary Kft. Tel.: +36 1 202 1980 |
|
Danmark Baxalta Denmark A/S Tlf: +45 32 70 12 00 |
Malta Baxalta UK Limited Tel.: +44 1 635 798 777 |
|
Deutschland Baxalta Deutschland GmbH Tel: +49 89 262077-011 |
Nederland Baxalta Netherlands B.V. Tel: +31 30 799 27 77 |
|
Eesti OÜ Baxter Estonia Tel.: +372 6 515 120 |
Norge Baxalta Norway AS Tlf: +47 22 585 000 |
|
EXlába Baxter (Hellas) E.n.E. TqL: +30 210 28 80 000 |
Osterreich Baxalta Osterreich GmbH Tel.: +43 1 20100-0 |
|
España Baxalta Spain S.L. Tel: +34 91 790 42 22 |
Polska Baxter Polska Sp. z o.o. Tel.: +48 22 4883 777 |
|
France Baxalta France S.A.S. Tél: +33 1 70 96 06 00 |
Portugal Baxalta Portugal, Unipessoal, Lda. Tel: +351 21 122 03 00 |
|
Hrvatska Baxter d.o.o. Tel: +386 1 420 16 80 |
Romania FARMACEUTICA REMEDIA SA Tel.: +40 21 321 16 40 |
|
Ireland Baxalta UK Limited Tel: +44 1 635 798 777 |
Slovenija Baxter d.o.o. Tel.: +386 1 420 16 80 |
Suomi/Finland
Italia
Baxalta Italy S.r.l.
Tel: +39 06 45224 600
Baxalta Finland Oy Puh/Tel: +358 201478200
Sverige
Baxalta Sweden AB Tel: +46 8 50 53 26 00
United Kingdom
Baxalta UK Limited Tel: +44 1 635 798 777
Fecha de la última revisión de este prospecto
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu/.
Instrucciones para la preparación y administración
ADVATE no se debe mezclar con otros medicamentos o disolventes.
Se recomienda encarecidamente registrar el nombre y el número de lote del producto cada vez que se
administre ADVATE.
Instrucciones para la reconstitución
• No usar después de la fecha de caducidad que aparece en las etiquetas y en el envase.
• No lo utilice si la tapa del blister no está perfectamente sellada.
• No refrigerar la preparación después de la reconstitución.
1. Si el medicamento se encuentra en la nevera, coger de la nevera el blister sellado (contiene viales de polvo y disolvente precargados con el sistema para su reconstitución) y dejarlo a temperatura ambiente (entre 15 °C y 25 °C).
2. Lavar las manos con jabón y agua templada.
3. Abrir el envoltorio de ADVATE quitando la tapa. Extraer el sistema BAXJECT III del blister.
4. Colocar ADVATE en una superficie plana con el vial de disolvente encima (Fig. 1). El vial de disolvente tiene una raya azul. No quitar el protector azul hasta que se le indique más adelante.
5. Mientras sujeta ADVATE con una mano en el sistema BAXJECT III, presionar con fuerza el vial de disolvente con la otra mano hasta que el sistema quede totalmente contraído y el disolvente entre dentro del vial de ADVATE (Fig. 2). No inclinar el sistema hasta que haya finalizado la transferencia.
6. Comprobar que ha finalizado la transferencia del disolvente. Agitar con suavidad hasta que todo el material se haya disuelto. Asegúrese de que el polvo ADVATE esté completamente disuelto, si no es así, toda la solución reconstituida no pasará a través del filtro del equipo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Después de la reconstitución la solución debe ser transparente, incolora y no presenta partículas extrañas.
Fig.l

Fig.2
Fig. 3

Instrucciones para la inyección
Se requiere una técnica aséptica durante la administración.
Para la administración se requiere el uso de una jeringa luer-lock.
Nota importante:
• No intente administrar la inyección a menos que haya recibido una formación especial de su médico o enfermera.
• Inspeccione la solución preparada para detectar partículas o decoloración antes de la administración (la solución debe ser transparente, incolora y libre de partículas extrañas).
No utilice ADVATE si la solución no está totalmente transparente o no está disuelta completamente.
1. Quitar el protector azul del sistema BAXJECT III. No introducir aire en la jeringa. Conectar la jeringa al sistema BAXJECT III.
2. Invertir el sistema (el vial con la solución reconstituida en la parte superior). Introducir la solución reconstituida en la jeringa, tirando del émbolo hacia atrás lentamente.
3. Desconectar la jeringa.
4. Conectar una aguja de perfusión con aletas a la jeringa. Inyectar intravenosamente. La solución se debe administrar lentamente, a una velocidad determinada de acuerdo al nivel de comodidad del paciente, que no exceda de 10 ml por minuto. Debe tomarse el pulso antes y durante la administración de ADVATE. Si observa un aumento significativo del pulso, debe reducir la velocidad de administración o interrumpir temporalmente la inyección, normalmente esto permite que los síntomas desaparezcan en seguida (ver sección 4 “Posibles efectos adversos”).
5. Elimine la solución no utilizada de forma apropiada.
Esta información está destinada únicamente a profesionales del sector sanitario:
Tratamiento bajo demanda
En el caso de los episodios hemorrágicos posteriores, la actividad de factor VIII no debe ser inferior al nivel de actividad plasmática dada (en % del normal o UI/dl) en el periodo correspondiente. Se puede utilizar la siguiente tabla como guía de dosificación en episodios hemorrágicos y cirugía.
La dosis y frecuencia de la administración se debe adaptar a la respuesta clínica en cada caso individual.
En determinadas circunstancias (ej.: presencia de un título bajo de inhibidor) pueden ser necesarias dosis mayores a las calculadas mediante la fórmula.
|
Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel de Factor VIII requerido (% o UI/dl) |
Frecuencia de dosis (horas) / duración de la terapia (días) |
|
Hemorragia | ||
|
Hemartrosis incipiente o hemorragia muscular u oral. |
20 - 40 |
Repetir la inyección cada 12 a 24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) al menos 1 día hasta que el episodio hemorrágico, según indique el dolor, se resuelva o se logre la curación. |
|
Hemartrosis más extensa, hemorragia muscular o hematoma. |
30 - 60 |
Repetir la inyección cada 12-24 horas (cada 8 a 24 horas en los pacientes menores de 6 años) durante 3 a 4 días o más, hasta que cese el dolor y la incapacidad aguda. |
|
Hemorragia con riesgo vital. |
60 - 100 |
Repetir la inyección cada 8 a 24 horas (cada 6 a 12 horas en los pacientes menores de 6 años) hasta superar el peligro. |
|
Cirugía | ||
|
Menor Incluyendo extracción dental. |
30 - 60 |
Cada 24 horas (cada 12 a 24 horas en los pacientes menores de 6 años), al menos 1 día, hasta lograr la curación. |
|
Mayor |
80 - 100 (pre y postoperatorio) |
Repetir la inyección cada 8-24 horas (cada 6 a 24 horas en los pacientes menores de 6 años) hasta que se consiga una curación adecuada de la herida, y luego al menos otros 7 días de terapia para mantener una actividad de factor VIII del 30% al 60% (UI/dl). |
274