Adcetris 50Mg Polvo Para Concentrado Para Solucion Para Perfusion
ANEXO I
FICHA TÉCNICA O RESUMEN DE LAS CARACTERÍSTICAS DEL PRODUCTO
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección 4.8, en la que se incluye información sobre cómo notificarlas.
1. NOMBRE DEL MEDICAMENTO
ADCETRIS 50 mg polvo para concentrado para solución para perfusión.
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada vial contiene 50 mg de brentuximab vedotina.
Tras la reconstitución (ver sección 6.6), cada ml contiene 5 mg de brentuximab vedotina.
ADCETRIS es un anticuerpo conjugado formado por un anticuerpo monoclonal dirigido contra CD30 (inmunoglobulina G1 [IgG1] quimérica recombinante, producida mediante tecnología de ADN recombinante en células de ovario de hámster chino) que se une de forma covalente al agente antimicrotúbulos monometil auristatina E (MMAE).
Excipientes con efecto conocido
Cada vial contiene aproximadamente 13,2 mg de sodio.
Para consultar la lista completa de excipientes, ver sección 6.1.
3. FORMA FARMACÉUTICA
Polvo para concentrado para solución para perfusión. Pasta o polvo de color blanco o blanquecino.
4. DATOS CLÍNICOS
4.1 Indicaciones terapéuticas
ADCETRIS está indicado para el tratamiento de pacientes adultos con linfoma de Hodgkin (LH) CD30+ en recaída o refractario:
1. después de trasplante autólogo de células madre o
2. después de al menos dos tratamientos previos cuando el trasplante autólogo de células madre o la poliquimioterapia no es una opción terapéutica.
ADCETRIS está indicado para el tratamiento de pacientes adultos con LH CD30+ con mayor riesgo de recaída o progresión después de un trasplante autólogo de células madre (ver sección 5.1).
ADCETRIS está indicado para el tratamiento de pacientes adultos con linfoma anaplásico de células grandes (LACG) sistémico en recaída o refractario.
4.2 Posología y forma de administración
Brentuximab vedotina debe administrarse bajo la supervisión de un médico con experiencia en el uso de fármacos antineoplásicos.
Posología
La dosis recomendada es de 1,8 mg/kg, administrados en perfusión intravenosa a lo largo de 30 minutos cada 3 semanas.
La dosis de inicio recomendada para el retratamiento de pacientes con LH en recaída o refractario o LACG sistémico que previamente respondieron al tratamiento con ADCETRIS es de 1,8 mg/kg administrados en perfusión intravenosa a lo largo de 30 minutos cada 3 semanas. De forma alternativa, el tratamiento se puede iniciar con la última dosis tolerada (véase sección 5.1).
Insuficiencia renal
La dosis recomendada de inicio de tratamiento en pacientes con insuficiencia renal grave es 1,2 mg/kg administrados en perfusión intravenosa a lo largo de 30 minutos cada 3 semanas. Los pacientes con insuficiencia renal deben ser vigilados estrechamente debido a los efectos adversos (ver sección 5.2).
Insuficiencia hepática
La dosis recomendada de inicio de tratamiento en pacientes con insuficiencia hepática grave es
1,2 mg/kg administrados en perfusión intravenosa a lo largo de 30 minutos cada 3 semanas. Los pacientes con insuficiencia hepática deben ser vigilados estrechamente debido a los efectos adversos (ver sección 5.2).
Si el paciente pesa más de 100 kg, la dosis debe calcularse basándose en un peso de 100 kg (ver sección 6.6).
Debe vigilarse el hemograma completo antes de administrar cada dosis de este tratamiento (ver sección 4.4).
Se debe vigilar a los pacientes durante y después de la perfusión (ver sección 4.4).
Debe continuarse el tratamiento hasta que se produzca una progresión de la enfermedad o una toxicidad inaceptable (ver sección 4.4).
Los pacientes con LH en recaída o refractario o LACG que logren un resultado de enfermedad estable
0 mejoría deben recibir un mínimo de 8 ciclos y un máximo de hasta 16 ciclos (aproximadamente
1 año) (ver sección 5.1).
En los pacientes con LH con mayor riesgo de recaída o progresión después de un trasplante autólogo de células madre, el tratamiento con ADCETRIS debe iniciarse tras recuperación del trasplante autólogo de células madre atendiendo al juicio clínico. Estos pacientes deben recibir hasta 16 ciclos (ver sección 5.1).
Ajustes de dosis
Neutropenia
Si aparece neutropenia durante el tratamiento, ésta debe controlarse mediante aplazamientos de la dosis. Véanse en la tabla 1 siguiente las recomendaciones posológicas adecuadas (ver también sección 4.4).
Tabla 1: Recomendaciones de administración en caso de neutropenia
|
Grado de intensidad de la neutropenia |
Modificación del régimen |
|
(signos y síntomas [descripción abreviada de los CTCAEa]) |
de administración |
|
Grado 1 (<LIN-1500/mm3 <LIN-1,5 x 109/l) o Grado 2 (<1500-1000/mm3 <1,5-1,0 x 109/l) |
Continuar con la misma dosis y régimen |
|
Grado 3 (<1000-500/mm3 <1,0-0,5 x 109/l) o Grado 4 (<500/mm3 <0,5 x 109/l) |
Aplazar la dosis hasta la disminución de la toxicidad a un grado <2 o al estado basal y reanudar luego el tratamiento con la misma dosis y régimenb. Considerar el apoyo con factores de crecimiento (G-CSF o GM-CSF) en ciclos posteriores en los pacientes que desarrollen neutropenia de grado 3 o 4. |
a' Gradación basada en los Criterios terminológicos comunes para acontecimientos adversos
(CTCAE) del National Cancer Institute (NCI), v3.0; véase Neutrófilos/granulocitos; LIN = límite inferior normal.
b. Los pacientes que desarrollen linfopenia de grado 3 ó 4 pueden continuar el tratamiento sin interrupción.
Neuropatía periférica
Si durante el tratamiento aparece o empeora una neuropatía sensorial o motora periférica, véanse las recomendaciones posológicas apropiadas en la tabla 2 siguiente (ver sección 4.4).
Tabla 2: Recomendaciones de administración en caso de neuropatía sensorial o motora periférica _nueva o agravada_
|
Gravedad de la neuropatía sensorial o motora periférica (signos y síntomas [descripción abreviada de los CTCAEa]) |
Modificación de la dosis y del régimen |
|
Grado 1 (parestesia y/o pérdida de reflejos sin pérdida de función) |
Continuar con la misma dosis y régimen |
|
Grado 2 (interfiere en la función pero no en las actividades de la vida diaria) o Grado 3 (interfiere en las actividades de la vida diaria) |
Aplazar la dosis hasta la disminución de la toxicidad a un grado <1 o al estado basal y reiniciar luego el tratamiento a una dosis reducida de 1,2 mg/kg cada 3 semanas |
|
Grado 4 (neuropatía sensorial incapacitante, o neuropatía motora que amenaza la vida u origina parálisis) |
Interrumpir el tratamiento |
a' Gradación basada en los Criterios terminológicos comunes para acontecimientos adversos (CTCAE) del National Cancer Institute (NCI), v3.0; véanse neuropatía: motora; neuropatía: sensorial, y dolor neuropático.
Pacientes de edad avanzada
No se han establecido la seguridad y la eficacia en pacientes de edad avanzada de 65 o más años de edad. No se dispone de datos.
Población pediátrica
No se han establecido aún la seguridad y la eficacia en niños menores de 18 años. No se dispone de datos.
Se ha observado depleción del timo en estudios no clínicos (ver sección 5.3).
Forma de administración
La dosis recomendada de ADCETRIS se perfunde en 30 minutos.
Para consultar las instrucciones de reconstitución y dilución del medicamento antes de la administración, ver sección 6.6.
Brentuximab vedotina no se debe administrar en inyección intravenosa rápida o en bolo. Brentuximab vedotina se debe administrar a través de una vía intravenosa específica y no debe mezclarse con otros medicamentos (ver sección 6.2).
4.3 Contraindicaciones
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
El uso combinado de bleomicina y brentuximab vedotina causa toxicidad pulmonar.
4.4 Advertencias y precauciones especiales de empleo Leucoencefalopatía multifocal progresiva
La reactivación del virus de John Cunningham (VJC), que provoca la aparición de leucoencefalopatía multifocal progresiva (LMP) y muerte, puede ocurrir en pacientes tratados con brentuximab vedotina. Se ha notificado LMP en pacientes que recibían este tratamiento después de varios regímenes quimioterapéuticos previos. La LMP es una enfermedad desmielinizante rara del sistema nervioso central originada por la reactivación del VJC latente y es a menudo mortal.
Se debe vigilar estrechamente a los pacientes en busca de la aparición o empeoramiento de signos o síntomas neurológicos, cognitivos o conductuales, que pueden sugerir la presencia de LMP. En cualquier caso con sospecha de LMP debe suspenderse la administración de brentuximab vedotina. La evaluación sugerida de la LMP comprende una consulta neurológica, resonancia magnética cerebral con gadolinio y análisis de ADN del VJC en el líquido cefalorraquídeo mediante reacción en cadena de la polimerasa o una biopsia cerebral con indicios del VJC. Una RCP del VJC negativa no descarta la LMP. Si no puede establecerse un diagnóstico alternativo, el seguimiento y la evaluación adicionales pueden estar justificados. La administración de brentuximab vedotina debe interrumpirse permanentemente si se confirma el diagnóstico de LMP.
El médico debe estar especialmente alerta a los síntomas indicativos de LMP que el paciente pueda no observar (p. ej., síntomas cognitivos, neurológicos o psiquiátricos).
Pancreatitis
Se ha observado pancreatitis aguda en pacientes tratados con brentuximab vedotina. Se ha notificado algún caso mortal.
Se debe vigilar de cerca a los pacientes que presenten o empeoren un dolor abdominal, que pueda sugerir pancreatitis aguda. La evaluación del paciente puede incluir un examen físico, análisis de laboratorio para evaluar la amilasa y la lipasa séricas, y estudios de imagen abdominal, como ecografía y otros métodos diagnósticos adecuados. Se debe evaluar el uso de brentuximab vedotina en cualquier caso de sospecha de pancreatitis aguda. Si se confirma el diagnóstico de pancreatitis aguda se debe suspender el tratamiento con brentuximab vedotina.
Toxicidad pulmonar
Se han notificado algunos casos de toxicidad pulmonar, entre los que se incluyen neumonitis, enfermedad pulmonar intersticial, síndrome de dificultad respiratoria aguda (SDRA), algunos con desenlace mortal, en pacientes tratados con brentuximab vedotina. Aunque no se ha establecido una asociación causal con el uso de brentuximab vedotina, no se puede descartar el riesgo de toxicidad pulmonar. En caso de aparición o empeoramiento de síntomas pulmonares (ej. tos, disnea), se debe evaluar rápidamente el diagnóstico y tratar de forma conveniente a los pacientes. Se debe considerar la posibilidad de continuar con la medicación con brentuximab vedotina durante la evaluación y hasta que haya mejoría sintomática.
Infecciones graves y oportunistas
Se han notificado infecciones graves como neumonía, bacteriemia estafilocócica, sepsis/shock séptico (incluyendo desenlaces mortales) y herpes zoster, e infecciones oportunistas como neumonía por Pneumocystis jiroveci y candidiasis oral, en pacientes tratados con brentuximab vedotina. Debe vigilarse atentamente a los pacientes durante el tratamiento en busca de posibles infecciones graves y oportunistas.
Reacciones relacionadas con la perfusión
Se han notificado reacciones relacionadas con la perfusión (RRP) inmediatas y retardadas, así como reacciones anafilácticas.
Debe vigilarse cuidadosamente a los pacientes durante y después de la perfusión. Si se produce una reacción anafiláctica, se debe interrumpir inmediata y permanentemente la administración de brentuximab vedotina y se debe administrar tratamiento médico apropiado.
Si se produce una RRP, se debe interrumpir la perfusión e instaurarse tratamiento médico apropiado. La perfusión puede reiniciarse a una velocidad menor tras la resolución de los síntomas. Los pacientes que hayan sufrido una RRP previa se deben premedicar antes de las perfusiones siguientes. La premedicación puede incluir paracetamol, un antihistamínico y un corticosteroide.
Las RRP son más frecuentes y más graves en los pacientes con anticuerpos frente a brentuximab vedotina (ver sección 4.8).
Síndrome de lisis tumoral
Se ha notificado síndrome de lisis tumoral (SLT) con brentuximab vedotina. Los pacientes con tumor en proliferación rápida y carga tumoral elevada corren riesgo de sufrir síndrome de lisis tumoral. Se debe vigilar estrechamente a estos pacientes y tratarlos de acuerdo con la mejor práctica médica. El tratamiento del SLT debería incluir hidratación intensiva, vigilancia de la función renal, corrección de las anomalías electrolíticas, tratamiento antihiperuricémico y cuidados de soporte.
Neuropatía periférica
El tratamiento con brentuximab vedotina puede causar una neuropatía periférica, tanto sensorial como motora. La neuropatía periférica inducida por brentuximab vedotina es un efecto característico de la exposición acumulada a este medicamento y es reversible en la mayoría de los casos.
En los ensayos clínicos pivotales de fase II (SG035-003 y SG035-004), la incidencia de neuropatía periférica preexistente fue del 24%. Un 56% de los pacientes fueron tratados de neuropatía emergente. En el momento de la última evaluación, la mayoría de los pacientes (83%) mostraron mejoría o resolución de sus síntomas de neuropatía periférica. Entre los pacientes que presentaron neuropatía periférica, el 17% abandonaron el tratamiento con brentuximab vedotina, un 13% redujo la dosis y en un 21% las dosis se retrasaron.
La incidencia de neuropatía periférica preexistente en pacientes con LH en recaída o refractario o con LACG sistémico retratados con brentuximab vedotina fue del 48%. Un 69% de los pacientes fueron tratados de neuropatía emergente. En el momento de la última evaluación, la mayoría de los pacientes retratados con neuropatía periférica relacionada con el tratamiento (80%) habían mejorado o habían dejado de presentar síntomas de neuropatía emergente. La neuropatía periférica emergente determinó la interrupción del tratamiento en un 21% y modificación de dosis en un 34% de los pacientes retratados.
En los ensayos clínicos de fase 3, en el momento de la última evaluación, la mayoría de los pacientes incluidos en el brazo brentuximab vedotina (85%) mostraron mejoría o resolución de sus síntomas de neuropatía periférica. Entre los pacientes que notificaron neuropatía periférica, el 23% interrumpieron el tratamiento con brentuximab vedotina, un 29% redujo la dosis y en un 22% las dosis se retrasaron.
Se debe vigilar a los pacientes en busca de síntomas de neuropatía como hipoestesia, hiperestesia, parestesia, molestias, sensación de quemazón, dolor neuropático o debilidad. En los pacientes que sufran neuropatía periférica nueva o agravada pueden precisarse un aplazamiento y una reducción de la dosis de brentuximab vedotina o la interrupción del tratamiento (ver sección 4.2).
Toxicidad hematológica
Con brentuximab vedotina pueden aparecer anemia de grado 3 ó 4, trombocitopenia y neutropenia de grado 3 ó 4 prolongada (>1 semana). Se debe vigilar el hemograma completo antes de la administración de cada dosis. Si aparece neutropenia de grado 3 ó 4, véase la sección 4.2.
Neutropenia febril
Se ha notificado neutropenia febril (fiebre de origen desconocido sin infección clínica o microbiológicamente comprobada con un recuento absoluto de neutrófilos <1,0 x 109/l, fiebre >38,5°C; véase CTCAE v3) con el tratamiento de brentuximab vedotina. Se debe vigilar el hemograma completo antes de la administración de cada dosis de este tratamiento. Si aparece neutropenia febril, se debe vigilar estrechamente a los pacientes en busca de fiebre y tratarlos según la práctica médica óptima.
Síndrome de Stevens-Johnson y necrólisis epidérmica tóxica
Se ha notificado la aparición del síndrome de Stevens-Johnson (SSJ) y de necrólisis epidérmica tóxica (NET) con brentuximab vedotina. Se han notificado casos mortales. Si se producen SSJ o NET, se debe interrumpir el tratamiento con brentuximab vedotina y administrarse el tratamiento médico apropiado.
Complicaciones gastrointestinales
En pacientes tratados con brentuximab vedotina se han notificado complicaciones gastrointestinales (GI), entre las que se incluyen obstrucción intestinal, íleo, enterocolitis, colitis neutropénica, erosiones, úlceras, perforaciones y hemorragias, algunas con desenlace mortal. En el caso de que se presenten nuevos síntomas, o empeoramiento de los síntomas de GI, se debe realizar una evaluación para un diagnóstico rápido y tratarse convenientemente.
Hepatotoxicidad
Se ha notificado hepatotoxicidad en forma de elevación en los niveles de alanino aminotransferasa (ALT) y aspartato aminotransferasa (AST) con brentuximab vedotina. También se han producido casos graves de hepatotoxicidad con desenlace mortal. La presencia previa de la enfermedad hepática, las comorbilidades y las medicaciones concomitantes también pueden aumentar el riesgo. Se deben realizar pruebas de la función hepática antes de iniciar el tratamiento y mantener una vigilancia de forma rutinaria en los pacientes tratados con brentuximab vedotina. Los pacientes que experimenten hepatotoxicidad pueden requerir un retraso o cambio en la dosis, o la interrupción del tratamiento con brentuximab vedotina.
Hiperglucemia
Se ha notificado hiperglucemia durante los ensayos clínicos en pacientes con un índice de masa corporal (IMC) elevado, con o sin antecedentes de diabetes mellitus. No obstante, se debe vigilar estrechamente la glucosa sérica de cualquier paciente que sufra un episodio de hiperglucemia. Debe administrarse tratamiento antidiabético según proceda.
Insuficiencia renal y hepática
La experiencia en pacientes con insuficiencia renal y hepática es limitada. Los datos disponibles indican que la insuficiencia renal grave, la insuficiencia hepática y las concentraciones séricas bajas de albúmina podrían afectar al aclaramiento de la MMAE (ver sección 5.2).
Contenido de sodio de los excipientes
Este medicamento contiene un máximo de 2,1 mmol (o 47 mg) de sodio por dosis, lo que debe tenerse en cuenta en el tratamiento de pacientes con dietas pobres en sodio.
4.5 Interacción con otros medicamentos y otras formas de interacción
Interacción con medicamentos metabolizados por la vía de la CYP3A4 (inhibidores o inductores de la CYP3A4)
La administración concomitante de brentuximab vedotina con el ketoconazol, un inhibidor potente de la CYP3A4 y de la P-gp, aumentó la exposición al fármaco antimicrotúbulos MMAE en alrededor del 73% y no alteró la exposición plasmática a brentuximab vedotina. En consecuencia, la administración concomitante de brentuximab vedotina con inhibidores potentes de la CYP3A4 y la P-gp puede aumentar la incidencia de neutropenia. Si aparece neutropenia, consúltese la tabla 1: Recomendaciones de administración en caso de neutropenia (ver sección 4.2).
La administración concomitante de brentuximab vedotina con la rifampicina, un inductor potente de la CYP3A4, no alteró la exposición plasmática a brentuximab vedotina. A pesar de que los datos escasos sobre PK, la administración concomitante de rifampicina parece reducir las concentraciones en plasma de metabolitos de MMAE que podían analizarse.
La administración concomitante de midazolam, un sustrato de la CYP3A4, con brentuximab vedotina no alteró el metabolismo del midazolam; en consecuencia, no se espera que brentuximab vedotina altere la exposición a los medicamentos metabolizados por enzimas CYP3A4.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil
Las mujeres en edad fértil deben utilizar dos métodos anticonceptivos eficaces durante el tratamiento con brentuximab vedotina y hasta 6 meses después del tratamiento.
Embarazo
No se dispone de datos sobre el uso de brentuximab vedotina en mujeres embarazadas. Los estudios en animales han mostrado toxicidad para la reproducción (ver sección 5.3).
Brentuximab vedotina no se debe utilizar durante el embarazo a menos que el beneficio para la madre supere al potencial riesgo para el feto. Si es necesario tratar a una mujer embarazada, debe advertírsele claramente del potencial riesgo para el feto.
Ver en el apartado siguiente sobre fertilidad los consejos para las mujeres cuyas parejas masculinas estén siendo tratadas con brentuximab vedotina.
Lactancia
No se dispone de datos sobre si brentuximab vedotina o sus metabolitos se excretan en la leche humana.
No se puede excluir el riesgo en recién nacidos/lactantes.
Debe decidirse si se interrumpe la lactancia, o se interrumpe este tratamiento, o se prescinde de él, teniendo en cuenta el posible riesgo de la lactancia para el niño y el efecto beneficioso del tratamiento para la madre.
Fertilidad
En estudios no clínicos, el tratamiento con brentuximab vedotina ha originado toxicidad testicular, y puede alterar la fertilidad masculina. Se ha demostrado que la MMAE tiene propiedades aneugénicas (ver sección 5.3). Por consiguiente, se aconseja a los hombres tratados con este medicamento que hagan congelar y conservar muestras de su semen antes del tratamiento. Se aconseja a los hombres tratados con este medicamento que no engendren un hijo durante el tratamiento ni hasta 6 meses después de la última dosis.
4.7 Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de brentuximab vedotina sobre la capacidad para conducir y utilizar máquinas es pequeña.
4.8 Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad de ADCETRIS está basado en los datos de los ensayos clínicos disponibles, en el programa de suministro de medicamentos en situaciones especiales (NPP) y en la experiencia poscomercialización hasta la fecha. Las frecuencias de las reacciones adversas que se describen a continuación y en la Tabla 3 se han determinado en base a los datos generados en estudios clínicos.
En dos ensayos clínicos de fase 2 realizados en pacientes con LH en recaída o refractario o LACG ADCETRIS se administró en monoterapia en 160 pacientes. La mediana del número de ciclos fue de 9 en pacientes con LH en recaída o refractario y de 7 en pacientes con LACG en recaída o refractario. ADCETRIS también se administró en monoterapia en 167 pacientes de los 329 pacientes con LH con mayor riesgo de recaída o progresión después de un trasplante autólogo de células madre, incluidos en un ensayo clínico aleatorio controlado por placebo de fase 3. La mediana del número de ciclos recibidos en ambos brazos fue de 15.
Las infecciones graves y oportunistas fueron muy frecuentes en pacientes tratados con este medicamento (ver sección 4.4). En la población de los estudios de fase 2 y fase 3, las infecciones oportunistas notificadas con más frecuencia fueron herpes zoster y herpes simple.
Las reacciones adversas graves al medicamento en la población de los estudios pivotales de fase 2 y fase 3 fueron: neumonía, síndrome de distress respiratorio agudo, cefalea, neutropenia, trombocitopenia, estreñimiento, diarrea, vómitos, náuseas, pirexia, neuropatía motora periférica, neuropatía sensorial periférica, hiperglucemia, polineuropatía desmielinizante, síndrome de lisis tumoral y síndrome de Stevens-Johnson.
Las reacciones adversas observadas con más frecuencia (>20%) en la población de los estudios pivotales de fase 2 y fase 3 fueron: neuropatía sensorial periférica, cansancio, náuseas, diarrea, infección respiratoria del tracto superior, neutropenia ytos. Además, también se observaron reacciones adversas en >20 % en forma de vómitos y pirexia en los ensayos clínicos de fase 2, y de neuropatía motora periférica en los ensayos clínicos de fase 3.
Las reacciones adversas originaron la interrupción del tratamiento en el 23% y el 32% de los pacientes que recibían brentuximab vedotina en los ensayos clínicos de fase 2 y de fase 3 respectivamente. Las reacciones adversas graves que originaron la interrupción del tratamiento en dos o más pacientes , tanto en el ensayo clínico de fase 2 como en el de fase 3, fueron neuropatía sensorial periférica, neuropatía motora periférica , polineuropatía desmielinizante y enfermedad de Hodgkin recurrente, vómitos y síndrome de distress respiratorio agudo. La parestesia también originó la interrupción del tratamiento en dos o más pacientes, tanto en el ensayo clínico de fase 2 como en el de fase 3.
Los datos de seguridad de los pacientes con LH en recaída o refractario que no habían recibido un trasplante autólogo de células madre y que habían sido tratados con la dosis recomendada de
1,8 mg/kg cada tres semanas en el ensayo fase I de escalado de dosis, en los estudios clínicos farmacológicos (n=15 pacientes), y en el NPP (n=26 pacientes) (ver sección 5.1) resultaron consistentes con el perfil de seguridad de los estudios pivotales.
Tabla de reacciones adversas
Las reacciones adversas de ADCETRIS se enumeran según el sistema de clasificación de órganos y término preferido del MedDRA (ver tabla 3). Dentro de cada sistema de clasificación de órganos, las reacciones adversas se enumeran en las categorías de frecuencia siguientes: muy frecuentes (>1/10); frecuentes (>1/100 a < 1/10); poco frecuentes (>1/1.000 a < 1/100); raras (>1/10.000 a <1/1.000); muy raras (<1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
|
Sistema de Clasificación de Órganos |
Reacciones adversas |
|
Infecciones e infestaciones | |
|
Muy frecuentes: |
Infección3, infección respiratoria del tracto superior |
|
Frecuentes: |
Sepsis/shock séptico, herpes zoster, neumonía, herpes simple |
|
Poco frecuentes: |
Candidiasis oral, neumonía por Pneumocystis jiroveci, bacteriemia estafilocócica |
|
Frecuencia no conocida |
Leucoencefalopatía multifocal progresiva |
|
Trastornos de la sangre y del sistema linfático | |
|
Muy frecuentes: |
Neutropenia |
|
Frecuentes: |
Anemia, trombocitopenia |
|
Frecuencia no conocida |
Neutropenia febril |
|
Trastornos del sistema inmunológico | |
|
Frecuencia no conocida |
Reacción anafiláctica |
|
Trastornos del metabolismo y de la nutrición | |
|
Frecuentes: |
Hiperglucemia |
|
Poco frecuentes: |
Síndrome de lisis tumoral |
|
Trastornos del sistema nervioso | |
|
Muy frecuentes: |
Neuropatía sensorial periférica, neuropatía motora periférica |
|
Frecuentes: |
Mareos, polineuropatía desmielinizante |
|
Trastornos respiratorios, torácicos y mediastínicos | |
|
Muy frecuentes: |
Tos, disnea |
|
Trastornos gastrointestinales | |
|
Muy frecuentes: |
Diarrea, náuseas, vómitos, estreñimiento, dolor abdominal |
|
Poco frecuentes |
Pancreatitis aguda |
|
Trastornos hepatobiliares | |
|
Frecuentes: |
Aumento de alanina aminotransferasa/aspartato aminotransferasa (ALT/AST) |
|
Trastornos de la piel y del tejido subcutáneo | |
|
Muy frecuentes: |
Alopecia, prurito |
|
Frecuentes: |
Exantema |
|
Raros: |
Síndrome de Stevens-Johnson / necrólisis epidérmica tóxica |
|
Trastornos musculoesqueléticos y del tejido conjuntivo | |
|
Muy frecuentes: |
Mialgia, artralgia |
|
Frecuentes: |
Dolor de espalda |
|
Trastornos generales y alteraciones en el lugar de administración | |
|
Muy frecuentes: |
Cansancio, escalofríos, pirexia, reacciones relacionadas con la perfusiónb |
|
Exploraciones complementarias: | |
|
Muy frecuentes |
Disminución de peso |
a' Los términos preferidos que se notificaron en el apartado Infecciones e Infestaciones son sepsis/shock séptico, infección respiratoria superior, herpes zoster y neumonía. b' Los términos preferidos asociados con las RRP fueroncefalea, exantema, dolor de espalda, vómitos, escalofríos , náuseas, disnea, prurito y tos .
La neutropenia causó el aplazamiento de la administración en el 14% y el 22% de los pacientes en los ensayos clínicos de fase 2 y de fase 3 respectivamente.
Con este tratamiento puede aparecer neutropenia grave y prolongada (>1 semana), que puede aumentar el riesgo de que los pacientes contraigan infecciones graves. En la población del ensayo clínico de fase 2, la mediana de duración de la neutropenia de grado 3 ó 4 fue limitada (1 semana); el 2% de los pacientes tuvieron neutropenia de grado 4 de >7 días de duración. Menos de la mitad de los pacientes de la población de los estudios pivotales de fase 2 con neutropenia de grado 3 ó 4 sufrieron temporalmente infecciones asociadas, de las cuales la mayoría fueron de grado 1 ó 2.
En la población del ensayo clínico de fase 3, se notificaron neutropenia de grado 3 en el 22% de los pacientes en el brazo con brentuximab vedotina y neutropenia de grado 4 en el 7% de los pacientes en el brazo con brentuximab vedotina. Ninguno de los pacientes requirió de una reducción de la dosis ni de interrupción del tratamiento por la neutropenia.
En la población del ensayo clínico de fase 3, se notificaron infecciones graves en el 9% de los pacientes en el brazo con brentuximab vedotina. No se notificaron casos de bacteriemia, sepsis o shock séptico en el brazo con brentuximab vedotina.
La neuropatía sensorial periférica causó el aplazamiento de la administración en el 13% y el 16% de los pacientes en los ensayos clínicos de fase 2 y de fase 3 respectivamente. Además, la neuropatía motoraperiférica y la infección del tracto respiratorio superior causaron aplazamientos en la administración de la dosis en el 6% de los pacientes en el ensayo de fase 3.
La neuropatía sensorial periférica causó el aplazamiento de la administración de la dosis en el 9% y el 22% de los pacientes en los ensayos clínicos de fase 2 y de fase 3 respectivamente. Además, la neuropatía motora periférica también causó reducciones de dosis en el 6% de los pacientes en el ensayo clínico de fase 3. El noventa por ciento (90%) y el sesenta y ocho por ciento (68%) de los pacientes en el ensayo clínico de fase 2 y de fase 3 respectivamente mantuvieron la dosis recomendada de 1,8 mg/kg durante el tratamiento.
En los pacientes que sufrieron neuropatía periférica en el ensayo clínico de fase 2, la mediana de seguimiento desde el final del tratamiento hasta la última evaluación fue de alrededor de 48,9 semanas. En el momento de la última evaluación, el 83% de los 89 pacientes que sufrieron neuropatía periférica mostraban resolución o mejoría de sus síntomas de neuropatía periférica. La media del tiempo desde el comienzo hasta la resolución o la mejoría de todos los episodios fue de 16 semanas (desde 0,3 a 106,6 semanas).
Entre los pacientes que experimentaron neuropatía periférica en el ensayo clínico de fase 3, la mediana de seguimiento desde el final de tratamiento hasta la última evaluación fue de alrededor de 98 semanas. En el momento de la última evaluación, el 85% de los pacientes con neuropatía periférica en el brazo con brentuximab vedotina ya no presentaban síntomas de neuropatía periférica o los síntomas habían mejorado. En términos generales, la mediana de tiempo hasta la resolución o mejora de los casos de neuropatía periférica en el brazo con brentuximab vedotina fue de 23,4 semanas (intervalo de 0,1 semanas a 138,3 semanas).
Se notificaron RRP en el 11% y el 15% de los pacientes en los ensayos clínicos de fase 2 y de fase 3 respectivamente. Tanto en el ensayo de fase 2 como en el de fase 3, las reacciones adversas más comúnmente asociadas a las RRP fueron de leves a moderadas (grado 2 y grado 2), e incluyeron cefalea, erupción, dolor de espalda, vómitos, escalofríos, náuseas, disnea, prurito y tos.
Se ha notificado la aparición de reacciones anafilácticas (ver sección 4.4). Los síntomas de una reacción anafiláctica pueden ser, entre otros, urticaria, angioedema, hipotensión y broncospasmo.
Se ha notificado la aparición de neutropenia febril (ver sección 4,2). Un paciente reclutado para un ensayo fase 1 de escalado de dosis sufrió neutropenia febril de grado 5 después de recibir una sola dosis de 3,6 mg/kg de brentuximab vedotina.
Inmunogenicidad
En dos estudios pivotales de fase 2, se hicieron análisis de anticuerpos contra brentuximab vedotina cada 3 semanas en pacientes con LH o LACGs en recaída o refractario empleando un inmunoensayo electroquimioluminiscente sensible. También se realizaron análisis a los pacientes con LH en mayor riesgo de recaída o progresión después de un trasplante autólogo de células madre en el estudio clínico de fase 3. Alrededor del 7% de los pacientes del estudio de fase 2 y el 6% de los pacientes en el brazo con brentuximab vedotina del estudio de fase 3 desarrollaron positividad persistente a anticuerpos anti tratamiento (AAT)Dos pacientes en los estudios de fase 2 y dos pacientes en el estudio de fase 3 sufrieron reacciones adversas congruentes con RRP que determinaron la interrupción del tratamiento.
La presencia de anticuerpos contra brentuximab vedotina no estuvo acompañada de una reducción clínicamente significativa de las concentraciones séricas de brentuximab vedotina, y no originó un descenso de la eficacia de brentuximab vedotina. Aunque la presencia de anticuerpos contra brentuximab vedotina no predice necesariamente la aparición de una RRP, se observó una mayor incidencia de RRP en los pacientes con AAT persistentemente positivos que en los que presentaban AAT positivos de forma temporal y en los que nunca los tuvieron .
Retratamiento
El retratamiento con Adcetris se administró a 21 pacientes con LH en recaída o refractario y en 8 pacientes con LACG sistémico en recaída. La mediana del número de ciclos fue de 7 (rango de 2 a 37 ciclos) (ver sección 5.1). Los tipos y grados de reacciones adversas notificados en pacientes retratados con Adcetris fueron consistentes con los observados en la combinación de los ensayos pivotales de fase II, con la excepción de la neuropatía periférica motora, cuya incidencia fue mayor (28% vs. 9% en los ensayos pivotales de fase II) y fue principalmente de grados 1 ó 2. Los pacientes también presentaron una incidencia mayor de artralgia, anemia de grado 3 y dolor de espalda en comparación con los pacientes observados en la combinación de los ensayos pivotales de fase II.
Notificación de sospechas de reacciones adversas
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación incluido en el Apéndice V.
4.9 Sobredosis
Se desconoce un antídoto para la sobredosis de brentuximab vedotina. En caso de sobredosis, debe vigilarse estrechamente al paciente en busca de reacciones adversas, especialmente neutropenia, y debe administrarse tratamiento de soporte (ver sección 4.4).
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: fármacos antineoplásicos; otros fármacos antineoplásicos; anticuerpos monoclonales, código ATC: L01XC12.
Mecanismo de acción
Brentuximab vedotina es un anticuerpo conjugado (ACC) que libera un fármaco antineoplásico que origina selectivamente la muerte celular apoptótica de las células tumorales que expresan CD30. Los datos preclínicos indican que la actividad biológica de brentuximab vedotina es resultado de un proceso de varias etapas. La unión del ACC a CD30 sobre la superficie celular inicia la incorporación del complejo ACC-CD30, que se desplaza luego al compartimento lisosomal. Dentro de la célula, se libera una sola especie activa definida, la MMAE, mediante escisión proteolítica. La unión de la MMAE a la tubulina altera la red de microtúbulos del interior de la célula, induce a la detención del ciclo celular y origina la muerte apoptótica de la célula tumoral que expresa CD-30.
El LH clásico y el LACG sistémico expresan CD30 como un antígeno de superficie de sus células malignas. Esta expresión es independiente del estadío de la enfermedad, de la línea de tratamiento o de la situación en relación al trasplante. Esos aspectos hacen que CD30 sea una diana para la intervención terapéutica. Debido al mecanismo de acción dirigido al CD30, brentuximab vedotina es capaz de superar la quimiorresistencia dado que CD30 se expresa de manera consistente en pacientes que son refractarios a poliquimioterapia, independientemente del estado previo en relación al trasplante. El mecanismo de acción dirigido al CD30 de brentuximab vedotina, la expresión persistente de CD30 en el LH clásico y en el LACG sistémico y el espectro terapéutico y la evidencia clínica en dos patologías tumorales CD30-positivas, tras múltiples líneas de tratamiento, proporcionan una base biológica para su uso en pacientes con LH clásico en recaída y refractario y en LACG sistémico con o sin previo trasplante autólogo de células madre.
No se han descartado contribuciones de otras funciones asociadas con anticuerpos al mecanismo de acción.
Efectos farmacodinámicos
Electrofisiología cardíaca
Cuarenta y seis (46) de los 52 pacientes con procesos hematológicos malignos que expresaban CD30 que recibieron 1,8 mg/kg de brentuximab vedotina cada 3 semanas como parte de un estudio de fase 1 de seguridad cardíaca multicéntrico abierto de un solo brazo fueron evaluables. El objetivo principal fue evaluar el efecto de brentuximab vedotina sobre la repolarización ventricular cardíaca, y el análisis principal predefinido fue el del cambio del QTc desde el momento basal hasta varios puntos temporales del ciclo 1.
El intervalo de confianza (IC) del 90% superior alrededor del efecto medio en el QTc fue <10 ms en cada uno de los puntos temporales post-basales de los ciclos 1 y 3. Estos datos indican la ausencia de prolongación del QT de importancia clínica debida a la administración de brentuximab vedotina en dosis de 1,8 mg/kg cada 3 semanas en pacientes con procesos malignos que expresan CD30.
Eficacia clínica
Linfoma de Hodgkin Estudio SG035-0003
Se evaluaron la eficacia y la seguridad de brentuximab vedotina como fármaco único en un estudio pivotal multicéntrico abierto de un solo brazo en 102 pacientes con LH en recaída o refractario. Véase un resumen de las características basales de los pacientes y de la enfermedad en la tabla 4 siguiente.
3 meses siguientes a la finalización del tratamiento de primera línea.
fase 2 en LH en recaída o refractario
|
Características de los pacientes |
N = 102 |
|
Mediana de edad, años (intervalo) |
31 años (15-77) |
|
Sexo |
48V (47%)/54M (53%) |
|
Estado funcional del ECOG | |
|
0 |
42 (41%) |
|
1 |
60 (59%) |
|
Trasplante autólogo de células madre previo |
102 (100%) |
|
Regímenes de quimioterapia previos |
3,5 (1-13) |
|
Tiempo desde el trasplante autólogo de células madre hasta la |
6,7 meses (0-131) |
|
primera recidiva post-trasplante | |
|
Enfermedad que expresa CD30 confirmada histológicamente |
102 (100%) |
|
Características de la enfermedad | |
|
Refractaria al tratamiento de primera línea3 |
72 (71%) |
|
Refractaria al tratamiento más reciente |
43 (42%) |
|
Síntomas B basales |
35 (33%) |
|
Estadío III en el diagnóstico inicial |
27 (26%) |
|
Estadío IV en el diagnóstico inicial |
20 (20%) |
a' Se define como LH refractario al que no experimenta remisión completa o progresa en los
Dieciocho (18) pacientes (18%) recibieron 16 ciclos de brentuximab vedotina, y la mediana del número de ciclos recibidos fue de 9 (entre 1 y 16).
La respuesta al tratamiento con brentuximab vedotina fue valorada por un Centro de Revisión Independiente (CRI) empleando los Criterios de Respuesta Revisados para el Linfoma Maligno (Cheson, 2007). La respuesta al tratamiento se valoró mediante TC helicoidal de tórax, cuello, abdomen y pelvis, AGPET y datos clínicos. Se hicieron valoraciones de la respuesta en los ciclos 2, 4, 7, 10, 13 y 16 con PET en los ciclos 4 y 7.
La tasa de respuesta objetiva (TRO) por valoración del CRI fue del 75% (76 de 102 pacientes del conjunto por intención de tratar [IT]), y se logró la reducción del tumor en el 94% de los pacientes. La tasa de remisión completa (RC) fue del 33% (34 de 102 pacientes del conjunto por IT). La mediana de supervivencia global (SG) es de 40,5 meses (la mediana del tiempo de observación (tiempo hasta la muerte o el último contacto) desde la primera dosis fue de 35,1 meses (intervalo de 1,8 a 72,9+ meses). La tasa de supervivencia global estimada a los 5 años fue del 41% (IC del 95% [31%, 51%]). Las valoraciones de los investigadores fueron en general congruentes con la revisión independiente de las exploraciones de imagen. De los pacientes tratados, 8 de los que respondían pasaron a recibir un TCM alogénico. Para más resultados de eficacia, véase la tabla 5.
con 1,8 mg/kg de brentuximab vedotina cada 3 semanas
|
Mejor respuesta clínica (N = 102) |
CRI N (%) |
IC del 95% |
|
Tasa de respuesta objetiva (RC + RP) |
76 (75) |
64,9, 82,6 |
|
Remisión completa (RC) |
34 (33) |
24,3, 43,4 |
|
Remisión parcial (RP) |
42 (41) |
NP |
|
Tasa de control de la enfermedad (RC + |
98 (96) |
90,3, 98,9 |
|
RP + EE) | ||
|
Duración de la respuesta |
Mediana según el CRI |
IC del 95% |
|
Tasa de respuesta objetiva (RC + RP)a |
6,7 meses |
3,6, 14,8 |
|
Remisión completa (RC) |
27,9 meses |
10,8, NCb |
|
Supervivencia global |
IC del 95% | |
|
Mediana |
40,5 meses |
28,7, 61,9 |
|
Tasa de SG estimada a los 5 años |
41% |
31%, 51% |
a' La duración de la respuesta osciló entre 1,2+ meses y 43+ meses, y la mediana de seguimiento desde la primera dosis de los pacientes que lograron respuesta objetiva (RO) según el CRI fue de 9,0 meses. b' No calculable.
Un análisis exploratorio intrapaciente mostró que aproximadamente el 64% de los pacientes con LH tratados con brentuximab vedotina dentro del ensayo clínicoSG035-0003, experimentaban una mejoría en el beneficio clínico medido por una supervivencia libre de progresión (SLP) más prolongada en comparación con su línea de tratamiento previo más reciente.
De los 35 pacientes (33%) que tenían síntomas B basales, 27 pacientes (77%) experimentaron resolución de todos los síntomas B a una mediana de tiempo de 0,7 meses desde la instauración de brentuximab vedotina.
Se recogieron datos de pacientes (n=15) incluidos en el ensayo fase I de escalado de dosis y de los estudios farmacológicos, así como de pacientes (n=26) del programa de suministro de medicamentos en situaciones especiales con LH en recaída o refractario que no habían recibido trasplante autólogo de células madre, y que fueron tratados con 1,8 mg/kg de brentuximab vedotina cada 3 semanas.
Las características basales de los pacientes mostraron fracaso de múltiples regímenes quimioterápicos previos (mediana de 3 con un intervalo entre 1 y 7) antes de la primera administración de brentuximab vedotina. El cincuenta y nueve por ciento (59%) de los pacientes tenían estadío avanzado de su enfermedad (estadíos III y IV) en el diagnóstico inicial.
Los resultados de estos estudios fase I y de la experiencia del NPP mostraron que, en pacientes con LH en recaída o refractario sin trasplante autólogo de células madre previo, se pueden alcanzar respuestas clínicas significativas evidenciadas por el análisis del investigador, una tasa de respuesta objetiva del 54% y una tasa de respuestas completas del 22% después de una mediana de cinco ciclos de brentuximab vedotina.
Estudio SGN35-005
Se evaluaron la eficacia y la seguridad de brentuximab vedotina en un estudio multicéntrico aleatorio, doble ciego, controlado con placebo y de dos brazos realizado en 329 pacientes con LH con riesgo de recaída o progresión después del trasplante autólogo de células madre. Se excluyeron del ensayo los pacientes con enfermedad cerebral/meníngea conocida, con inclusión de MLP. Ver tabla 6 para conocer las características de esos pacientes. De los 329 pacientes, 165 pacientes se asignaron de forma aleatoria al brazo de tratamiento y 164 pacientes se asignaron al azar al brazo de placebo. En el estudio, los pacientes debían recibir la primera dosis tras recuperación del trasplante autólogo de células madre (entre 30 y 45 días después del trasplante autólogo de células madre). Los pacientes fueron tratados con 1,8 mg/kg de ADCETRIS o con el placebo correspondiente por vía intravenosa durante 30 minutos cada 3 semanas hasta un total de 16 ciclos.
Los pacientes elegibles debían presentar al menos uno de los factores de riesgo siguientes:
• LH refractario al tratamiento de primera línea
• LH en recaída o progresión producido <12 meses después del final del tratamiento de primera
línea
• Compromiso extraganglionar en el momento previo a la recaída después del trasplante autólogo
de células madre, con inclusión de extensión extraganglionar de masas ganglionares adyacentes a órganos vitales.
Tabla 6: Resumen de las características basales de los pacientes y de la enfermedad en el ensayo de fase 3 tras trasplante autólogo de células madre.
|
Características de los pacientes |
Brentuximab vedotina N = 165 |
Placebo N = 164 |
|
Mediana de edad, años (intervalo) |
33 años (18-71) |
32 años (18-76) |
|
Sexo |
76V (46%)/89F (54%) |
97V (59%)/67F (41%) |
|
Estado funcional de ECOG | ||
|
0 |
87 (53%) |
97 (59%) |
|
1 |
77 (47%) |
67 (41%) |
|
2 |
1 (1%) |
0 |
|
Características de la enfermedad | ||
|
Mediana de número de regímenes de |
2 (2-8) |
2 (2-7) |
|
quimioterapia previa (intervalo) | ||
|
Mediana de tiempo desde diagnóstico de |
18.7 meses (6.1-204.0) |
18.8 meses (7.4-180.8) |
|
LH hasta primera dosis (intervalo) Estadío de la enfermedad al inicio del diagnóstico de HL | ||
|
Estadío I |
1 (1%) |
5 (3%) |
|
Estadío II |
73 (44%) |
61 (37%) |
|
Estadío III |
48 (29%) |
45 (27%) |
|
Estadío IV |
43 (26%) |
51 (31%) |
|
Desconocido |
0 |
2 (1%) |
|
PET-CT Estadío previo a trasplante autólogo de células madre | ||
|
FDG-POSITIVO |
64 (39%) |
51 (31%) |
|
FDG-NEGATIVO |
56 (34%) |
57 (35%) |
|
NO REALIZADO |
45 (27%) |
56 (34%) |
a
|
Compromiso extraganglionar en el momento previo a la recaída después de trasplante autólogo de células madre |
54 (33%) |
53 (32%) |
|
Síntomas Ba |
47 (28%) |
40 (24%) |
|
Mejor respuesta a tratamiento de rescate antes de trasplante autólogo de células madre b | ||
|
Respuesta completa |
61 (37%) |
62 (38%) |
|
Respuesta parcial |
57 (35%) |
56 (34%) |
|
Respuesta estable Estado LH tras el final del tratamiento estándar de quimioterapia de primera líneab |
47 (28%) |
46 (28%) |
|
Refractario |
99 (60%) |
97 (59%) |
|
Refractario <12 meses |
53 (32%) |
54 (33%) |
|
Recaída >=12 meses |
13 (8%) |
13 (8%) |
En enfermedad refractaria o hasta progresión o recaída después de tratamiento de primera línea. Factores de estratificación en asignación aleatoria.
b
Los resultados de la eficacia se muestran en la Tabla 7. La variable principal de evaluación de SLP se cumplió y mostró una diferencia en la mediana de SLP de 1,8 meses a favor del brazo de tratamiento.
Tabla 7: Resultados de la eficacia en pacientes LH con mayor riesgo de recaída o progresión después de trasplante autólogo de células madre tratados con 1,8 mg/kg de brentuximab vedotina cada 3 semanas
|
Brentuximab vedotina N = 165 |
Placebo N = 164 |
Estratificación de cociente de riesgo | |
|
Mediana por CRI | |||
|
0.57 | |||
|
Supervivencia libre de progresión3 |
42,9 meses (IC del 95% [30,4, 42.9]) |
24,1 meses (IC del 95% [11,5, -]) |
(IC del 95% [0,40, 0,81]) Test log-rank estratificado P=0,001 |
|
Mediana por investigador | |||
|
No alcanzada (IC del 95% [26,4, -]) |
15,8 meses (IC del 95% [8,5, -]) |
0,5 (IC del 95% [0,36, 0,70])b | |
|
Número de muertes (%) | |||
|
Supervivencia general |
28 (17) |
25 (15) |
1.15 (IC del 95% [0,67, 1,97] |
a' En el momento del primer análisis, la mediana de tiempo de seguimiento en ambos brazos era de
30 meses [intervalo 0 a 50].
b' No se realizó el test de log-rank estratificado de SLP por investigador.
Se realizaron análisis de SLP de subgrupo previamente especificado por mejor respuesta de paciente hasta terapia de rescate antes de trasplante autólogo de células madre, estado de LH después del tratamiento de primera línea, edad, sexo, peso basal, escala ECOG, número de tratamientos antes de trasplante autólogo con células madre, región geográfica, estado PET antes de trasplante autólogo con células madre, estado de síntoma B después del fracaso del tratamiento de primera línea y estado de enfermedad extraganglionar antes de trasplante autólogo de células madre. Los análisis mostraron una tendencia sólida en beneficio de los pacientes a los que se había administrado brentuximab vedotina en comparación con pacientes a los que se había administrado placebo, con excepción de pacientes > 65 años de edad (n=8).
No se observaron diferencias en la calidad de vida entre los brazos de tratamiento y placebo. Los análisis del uso racional de medicamentos mostraron que las hospitalizaciones y las visitas ambulatorias, así como los días laborables/otras actividades perdidos por los pacientes y cuidadores fueron menores con brentuximab vedotina en comparación con placebo en pacientes con LH con mayor riesgo de recaída.
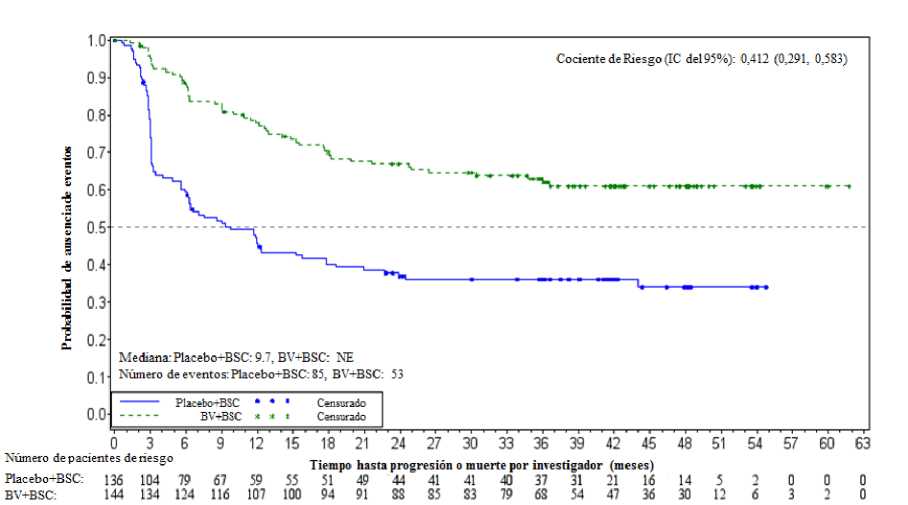
Un análisis actualizado realizado tres años después del seguimiento mostró una mejora de la SLP sostenible por CRI (HR = 0,58 [IC del 95% (0,41, 0,81)]).
Análisis _post hoc del_ factor de riesgo
Se realizaron análisis post hoc para evaluar el impacto del mayor riesgo (número de factores de riesgo) sobre el beneficio clínico (Tabla 8). Los factores de riesgo representativos para estos análisis fueron:
• LH producido <12 meses o LH refractario al tratamiento de primera línea
• Mejor respuesta RP o SD al tratamiento de rescate reciente determinada por TC y/o PET
• Enfermedad extraganglionar en el momento de la recaída previa al trasplante autólogo de
células madre
• Síntomas B en el momento de la recaída previa al trasplante autólogo con células madre
• Dos o más tratamientos de rescate previos
Los resultados de estos análisis post hoc sugieren un mayor beneficio clínico para pacientes con dos o más factores de riesgo, pero ninguna diferencia sobre ninguno de los factores de riesgo individuales. No se ha observado beneficio en términos de SLP o SG en pacientes con un factor de riesgo de recaída o progresión.
Tabla 8: Resumen de SLP por CRI y SG por número de factores de riesgo en el ensayo clínico de fase
3 de LH tras trasplante autólogo de células madre
|
Supervivencia libre de progresión por CRI | |||||||||
|
Número de factores de riesgo = 1 |
Número de factores de riesgo > 2 |
Número de factores de riesgo > 3 | |||||||
|
Brentuximab vedotina N = 21 |
Placebo N = 28 |
Brentuximab vedotina N = 144 |
Placebo N = 136 |
Brentuximab vedotina N = 82 |
Placebo N = 84 | ||||
|
Número de pacientes con progresión de la enfermedad o muerte3 (%) |
9 (43) |
7 (25) |
51 (35) |
68 (50) |
32 (39) |
49 (58) | |||
|
Cociente de riesgo estratificado |
1,65 (IC del 95% [0,60, 4,55])b |
0,49 (IC del 95% [0,34, 0,71]) |
0,43 (IC del 95% [0,27, 0,68]) | ||||||
|
Supervivencia general | |||||||||
|
Número de factores de riesgo = 1 |
Número de factores de riesgo > 2 |
Número de factores de riesgo > 3 | |||||||
|
Brentuximab vedotina N = 21 |
Placebo N = 28 |
Brentuximab vedotina N = 144 |
Placebo N = 136 |
Brentuximab vedotina N = 82 |
Placebo N = 84 | ||||
|
Número de muertes3 (%) |
5 (24) |
1 (4) |
23 (16) |
24 (18) |
15 (18) |
16 (19) | |||
|
Cociente de riesgo estratificado |
7,94 (IC del 95% [0,93, 68,06])b |
0,94 (IC del 95% [0,53, 1,67]) |
0,92 (IC del 95% [0,45, 1,88]) | ||||||
a' Muerte sin progresión previa o más de una visita de evaluación perdida b' Indica resultados del análisis no estratificado. c' Los casos son muertes debido a cualquier otra causa.
En el momento del análisis de actualización (3 años de seguimiento) de pacientes con dos o más factores de riesgo, la hazard ratio de la SLP por CRI fue de 0,49 (IC del 95% [0,34, 0,71]) y la hazard ratio de SLP por investigador fue de 0,41 (IC del 95% [0,29, 0,58]) (ver Figuras 1 y 2).
Figura 1: Estimación Kaplan-Meier de la SLP por CRI en pacientes con >2 factores de riesgo
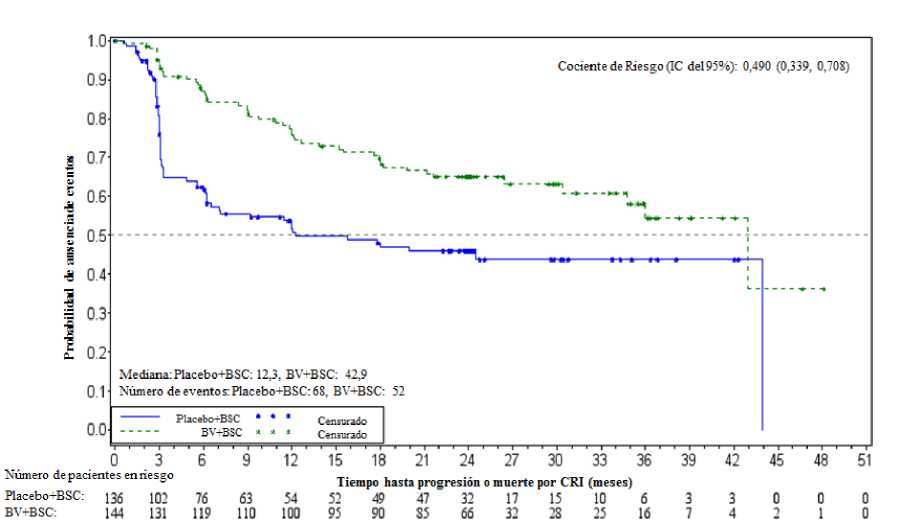
Figura 2: Estimación Kaplan-Meier de la SLP por investigador en pacientes con >2 factores de riesgo
Estudio SGN35-006 (Ensayo del retratamiento)
La eficacia del retratamiento en pacientes que con anterioridad habían respondido (RC o RP) al tratamiento con brentuximab vedotina se evaluó en un ensayo multicéntrico abierto de fase II. Se administró una dosis de inicio de 1,8 mg/kg de Adcetris a 20 pacientes con LH en recaída o refractario y una dosis de inicio de 1,2 mg/kg de Adcetris a un paciente de forma intravenosa durante 30 minutos cada 3 semanas. La mediana del número de ciclos fue de 7 (rango de 2 a 37 ciclos). De los 20 pacientes evaluables con LH, 6 pacientes (30%) lograron una RC y 6 pacientes (30%) alcanzaron una RP con el retratamiento con brentuximab vedotina, en una TRO del 60%. La mediana de duración de la respuesta fue de 9,2 y 9,4 meses en pacientes que habían alcanzado una RO (RC + RP) y RC, respectivamente.
Linfoma anaplásico de células grandes sistémico Estudio SG035-0004
Se evaluaron la eficacia y la seguridad de brentuximab vedotina como fármaco único en un estudio multicéntrico, abierto, de un solo brazo en 58 pacientes con LACGs en recaída o refractario. Véase un resumen de las características basales de los pacientes y de la enfermedad en la tabla 9 siguiente.
Tabla 9: Resumen de las características basales de los pacientes y de la enfermedad en el estudio de
fase 2 en LACGs en recaída o refractario
|
Características de los pacientes |
z II 'Jl 00 |
|
Mediana de edad, años (intervalo) |
52 años (14-76) |
|
Sexo |
33V (57%)/25M (43%) |
|
Estado funcional del ECOGa | |
|
0 |
19 (33%) |
|
1 |
38 (66%) |
|
Trasplante autólogo de células madre previo |
15 (26%) |
|
Regímenes de quimioterapia previos (intervalo) |
2 (1-6) |
|
Enfermedad que expresa CD30 confirmada |
57 (98%) |
|
histológicamente | |
|
Enfermedad negativa para cinasa de linfoma anaplásico (EN |
42 (72%) |
|
ALK) | |
|
Características de la enfermedad | |
|
Refractaria al tratamiento de primera líneab |
36 (62%) |
|
Refractaria al tratamiento más reciente |
29 (50%) |
|
Recidiva tras el tratamiento más reciente |
29 (50%) |
|
Síntomas B basales |
17 (29%) |
|
Estadio III en el diagnóstico inicial |
8 (14%) |
|
Estadio IV en el diagnóstico inicial |
21 (36%) |
a. Un paciente tenía un estado del ECOG basal de 2, lo que estaba prohibido por el protocolo, y se recoge como incumplidor de los criterios de inclusión.
b. Se define como LACGs primario refractario al que no experimenta remisión completa o progresa en los 3 meses siguientes a la finalización del tratamiento de primera línea.
La mediana del tiempo desde el diagnóstico inicial del LACGs hasta la primera administración de brentuximab vedotina fue de 16,8 meses.
Diez (10) pacientes (17%) recibieron 16 ciclos de brentuximab vedotina, y la mediana del número de ciclos recibidos fue de 7 (entre 1 y 16).
La respuesta al tratamiento con brentuximab vedotina fue valorada por un Centro de Revisión Independiente (CRI) empleando los Criterios de Respuesta Revisados para el Linfoma Maligno (Cheson, 2007). La respuesta al tratamiento se valoró mediante TC helicoidal de tórax, cuello, abdomen y pelvis, PET y datos clínicos. Se hicieron valoraciones de la respuesta en los ciclos 2, 4, 7, 10, 13 y 16 con PET en los ciclos 4 y 7.
La valoración de la TRO por el CRI fue del 86% (50 de 58 pacientes del conjunto IT). La tasa de RC fue del 59% (34 de 58 pacientes del conjunto por IT), y se logró una reducción del tumor en el 97% de los pacientes. La supervivencia global estimada a los 36 meses fue del 63% (la mediana del tiempo de observación (tiempo hasta la muerte o el último contacto) desde la primera dosis fue de 33,4 meses). Las valoraciones de los investigadores fueron en general congruentes con la revisión independiente de las exploraciones de imagen. De los pacientes tratados, 9 de los que respondieron pasaron a recibir un trasplante de células madre (TCM) alogénico, y otros 7 que respondieron recibieron TCM autólogo. Para más resultados de eficacia, véase la tabla 10.
Tabla 10: Resultados de eficacia en pacientes con LACGs en recaída o refractario tratados con
1,8 mg/kg de brentuximab vedotina cada 3 semanas
|
Mejor respuesta clínica (N = 58) |
CRI N (%) |
IC del 95% |
|
Tasa de respuesta objetiva (RC+RP) |
50 (86) |
74,6, 93,9 |
|
Remisión completa (RC) |
34 (59) |
44,9, 71,4 |
|
Remisión parcial (RP) |
16 (28) |
NP |
|
Tasa de control de la enfermedad |
52 (90) |
78,8, 96,1 |
|
(RC + RP + EE) | ||
|
Duración de la respuesta |
Mediana según el CRI |
IC del 95% |
|
Respuesta objetiva (RC + RP)a |
13,2 |
5,7, NCb |
|
Remisión completa (RC) |
No alcanzada |
13,0, NC |
|
Supervivencia global |
Mediana |
IC del 95% |
|
Mediana |
No alcanzada0 |
21,3, NC |
a' La duración de la respuesta fue de 0,1+ meses a 21,7+ meses, y la mediana de seguimiento desde la primera dosis de los pacientes que lograron respuesta objetiva (RO) según el CRI fue de 11,8 meses. b' No calculable.
c' La supervivencia global estimada a los 36 meses fue del 63% (la mediana del tiempo de observación (tiempo hasta la muerte o el último contacto) desde la primera dosis fue de 33,4 meses).
Un análisis exploratorio intrapaciente mostró que aproximadamente el 69% de los pacientes con LACG sistémico tratados con brentuximab vedotina dentro del estudio clínico SGO35- 0004, experimentaban una mejoría del efecto clínico beneficioso medido por la supervivencia libre de progresión (SLP) más prolongada en comparación con su línea de tratamiento previo más reciente.
De los 17 pacientes (29%) que tenían síntomas B basales, 14 (82%) experimentaron resolución de todos los síntomas B a una mediana de tiempo de 0,7 meses desde la instauración de brentuximab vedotina.
Estudio SGN36-006 (Estudio del retratamiento)
La eficacia del retratamiento en pacientes que con anterioridad habían respondido (RC o RP) al tratamiento con brentuximab vedotina se evaluó en un ensayo multicéntrico abierto de fase II. Se administró una dosis de inicio de 1,8 mg/kg a 7 pacientes con LACG en recaída y una dosis de inicio de 1,2 mg/kg de Adcetris a un paciente de forma intravenosa durante 30 minutos cada 3 semanas. La mediana del número de ciclos fue de 8,5 (de 2 a 30 ciclos). De los 8 pacientes con LACG sistémico,
3 se retrataron dos veces durante un total de 11 ciclos de retratamiento, El retratamiento con brentuximab vedotina resultó en 6 RC (55%) y 4 (RP) 36%), en una TRO del 91%. La mediana de duración de la respuesta fue de 8,8 y 12,3 meses en pacientes que habían logrado una RO (RC + RP) y RC, respectivamente.
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Adcetris en uno o más grupos de la población pediátrica en el tratamiento del Linfoma de Hodgkin y en el tratamiento del linfoma anaplásico de células grandes (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
Este medicamento se ha autorizado con una “aprobación condicional”.
Esta modalidad de aprobación significa que se espera obtener más información de este medicamento. La Agencia Europea de Medicamentos revisará anualmente la información nueva de este medicamento, y esta Ficha Técnica o Resumen de las Características del Producto (RCP) se actualizará cuando sea necesario.
5.2 Propiedades farmacocinéticas
La farmacocinética de brentuximab vedotina se evaluó en estudios de fase 1 y en un análisis de datos de farmacocinética poblacional de datos de 314 pacientes. En todos los ensayos clínicos, brentuximab vedotina se administró en perfusión intravenosa.
Se observaron habitualmente concentraciones máximas del ACC brentuximab vedotina al final de la perfusión o en el momento de la recogida de muestra más próxima al final de la perfusión. Se observó un descenso multiexponencial de las concentraciones séricas del ACC, con una semivida terminal aproximada de 4 a 6 días. Las exposiciones fueron aproximadamente proporcionales a la dosis. Con las dosis múltiples del régimen cada 3 semanas se observó una acumulación mínima o nula del ACC, compatible con la estimación de la semivida terminal. La Cmáx y el AUC característicos del ACC tras una sola dosis de 1,8 mg/kg en un estudio de fase 1 fueron de alrededor de 31,98 ^g/ml y 79,41 ^g/ml al día, respectivamente.
La MMAE es el principal metabolito de brentuximab vedotina. Las medianas aproximadas de la Cmáx, el AUC y el Tmáx de la MMAE tras una sola dosis de 1,8 mg/kg del ACC en el estudio de fase 1 fueron de 4,97 ng/ml, 37,03 ng/ml al día y 2,09 días, respectivamente. Las exposiciones a la MMAE disminuyeron después de dosis múltiples de brentuximab vedotina, observándose alrededor del 50 al 80% de la exposición de la primera dosis en las dosis posteriores. La MMAE se metaboliza aún más como un metabolito igualmente potente; sin embargo, su exposición es inferior en un orden de magnitud a la de la MMAE. Por consiguiente, no es probable que haya ninguna contribución sustancial a los efectos sistémicos de la MMAE.
En el primer ciclo, la mayor exposición a MMAE se asoció con un descenso absoluto de la cifra de neutrófilos.
Distribución
In vitro, la unión de la MMAE a las proteínas plasmáticas del suero humano osciló entre el 68 y el 82%. No es probable que la MMAE desplace los medicamentos que se unen intensamente a las proteínas o sea desplazada por ellos. In vitro, la MMAE fue un sustrato de la P-gp y no fue un inhibidor de la P-gp en concentraciones clínicas.
En los seres humanos, el volumen de distribución en estado de equilibrio medio del ACC fue de 6-10 l, aproximadamente. Basándose en la estimación de la FC poblacional, los volúmenes de distribución aparentes típicos (VM y VMP) de la MMAE fueron de 7,37 y 36,4 l, respectivamente.
Biotransformación
Se espera que el ACC se catabolice como proteína con reciclado o eliminación de los aminoácidos componentes.
Los datos in vivo en animales y seres humanos indican que sólo se metaboliza una pequeña fracción de la MMAE liberada de brentuximab vedotina. No se han determinado las concentraciones de metabolitos de MMAE en el plasma humano. Se ha demostrado que al menos un metabolito de la MMAE es activo in vitro.
La MMAE es un sustrato de la CYP3A4 y, posiblemente, de la CYP2D6. Datos in vitro indican que el metabolismo de la MMAE que se produce es primordialmente debido a oxidación por la CYP3A4/5. Estudios in vitro en los que se utilizaron microsomas hepáticos indican que la MMAE sólo inhibe la CYP3A4/5 en concentraciones mucho más altas que las alcanzadas durante la aplicación clínica. La MMAE no inhibe otras isoformas.
La MMAE no indujo ninguna enzima importante del CYP450 en cultivos primarios de hepatocitos humanos.
Eliminación
El ACC se elimina por catabolismo, con un aclaramiento y una semivida característicos estimados de 1,457 l/día y de 4-6 días, respectivamente.
La eliminación de la MMAE estuvo limitada por su tasa de liberación del ACC; el aclaramiento aparente típico y la semivida de la MMAE fueron de 19,99 l/día y de 3-4 días, respectivamente.
Se realizó un estudio de excreción en pacientes que recibieron una dosis de 1,8 mg/kg de brentuximab vedotina. Alrededor del 24% de la MMAE total administrada como parte del ACC durante una perfusión de brentuximab vedotina se recuperó tanto en la orina como en las heces durante un período de una semana. De la MMAE recuperada, alrededor del 72% se recuperó en las heces. En la orina se excretó una cantidad menor de la MMAE (28%).
Farmacocinética en poblaciones especiales
El análisis FC poblacional mostró que la concentración sérica de albúmina basal era una covariable importante del aclaramiento de la MMAE. El análisis indicó que el aclaramiento de la MMAE fue 2 veces menor en los pacientes con concentraciones séricas de albúmina bajas de <3,0 g/dl, que en los que tenían concentraciones séricas de albúmina dentro del intervalo normal.
Insuficiencia hepática
Un estudio evaluó la farmacocinética de brentuximab vedotina y de la MMAE después de la administración de 1,2 mg/kg de ADCETRIS en pacientes con insuficiencia hepática leve (Child-Pugh A; n=1), moderada (Child-Pugh B; n=5) y grave (Child-Pugh C; n=1). En comparación con los pacientes con función hepática normal, la exposición de la MMAE aumentó aproximadamente
2.3 veces (IC 90% 1,27 a 4,12 veces) en pacientes con insuficiencia hepática.
Insuficiencia renal
Un estudio evaluó la farmacocinética de brentuximab vedotina y de la MMAE después de la administración de 1,2 mg/kg de ADCETRIS en pacientes con insuficiencia renal leve (n=4), moderada (n=3) y grave (n=3). En comparación con los pacientes con función renal normal, la exposición de la MMAE aumentó aproximadamente 1,9 veces (IC 90% 0,85 a 4,21 veces) en los pacientes con insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min). No se observó efecto en pacientes con insuficiencia renal leve o moderada.
Pacientes de edad avanzada
En los estudios clínicos de brentuximab vedotina no se incluyó un número suficiente de pacientes de 65 o más años de edad para poder determinar si responden de forma diferente a los pacientes más jóvenes.
Población pediátrica
En los estudios clínicos de brentuximab vedotina no se incluyó un número suficiente de pacientes de menos de 18 años de edad para poder determinar si su perfil FC difiere del de los pacientes adultos.
5.3 Datos preclínicos sobre seguridad
En un estudio in vivo de micronúcleos de médula ósea de rata se ha demostrado que la MMAE tiene propiedades aneugénicas. Estos resultados fueron congruentes con el efecto farmacológico de la MMAE sobre el aparato mitótico (alteración de la red de microtúbulos) de las células.
No se han estudiado los efectos de brentuximab vedotina sobre la fertilidad masculina y femenina humana. No obstante, los resultados de estudios de toxicidad con dosis repetidas en ratas indican el potencial de brentuximab vedotina para alterar la función reproductora y la fertilidad de los machos.
La atrofia y la degeneración testiculares fUeron parcialmente reversibles tras un período sin tratamiento de 16 semanas.
Brentuximab vedotina causó mortalidad embriofetal en ratas hembra preñadas.
En estudios no clínicos se observaron depleción linfoide y reducción de peso del timo, congruentes con la alteración farmacológica de los microtúbulos causada por la MMAE procedente de brentuximab vedotina.
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Ácido cítrico monohidrato Citrato de sodio dihidrato a,a-trehalosa dihidrato Polisorbato 80
6.2 Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros excepto aquellos mencionados en la sección 6.6.
6.3 Periodo de validez
4 años.
Tras la reconstitución/dilución, desde el punto de vista microbiológico, el producto debe ser utilizado inmediatamente. Sin embargo, se ha demostrado estabilidad química y física en uso durante 24 horas a 2°C-8° C.
6.4 Precauciones especiales de conservación
Conservar en nevera (entre 2°C y 8°C).
No congelar.
Conservar el vial en el embalaje original para protegerlo de la luz.
Para las condiciones de conservación tras la reconstitución y la dilución del medicamento, ver sección 6.3.
6.5 Naturaleza y contenido del envase
Vial de vidrio tipo I con tapón de caucho butílico y precinto de aluminio/plástico con 50 mg de polvo. Envase de 1 vial.
6.6 Precauciones especiales de eliminación y otras manipulaciones
Precauciones generales
Deben considerarse los procedimientos de manipulación y eliminación adecuados de medicamentos contra el cáncer.
Debe seguirse una técnica aséptica adecuada durante toda la manipulación de este medicamento. Instrucciones para la reconstitución
Cada vial de un solo uso debe reconstituirse con 10,5 ml de agua para preparaciones inyectables hasta una concentración final de 5 mg/ml. Cada vial contiene un sobrellenado del 10%, es decir, hay 55 mg de ADCETRIS por vial y un volumen reconstituido total de 11 ml.
1. Dirija el chorro hacia la pared del vial, no directamente a la pasta o polvo.
2. Gire suavemente el vial para facilitar la disolución. NO AGITE.
3. La solución reconstituida en el vial es una solución incolora transparente o ligeramente opalescente con un pH final de 6,6.
4. La solución reconstituida debe inspeccionarse visualmente en busca de partículas extrañas y/o decoloración. Si se observan partículas o decoloración, desechar el medicamento.
Preparación de la solución para perfusión
Debe extraerse del vial o viales la cantidad apropiada de ADCETRIS reconstituido y añadirse a una bolsa de perfusión que contenga solución para preparaciones inyectables de cloruro sódico a 9 mg/ml (0,9%) a fin de lograr una concentración final de 0,4-1,2 mg/ml de ADCETRIS. El volumen de diluyente recomendado es de 150 ml. Una vez reconstituido, ADCETRIS también puede diluirse en dextrosa al 5% para preparaciones inyectables o en solución de Ringer con lactato para preparaciones inyectables.
Invierta suavemente la bolsa para mezclar la solución que contiene ADCETRIS. NO AGITE.
Cualquier resto que permanezca en el vial, después de extraer el volumen que se va a diluir, debe ser desechado de acuerdo con la normativa legal.
No añada otros medicamentos a la solución de ADCETRIS preparada para perfusión o al equipo para perfusión intravenosa. La vía de perfusión debe lavarse después de la administración con solución para preparaciones inyectables de cloruro sódico a 9 mg/ml (0,9%), dextrosa al 5% para preparaciones inyectables o solución de Ringer con lactato para preparaciones inyectables.
Tras la dilución, perfunda la solución de ADCETRIS inmediatamente a la velocidad de perfusión recomendada.
El tiempo total de conservación de la solución, desde la reconstitución a la perfusión, no debe exceder las 24 horas.
Determinación de la dosis:
Cálculo para determinar la dosis total de ADCETRIS (ml) para su posterior dilución (ver sección 4.2):
Dosis de ADCETRIS (mg/kg) x peso del paciente (kg) ^ ,.............. , .......
-:-, ,vs, F F= Dosis total de ADCETRIS (ml) para dilución ulterior
Concentración del vial reconstituido (5 mg/ml)
Nota: Si el paciente pesa más de 100 kg, la dosis debe calcularse basándose en un peso de 100 kg. La dosis máxima recomendada es de 180 mg.
Cálculo para determinar el número total de viales de ADCETRIS necesarios:
Dosis de ADCETRIS (ml) que debe administrarse Número de viales de
Volumen total por vial (10 ml/vial) ADCETRIS necesarios
Tabla 11: Ejemplos de cálculo para pacientes que reciban la dosis recomendada de 1,8 mg/kg de _ADCETRIS con pesos de entre 60 y 120 kg_
|
Peso del paciente (kg) |
Dosis total = peso del paciente multiplicado por la dosis recomendada (1,8 mg/kga) |
Volumen total a diluirb = dosis total dividida entre la concentración del vial reconstituido (5 mg/ml) |
Número de viales necesarios = volumen total a diluir dividido entre el volumen total por vial (10 ml/vial) |
|
60 kg |
108 mg |
21,6 ml |
2,16 viales |
|
80 kg |
144 mg |
28,8 ml |
2,88 viales |
|
100 kg |
180 mg |
36 ml |
3,6 viales |
|
120 kgc |
180 mgd |
36 ml |
3,6 viales |
a. Para una dosis reducida, utilice 1,2 mg/kg para el cálculo.
b. Para diluir en 150 ml de diluyente y administrar por perfusión intravenosa en 30 minutos cada
3 semanas.
c. Si el paciente pesa más de 100 kg, la dosis debe calcularse basándose en un peso de 100 kg.
d. La dosis máxima recomendada es de 180 mg.
Eliminación
ADCETRIS es para un solo uso.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
7. TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Takeda Pharma A/S Dybendal Alle 10 2630 Taastrup Dinamarca
8. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/794/001
9. FECHA DE LA PRIMERA AUTORIZACIÓN/RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 25 de octubre 2012.
Fecha de la renovación de la autorización: 10 de septiembre 2015.
10. FECHA DE LA REVISIÓN DEL TEXTO
La información detallada de este medicamento está disponible en la página de internet de la Agencia Europea del Medicamento http://www.ema.europa.eu
A. FABRICANTES DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
E. OBLIGACIÓN ESPECÍFICA DE LLEVAR A CABO MEDIDAS POSTAUTORIZACIÓN EN RELACIÓN CON UNA AUTORIZACIÓN DE COMERCIALIZACIÓN CONDICIONAL
A. FABRICANTES DEL PRINCIPIO ACTIVO BIOLÓGICO Y FABRICANTES RESPONSABLES DE LA LIBERACIÓN DE LOS LOTES
Nombre y dirección de los fabricantes del principio activo biológico.
Piramal Healthcare UK Ltd.
Earls Road, Grangemouth Stirlingshire, Scotland FK3 8XG Reino Unido
Lonza AG Lonzastrasse 3930 Visp Suiza
Nombre y dirección de los fabricantes responsables de la liberación de los lotes
Takeda Austria GmbH St. Peter-StraPe 25 A-4020 Linz Austria
Takeda Italia S.p.A.
Via Crosa, 86 28065 Cerano (NO)
Italia
El prospecto impreso del medicamento debe especificar el nombre y dirección del fabricante responsable de la liberación del lote en cuestión.
B. CONDICIONES O RESTRICCIONES DE SUMINISTRO Y USO
Medicamento sujeto a prescripción médica restringida (ver Anexo I: Ficha Técnica o Resumen de las Características del Producto, sección 4.2).
C. OTRAS CONDICIONES Y REQUISITOS DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
• Informes Periódicos de Seguridad (IPS)
Los requerimientos para la presentación de los informes periódicos de seguridad para este medicamento se establecen en la lista de fechas de referencia de la Unión (lista EURD) prevista en el artículo 107quater, apartado 7, de la Directiva 2001/83/CE y publicada en el portal web europeo sobre medicamentos.
D. CONDICIONES O RESTRICCIONES EN RELACIÓN CON LA UTILIZACIÓN SEGURA Y EFICAZ DEL MEDICAMENTO
• Plan de Gestión de Riesgos (PGR)
El TAC realizará las actividades e intervenciones de farmacovigilancia necesarias según lo acordado en la versión del PGR incluido en el Módulo 1.8.2. de la Autorización de Comercialización y en cualquier actualización del PGR que se acuerde posteriormente.
Se debe presentar un PGR actualizado:
• A petición de la Agencia Europea de Medicamentos.
• Cuandose modifique el sistema de gestión de riesgos, especialmente como resultado de nueva información disponible que pueda conllevar cambios relevantes en el perfil beneficio/riesgo, o como resultado de la consecución de un hito importante (farmacovigilancia o minimización de riesgos)
E. OBLIGACIÓN ESPECÍFICA DE LLEVAR A CABO MEDIDAS POST
AUTORIZACIÓN EN RELACIÓN CON UNA AUTORIZACIÓN DE COMERCIALIZACIÓN CONDICIONAL
Al ser esta una autorización de comercialización condicional y de según lo que establece el
Artículo 14(7) del Reglamento (CE) 726/2004, el TAC deberá llevar a cabo, dentro del plazo
establecido, las siguientes medidas:
|
Descripción |
Fecha límite |
|
Debe proporcionarse información adicional sobre el seguimiento de supervivencia global de los pacientes incluidos en el estudio SG035-004, incluyendo un subanálisis de los pacientes >100kg. Los datos deben ser presentados en el contexto de controles históricos |
Informe anual del estudio SG035-004 hasta 2016 o cuando los datos de supervivencia global sean suficientemente maduros (al menos 50% de SG observado), lo que ocurra antes |
|
Debe realizarse un estudio de seguridad post-autorización no intervencional (PASS) en la población estudiada tanto con LH como con LACGs (n=500), incluyendo un número suficiente de pacientes con LACG (a saber al menos n=50, Estudio MA25101) |
Informe final del estudio: 31/12/2018 |
|
Realizar un estudio de un solo brazo en una población de pacientes similar a la población de LACGs, investigando la tasa de respuesta, duración de la respuesta, tasa de un (segundo) trasplante autólogo de células madre y datos en subpoblaciones (incluyendo pero no necesariamente restringido al estatus ALK y edad) en base a un protocolo acordado con el CHMP (Estudio C25006) |
Informe final del estudio antes de Q1 2021 |
|
Realizar un estudio de un solo brazo estudiando la población con LH r/r no elegible para trasplante autólogo de células madre, investigando la tasa de respuesta, SLP, SG, proporción de pacientes que proceden a trasplante y seguridad (n= aprox. 60 pacientes) en base a un protocolo acordado con el CHMP |
Informe final del estudio antes de Q2 2017 |
ETIQUETADO Y PROSPECTO
A. ETIQUETADO
INFORMACIÓN QUE DEBE FIGURAR EN EL EMBALAJE EXTERIOR ESTUCHE
1. NOMBRE DEL MEDICAMENTO
ADCETRIS 50 mg polvo para concentrado para solución para perfusión brentuximab vedotina
2. PRINCIPIO(S) ACTIVO(S)
Cada vial contiene 50 mg de brentuximab vedotina
Tras la reconstitución, cada vial contiene 5 mg/ml de brentuximab vedotina
3. LISTA DE EXCIPIENTES
Excipientes: Ácido cítrico monohidrato, citrato de sodio dihidrato, a,a-trehalosa dihidrato, polisorbato 80.
Para mayor información consultar el prospecto.
4. FORMA FARMACÉUTICA Y CONTENIDO DEL ENVASE
Polvo para concentrado para solución para perfusión. 1 vial
5. FORMA Y VÍA(S) DE ADMINISTRACIÓN
Para vía intravenosa tras reconstitución y dilución Leer el prospecto antes de utilizar este medicamento.
6. ADVERTENCIA ESPECIAL DE QUE EL MEDICAMENTO DEBE MANTENERSE FUERA DE LA VISTA Y DEL ALCANCE DE LOS NIÑOS
Mantener fuera de la vista y del alcance de los niños.
7. OTRA(S) ADVERTENCIA(S) ESPECIAL(ES), SI ES NECESARIO
8. FECHA DE CADUCIDAD
CAD
9. CONDICIONES ESPECIALES DE CONSERVACIÓN
Conservar en nevera No congelar
Conservar el vial en el embalaje exterior para protegerlo de la luz.
10. PRECAUCIONES ESPECIALES DE ELIMINACIÓN DEL MEDICAMENTO NO UTILIZADO Y DE LOS MATERIALES DERIVADOS DE SU USO (CUANDO CORRESPONDA)
Para un solo uso
11. NOMBRE Y DIRECCIÓN DEL TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Takeda Pharma A/S Dybendal Alle 10 2630 Taastrup Dinamarca
12. NÚMERO(S) DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU/1/12/794/001
13. NÚMERO DE LOTE
Lote
14. CONDICIONES GENERALES DE DISPENSACIÓN
Medicamento sujeto a prescripción médica
15. INSTRUCCIONES DE USO
16. INFORMACIÓN EN BRAILLE
Se acepta la justificación para no incluir la información en Braille
INFORMACIÓN MÍNIMA QUE DEBE INCLUIRSE EN PEQUEÑOS ACONDICIONAMIENTOS PRIMARIOS
ETIQUETA DEL VIAL
1. NOMBRE DEL MEDICAMENTO Y VÍA(S) DE ADMINISTRACIÓN
ADCETRIS 50 mg, polvo para concentrado para solución para perfusión brentuximab vedotina Vía IV
2. FORMA DE ADMINISTRACIÓN
Para vía intravenosa tras reconstitución y dilución
3. FECHA DE CADUCIDAD
CAD
4. NÚMERO DE LOTE
Lote
5. CONTENIDO EN PESO, EN VOLUMEN O EN UNIDADES
50 mg
6. OTROS
B. PROSPECTO
Prospecto: información para el paciente
Adcetris 50 mg polvo para concentrado para solución para perfusión
brentuximab vedotina
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección 4 incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico.
- Si experimenta efectos adversos, consulte a su médico, incluso si se trata de efectos adversos que no aparecen en este prospecto. Ver sección 4.
Contenido del prospecto
1. Qué es Adcetris y para qué se utiliza
2. Qué necesita saber antes de empezar a usar Adcetris
3. Cómo usar Adcetris
4. Posibles efectos adversos
5. Conservación de Adcetris
6. Contenido del envase e información adicional
1. Qué es Adcetris y para qué se utiliza
Adcetris contiene el principio activo brentuximab vedotina, un medicamento contra el cáncer que está formado por un anticuerpo monoclonal unido a una sustancia destinada a destruir las células cancerosas. El anticuerpo monoclonal transporta esta sustancia a las células cancerosas. Un anticuerpo monoclonal es una proteína que reconoce determinadas células cancerosas.
Adcetris se utiliza para tratar el linfoma de Hodgkin clásico que:
- ha reaparecido o no ha respondido tras una perfusión en su cuerpo de sus propias células madre (trasplante autólogo de células madre), o
- ha reaparecido o nunca ha respondido a al menos dos tratamientos previos, y cuando no puede recibir ninguna otra combinación de tratamientos para el cáncer o no puede someterse a trasplante autólogo de células madre.
El linfoma de Hodgkin clásico expresa sobre la superficie de las células proteínas específicas diferentes de las del linfoma de Hodgkin no clásico.
Adcetris también se utiliza para reducir la probabilidad de reaparición de linfoma de Hodgkin clásico tras un trasplante autólogo de células madre en pacientes con determinados factores de riesgo.
Adcetris se utiliza para tratar el linfoma anaplásico de células grandes sistémico presente en sus ganglios linfáticos y/o en otras partes de su cuerpo que:
- no ha respondido a otros tipos de tratamientos contra el cáncer o
- ha reaparecido tras un tratamiento previo contra el cáncer.
Tanto el linfoma de Hodgkin como el linfoma anaplásico de células grandes sistémico son tipos de cáncer de los glóbulos blancos.
2. Qué necesita saber antes de empezar a usar Adcetris
NO use Adcetris:
- si es alérgico a brentuximab vedotina o a cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- si recibe actualmente bleomicina, un fármaco contra el cáncer.
Advertencias y precauciones
Cuando reciba este medicamento por primera vez y durante el curso del tratamiento, informe a su
médico si:
- sufre confusión, dificultad para pensar, pérdida de memoria, visión borrosa o pérdida de visión, pérdida de fuerza, disminución del control o la sensación en un brazo o pierna, cambio del modo de andar o pérdida de equilibrio, ya que pueden ser síntomas de una dolencia cerebral grave y potencialmente mortal denominada leucoencefalopatía multifocal progresiva (LMP). Si sufre estos síntomas antes del tratamiento con este medicamento, informe inmediatamente a su médico de cualquier cambio de tales síntomas. También debe informar a su pareja o cuidadores sobre su tratamiento, ya que pueden observar síntomas de los que usted no sea consciente.
- sufre dolor de estómago fuerte y persistente, con o sin náuseas y vómitos, ya que pueden ser síntomas de una enfermedad grave y mortal conocida como pancreatitis (inflamación del páncreas).
- le aparece o siente por primera vez dificultad para respirar o tos, o si éstas empeoran, ya que podrían ser síntomas de una complicación pulmonar grave y potencialmente mortal (toxicidad pulmonar).
- está tomando o ha tomado previamente medicamentos que puedan afectar a su sistema inmunológico, como quimioterapia o fármacos inmunodepresores.
- sufre o cree que sufre una infección. Algunas infecciones pueden ser graves y deberse a virus, bacterias u otras causas que pueden ser peligrosas para la vida.
- nota un sonido silbante durante la respiración (sibilancia) o dificultad para respirar, habones, picores o hinchazón (signos de una reacción a la perfusión). Para información más detallada, véase “Reacciones a la perfusión” en la sección 4.
- tiene problemas por un cambio de sensibilidad de la piel, sobre todo en las manos o los pies, como entumecimiento, hormigueo, sensación de quemazón, dolor, molestias o debilidad (neuropatía).
- sufre dolores de cabeza, se siente cansado, tiene mareo, está pálido (anemia) o sufre sangrado o moretones inusuales bajo la piel, pérdida de sangre más prolongada de lo habitual después de haberle extraído sangre o sangrado en las encías (trombocitopenia).
- sufre escalofríos o tiritona, o sensación de calor; debe tomarse la temperatura, ya que puede tener fiebre. La fiebre con un recuento bajo de glóbulos blancos puede ser un signo de infección grave.
- sufre mareo, descenso del volumen de orina, confusión, vómitos, náuseas, hinchazón, falta de aire u otras alteraciones del ritmo cardíaco (ésta puede ser una complicación potencialmente peligrosa para la vida denominada síndrome de lisis tumoral).
- sufre síntomas de tipo gripal seguidos de una erupción roja o purpúrea dolorosa que se extiende y forma ampollas, llegando a un amplio desprendimiento de la piel que puede resultar potencialmente mortal (puede ser una reacción grave de la piel denominada síndrome de Stevens-Johnson o necrólisis epidérmica tóxica).
- sufre dolor estomacal de nueva aparición o un empeoramiento de ese cuadro, náuseas, vómitos, estreñimiento, ya que pueden ser síntomas de complicación intestinal o estomacal grave y potencialmente mortal (complicaciones gastrointestinales).
- presenta resultados anormales en un análisis hepático, ya que puede estar relacionado con un daño grave y potencialmente mortal en el hígado (hepatotoxicidad). La hepatopatía, así como otras afecciones, que puedan haber estado presentes antes de iniciar el tratamiento con Adcetris y algunos medicamentos que esté tomando actualmente podrían aumentar el riesgo de lesión hepática.
- se siente cansado, orina con frecuencia, nota aumento de la sed y del apetito con pérdida de peso no deseada o está irritable (hiperglucemia).
- tiene problemas de riñón o de hígado.
Su médico le hará análisis de sangre periódicos para asegurarse de que recibe este medicamento con seguridad.
Uso de Adcetris con otros medicamentos
Informe a su médico si está tomando otros medicamentos, si ha tomado alguno recientemente o si empieza a tomar medicamentos nuevos. Esto incluye las plantas medicinales y otros medicamentos que pueda obtener sin receta.
Embarazo, lactancia y fertilidad
Usted y su pareja deben utilizar dos métodos anticonceptivos eficaces durante su tratamiento con este medicamento. Las mujeres deben seguir usando métodos anticonceptivos durante 6 meses después de la última dosis de Adcetris.
No debe utilizar este medicamento si está embarazada, a menos que usted y su médico decidan que el beneficio para usted supera al riesgo potencial para el feto.
Es importante que informe a su médico antes del tratamiento y durante el tratamiento si está embarazada, cree que podría estar embarazada o tiene intención de quedarse embarazada.
Si está dando el pecho a su hijo, debe hablar con su médico sobre si debe recibir este medicamento.
Se aconseja a los hombres tratados con este medicamento que pidan que se congelen y conserven muestras de su semen antes del tratamiento. Se aconseja a los hombres que no engendren un hijo durante el tratamiento con este medicamento ni en los 6 meses siguientes a la última dosis del medicamento.
Conducción y uso de máquinas
El tratamiento puede influir en su capacidad para conducir o utilizar máquinas. Si se siente mal durante el tratamiento, no conduzca ni utilice máquinas.
Adcetris contiene sodio
Este medicamento contiene un máximo de 2,1 mmol (o 47 mg) de sodio por dosis, algo que deben tener en cuenta en los pacientes que sigan una dieta con control de sodio.
3. Cómo usar Adcetris
En caso de duda sobre el uso de este medicamento, pregunte al médico o a la enfermera que le administre la perfusión.
Dosis y frecuencia
La dosis de este medicamento depende de su peso corporal. La dosis habitual de inicio de tratamiento con Adcetris es de 1,8 mg/kg, administrados una vez cada 3 semanas durante no más de un año. Su médico puede disminuir su dosis de inicio de tratamiento hasta 1,2 mg/kg si usted padece problemas hepáticos o renales.
Adcetris sólo debe administrarse a adultos. No está previsto su uso en niños.
Cómo se administra Adcetris
Este medicamento se le administrará en vena (por vía intravenosa) mediante perfusión. El médico o la enfermera lo administrarán en 30 minutos. El médico o la enfermera le vigilarán además durante y después de la perfusión.
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.
4. Posibles efectos adversos
Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Reacciones a la perfusión
Los medicamentos de este tipo (anticuerpos monoclonales) pueden causar reacciones a la perfusión como:
- una erupción
- falta de aire
- dificultad para respirar
- tos
- opresión en el pecho
- fiebre
- dolor de espalda
- escalofríos
- dolor de cabezasensación de malestar (náusea) o de estar enfermo (vómitos)
Las reacciones a la perfusión de este medicamento afectan a más de 1 de cada 10 personas.
En general, estas reacciones se producen entre unos minutos y varias horas después de completarse la perfusión. No obstante, en casos poco frecuentes pueden aparecer más de varias horas después de completada la perfusión. Estas reacciones a la perfusión pueden ser graves o incluso mortales (conocidas como reacciones anafilácticas). Se desconoce con qué frecuencia las reacciones a la perfusión de este medicamento son graves o mortales.
Pueden administrársele otros medicamentos como
- antihistamínicos, corticosteroides o paracetamol
para ayudar a reducir cualquiera de las reacciones anteriores si ya las ha sufrido al recibir este tipo de medicamento.
Si cree que ha sufrido previamente una reacción parecida, informe al médico ANTES de que le administren este medicamento.
Si desarrolla reacciones a la perfusión (como las antes indicadas), el médico puede dejar de administrar este medicamento e iniciar un tratamiento de apoyo.
Si se le reanuda la perfusión, el médico puede alargar el tiempo durante el que se administre para que pueda tolerarla mejor.
Informe inmediatamente al médico si observa cualquiera de los síntomas siguientes, ya que algunos de ellos pueden ser indicativos de un proceso grave o posiblemente mortal:
- síntomas de leucoencefalopatía multifocal progresiva (LMP) como confusión, dificultad para pensar, pérdida de memoria, visión borrosa o pérdida de visión, pérdida de fuerza, disminución del control o la sensación en un brazo o pierna, cambio del modo de andar o pérdida de equilibrio (para información más detallada, véase la sección 2). La frecuencia de este proceso no puede estimarse a partir de los datos disponibles.
- síntomas de inflamación del páncreas (pancreatitis) tales como dolor de estómago fuerte y persistente, con o sin náuseas y vómitos (afecta a menos de 1 de cada 100 personas).
- disnea o tos (afecta a más de 1 de cada 10 personas)
- síntomas de tipo gripal seguidos de una erupción roja o purpúrea dolorosa que se extiende y forma ampollas, llegando a un amplio desprendimiento de la piel (afecta a menos de 1 de cada 1000 personas).
- un cambio de la sensación o la sensibilidad, sobre todo en la piel, entumecimiento, hormigueo, molestias, sensación de quemazón, debilidad o dolor en manos o pies (neuropatía; afecta a más de 1 de cada 10 personas).
- sensación de debilidad (afecta a más de 1 de cada 10 personas).
- estreñimiento (afecta a más de 1 de cada 10 personas).
- diarrea, vómitos (afectan a más de 1 de cada 10 personas).
- escalofríos o tiritona (afectan a más de 1 de cada 10 personas).
- sensación de cansancio, micción frecuente, aumento de la sed y del apetito con pérdida de peso no deseada e irritabilidad (pueden ser signos de hiperglucemia, que afecta a menos de 1 de cada 10 personas).
- sangrado o moretones inusuales bajo la piel, sangrado más prolongado de lo habitual tras la extracción de sangre o sangrado de las encías (pueden ser signos de trombocitopenia, que afecta a menos de 1 de cada 10 personas).
- dolores de cabeza, mareo, palidez (pueden ser signos de anemia, que afecta a menos de 1 de cada 10 personas).
Puede sufrir los efectos adversos siguientes:
Efectos adversos muy frecuentes (afectan a más de 1 de cada 10 personas)
- reducción del número de glóbulos blancos
- infección del tracto respiratorio superior
- pérdida de peso
- infección
- náuseas
- dolor abdominal
- picor
- caída o debilitamiento inusual del pelo
- dolor muscular
- dolor articular o articulaciones hinchadas y dolorosas
Efectos adversos frecuentes (afectan hasta 1 de cada 10 personas)
- infección en la sangre (sepsis) y/o shock séptico (una forma de sepsis potencialmente mortal); neumonía
- disminución del número de plaquetas en la sangre
- mareo
- ampollas que pueden secarse o formar costras
- aumento del azúcar en sangre
- aumento de los niveles de enzimas hepáticas
Efectos adversos poco frecuentes (afectan hasta 1 de cada 100 personas)
- Síndrome de lisis tumoral - un proceso que puede poner en peligro la vida en el que puede sufrir mareo, reducción de la micción, confusión, vómitos, náuseas, hinchazón, falta de aire u otras alteraciones del ritmo cardíaco.
- formación en la boca de placas elevadas dolorosas de color amarillento (aftas).
Efectos adversos raros (afectan a menos de 1 de cada 1000 personas)
- Síndrome de Stevens-Johnson y necrólisis epidérmica tóxica - un trastorno grave raro en el que puede tener síntomas de tipo gripal seguidos de una erupción roja o purpúrea dolorosa que se extiende y forma ampollas, llegando a un amplio desprendimiento de la piel.
Frecuencia no conocida (no puede estimarse a partir de los datos disponibles)
- disminución del número de leucocitos, con fiebre.
Comunicación de efectos adversos
Si experimenta cualquier tipo de efecto adverso, consulte a su médico, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente a través del sistema nacional de notificación incluido en el Apéndice V. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
5. Conservación de Adcetris
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta del vial y en la caja después de CAD. La fecha de caducidad es el último día del mes que se indica.
Vial no abierto: Conservar en nevera (entre 2 °C y 8°C). No congelar.
Conservar el vial en el embalaje original para protegerlo de la luz.
Solución reconstituida/diluida: Usarla inmediatamente o conservarla en nevera (entre 2 °C y 8°C) y usarla en un plazo de 24 horas.
No utilice este medicamento si observa partículas o decoloración antes de la administración.
Los medicamentos no se deben tirar por los desagües ni a la basura. El médico o la enfermera desecharán este medicamento. De esta forma, ayudará a proteger el medio ambiente.
6. Contenido del envase e información adicional Composición de Adcetris
- El principio activo es brentuximab vedotina. Cada vial contiene 50 mg de brentuximab vedotina. Tras la reconstitución, cada ml de solución contiene 5 mg de Adcetris.
- Los demás componentes son ácido cítrico monohidrato, citrato de sodio dihidrato, a,a-trehalosa dihidrato y polisorbato 80. Véase más información sobre el sodio en la sección 2.
Aspecto del producto y contenido del envase
Adcetris es una pasta o polvo blanco o blanquecino para concentrado para solución para perfusión suministrado en un vial de vidrio.
Cada envase de Adcetris consta de un vial.
Titular de la autorización de comercialización:
Takeda Pharma A/S Dybendal Alle 10 2630 Taastrup Dinamarca
Responsable de la fabricación
Takeda Austria GmbH St. Peter-StraPe 25 A-4020 Linz Austria
Takeda Italia S.p.A.
Vía Crosa, 86 28065 Cerano (NO)
Italia
Pueden solicitar más información respecto a este medicamento dirigiéndose al representante local del titular de la autorización de comercialización:
|
Belgie/Belgique/Belgien Takeda Belgium Tel/Tél: +32 2 464 06 11 takeda-belgium@takeda.com |
Lietuva Takeda, UAB Tel: +370 521 09 070 |
|
Etarapna TaKega Etarapna Tea.: + 359 2 958 27 36; + 359 2 958 15 29 |
Luxembourg/Luxemburg Takeda Belgium Tel/Tél: +32 2 464 06 11 takeda-belgium@takeda.com |
|
Ceská republika Takeda Pharmaceuticals Czech Republic s.r.o. Tel: + 420 234 722 722 |
Magyarország Takeda Pharma Kft. Tel: +361 2707030 |
|
Danmark Takeda Pharma A/S Tlf: +45 46 77 11 11 |
Malta Takeda Italia S.p.A. Tel: +39 06 502601 |
|
Deutschland Takeda GmbH Tel: 0800 825 3325 medinfo@takeda.de |
Nederland Takeda Nederland bv Tel: +31 23 56 68 777 nl.medical.info@takeda.com |
|
Eesti Takeda Pharma AS Tel: +372 6177 669 |
Norge Takeda Nycomed AS Tlf: +47 6676 3030 infonorge@takeda.com |
|
EXláSa TAKEDA EAAAI A.E Tel: +30 210 6729570 gr.info@takeda.com |
Osterreich Takeda Pharma Ges.m.b.H. Tel: +43 (0) 800-20 80 50 |
|
España Takeda Farmacéutica España S.A Tel: +34 917 14 99 00 spain@takeda.com |
Polska Takeda Polska Sp. z o.o tel. + 48 22 608 13 00 |
|
France Takeda France Tel. +33 1 46 25 16 16 |
Portugal Takeda Farmacéuticos Portugal, Lda. Tel: + 351 21 120 1457 |
|
Hrvatska Takeda Pharmaceuticals Croatia d.o.o. Tel: +385 1 377 88 96 |
Romania Takeda Pharmaceuticals SRL Tel: +40 21 335 03 91 |
|
Ireland Takeda Products Ireland Limited Tel: +44 (0)1628 537 900 |
Slovenija Takeda GmbH, Podruznica Slovenija Tel: + 386 (0) 59 082 480 |
|
Ísland Vistor hf. tel: +354 535 7000 vistor@vistor.is |
Slovenská republika Takeda Pharmaceuticals Slovakia s.r.o Tel: +421 (2) 20 602 600 |
|
Italia Takeda Italia S.p.A. Tel: +39 06 502601 |
Suomi/Finland Takeda Oy Tel. +358 20 746 5000 infoposti@takeda.com |
|
Kúnpoq Takeda Pharma A/S T^: +45 46 77 11 11 |
Sverige Takeda Pharma AB Tel: +46 8 731 28 00 infosweden@takeda.com |
|
Latvija Takeda Latvia SIA Tel: +371 67840082 |
United Kingdom Takeda UK Ltd Tel: +44 (0)1628 537 900 |
Fecha de la última revisión de este prospecto:
Este medicamento se ha autorizado con una “aprobación condicional”. Esta modalidad de aprobación significa que se espera obtener más información de este medicamento.
La Agencia Europea de Medicamentos revisará anualmente la información nueva de este medicamento y este prospecto se actualizará cuando sea necesario.
La información detallada de este medicamento está disponible en la página web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu .
Esta información está destinada únicamente a profesionales del sector sanitario:
Instrucciones para la reconstitución
Cada vial de un solo uso debe reconstituirse con 10,5 ml de agua para preparaciones inyectables para alcanzar una concentración final de 5 mg/ml. Cada vial contiene un sobrellenado del 10%, es decir, hay 55 mg de Adcetris por vial y un volumen reconstituido total de 11 ml.
1. Dirija el chorro hacia la pared del vial, no directamente a la pasta o polvo.
2. Gire suavemente el vial para facilitar la disolución. NO AGITE.
3. La solución reconstituida en el vial es una solución incolora transparente o ligeramente opalescente con un pH final de 6,6.
4. La solución reconstituida debe inspeccionarse visualmente en busca de partículas extrañas y/o cambios de color. En el caso de que se observe partículas extrañas y/o cambios de color, debe desecharse el medicamento.
Preparación de la solución para perfusión
Debe extraerse del vial o viales la cantidad apropiada de Adcetris reconstituido y añadirse a una bolsa de perfusión que contenga solución para preparaciones inyectables de cloruro sódico a 9 mg/ml (0,9%) a fin de lograr una concentración final de 0,4-1,2 mg/ml de Adcetris.
El volumen de diluyente recomendado es de 150 ml. Una vez reconstituido, Adcetris también puede diluirse en solución para preparaciones inyectables de dextrosa al 5% o en solución para preparaciones inyectables de Ringer con lactato.
Invierta suavemente la bolsa para mezclar la solución que contiene Adcetris. NO AGITE.
Cualquier resto de medicamento en el vial, después de extraer el volumen a diluir, debe ser desechado de acuerdo con la normativa local.
No añada otros medicamentos a la solución para perfusión de Adcetris preparada o al equipo para perfusión intravenosa. La vía de perfusión debe lavarse después de la administración con solución para preparaciones inyectables de cloruro sódico a 9 mg/ml (0,9%), dextrosa al 5% para preparaciones inyectables o solución de Ringer con lactato para preparaciones inyectables.
Tras la dilución, perfunda la solución de Adcetris inmediatamente a la velocidad de perfusión recomendada.
El tiempo total de conservación de la solución, desde la reconstitución a la perfusión, no debe exceder de 24 horas.
Eliminación
Adcetris es para un solo uso.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
46